
Submitted:
03 September 2024
Posted:
03 September 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The COVID-19 pandemic caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus has exposed the vulnerabilities and unpreparedness of the global healthcare system in dealing with emerging zoonoses. In the past two decades, coronaviruses (CoV) have been responsible for three major viral outbreaks, and the likelihood of future outbreaks caused by these viruses is high and nearly inevitable. Therefore, effective prophylactic universal vaccines targeting multiple circulating and emerging coronavirus strains are warranted. This study utilized an immunoinformatic approach to identify evolutionarily conserved CD4+ (HTL) and CD8+ (CTL) T cells, and B-cell epitopes in the coronaviral spike (S) glycoprotein. A total of 132 epitopes were identified, with the majority of them found conserved in the bat CoV, pangolins CoV, endemic coronaviruses, SARS-CoV-2, and Middle East respiratory syndrome coronavirus (MERS-CoV). Their peptide sequences were then aligned and assembled to identify the overlapping regions. Eventually, two major peptide assemblies were derived based on their promising immune-stimulating properties. In this light, they can serve as lead candidates for universal coronavirus vaccine development, particularly in the search for pan-coronavirus multi-epitope universal vaccines that can confer protection against current and novel coronaviruses.
Keywords:
SARS-CoV-2
; coronavirus
; universal vaccine
; peptide
; cell-mediated immunity
1. Introduction
Coronaviruses (CoVs) were thought to only cause common cold in humans until 2002-2003 when severe acute respiratory syndrome-coronavirus (SARS-CoV) struck the world health system. The SARS-CoV outbreak caused 8,098 infections and 774 deaths globally (~10% mortality) [1]. Nearly 10 years after the SARS outbreak, another coronavirus outbreak took place in the Middle East and the etiological agent was identified as Middle East respiratory syndrome coronavirus (MERS-CoV). The outbreak then spread to South Korea with the first case reported in 2015 involving a 65-year-old man who had recently travelled to the Middle East [2]. Compared to SARS-CoV, MERS-CoV caused the highest mortality rate of ~35% among the three outbreak strains with a total of 2,458 infections and 848 reported deaths. In December 2019, a novel coronavirus strain namely SARS-CoV-2 caused a large-scale viral outbreak in WuHan, China. Since then, it has spread rampantly throughout the world and caused respiratory distress in humans. The disease associated with SARS-CoV-2 was coined COVID-19 and resulted in the breakdown of the healthcare system in many countries. The outbreak was eventually announced as a global pandemic by WHO on 11 March 2020 [3]. As of July 2024, COVID-19 has affected more than 775 million people and caused more than 7 million deaths worldwide [4].
As the coronaviral spike (S) glycoprotein is located outside the viral particle and mediates the viral entry into the host epithelial cells, it is undoubtedly the main target of neutralizing antibodies (NAbs) upon infection, therefore, making it the most important therapeutic target and in vaccine design. However, the emergence of new SARS-CoV-2 Omicron variants has rendered the vaccines ineffective, with ChAdOx1 nCoV-19 (Vaxzevria, AstraZeneca) conferring almost no protection from 20-24 weeks after the second dose of vaccination [5]. It is also notable that the emergence of new variants of concern (VOC), such as Omicron has raised attention globally as the new variants can escape the neutralizing antibodies and have increased transmissibility due to the presence of more than 30 mutations as compared to the parental strain, SARS-CoV-2-Wuhan-Hu-1 [6,7,8,9]. In view of the rising concerns regarding the increased infectivity of the new variants and controversies about the effectiveness of the vaccines, there is an urgent need for a pan-coronavirus vaccine that can induce the synthesis of neutralizing antibodies and is more comprehensive in conferring protection against the newly emerging variants as well as future coronavirus outbreaks While many groups predicted and identified evolutionarily conserved epitopes in-silico [10,11,12,13,14], and some of them were validated in-vitro and in-vivo [15,16,17,18,19,20,21], this study scrutinized the conserved epitopes further. Many predicted CTL, HTL, and LBL epitopes were found in the close vicinity in the S glycoprotein. Instead of studying them individually, they were aligned into single and relatively long peptide sequences. This novel strategy of having multi-epitopes is expected to stimulate a stronger and multi-faceted immune response against coronaviruses, addressing the limitations of the current vaccines against the emerging variants.
In this study, the evolutionarily conserved epitopes in both human and animal coronaviruses were identified using unique immunoinformatics approaches. After a stringent scrutiny and selection, 52 CTL epitopes, 11 HTL epitopes, and 68 linear B-lymphocyte (LBL) epitopes were identified from 30 coronavirus sequences derived from human CoVs (hCoVs) responsible for the common cold, SARS-CoV, MERS-CoV and SARS-CoV-2. Subsequently, the predicted epitopes were aligned and assembled into 2 final composite peptide sequences that were found to be evolutionarily conserved in SARS-CoVs, bats, and pangolin coronaviruses. These 2 assembled epitopes are not only found to be conserved in many of the coronavirus strains, but they also possess HTL, CTL, and B-cell antigen binding sites, and they match a diverse array of HLA class-I and -II supertypes prevalent in the human population, indicating their potential to activate both T and B cells effectively. Although the epitopes were only found to be mostly conserved in SARS-CoVs, bats, and pangolin coronaviruses, their distinctive compatibility with human HTL and CTL, and B cells renders them high potential in vaccine development. Altogether, the discoveries not only pave the way for the development of a pan-coronavirus multiepitope vaccine to combat existing and novel coronavirus strains but immunoinformatics are highly applicable in universal vaccine development, especially in identifying immunogenic conserved epitopes in target antigens.
2. Materials and Methods
2.1. Coronaviral S Gene Sequence Retrieval and Sequence Conservation Analysis
Forty-two coronaviral peptide sequences of the S gene were retrieved from the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) as listed in Table 1, Table 2 and Table 3. A total of 24 sequences encompassing SARS-CoV-2 and its variants were retrieved from the Genbank (Table 1.) with the latest variant being XBB.1.5. Six sequences of the other coronaviruses causing SARS, MERS and common flu in humans are listed in Table 2. Table 3., on the other hand, tabulates twelve sequences of coronaviruses isolated in bats, pangolins and birds.
In order to identify the conserved regions in the coronaviral S glycoproteins, the amino acid sequence of the S glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Table 1.) was used as a reference sequence to perform Clustal Omega multiple sequence alignments in the EMBL-EBI (https://www.ebi.ac.uk/). The alignment was based on the Percentage Identity Threshold of 80% in the amino acid sequences using Jalview 2.11.2.6 (https://www.jalview.org/). The evolutionarily conserved regions of the S glycoproteins were identified and subjected to antigenicity screening, selection, and assembly.
2.2. The flow of Prediction of Conserved HTL, CTL and Linear B-lymphocyte (LBL) Epitopes of Coronaviral S glycoproteins
The prediction was performed separately for (i) CTL, (ii) HTL, and (iii) LBL epitopes by referring to their respective databases. The flow of prediction of conversed epitopes is depicted in Figure 1. The conserved epitopes were individually screened and identified, and their antigenicity and toxigenicity were predicted using VaxiJen 2.0 and ToxinPred, respectively.
2.2.1. Prediction of Conserved CTL Epitopes
The conserved CTL epitopes were identified using NetCTL-1.2 (https://services.healthtech.dtu.dk/services/NetCTL-1.2/). A total of 30 amino acid sequences of human coronaviral S glycoproteins were uploaded to NetCTL-1.2 by following the default criteria, which entailed 9 amino acids in length with a minimum threshold of 0.75. The available HLA class-I supertypes provided by NetCTL-1.2 included A1, A2, A24, A26, B7, B8, B27, B39, B44, B58, and B62. The redundant epitope sequences were filtered and subjected to the subsequent screening. The selected epitopes were then subjected to in-silico antigenicity screening using VaxiJen 2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). The screening was performed using the default settings and “Virus” was selected as the target organism as part of the prediction criteria. Antigens labelled as “Probable Antigen” were sorted and selected. Subsequently, the selected antigens were screened for their immunogenicity using the IEDB Class I immunogenicity web-based prediction tool (http://tools.iedb.org/immunogenicity/). The non-immunogenic epitopes were indicated with negative scores and removed, the remaining epitopes were later examined using ToxinPred (https://webs.iiitd.edu.in/raghava/toxinpred/protein.php) to eliminate the probable toxic CTL epitopes. Following that, the epitopes were selected based on their high frequencies across different strains and supertypes. Table 4. summarizes the prediction of conserved CTL epitopes.
2.2.2. Prediction of Conserved HTL Epitopes
In the conserved HTL epitope prediction, the amino acid sequences of the coronaviruses were screened using IEDB MHC-II (http://tools.iedb.org/mhcii/) with the following prediction conditions: (i) Percentile rank: 25%, 15-mers amino acid; (ii) prediction method: Consensus 2.22; (iii) HLA supertypes: HLA-DR, HLA-DQ, and HLA-DP. Table 5. summarizes the prediction conditions for the conserved HTL epitopes. Epitopes with a percentile rank lower than 20.0 were eliminated as it indicates that those epitopes capture less than 50% immune response [22].
Similar to the conserved CTL epitope prediction, the antigenicity and toxigenicity of the epitopes were analysed using the same methods as shown in Table 4. In addition, the HTL epitopes were screened using IFNepitope (https://webs.iiitd.edu.in/raghava/ifnepitope/predict.php) for their abilities to induce interferon synthesis. The prediction criteria included “Motif and SVM hybrid” as the prediction approach and “IFN-gamma versus Non IFN-gamma” as the prediction model. The “NEGATIVE” HTL epitopes were removed.
2.2.3. Prediction of Conserved LBL Epitopes
ABCPred (https://webs.iiitd.edu.in/raghava/abcpred/ABC_submission.html) and SVMTriP (http://sysbio.unl.edu/SVMTriP/prediction.php) were used in predicting the conserved LBL epitopes with criteria such as 16-mers amino acids in length for both tools and the threshold of 0.51 and 0.50, respectively as shown in Table 6. All the predicted epitopes were selected for further antigenicity and toxigenicity prediction. The antigenicity and toxigenicity of the epitopes were screened as described in Table 4.
2.3. Alignment of the Predicted Conserved CTL, HTL and LBL Epitopes and Allergenicity Prediction
To identify the locations of the epitopes identified in Section 2.2, SARS-CoV-2-Wuhan-Hu-1 S glycoprotein was used as a reference sequence for multiple sequence alignment. The overlapped regions of the epitopes were aligned, they were then assembled into long amino acid sequences containing the conserved CTL, HTL, and LBL epitopes. Subsequently, the assembled peptide sequences were screened for allergenicity using AllergenFP v1.0 (https://www.ddg-pharmfac.net/AllergenFP/) to identify the probable allergens in the assembled sequences. The probable allergenic sequences, if any, were eliminated.
2.4. Structural Visualisation of Assembled Epitopes
The structural information of the close (PDB: 6VXX) and open states (PBD: 6VYB) of the S glycoprotein were retrieved from the RCSB Protein Data Bank (https://www.rcsb.org/). By using the retrieved information, the structures of the assembled conserved epitopes were then visualized using the UCSF ChimeraX (https://www.cgl.ucsf.edu/chimerax/).
3. Results
3.1. Conserved Regions in the S glycoproteins of Bat and Pangolin CoV, hCoVs, SARS-CoV-2, SARS-CoV, and MERS-CoV
The S glycoproteins of coronaviruses consist of a signal peptide, a receptor binding subunit (S1) and a fusion subunit (S2) with a length ranging from 1105-1351 amino acids [23,24,25,26]. With reference to the SARS-CoV-2-Wuhan-Hu-1, in the S1 subunit, there is an N-terminal domain (NTD, 14–305 residues) and receptor-binding domain (RBD, 319–541 residues), whereas, in the S2 subunit, there is a fusion peptide (FP, 788–806 residues), heptapeptide repeat sequence 1 (HR1) (912–984 residues), HR2 (1163–1213 residues), transmembrane domain (TM, 1213–1237 residues), and cytoplasmic domain (1237–1273 residues) [27]. As the coronavirus S glycoprotein is located outside of the viral particle and mediates the viral entry into the host epithelial cells, it is undoubtedly the main target of neutralizing antibodies (NAbs) upon infection, therefore, making it the most important therapeutic target and essential in vaccine design.
Given the importance of coronaviral S glycoprotein in vaccine development, this study aimed to screen and identify the evolutionarily conserved sequences in animal and human coronaviral S glycoproteins. The amino acid sequence of SARS-CoV-2-Wuhan-Hu-1 S glycoprotein served as the reference sequence to all retrieved coronavirus sequences. Upon screening and alignment, the results revealed that bat coronavirus strains such as Bat CoV RATG13, Bat CoV ZXC21, and Bat CoV YN02, and pangolin coronavirus strains such as Pangolin CoV GX-P2V, Pangolin CoV GX-P5E, Pangolin CoV GX-P5L, Pangolin CoV GX-P1E, Pangolin CoV GX-P4L, and Pangolin CoV MP789 showed some level of evolutionary divergence compared to that of SARS-CoV-2-Wuhan-Hu-1 with an identity threshold above 80% (Supplementary Figure S1). The results also explain why bats or pangolins are deduced as the most likely reservoirs of SARS-CoV-2. In contrast, three selected avian coronaviral S glycoproteins were distantly related to the reference sequence due to their relatively high evolutionary divergence.
The amino acid sequences of SARS-CoV-2 and hCoVs causing the common cold were also aligned with the reference sequence (Supplementary Figure S2). The S1 regions of hCoVs, i.e., H-CoV-HKU1–genotype B, CoV-OC43, CoV-NL63, and CoV-229E showed insignificant similarities to the reference sequence. Interestingly, their S2 regions were relatively conserved particularly at residues S815 - S874, S897 - S934, S944 - S1069, and S1207 - S1218. The residues S897 - S1069 corresponded to the HR1 and HR2 regions of the S2 subunit, whereas the S1207 - S1218 region was part of the HR2 and TM domain. The conservation of the HR1 and HR2 regions was documented previously and suggested as the targets for the development of fusion inhibitor agents [28,29]. Furthermore, the alignment of S glycoprotein sequences of SARS-CoV-2 and its variants, MERS-CoV, and SARS-CoV revealed that SARS-CoV, SARS-CoV-2 and its variants were highly similar to each other (Supplementary Figure S3). This finding suggests that the emergence of SARS-CoV and SARS-CoV-2 might be due to the recombination of viral genomes between bat coronaviruses in their natural reservoir (bats) or the intermediate host (pangolin), or both. There were no observable evolutionarily conserved regions in the S glycoprotein sequence of MERS-CoV in relative to that of SARS-CoV. Altogether, the alignment of coronaviral S glycoproteins with the reference sequence revealed a high evolutionary relationship between SARS-CoV (Urbani), bat CoVs, and pangolin CoVs. It is suggested that the emergence of the highly contagious and pandemic-causing SARS-CoV-2 is highly attributable to genome recombination or mutations of the coronavirus in animal hosts such as bats [30,31,32]. The high evolutionary relationship among coronaviruses sheds light into the development of universal vaccines using conserved epitopes.
3.2. Prediction and Screening of Conserved CTL Epitopes of S glycoprotein
The conserved CTL epitopes of S glycoprotein were first screened and predicted based on 30 coronaviral S glycoprotein sequences. The NetCTL-1.2 of DTU Health Tech provides high sensitivity and specificity among the publicly available bioinformatics tools [33,34]. This web-based bioinformatics tool utilizes a combination of predictive algorithms including proteasomal cleavage, TAP transport efficiency, and MHC class-I affinity to acquire highly probable CTL epitopes in a given sequence. Given the easy accessibility, the epitopes were selected based on the available human leucocyte antigen (HLA) class-I supertypes provided by the algorithms, such as A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58, and B62 supertypes.
HLA class-I is known to be responsible for presenting processed antigens to T-cell receptors. Generally, there are three classical HLA-class-I encoding genes (HLA-A, HLA-B, and HLA-C) and all of them are extremely polymorphic. The number of identified HLA alleles has grown exponentially over the past decades and is likely to increase with time. To date, there are over 36,000 sequences of highly curated HLA alleles deposited into the IPD-IMGT/HLA Database (https://www.ebi.ac.uk/ipd/imgt/hla/). Undoubtedly, the vast number of HLA alleles makes the epitope prediction significantly complex and impractical. Thus, in the mid-1990s, an allele-specific classification called HLA Supertype, in which the first 9 HLA class-I supertypes were described [35] and 3 more HLA class-I supertypes were added later hence the 12 HLA class-I supertypes in the latest update [36]. dos Santos Francisco et al. (2015) investigated HLA class-I supertype frequencies among 55 human populations and found that HLA supertypes A2, A3, B7, B27, and B44 were evenly distributed and not specific to only certain populations [37]. Half of the populations showed frequencies at 14-29% for A2, 14-32% for A3, 18-31% for B7, and 21-32% for B44. In contrast, HLA supertypes A1, A24, B58, and B62 had greater frequency variations among the studied populations. It is also worth mentioning that the A24 supertype was found at higher frequencies (40% on average) in SEA, PAC, AUS, NEA and AME meanwhile the A1 supertype had an average frequency of 21% in Africa, Europe, and Southwest Asia [37].
The prediction of conserved CTL epitopes was based on the HLA class-I supertypes to cover as many human populations as possible. The initial screening yielded 1,048,575 potential CTL epitopes that matched the 12 HLA class-I supertypes. The large number of epitopes was then streamed down based on their antigenicity, immunogenicity, and toxigenicity. The elimination was performed using VaxiJen 2.0, IEDB MHC Class I immunogenicity and ToxinPred, respectively. The remaining 2,114 epitopes were subjected to another round of screening based on the frequency of appearance in 30 coronavirus strains and 12 HLA class-I supertypes. After stringent selection and removal of redundant epitopes, 12 epitopes (Table 7) were chosen for further analysis during the epitope’s alignment step.
3.3. Prediction and Screening of Conserved HTL Epitopes
The IEDB MHC-II prediction tool was used to predict and identify HTL epitopes because it provides a remarkable performance score owing to the embedded IEDB consensus 2.22 method [38,39]. Thirty coronavirus S glycoprotein sequences were mapped to 27 most widely distributed HLA class-II alleles as described by Greenbaum et al. (2011) [40]. A total of 108,767 HTL epitopes were identified and selected based on their being in the top 20% of the consensus percentile rank, corresponding to their abilities to capture 50% of the total immune response [41].
The epitopes were subjected to antigenicity, IFN-inducing, and toxigenicity predictions. The total number of remaining peptides was 1,377, which rendered difficulties in epitope selection. Consequently, peptides that matched 50% or greater of the HLA class-II alleles and coronavirus strains were chosen. There were 52 epitopes retained (Supplementary Table S1) and they were subjected to sequence alignment with that of SARS-CoV-2-Wuhan-Hu-1 to locate their positions. All of them were highly related to SARS-CoV-2 and its variants. Among them, 3 epitopes including HTL3, HTL6 and HTL26 were also related to SARS-CoV (Urbani).
3.4. Prediction and Screening of Conserved LBL Epitopes
The identified conserved LBL epitopes represented potential antigen candidates for stimulating the humoral immune response. Generally, B-cell epitopes are divided into (i) linear and continuous or (ii) conformational and non-continuous (Figure 2.). Although the vast majority of B-cell epitopes are conformational (approximately 90%) [42,43], the prediction of conformational B-cell epitopes is not as established as the LBL epitopes. Thus, the LBL epitope prediction has gained the most attention, especially in epitope-based vaccine development.
In contrast to the conserved HTL and CTL epitope predictions, screening and prediction of LBL are exclusive of HLA class-I and -II alleles. This is because B cells recognize antigens via B-cell receptors (BCR), known as membrane-bound immunoglobulins (Ig). Immunoglobulins consist of a constant fragment (Fc) region at the stalk and a variable (V) domain at the top. Given its functions in antigen binding, the V domain is responsible for the enormous theoretical diversity (1013−15) of the BCR repertoire [44,45]. Despite the high plasticity and diversity of the BCR repertoire, several lines of evidence demonstrated high frequencies of shared BCR clonotypes or elements in human BCR [46,47].
On this account, the conserved LBL epitopes were screened and identified using the ABCPred and SVMTriP prediction tools. These tools are considerably accurate in their predictions [48,49] and the results are analyzable. A total of 4,238 peptides with thresholds of 0.5 and greater were obtained after eliminating the duplicated sequences. The antigenicity and toxicity predictions were performed as described earlier to exclude non-antigenic and toxigenic epitopes, leaving only 621 peptide sequences for further analysis. Subsequently, the number of epitopes was narrowed to 68 by retaining the peptides similar to the sequences or found in 50% or greater of the coronavirus strains (Table 8.).
3.5. Alignment and Assembly of the Identified HTL, CTL T, and LBL Epitopes
In this study, a total of 131 epitopes (12 CTL epitopes, 52 HTL epitopes, and 68 LBL epitopes) were identified. Generally, the selection criteria included (i) within the S1 or S2 region, (ii) conserved regions, and (iii) matching most HLA class-I and -II supertypes. The identified epitopes were aligned to the SARS-CoV-2-Wuhan-Hu-1 S glycoprotein to identify their positions (Supplementary Figure S4). They were then assembled and combined into 2 peptide sequences encompassing 39 and 34 amino acid residues, respectively (Table 9.). Interestingly, the sequences of both assemblies, i.e., Epi1 and Epi2 corresponded to the epitopes located within the S1 region of the S glycoprotein. Epi1 was located at the N-terminal domain (NTD) (S256-294) while Epi2 was found in the RBD (S492-525). In addition, they were relatively conserved among the pangolin and bat coronaviral S glycoproteins (Table 3.) except for avian coronavirus strains. Notably, Epi1 was 66.7% (26/39) and 43.6% (17/39) similar to that of SARS-CoV and MERS-CoV, respectively (Supplementary Figure S3), suggesting that the conservation of Epi1 renders it a promising candidate of a broad-spectrum, cross-protective vaccine that potentially offers prophylactic protection against multiple coronavirus strains.
Next, the allergenicity of Epi1 and Epi2 was determined by using AllergenFP v1.0. The results showed that Epi1 was a potential allergen whereas Epi2 was a non-allergen. Epi1 had the highest Tanimoto similarity index to a major allergen Pru av 1 (UniProtKB/Swiss-Prot ID: O24248) that causes birch pollinosis and oral allergy in patients allergic to cherry. Epi1 shared 9 amino acids with that of Pru av 1 peptide albeit scattered throughout the latter’s peptide sequence (Supplementary Figure S5). Nonetheless, the overlapping residues in the Epi1 and Pru av 1 peptides do not coincide with the known IgE binding regions of the Pru av 1 peptide, i.e., the P-loop region (44LEGDGGPGT52) [50,51], suggesting that the allergenic potential of Epi1 is mostly negligible. Furthermore, due to its relatively smaller molecular size, the 3D structure and physiochemical properties of Epi1 are not definitive, thus it is inconclusive to serve as an allergen after modification for vaccine development.
The assembled peptides matched a wide range of HLA class-I and HLA class-II supertypes (Table 9.). In terms of the HLA class-I supertypes, the peptides matched 7 out of 12 (58.3%) HLA class-I supertypes mainly the supertypes A and B, which are globally distributed in all human populations [52]. Of the 27 available HLA class-II supertypes, Epi1 and Epi2 matched 23 (85.2%) and 20 (74.1%) HLA class-II alleles, respectively. In terms of the conserved LBL epitopes, Epi1 and Epi2 matched 3 and 2 LBL epitope sequences, respectively.
3.6. Identification of the Locations of the Conserved Epitopes in Coronaviral S glycoprotein
As mentioned previously, the Epi1 and Epi2 are located at the NTD (S256-294) and RBD (S492-525), respectively (Figure 3a). Figure 3. indicates the close-state and open-state positions of Epi1 and Epi2 on the S glycoprotein.
The position of epitopes on an antigen contributes to its antigenicity and immunogenicity. The findings showed that Epi1 (cyan) was slightly embedded within the NTD (grey) and, therefore, was relatively less exposed than Epi2 (orange) located within the RBD domain (S319-541). This justifies the conservation of Epi1. Nonetheless, the conservation of epitopes is not solely determined by the exposure to immune cells or antibodies, it also depends on the functional importance of the epitopes. Mutations in conserved epitopes possibly disrupt key processes, such as viral attachment, entry, and immune evasion, thereby compromising the viral infectivity and replication in host cells [53,54,55]. In this light, conserved epitopes are important to ensure the structural and functional integrity of viral particles.
4. Discussion
The 2019 SARS-CoV-2 pandemic revealed the unpreparedness of global healthcare systems to effectively respond to such a crisis, eventually leading to the breakdown of healthcare systems. The evolution and natural selection of coronaviruses are believed to contribute to the emergence of various VOCs with exceptional abilities to escape vaccine- and infection-induced immunity. The COVID-19 prophylactic vaccines are mainly based on the whole S glycoprotein subunit of SARS-CoV-2-Wuhan-1 due to its high antigenicity and immunogenicity [56,57,58,59,60,61,62]. However, the effectiveness of the COVID-19 vaccines is becoming less pronounced following the existence of immune-evading variants due to the perpetual gene mutations [63,64,65,66]. In addition, the immune imprinting induced by immunization and previous infections also reduces the efficaciousness of the vaccines against newly emerged variants [67,68]. In this light, a conserved multi-epitope approach has been adopted to develop pre-emptive vaccines against highly mutable coronaviruses by targeting the critical functional viral antigens. This strategy not only induces broad and long-lasting immune responses, it also prevents the constant review of vaccine formulations due to viral mutations. To achieve this, epitope identification and characterization are entailed to generate epitope maps depicting their antibody specificities in-silico prior to rigorous in-vitro and in-vivo empirical investigations [13,14,18,19,20,21,69,70,71].
Phylogenetically related zoonotic coronaviruses including distantly related avian coronaviruses were included in this study to identify and analyze the conserved regions. A significant genetic divergence was observed in avian and human coronaviruses compared to SARS-CoV-2. This divergence is especially notable when comparing avian coronaviruses, which belong to the Gammacoronavirus genus with human coronaviruses and SARS-CoV-2, which belong to the Alphacoronavirus and Betacoronavirus genera, respectively. This observation is consistent with the phylogenetic data reported by Gilbert & Tengs (2021) [72]. It is noteworthy that none of the avian coronaviruses has been reported to infect humans to date. In this light, the prediction of conserved epitopes of coronaviruses prioritizes those and their close zoonotic counterparts causing diseases in humans (hCoVs, MERS-CoV, SARS-CoV, and SARS-CoV-2).
Initially, 12 HTL epitopes, 52 CTL epitopes, and 68 LBL epitopes were identified and the majority of them were conserved across the coronavirus strains. The avian coronaviruses, hCoVs, and MERS-CoV were distantly related to SARS-CoV, SARS-CoV-2, bat CoVs, and pangolin CoVs. The evolutionary convergence among those coronaviruses is likely due to the different natural and/or intermediate hosts [73]. In addition, it is noteworthy that the evolutionary convergence also results in the host-cell receptor variations as observed in hCoVs of which the surface receptors responsible for viral adsorption are mainly surface peptidases and sialic acid-rich glycan-based receptors [74].
Many of the identified epitope sequences overlapped with one another, therefore, they were aligned and assembled into single peptide sequences. Two peptide assemblies, Epi1 and Epi2, were produced, consisting of HTL, CTL, and LBL epitopes. Those sequences represented residues S256-294 (SGWTAGAAAYYVGYLQPRTFLLKYNENGTITDAVDCALD) and S492-525 (LQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVC) of the S glycoprotein. Epi1 was located in the S1 region, particularly the NTD; Epi2, on the other hand, is found in the RBD region. Coincidentally, Epi1 was similar to the peptide sequence reported by Meyer et al. (2023) [17]. The study reported that the S369-277 epitope could induce a high magnitude of CTL immune response after in-vitro stimulation [17]. Furthermore, an in-vivo study confirmed that the Epi1 sequence (YYVGYLQPRTFLLKY) could induce a robust antigen-specific IFN-γ-producing CTL response [15] meanwhile an in-silico study identified the SGWTAGAAAYYV motif of Epi1 as the immunodominant site for T-cell and humoral responses [12]. Collectively, Epi1 is a robust candidate for the development of multi-epitope vaccines. Epi2 (S492-525), on the other hand, has not been reported in any previous studies but its PYRVVVLSF motif was hypothesized to induce adaptive immunity such as the production of neutralizing antibodies [11].
Human leukocyte antigen (HLA) alleles are among the most gene-dense and polymorphic regions in the human genome [75]. HLA molecules are responsible for antigen presentation to T-cell receptors (TcR) on CTL and HTL, and therefore, can readily affect the vaccine-induced immune response [34,76,77,78,79,80,81,82,83,84]. In this light, it is important to retain antigenic epitopes that can interact and bind to HLA class-I and class-II molecules in vaccine design and development. Epi1 and Epi2 fulfill the characteristics of immunogenic vaccine candidates given their multiple T-cell and B-cell epitopes. Among the matched HLA class-I supertypes, the A*02 supertype in both Epi1 and Epi2 is prevalently found in almost all human populations [52]. In regard to the HLA class-II supertypes, more than 20 HLA class-II supertypes were identified, together with the LBL epitopes, Epi1 and Epi2 are expected to trigger cellular and humoral immune responses, thereby providing more comprehensive protection against coronaviruses [43,85].
Given the conservation of Epi1 and Epi2, they hold promises as lead antigens in universal multi-epitope vaccine development, particularly in fighting the upcoming mutants. This helps address issues concerning the constant gene mutations and immune evasion seen in coronaviruses. Epi2 consists of most of the important residues required to form tight binding with ACE2 receptors [86,87,88]. It also encompasses well-known mutation sites found in the currently circulating Omicron variant, i.e., N501 and Y505. The N501Y mutation can lower neutralizing antibody binding in-vitro [89], while the Y505H mutation reduces viral protein stability, affects viral infectivity and promotes immune evasion [90,91]. Given the importance of the mutations, including them in a vaccine formulation is likely to add to the relevance of the multi-epitope vaccine with the circulating coronavirus variants hence greater immune protection.
Incorporating multiple epitopes in a vaccine formulation can offer broader and more durable protection against a wider range of viral variants. The multi-epitope sequences identified in this study shed light on the ongoing development and applications of coronavirus vaccines. To better model the binding affinity and stability between the epitopes with their counterparts such as TLR 3 and TLR4 receptors, docking analysis and dynamics simulation can be employed, thereby improving their efficacy in in vitro and in vivo studies [92]. Additionally, the epitopes also can be incorporated into vaccine formulations by conjugating them with virus-like particles (VLPs) or nanoparticle-based delivery systems to enhance their abilities in inducing humoral and cellular responses [93,94]. Furthermore, the epitopes can be developed into multivalent vaccines consisting of promising flu antigens such as nucleoprotein (NP) of influenza A virus (IAV), which assembles into virus-like particles (VLP) for vaccine delivery [23,95]. To strengthen the multivalency of the vaccine, highly conserved matrix 2 ectodomain protein (M2e) of IAV can be added into the vaccine formulation. The IAV M2e is known for conferring partial protection in animal models against IAV [96,97]. Collectively, the aforementioned prospective applications highlight the versatility and the potential of the multi-epitope peptides in curbing coronavirus infections.
This study revealed the evolutionarily conserved regions in SARS-CoV-2, SARS-CoV, and some animal coronaviruses while highlighting the genetic divergence in MERS-CoV and hCoVs that exhibited nearly no sequence similarity in the S1 subunit. This finding underscores a distinctive genetic difference among coronaviruses. Despite the genetic variations, the HR1 and HR2 regions of the S2 subunit displayed some degree of sequence similarities across the coronaviruses, indicating their potentials as the therapeutic targets.
5. Conclusions
In conclusion, the evolutionarily conserved epitopes are present among animal and human coronaviral S glycoproteins. Overall, 132 candidates representing HTL, CTL and LBL epitopes with relatively low evolutionary divergence were identified. They were screened and filtered into two final peptide assemblies: Epi1 is composed of 4 HLA class-I, 5 HLA class-II, and 3 LBL epitopes, meanwhile, Epi2 consists of 2 HLA class-I, 8 HLA class-II, and 2 LBL epitopes. These two peptide sequences, located within the S1 subunit, retain high population coverage and conservation properties, hence broad applicability and high effectiveness as lead universal vaccine candidates. Notably, Epi1 also contains immunodominant CTL epitopes, which adds to its potential as a vaccine candidate. Collectively, the conserved epitopes provide a robust foundation for vaccine development. They are expected to stimulate broad-spectrum immunity to mitigate the impact of infections of coronaviruses.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Table S1: Final selected HTL epitopes; Supplementary Figure S1: Animal coronaviruses alignment result with SARS-CoV-2-Wuhan-Hu-1 strain as the reference sequence; Supplementary Figure S2: Human coronaviruses alignment result with SARS-CoV-2-Wuhan-Hu-1 strain as the reference sequence; Supplementary Figure S3: Alignment result of SARS-CoV, MERS-CoV, and all SARS-CoV-2 variants with SARS-CoV-2-Wuhan-Hu-1 strain as the reference sequence; Supplementary Figure S4: Alignment result of assembled epitopes, Sepi1 and Sepi2, to SARS-CoV-2-Wuhan-Hu-1 sequence; Supplementary Material S5: Alignment result of Sepi1 to Pru av 1 allergen peptide sequence.
Author Contributions
Conceptualization, Y.C.O, B.A.T. and W.B.Y.; methodology & investigation, Y.C.O and W.B.Y.; data analysis and validation, Y.C.O, B.A.T. and W.B.Y.; writing—original draft preparation, Y.C.O.; writing—review and editing, Y.C.O, B.A.T. and W.B.Y.; supervision, B.A.T. and W.B.Y.; project administration, W.B.Y.; funding acquisition, W.B.Y. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by the Fundamental Research Grant Scheme (FRGS/1/2021/STG03/UKM/02/2) from Ministry of Higher Education, Malaysia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The postgraduate student and partial publication fee are sponsored by the United States Agency for International Development (USAID) through SEAOHUN 2024 Scholarship Program. The contents are the responsibility of the author(s) and do not necessarily reflect the views of USAID or the United States Government.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pormohammad, A., Ghorbani, S., Khatami, A., Farzi, R., Baradaran, B., Turner, D.L., Turner, R.J., Bahr, N.C., Idrovo, J.P.: Comparison of confirmed COVID-19 with SARS and MERS cases - Clinical characteristics, laboratory findings, radiographic signs and outcomes: A systematic review and meta-analysis. Rev Med Virol. 30, (2020). [CrossRef]
- Hui, D.S., Perlman, S., Zumla, A.: Spread of MERS to South Korea and China. Lancet Respir Med. 3, 509–510 (2015). [CrossRef]
- WHO Director-General’s opening remarks at the media briefing on COVID-19 - 11 March 2020, https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020.
- COVID-19 cases | WHO COVID-19 dashboard, https://data.who.
- Andrews, N., Stowe, J., Kirsebom, F., Toffa, S., Rickeard, T., Gallagher, E., Gower, C., Kall, M., Groves, N., O’Connell, A.-M., Simons, D., Blomquist, P.B., Zaidi, A., Nash, S., Iwani Binti Abdul Aziz, N., Thelwall, S., Dabrera, G., Myers, R., Amirthalingam, G., Gharbia, S., Barrett, J.C., Elson, R., Ladhani, S.N., Ferguson, N., Zambon, M., Campbell, C.N.J., Brown, K., Hopkins, S., Chand, M., Ramsay, M., Lopez Bernal, J.: Covid-19 Vaccine Effectiveness against the Omicron (B.1.1.529) Variant. New England Journal of Medicine. 386, 1532–1546 (2022). [CrossRef]
- Greaney, A.J., Starr, T.N., Gilchuk, P., Zost, S.J., Binshtein, E., Loes, A.N., Hilton, S.K., Huddleston, J., Eguia, R., Crawford, K.H.D., Dingens, A.S., Nargi, R.S., Sutton, R.E., Suryadevara, N., Rothlauf, P.W., Liu, Z., Whelan, S.P.J., Carnahan, R.H., Crowe, J.E., Bloom, J.D.: Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe. 29, 44-57.e9 (2021). [CrossRef]
- Harvey, W.T., Carabelli, A.M., Jackson, B., Gupta, R.K., Thomson, E.C., Harrison, E.M., Ludden, C., Reeve, R., Rambaut, A., Peacock, S.J., Robertson, D.L.: SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 19, 409–424 (2021). [CrossRef]
- Malik, J.A., Ahmed, S., Mir, A., Shinde, M., Bender, O., Alshammari, F., Ansari, M., Anwar, S.: The SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to new challenges. J Infect Public Health. 15, 228–240 (2022). [CrossRef]
- McLean, G., Kamil, J., Lee, B., Moore, P., Schulz, T.F., Muik, A., Sahin, U., Türeci, Ö., Pather, S.: The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio. 13, e02979-21 (2022). [CrossRef]
- Prakash, S., Srivastava, R., Coulon, P.-G., Dhanushkodi, N.R., Chentoufi, A.A., Tifrea, D.F., Edwards, R.A., Figueroa, C.J., Schubl, S.D., Hsieh, L., Buchmeier, M.J., Bouziane, M., Nesburn, A.B., Kuppermann, B.D., BenMohamed, L.: Genome-Wide B Cell, CD4+, and CD8+ T Cell Epitopes That Are Highly Conserved between Human and Animal Coronaviruses, Identified from SARS-CoV-2 as Targets for Preemptive Pan-Coronavirus Vaccines. Journal of immunology (Baltimore, Md. 206, 2566–2582 (2021). [CrossRef]
- Bagherzadeh, M.A., Izadi, M., Baesi, K., Jahromi, M.A.M., Pirestani, M.: Considering epitopes conservity in targeting SARS-CoV-2 mutations in variants: a novel immunoinformatics approach to vaccine design. Sci Rep. 12, 14017 (2022). [CrossRef]
- Jaiswal, V., Lee, H.J.: Conservation and Evolution of Antigenic Determinants of SARS-CoV-2: An Insight for Immune Escape and Vaccine Design. Front Immunol. 13, (2022). [CrossRef]
- Ahmed, S.F., Quadeer, A.A., McKay, M.R.: Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses. 12, 254 (2020). [CrossRef]
- Ismail, S., Ahmad, S., Azam, S.S.: Immunoinformatics characterization of SARS-CoV-2 spike glycoprotein for prioritization of epitope based multivalent peptide vaccine. J Mol Liq. 314, 113612 (2020). [CrossRef]
- Jiang, S., Wu, S., Zhao, G., He, Y., Guo, X., Zhang, Z., Hou, J., Ding, Y., Cheng, A., Wang, B.: Identification of a promiscuous conserved CTL epitope within the SARS-CoV-2 spike protein. Emerg Microbes Infect. 11, 730 (2022). [CrossRef]
- Smith, T.R.F., Patel, A., Ramos, S., Elwood, D., Zhu, X., Yan, J., Gary, E.N., Walker, S.N., Schultheis, K., Purwar, M., Xu, Z., Walters, J., Bhojnagarwala, P., Yang, M., Chokkalingam, N., Pezzoli, P., Parzych, E., Reuschel, E.L., Doan, A., Tursi, N., Vasquez, M., Choi, J., Tello-Ruiz, E., Maricic, I., Bah, M.A., Wu, Y., Amante, D., Park, D.H., Dia, Y., Ali, A.R., Zaidi, F.I., Generotti, A., Kim, K.Y., Herring, T.A., Reeder, S., Andrade, V.M., Buttigieg, K., Zhao, G., Wu, J.-M., Li, D., Bao, L., Liu, J., Deng, W., Qin, C., Brown, A.S., Khoshnejad, M., Wang, N., Chu, J., Wrapp, D., McLellan, J.S., Muthumani, K., Wang, B., Carroll, M.W., Kim, J.J., Boyer, J., Kulp, D.W., Humeau, L.M.P.F., Weiner, D.B., Broderick, K.E.: Immunogenicity of a DNA vaccine candidate for COVID-19. Nat Commun. 11, 2601 (2020). [CrossRef]
- Meyer, S., Blaas, I., Bollineni, R.C., Delic-Sarac, M., Tran, T.T., Knetter, C., Dai, K.-Z., Madssen, T.S., Vaage, J.T., Gustavsen, A., Yang, W., Nissen-Meyer, L.S.H., Douvlataniotis, K., Laos, M., Nielsen, M.M., Thiede, B., Søraas, A., Lund-Johansen, F., Rustad, E.H., Olweus, J.: Prevalent and immunodominant CD8 T cell epitopes are conserved in SARS-CoV-2 variants. Cell Rep. 42, 111995 (2023). [CrossRef]
- Mishra, N., Huang, X., Joshi, S., Guo, C., Ng, J., Thakkar, R., Wu, Y., Dong, X., Li, Q., Pinapati, R.S., Sullivan, E., Caciula, A., Tokarz, R., Briese, T., Lu, J., Lipkin, W.I.: Immunoreactive peptide maps of SARS-CoV-2. Commun Biol. 4, 225 (2021). [CrossRef]
- Wang, H., Wu, X., Zhang, X., Hou, X., Liang, T., Wang, D., Teng, F., Dai, J., Duan, H., Guo, S., Li, Y., Yu, X.: SARS-CoV-2 Proteome Microarray for Mapping COVID-19 Antibody Interactions at Amino Acid Resolution. ACS Cent Sci. 6, 2238–2249 (2020). [CrossRef]
- Sikora, M., von Bülow, S., Blanc, F.E.C., Gecht, M., Covino, R., Hummer, G.: Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput Biol. 17, e1008790 (2021). [CrossRef]
- Schwarz, T., Heiss, K., Mahendran, Y., Casilag, F., Kurth, F., Sander, L.E., Wendtner, C.-M., Hoechstetter, M.A., Müller, M.A., Sekul, R., Drosten, C., Stadler, V., Corman, V.M.: SARS-CoV-2 Proteome-Wide Analysis Revealed Significant Epitope Signatures in COVID-19 Patients. Front Immunol. 12, 629185 (2021). [CrossRef]
- Paul, S., Arlehamn, C.S.L., Scriba, T.J., Dillon, M.B.C., Oseroff, C., Hinz, D., McKinney, D.M., Pro, S.C., Sidney, J., Peters, B., Sette, A.: Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J Immunol Methods. 422, 28–34 (2015). [CrossRef]
- Chenavas, S., Estrozi, L.F., Slama-Schwok, A., Delmas, B., Primo, C. Di, Baudin, F., Li, X., Crépin, T., Ruigrok, R.W.H.: Monomeric Nucleoprotein of Influenza A Virus. PLoS Pathog. 9, e1003275 (2013). [CrossRef]
- Kirchdoerfer, R.N., Cottrell, C.A., Wang, N., Pallesen, J., Yassine, H.M., Turner, H.L., Corbett, K.S., Graham, B.S., McLellan, J.S., Ward, A.B.: Pre-fusion structure of a human coronavirus spike protein. Nature. 531, 118–121 (2016). [CrossRef]
- Li, Z., Tomlinson, A.C.A., Wong, A.H.M., Zhou, D., Desforges, M., Talbot, P.J., Benlekbir, S., Rubinstein, J.L., Rini, J.M.: The human coronavirus HCoV-229E S-protein structure and receptor binding. Elife. 8, e51230 (2019). [CrossRef]
- Wang, C., Hesketh, E.L., Shamorkina, T.M., Li, W., Franken, P.J., Drabek, D., van Haperen, R., Townend, S., van Kuppeveld, F.J.M., Grosveld, F., Ranson, N.A., Snijder, J., de Groot, R.J., Hurdiss, D.L., Bosch, B.-J.: Antigenic structure of the human coronavirus OC43 spike reveals exposed and occluded neutralizing epitopes. Nat Commun. 13, 2921 (2022). [CrossRef]
- Huang, Y., Yang, C., Xu, X., Xu, W., Liu, S.: Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 41, 1141–1149 (2020). [CrossRef]
- Xia, X.: Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design. Viruses. 13, (2021). [CrossRef]
- Zheng, Z., Monteil, V.M., Maurer-Stroh, S., Yew, C.W., Leong, C., Mohd-Ismail, N.K., Arularasu, S.C., Chow, V.T.K., Lin, R.T.P., Mirazimi, A., Hong, W., Tan, Y.J.: Monoclonal antibodies for the S2 subunit of spike of SARS-CoV-1 cross-react with the newly-emerged SARS-CoV-2. Eurosurveillance. 25, 19–28 (2020). [CrossRef]
- Boni, M.F., Lemey, P., Jiang, X., Lam, T.T.-Y., Perry, B.W., Castoe, T.A., Rambaut, A., Robertson, D.L.: Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 5, 1408–1417 (2020). [CrossRef]
- Li, X., Giorgi, E.E., Marichannegowda, M.H., Foley, B., Xiao, C., Kong, X.-P., Chen, Y., Gnanakaran, S., Korber, B., Gao, F.: Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci Adv. 6, eabb9153 (2020). [CrossRef]
- Sajini, A.A., Alkayyal, A.A., Mubaraki, F.A.: The Recombination Potential between SARS-CoV-2 and MERS-CoV from Cross-Species Spill-over Infections. J Epidemiol Glob Health. 11, 155–159 (2021). [CrossRef]
- Larsen, M.V., Lundegaard, C., Lamberth, K., Buus, S., Lund, O., Nielsen, M.: Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 8, 424 (2007). [CrossRef]
- Larsen, M.V., Lundegaard, C., Lamberth, K., Buus, S., Brunak, S., Lund, O., Nielsen, M.: An integrative approach to CTL epitope prediction: A combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol. 35, 2295–2303 (2005). [CrossRef]
- Sidney, J., Grey, H.M., Kubo, R.T., Sette, A.: Practical, biochemical and evolutionary implications of the discovery of HLA class I supermotifs. Immunol Today. 17, 261–266 (1996). [CrossRef]
- Sidney, J., Peters, B., Frahm, N., Brander, C., Sette, A.: HLA class I supertypes: a revised and updated classification. BMC Immunol. 9, 1 (2008). [CrossRef]
- dos Santos Francisco, R., Buhler, S., Nunes, J.M., Bitarello, B.D., França, G.S., Meyer, D., Sanchez-Mazas, A.: HLA supertype variation across populations: new insights into the role of natural selection in the evolution of HLA-A and HLA-B polymorphisms. Immunogenetics. 67, 651–663 (2015). [CrossRef]
- Andreatta, M., Trolle, T., Yan, Z., Greenbaum, J.A., Peters, B., Nielsen, M.: An automated benchmarking platform for MHC class II binding prediction methods. Bioinformatics. 34, 1522–1528 (2018). [CrossRef]
- Wang, P., Sidney, J., Dow, C., Mothé, B., Sette, A., Peters, B.: A Systematic Assessment of MHC Class II Peptide Binding Predictions and Evaluation of a Consensus Approach. PLoS Comput Biol. 4, e1000048 (2008). [CrossRef]
- Greenbaum, J., Sidney, J., Chung, J., Brander, C., Peters, B., Sette, A.: Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics. 63, 325–335 (2011). [CrossRef]
- Paul, S., Arlehamn, C.S.L., Scriba, T.J., Dillon, M.B.C., Oseroff, C., Hinz, D., McKinney, D.M., Pro, S.C., Sidney, J., Peters, B., Sette, A.: Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J Immunol Methods. 422, 28–34 (2015). [CrossRef]
- Regenmortel, M.H. V: What Is a B-Cell Epitope? In: Schutkowski, M. and Reineke, U. (eds.) Epitope Mapping Protocols. pp. 3–20. Humana Press, Totowa, NJ (2009).
- Sanchez-Trincado, J.L., Gomez-Perosanz, M., Reche, P.A.: Fundamentals and Methods for T- and B-Cell Epitope Prediction. J Immunol Res. 2017, 2680160 (2017). [CrossRef]
- Chaudhary, N., Wesemann, D.R.: Analyzing Immunoglobulin Repertoires. Front Immunol. 9, (2018).
- Raybould, M.I.J., Rees, A.R., Deane, C.M.: Current strategies for detecting functional convergence across B-cell receptor repertoires. MAbs. 13, 1996732 (2021). [CrossRef]
- Briney, B., Inderbitzin, A., Joyce, C., Burton, D.R.: Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature. 566, 393–397 (2019). [CrossRef]
- Soto, C., Bombardi, R.G., Branchizio, A., Kose, N., Matta, P., Sevy, A.M., Sinkovits, R.S., Gilchuk, P., Finn, J.A., Crowe, J.E.: High frequency of shared clonotypes in human B cell receptor repertoires. Nature. 566, 398–402 (2019). [CrossRef]
- Galanis, K.A., Nastou, K.C., Papandreou, N.C., Petichakis, G.N., Iconomidou, V.A.: Linear B-cell epitope prediction: a performance review of currently available methods, https://www.biorxiv.org/content/10.1101/833418v1, (2019).
- Yao, B., Zhang, L., Liang, S., Zhang, C.: SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS One. 7, e45152 (2012). [CrossRef]
- Scheurer, S., Son, D.Y., Boehm, M., Karamloo, F., Franke, S., Hoffmann, A., Haustein, D., Vieths, S.: Cross-reactivity and epitope analysis of Pru a 1, the major cherry allergen. Mol Immunol. 36, 155–167 (1999). [CrossRef]
- Neudecker, P., Lehmann, K., Nerkamp, J., Haase, T., Wangorsch, A., Fötisch, K., Hoffmann, S., Rösch, P., Vieths, S., Scheurer, S.: Mutational epitope analysis of Pru av 1 and Api g 1, the major allergens of cherry (Prunus avium) and celery (Apium graveolens): correlating IgE reactivity with three-dimensional structure. Biochemical Journal. 376, 97–107 (2003). [CrossRef]
- Arrieta-Bolaños, E., Hernández-Zaragoza, D.I., Barquera, R.: An HLA map of the world: A comparison of HLA frequencies in 200 worldwide populations reveals diverse patterns for class I and class II. Front Genet. 14, 866407 (2023). [CrossRef]
- Zhang, B., Xu, S., Liu, M., Wei, Y., Wang, Q., Shen, W., Lei, C.Q., Zhu, Q.: The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy. 19, 1916 (2023). [CrossRef]
- Jiao, C., Wang, B., Chen, P., Jiang, Y., Liu, J.: Analysis of the conserved protective epitopes of hemagglutinin on influenza A viruses. Front Immunol. 14, 1086297 (2023). [CrossRef]
- Corti, D., Lanzavecchia, A.: Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 31, 705–742 (2013). [CrossRef]
- Krammer, F. : SARS-CoV-2 vaccines in development. Nature. 586, 516–527 (2020). [CrossRef]
- Kyriakidis, N.C., López-Cortés, A., González, E.V., Grimaldos, A.B., Prado, E.O.: SARS-CoV-2 vaccines strategies: a comprehensive review of phase 3 candidates. NPJ Vaccines. 6, 28 (2021). [CrossRef]
- Martínez-Flores, D., Zepeda-Cervantes, J., Cruz-Reséndiz, A., Aguirre-Sampieri, S., Sampieri, A., Vaca, L.: SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front Immunol. 12, 701501 (2021). [CrossRef]
- Samrat, S.K., Tharappel, A.M., Li, Z., Li, H.: Prospect of SARS-CoV-2 spike protein: Potential role in vaccine and therapeutic development. Virus Res. 288, 198141 (2020). [CrossRef]
- Sulbaran, G., Maisonnasse, P., Amen, A., Effantin, G., Guilligay, D., Dereuddre-Bosquet, N., Burger, J.A., Poniman, M., Grobben, M., Buisson, M., Dergan Dylon, S., Naninck, T., Lemaître, J., Gros, W., Gallouët, A.-S., Marlin, R., Bouillier, C., Contreras, V., Relouzat, F., Fenel, D., Thepaut, M., Bally, I., Thielens, N., Fieschi, F., Schoehn, G., van der Werf, S., van Gils, M.J., Sanders, R.W., Poignard, P., Le Grand, R., Weissenhorn, W.: Immunization with synthetic SARS-CoV-2 S glycoprotein virus-like particles protects macaques from infection. Cell Rep Med. 3, 100528 (2022). [CrossRef]
- Wrapp, D., Wang, N., Corbett, K.S., Goldsmith, J.A., Hsieh, C.-L., Abiona, O., Graham, B.S., McLellan, J.S.: Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 367, 1260–1263 (2020). [CrossRef]
- Sternberg, A., Naujokat, C.: Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 257, 118056 (2020). [CrossRef]
- Cao, Y., Yisimayi, A., Jian, F., Song, W., Xiao, T., Wang, L., Du, S., Wang, J., Li, Q., Chen, X., Yu, Y., Wang, P., Zhang, Z., Liu, P., An, R., Hao, X., Wang, Y., Wang, J., Feng, R., Sun, H., Zhao, L., Zhang, W., Zhao, D., Zheng, J., Yu, L., Li, C., Zhang, N., Wang, R., Niu, X., Yang, S., Song, X., Chai, Y., Hu, Y., Shi, Y., Zheng, L., Li, Z., Gu, Q., Shao, F., Huang, W., Jin, R., Shen, Z., Wang, Y., Wang, X., Xiao, J., Xie, X.S.: BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature. 608, 593–602 (2022). [CrossRef]
- Greaney, A.J., Loes, A.N., Crawford, K.H.D., Starr, T.N., Malone, K.D., Chu, H.Y., Bloom, J.D.: Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 29, 463-476.e6 (2021). [CrossRef]
- Greaney, A.J., Starr, T.N., Barnes, C.O., Weisblum, Y., Schmidt, F., Caskey, M., Gaebler, C., Cho, A., Agudelo, M., Finkin, S., Wang, Z., Poston, D., Muecksch, F., Hatziioannou, T., Bieniasz, P.D., Robbiani, D.F., Nussenzweig, M.C., Bjorkman, P.J., Bloom, J.D.: Mutational escape from the polyclonal antibody response to SARS-CoV-2 infection is largely shaped by a single class of antibodies, https://www.biorxiv.org/content/10.1101/2021.03.17.435863v1, (2021).
- Weisblum, Y., Schmidt, F., Zhang, F., DaSilva, J., Poston, D., Lorenzi, J.C.C., Muecksch, F., Rutkowska, M., Hoffmann, H.-H., Michailidis, E., Gaebler, C., Agudelo, M., Cho, A., Wang, Z., Gazumyan, A., Cipolla, M., Luchsinger, L., Hillyer, C.D., Caskey, M., Robbiani, D.F., Rice, C.M., Nussenzweig, M.C., Hatziioannou, T., Bieniasz, P.D.: Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife. 9, e61312 (2020). [CrossRef]
- Röltgen, K., Nielsen, S.C.A., Silva, O., Younes, S.F., Zaslavsky, M., Costales, C., Yang, F., Wirz, O.F., Solis, D., Hoh, R.A., Wang, A., Arunachalam, P.S., Colburg, D., Zhao, S., Haraguchi, E., Lee, A.S., Shah, M.M., Manohar, M., Chang, I., Gao, F., Mallajosyula, V., Li, C., Liu, J., Shoura, M.J., Sindher, S.B., Parsons, E., Dashdorj, N.J., Dashdorj, N.D., Monroe, R., Serrano, G.E., Beach, T.G., Chinthrajah, R.S., Charville, G.W., Wilbur, J.L., Wohlstadter, J.N., Davis, M.M., Pulendran, B., Troxell, M.L., Sigal, G.B., Natkunam, Y., Pinsky, B.A., Nadeau, K.C., Boyd, S.D.: Immune imprinting, breadth of variant recognition, and germinal center response in human SARS-CoV-2 infection and vaccination. Cell. 185, 1025-1040.e14 (2022). [CrossRef]
- Wheatley, A.K., Fox, A., Tan, H.-X., Juno, J.A., Davenport, M.P., Subbarao, K., Kent, S.J.: Immune imprinting and SARS-CoV-2 vaccine design. Trends Immunol. 42, 956–959 (2021). [CrossRef]
- Li, Y., Ma, M., Lei, Q., Wang, F., Hong, W., Lai, D., Hou, H., Xu, Z., Zhang, B., Chen, H., Yu, C., Xue, J., Zheng, Y., Wang, X., Jiang, H., Zhang, H., Qi, H., Guo, S., Zhang, Y., Lin, X., Yao, Z., Wu, J., Sheng, H., Zhang, Y., Wei, H., Sun, Z., Fan, X., Tao, S.: Linear epitope landscape of the SARS-CoV-2 Spike protein constructed from 1,051 COVID-19 patients. Cell Rep. 34, 108915 (2021). [CrossRef]
- Zhang, B., Hu, Y., Chen, L., Yau, T., Tong, Y., Hu, J., Cai, J., Chan, K.-H., Dou, Y., Deng, J., Wang, X., Hung, I.F.-N., To, K.K.-W., Yuen, K.Y., Huang, J.-D.: Mining of epitopes on spike protein of SARS-CoV-2 from COVID-19 patients. Cell Res. 30, 702–704 (2020). [CrossRef]
- Zhang, B.-Z., Chu, H., Han, S., Shuai, H., Deng, J., Hu, Y., Gong, H., Lee, A.C.-Y., Zou, Z., Yau, T., Wu, W., Hung, I.F.-N., Chan, J.F.-W., Yuen, K.-Y., Huang, J.-D.: SARS-CoV-2 infects human neural progenitor cells and brain organoids. Cell Res. 30, 928–931 (2020). [CrossRef]
- Gilbert, C., Tengs, T.: No species-level losses of s2m suggests critical role in replication of SARS-related coronaviruses. Sci Rep. 11, 16145 (2021). [CrossRef]
- Corman, V.M., Muth, D., Niemeyer, D., Drosten, C.: Hosts and Sources of Endemic Human Coronaviruses. Adv Virus Res. 100, 163–188 (2018). [CrossRef]
- Flerlage, T., Boyd, D.F., Meliopoulos, V., Thomas, P.G., Schultz-Cherry, S.: Influenza virus and SARS-CoV-2: pathogenesis and host responses in the respiratory tract. Nat Rev Microbiol. 19, 425–441 (2021). [CrossRef]
- Traherne, J.A.: Human MHC architecture and evolution: implications for disease association studies. Int J Immunogenet. 35, 179–192 (2008). [CrossRef]
- Gartland, A.J., Li, S., McNevin, J., Tomaras, G.D., Gottardo, R., Janes, H., Fong, Y., Morris, D., Geraghty, D.E., Kijak, G.H., Edlefsen, P.T., Frahm, N., Larsen, B.B., Tovanabutra, S., Sanders-Buell, E., deCamp, A.C., Magaret, C.A., Ahmed, H., Goodridge, J.P., Chen, L., Konopa, P., Nariya, S., Stoddard, J.N., Wong, K., Zhao, H., Deng, W., Maust, B.S., Bose, M., Howell, S., Bates, A., Lazzaro, M., O’Sullivan, A., Lei, E., Bradfield, A., Ibitamuno, G., Assawadarachai, V., O’Connell, R.J., deSouza, M.S., Nitayaphan, S., Rerks-Ngarm, S., Robb, M.L., Sidney, J., Sette, A., Zolla-Pazner, S., Montefiori, D., McElrath, M.J., Mullins, J.I., Kim, J.H., Gilbert, P.B., Hertz, T.: Analysis of HLA A*02 Association with Vaccine Efficacy in the RV144 HIV-1 Vaccine Trial. J Virol. 88, 8242–8255 (2014). [CrossRef]
- Liu, Y., Guo, T., Yu, Q., Zhang, H., Du, J., Zhang, Y., Xia, S., Yang, H., Li, Q.: Association of human leukocyte antigen alleles and supertypes with immunogenicity of oral rotavirus vaccine given to infants in China. Medicine. 97, (2018). [CrossRef]
- Mentzer, A.J., O’Connor, D., Bibi, S., Chelysheva, I., Clutterbuck, E.A., Demissie, T., Dinesh, T., Edwards, N.J., Felle, S., Feng, S., Flaxman, A.L., Karp-Tatham, E., Li, G., Liu, X., Marchevsky, N., Godfrey, L., Makinson, R., Bull, M.B., Fowler, J., Alamad, B., Malinauskas, T., Chong, A.Y., Sanders, K., Shaw, R.H., Voysey, M., Snape, M.D., Pollard, A.J., Lambe, T., Knight, J.C.: Human leukocyte antigen alleles associate with COVID-19 vaccine immunogenicity and risk of breakthrough infection. Nat Med. 29, 147–157 (2023). [CrossRef]
- Milich, D.R., Leroux-Roels, G.G.: Immunogenetics of the response to HBsAg vaccination. Autoimmun Rev. 2, 248–257 (2003). [CrossRef]
- Nielsen, C.M., Vekemans, J., Lievens, M., Kester, K.E., Regules, J.A., Ockenhouse, C.F.: RTS,S malaria vaccine efficacy and immunogenicity during Plasmodium falciparum challenge is associated with HLA genotype. Vaccine. 36, 1637–1642 (2018). [CrossRef]
- Nishida, N., Sugiyama, M., Sawai, H., Nishina, S., Sakai, A., Ohashi, J., Khor, S., Kakisaka, K., Tsuchiura, T., Hino, K., Sumazaki, R., Takikawa, Y., Murata, K., Kanda, T., Yokosuka, O., Tokunaga, K., Mizokami, M.: Key HLA-DRB1-DQB1 haplotypes and role of the BTNL2 gene for response to a hepatitis B vaccine. Hepatology. 68, 848–858 (2018). [CrossRef]
- O’Connor, D., Png, E., Khor, C.C., Snape, M.D., Hill, A.V.S., Klis, F. van der, Hoggart, C., Levin, M., Hibberd, M.L., Pollard, A.J.: Common Genetic Variations Associated with the Persistence of Immunity following Childhood Immunization. Cell Rep. 27, 3241-3253.e4 (2019). [CrossRef]
- Posteraro, B., Pastorino, R., Di Giannantonio, P., Ianuale, C., Amore, R., Ricciardi, W., Boccia, S.: The link between genetic variation and variability in vaccine responses: Systematic review and meta-analyses. Vaccine. 32, 1661–1669 (2014). [CrossRef]
- Ovsyannikova, I.G., Haralambieva, I.H., Vierkant, R.A., O’Byrne, M.M., Jacobson, R.M., Poland, G.A.: The Association of CD46, SLAM and CD209 Cellular Receptor Gene SNPs with Variations in Measles Vaccine-Induced Immune Responses: A Replication Study and Examination of Novel Polymorphisms. Hum Hered. 72, 206 (2011). [CrossRef]
- Potocnakova, L., Bhide, M., Pulzova, L.B.: An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction. J Immunol Res. 2016, 6760830 (2016). [CrossRef]
- Jawad, B., Adhikari, P., Podgornik, R., Ching, W.Y.: Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J Chem Inf Model. 61, 4425–4441 (2021). [CrossRef]
- Borkotoky, S., Dey, D., Hazarika, Z.: Interactions of angiotensin-converting enzyme-2 (ACE2) and SARS-CoV-2 spike receptor-binding domain (RBD): a structural perspective. Mol Biol Rep. 50, 2713 (2023). [CrossRef]
- Yi, C., Sun, X., Ye, J., Ding, L., Liu, M., Yang, Z., Lu, X., Zhang, Y., Ma, L., Gu, W., Qu, A., Xu, J., Shi, Z., Ling, Z., Sun, B.: Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cellular & Molecular Immunology 2020 17:6. 17, 621–630 (2020). [CrossRef]
- Sun, C., Kang, Y.F., Liu, Y.T., Kong, X.W., Xu, H.Q., Xiong, D., Xie, C., Liu, Y.H., Peng, S., Feng, G.K., Liu, Z., Zeng, M.S.: Parallel profiling of antigenicity alteration and immune escape of SARS-CoV-2 Omicron and other variants. Signal Transduction and Targeted Therapy 2022 7:1. 7, 1–10 (2022). [CrossRef]
- Verkhivker, G., Alshahrani, M., Gupta, G.: Balancing Functional Tradeoffs between Protein Stability and ACE2 Binding in the SARS-CoV-2 Omicron BA.2, BA.2.75 and XBB Lineages: Dynamics-Based Network Models Reveal Epistatic Effects Modulating Compensatory Dynamic and Energetic Changes. Viruses. 15, 1143 (2023). [CrossRef]
- Kumar, S., Thambiraja, T.S., Karuppanan, K., Subramaniam, G.: Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J Med Virol. 94, 1641–1649 (2022). [CrossRef]
- Singh, A., Thakur, M., Sharma, L.K., Chandra, K.: Designing a multi-epitope peptide based vaccine against SARS-CoV-2. Sci Rep. 10, 16219 (2020). [CrossRef]
- Wu, X., Li, W., Rong, H., Pan, J., Zhang, X., Hu, Q., Shi, Z.-L., Zhang, X.-E., Cui, Z.: A Nanoparticle Vaccine Displaying Conserved Epitopes of the Preexisting Neutralizing Antibody Confers Broad Protection against SARS-CoV-2 Variants. ACS Nano. 18, 17749–17763 (2024). [CrossRef]
- He, L., Lin, X., Wang, Y., Abraham, C., Sou, C., Ngo, T., Zhang, Y., Wilson, I.A., Zhu, J.: Single-component, self-assembling, protein nanoparticles presenting the receptor binding domain and stabilized spike as SARS-CoV-2 vaccine candidates. Sci Adv. 7, (2021). [CrossRef]
- Ng, A.K.-L., Zhang, H., Tan, K., Li, Z., Liu, J., Chan, P.K.-S., Li, S.-M., Chan, W.-Y., Au, S.W.-N., Joachimiak, A., Walz, T., Wang, J.-H., Shaw, P.-C.: Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. The FASEB Journal. 22, 3638 (2008). [CrossRef]
- Wu, F., Huang, J.-H., Yuan, X.-Y., Huang, W.-S., Chen, Y.-H.: Characterization of immunity induced by M2e of influenza virus. Vaccine. 25, 8868–8873 (2007). [CrossRef]
- Gao, X., Wang, W., Li, Y., Zhang, S., Duan, Y., Xing, L., Zhao, Z., Zhang, P., Li, Z., Li, R., Wang, X., Yang, P.: Enhanced Influenza VLP vaccines comprising matrix-2 ectodomain and nucleoprotein epitopes protects mice from lethal challenge. Antiviral Res. 98, 4–11 (2013). [CrossRef]
Figure 1.
Flow of in-silico prediction of conserved epitopes of the coronaviral S proteins. The orange-, green- and blue-coloured lines represent the CTL, LBL and HTL prediction steps, respectively.
Figure 1.
Flow of in-silico prediction of conserved epitopes of the coronaviral S proteins. The orange-, green- and blue-coloured lines represent the CTL, LBL and HTL prediction steps, respectively.
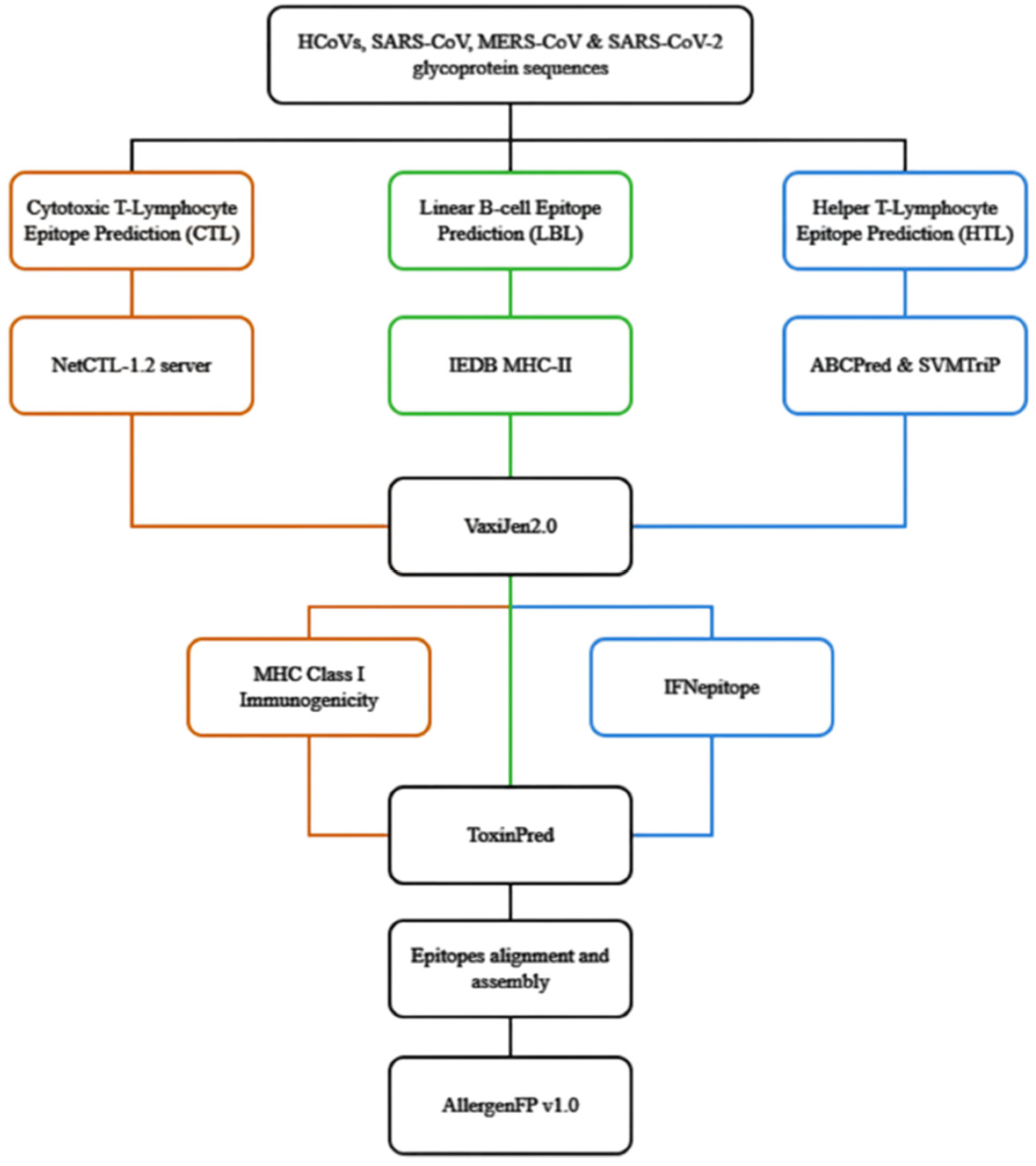
Figure 2.
Schematic diagram of linear and conformational B-cell epitopes. Panels (i) linear or continuous B-cell epitopes composed of amino acid residue that are sequential to one another; (ii) conformational or non-continuous B-cell epitopes composed of amino acids that are non-sequential and scattered along the peptide sequence.
Figure 2.
Schematic diagram of linear and conformational B-cell epitopes. Panels (i) linear or continuous B-cell epitopes composed of amino acid residue that are sequential to one another; (ii) conformational or non-continuous B-cell epitopes composed of amino acids that are non-sequential and scattered along the peptide sequence.
Figure 3.
(a) Schematic diagram of SARS-CoV-2 S glycoprotein with different colours representing the S1 subunit (S14-685) (magenta) and S2 subunit (S686-1273) (cornflower blue). (b) Ribbon and 3D structures of the close state of SARS-CoV-2 S glycoprotein (PDB: 6VXX). (b1) The ribbon structure of close-state S glycoprotein. The locations of Epi1 and Epi2 were in cyan and orange colours, respectively. (b2) The orthogonal view of the close state of S glycoprotein with Epi1 (cyan) and Epi2 (orange). (b3) The top-down view of the close state of S glycoprotein displaying Epi1 (cyan) and Epi2 (orange). (c) Ribbon and 3D structures of the open state of SARS-CoV-2 S glycoprotein (PBD: 6VYB); (c1) The ribbon structure of open-state S glycoprotein and the locations of Epi1 and Epi2 were in cyan and orange colours, respectively. (c2) The orthogonal view of the open state of S glycoprotein displaying Epi1 (cyan) and Epi2 (orange). (c3) The top-down view of the open state of S glycoprotein with Epi1 and Epi2 in cyan and orange colours, respectively.
Figure 3.
(a) Schematic diagram of SARS-CoV-2 S glycoprotein with different colours representing the S1 subunit (S14-685) (magenta) and S2 subunit (S686-1273) (cornflower blue). (b) Ribbon and 3D structures of the close state of SARS-CoV-2 S glycoprotein (PDB: 6VXX). (b1) The ribbon structure of close-state S glycoprotein. The locations of Epi1 and Epi2 were in cyan and orange colours, respectively. (b2) The orthogonal view of the close state of S glycoprotein with Epi1 (cyan) and Epi2 (orange). (b3) The top-down view of the close state of S glycoprotein displaying Epi1 (cyan) and Epi2 (orange). (c) Ribbon and 3D structures of the open state of SARS-CoV-2 S glycoprotein (PBD: 6VYB); (c1) The ribbon structure of open-state S glycoprotein and the locations of Epi1 and Epi2 were in cyan and orange colours, respectively. (c2) The orthogonal view of the open state of S glycoprotein displaying Epi1 (cyan) and Epi2 (orange). (c3) The top-down view of the open state of S glycoprotein with Epi1 and Epi2 in cyan and orange colours, respectively.
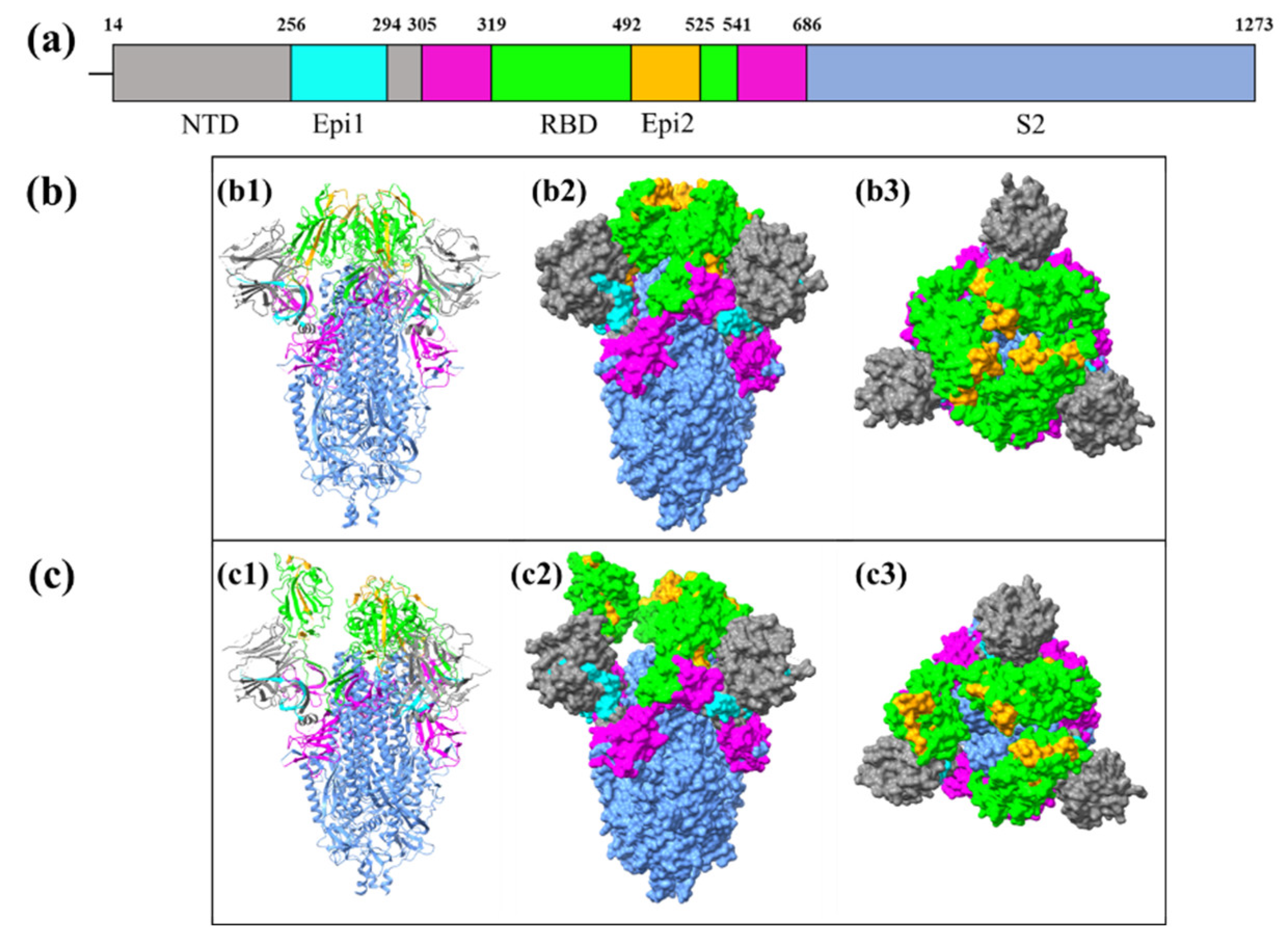

Table 1.
SARS-CoV-2 and its variants.
| Nomenclature | Lineage | Accession number |
|---|---|---|
| SARS-CoV-2-Wuhan-Hu-1 strain | NC_045512.2 | |
| Alpha | B.1.1.7 | OK340744.1 |
| Beta | B.1.351 | OQ341818.1 |
| Delta | B.1.617.2 | OQ314763.1 |
| Gamma | P.1 | OQ316323.1 |
| Omicron | B.1.1.529 | OQ344199.1 |
| Omicron | BA.1 | OQ355083.1 |
| Omicron | BA.1.1 | OQ352636.1 |
| Omicron | BA.2 | OQ341824.1 |
| Omicron | BA.2.12.1 | OQ355080.1 |
| Omicron | BA.2.75 | OQ215893.1 |
| Omicron | BA.2.75.2 | OQ346937.1 |
| Omicron | BA.4 | OQ333888.1 |
| Omicron | BA.4.6 | OQ349323.1 |
| Omicron | BA.5 | OQ343976.1 |
| Omicron | BA.5.2.6 | OQ346806.1 |
| Omicron | BF.11 | OQ347094.1 |
| Omicron | BF.7 | OQ346784.1 |
| Omicron | BN.1 | OQ346744.1 |
| Omicron | BQ.1 | OQ346454.1 |
| Omicron | BQ.1.1 | OQ346605.1 |
| Omicron | CH.1.1 | OQ346876.1 |
| Omicron | XBB | OQ347865.1 |
| Omicron | XBB.1.5 | XBB.1.5 is a sub-lineage of XBB with an additional spike RBD mutation S486P |
Table 2.
Sequences of hCoVs, SARS and MERS with their accession numbers.
| Nomenclature | Accession number |
|---|---|
| MERS-CoV | NC_019843 |
| SARS-CoV (Urbani) | AY278741.1 |
| HCoV-HKU1–genotype B | AY884001 |
| HCoV-OC43 | KF923903 |
| HCoV-NL63 | NC_005831 |
Table 3.
Coronaviruses infecting bats, pangolins and birds.
| Strain name | Accession number |
|---|---|
| Bat CoV RATG13 | MN996532.2 |
| Bat CoV ZXC21 | MG772934.1 |
| Bat CoV YN02 | MW201982.1 |
| Pangolin CoV GX-P2V | MT072864.1 |
| Pangolin CoV GX-P5E | MT040336.1 |
| Pangolin CoV GX-P5L | MT040335.1 |
| Pangolin CoV GX-P1E | MT040334.1 |
| Pangolin CoV GX-P4L | MT040333.1 |
| Pangolin CoV MP789 | MT121216.1 |
| Avian CoV Ind-TN92-03 | NC_048213.1 |
| Avian CoV DK/GD/27/2014 | NC_048214.1 |
| Avian CoV MG10 | NC_010800.1 |
Table 4.
CTL prediction tools and their prediction criteria.
| CTL Prediction tools | Prediction tool’s criteria |
|---|---|
| NetCTL-1.2 |
|
| VaxiJen 2.0 |
|
| IEDB MHC Class I immunogenicity |
|
| ToxinPred |
|
Table 5.
Prediction flow and criteria of conserved HTL epitopes.
| HTL Prediction tools | Prediction tool’s criteria |
|---|---|
| IEDB MHC-II | 1. Percentile rank: 20%, 15-mers. 2. Method: Consensus 2.22. 3. HLA Supertype: HLA-DR, HLA-DQ, HLA-DP. i. HLA-DR: • DRB1*01:01 • DRB1*07:01 • DRB1*09:01 • DRB3-01:01 • DRB4*01:01 ii. HLA-DQ: • DQA1*01:01/ DQB1*05:01 • DQA1*01:02/ DQB1*06:02 • DQA1*03:01/ DQB1*03:02 • DQA1*04:01/ DQB1*04:02 • DQA1*05:01/ DQB1*02:01 • DQA1*05:01/ DQB1*03:01 iii. HLA-DP: • DPA1*01/ DPB1*04:01 • DPA1*01:03/ DPB1*02:01 • DPA1*02:01/ DPB1*01:01 • DPA1*02:01/ DPB1*05:01 • DPA1*03:01/ DPB1*04:02 4. Exclude epitopes with percentile rank higher than 20.0 |
| IFNepitope | 1. Prediction approach: Motif and SVM hybrid. 2. Model for prediction: IFN-gamma versus Non IFN-gamma. 3. Exclude “NEGATIVE” epitopes. |
Table 6.
Prediction flow and criteria of conserved LBL epitopes.
| LBL Prediction tools | Prediction tool’s condition |
|---|---|
| ABCPred | Length of epitope: 16-mers Threshold: 0.51 and above Overlapping filter: ON |
| SVMTriP | Length of epitope: 16-mers Select epitopes with a score of 0.5 and above |
Table 7.
Final selected CTL epitopes.
| Epitopes | Number of coronavirus strains in which the epitope is found (out of 30) | Location in the S glycoprotein* | Assigned name |
|---|---|---|---|
| RVVVLSFEL | 25 | 509-517 | CTL1 |
| STQDLFLPF | 24 | 50-59 | CTL2 |
| WTAGAAAYY | 24 | 258-266 | CTL3 |
| YLQPRTFLL | 24 | 269-277 | CTL4 |
| QIITTDNTF | 24 | 1113-1121 | CTL5 |
| GAAAYYVGY | 24 | 261-269 | CTL6 |
| ITDAVDCAL | 24 | 284-293 | CTL7 |
| FTISVTTEI | 24 | 718-726 | CTL8 |
| FVFLVLLPL | 23 | 2-9 | CTL9 |
| QSYGFRPTY | 15 | 493-501 | CTL10 |
| SVLYNFAPF | 13 | 366-374 | CTL11 |
| YQPYRVVVL | 6 | 505-513 | CTL12 |
*Reference sequence: SARS-CoV-2-Wuhan-Hu-1 sequence.
Table 8.
Final selected LBL epitopes.
| Peptide sequence | Number of matched coronavirus strains | Location in S glycoprotein | Assigned Name |
|---|---|---|---|
| CVLGQSKRVDFCGKGY | 25 | 1045-1060 | LBL1 |
| DKYFKNHTSPDVDLGD | 25 | 1166-1181 | LBL2 |
| DEDDSEPVLKGVKLHY | 25 | 1270-1285 | LBL3 |
| AMQMAYRFNGIGVTQN | 25 | 899-914 | LBL4 |
| AGAALQIPFAMQMAYR | 25 | 903-918 | LBL5 |
| FAMQMAYRFNGIGVTQ | 25 | 911-926 | LBL6 |
| ASANLAATKMSECVLG | 24 | 1033-1048 | LBL7 |
| ATKMSECVLGQSKRVD | 24 | 1039-1054 | LBL8 |
| HGVVFLHVTYVPAQEK | 24 | 1071-1086 | LBL9 |
| HVTYVPAQEKNFTTAP | 24 | 1077-1092 | LBL10 |
| FVSGNCDVVIGIVNNT | 24 | 1134-1149 | LBL11 |
| VIGIVNNTVYDPLQPE | 24 | 1142-1157 | LBL12 |
| HTSPDVDLGDISGINA | 24 | 1172-1187 | LBL13 |
| LGDISGINASVVNIQK | 24 | 1179-1194 | LBL14 |
| GTTLDSKTQSLLIVNN | 24 | 120-135 | LBL15 |
| ESLIDLQELGKYEQYI | 24 | 1208-1223 | LBL16 |
| YVGYLQPRTFLLKYNE | 24 | 279-294 | LBL17 |
| NENGTITDAVDCALDP | 24 | 293-308 | LBL18 |
| AVDCALDPLSETKCTL | 24 | 301-316 | LBL19 |
| DPLSETKCTLKSFTVE | 24 | 307-322 | LBL20 |
| TVEKGIYQTSNFRVQP | 24 | 320-335 | LBL21 |
| VQPTESIVRFPNITNL | 24 | 333-348 | LBL22 |
| NDLCFTNVYADSFVIR | 24 | 388-403 | LBL23 |
| PTKLNDLCFTNVYADS | 24 | 397-412 | LBL24 |
| VVLSFELLHAPATVCG | 24 | 524-539 | LBL25 |
| FRSSVLHSTQDLFLPF | 24 | 56-71 | LBL26 |
| TDAVRDPQTLEILDIT | 24 | 586-601 | LBL27 |
| EILDITPCSFGGVSVI | 24 | 596-611 | LBL28 |
| GVSVITPGTNTSNQVA | 24 | 607-622 | LBL29 |
| HSTQDLFLPFFSNVTW | 24 | 62-77 | LBL30 |
| YSTGSNVFQTRAGCLI | 24 | 649-664 | LBL31 |
| TISVTTEILPVSMTKT | 24 | 732-747 | LBL32 |
| TECSNLLLQYGSFCTQ | 24 | 760-775 | LBL33 |
| RALTGIAVEQDKNTQE | 24 | 778-793 | LBL34 |
| AVEQDKNTQEVFAQVK | 24 | 784-799 | LBL35 |
| EMIAQYTSALLAGTIT | 24 | 881-896 | LBL36 |
| AGTITSGWTFGAGAAL | 24 | 892-907 | LBL37 |
| IGKIQDSLSSTASALG | 24 | 944-959 | LBL38 |
| FKCYGVSPTKLNDLCF | 24 | 374-389 | LBL39 |
| FVTQRNFYEPQIITTD | 23 | 1116-1131 | LBL40 |
| YEQYIKWPWYIWLGFI | 23 | 1219-1234 | LBL41 |
| PWYIWLGFIAGLIAIV | 23 | 1226-1241 | LBL42 |
| EPLVDLPIGINITRFQ | 23 | 237-252 | LBL43 |
| QTLLALHRSYLTPGDS | 23 | 239-254 | LBL44 |
| TRFQTLLALHRSYLTP | 23 | 249-264 | LBL45 |
| NQVAVLYQGVNCTEVP | 23 | 606-621 | LBL46 |
| YQGVNCTEVPVAIHAD | 23 | 612-627 | LBL47 |
| NNSIAIPTNFTISVTT | 23 | 722-737 | LBL48 |
| RDLICAQKFNGLTVLP | 23 | 860-875 | LBL49 |
| VFLVLLPLVSSQCVNL | 22 | 16-31 | LBL50 |
| TGTGVLTESNKKFLPF | 22 | 560-575 | LBL51 |
| NNSYECDIPIGAGICA | 22 | 670-685 | LBL52 |
| SQSIIAYTMSLGAENS | 22 | 702-717 | LBL53 |
| YTMSLGAENSVAYSNN | 22 | 708-723 | LBL54 |
| GDCLGDIAARDLICAQ | 22 | 851-866 | LBL55 |
| DIPIGAGICASYQTQT | 21 | 663-678 | LBL56 |
| PFLMDLEGKQGNFKNL | 20 | 187-202 | LBL57 |
| GWTAGAAAYYVGYLQP | 20 | 270-285 | LBL58 |
| HRSYLTPGDSSSGWTA | 19 | 258-273 | LBL59 |
| YGVGHQPYRVVVLSFE | 19 | 501-516 | LBL60 |
| SYQTQTKSHRRARSVA | 19 | 673-688 | LBL61 |
| TASALGKLQDVVNHNA | 19 | 941-956 | LBL62 |
| KQLSSKFGAISSVLND | 19 | 964-979 | LBL63 |
| PVLPFNDGVYFASTEK | 18 | 95-110 | LBL64 |
| PGQTGNIADYNYKLPD | 17 | 412-427 | LBL65 |
| RKSNLKPFERDISTEI | 17 | 470-485 | LBL66 |
| GSFCTQLKRALTGIAV | 17 | 757-772 | LBL67 |
| LQSYGFRPTYGVGHQP | 15 | 492-507 | LBL68 |
Table 9.
Final assembled epitopes.
| Combination of Peptide | Peptide Sequence | Peptide location* | Peptide Length | Matched HLA class-I Supertype | Matched HLA class-II Supertype | Assigned Name |
|---|---|---|---|---|---|---|
| CTL3+ CTL4+ CTL6+ CTL7+ HTL50+ HTL42+ HTL30+ HTL31+ HTL43+ LBL59+ LBL58+ LBL17 | SGWTAGAAAYYVGYLQPRTFLLKYNENGTITDAVDCALD | 256-294 (N-terminal domain) | 39 | A1, A2, A26, B8, B39, B58, B62 | HLA-DPA1*01/DPB1*04:01; HLA-DPA1*01:03/DPB1*02:01; HLA-DPA1*02:01/DPB1*01:01; HLA-DPA1*02:01/DPB1*05:01; HLA-DPA1*03:01/DPB1*04:02; HLA-DQA1*01:01/DQB1*05:01; HLA-DQA1*01:02/DQB1*06:02; HLA-DQA1*04:01/DQB1*04:02; HLA-DQA1*05:01/DQB1*02:01; HLA-DQA1*05:01/DQB1*03:01; HLA-DRB1*01:01; HLA-DRB1*07:01; HLA-DRB1*09:01 | Epi1 |
| CTL1+ CTL10+ HTL51+ HTL25+ HTL14+ HTL22+ HTL23+ HTL15+ HTL16+ HTL45+ LBL60+ LBL68 | LQSYGFQPTNGVGYQPYRVVVLSFELLHAPATVC | 492-525 (RBD) | 34 | A1, A2, A3, B7, B27, B58, B62 | HLA-DPA1*01/DPB1*04:01; HLA-DPA1*01:03/DPB1*02:01; HLA-DPA1*02:01/DPB1*01:01; HLA-DPA1*02:01/DPB1*05:01; HLA-DPA1*03:01/DPB1*04:02; HLA-DQA1*01:01/DQB1*05:01; HLA-DQA1*03:01/DQB1*03:02; HLA-DQA1*05:01/DQB1*02:01; HLA-DRB1*01:01; HLA-DRB1*07:01; HLA-DRB1*09:01; HLA-DRB4*01:01 | Epi2 |
* Reference sequence: SARS-CoV-2-Wuhan-Hu-1 sequence.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



