
Submitted:
03 September 2024
Posted:
03 September 2024
You are already at the latest version
Abstract
The microvessel neurovascular unit with its brain endothelial cell (BEC) and blood-brain barrier remodeling is important in the development of impaired cognition in sporadic or late-onset Alzheimer’s disease (LOAD) and vascular dementia or vascular cognitive impairment and dementia including cerebral amyloid angiopathy in neurodegeneration. LOAD is considered to be the number one cause of dementia globally; however, when one considers the role of mixed dementia (MD) (the combination of both the amyloid cascade hypothesis and the vascular hypothesis of LOAD), it becomes apparent that MD is the number one cause. The microvessel BECs are the first cells in the brain to be exposed to the peripheral neurotoxins of the systemic circulation and are therefore the brain cells at the highest risk for early and chronic injury. Therefore, it is not surprising that microvessel BECs are the first cells to undergo injury and the response to injury wound healing mechanism and remodeling that is excessive and recurrent in the aging process plus other age-related diseases such as cerebro-cardiovascular disease, hypertension, type 2 diabetes mellitus, Parkinson’s disease, plus others. This narrative review explores the intricate relationship between microvessel remodeling, cerebral small vessel disease (SVD), and neurodegeneration as occurs in LOAD. Further, it discusses the current understanding of how microvessel dysfunction, disruption, and pathology contribute to the pathogenesis of LOAD and thus, highlights potential avenues for therapeutic interventions.
Keywords:
Brain endothelial cells
; Blood-brain barrier
; Dementia
; Late-onset Alzheimer's disease
; Microvessel remodeling
; Neurodegeneration
; Neurovascular unit
; Small vessel disease
Introduction
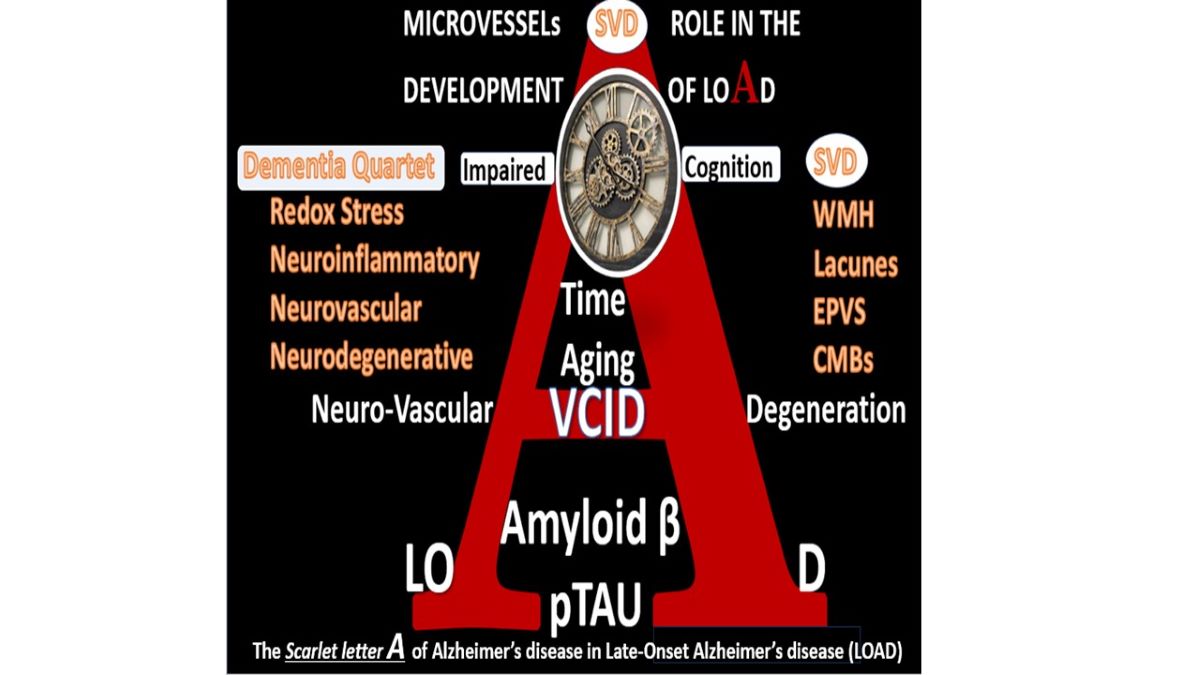
The role of macrovascular and microvascular remodeling in the development of neurodegeneration and dementia of sporadic, aging-related late-onset Alzheimer’s disease (LOAD) as compared to genetic-related early onset Alzheimer’s disease (EOAD) has undergone numerous historic pendulum swings since the early 1900s [1,2,3,4]. Numerous descriptive hypotheses regarding the causes of LOAD have been considered over the years, which include the aging-related hypothesis, the cholinergic hypothesis, amyloid cascade hypothesis, tau propagation hypothesis, mitochondrial cascade hypothesis, calcium homeostasis hypothesis, neurovascular hypothesis, inflammatory hypothesis, metal ion hypothesis, genetic/environmental hypothesis, vascular hypothesis and lymphatic system hypothesis (Box 1) [5,6,7].
Box.
Major existing and emerging hypotheses for late-onset Alzheimer’s disease (LOAD). In regard to number III. Aβ cascade hypothesis or the amyloid cascade hypothesis, the comment that Aβ may be necessary but not sufficient was introduced by Musiek and Holtzman [6]. Image provided with permission by CC 4.0 [6]. AD, Alzheimer’s disease; aMt, aberrant mitochondria; aMGCs, aberrant microglia cells; APOE-ε4, apolipoprotein E epsilon 4; BOLD, blood-oxygen-level-dependent imaging; CBF, cerebral blood flow; LOAD, late-onset Alzheimer’s disease; MGC, microglia cell; Mt, mitochondria; PET, positron emission tomography; ROS, reactive oxygen species; T1DM, type 1 diabetes mellitus
Box.
Major existing and emerging hypotheses for late-onset Alzheimer’s disease (LOAD). In regard to number III. Aβ cascade hypothesis or the amyloid cascade hypothesis, the comment that Aβ may be necessary but not sufficient was introduced by Musiek and Holtzman [6]. Image provided with permission by CC 4.0 [6]. AD, Alzheimer’s disease; aMt, aberrant mitochondria; aMGCs, aberrant microglia cells; APOE-ε4, apolipoprotein E epsilon 4; BOLD, blood-oxygen-level-dependent imaging; CBF, cerebral blood flow; LOAD, late-onset Alzheimer’s disease; MGC, microglia cell; Mt, mitochondria; PET, positron emission tomography; ROS, reactive oxygen species; T1DM, type 1 diabetes mellitus

Initially and for many decades, the aging and vascular hypothesis held strong; however, once the amyloid cascade hypothesis was discovered in 1991 the pendulum rather quickly swung to this hypothesis as the most likely leading cause for LOAD [8,9]. Additionally, Swerdlow and Khan importantly proposed a mitochondrial cascade hypothesis with redox stress as a leading cause for sporadic Alzheimer’s disease or LOAD in 2004 [10]. Notably the amyloid cascade hypothesis may be somewhat flawed, in that, amyloid beta (Aβ) by itself may be necessary but not sufficient to be the primary cause for the development of LOAD [11].
Just as many things in life occur, the ol adage that ‘what goes around comes around’ has taken hold and now the current terminology regarding vascular microvessel remodeling is quite active in the literature and is referred to as the vascular contributions to cognitive impairment and dementia (VCID). VCID encompasses all types of cerebrovascular cardiovascular disease-related conditions associated with impaired cognition and cognitive decline.
Microvessel cerebral small vessel disease (SVD) is considered to be the most important vascular contributor to cognitive decline and dementia [12]. Importantly, Jacob et al., were able to share that SVD baseline severity and progression were independently associated with an increase in risk of all-cause dementia over a follow-up of 14 years with white matter hyperintensities (WMH) progression predicting incident all-cause dementia [12].
Because of the proven ability to prevent and treat cardiovascular disease including hypertension, the NIH has recently designated VCID as a critical focus of research [13]. Moreover, the recent MarkVCID Biomarker Development and Validation Consortium has been designed to better predict, study, and diagnose SVD in the brain and its role in VCID. The National Institutes of Health (NIH) has launched MarkVCID, which is a national consortium designed to accelerate the development of new and existing biomarkers for small vessel VCID. The overall goal of the consortium is to deliver high-quality biomarkers ready for use in clinical trials aimed at generating scientific breakthroughs in our understanding and treatment of VCID [13].
Brain Microvessels may be defined as those arterioles, precapillary arterioles, capillaries, postcapillary venules and venules that are ≤ 500 μm that may have only one-two layers VSMC in their media or just a single layer of pericyte(s) (Pcs) (Figure 1) [1,2].
Thus, they are structurally designed to function both as resistance and transport microvessels, which allow for the regulation of hydrodynamics and the exchange of nutrients and waste products, oxygen and carbon monoxide, and water within the central nervous system (CNS) [1,2,3,4,5,6,7,8,9,10,11,12,13,14].
Box 2.
Comparing the four major identifiable structures of cerebral small vessel disease (SVD) by MRI observations. Lacunes (a footprint of stroke); 2. enlarged perivascular spaces (EPVS) (a biomarker of glymphatic system pathway dysfunction (GLY Dys)); 3. white matter hyperintensities (WMH) (footprint of ischemia); 4. cerebral microbleeds (CMBs) (biomarkers of SVD/stroke with hemorrhage or ischemic infarct); 5. recent small subcortical infarcts (MRI representative findings of recent infarction similar to lacune parameters but with greater flair suggesting recent occurrence, not presented in this figure). The location of CMBs has further clinical importance in that lobar/cortical CMBs are CAA-related and deep, basal, infratentorial CMBs are hypertension-related. Modified Figure image provided with permission by CC 4.0 [1ShulyWhy]. CAA, cerebral amyloid angiopathy; mm, millimeter; MRI, magnetic resonance imaging.
Box 2.
Comparing the four major identifiable structures of cerebral small vessel disease (SVD) by MRI observations. Lacunes (a footprint of stroke); 2. enlarged perivascular spaces (EPVS) (a biomarker of glymphatic system pathway dysfunction (GLY Dys)); 3. white matter hyperintensities (WMH) (footprint of ischemia); 4. cerebral microbleeds (CMBs) (biomarkers of SVD/stroke with hemorrhage or ischemic infarct); 5. recent small subcortical infarcts (MRI representative findings of recent infarction similar to lacune parameters but with greater flair suggesting recent occurrence, not presented in this figure). The location of CMBs has further clinical importance in that lobar/cortical CMBs are CAA-related and deep, basal, infratentorial CMBs are hypertension-related. Modified Figure image provided with permission by CC 4.0 [1ShulyWhy]. CAA, cerebral amyloid angiopathy; mm, millimeter; MRI, magnetic resonance imaging.
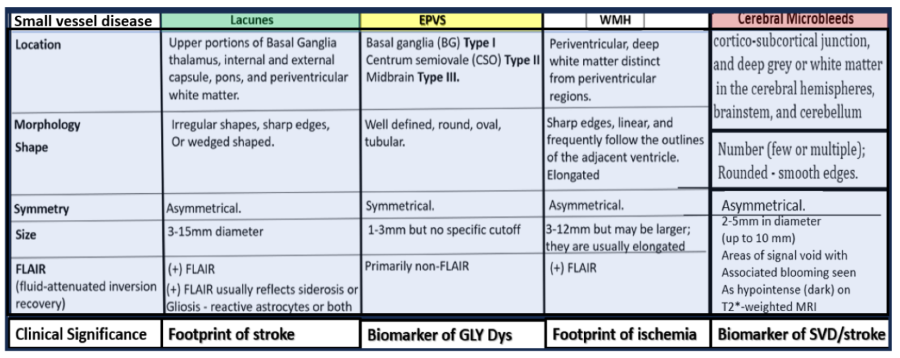
Microvessel SVD refers to a variety of structural and functional changes involving small perforating arterioles, capillaries, and venules of the brain [14]. Further, these SVDs are broken down into at least 4 main categories such as WMH, lacunes, enlarged perivascular spaces (EPVS), and cerebral microbleeds (CMBs) (Box 2) [1,14].
Indeed, SVD is a group of microvessel diseases that affect small arteries, precapillary arterioles, and postcapillary venules in the brain, and can manifest as lacunes, EPVS, WMH, CMBs capable of promoting the development of neurodegeneration [14,15].
It is important to note here that when utilizing TEM microscopy, one cannot visually observe how the protoplasmic perivascular astrocytes endfeet (pvACef) connect with regional neurons to create neurovascular unit (NVU) coupling. The NVU (Figure 2) consists of the BEC that line the vascular lumen, the pericytes that are formed within the basement membrane (BM), which supports the BEC and contributes to the synthesis and maintenance of the tight and adherens junctions (TJ/AJ), junctional adhesion molecules (JAMs) and vascular endothelial cadherin(s) (VE-Cads or cluster of differentiation CD144) responsible for creating the blood-brain barrier (BBB), and the adjacent protoplasmic perivascular astrocyte endfeet that adheres tightly to the combined BEC and pericyte BM that communicates via astrocyte cytoplasmic processes to regional neurons, neuronal dendritic synapses, and terminal axon synapses that form a tripartite synapse (Figure 2) [16,17,18,19,20].
Notebly, pvACs with their endfeet are the master connecting cells of the CNS [3, 16,
While this narrative focuses on microvessel remodeling, it is important to briefly discuss the role of macrovessel remodeling in both the extracranial and intracranial macrovessels (≥ 5 μm with at least 2 layers of vascular smooth muscle cell(s) (VSMCs) within their media). Macrovessels are capable of undergoing primarily atherosclerotic and arteriolosclerotic remodeling that result in the development of stroke prone regions of involvement that result in primarily decreased cerebral blood flow (CBF) ischemic strokes via not only decreased CBF but also thromboembolic phenomenon to result in ischemic stroke, which is the number one cause for stroke and impaired remodeling to result in increased redox stress, neuroinflammation, neurodegeneration and impaired cognition of vascular dementia (VaD) or VCID [22,23,24].
Just as LOAD is a multifactorial and heterogenous aging-related neurological disease [23,25], neurodegeneration (ND) is also a complicated multifactorial process due to the slow development of CNS neuronal atrophy and loss as a result of programmed cell death that is an important final step in the development of dementia [26]. In this narrative we will focus on the ND causation due to misfolded proteins, which include the accumulation of toxic oligomers of extracellular Aβ (CNS Aβ (1-42)) as interstitial senile or Aβ plaques (LOAD) and primarily Aβ 1-40 within precapillary arterioles in the interstitial and perivascular adventitia basement membranes and extracellular matrix (ECM) associated cerebral amyloid angiopathy (CAA)). While not a focus, it is important to note that intracellular misfolded tau proteins form misfolded neurofibrillary tangles (NFTs) due to aberrant hyperphosphorylation, which promote neuronal dysfunction, impaired cognition, and neurodegeneration [5].
Notably, Iturria-Medina et al. (2016) have stated based upon multifactorial data-driven analysis that vascular dysregulation and microvessel remodeling might be the earliest/strongest brain pathologic factor associated with LOAD development that is followed by Aβ deposition, misfolded tau formation with neurofibrillary tangles, glucose metabolism dysregulation, functional impairment, neuronal apoptosis, and grey matter atrophy [3].
Dementia is an umbrella more generalized term, which consists of four types of dementia including i) Alzheimer’s - LOAD dementia, ii) VaD, co-occurring or mixed dementias (MD), frontal temporal dementia, and Lewy body dementia [23]. Herein, the discussion will primarily focus on SVD, VaD/VCID, and Alzheimers disease or LOAD. Indeed, sporadic and age-related neurodegenerative disorders such as LOAD are the consequence of aging-associated multifactorial biological dysfunction with a heterogenous background [3,4,27,28].
When studying chronic age-related diseases such as co-occurring VaD with LOAD, which includes both arterial macrovessel disease and microvessel disease (SVD), it is important to understand structural remodeling [29]. Microvessel remodeling as a result of chronic peripherally-derived neurotoxic metabolic alterations as it relates to concurrent remodeling of arterioles, precapillary arterioles, the true capillary, the postcapillary venules and veins in development of vascular remodeling and small vessel disease and neurodegeneration is very complicated. Thus, it seems appropriate to pay homage to the microvessels and their component cells consisting of BECs, Pcs, and connecting astrocyte end feet that result in the formation of the NVU of the brain to control CBF providing nourishment and delivery of oxygen to neuronal cells for proper function.
The widely accepted current hypothesis of neurodegeneration in LOAD has previously been widely centered around abnormal misfolded amyloid and tau protein aggregation has been undergoing some changes recently and more are now pointing to the importance of the macro- and microvascular contributions to neurodegeneration [14,15,22,23,24,29,30,31,32,33]. Recently, emerging evidence suggests that endothelial dysfunction is an early and primary event in AD pathogenesis that may precede abnormal protein aggregation and directly contribute to neurodegeneration and synaptic injury. Importantly, microvessel SVD has been increasingly recognized as a significant contributor to the development of neurodegeneration via Microvessel SVD dysfunction and damage [14]; WMH Changes and Neurodegeneration [34Rizvi]; Capillary NVU Blood-Brain Barrier dysfunction [35]; Inflammation and Neurodegeneration [23]; Cerebral Microbleeds and Neurodegeneration [36].
Neurovascular Hypothesis: Microvessel Remodeling and Cerebral Small Vessel Disease (SVD) Contributions to Neurodegeneration
Cerebral microvessel remodeling may be simply defined as a structural rearranging and an adaptive process that occurs in a long-term, chronic response to changes in hemodynamic conditions or the effects of injurious neurotoxic stimuli as in the response to injury wound healing mechanism [29]
2.Neurovascular Unit Brain Endothelial Cells and Brain Endothelial Cell Activation and
BECs are the first cells to be exposed to multiple injurious species (neurotoxins) from the peripheral systemic circulation and are the first cells in the brain to undergo functional and structural remodeling (Figure 3) [29,37,39].
Box 3.
10 major transmission electron micrograph (TEM) aberrant remodeling changes associated with brain endothelial cell activation and dysfunction (BECact/dys) to better understand the various brain endothelial cell response to injury wound healing phenotypes. Figure image reproduced with permission by CC 4.0 [1,40].
Box 3.
10 major transmission electron micrograph (TEM) aberrant remodeling changes associated with brain endothelial cell activation and dysfunction (BECact/dys) to better understand the various brain endothelial cell response to injury wound healing phenotypes. Figure image reproduced with permission by CC 4.0 [1,40].

Over the past decade author has been able to discern at least 10 major transmission electron microscopic (TEM) ultrastructural remodeling changes microvessel brain endothelial cell activation and dysfunction (BECact/dys) phenotypes (Box 3) [1,40].
BECact/dys and blood-brain barrier dysfunction and disruption (BBBdd) with increased permeability either simultaneously co-occurs or occurs immediately following BECact/dys (Figure 4) [2,39,40,41].
2.Neurovascular Unit Pericyte (Pc) Remodeling
Brain NVU Pcs are known to interact with BECs, basal lamina (basement membrane), and glial cells and play a critical supportive role in synthesis and maintenance of BBB-TJ/AJ providing stability, local control of regional capillary blood flow via NVU coupling via ACs, angiogenesis, and immune responses since the are known to be antigen presenting cells [44,45,46,47]. Brain NVU Pcs are indeed mural vascular supportive cells of (BBB) and are partially responsible for synthesis and maintenance of the BBB TJ/AJ formation that undergo degeneration in the neurodegenerative disease LOAD that is characterized by early neurovascular dysfunction and uncoupling that associate with the elevation of amyloid β-peptide (Aβ), tau pathology and neuronal apoptosis and loss. These changes lead to progressive cognitive impairment, neurodegeneration, and dementia [46]. BEC and Pc interactions and connections are crucial for proper NVU function and maintain CBF to regional active neurons to result in NVU coupling along with the necessary function of NVU protoplasmic perivascular astrocyte endfeet (pvACef) (Box 4) [44,45,46,47,48,49,50,51,52,53,54,55].
Box 4.
Pericyte and Brain Endothelial Cell Protein Interactions and Interdependent Biomarkers and Signaling Proteins. Image modified with permission by CC 4.0 [44,45]. a -SMA, alpha-smooth muscle actin; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; LDL-C R, low-density lipoprotein-C receptor; NG2, neuroglia 2; PDGF-B, platelet-derived growth factor-B; PDGF-B (R), receptor for PDGF-B, VEGF A (Pc positive (+), B (BEC +), vascular endothelial growth factor A, B; vWf, von Willebrand factor.
Box 4.
Pericyte and Brain Endothelial Cell Protein Interactions and Interdependent Biomarkers and Signaling Proteins. Image modified with permission by CC 4.0 [44,45]. a -SMA, alpha-smooth muscle actin; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; LDL-C R, low-density lipoprotein-C receptor; NG2, neuroglia 2; PDGF-B, platelet-derived growth factor-B; PDGF-B (R), receptor for PDGF-B, VEGF A (Pc positive (+), B (BEC +), vascular endothelial growth factor A, B; vWf, von Willebrand factor.

Brain Pcs are known to be able to integrate, coordinate, and process signals from their neighboring cells of the NVU to generate diverse functional responses that are critical for CNS homeostatic functions in health and pathologic remodeling and dysfunction in disease. These responses in health include regulation of the blood-brain barrier permeability, angiogenesis, clearance of toxic metabolites, capillary hemodynamic responses (vasodilation and contraction), neuroinflammation (Pc and BEC both are capable are antigen presenting cells (APCs), and also have the potential to become mesenchymal stem cell due to their pleiotropic potential [44,45,46,47,48,49,50,51,52,53,54,55,56]. Further, brain NVU pericytes not only provide structural support of the NVU BECs but are also perfectly positioned within the NVU to facilitate peripheral systemic metainflammation that may result in BECact/dys and BBBdd (Fig 7) [44].
Figure 7.
Brain Pericyte(s) (Pcs) and Brain Endothelial cell(s) (BECs) are closely interconnected and are interdependent on one another for paracrine signaling making the Pc a critical cell for proper homeostatic function of the neurovascular unit(s) (NVU) role in the control of regional cerebral blood flow (CBF). Panel A demonstrates the normal arrangement of the BEC, Pc, and perivascular astrocyte endfeet (pvACef). Note how the basement membrane (black open arrows) is interspersed and separates the luminal BEC, Pc, and the more abluminal pvACef. Also, note the interrogating microglia cell (iMGC) (white closed arrow). Panel B illustrates with better clarity the close interactions between the Pc and the BEC (EC), which importantly allows for paracrine signaling as well as structural and functional support by the NVUs Pc of the BEC. Panel C illustrates the paracrine signaling of nitric oxide (NO) synthesized by the eNOS enzyme to signal the Pc cell to relax to allow for vasodilation and the supportive connection by platelet derived growth factor beta (PDGFβ) of the EC to interact with the PDGFβ receptor (PDGFβ R) of the adjacent Pc. Images provided with permission by CC 4.0 [44,45]. ACef, astrocyte endfeet; AQP4, aquaporin 4; PcP, N, nucleus; pericyte processes-endfeet-foot processe; TJ/AJ, tight and adherens junctions; VEGF A-B, vascular endothelial growth factor A-B; VEGF R, receptor for VEGF. .
Figure 7.
Brain Pericyte(s) (Pcs) and Brain Endothelial cell(s) (BECs) are closely interconnected and are interdependent on one another for paracrine signaling making the Pc a critical cell for proper homeostatic function of the neurovascular unit(s) (NVU) role in the control of regional cerebral blood flow (CBF). Panel A demonstrates the normal arrangement of the BEC, Pc, and perivascular astrocyte endfeet (pvACef). Note how the basement membrane (black open arrows) is interspersed and separates the luminal BEC, Pc, and the more abluminal pvACef. Also, note the interrogating microglia cell (iMGC) (white closed arrow). Panel B illustrates with better clarity the close interactions between the Pc and the BEC (EC), which importantly allows for paracrine signaling as well as structural and functional support by the NVUs Pc of the BEC. Panel C illustrates the paracrine signaling of nitric oxide (NO) synthesized by the eNOS enzyme to signal the Pc cell to relax to allow for vasodilation and the supportive connection by platelet derived growth factor beta (PDGFβ) of the EC to interact with the PDGFβ receptor (PDGFβ R) of the adjacent Pc. Images provided with permission by CC 4.0 [44,45]. ACef, astrocyte endfeet; AQP4, aquaporin 4; PcP, N, nucleus; pericyte processes-endfeet-foot processe; TJ/AJ, tight and adherens junctions; VEGF A-B, vascular endothelial growth factor A-B; VEGF R, receptor for VEGF. .
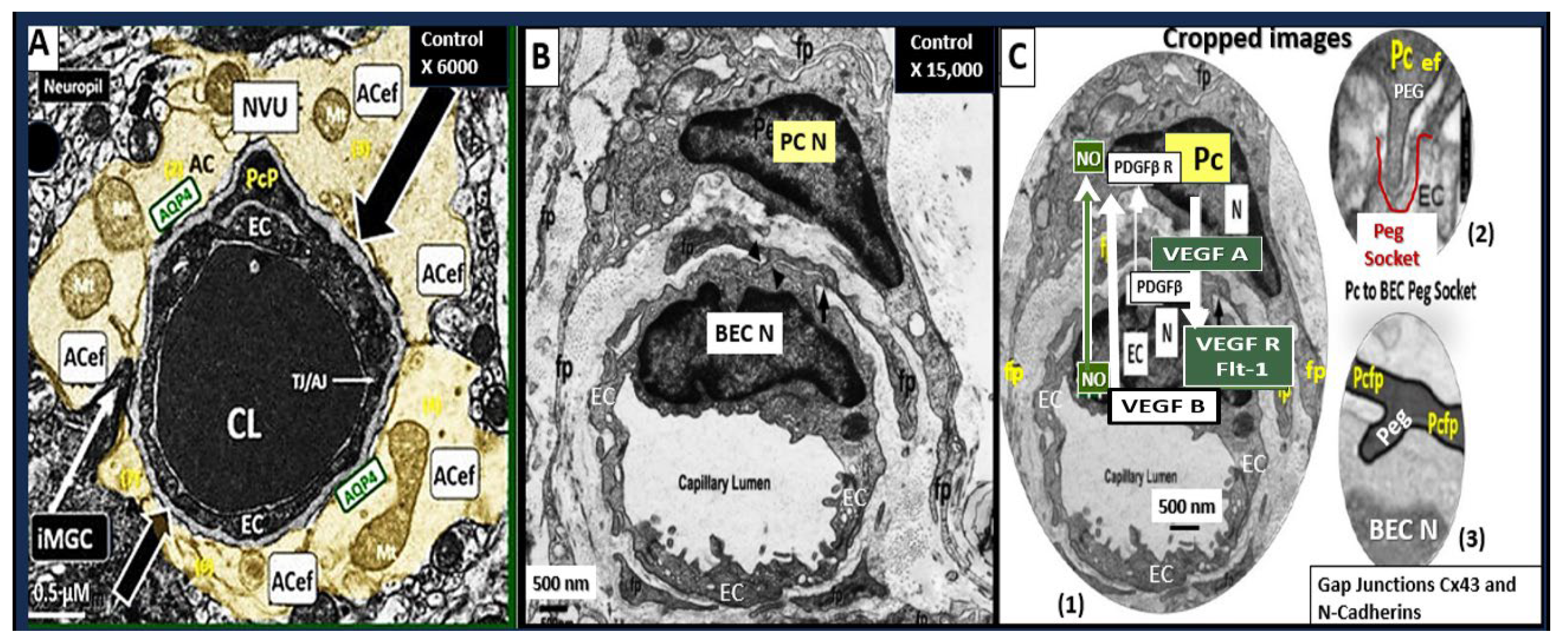
Notebly, vascular endothelial growth factor A (VEGFA) is secreted by Pcs and signals BEC VEGFR1 or Flt-1 to result in BEC migration, proliferation, remodeling of its structure to result in angiogenesis [57]. In contrast, VEGFB is secreted by BECs and has a potent survival/anti-apoptotic effect, while lacking a general angiogenic activity [58].
NVU Pc’s are know to be very sensitive to oxidative-redox stress in addition to pCC due to leaky BECact/dys and BBBdd [46,50,51,58]. Pc sensitivity to redox stress and neuroinflammation contributes to aberrant structural remodeling, dysfunction, degenerative remodeling changes and loss per apoptosis, formation of ghost cells and contribute to NVU dysfunction with NVU uncoupling and decreased regional CBF with ensuing hypometabolism, hypoxia and neurodegeneration (Figure 8) [6,41,59,60,61].
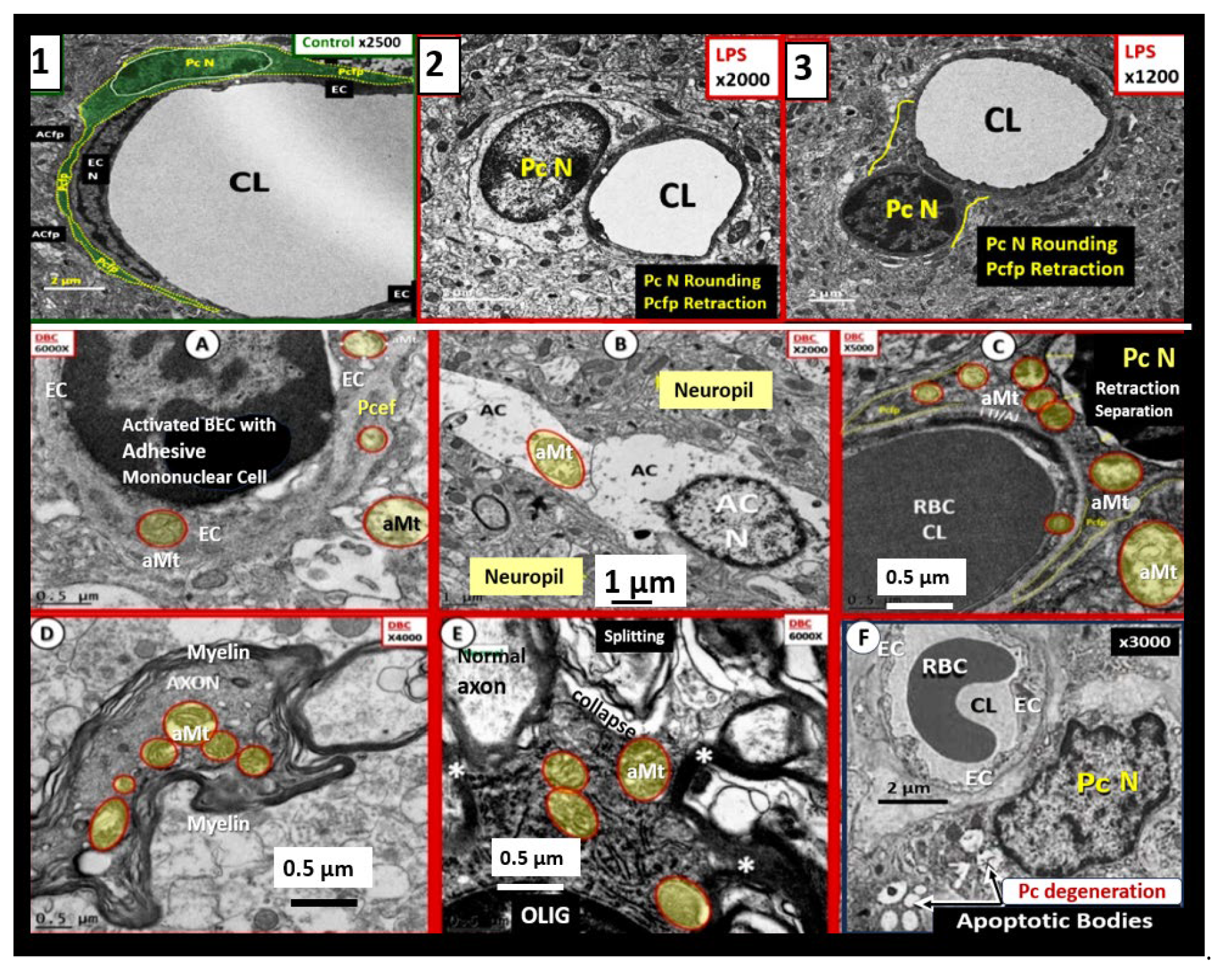
Figure 7.
Pericyte endfeet (Pcef) retraction and pericyte nucleus (Pc N) rounding are early signs of Pc dysfunction along with the formation of aberrant mitochondria (aMt) that are found in brain endothelial cells (ECs), Pcs, astrocyte(s) (ACs), neuronal axons, and oligodendrocytes (OLIG) along with Pc cytoplasmic apoptotic changes with apoptotic bodies. Panels 1-3 illustrate Pc N rounding and retraction of Pcef in panels 2 and 3 as compared to control model Pc N elongation and elongation of Pcef (pseudo-colored green) in panel Panels A-E depict aberrant mitochondria (aMt) in BECs, ACs, Pcef, neuronal axons, and oligodendrocytes (OLIG) respectively. Panel F depicts an apoptotic Pc with numerous apoptotic bodies observed in this Pc cytoplasm (arrows) that are indicative of Pc degeneration and death. Scale bars vary and are included in images presented. Modified images provided with permission by CC 4.0 [59,60,61]. BEC, brain endothelial cell; CL, capillary lumen; N, nucleus; RBC, red blood cell.
Figure 7.
Pericyte endfeet (Pcef) retraction and pericyte nucleus (Pc N) rounding are early signs of Pc dysfunction along with the formation of aberrant mitochondria (aMt) that are found in brain endothelial cells (ECs), Pcs, astrocyte(s) (ACs), neuronal axons, and oligodendrocytes (OLIG) along with Pc cytoplasmic apoptotic changes with apoptotic bodies. Panels 1-3 illustrate Pc N rounding and retraction of Pcef in panels 2 and 3 as compared to control model Pc N elongation and elongation of Pcef (pseudo-colored green) in panel Panels A-E depict aberrant mitochondria (aMt) in BECs, ACs, Pcef, neuronal axons, and oligodendrocytes (OLIG) respectively. Panel F depicts an apoptotic Pc with numerous apoptotic bodies observed in this Pc cytoplasm (arrows) that are indicative of Pc degeneration and death. Scale bars vary and are included in images presented. Modified images provided with permission by CC 4.0 [59,60,61]. BEC, brain endothelial cell; CL, capillary lumen; N, nucleus; RBC, red blood cell.
Aβ (1-42) extracellular-interstitial plaques and toxic oligomers result in BECact/dys with increased BEC-derived ROS (primarily from membraneous NADPH oxidases (Nox1) and mitochondrial ROS with subsequent Pc activation, dysfunction, and or degeneration due to redox stress provided by the BECact/dys with increased local paracrine signaling to adjacent Pcs via the powerful BEC-derived vasoconstrictor endothelin 1 (ET-1) as a result of redox stress and ROS (Figure 9) [62].
Additionally, BEC-derived ET-1 signaling results in Pc vasoconstriction and ensuing decreased regional NVU decreased CBF with hypometabolism, hypoperfusion, and ischemia/hypoxia that are associated with degenerative neurites as observed in figure 9 that are indicative of neurodegeneration and impaired cognition [62].
Pc’s independently, and Pc-BEC interactions play critical roles in maintaining normal vascular (NVU coupling) function [63]. Pcs are mural cells in the brain and this provides a unique location for BEC/Pc interactions with paracrine signaling in cerebral small vessels (arteries, arterioles, precapillary arterioles, capillaries, postcapillary venules, and veins) of NVU that also allow them to occupy nearly 80% coverage of the abluminal side of BECs [64]. Pc’s also act as an intermediary cell sandwiched between the luminal BECs and pvACef within a shared BM and more luminal to the abluminal parenchymal neuropil that allow maintenance of the NVU BBB by regulating endothelial activity such as TJ/AJ formation and transcytosis, in addition to guiding the astrocytic endfeet polarization and attachment to the outer BM, which leads to a great segway into the following section regarding the NVU pvACef [65,66].
2.Brief Overview of Neurovascular Unit (NVU) Reactive Perivascular Astrocyte(s) (rpvACs) and Reactive Microglia cells (rMGCs) that Contribute to Neurodegeneration (ND)
Glial astrocyte(s) (ACs) are considered to be one of the most abundant cells within the CNS and via their innate connecting and communicating functions and they play an important and essential role in maintaining homeostasis and structural support as also depicted in previous figure 2 (Figure 10) [16].
ACs are also known to be master communicating/connecting cells in the brain and connect the NVU to regional neurons and synapses to provide homeostatic coupling, provision for synapse formation, maintenace, and support via their cradling effect in addition to their pruning effects along with MGCs as in previous figure 2 (Figure 11). [16,17,18,19,20,21].
Additionally, ACs are a major supplier of energy to regional neurons in the form of glucose and lactate since they are able to store glycogen and can undergo glycolysis. Additionally, ACs are a major source of antioxidant reserve via glutathione and superoxide dismutase. Also, they have the capability to provide necessary growth factors such as brain-derived growth factor (BDGF), transforming growth factor-beta (TGF-β), nerve growth fact (NGF), insulin-like growth factor 1 (IGF-1), basic fibroblast growth factor (bFGF), transforming growth factors alpha (TGF)-α and TGF-β, brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF); all of which have been shown to exert neuroprotection [67,68,69,70].
While this narrative is focused primarily on NVU BEC microvessel remodeling and their contribution to SVD, the reactive pvACef (rpvACef) and rMGCs remodeling are also known to be associated with NVU uncoupling (Figure 12).
In addition to the rpvACs and rMGCs as in figure 12 there is also another proinflammatory cell that is now known to reside within the perivascular spaces of the perivascular unit when BECact/dys and BBBdd occur. This cell is the reactive perivascular macrophage (rPVMΦ) that has been identified within the perivascular spaces of microvessels in neuroinflammatory-induced lipopolysaccharide (LPS) treated models (Figure 13) [45,59,71,72].
NVU uncoupling is associated with decreased CBF with hypoperfusion, hypometabolism, hypoxia, and ischemia to regional neurons resulting in impaired cognition and neurodegeneration. Further, aberrant cerebrovascular reactivity is associated with NVU uncoupling, which is a common and prominent feature in the human brain during the early stages of the aging mild cognitive impairment (MCI)-LOAD spectrum [73].
rACs are capable of multipotential remodeling phenotypes in LOAD. Individuals with LOAD are exposed to multiple injurious stimuli such as ischemic and or hemorrhagic stroke, which result in rACs, rMGCs, central nervous system cytokines/chemokines (cnsC/C), ROS, and activation of matrix metalloproteinases 2, 9 (MMP 2, 9) [29]. Importantly a protective astrogliosis scar formation with proximal palisading and more distal hypertrophic rACs develops following this injury to the brain [29]. Additionally, it is known that Aβ oligomers and Aβ extracellular plaques are neurotoxic and are capable of inducing a rAC atrophic phenotype that is capable of causing synaptic connection disruptions, imbalance of neurotransmitter homeostasis and even neuronal death via increased excitotoxicity [74]. Neurotoxic Aβ oligomers and plaques induce primarily rACs with an atrophic phenotype in contrast to the palisading and hypertrophic rACs in glia scar formation (Figure 14) [74].
Importantly, it has been shown that the soluble Aβ oligomers are more important in the induction of neurotoxicity than the histopathologic mature footprint of Aβ plaques [77]. Indeed, Zlokovic has illustrated in a schematic the involvement of NVU BBB loss of integrity with impaired clearance of Aβ soluble oligomers that result in their accumulation and the increase in the early phase of neurodegeneration in LOAD development that associate with the importance of neurotoxic effects of soluble Aβ accumulation [78]. Thus, this schematic emphasizes the neurotoxicity of both reduced CBF and the accumulated soluble Aβ oligomers as they negatively affect synaptic impairment and promote neuronal loss due to apoptosis with neurodegeneration and impaired cognition.
Notably, Verkhratsky and Butt have pointed to the importance that human studies may not always conform and translate from rodent model studies, in that, human ACs in the neocortical regions are much larger and their cytoplasmic processes extend much further as compared to rodent models [17].
Neural Oxidative – Redox Stress (OxRS) Including: ROS, Reactive Oxygen, Nitrogen, Sulfur Species (RONSS), Iron Sulfur Clusters (ISCs) of the Reactive Species Interactome (RSI)
LOAD and/or MD may be defined by the accumulation of two types of insoluble misfolded, fibrous proteins, i.e., extracellular amyloid-β peptide deposited in senile Aβ plaques, and intracellular NFTs composed primarily of abnormal and hyperphosphorylated tau protein. In addition to ECM Aβ plaques and intracellular NFT (hyperphosphorylated tau), LOAD may be further pathologically characterized by regional neuronal death and atrophy that is associated with dysfunctional synapses or their loss of synaptic connections [1,2,3,4,79]
Redox homeostasis describes the normal physiologic process of reduction and oxidation in order to re-pair unstable, damaging, reduced ROS, which will include the following oxygen free radical’s: superoxide, (O2●−); (hydrogen peroxide (H2O2); hydroxyl radical (●OH), singlet oxygen (1O2) and reactive nitric oxide (•NO) along with organic analogues which include reactive nitrogen species (RNS) primarily peroxynitrite (ONOO-) [80,81,82]. Oxidative Stress implies a loss of redox homeostasis (imbalance) with an excess of ROS by the singular process of oxidation. Both redox and oxidative stress may be associated with an impairment of antioxidant defensive capacity as well as an overproduction of ROS. Oxidative redox stress (OxRS) implies a loss of this unique homeostasis resulting in an excess production of ROS either through the process of oxidation or reduction. Also, reactive iron and sulfur clusters (ISCs) are important regarding OxRS including ROS, RONSS, and the entire reactive species interactome in the brain and are generated by mitochondria as ISCs and mitochondria ROS (mtROS) (Figure 15) [83,84,85].
Notably, OxRS is known to result in dysfunctional and damaged proteins, lipids, and nucleic acids (RNA and DNA) that result in multiple biomarkers of OxRS in the brain as oxidatively modified proteins such as protein carbonyls and 3-nitrotyrosine; lipids such as 4-hydroxynonenal (HNE); nucleic acids such as 8-hydroxyguanine (8OHG) [86].
Each of the component cells of the microvascular NVU (BECs, Pcs, pvACef, and MGCs) are capable of generating large amounts of ROS and proinflammatory cytokines/chemokines (cnsC/C) when they become activated as in BECact/dys, or reactive as in rPcs, rACs, or rMGCs, due to various injurious stimuli with the formation of aMt (see figure 8A-E and figure 15 wherein aMt generate not only ROS but also redox reactive prooxidative iron and sulfur species (ISCs) and increased NOX activity [29,37]. Additionally, ROS and/or OxRS are known to form a vicious cycle with neuroinflammation and ROS begets ROS (Figure 16) [29,37,87].
Neuroinflammation and Neurodegeneration
Since OxRS/oxidative stress is involved in a vicious cycle with neuroinflammation as in figure 16 that contributes to neurodegeneration, this cycle can lead to sustained inflammation that can be detrimental long after the initial trigger and re-signal the neuroinflammation-OxRS vicious cycle that not only contributes to microvessel SVD but also neurodegeneration and impaired cognition as in LOAD (Figure 17) [37].
Neuroinflammation is one of the central features of LOAD and pro-inflammatory CNS cytokine/chemokine cnsCC signaling plays multiple roles in neurodegeneration. Further, neuroinflammation may be characterized by multiple cnsCCs that not only participate with OxRS in vicious cycles and hits as in figures 16 and 17 but also are capable of inducing neurodegeneration singularly (Figure 18) [88,89].
Indeed, chronic CNS neuroinflammation induces neurodegeneration.
Neurodegeneration
LOAD and/or MD (LOAD plus VaD) are the leading causes of sporadic dementia in the aging population (23, 91, 92). Individuals with LOAD/MD experience clinical symptoms of cognitive impairments, memory loss (especially recent memory loss, and behavioral changes that interfere with activities of daily living [93,94]. The dementia in LOAD/MD is associated with neurodegeneration that is characterized initially by synaptic injury, dysfunction and/or loss [95,96] followed by neuronal loss [97]. Further, these findings are also, accompanied by rACs and astrogliosis [98], rMGC proliferation [99,100], and the presence of neurofibrillary tangles composed of dystrophic neurites and hyperphosphorylated tau [95,101,102,103,104,105].
Multiple defective cellular processes such as aMt remodeling as in figure 8A-E with dysfunctional leakage of mtROS, increased OxRS from all cells of the NVU (BECs, Pcs, and reactive ACs and MGCs and neurons, lysosomal dysfunction with decreased disposal of Aβ aggregates and oligomers, and increased inflammation (cnsCC) with subsequent associated apoptotic loss and neurodegeneration as in figures 8F and 18 [92].
In regards to neurodegeneration and neuronal death there are four major cellular death pathways including necrosis, apoptosis, excitatory, and autophagic. In MD there would be at least two different neuronal cell death pathways namely apoptosis in LOAD and ischemic necrosis in VaD [106].
Mitochondria are an important neuronal organelle producing reactive oxygen species (ROS) in addition to plasma membraneous NADPH oxidase (Nox1) in LOAD [107,108]. According to Xie et al., leaky aMt could be considered the central pawns in the development of neurodegeneration and loss via neuronal apoptosis and LOAD as illustrated in figure 8E-F. [108]. Increased and accelerated accumulation of Aβ plaque aggregation and deposition within the CNS ECM plaques in LOAD associate with increased soluble Aβ oligomers as in figure 8 and 9A. Indeed, these findings are result from an imbalance in Aβ production, aggregation and impaired clearance [92] that associate with the development of SVD and identification of EPVS by MRI. Aβ clearance is mediated by proteolytic enzymes such as neprilysin [109], chaperone molecules such as ApoE in health [110] and lysosomal autophagy [111], and non-lysosomal pathways via the proteasome [112] in health, which is impaired in LOAD/MD. It is currently thought that soluble Aβ oligomers rather than the fibrils within the Aβ plaques are responsible for the neurotoxic effect on synapses and neurons associated with neurodegeneration [77,113,114].
It is important to note here that SVD is capable of promoting and contributing to the development of neurodegeneration (Figure 19) [14,15].
Further, Crews and Masliah have put for the following sequences for neurogenic development with impaired cognition that is observed in LOAD as follows. Neurotoxic effects of Aβ plaque and its oligomers result in signaling alteration in neuronal neurodegenerative kinases including Fyn, FAK, GSK3β, and CDK5. These alterations result in alterations in cytoskeletal and synaptic proteins resulting in tau-derived neurofibrillary tangles (NFTs). aMt result in leaky neurotoxic mtROS; neuronal oxidative/redox stress; impaired lysosomal uptake and proteolysis and are associated withcnsCCs, which proceed to neurodegeneration consisting of synaptic injury and dysfunction; defects in neurogenesis that lead to impaired cognition LOAD along with neuronal apoptosis and loss with cerebral atrophy [92].
Conclusions
While dementia is a more generalized term that describes the loss of the ability to think, remember, and reason to levels that interfere with daily life and activities, it can then be broken down to its specific causes or disease. These specific diseases consist of four types of dementia including i) Alzheimer’s – LOAD-type dementia, ii) VaD or co-occurring LOAD and VaD resulting in mixed dementias) (MDs), iii) frontal temporal dementia, and iv) Lewy body dementia [23].
Specifically, Dr. Alois Alzheimer described a 55-year-old female individual with dementia and this is typically now referred to as EOAD [123,124,125] in contrast to the global pandemic form of LOAD that has been the main focus of this narrative review. Thus, LOAD is expected to continue to grow nearly exponentially as the growing global population continues to age and experience longer life cycles especially, in the global post-world-war II baby boom generation for the next 1.5 decades with serious costs to the nations and suffering individuals and their family and professional care providers and long-term care facilities [6].
Importantly, VCID incorporates multiple cardio-cerebrovascular diseases the include cardiac disease, micro and macrovessel disease both within the CNS and extracranial locations such as micro-infarcts/hemorrhages and CMBs, which may be silent; TIAs with brief clinical episodes, small vessel ischemic and hemorrhagic stroke; CAA; macrovessel ischemic and hemorrhagic stroke; cardiac disease (arrhythmia’s such as atrial fibrillation with thromboembolic stroke, decreased cardiac output such as myocardial infarction and congestive heart failure, and cardiac surgeries that are associated with decreased CBF and chronic cerebral hypoxia (CCH) to name a few). This places VCID centrally in the overlapping Venn diagrams (Figure 21) [126].
The original amyloid cascade hypothesis has garnered the interest of most researchers, clinicians, and pharmaceutical companies during the past 2 decades [8,9]. Zlokovic initially proposed both a 2-hit hypothesis [23], and a vasculo-neuronal-inflammatory triad model of neurodegenerative disorders in 2011 [127], which contributes to the development of neurodegeneration as found in LOAD and MD. Herein, author currently proposes an ‘quartet of mechanisms’ that are importantly involved in the development of ND and MD in aging individuals that additionally emphasizes redox stress in addition to neurovascular mechanisms and include: i) oxidative redox stress (OxRS), ii) neuroinflammation, iii) neurovascular, and iv) neurodegenerative mechanisms in the development of LOAD and MD as proposed when figures 17, 19, and 21 are combined (Figure 22).
Recently, Ter Telgte et al., [128] have shown that there is a penumbra effect that may result in diffusion defects that associate with SVD phenotypes as observed on MRI images that is comparable to the penumbra effect associated with acute stroke and remodeling [129,130]. The post stroke penumbra is the viable tissue around the irreversibly damaged ischemic core as in figure 14C [130]. Further, they found that these focal effects were capable of spreading to become global effects at remote more distal regions within the CNS from the subcortical white matter regions to the cortical grey matter regions. As a result of their findings, it is now thought that the structural integrity may become dysfunctional and disrupted. This could interrupt the brains network informational highway that is responsible for the information transference and processing of information transfer, which could result in impaired cognition. This suggests that what we observe on MRI studies may only be the tip of the iceberg in regards to the loss of function, impaired cognition, and the aberrant motor skills that may be affected in a concurrent manner as depicted in figures 21 and 22 in the development of neurodegeneration, LOAD, VaD, and MD [128].
In summary, when studying chronic age-related diseases such as co-occurring VaD (including both arterial macrovessel and microvessel disease SVD), VaD, LOAD, and MD it is important to understand microvessel SVD structural remodeling as a result of chronic peripherally-derived neurotoxic multiple injurious species as it relates to concurrent remodeling of arterioles, precapillary arterioles, the true capillary, the postcapillary venules and veins in development of vascular remodeling and microvessel SVD and neurodegeneration. Thus, it seems appropriate to pay homage to the microvessel SVD and specifically the component NVU cells in SVD. Since the development of both microvessel SVD and LOAD are multifactorial it is felt that it will require a multifactorial approach in order to better understand this association.
The initiation of the microvessel SVD remodeling and neurodegeneration seems to continuously come back and point to the importance of the early development of BECact/dys and BBBdd. The combination of increased BEC inflammatory signaling with the production of cnsCC, decreased eNOS function with eNOS uncoupling, decreased NO, increased OxRS provided by increased BEC NOX production and mt ROS leakage and eNOS uncoupling, along with the brain endothelial-mediated neurotoxicity proposed by Grammas [131] as in figure 2 contribute to decreased CBF and neurodegeneration [55].
This narrative review has examined the background of microvessel SVD development and not only its association to VaD but also LOAD that combines to develop mixed dementia in the introduction. Next the role of microvascular NVU and each of its constituent cells especially, the BECact/dys and BBBdd in section This allows leakage of peripheral neurotoxins into the CNS to result in not only the entry of pCC into the CNS but also the subsequent activation of cnsCC promoting a state of neural OxRS in section 3. This contributes to ongoing neuroinflammation in section 4 and its relationship to neurodegeneration discussed in section 5.
Future Directions
SVD not only contributes to the development of VAD but also contributes to the pathogenesis of LOAD through various mechanisms as discussed throughout this narrative review via the quartet of i). Neurovascular, ii). OxRS, iii). Neuroinflammation, and iv). Neurodegenerative mechanisms. The developmental and pathobiological mechanisms of microvessel SVD and LOAD are both multifactorial and therefore the approach to treatment and future directions will most likely require a multifactorial and multifunctional treatment approach (Figure 23) [127,132,133].
Funding
Author has not received grants from any funding agency in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
The tissues provided for the representative electron microscopic images utilized in this manuscript were all approved in advance by the University of Missouri Institutional Animal Care and Use Committee (No. 190). The animals were cared for in accordance with National Institutes of Health guidelines and by the Institutional Animal Care and Use Committees at the Harry S. Truman Memorial Veterans Hospital and the University of Missouri, Columbia, MO, USA, which conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data and materials can be provided upon reasonable request.
Acknowledgments
The author would like to acknowledge Tatyana Shulyatnikova for the contribution of many artistic illustrations and editing of this manuscript. The author would also like to acknowledge DeAna Grant Research Specialist of the Electron Microscopy Core Facility at the Roy Blunt NextGen Precision Health Research Center, University of Missouri, Columbia, Missouri. The author also acknowledges the kind support of the William A. Banks Lab and Michelle Erickson at the VA Medical Center, Seattle, Washington.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
Aβ, amyloid beta; AC, astrocyte; ACef, astrocyte endfeet; BBB, blood–brain barrier; BEC(s), brain endothelial cell(s); BECact/dys, brain endothelial cell activation/dysfunction; BBBdd, blood-brain barrier dysfunction disruption; BDGF, brain-derived growth factor; bFGF, basic fibroblast growth factor; BM, basement membrane; CAA, cerebral amyloid angiopathy; CBF, cerebral blood flow; CL, capillary lumen; CMB(s), cerebral microbleeds; CNS, central nervous system; cnsCC, central nervous cytokines chemokines; ELOAD, early-onset Alzheimer’s disease; EPVS, enlarged perivascular spaces; ET-1, endothelin 1; GDNF, glia cell-derived growth factor; GS, glymphatic space; ISC, iron sulfur clusters; JAMs, junctional adhesion molecules; LOAD, late-onset Alzheimer’s disease; MCI, mild or minimal cognitive impairment; MD(s), mixed dementias; MGCs, microglia cells; MMP-2,-9, matrix metalloproteinase-2,-9; MRI, magnetic resonance imaging; mtROS, mitochondrial ROS; NADPH Ox, nicotine adenine diphosphate reduced oxidase; ND, neurodegeneration; NGF, nerve growth factor; NIH, National Institute of Health; NGTs, neurofibrillary tangles; NO, nitric oxide; NOX, NADPH oxidase, NVU, neurovascular unit; OxRS, oxidative redox stress; Pc, pericyte; pCC, peripheral cytokines chemokines; Pcfp, pericyte foot process; pvACfp/ef, perivascular astrocyte foot processes/endfeet; PVS, perivascular spaces; PVS/EPVS, perivascular space/enlarged perivascular space; ROS, reactive oxygen species, RONS, reactive oxygen, nitrogen species; RONSS, reactive oxygen, nitrogen, sulfur species; rPVACfp/ef, reactive perivascular astrocyte foot processes/endfeet; rPVMΦ, reactive resident perivascular macrophages; RSI, reactive species interactome; SVD, cerebral small vessel disease; TEM, transmission electron microscopy; TGFβ, transforming growth factor beta; TI/AJs, tight and adherens junctions; TNFα, tumor necrosis factor alpha; VaD, vascular dementia; VCAM-1, vascular cell adhesion molecule-1; VCID, vascular contributions to cognitive impairment and dementia; VEGF A or B, vascular endothelial growth factor A or B; VSMC(s), vascular smooth muscle cells; WMH, white matter hyperintensities.
References
- Shulyatnikova T, Hayden MR. Why Are Perivascular Spaces Important? Medicina (Kaunas). 2023, 59, 917. [Google Scholar] [CrossRef]
- Hayden, MR. A Closer Look at the Perivascular Unit in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Biomedicines. 2024, 12, 96. [Google Scholar] [CrossRef]
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC. Alzheimer's Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nature Commun. 2016, 7, 11934. [Google Scholar] [CrossRef]
- Iadecola, C. The Pathobiology of Vascular Dementia. Neuron. 2013, 80, 844–866. [Google Scholar] [CrossRef]
- Liu PP, Xie Y, Meng XY, JS. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther. Signal Transduct Target Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Hayden, MR. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef]
- Musiek ES, Holtzman DM. Three Dimensions of the Amyloid Hypothesis: Time, Space, and “Wingmen”. Nat Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991, 6, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses. 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Musiek ES, Holtzman DM. Three Dimensions of the Amyloid Hypothesis: Time, Space, and “Wingmen”. Nat Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Jacob MA, Cai M, van de Donk V, Bergkamp M, Marques J, Norris DG, Kessels RPC, et al. Cerebral Small Vessel Disease Progression and the Risk of Dementia: A 14-Year Follow-Up Study. Am J Psychiatry. 2023, 180, 508–518. [Google Scholar] [CrossRef] [PubMed]
- NIH: National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/ last visited on July 18, 2024.
- Wardlaw JM, Smith C, Dichgans M. Small vessel disease: Mechanisms and clinical implications. Lancet Neurol. 2019, 18, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Paoletti FP, Simoni S, Parnetti L, Gaetani L. The Contribution of Small Vessel Disease to Neurodegeneration: Focus on Alzheimer's Disease, Parkinson's Disease and Multiple Sclerosis. I J Mol Sci. 2021, 22, 4958. [Google Scholar] [CrossRef]
- Hayden, MR. Protoplasmic Perivascular Astrocytes Play a Crucial Role in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Neuroglia. 2023, 4, 307–328. [Google Scholar] [CrossRef]
- Verkhratsky A, Butt AM. Neuroglia: Function and Pathology, 1st ed.; Academic Press: London, UK, 2023. [Google Scholar]
- Verkhratsky A, Nedergaard M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130595. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Verkhratsky A, Parpura V, LI B, Sucder C. Astrocytes: The housekeepers of the CNS. Adv Neurobiol. 2021, 26, 21–53. [Google Scholar] [CrossRef]
- Hayden, MR. Hypothesis: Astrocyte Foot Processes Detachment from the Neurovascular Unit in Female Diabetic Mice May Impair Modulation of Information Processing—Six Degrees of Separation. Brain Sci. 2019, 9, 83. [Google Scholar] [CrossRef]
- de la Torre, JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002, 33, 1152–62. [Google Scholar] [CrossRef]
- Zlokovic, BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, Grossman M, Trojanowski JQ. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer's Coordinating Centre. Brain 2013, 136, 2697–2706. [Google Scholar] [CrossRef]
- Avelar-Pereira B, Belloy ME, O'Hara R, Hadi Hosseini SM. Decoding the heterogeneity of Alzheimer's disease diagnosis and progression using multilayer networks. Mol Psychiatry. 2023, 28, 2423–2432. [Google Scholar] [CrossRef]
- Jellinger, A. Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med. 2010, 14, 457–487. [Google Scholar] [CrossRef]
- Iturria-Medina Y, Hachinski V, Evans AC. The vascular facet of late-onset Alzheimer's disease: an essential factor in a complex multifactorial disorder. Curr Opin Neurol. 2017, 30, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction-The disregarded partner of Alzheimer's disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef]
- Hayden, MR. Brain Injury: Response to Injury Wound-Healing Mechanisms and Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Medicina (Kaunas). 2023, 59, 1337. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C. Alzheimer disease as a vascular disorder: Nosological 9. de la Torre J.C. Cerebromicrovascular pathology in Alzheimer’s disease compared to normal aging. Gerontology. 1997, 43, 26–43. [Google Scholar] [CrossRef]
- O’Brien, J.T. , Markus H. S. Vascular risk factors and Alzheimer’s disease. BMC Med. 2014, 12, 218. [Google Scholar] [CrossRef]
- Grammas, P. Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 26. [Google Scholar] [CrossRef]
- de la Torre, J.C. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet. Neurol. 2004, 3, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Rizvi B, Lao PJ, Chesebro AG, Dworkin JD, Amarante E, Beato JM, et al. Association of Regional White Matter Hyperintensities With Longitudinal Alzheimer-Like Pattern of Neurodegeneration in Older Adults. JAMA Netw Open. 2021, 4, e2125166. [Google Scholar] [CrossRef] [PubMed]
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhen Zhao Z, et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron. 2015, 85, 296–302. [Google Scholar] [CrossRef]
- Gyanwali B, Shaik MA, Tan CS, Vrooman H, Venketasubramanian N, Chen C, Hilal S. Mixed-location cerebral microbleeds as a biomarker of neurodegeneration in a memory clinic population. Aging (Albany NY). 2019, 11, 36. 10581–10596. [Google Scholar] [CrossRef]
- Hayden, MR. Cerebral Microbleeds Associate with Brain Endothelial Cell Activation-Dysfunction and Blood–Brain Barrier Dysfunction/Disruption with Increased Risk of Hemorrhagic and Ischemic Stroke. Biomedicines. 2024, 12, 1463. [Google Scholar] [CrossRef] [PubMed]
- Han, F. Cerebral microvascular dysfunction and neurodegeneration in dementia. Stroke Vasc Neurol. 2019, 4, 105–107. [Google Scholar] [CrossRef]
- Hayden, MR. Brain Endothelial Cells Play a Central Role in the Development of Enlarged Perivascular Spaces in the Metabolic Syndrome. Medicina. 2023, 59, 1124. [Google Scholar] [CrossRef]
- Hayden, MR. Brain endothelial cell activation and dysfunction associate with and contribute to the development of enlarged perivascular spaces and cerebral small vessel disease. Histol Histopathol 2024, 10, 18792. [Google Scholar] [CrossRef]
- Hayden MR, Grant DG, Aroor AR, DeMarco VG. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia. 2018, 1, 220–244. [Google Scholar] [CrossRef]
- Hayden, MR. The Brain Endothelial Cell Glycocalyx Plays a Crucial Role in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Life (Basel). 2023, 13, 1955. [Google Scholar] [CrossRef]
- Salameh TS, Shah GN, Price TO, Hayden MR, Banks WA. Blood-Brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate. J Pharmacol Exp Ther. 2016, 359, 452–459. [CrossRef] [PubMed]
- Hayden MR, Yang Y, Habibi J, Bagree SV, Sowers JR. Pericytopathy: oxidative stress and impaired cellular longevity in the pancreas and skeletal muscle in metabolic syndrome and type 2 diabetes. Oxid Med Cell Longev. 2010, 3, 290–303. [Google Scholar] [CrossRef]
- Hayden, MR. Pericytes and Perivascular Macrophages Play a Key Role in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome and Type 2 Diabetes Mellitus. J Alzheimers & Neurodegenerative Diseases. 2023, 9, 062. [Google Scholar] [CrossRef]
- Winkler EA, Sagare AP, Zlokovic BV. The Pericyte: A Forgotten Cell Type with Important Implications for Alzheimer's Disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef]
- Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat Neurosci. 2016, 19, 771–783. [CrossRef] [PubMed]
- Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells Tissues Organs. 2001, 169, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T. What is a pericyte? J Cereb Blood Flow Metab. 2016, 36, 451–455. [Google Scholar] [CrossRef]
- Uemura Mt, Maki T, Ihara M, Lee VMY, Trojanowski JQ. Brain Microvascular Pericytes in Vascular Cognitive Impairment and Dementia. 2020, 12, 80. [Google Scholar] [CrossRef]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O'Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Cai W, Liu H, Zhao J, Chen LY, Chen J, Zhengqi Lu, Hu X. Pericytes in brain injury and repair after ischemic stroke. Transl Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Korte N, Nortley R, Attwell D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer's disease. Acta Neuropathol. 2020, 140, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy P, Katychev A, Wang X, Van Buren E. CNS microvascular pericytes exhibit multipotential stem cell activity. J Cereb Blood Flow Metab. 2006, 26(5), 613–624. [Google Scholar] [CrossRef]
- Darden J, Payne LB, Zhao H, Chappell JC. Excess Vascular Endothelial Growth Factor-A Disrupts Pericyte Recruitment during Blood Vessel Formation. Angiogenesis. 2019, 22, 167–183. [Google Scholar] [CrossRef]
- Li X, Lee C, Tang Z, Zhang F, Arjunan P, Li Y, Hou X, Kumar A, Dong L. VEGF-B: a survival, or an angiogenic factor? Cell Adh Migr. 2009, 3, 322–7. [Google Scholar] [CrossRef]
- Erickson MA, Shulyatnikova T, Banks WA. Hayden MR. Ultrastructural Remodeling of the Blood-Brain Barrier and Neurovascular Unit by Lipopolysaccharide-Induced Neuroinflammation. Int J Mol Sci. 2023, 24, 1640. [Google Scholar] [CrossRef]
- Hayden MR, Grant DG, Aroor AR, DeMarco VG. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model–Part II: Microglia and Mitochondria. Neuroglia. 2018, 1, 311–326. [Google Scholar] [CrossRef]
- Hayden, MR. The Mighty Mitochondria Are Unifying Organelles and Metabolic Hubs in Multiple Organs of Obesity, Insulin Resistance, Metabolic Syndrome, and Type 2 Diabetes: An Observational Ultrastructure Study. 2022, 23, 4820. [Google Scholar] [CrossRef]
- Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, et al. Amyloid β oligomers constrict human capillaries in Alzheimer's disease via signaling to pericytes. Science. 2019, 365, eaav9518. [Google Scholar] [CrossRef]
- Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef]
- Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer's disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Chen S, Guo D, Zhu Y, Xiao S, Xie J, Zhang Z, et al. Amyloid β oligomer induces cerebral vasculopathy via pericyte-mediated endothelial dysfunction. Alzheimers Res Ther. 2024, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Duenas M, Luquin S, Chowen JA, Torres-Aleman I, Naftolin F, Garcia-Segura LM. Gonadal hormone regulation of insulin-like growth factor-I-like immunoreactivity in hypothalamic astroglia of developing and adult rats. Neuroendocrinology. 1994, 59, 528–538. [Google Scholar] [CrossRef]
- Buchanan CD, Mahesh VB, Brann DW. Estrogen-astrocyte-luteinizing hormone-releasing hormone signaling: a role for transforming growth factor-beta (1). Biol Reprod. 2000, 62, 1710–1721. [Google Scholar] [CrossRef]
- Flores C, Salmaso N, Cain S, Rodaros D, Stewart J. Ovariectomy of adult rats leads to increased expression of astrocytic basic fibroblast growth factor in the ventral tegmental area and in dopaminergic projection regions of the entorhinal and prefrontal cortex. J Neurosci. 1999, 19, 8665–8673. [Google Scholar] [CrossRef]
- Karki P, Smith K, Johnson J Jr. , Lee E. Astrocyte-derived growth factors and estrogen neuroprotection: Role of transforming growth factor-α in estrogen-induced upregulation of glutamate transporters in astrocytes. Mol Cell Endocrinol. 2014, 389, 58–64. [Google Scholar] [CrossRef]
- Owens T, Bechmann I, Engelhardt B. Perivascular Spaces and the Two Steps to Neuroinflammation. Journal of Neuropathology & Experimental Neurology. 2008, 67, 1113–1121. [Google Scholar] [CrossRef]
- Trolli F, Cipollini V, Moci M, Morena E, Palotai M, Rinaldi V, et al. Perivascular Unit: This Must Be the Place. The Anatomical Crossroad Between the Immune Vascular and Nervous System. Front Neuroanat. 2020, 14, 17. [Google Scholar] [CrossRef]
- Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer's disease. Nat Rev Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Verkratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes in Alzheimer's Disease. Neurotherapeutics. 2010, 7, 399–412. [Google Scholar] [CrossRef]
- Walker, LC. Aβ plaques. Free Neuropathol. 2020, 1, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Shulyatnikova T, Verkhratsky A. Astroglia in Sepsis Associated Encephalopathy. Neurochem Res. 2020, 45, 83–99. [CrossRef] [PubMed]
- Tolar M, Hey J, Power A, Abushakra S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int J Mol Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Zlokovic, BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008, 57, 178–201. [Google Scholar] [CrossRef]
- Rabinovici, GD. Late-onset Alzheimer Disease. Continuum (Minneap Minn) 2019, 25, 14–33. [Google Scholar] [CrossRef]
- Hayden MR, Tyagi SC. Intimal redox stress: accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc Diabetol. 2002, 17, 3. [Google Scholar] [CrossRef]
- Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer's disease brain: New insights from redox proteomics. European Journal of Pharmacology. 2006, 545, 39–50. [Google Scholar] [CrossRef]
- Sbodio JI, Snyder SH, Bindu DP. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid Redox Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef]
- Shi R, Hou W, Wang ZQ, Xu X. Biogenesis of Iron–Sulfur Clusters and Their Role in DNA Metabolism. Front Cell Dev Biol. 2021, 9, 735678. [Google Scholar] [CrossRef]
- Isaya, G. Mitochondrial iron-sulfur cluster dysfunction in neurodegenerative disease. Front Pharmacol. 2014, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Selvanathan A, Sankaran BP. Mitochondrial iron-sulfur cluster biogenesis and neurological disorders. Mitochondrion. 2022, 62, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer's disease brain: New insights from redox proteomics. Eur J Pharmacol. 2006, 545, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Hayden, MR. Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease. Medicina (Kaunas). 2023, 59, 561. [Google Scholar] [CrossRef] [PubMed]
- Kiraly M, Foss JF, Giordano T. Neuroinflammation, Its Role in Alzheimer’s Disease and Therapeutic strategies. J Prev Alzheimer’s Dis. 2023, 10, 686–698. [Google Scholar] [CrossRef]
- Thakur S, Dhapola R, Sarma P, Medhi B, Reddy DH. Neuroinflammation in Alzheimer's Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation. 2023, 46, 1–17. [Google Scholar] [CrossRef]
- Walker, LC. Aβ Plaques. Free Neuropathol. 2020, 1, 1–31. [Google Scholar] [CrossRef]
- Ashford, J.W. APOE genotype effects on Alzheimer's disease onset and epidemiology. J. Mol. Neurosci. 2004, 23, 157–165. [Google Scholar] [CrossRef]
- Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer's disease-. Hum Mol Genet. 2010, 19, R12–20. [Google Scholar] [CrossRef]
- Katzman, R. Alzheimer's disease. N. Engl. J. Med. 1986, 314, 964–973. [Google Scholar] [CrossRef]
- Budson AE, Price BH. Memory dysfunction. N. Engl. J. Med. 2005, 352, 692–699. [CrossRef]
- Masliah E, Mallory M, Alford M, DeTeresa R, Iwai A, Saitoh T. Molecular mechanisms of synaptic disconnection in Alzheimer's disease. In: Hyman B, Duyckaerts C, Christen Y, editors. Connections, Cognition and Alzheimer's Disease. Berlin: Springer; 1997. pp. 121–140.
- DeKosky S, Scheff S. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Terry, R. , Peck A., DeTeresa R., Schechter R., Horoupian D. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann. Neurol. 1981, 10, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Beach T, Walker R, McGeer E. Patterns of gliosis in Alzheimer's disease and aging cerebrum. Glia. 1989, 2, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Rogers J, Luber-Narod J, Styren S, Civin W. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol. Aging. 1988, 9, 339–349. [Google Scholar] [CrossRef]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer disease. Acta Neuropathol. 1991, 83, 12–20. [Google Scholar] [CrossRef]
- Trojanowski JQ, Lee VM. ‘Fatal attractions’ of proteins. A comprehensive hypothetical mechanism underlying Alzheimer's disease and other neurodegenerative disorders. Ann. N. Y. Acad. Sci. 2000, 924, 62–67. [Google Scholar] [CrossRef]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Iqbal K, Grundke-Iqbal I. Neurofibrillary pathology leads to synaptic loss and not the other way around in Alzheimer disease. J. Alzheimers Dis. 2002, 4, 235–238. [Google Scholar] [CrossRef]
- Mandelkow EM, Mandelkow E. Tau in Alzheimer's disease. Trends Cell. Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Crews L, Rockenstein E, Masliah E. APP transgenic modeling of Alzheimer's disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct Funct. 2010, 214, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Gorman, AM. Neuronal cell death in neurodegenerative diseases: recurring themes around protein handling. J Cell Mol Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef]
- Padurariu M, Ciobica A, Lefter R, Serban IL, Stefanescu C, Chirita R. “The oxidative stress hypothesis in Alzheimer’s disease,”. Psychiatria Danubina. 2013, 25, 401–409. [Google Scholar]
- Xie A, Gao J, Xu L, Meng D. Shared Mechanisms of Neurodegeneration in Alzheimer’s Disease and Parkinson’s Disease. Biomed Res Int. 2014, 2014, 648740. [Google Scholar] [CrossRef]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee H J, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science. 2001, 292, 1550–1552. [Google Scholar] [CrossRef]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Bendiske J, Bahr BA. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis—an approach for slowing Alzheimer disease? J Neuropathol. Exp. Neurol. 2003, 62, 451–463. [Google Scholar] [CrossRef]
- Marambaud P, Zhao H, Davies P. Resveratrol promotes clearance of Alzheimer's disease amyloid-beta peptides. J. Biol. Chem. 2005, 280, 37377–37382. [Google Scholar] [CrossRef]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept. Lett. 2004, 11, 213–228. [Google Scholar] [CrossRef]
- Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef] [PubMed]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack Jr CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Jack CR Jr, Petersen RC, Xu YC, Waring SC, O'Brien PC, Tangalos EG, Smith GE, Ivnik RJ, Kokmen E. Medial temporal atrophy on MRI in normal aging and very mild Alzheimer's disease. Neurology. 1997, 49, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Banks WA, Hayden MR. Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. Int J Mol Sci. 2021, 22, 5427. [Google Scholar] [CrossRef]
- Ramos-Rodriguez JJ, Ortiz O, Jimenez-Palomares M, Kay KR, Berrocoso E, Murillo-Carretero MI, et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology. 2013, 38, 2462–2475. [Google Scholar] [CrossRef]
- Meftah S, Gan J. Alzheimer’s disease as a synaptopathy: Evidence for dysfunction of synapses during disease progression. Front Synaptic Neurosci. 2023, 15, 1129036. [Google Scholar] [CrossRef]
- Selkoe, DJ. Alzheimer's disease is a synaptic failure. Science. 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Jackson J, Jambrina E, Li J, Marston H, Menzies F, Phillips K, et al. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef]
- Schirinzi T, Canevelli M, Suppa A, Bologna M, Marsili L. The continuum between neurodegeneration, brain plasticity, and movement: A critical appraisal. Rev. Neurosci. 2020, 31, 723–742. [Google Scholar] [CrossRef]
- Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer's 1907 paper, "Uber eine eigenartige Erkankung der Hirnrinde".
- Clin Anat. 1995, 8, 429-431. [CrossRef]
- Alzheimer A, Förstl H, Levy R. On certain peculiar diseases of old age. Hist Psychiatry. 1991, 2, 71–101. [Google Scholar] [CrossRef]
- Alzheimer, A. A contribution concerning the pathological anatomy of mental disturbances in old age. Alzheimer Dis Assoc Disord. 1991, 5, 69–70. [Google Scholar]
- Corriveau RA, Bosetti F, Emr M, Gladman JT, Koenig JI, Moy CS, et al. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): A Framework for Advancing Research Priorities in the Cerebrovascular Biology of Cognitive Decline. Cell Mol Neurobiol. 2016, 36, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic BV, Griffin JH. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci. 2011, 34, 198–209. [Google Scholar] [CrossRef]
- Ter Telgte A, van Leijsen EMC, Wiegertjes K, Klijn CJM, Tuladhar AM, de Leeuw FE. Cerebral small vessel disease: from a focal to a global perspective. Nat Rev Neurol. 2018, 14, 387–398. [Google Scholar] [CrossRef]
- Fischer M, Garcia JH. The Ischemic Penumbra: Identification, Evolution and Treatment Concepts. Cerebrovascular diseases. 2004, 17, 1–6. [Google Scholar] [CrossRef]
- Liu S, Levine SR, Winn HR. Targeting ischemic penumbra: part I - from pathophysiology to therapeutic strategy. Exp Stroke Transl Med. 2010, 3, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Grammas P, Moore P, Weigel PH. Microvessels from Alzheimer’s Disease Brains Kill Neurons in Vitro. Am J Pathol. 1999, 154, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Markus HS, de Leeuw FE. Cerebral small vessel disease: Recent advances and future directions. Int J Stroke. 2023, 18, 4–14. [Google Scholar] [CrossRef]
- Kim HW, Hong J, Jeon JC. Cerebral Small Vessel Disease and Alzheimer's Disease: A Review. Front Neurol. 2020, 11, 927. [Google Scholar] [CrossRef]
Figure 1.
Three transition electron microscopy (TEM) cross section images of microvessels from various animal models from layer III in the frontal cortex at various magnifications that represent microvessels. These images are in contrast to those macrovessels that have a diameter of ≥ 5 μm with at least 2 layers of vascular smooth muscle cell (s) (VSMCs) within their media. Magnification 3μm, 0.5 μm, 5 μm (far-left, middle, and far-right respectively). Images provided with permission by CC 4.0 [1,2]. AC, astrocytes pseudo-colored gold in; far-left and pseudo-colored blue far-right images. AQP-4, aquaporin 4; AC, perivascular astrocyte; AC1, AC2, astrocyte endfeet numbers 1 and 2 ACef, perivascular astrocyte endfeet; CL, capillary lumen; EC, brain endothelial cell; gs, glymphatic space; lys, lysosome; Mt, mitochondria; N, nucleus; NVU, neurovascular unit; Pc, pericyte; PcN, pericyte nucleus; Pcp, pericyte endfeet processes; PVS, perivascular space; rMGC, interrogating or reactive microglia; rMФ, reactive macrophage; TJ/AJ, tight junctions/adherens junctions.
Figure 1.
Three transition electron microscopy (TEM) cross section images of microvessels from various animal models from layer III in the frontal cortex at various magnifications that represent microvessels. These images are in contrast to those macrovessels that have a diameter of ≥ 5 μm with at least 2 layers of vascular smooth muscle cell (s) (VSMCs) within their media. Magnification 3μm, 0.5 μm, 5 μm (far-left, middle, and far-right respectively). Images provided with permission by CC 4.0 [1,2]. AC, astrocytes pseudo-colored gold in; far-left and pseudo-colored blue far-right images. AQP-4, aquaporin 4; AC, perivascular astrocyte; AC1, AC2, astrocyte endfeet numbers 1 and 2 ACef, perivascular astrocyte endfeet; CL, capillary lumen; EC, brain endothelial cell; gs, glymphatic space; lys, lysosome; Mt, mitochondria; N, nucleus; NVU, neurovascular unit; Pc, pericyte; PcN, pericyte nucleus; Pcp, pericyte endfeet processes; PVS, perivascular space; rMGC, interrogating or reactive microglia; rMФ, reactive macrophage; TJ/AJ, tight junctions/adherens junctions.
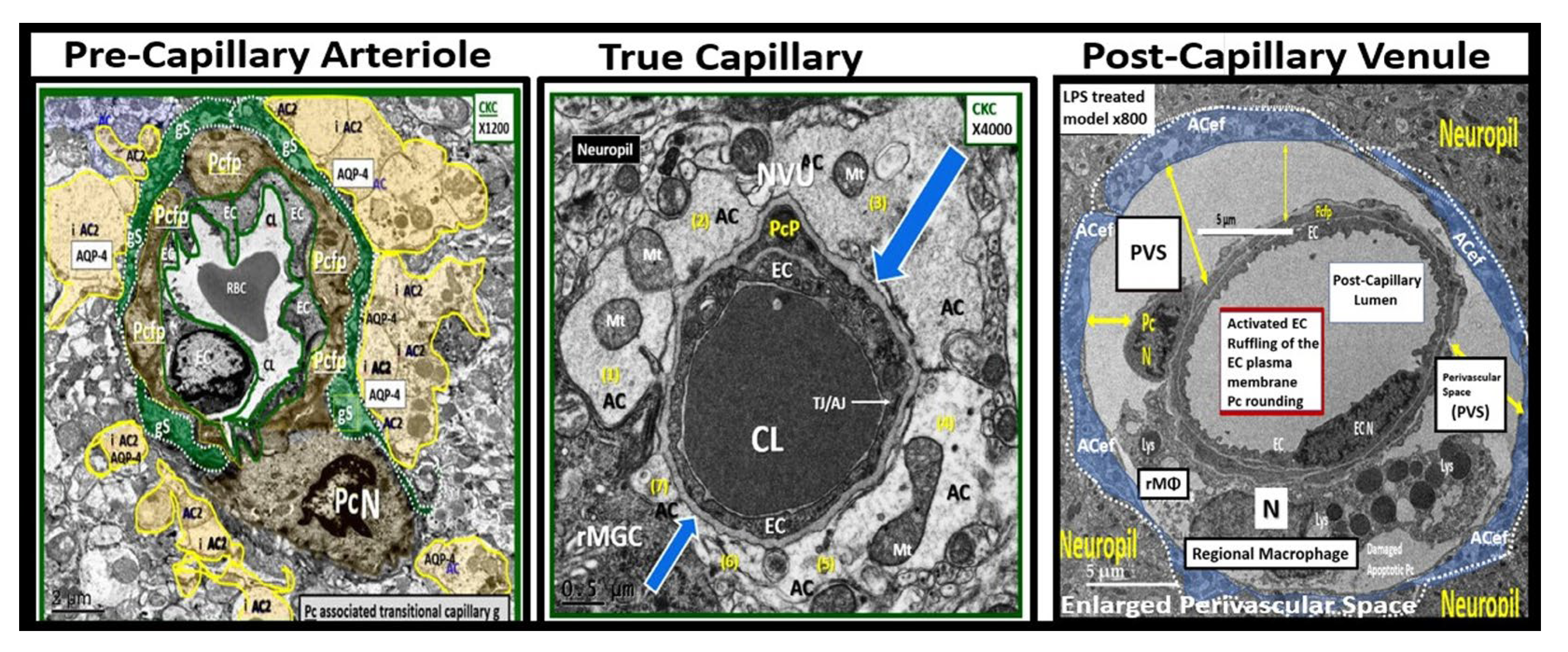
Figure 2.
perivascular astrocyte endfeet (pvACef) provide the connections between the capillary and regional neurons to form the neurovascular unit (NVU). This combined cartoon illustration and transmission electron micrographs allows one to better visualize how the pvACef connect the Mural capillary to the regional neurons (Panel A). Note that in panel A that there is detachment and retraction of pvACef that are noted in the diabetic db/db models. Other pvACef connect with the synapses (Panel B). Images in panel A and B were provided with permission by CC 4.0 [16OK]. AC, astrocyte; ACef, astrocyte endfeet; AQP-4, aquaporin four; BDGF, brain derived growth factor; Ca++, calcium ion; CKC, control model; DBC, diabetic db/db model; DBE, diabetic db/db treated with empagliflozin; GDGF, glioma-derived growth factor; PSD, post synaptic density; S, synapse; TGF-β, transforming growth factor beta.
Figure 2.
perivascular astrocyte endfeet (pvACef) provide the connections between the capillary and regional neurons to form the neurovascular unit (NVU). This combined cartoon illustration and transmission electron micrographs allows one to better visualize how the pvACef connect the Mural capillary to the regional neurons (Panel A). Note that in panel A that there is detachment and retraction of pvACef that are noted in the diabetic db/db models. Other pvACef connect with the synapses (Panel B). Images in panel A and B were provided with permission by CC 4.0 [16OK]. AC, astrocyte; ACef, astrocyte endfeet; AQP-4, aquaporin four; BDGF, brain derived growth factor; Ca++, calcium ion; CKC, control model; DBC, diabetic db/db model; DBE, diabetic db/db treated with empagliflozin; GDGF, glioma-derived growth factor; PSD, post synaptic density; S, synapse; TGF-β, transforming growth factor beta.

Figure 3.
Multiple peripheral-systemic injurious species (neurotoxins) affect the brain endothelial cells (BECs) of the brain. These injurious species active the BECs of the neurovascular unit (NVU) to result in BEC activation and dysfunction (BECact/dys) and blood-brain barrier dysfunction/disruption (BBBdd). BECact/dys and BBBdd are biomarkers for the development of cerebral small vessel disease (SVD). Note the red-dashed line at the top of this image since it demarks the location of the multiple injurious species that are responsible for the initial brain endothelial cell injury in multiple clinical diseases and structural abnormalities, which importantly include SVD. Image provided with permission by CC 4.0 [37,39]. Ang II, angiotensin two; BBB, blood–brain barrier; BEC, brain endothelial cell; BECact/dys, brain endothelial cell activation/dysfunction; BH4, tetrahydrobiopterin; CCL2, chemokine (C-C motif) ligand 2; Cox-2, cyclo-oxygenase-2; Cox-2/PGE2 axis, cyclo-oxygenase-2; Prostaglandin E2; ecGCx, endothelial glycocalyx; intercellular adhesion molecule-1; ICAM-1; IL-1β; interleukin-1β; IL-6; interleukin-6; JAMs, junctional adhesion molecules; LDL, low density lipoprotein cholesterol; LPa, lipoprotein little a; MCP-1, monocyte chemotactic protein-1; NO, nitric oxide; Nox2, (NADPH Ox (nicotinamide adenine dinucleotide phosphate oxidase); ONOO, peroxinitrite; pnsCC, peripheral nervous system cytokines and chemokines; peroxinitrite; NVU, neurovascular unit; peroxinitrite; RBC, red blood cell; RONSS, reactive oxygen, nitrogen, sulfur species; ROS, reactive oxygen species; RSI, reactive species interactome; T, transcytosis; TNFα, tumor necro.
Figure 3.
Multiple peripheral-systemic injurious species (neurotoxins) affect the brain endothelial cells (BECs) of the brain. These injurious species active the BECs of the neurovascular unit (NVU) to result in BEC activation and dysfunction (BECact/dys) and blood-brain barrier dysfunction/disruption (BBBdd). BECact/dys and BBBdd are biomarkers for the development of cerebral small vessel disease (SVD). Note the red-dashed line at the top of this image since it demarks the location of the multiple injurious species that are responsible for the initial brain endothelial cell injury in multiple clinical diseases and structural abnormalities, which importantly include SVD. Image provided with permission by CC 4.0 [37,39]. Ang II, angiotensin two; BBB, blood–brain barrier; BEC, brain endothelial cell; BECact/dys, brain endothelial cell activation/dysfunction; BH4, tetrahydrobiopterin; CCL2, chemokine (C-C motif) ligand 2; Cox-2, cyclo-oxygenase-2; Cox-2/PGE2 axis, cyclo-oxygenase-2; Prostaglandin E2; ecGCx, endothelial glycocalyx; intercellular adhesion molecule-1; ICAM-1; IL-1β; interleukin-1β; IL-6; interleukin-6; JAMs, junctional adhesion molecules; LDL, low density lipoprotein cholesterol; LPa, lipoprotein little a; MCP-1, monocyte chemotactic protein-1; NO, nitric oxide; Nox2, (NADPH Ox (nicotinamide adenine dinucleotide phosphate oxidase); ONOO, peroxinitrite; pnsCC, peripheral nervous system cytokines and chemokines; peroxinitrite; NVU, neurovascular unit; peroxinitrite; RBC, red blood cell; RONSS, reactive oxygen, nitrogen, sulfur species; ROS, reactive oxygen species; RSI, reactive species interactome; T, transcytosis; TNFα, tumor necro.
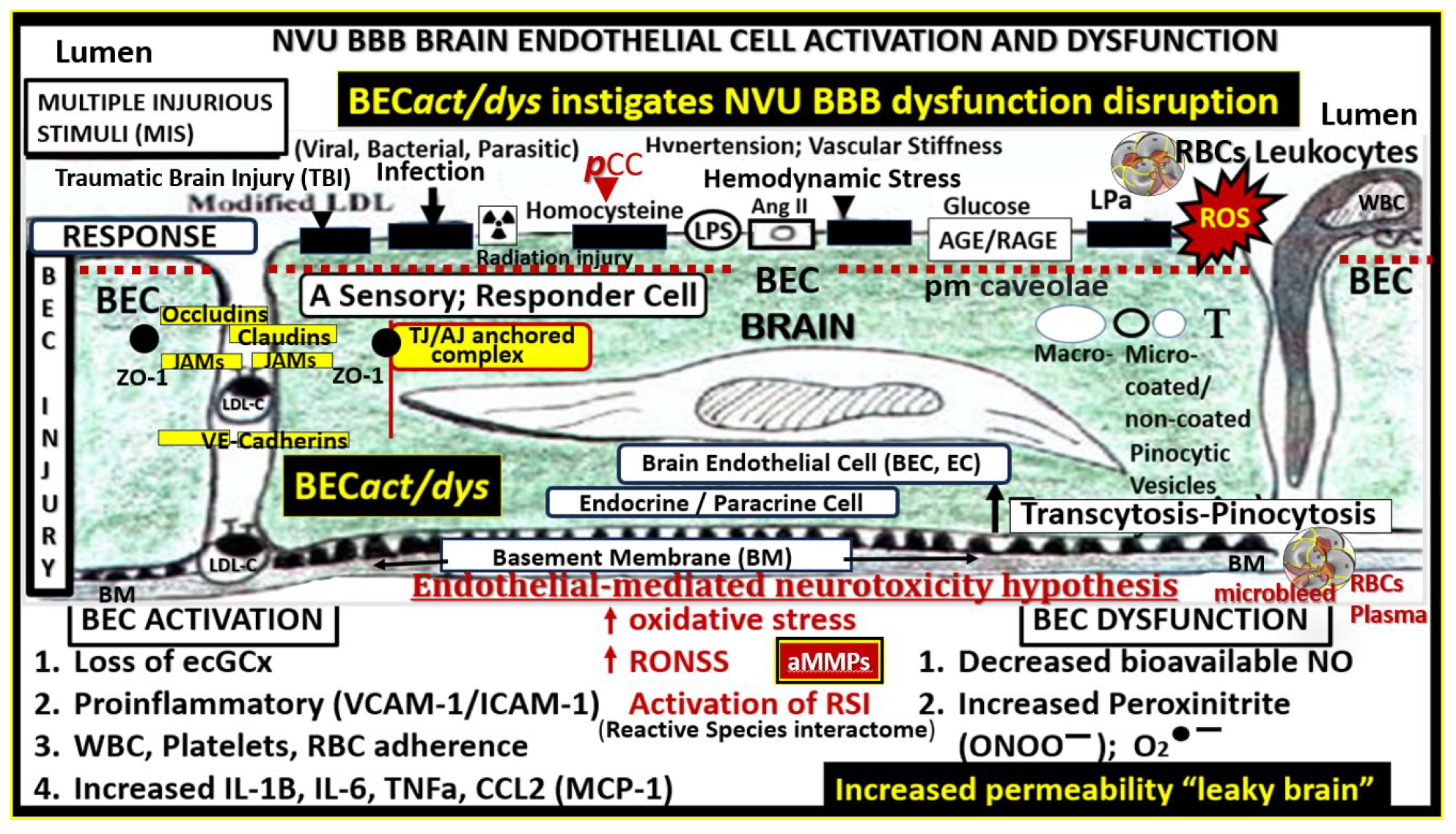
Figure 4.
Brain endothelial cell activation and dysfunction (BECact/dys) phenotypes in obese 20-week-old female diabetic db/db models. Panels A and B demonstrate normal control phenotypes of microvessel neurovascular unit (NVU) BEC from control C57B5J model at 20-weeks in the frontal cortical layer III in cross and longitudinal sections panel A and B respectively. Note that the perivascular astrocytes are pseudo-colored golden with normal appearing electron dense mitochondria and additionally note the cyan-colored line that demarcates the glia limitans in panels A and B. Panel C demonstrates the control normal phenotype in the C57B6J model at 20-weeks. Panel D depicts an activated BEC with marked abrupt swelling of the BEC that is hyperlucent as compared to the adjacent normal thickness of the BEC, which depicts BECact/dys phenotype in the 20-week-old db/db model from the frontal cortex of layer III. Panel E depicts the BM thickening of BECs in panel D and note the vacuole-like structures (V) within the BM. Panels F - I depict the adhesion of monocyte panel F, a lymphocyte panel G, a platelet panel H, and a red blood cell (RBC) panel I. Modified Images provided with permission by CC 4.0 [2,39,40,41]. AC, astrocyte; ACfp, astrocyte foot process-endfeet; BM, basement membrane; Cap L, capillary lumen; CL, capillary lumen; ECact, brain endothelial cell activation; iMGC, interrogating microglia cell; Mt, mitochondria; Mp, microparticles; Pc, pericyte. .
Figure 4.
Brain endothelial cell activation and dysfunction (BECact/dys) phenotypes in obese 20-week-old female diabetic db/db models. Panels A and B demonstrate normal control phenotypes of microvessel neurovascular unit (NVU) BEC from control C57B5J model at 20-weeks in the frontal cortical layer III in cross and longitudinal sections panel A and B respectively. Note that the perivascular astrocytes are pseudo-colored golden with normal appearing electron dense mitochondria and additionally note the cyan-colored line that demarcates the glia limitans in panels A and B. Panel C demonstrates the control normal phenotype in the C57B6J model at 20-weeks. Panel D depicts an activated BEC with marked abrupt swelling of the BEC that is hyperlucent as compared to the adjacent normal thickness of the BEC, which depicts BECact/dys phenotype in the 20-week-old db/db model from the frontal cortex of layer III. Panel E depicts the BM thickening of BECs in panel D and note the vacuole-like structures (V) within the BM. Panels F - I depict the adhesion of monocyte panel F, a lymphocyte panel G, a platelet panel H, and a red blood cell (RBC) panel I. Modified Images provided with permission by CC 4.0 [2,39,40,41]. AC, astrocyte; ACfp, astrocyte foot process-endfeet; BM, basement membrane; Cap L, capillary lumen; CL, capillary lumen; ECact, brain endothelial cell activation; iMGC, interrogating microglia cell; Mt, mitochondria; Mp, microparticles; Pc, pericyte. .
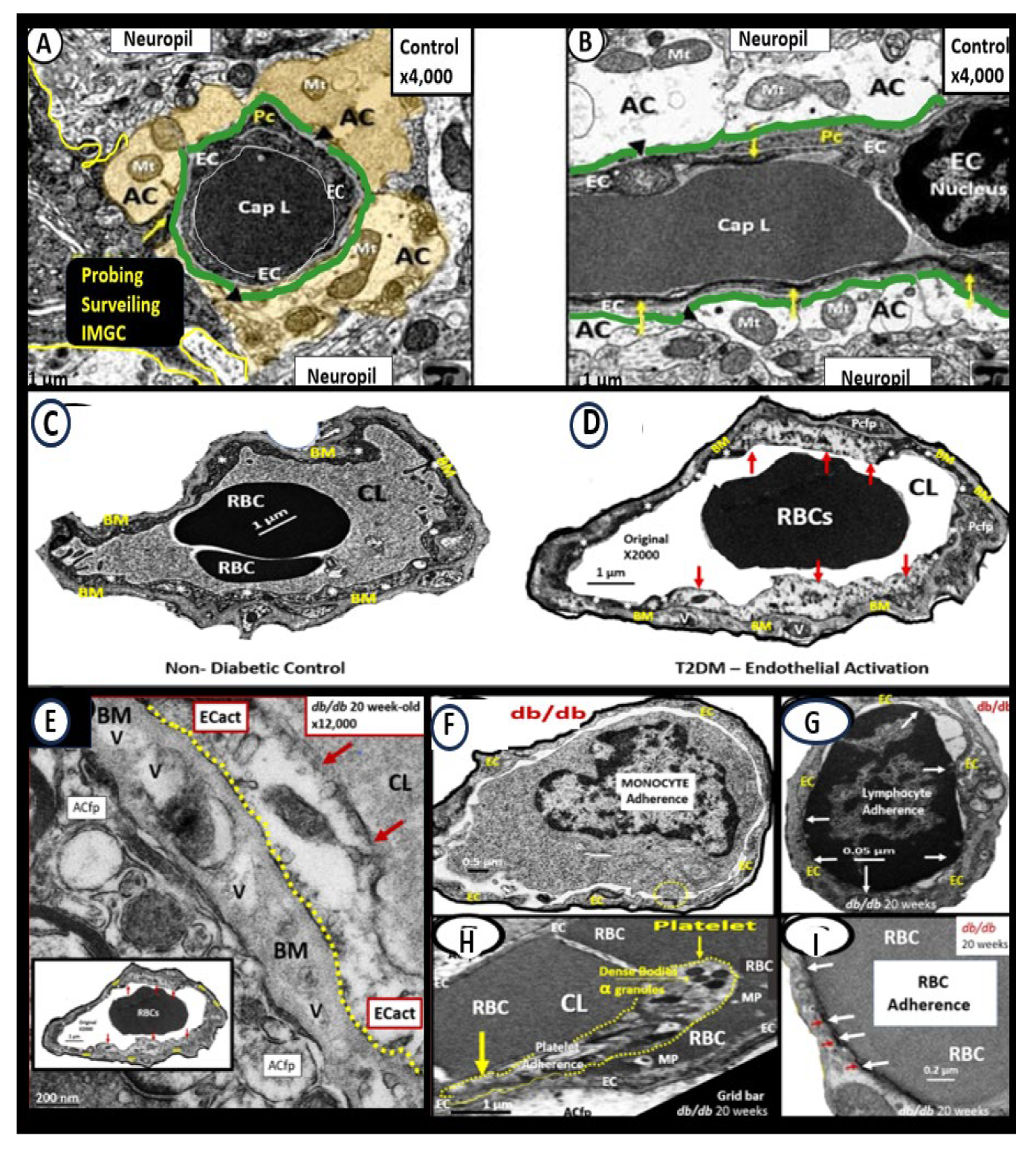
Figure 5.
Proinflammatory LPS results in attenuation and discontinuous endothelial glycocalyx (ecGCx) and increased pinocytosis/transcytosis in activated brain endothelial cell activation/dysfunction (BECact/dys). Far-left panels B and D depict an attenuation and discontinuous ecGCx with large naked gaps of BECs as compared to controls with an intact and continuous ecGCx as in panels A and C. Far-right panels B and D depict increased pinocytosis/transcytosis of BEC as compare to control panels A and C. Panels C and D are illustrations to improve and highlight the findings of the TEMs depicted in panels A and D. Images provided with permission by CC 4.0 [42]. ACfp, astrocyte foot process endfeet; atMGC, attracted microglia cell(s); BECact/dys, brain endothelial cell activation/dysfunction; CL, capillary lumen; ecGCx, brain endothelial cell glycocalyx; EC N, brain endothelial cell nucleus; LPS, lipopolysaccharide; Pc N, pericyte nucleus; PVS, perivascular space;.
Figure 5.
Proinflammatory LPS results in attenuation and discontinuous endothelial glycocalyx (ecGCx) and increased pinocytosis/transcytosis in activated brain endothelial cell activation/dysfunction (BECact/dys). Far-left panels B and D depict an attenuation and discontinuous ecGCx with large naked gaps of BECs as compared to controls with an intact and continuous ecGCx as in panels A and C. Far-right panels B and D depict increased pinocytosis/transcytosis of BEC as compare to control panels A and C. Panels C and D are illustrations to improve and highlight the findings of the TEMs depicted in panels A and D. Images provided with permission by CC 4.0 [42]. ACfp, astrocyte foot process endfeet; atMGC, attracted microglia cell(s); BECact/dys, brain endothelial cell activation/dysfunction; CL, capillary lumen; ecGCx, brain endothelial cell glycocalyx; EC N, brain endothelial cell nucleus; LPS, lipopolysaccharide; Pc N, pericyte nucleus; PVS, perivascular space;.
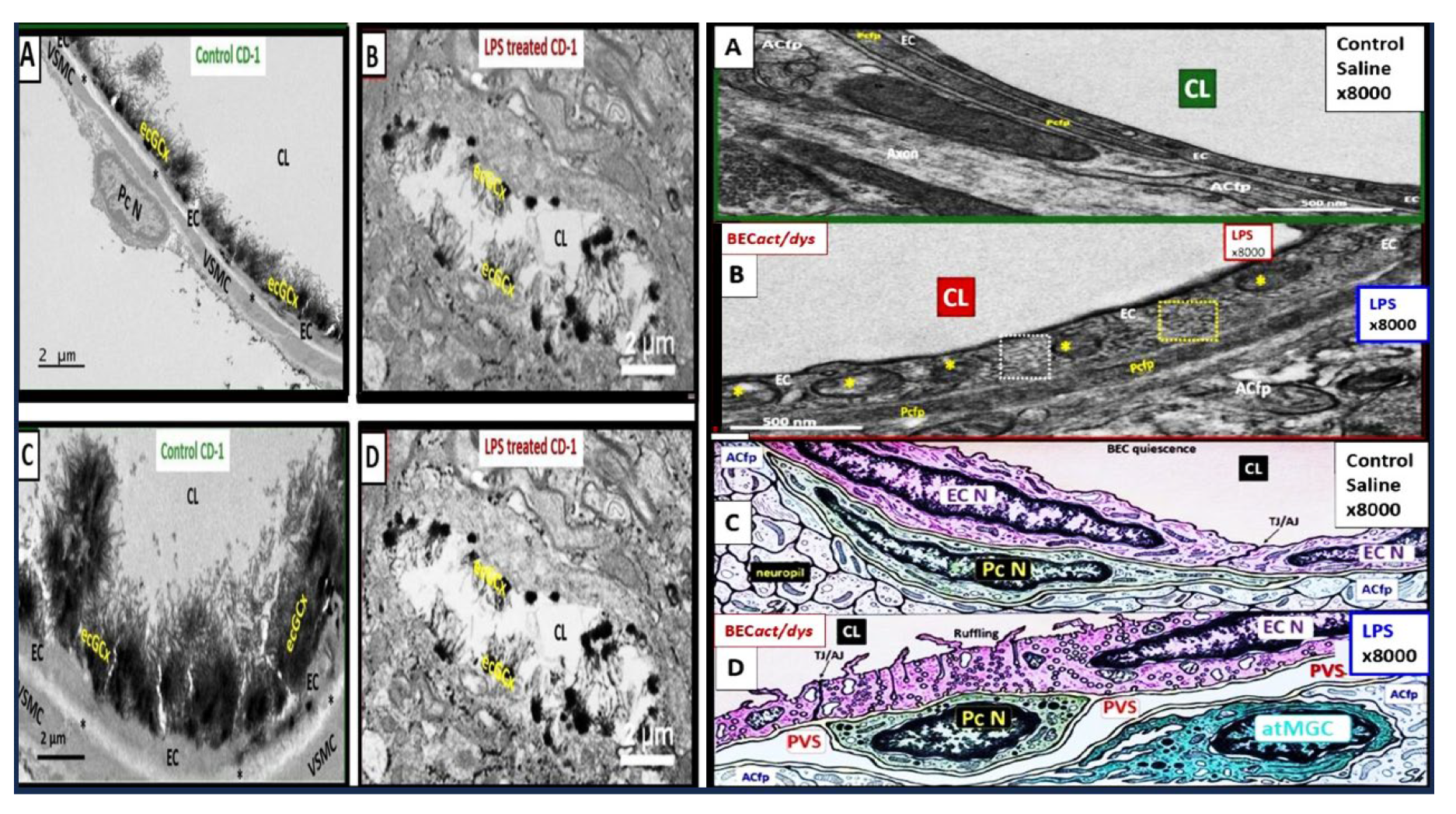
Figure 6.
Loss and/or disruption of the normal continuous tight and adherence junction(s) (TJ/AJs) paracellular blood–brain barrier (BBB) in male CD-1 streptozotocin-induced (STZ) diabetic preclinical mice models resulting in blood–brain barrier dysfunction and disruption (BBBdd) protected by the carbonic anhydrase inhibitor (Topiramate a mitochondria antioxidant) in the mid brain as compared to the cerebellum. STZ-induced diabetic mice revealed disruption of the BBB by 14C-sucrose measurements. Panel (A) displays three prominent elongated and continuous highly electron-dense TJ/AJ (white and black arrows). Panel (B) depicts a discontinuous and disrupted TJ/AJ (black arrows) into three distinct segments in the midbrain and note how TJ/AJs tend to form at the BEC-BEC overlap junctions in panels A, B, C. Panel (C) illustrates that treatment with Topiramate (TOP) prevented this disruption in the brain endothelial cell BBB (yellow and black arrows and yellow dashed line below the intact BBB TJ/AJ) in the midbrain. Revised figure images provided with permission by CC 4.0 [39,42]. Magnification ×3000; scale bar = 0.5 μm (A); ×10,000; scale bar = 0.2 μm in (B, C). BEC, brain endothelial cell; EC, brain endothelial cell; CL, capillary lumen; Pc, pericyte; RBC, red blood cell; Rx, treatment; T1DM, type 1 diabetes mellitus; TOP, topiramate.
Figure 6.
Loss and/or disruption of the normal continuous tight and adherence junction(s) (TJ/AJs) paracellular blood–brain barrier (BBB) in male CD-1 streptozotocin-induced (STZ) diabetic preclinical mice models resulting in blood–brain barrier dysfunction and disruption (BBBdd) protected by the carbonic anhydrase inhibitor (Topiramate a mitochondria antioxidant) in the mid brain as compared to the cerebellum. STZ-induced diabetic mice revealed disruption of the BBB by 14C-sucrose measurements. Panel (A) displays three prominent elongated and continuous highly electron-dense TJ/AJ (white and black arrows). Panel (B) depicts a discontinuous and disrupted TJ/AJ (black arrows) into three distinct segments in the midbrain and note how TJ/AJs tend to form at the BEC-BEC overlap junctions in panels A, B, C. Panel (C) illustrates that treatment with Topiramate (TOP) prevented this disruption in the brain endothelial cell BBB (yellow and black arrows and yellow dashed line below the intact BBB TJ/AJ) in the midbrain. Revised figure images provided with permission by CC 4.0 [39,42]. Magnification ×3000; scale bar = 0.5 μm (A); ×10,000; scale bar = 0.2 μm in (B, C). BEC, brain endothelial cell; EC, brain endothelial cell; CL, capillary lumen; Pc, pericyte; RBC, red blood cell; Rx, treatment; T1DM, type 1 diabetes mellitus; TOP, topiramate.
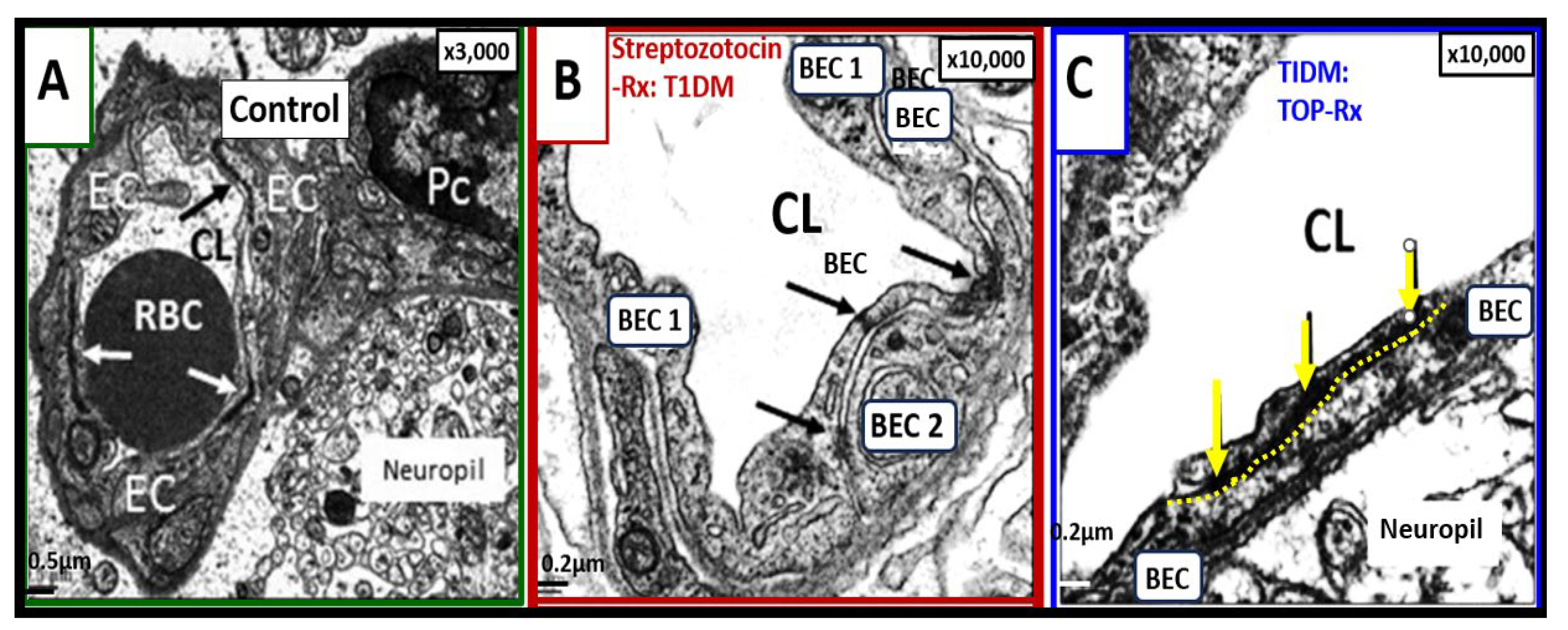
Figure 8.
Neurotoxicity of amyloid beta in close proximity to the neurovascular unit (NVU) brain endothelial cell(s) (BECs) may result in blood-brain barrier dysfunction and disruption (BBBdd). These images are from a nine-month-old 5xFAD male mouse model from an old slide-deck not previously studied or published. Panel A depicts an interstitial extracellular matrix-interstitial amyloid beta (Aβ) (pseudo-colored red) in close proximity to the NVU that appears to nearly touch the outer basement membrane (BM) of the NVU depicted. Importantly, note that this image depicts degenerative neurites (yellow arrows and outlined in yellow dashed-lines) within the adjacent interstitial neuropil. Panel B depicts an exploded image in Microsoft paint with intact scale bar. Insert C is a further exploded image of how Aβ is closely adjacent to the microvessel NVU BEC basement membrane (BM) and appears to actually be in direct contact with the BM. Indeed, this structural arrangement contributes to Aβ being neurotoxic to BECs and creating a vicious cycle of BEC injury by Aβ and BECact/dys that that contributes to a further increase in ROS and damage to the NVU with eventual NVU uncoupling and decrease in regional cerebral blood flow that will ultimately increase neurodegeneration and increased or accelerated amyloid beta deposition. This image has not been previously published. .
Figure 8.
Neurotoxicity of amyloid beta in close proximity to the neurovascular unit (NVU) brain endothelial cell(s) (BECs) may result in blood-brain barrier dysfunction and disruption (BBBdd). These images are from a nine-month-old 5xFAD male mouse model from an old slide-deck not previously studied or published. Panel A depicts an interstitial extracellular matrix-interstitial amyloid beta (Aβ) (pseudo-colored red) in close proximity to the NVU that appears to nearly touch the outer basement membrane (BM) of the NVU depicted. Importantly, note that this image depicts degenerative neurites (yellow arrows and outlined in yellow dashed-lines) within the adjacent interstitial neuropil. Panel B depicts an exploded image in Microsoft paint with intact scale bar. Insert C is a further exploded image of how Aβ is closely adjacent to the microvessel NVU BEC basement membrane (BM) and appears to actually be in direct contact with the BM. Indeed, this structural arrangement contributes to Aβ being neurotoxic to BECs and creating a vicious cycle of BEC injury by Aβ and BECact/dys that that contributes to a further increase in ROS and damage to the NVU with eventual NVU uncoupling and decrease in regional cerebral blood flow that will ultimately increase neurodegeneration and increased or accelerated amyloid beta deposition. This image has not been previously published. .
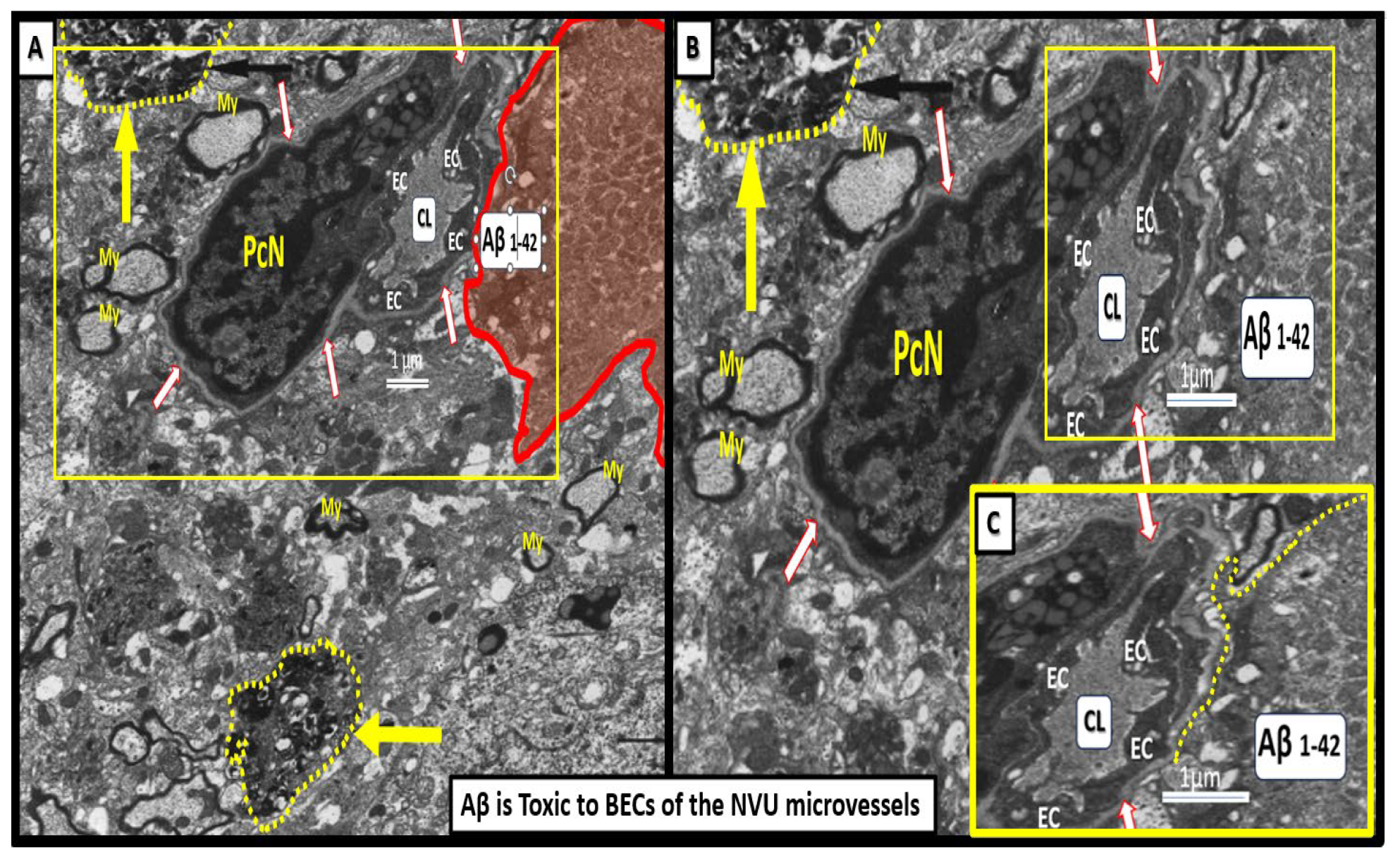
Figure.
Astrocyte(s) (ACs) are the master communication/connecting cell(s) (CCCs) within the brain universe. This collection of illustrations and transmission electron microscopy (TEM) images demonstrates the CCC functions of ACs via their various perivascular, perisynaptic, perineuronal endfeet, and cell-cell junctions (inserts 1-5). Insert 1 demonstrates the important role of perivascular astrocyte endfeet (pvACef; pseudo-colored golden) communication/connection. Note how this communicating/connecting AC allows for neurovascular coupling with regional neurons (insert 3) in frontal cortex layer III in control mice at 20-weeks of age. Insert 2 illustrates the communication/connection of the astrocyte perisynaptic endfeet (psACef) (pseudo-colored yellow). Insert 3 illustrates the communication/connection of ACs to myelinated and unmyelinated neurons. Insert 4 depicts the lost connections between a reactive microglial cell (rMGC) (pseudo-colored blue with nuclear chromatin condensation) and multiple reactive, detached, and separated ACs (pseudo-colored red) adjacent to a neurovascular unit (NVU) with a single intact non-reactive ACs (pseudo-colored yellow) in diabetic db/db model cortical layer III at 20-weeks of age. Insert 5 illustrates AC-to-AC connections in cortical layer III in control models (hand-drawn computer-assisted illustration of light microscopic toluidine blue stained images from control C57BL/6J models) via gap junction connexins (Cx43). Inserts 1–4 have scale bars of 0.5 μm, 100 nm, 1 μm, and 2 μm, respectively. The background image represents a hand-drawn computer-assisted image derived from control C57BL/6J toluidine blue stained models and does not have a scale bar. This highly modified image is provided with permission by CC 4.0 [16]. ACfp, protoplasmic astrocyte endfeet; ACPVef, astrocyte perivascular endfeet; Cap L, capillary lumen; EC, brain endothelial cell; iMGC, interrogating microglial cell; Mt, mitochondria; N, nucleus; Pc, pericyte; PSD, post-synaptic density; PVACef, perivascular astrocyte endfeet; rMGC, reactive microglia cell; psACef, perisynaptic astrocyte endfeet.
Figure.
Astrocyte(s) (ACs) are the master communication/connecting cell(s) (CCCs) within the brain universe. This collection of illustrations and transmission electron microscopy (TEM) images demonstrates the CCC functions of ACs via their various perivascular, perisynaptic, perineuronal endfeet, and cell-cell junctions (inserts 1-5). Insert 1 demonstrates the important role of perivascular astrocyte endfeet (pvACef; pseudo-colored golden) communication/connection. Note how this communicating/connecting AC allows for neurovascular coupling with regional neurons (insert 3) in frontal cortex layer III in control mice at 20-weeks of age. Insert 2 illustrates the communication/connection of the astrocyte perisynaptic endfeet (psACef) (pseudo-colored yellow). Insert 3 illustrates the communication/connection of ACs to myelinated and unmyelinated neurons. Insert 4 depicts the lost connections between a reactive microglial cell (rMGC) (pseudo-colored blue with nuclear chromatin condensation) and multiple reactive, detached, and separated ACs (pseudo-colored red) adjacent to a neurovascular unit (NVU) with a single intact non-reactive ACs (pseudo-colored yellow) in diabetic db/db model cortical layer III at 20-weeks of age. Insert 5 illustrates AC-to-AC connections in cortical layer III in control models (hand-drawn computer-assisted illustration of light microscopic toluidine blue stained images from control C57BL/6J models) via gap junction connexins (Cx43). Inserts 1–4 have scale bars of 0.5 μm, 100 nm, 1 μm, and 2 μm, respectively. The background image represents a hand-drawn computer-assisted image derived from control C57BL/6J toluidine blue stained models and does not have a scale bar. This highly modified image is provided with permission by CC 4.0 [16]. ACfp, protoplasmic astrocyte endfeet; ACPVef, astrocyte perivascular endfeet; Cap L, capillary lumen; EC, brain endothelial cell; iMGC, interrogating microglial cell; Mt, mitochondria; N, nucleus; Pc, pericyte; PSD, post-synaptic density; PVACef, perivascular astrocyte endfeet; rMGC, reactive microglia cell; psACef, perisynaptic astrocyte endfeet.
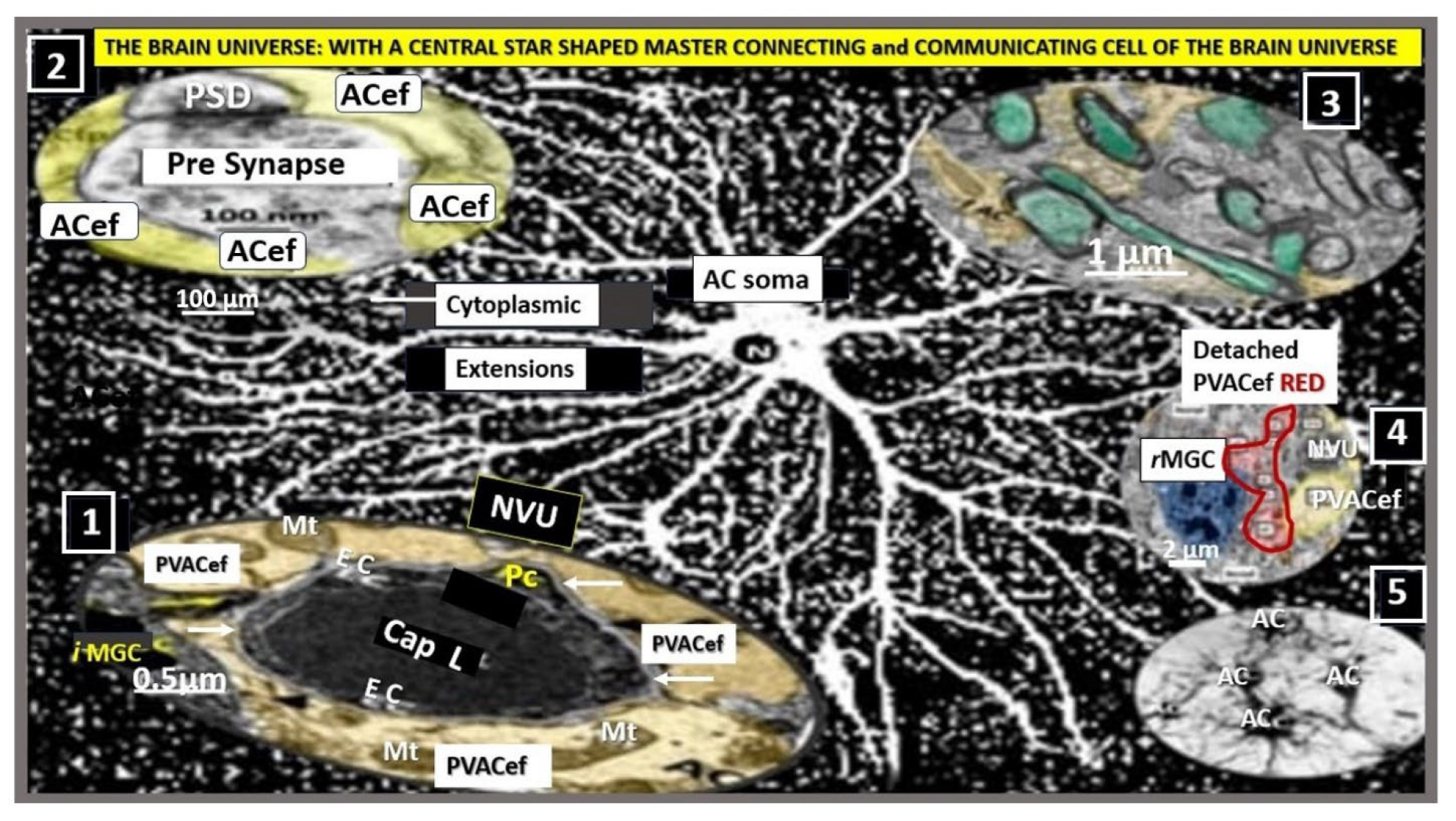
Figure 9.
A Master Communicating, Connecting, and Signaling Cell of the Brain Universe Providing Homeostasis. Astrocytes provide molecular, cellular and network communication, systemic, metabolic and whole organ homeostasis in addition to being neuroprotective. Centrally-located modified image of astrocyte from figure 10 provided with permission by CC 4.0 [16]. AC, astrocyte; ACef, astrocyte endfeet; AQP4, aquaporin-4; BBB, blood-brain barrier; Ca++, calcium; Cap L, capillary lumen; CBF, cerebral blood flow; Cl-, chloride; CNS, central nervous system; CO2, carbon monoxide; EC, brain endothelial cell; GABA, gamma aminobutyric acid; K+, potassium; Na+, sodium; NVU, neurovascular unit; PVACef, perivascular astrocyte endfeet; rMGC, reactive microglia cell.
Figure 9.
A Master Communicating, Connecting, and Signaling Cell of the Brain Universe Providing Homeostasis. Astrocytes provide molecular, cellular and network communication, systemic, metabolic and whole organ homeostasis in addition to being neuroprotective. Centrally-located modified image of astrocyte from figure 10 provided with permission by CC 4.0 [16]. AC, astrocyte; ACef, astrocyte endfeet; AQP4, aquaporin-4; BBB, blood-brain barrier; Ca++, calcium; Cap L, capillary lumen; CBF, cerebral blood flow; Cl-, chloride; CNS, central nervous system; CO2, carbon monoxide; EC, brain endothelial cell; GABA, gamma aminobutyric acid; K+, potassium; Na+, sodium; NVU, neurovascular unit; PVACef, perivascular astrocyte endfeet; rMGC, reactive microglia cell.
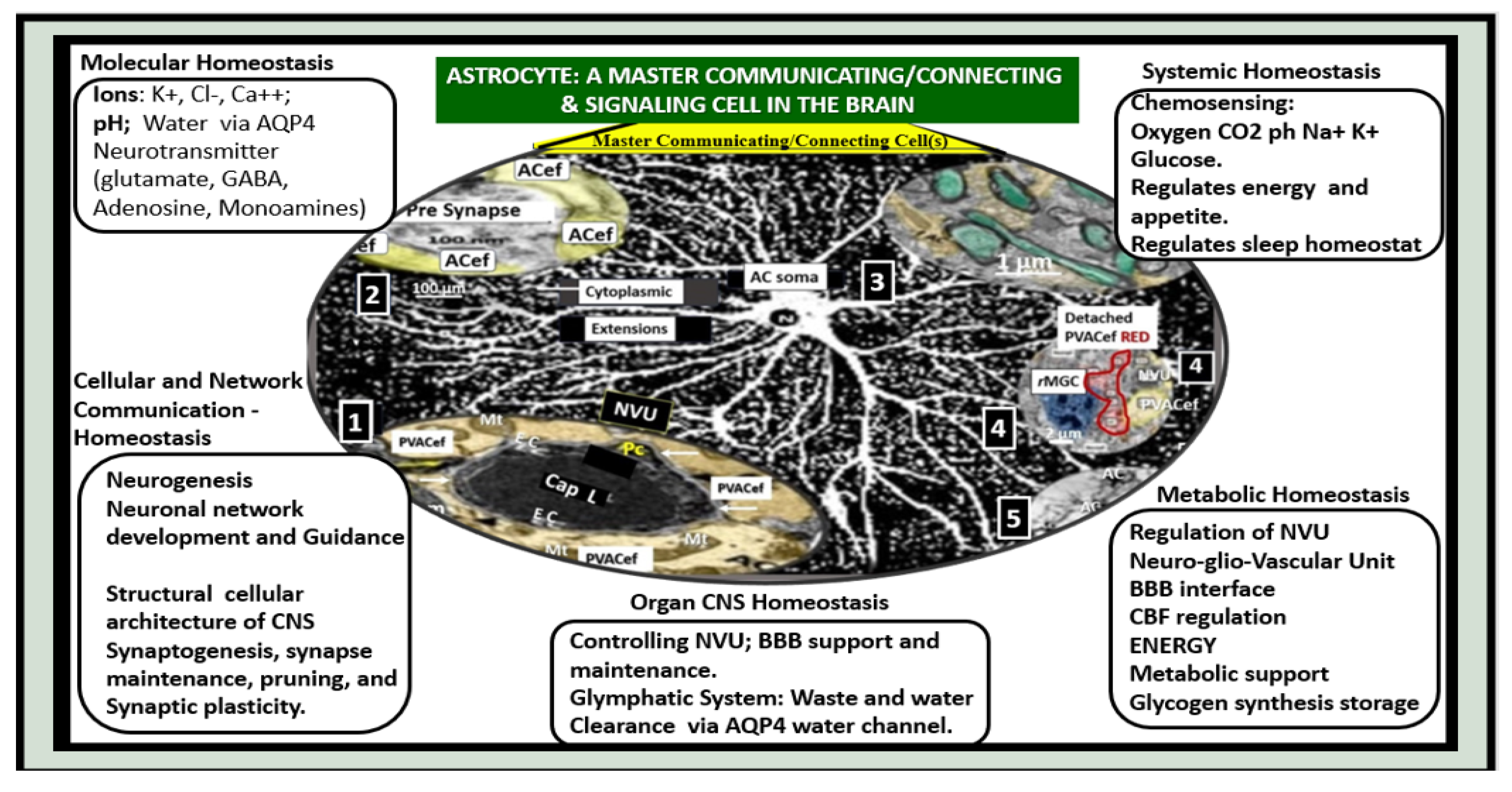
Figure 12.
Reactive Microglia Cell(s) (rMGCs) and Astrocyte(s) (rACs) Contribute to Decreased Cerebral Blood Flow and Neurodegeneration (ND). Panel A demonstrates a normal neurovascular unit (NVU) with its interrogating MGC (iMGC) (pseudo-colored green) and its intact astrocytes (iAC) (pseudo-colored golden). Panel B depicts a prominent rMGC invading the NVU (pseudo-colored red) and note the aberrant mitochondria (aMt) (pseudo-colored yellow and outlined in red). Notably the normally iAC have become detached and retracted, separated (drAC, pseudo-colored light blue) from the NVU outer basement membrane and results in NVU uncoupling with ensuing decreased cerebral blood flow to regional neurons resulting in hypometabolism, hypoperfusion, regional neuronal hypoxia/ischemia with subsequent neurodegeneration and impaired cognition. Modified images provided with permission by CC 4.0 [6,60]. CL, capillary lumen; drAC, detached reactive, retracted astrocyte endfeet; N, nucleus; RBC, red blood cell.
Figure 12.
Reactive Microglia Cell(s) (rMGCs) and Astrocyte(s) (rACs) Contribute to Decreased Cerebral Blood Flow and Neurodegeneration (ND). Panel A demonstrates a normal neurovascular unit (NVU) with its interrogating MGC (iMGC) (pseudo-colored green) and its intact astrocytes (iAC) (pseudo-colored golden). Panel B depicts a prominent rMGC invading the NVU (pseudo-colored red) and note the aberrant mitochondria (aMt) (pseudo-colored yellow and outlined in red). Notably the normally iAC have become detached and retracted, separated (drAC, pseudo-colored light blue) from the NVU outer basement membrane and results in NVU uncoupling with ensuing decreased cerebral blood flow to regional neurons resulting in hypometabolism, hypoperfusion, regional neuronal hypoxia/ischemia with subsequent neurodegeneration and impaired cognition. Modified images provided with permission by CC 4.0 [6,60]. CL, capillary lumen; drAC, detached reactive, retracted astrocyte endfeet; N, nucleus; RBC, red blood cell.
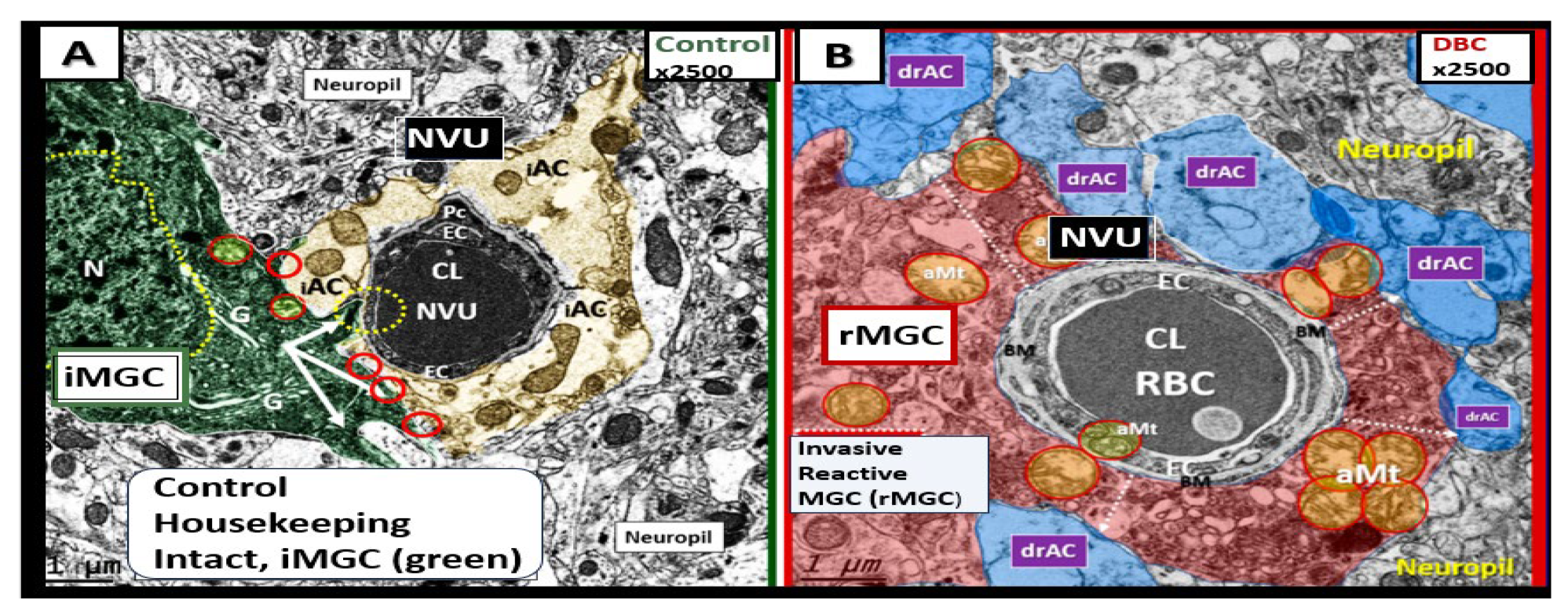
Figure 13.
Enlarged perivascular space (EPVS) and resident-reactive perivascular macrophage (rPVMΦs) in a postcapillary venule compared to a true capillary. Panel A demonstrates a normal true capillary in a 20-week-old female C57B6/J control model and note how the astrocyte endfeet (ACef) tightly abut the shared basement membrane (open black arrows) of the brain endothelial cell (BEC) and pericyte foot process (PcP). Panel B depicts an EPVS with a prominent rPVMΦ (pseudo-colored red) in a 20-week-old lipopolysaccharide (LPS)-treated CD-1 male model and importantly note how the ACfp are markedly separated from the capillary mural cells (BEC and Pc) (red double arrows). Panel C depicts the rPVMΦs in an exploded image with intimate contact with the Pcfps basal lamina and the rPVMΦ intimate contact with basal lamina of the ACef (outermost boundary of the EPVS abluminal lining) (dashed blue circles). Modified images provided with permission by CC 4.0 [45]. AQP4, aquaporin 4; Lys, lysosomes; Mt, mitochondria; N, nucleus; NVU, neurovascular unit; V, vacuoles; ves, vesicles.
Figure 13.
Enlarged perivascular space (EPVS) and resident-reactive perivascular macrophage (rPVMΦs) in a postcapillary venule compared to a true capillary. Panel A demonstrates a normal true capillary in a 20-week-old female C57B6/J control model and note how the astrocyte endfeet (ACef) tightly abut the shared basement membrane (open black arrows) of the brain endothelial cell (BEC) and pericyte foot process (PcP). Panel B depicts an EPVS with a prominent rPVMΦ (pseudo-colored red) in a 20-week-old lipopolysaccharide (LPS)-treated CD-1 male model and importantly note how the ACfp are markedly separated from the capillary mural cells (BEC and Pc) (red double arrows). Panel C depicts the rPVMΦs in an exploded image with intimate contact with the Pcfps basal lamina and the rPVMΦ intimate contact with basal lamina of the ACef (outermost boundary of the EPVS abluminal lining) (dashed blue circles). Modified images provided with permission by CC 4.0 [45]. AQP4, aquaporin 4; Lys, lysosomes; Mt, mitochondria; N, nucleus; NVU, neurovascular unit; V, vacuoles; ves, vesicles.
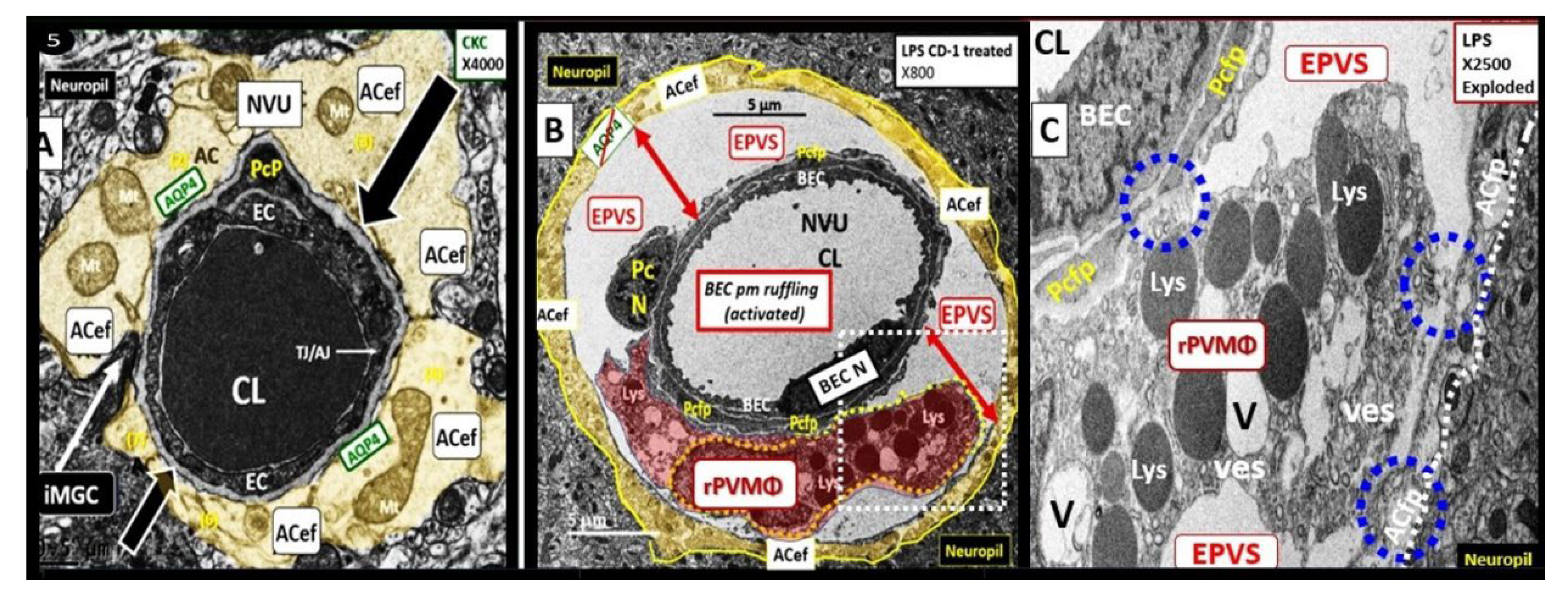
Figure 14.
Multipotential reactive astrocyte (rAC) phenotype remodeling in late-onset alzheimer’s disease (LOAD): From hypertrophy to atrophy. Panel A depicts extracellular matrix interstitial space amyloid beta (Aβ) plaques that are increased in LOAD and co-occur with soluble Aβ oligomers that are actually neurotoxic more than the Aβ mature plaques. Panel B lists the such as ischemic or hemorrhagic stroke with palisading and hypertrophic phenotypes that are in contrast to the rAC phenotype of atrophy that is associated with AC remodeling due to the neurotoxicity of Aβ oligomers and Aβ extracellular plaque aggregation discussed in C2. Modified images of Aβ in panel A are provided with permission by CC 4.0 [75] and reproduced image in C1 is provided with permission by CC 4.0 [29,76]. ApoE, apolipoprotein E; CNS, central nervous system; cnsCC, central nervous system cytokines/chemokines; DNA, deoxyribonucleic acid; GAGS, glycosaminoglycans; LOAD, late-onset Alzheimer’s disease; MMP(s), matrix metalloproteinases; PGN, proteoglycan(s); rAC(s), reactive astrocytes; ROI, region of interest; rMGC(s), reactive microglia cells; ROS, reactive oxygen species; RNA, ribonucleic acid.
Figure 14.
Multipotential reactive astrocyte (rAC) phenotype remodeling in late-onset alzheimer’s disease (LOAD): From hypertrophy to atrophy. Panel A depicts extracellular matrix interstitial space amyloid beta (Aβ) plaques that are increased in LOAD and co-occur with soluble Aβ oligomers that are actually neurotoxic more than the Aβ mature plaques. Panel B lists the such as ischemic or hemorrhagic stroke with palisading and hypertrophic phenotypes that are in contrast to the rAC phenotype of atrophy that is associated with AC remodeling due to the neurotoxicity of Aβ oligomers and Aβ extracellular plaque aggregation discussed in C2. Modified images of Aβ in panel A are provided with permission by CC 4.0 [75] and reproduced image in C1 is provided with permission by CC 4.0 [29,76]. ApoE, apolipoprotein E; CNS, central nervous system; cnsCC, central nervous system cytokines/chemokines; DNA, deoxyribonucleic acid; GAGS, glycosaminoglycans; LOAD, late-onset Alzheimer’s disease; MMP(s), matrix metalloproteinases; PGN, proteoglycan(s); rAC(s), reactive astrocytes; ROI, region of interest; rMGC(s), reactive microglia cells; ROS, reactive oxygen species; RNA, ribonucleic acid.
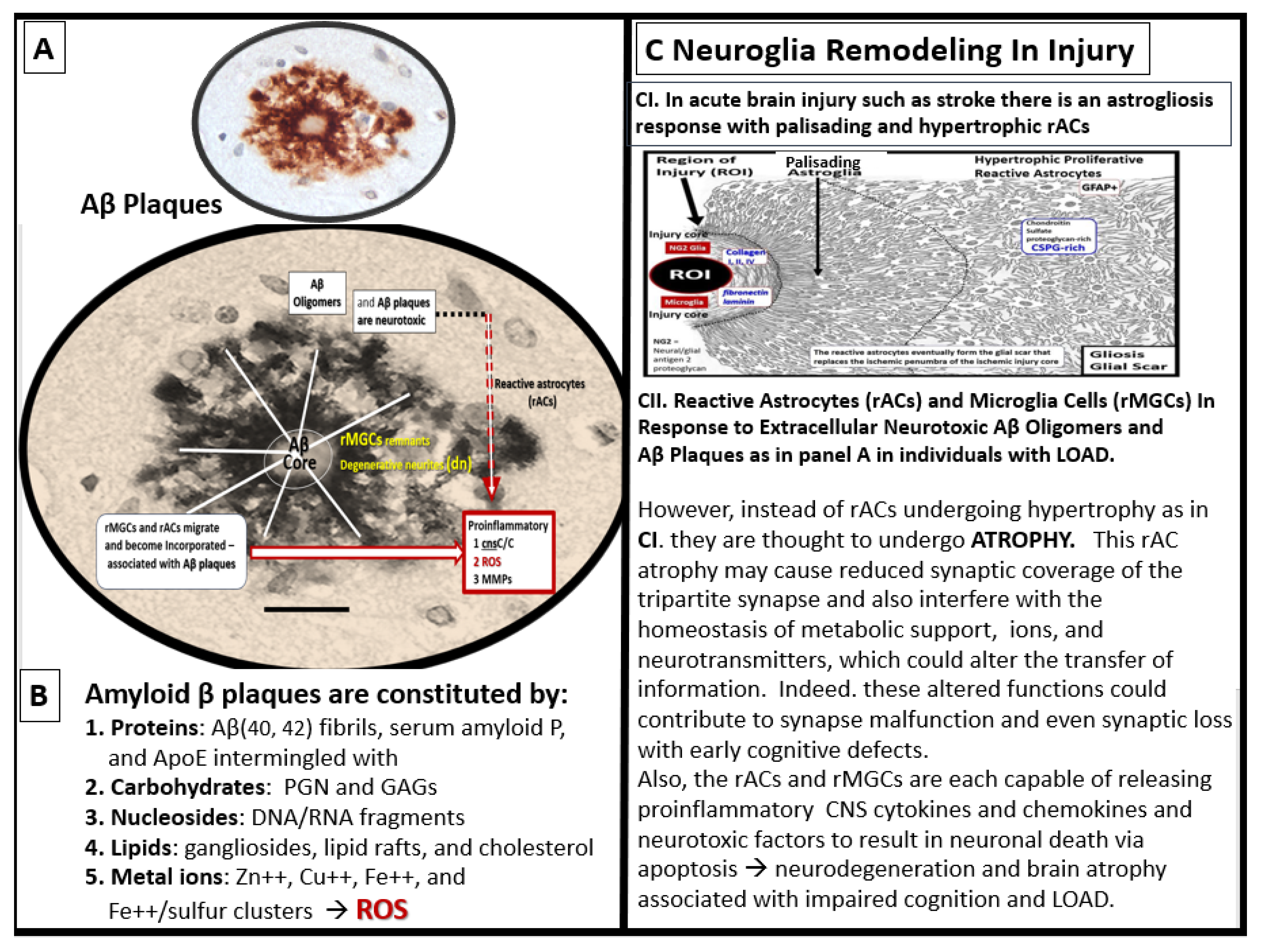
Figure 15.
Aberrant mitochondria (aMt) allow for leaky prooxidative ROS of the reactive species interactome (RSI) and prooxidative iron sulfur cluster(s) (ISCs) to enter the cytosol with dysfunction and damage to the cell and surrounding cells. Note how the aMt remodeling appears as compared to the normal control insert upper left with its hyperlucency, crista fragmentation and loss, loss of electron dense mitochondrial matrix, and permeabilization of the outer mitochondria membrane (asterisk). This permeabilization allows the redox-active and prooxidative mtROS and iron sulfur clusters to escape into the cellular cytosol resulting in cellular dysfunction, damage and even apoptosis. This prooxidant mechanism has an increased impact if the antioxidant reserves of super oxide dismutase (SOD), catalase, and glutathione (GSH) have previously been or are currently depleted. Revised background image of the aMt is provided with permission by CC 4.0 [61]. aMt, aberrant mitochondria; Cys, cysteine; ETC, electron transport chain; Fe, iron; ISC, iron sulfur cluster(s); MOMP, mitochondrial outer membrane permeabilization; Mt, mitochondria; mtROS, mitochondrial-derived reactive oxygen species; RONSS, reactive oxygen, nitrogen, sulfur species; S, sulfur.
Figure 15.
Aberrant mitochondria (aMt) allow for leaky prooxidative ROS of the reactive species interactome (RSI) and prooxidative iron sulfur cluster(s) (ISCs) to enter the cytosol with dysfunction and damage to the cell and surrounding cells. Note how the aMt remodeling appears as compared to the normal control insert upper left with its hyperlucency, crista fragmentation and loss, loss of electron dense mitochondrial matrix, and permeabilization of the outer mitochondria membrane (asterisk). This permeabilization allows the redox-active and prooxidative mtROS and iron sulfur clusters to escape into the cellular cytosol resulting in cellular dysfunction, damage and even apoptosis. This prooxidant mechanism has an increased impact if the antioxidant reserves of super oxide dismutase (SOD), catalase, and glutathione (GSH) have previously been or are currently depleted. Revised background image of the aMt is provided with permission by CC 4.0 [61]. aMt, aberrant mitochondria; Cys, cysteine; ETC, electron transport chain; Fe, iron; ISC, iron sulfur cluster(s); MOMP, mitochondrial outer membrane permeabilization; Mt, mitochondria; mtROS, mitochondrial-derived reactive oxygen species; RONSS, reactive oxygen, nitrogen, sulfur species; S, sulfur.
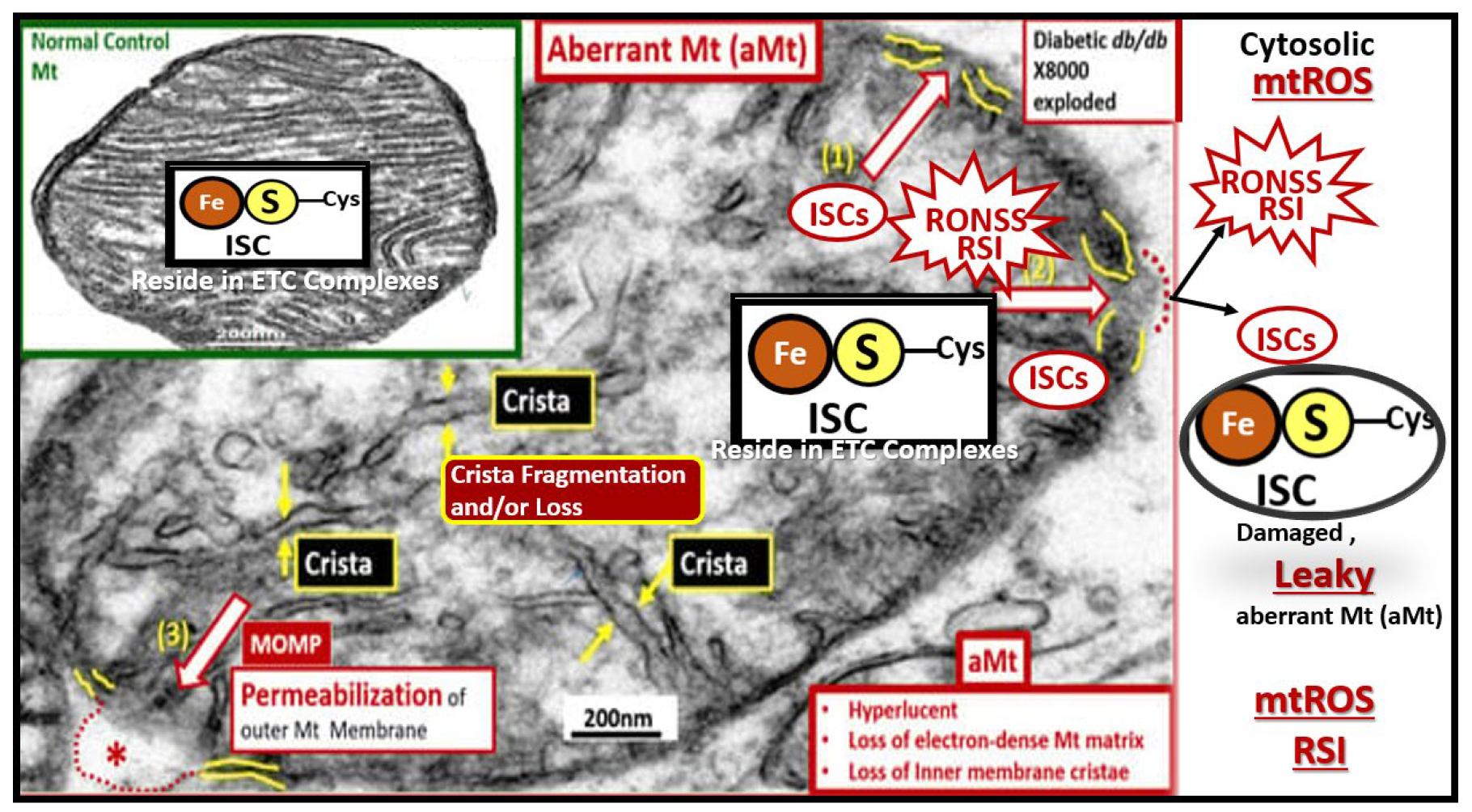
Figure 16.
Reactive oxygen species (ROS) begets ROS and Oxidative-Redox Stress (OxRS) is in a vicious cycle with neuroinflammation wherein this vicious cycle contributes to impaired cognition and neurodegeneration. Metabolic and hormonal excesses in addition to injury-trauma and the vicious cycle of neuroinflammation and ROS – OxRS lead to blood-brain barrier dysfunction and disruption with NVU uncoupling and regional hypometabolism and Ischemia/ischemia reperfusion injury results in a cascade to neurodegeneration and brain atrophy with impaired cognition that is associated with late-onset Alzheimer’s disease (LOAD). Modified figure provided with permission by CC 4.0 [29,37,87]. AGE/RAGE, advanced glycation end products/ receptor for advanced glycation end products; Ang II, angiotensin II; AT1R, angiotensin type 1 receptor; BH4, tetrahydrobiopterin; eNOS, endothelial nitric oxide synthase; FFA, free fatty acids; HPA, hypothalamic pituitary adrenal; I-CAM, intercellular adhesion molecule; MC, mast cell; MGCs, microglia cells; NADPH – NADPH Ox, reduced nicotinamide adenine dinucleotide phosphate oxidase; NVU. neurovascular unit; NF-kB, nuclear factor- kappa B; RAS, renin angiotensin system; RAAS, renin angiotensin aldosterone system; ROS/RNS, reactive oxygen species/reactive nitrogen species. UV, ultraviolet; VE-CAM, vascular endothelial cellular adhesion molecule.
Figure 16.
Reactive oxygen species (ROS) begets ROS and Oxidative-Redox Stress (OxRS) is in a vicious cycle with neuroinflammation wherein this vicious cycle contributes to impaired cognition and neurodegeneration. Metabolic and hormonal excesses in addition to injury-trauma and the vicious cycle of neuroinflammation and ROS – OxRS lead to blood-brain barrier dysfunction and disruption with NVU uncoupling and regional hypometabolism and Ischemia/ischemia reperfusion injury results in a cascade to neurodegeneration and brain atrophy with impaired cognition that is associated with late-onset Alzheimer’s disease (LOAD). Modified figure provided with permission by CC 4.0 [29,37,87]. AGE/RAGE, advanced glycation end products/ receptor for advanced glycation end products; Ang II, angiotensin II; AT1R, angiotensin type 1 receptor; BH4, tetrahydrobiopterin; eNOS, endothelial nitric oxide synthase; FFA, free fatty acids; HPA, hypothalamic pituitary adrenal; I-CAM, intercellular adhesion molecule; MC, mast cell; MGCs, microglia cells; NADPH – NADPH Ox, reduced nicotinamide adenine dinucleotide phosphate oxidase; NVU. neurovascular unit; NF-kB, nuclear factor- kappa B; RAS, renin angiotensin system; RAAS, renin angiotensin aldosterone system; ROS/RNS, reactive oxygen species/reactive nitrogen species. UV, ultraviolet; VE-CAM, vascular endothelial cellular adhesion molecule.
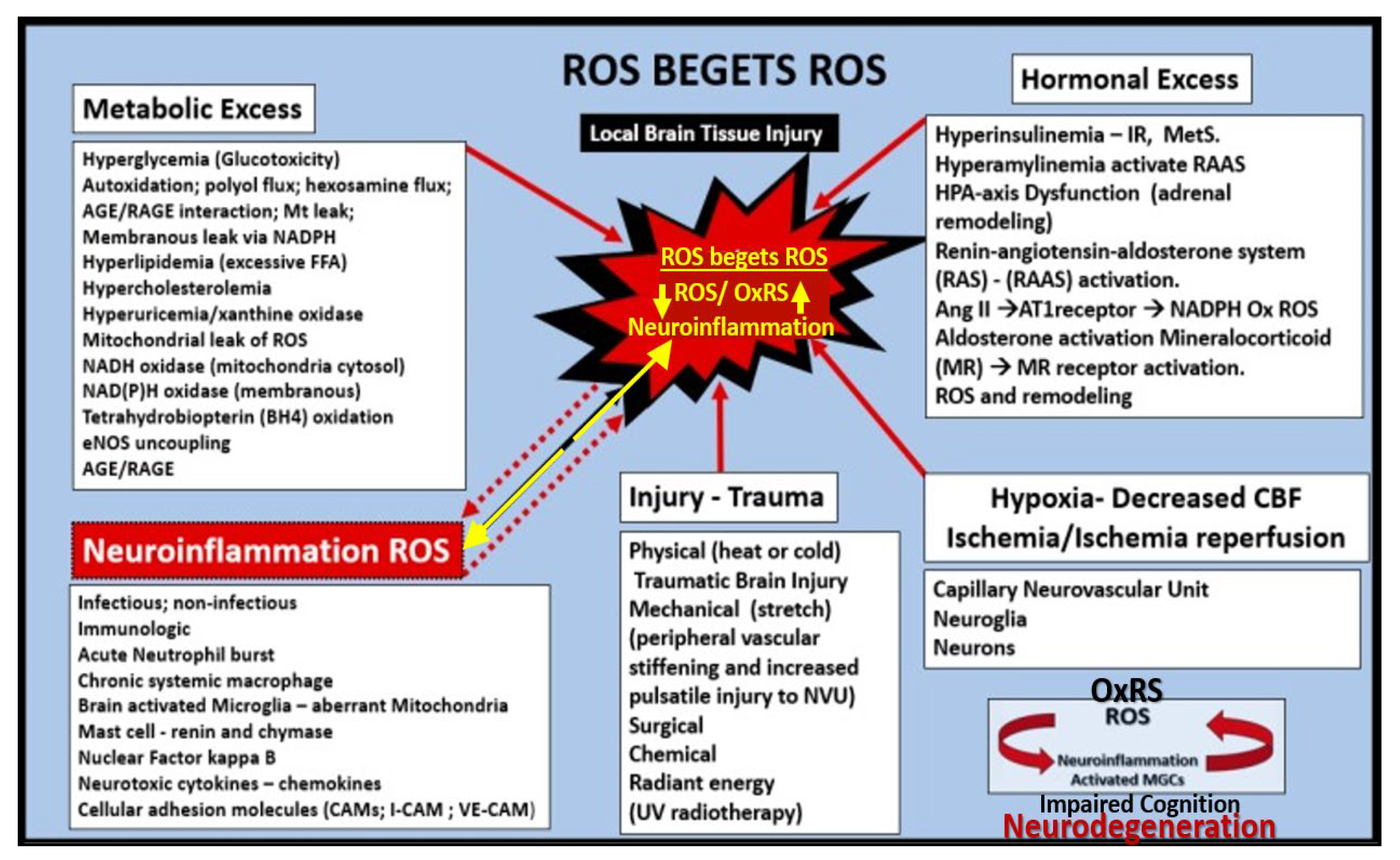
Figure 17.
Vicious cycles and multiple hits combine to result in small vessel disease (SVD), neuronal injury, neurodegeneration, brain atrophy, impaired cognition, and LOAD. Panel A depicts the importance of the vicious cycle between oxidative-redox stress (OxRS) and inflammation that result in microvessel remodeling and cerebral small vessel disease (SVD), which contribute to impaired cognition and the development of neurodegeneration. Panel B depicts the multiple hits that trigger the OxRS and neuroinflammation that are involved in the development of neuronal injury and neurodegeneration. Panel A is reproduced with permission by 4.0 [37]. Act, activation; BBB, blood-brain barrier; BBBdd, blood-brain barrier dysfunction and disruption; BEC, brain endothelial cell; CMBs, cerebral microbleeds; cnsCC, central nervous system cytokines/chemokines; dys, dysfunction; EPVS, enlarged perivascular spaces; MMP, matrix metalloproteinases; NFkappaB, nuclear factor kappa beta; pCC, peripheral cytokines/chemokines; ROS, reactive oxygen species; RONSS, reactive oxygen, nitrogen, sulfur species; RSI, reactive species interactome; TJ/AJ, tight and adherens junctions; VE, vascular endothelial.
Figure 17.
Vicious cycles and multiple hits combine to result in small vessel disease (SVD), neuronal injury, neurodegeneration, brain atrophy, impaired cognition, and LOAD. Panel A depicts the importance of the vicious cycle between oxidative-redox stress (OxRS) and inflammation that result in microvessel remodeling and cerebral small vessel disease (SVD), which contribute to impaired cognition and the development of neurodegeneration. Panel B depicts the multiple hits that trigger the OxRS and neuroinflammation that are involved in the development of neuronal injury and neurodegeneration. Panel A is reproduced with permission by 4.0 [37]. Act, activation; BBB, blood-brain barrier; BBBdd, blood-brain barrier dysfunction and disruption; BEC, brain endothelial cell; CMBs, cerebral microbleeds; cnsCC, central nervous system cytokines/chemokines; dys, dysfunction; EPVS, enlarged perivascular spaces; MMP, matrix metalloproteinases; NFkappaB, nuclear factor kappa beta; pCC, peripheral cytokines/chemokines; ROS, reactive oxygen species; RONSS, reactive oxygen, nitrogen, sulfur species; RSI, reactive species interactome; TJ/AJ, tight and adherens junctions; VE, vascular endothelial.
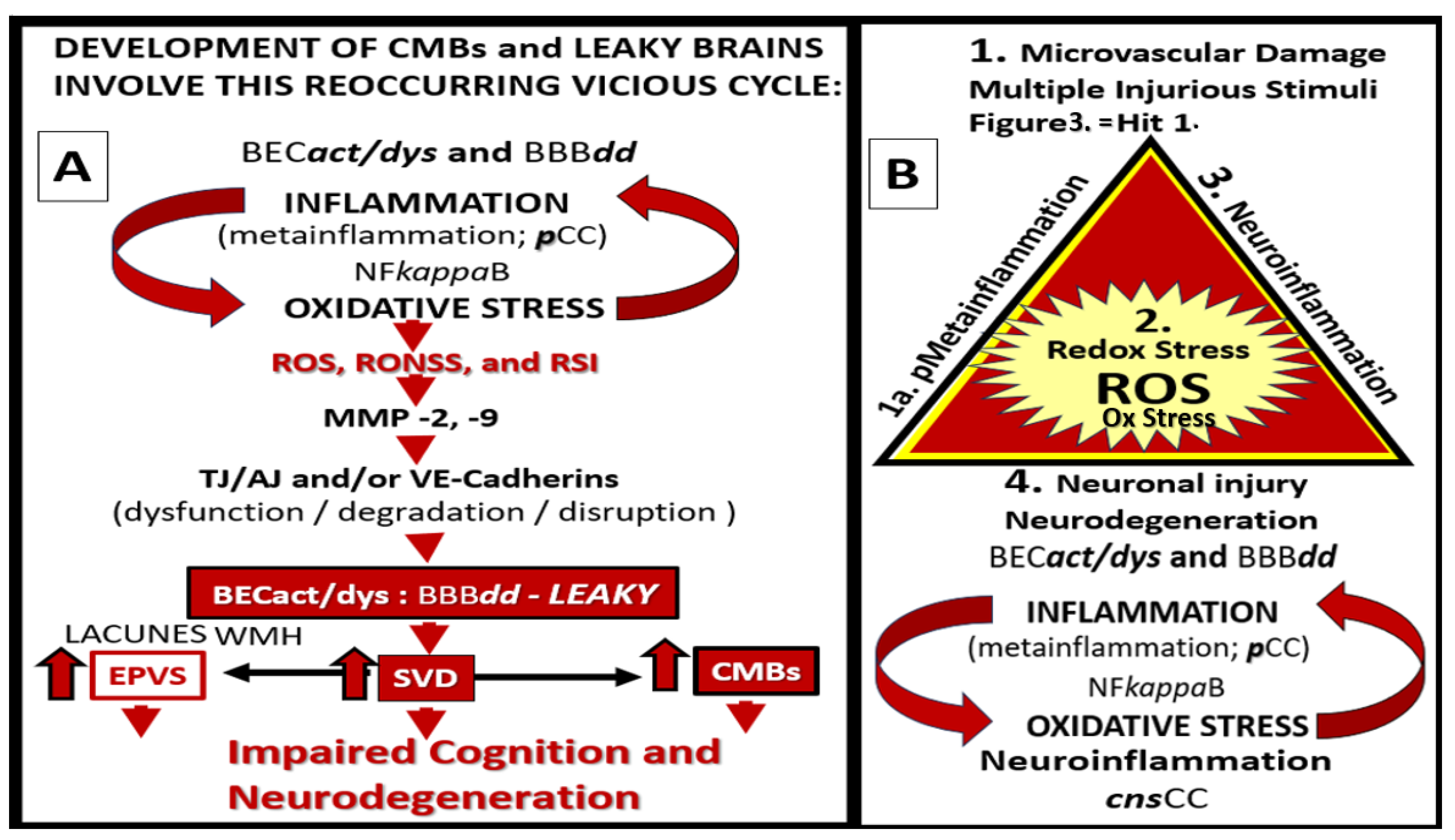
Figure 18.
Neuroinflammation induces neurodegeneration via proinflammatory central nervous system cytokines and chemokines (cnsCC), amyloid beta (Aβ), and tau. Image A reveals a cleaned transmission electron microscopy (TEM) image of a reactive microglia (rMGC). Image B depicts an illustration of a reactive astrocyte (rAC). Image C represents an illustration of a myelinated neuron. Image D depicts a portion of a myelinated neuronal axon that importantly depicts aberrant mitochondria (aMt) capable of leaking mitochondria reactive oxygen species (mtROS) and splitting of myelin that could impair informational transfer. Image E depicts a TEM image of a dendritic neurite (dn) outlined with yellow-dashed lines. Images F and G depict a TEM image of an Aβ plaque outlined by yellow-dashed lines. Images H and I depict immunohistologic light microscope images of Aβ. Image G scale bar = 5 μm. Image I scale bar = 20μm. Modified images A and D provided with permission by CC 4.0 [60,61]. Images H and I provided with permission by CC 4.0 [90]. aMt, aberrant mitochondria; CC, chromatin condensation; COX, cyclooxygenase; cnsCC, central nervous system cytokines chemokines; dn or dN, dendritic neurite; IL-1, interleukin 1; IL-1β, interleukin 1 beta; IL-6, interleukin 6; ISC, iron sulfur clusters; Lys, lysosome; mtROS, mitochondria reactive oxygen species; N, nucleus; NADPH Ox, nicotine adenine diphosphate oxidase; NFTs, neuro fibrillary tangles; NO, nitric oxide; NOX, NADPH Ox; OxRS, oxidative redox stress; rACs, reactive astrocytes; rMGCs, reactive microglia cells; TNFα, tumor necrosis alpha.
Figure 18.
Neuroinflammation induces neurodegeneration via proinflammatory central nervous system cytokines and chemokines (cnsCC), amyloid beta (Aβ), and tau. Image A reveals a cleaned transmission electron microscopy (TEM) image of a reactive microglia (rMGC). Image B depicts an illustration of a reactive astrocyte (rAC). Image C represents an illustration of a myelinated neuron. Image D depicts a portion of a myelinated neuronal axon that importantly depicts aberrant mitochondria (aMt) capable of leaking mitochondria reactive oxygen species (mtROS) and splitting of myelin that could impair informational transfer. Image E depicts a TEM image of a dendritic neurite (dn) outlined with yellow-dashed lines. Images F and G depict a TEM image of an Aβ plaque outlined by yellow-dashed lines. Images H and I depict immunohistologic light microscope images of Aβ. Image G scale bar = 5 μm. Image I scale bar = 20μm. Modified images A and D provided with permission by CC 4.0 [60,61]. Images H and I provided with permission by CC 4.0 [90]. aMt, aberrant mitochondria; CC, chromatin condensation; COX, cyclooxygenase; cnsCC, central nervous system cytokines chemokines; dn or dN, dendritic neurite; IL-1, interleukin 1; IL-1β, interleukin 1 beta; IL-6, interleukin 6; ISC, iron sulfur clusters; Lys, lysosome; mtROS, mitochondria reactive oxygen species; N, nucleus; NADPH Ox, nicotine adenine diphosphate oxidase; NFTs, neuro fibrillary tangles; NO, nitric oxide; NOX, NADPH Ox; OxRS, oxidative redox stress; rACs, reactive astrocytes; rMGCs, reactive microglia cells; TNFα, tumor necrosis alpha.
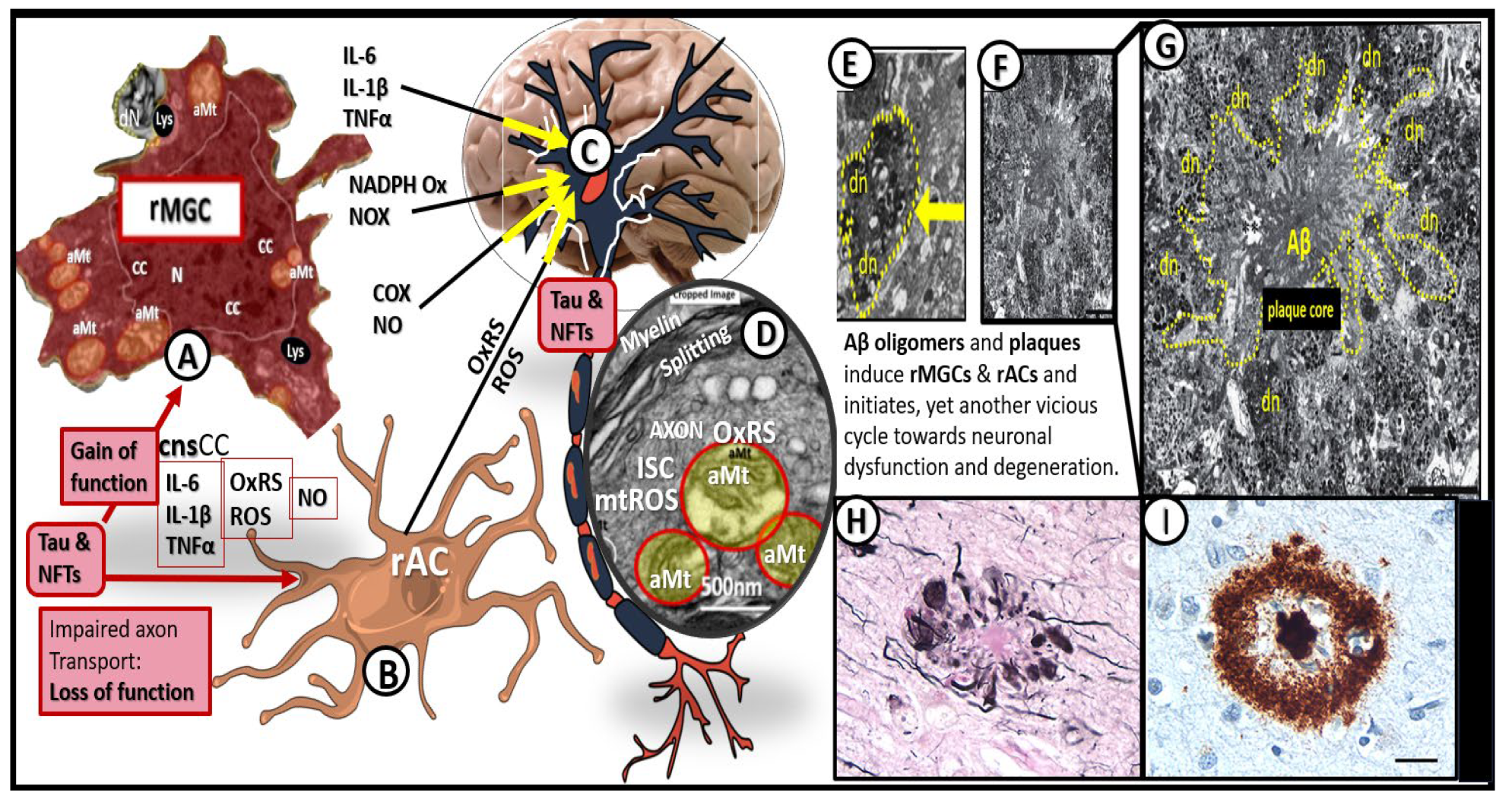
Figure 19.
Microvessel SVD definitely contributes to and are linked to the development of neurodegeneration. At least a half dozen mechanisms are involved. Importantly, note that number 5 points to the quartet of mechanisms involved in this linkage of SVD to neurodegeneration. Importantly these six mechanisms of linage seem to be initiated by brain endothelial cell activation and dysfunction (BECact/dys) with concurrent blood-brain barrier dysfunction and/or disruption (BBBdd). Aβ, amyloid beta; CBF, cerebral blood flow; EPVS, enlarged perivascular spaces, LOAD, late-onset Alzheimer’s disease; SVD, small vessel disease.
Figure 19.
Microvessel SVD definitely contributes to and are linked to the development of neurodegeneration. At least a half dozen mechanisms are involved. Importantly, note that number 5 points to the quartet of mechanisms involved in this linkage of SVD to neurodegeneration. Importantly these six mechanisms of linage seem to be initiated by brain endothelial cell activation and dysfunction (BECact/dys) with concurrent blood-brain barrier dysfunction and/or disruption (BBBdd). Aβ, amyloid beta; CBF, cerebral blood flow; EPVS, enlarged perivascular spaces, LOAD, late-onset Alzheimer’s disease; SVD, small vessel disease.
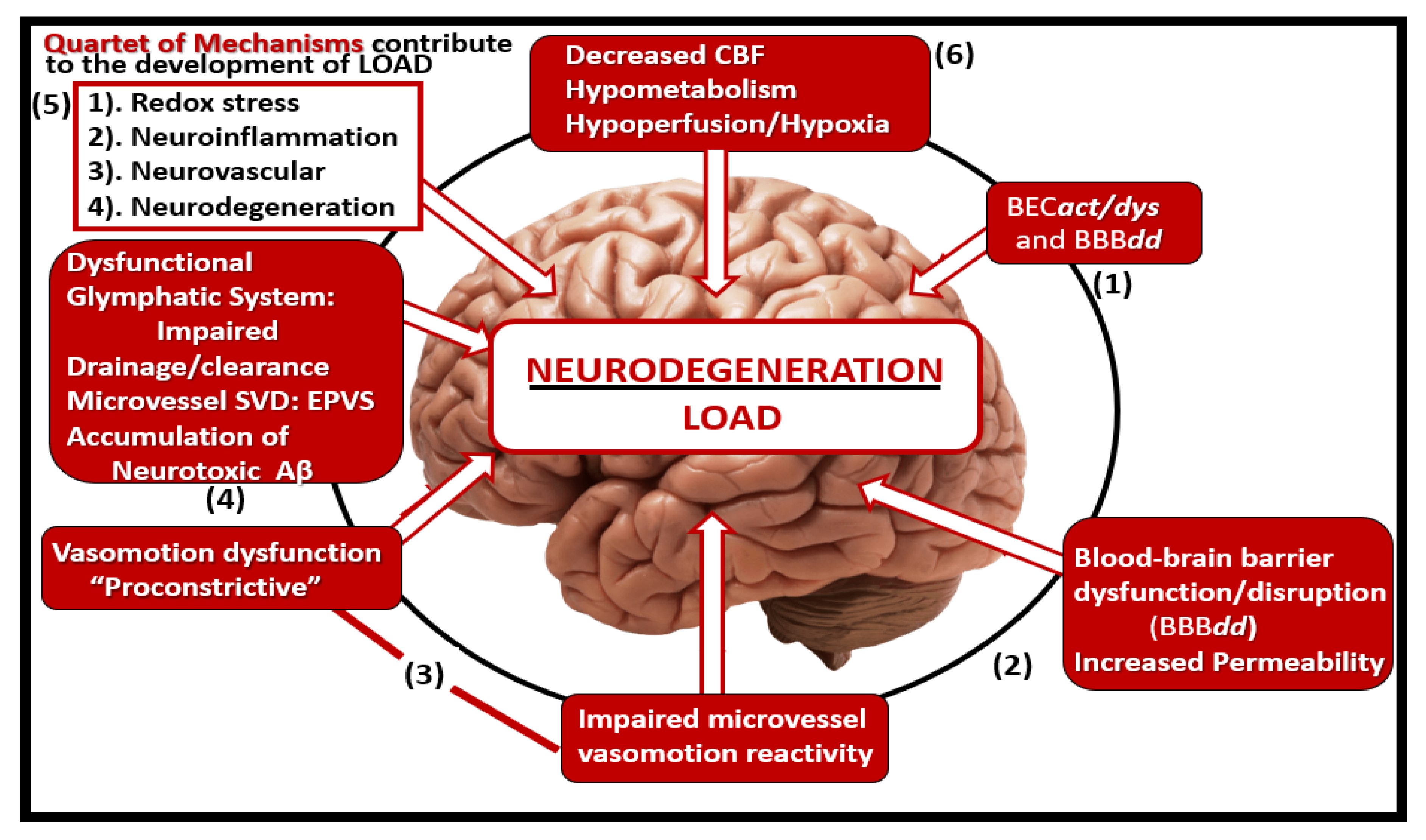
Figure 20.
Gross brain atrophy in the db/db obese diabetic model but not in the obese diabetic BTBR ob/ob model or db/db models treated with empagliflozin. Panel A demonstrates the normal gross brain in the control male model at 20-weeks of age. Panel B depicts marked brain atrophy at the time of surgical removal of the diabetic db/db models as compared to control models in Panel A. Note the marked atrophy or loss in the cortical-parietal-hippocampal regions (outlined by the yellow dashed lines) in comparison to the control in Panel A at 26-weeks of age and these remodeling changes were associated with a decrease in the brain wet weights upon removal. Panel C demonstrates the absence of remodeling atrophy in the ob/ob diabetic obese model at 20-weeks of age and also those db/db diabetic models treated with empagliflozin at 20-weeks of age. Modified panels A and B were provided with permission by CC 4.0 [117,118].
Figure 20.
Gross brain atrophy in the db/db obese diabetic model but not in the obese diabetic BTBR ob/ob model or db/db models treated with empagliflozin. Panel A demonstrates the normal gross brain in the control male model at 20-weeks of age. Panel B depicts marked brain atrophy at the time of surgical removal of the diabetic db/db models as compared to control models in Panel A. Note the marked atrophy or loss in the cortical-parietal-hippocampal regions (outlined by the yellow dashed lines) in comparison to the control in Panel A at 26-weeks of age and these remodeling changes were associated with a decrease in the brain wet weights upon removal. Panel C demonstrates the absence of remodeling atrophy in the ob/ob diabetic obese model at 20-weeks of age and also those db/db diabetic models treated with empagliflozin at 20-weeks of age. Modified panels A and B were provided with permission by CC 4.0 [117,118].
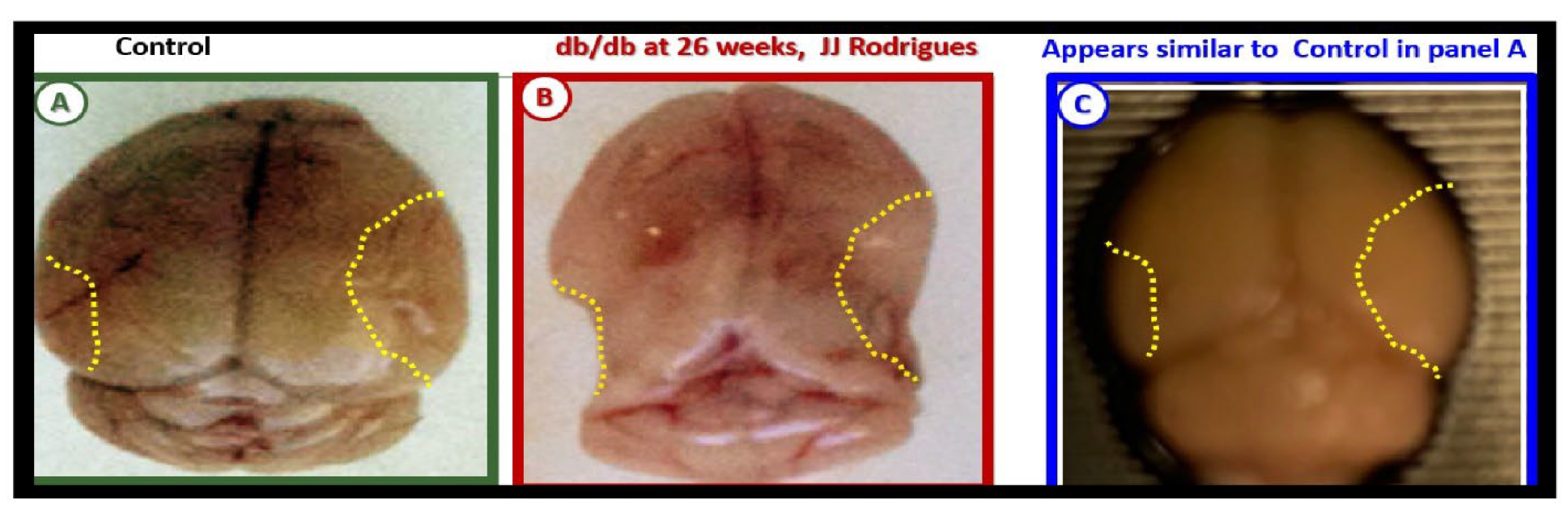
Figure 21.
Vascular contributions to cognitive impairment and dementia (VCID) is central to the development of neurodegeneration and late-onset Alzheimer;s disease (LOAD) and mixed dementia (MD). Cardio-cerebrovascular diseases including stroke (ischemic and hemorrhagic) contribute to brain endothelial cell activation and dysfunction (BECact/dys) with concurrent blood-brain barrier dysfunction and disruption (BBBdd) induce ischemia, hypoxia and chronic cerebral hypoperfusion (CCH) to result in neurodegeneration and dementia in LOAD and MD.
Figure 21.
Vascular contributions to cognitive impairment and dementia (VCID) is central to the development of neurodegeneration and late-onset Alzheimer;s disease (LOAD) and mixed dementia (MD). Cardio-cerebrovascular diseases including stroke (ischemic and hemorrhagic) contribute to brain endothelial cell activation and dysfunction (BECact/dys) with concurrent blood-brain barrier dysfunction and disruption (BBBdd) induce ischemia, hypoxia and chronic cerebral hypoperfusion (CCH) to result in neurodegeneration and dementia in LOAD and MD.
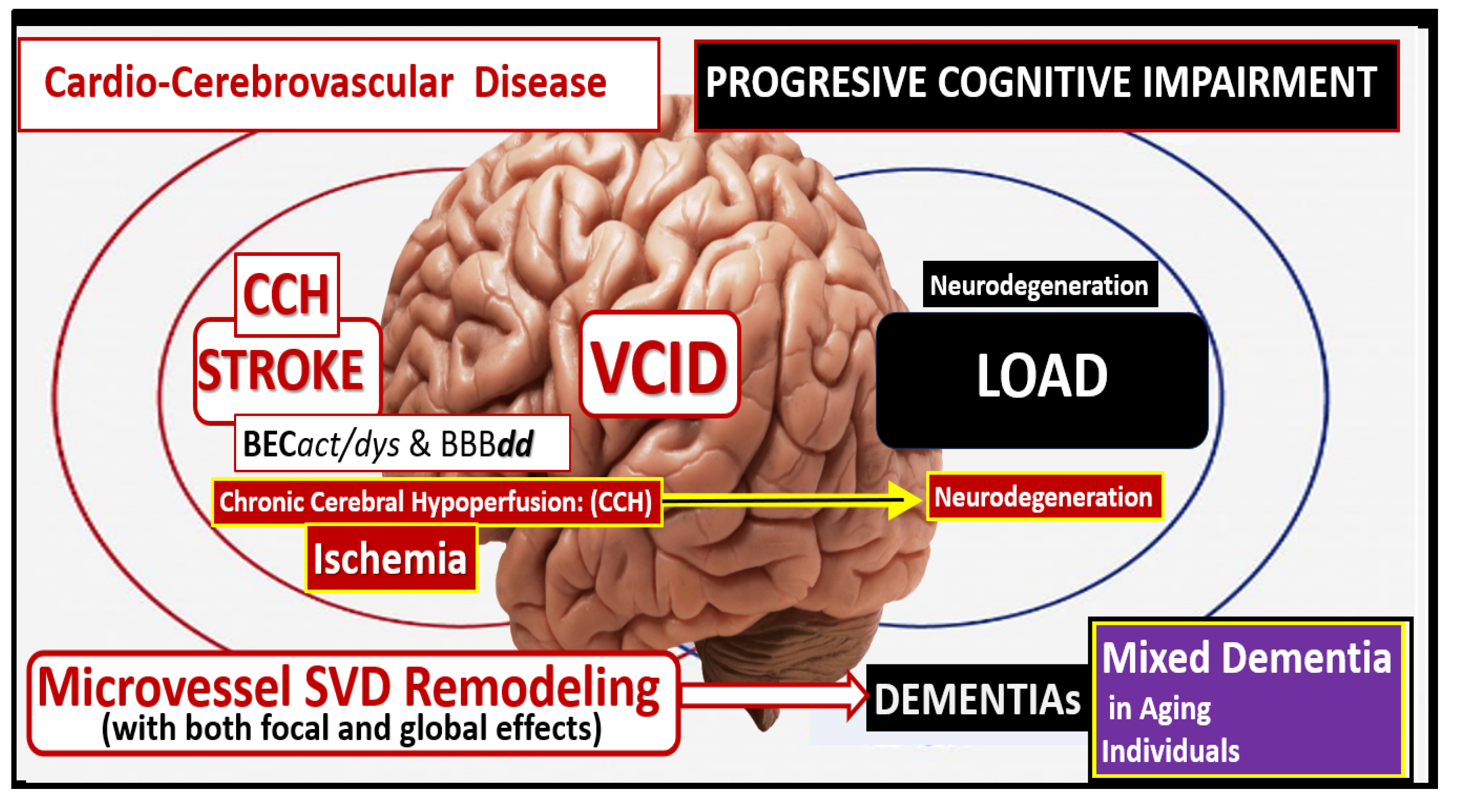
Figure 22.
The dementia quartet mechanisms intersect and contribute to neurodegeneration with neuronal synapse and neuron dysfunction and or loss with regional brain atrophy in LOAD and mixed dementia (MD) in addition to the small vessel disease (SVD) intersection and contribution to Neurodegeneration and MD.
Figure 22.
The dementia quartet mechanisms intersect and contribute to neurodegeneration with neuronal synapse and neuron dysfunction and or loss with regional brain atrophy in LOAD and mixed dementia (MD) in addition to the small vessel disease (SVD) intersection and contribution to Neurodegeneration and MD.
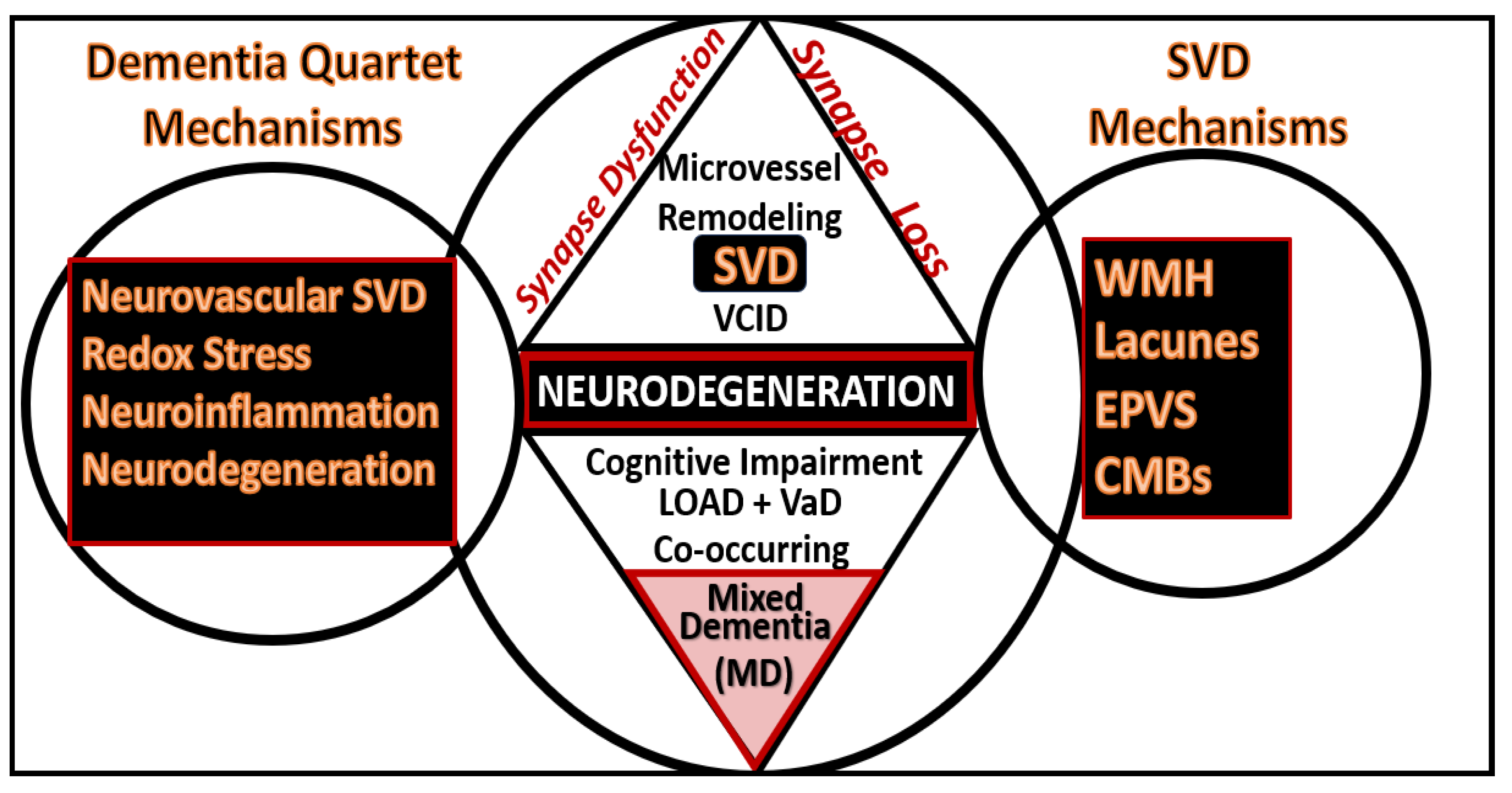
Figure 23.
Future directions in microvessel small vessel disease that contribute to late-onset Alzheimer’s disease. BECact/dys, brain endothelial cell activation and dysfunction; BBBdd, blood-brain barrier dysfunction and disruption; BH4, tetrahydrobiopterin; cnsCC, central nervous system cytokines chemokines; eNOS, endothelial nitric oxide synthase; H2S, hydrogen sulfide; LOAD, late-onset Alzheimer’s disease; MMP, matrix metalloproteinase; NO, nitric oxide; OxRS, oxidative redox stress; pCC, peripheral cytokines chemokines; SVD, small vessel disease; T, tesla; tPA, tissue-type plasminogen activator; VaD/VAD, vascular dementia. .
Figure 23.
Future directions in microvessel small vessel disease that contribute to late-onset Alzheimer’s disease. BECact/dys, brain endothelial cell activation and dysfunction; BBBdd, blood-brain barrier dysfunction and disruption; BH4, tetrahydrobiopterin; cnsCC, central nervous system cytokines chemokines; eNOS, endothelial nitric oxide synthase; H2S, hydrogen sulfide; LOAD, late-onset Alzheimer’s disease; MMP, matrix metalloproteinase; NO, nitric oxide; OxRS, oxidative redox stress; pCC, peripheral cytokines chemokines; SVD, small vessel disease; T, tesla; tPA, tissue-type plasminogen activator; VaD/VAD, vascular dementia. .
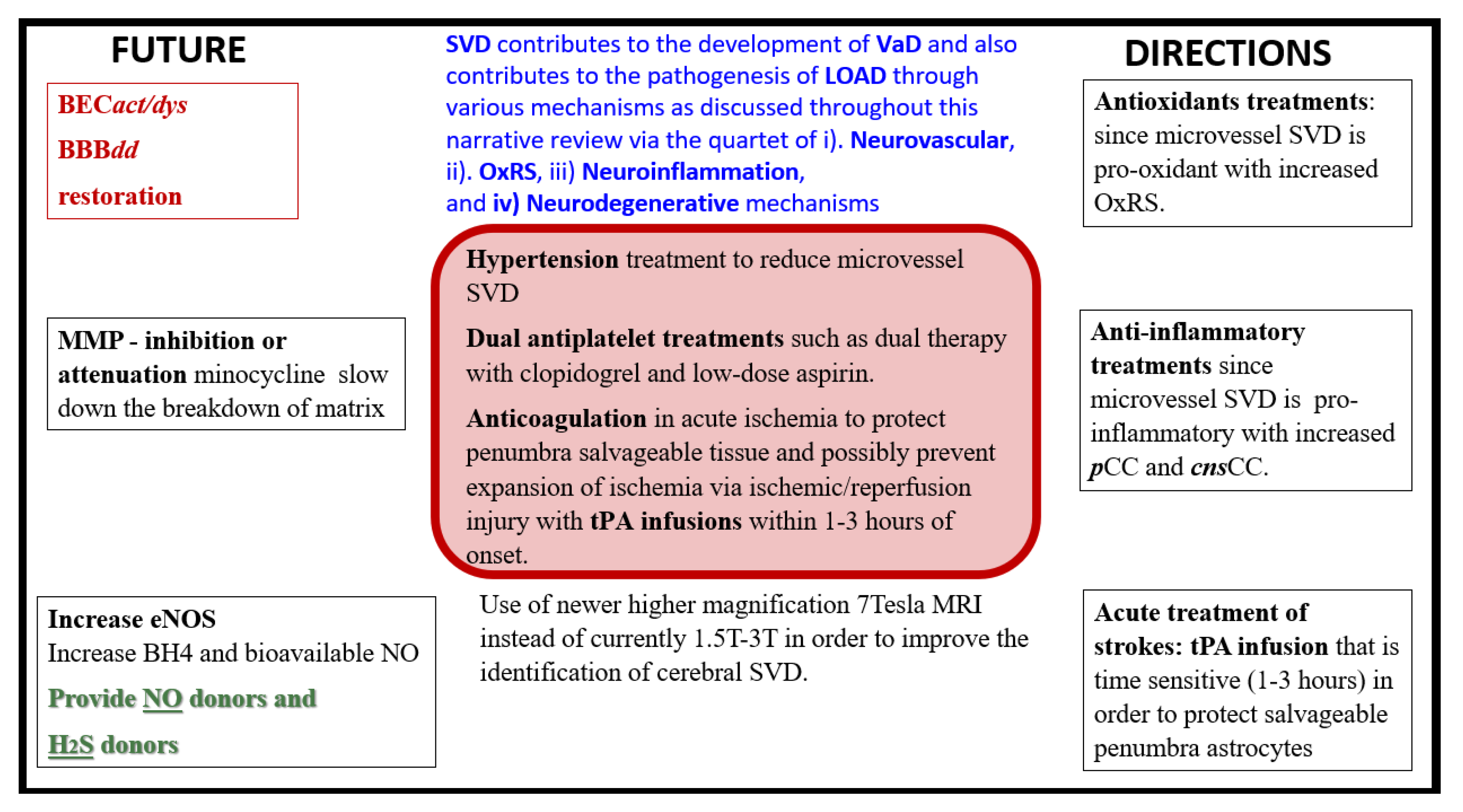
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



