
Submitted:
03 September 2024
Posted:
04 September 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
This study explores the integration of genome sequencing and polymerase chain reaction (PCR) based methods for tracking the diversity of Covid-19 variants in wastewater. The research focuses on monitoring various Omicron subvariants during a period of significant viral evolution. Genome sequencing, particularly using Oxford Nanopore Technology (ONT), provides a detailed view of emerging variants, surpassing the limitations of PCR-based detection kits that rely on known sequences. This study highlights the effectiveness of combining these methodologies to enhance early detection and inform public health strategies, especially in regions with limited clinical sequencing capabilities.
Keywords:
keyword 1
; Covid-19 2
; PCR 3
; Whole genome sequencing 4
; Wastewater-based epidemiology
1. Introduction
The Covid-19 pandemic, caused by the SARS-CoV-2 virus, highlighted the critical need for robust epidemiological tools to monitor and mitigate the spread of infectious diseases. One of the most significant challenges in mitigating the pandemic has been the emergence and spread of viral variants, which can have altered transmissibility, virulence, and vaccine efficacy [1,2]. Genome sequencing has emerged as a pivotal technology in tracking the temporal diversity of SARS-CoV-2 variants, providing invaluable insights into the evolutionary dynamics of the virus and informing public health responses.
Genome sequencing allows for the characterization of the detailed genetic makeup of SARS-CoV-2, enabling the identification of mutations and the classification of viral lineages. This technology has been instrumental in detecting variants of concern (VOCs) and variants of interest (VOIs), such as Alpha (B.1.1.7), Delta (B.1.617.2), and Omicron (B.1.1.529), which have had significant impacts on the trajectory of the pandemic [3,4]. By analyzing the viral genome, researchers can track the emergence of these variants and their spread across different regions and time periods, providing a real-time map of viral evolution [5,6,7].
Wastewater-based epidemiology (WBE) has emerged as a complementary approach to clinical testing, offering a non-invasive and cost-effective means to monitor community-level infections. This method involves the collection and analysis of wastewater samples to detect the presence of SARS-CoV-2 genetic material, allowing for the early detection of outbreaks and the assessment of variant prevalence in the population [8,9]. The integration of genome sequencing with WBE has proven particularly powerful, enabling the detection of low-frequency variants and providing a comprehensive view of viral diversity in a community [6,10,11,12].
Oxford Nanopore Technology (ONT) has been widely adopted for sequencing SARS-CoV-2 due to its portability, rapid turnaround time, and ability to generate long reads, which are beneficial for detecting complex variants and reconstructing viral genomes from mixed samples [9,13]. Studies employing ONT for wastewater sequencing have successfully identified and tracked the temporal diversity of SARS-CoV-2 variants, demonstrating its utility in public health surveillance and outbreak management [9,14,15,16].
Genome sequencing offers a more comprehensive approach to identifying COVID-19 variants compared to PCR testing, which was the pioneering tool developed to detect Covid-19 in wastewater early in the pandemic [8,17]. While PCR tests are effective in detecting the presence of the virus, they do not provide detailed information about the genetic sequence, which is crucial for identifying and tracking specific variants [18]. PCR-based identification of Covid-19 variants is also limited to the detection of specific, known mutations, whereas genome sequencing deciphers the entire genetic code of the virus, allowing researchers to detect new and emerging mutations and understand how the virus is evolving [6,16,19,20].
Few studies have compared the usefulness and accuracy of the two methods side-by-side [21], and the majority that have were using clinical nasal swabs [4,22,23], or a combination of clinical and wastewater samples [24,25]. In a review of 80 studies on Covid-19 variant determination, only two compared sequencing and PCR variant detection in wastewater [21,26,27]. This study aims to compare the application of genome sequencing in tracking the temporal diversity of Covid-19 variants with a commercially available quantitative polymerase chain reaction wastewater variant kit. The study occurred during a period of transition between various Omicron subvariants: BA.1, BA.2, BA.4, BA.5, XBB, and BQ.1. Additionally, the study evaluates the effectiveness of these combined methodologies in offering early warning signs for public health interventions and in understanding the geographical spread and persistence of different variants. This integrated approach is particularly relevant for informing targeted public health strategies, especially in rural areas with limited clinical sequencing capabilities. The insights gained from this study will contribute to the optimization of wastewater-based epidemiology as a valuable tool in managing current and future pandemics.
2. Materials and Methods
2.1. Study Location
Samples were gathered weekly, beginning June 2021, from 16 sites across Michigan’s Eastern Upper Peninsula (EUP). The EUP includes Alger, Chippewa, Luce, Mackinac, and Schoolcraft Counties, totaling about 70,000 residents over 5,566 square miles, with an average density of 11.3 people per square mile. Only 13 of the sites were included in this study: two did not sample during the winter due to prohibitive ice and snow cover; and one did not have any samples during the study period, January 9, 2023 - April 27, 2023, that met the minimum criteria for genome sequencing (discussed later). Figure 1 shows the sampling site locations included in this study.
2.2. Wastewater Sampling
Wastewater grab samples (250 mL) were collected from wastewater influent streams once per week. Samples were refrigerated or kept on ice until processed (up to 48 hours).
2.3. Viral Concentration
Each sample of raw sewer water (100 mL) was combined with 8% (w/vol) molecular grade polyethylene glycol (PEG) 8000 (Fisher Scientific) and 0.2 M NaCl (w/v) (Fisher Scientific). After mixing for two hours at 230 rpm and 4°C, samples were centrifuged at 4200 x g for 45 minutes at 4°C. The supernatant was removed using a sterile serological pipet, and the pellet was resuspended in the residual liquid.
2.4. RNA Extraction
Viral ribonucleic acid (RNA) was extracted from concentrated samples using the Qiagen QiAmp Viral RNA Minikit following the manufacturer’s custom protocol for the QIACube Connect (Qiagen, Germany). RNA extraction resulted in a final elution volume of 80 µL. Extracted RNA was used immediately for viral detection and quantification or stored at -80℃ for later use.
2.5. Initial Virus Detection and Quantification
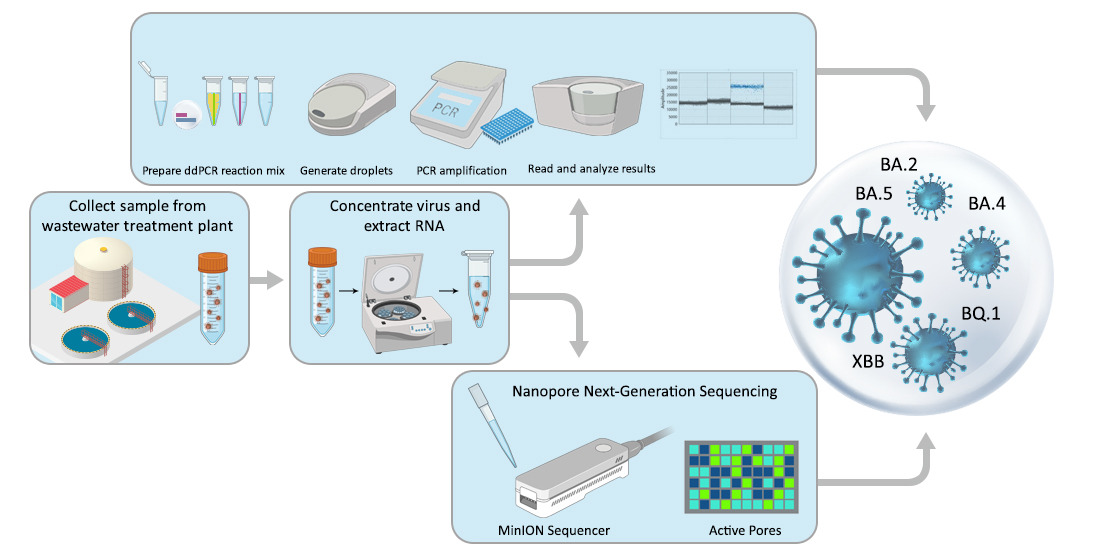
Bio-Rad’s One-step RT-ddPCR Advanced kit was used with the Bio-Rad Automated Droplet Generator and the QX200 ddPCR system to quantify N1, N2, and Phi6 RNA (Bio-Rad, USA). Each reaction contained a final concentration of 1x Supermix (Bio-Rad, USA), 20 U/µl reverse transcriptase (RT) (Bio-Rad, USA), 15 nM DTT (Bio-Rad, USA), 900 nmol of each primer (BioSearch Tech), 250 nmol of each probe (BioSearch Tech), 1 µL of nuclease-free water, and 5.5 µL of template RNA. The final reaction volume was 22 µL. Quality control samples on each plate included a non-template control, extraction control, and processing blank. Samples, controls, and blanks were analyzed in triplicate.
Droplets were generated in the Bio-Rad Automated Droplet Generator (ADG) by combining 20 µL of reaction volume with 70 µL of droplet generator oil (Bio-Rad, USA), resulting in a reaction mixture-oil emulsion of 40 µL containing up to 20,000 droplets. The droplets were transferred, via the ADG, to a 96-well PCR plate that was then heat-sealed with foil and put in a Bio-Rad C1000 deep-well thermal cycler for PCR amplification under the following conditions: 25℃ for 3 minutes, 50℃ for 60 minutes, 95℃ for 10 minutes, 40 cycles of 95℃ for 30 seconds and 55℃ for 1 minute, 98℃ for 10 minutes, and hold at 4℃. After thermal cycling, the plate was transferred to the Bio-Rad QX200 Droplet Reader for concentration determination via spectrophotometric detection of fluorescent probe signal in gene-target positive droplets. Amplitude thresholding was performed manually for each analysis using the QuantaSoft (BioRad) software. Lower limit of detection, N1, N2, and Phi6 gene copies for each sample were then determined using the QuantaSoft output.
2.6. Variant Determination using ddPCR
Samples that were N1 or N2-positive in the initial detection were then tested for variant detection using the BA.1 (A67V; del69-70 mutations) and BA.2 (R408S mutation) discrimination assay kit (GT Molecular). Each reaction contained 5.5 µL Supermix (Bio-Rad, USA), 2.2 µL reverse transcriptase (RT) (Bio-Rad, USA), 1.1 µL DTT (Bio-Rad, USA), 1 µL GT primer-probe solution (GT Molecular), 6.7 µL of nuclease-free water, and 5.5 µL of template RNA for a total reaction volume of 22 µL. Droplet generation was performed in the same manner as previously described. Thermal cycling conditions were as follows: 50℃ for 60 minutes, 95℃ for 10 minutes, 45 cycles of 94℃ for 30 seconds and 60℃ for 1 minute, 98℃ for 10 minutes, and hold at 4℃ for 30 minutes. Concentration and target gene copies were determined in the same manner as described above for N1/N2 determination.
2.7. Variant Determination using Genome Sequencing
Previously-extracted RNA was used for genome sequencing. Sequencing was performed, retrospectively, several months after ddPCR (digital droplet polymerase chain reaction) variant detection. Only samples with an N1 and N2 combined concentration of ≥9000 gene copies (GC) per 100 mL were sequenced [28]. Forty-three samples during the study period (January 9, 2023 - April 27, 2023) met this criteria for sequencing.
Reverse transcription was performed using the Midnight RT PCR Expansion kit (EXP-MRT001, Oxford Nanopore Technologies). An input volume of 8 µL of sample RNA was mixed with 2 µL LunaScript RT Supermix, then thermal cycled in the following conditions: 25℃ for 2 minutes, 55℃ for 10 minutes, 95℃ for 1 minute, and hold at 4℃. Midnight Primer pools A and B were then mixed according to manufacturer protocol and aliquoted 10 µL each into a clean 96-well plate. To each primer pool, 2.5 µL of RT reaction was added. Thermal cycling was performed under the following conditions: 98℃ for 30 seconds, 35 cycles of 98℃ for 30 seconds, 61℃ for 2 minutes, and 65℃ for 3 minutes, and hold at 4℃.
Addition of barcodes was performed according to manufacturer protocol using the Rapid Barcoding kit (SQK-RBK110.96, Oxford Nanopore Technologies). For each sample, 2.5 µL from primer pools A and B were combined with 2.5 µL or nuclease-free water in a clean 96-well plate. Then, 2.5 µL of Rapid Barcode were added to the corresponding sample wells. The barcoded plate was incubated at 39℃ for 2 minutes, then 88℃ for 2 minutes. Barcoded samples were then pooled in a clean Eppendorf DNA LoBind tube. Half of the pooled sample was used and half was stored at 4℃ in case needed for reloading during the sequencing run. To the pooled, barcoded sample, an equal volume of AMPure XP Beads were added, then mixed on a rotator mixer at room temperature for five minutes. After mixing, the tube was put on a magnet and the beads were washed twice with 1 mL of 80% ethanol. Residual ethanol was discarded and the pellet was resuspended with 15 µL of elution buffer and incubated at room temperature for 10 minutes. Up to 800 ng of DNA library was transferred to a clean tube, and combined with 1 µL of Rapid Adaptor F.
The prepared DNA library was combined with 37.5 µL Sequencing Buffer and 25.5 µL Loading Beads and loaded into a MinION R9.4.1 flow cell which was placed onto a MinION Mk1C sequencer (Oxford Nanopore Technologies) for 24-72 hours using the MinKNOW software. Data was analyzed using EPI2ME’s (Oxford Nanopore Technologies) FastqQC+ARTIC+NextClade workflow with the ARTIC nCoV-2019 protocol.
3. Results and Discussion
During the study period, N1+N2 gene copies fluctuated between 1163 and 2.8 million, with a mean of 32,127, and a median of 3607. Of the 210 samples tested from the 13 included sites, 43 met the minimum criteria of 9000 N1+N2 combined gene copies. Figure 2 shows the normalized N1+N2 gene copies by site over the study period.
All of the samples sequenced contained genetic markers from the Omicron family of subvariants. Of the 43 samples compared, 39.5% of samples had matching results between the GT Molecular PCR kits and ONT sequencing. Of these, 4% were an exact match, and 33.5% were an “assumed” match, meaning that if the GT kit gave positive results for both BA.1 and BA.2 it was possible that BA.4 or BA.5 were present, based on shared mutations among the four subvariants [29,30]. Seven percent of the samples did not register a positive result in the GT Molecular kit (<LOD) but were assigned a clade (a group of similar viruses based on genetics) using sequencing. Two of these samples were identified as BA.2 and one as recombinant. The remaining 53.5% of samples were not matched with the two methods utilized. The majority of non-matches were assigned as “recombinant” using sequencing (35%). Others were BA.3, XBB, and BQ.1, for which there existed no markers in the GT Molecular kit being used (see Table 1).
The period of January through April 2023 saw several Omicron subvariants circulating in the Midwest region of the United States, including BA.2, BA.4, BA.5, XBB, and BQ.1, with the dominant subvariant being XBB [31]. Although BA.4/5 variant PCR kits were available at the time of initial analysis, BA.1/2 kits were still being used while transitioning to genome sequencing. Furthermore, the frequency of BA.2, BA.2.12.1, BA.4, and BA.5, all BA.2 relatives, fluctuated during this transition period, making the BA.2 kits still relevant [30]. The paired sequencing and PCR data illustrate the temporal limitations of using PCR variant kits alone to determine current circulating Covid-19 variants. While BA.2 was a common subvariant in circulation in the study region during the study period, early instances of XBB (1/18/23) and BQ.1 (1/30/23) in the region were missed during initial PCR analysis (see Table 2). These results reflect that even though BA.4/BA.5 PCR kits were available, incidence of newer Omicron subvariants like XBB and BQ.1 would still have been missed because the PCR kits were not capable of detecting those variants at that time. Figure 3 shows the temporal occurrence of subvariants determined by PCR and ONT methods.
Although more sensitive to detecting key spike protein mutations [16], PCR kits for detecting COVID-19 variants have a significant limitation: they rely on the specific genetic sequences of known variants. Consequently, these kits require that a variant is already in circulation before materials can be created to detect it. This limitation means that PCR kits might not be effective in identifying new variants immediately as they emerge, potentially delaying the detection and tracking of these new strains [32,33]. Furthermore, recent variants of concern contain more than 30 mutations in the spike protein, complicating the detection of these variants and enhancing their ability to evade detection by standard testing methods like ddPCR [34]. In contrast, genome sequencing incorporates data from global sequencing efforts, often within days of new sequences being submitted [35]. This allows for real-time tracking and analysis of SARS-CoV-2 variants, providing insights into the virus’s spread and evolution without the delay for development of new detection materials [6,16].
Another advantage of sequencing wastewater samples is that it can provide data at a population scale in places where sequencing clinical samples is limited by resources [12,36], especially when overall clinical testing has declined significantly [37,38]. During the study period, the Michigan department of Health and Human Services reports that only two clinical samples from the entire region were sequenced (S.S., personal communication, 6/18/2024). Given that there were at least five different subvariants circulating at the time, each with potentially differing transmission and virulence characteristics, sequencing only two samples would provide little information about the distribution pattern and evolution of the virus across an expansive geographic region like the Eastern Upper Peninsula of Michigan.
In summary, using PCR for the initial detection and quantification of COVID-19 virus particles in wastewater remains one of the most effective, time-efficient, and cost-efficient methods for monitoring the virus within a population [39,40]. While PCR-based variant detection kits are highly sensitive to spike protein mutations [16], they depend on known genetic sequences, resulting in delays in identifying emerging variants [32,33]. In contrast, genome sequencing technologies like ONT offer early insights into new and emerging variants spreading within communities, surpassing the capabilities of variant-specific PCR tests [16]. This study underscores the value of ONT sequencing of wastewater in providing real-time information about dominant circulating variants, equipping health officials with critical data for making targeted and effective public health decisions. Real-time data is particularly crucial in regions like the Upper Peninsula of Michigan, where limited clinical samples are sequenced. Both methods provide data about circulating variants, but ONT provides a more complete picture in rapidly evolving Covid-19 scenarios by detecting individual mutations, which allows for the identification of any current variant as well as emerging or yet-unknown variants or subvariants. This is especially helpful during transitions between highly related subvariants like the BA.2 / BA.4 / BA.5 family.
Author Contributions
Conceptualization, M.J., T.N., B.S., and D.W.; formal analysis, M.J..; writing—original draft preparation, M.J.; writing—review and editing, M.J., T.N., B.S., and D.W.; project administration, T.N., B.S., and D.W. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was provided by the Michigan Department of Health and Human Services via the Epidemiology and Laboratory Capacity: Enhancing Detection Expansion through Coronavirus Response and Relief Supplemental Appropriations Act of 2021 (P.L. 116-260).
Data Availability Statement
Data available upon request from corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kaku, Y.; Okumura, K.; Padilla-Blanco, M.; Kosugi, Y.; Uriu, K.; A Hinay, A.; Chen, L.; Plianchaisuk, A.; Kobiyama, K.; Ishii, K.J.; et al. Virological characteristics of the SARS-CoV-2 JN.1 variant. Lancet Infect. Dis. 2024, 24, e82. [Google Scholar] [CrossRef] [PubMed]
- Kunal, S. , Ish, P., Aditi, & Gupta, K. (2022). Emergence of COVID-19 variants and its global impact. In S. Adibi, P. Griffin, M. Sanicas, M. Rashidi, & F. Lanfranchi (Eds.), Frontiers of COVID-19: Scientific and Clinical Aspects of the Novel Coronavirus 2019 (pp. 183–201). Springer International Publishing. [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Wang R, Chen J, Gao K, Hozumi Y, Yin C, Wei GW. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun Biol. 2021 Feb 15;4(1):228. 10.1038/s42003-021-01754-6. Erratum in: Commun Biol. 2021 Mar 3;4(1):311. 10.1038/s42003-021-01867-y.
- Davis, C.; Logan, N.; Tyson, G.; Orton, R.; Harvey, W.T.; Perkins, J.S.; Mollett, G.; Blacow, R.M.; Peacock, T.P.; Barclay, W.S.; et al. Reduced neutralisation of the Delta (B.1.617.2) SARS-CoV-2 variant of concern following vaccination. PLOS Pathog. 2021, 17, e1010022. [Google Scholar] [CrossRef] [PubMed]
- Pilapil, J.D.; Notarte, K.I.; Yeung, K.L. The dominance of co-circulating SARS-CoV-2 variants in wastewater. Int. J. Hyg. Environ. Heal. 2023, 253, 114224. [Google Scholar] [CrossRef] [PubMed]
- Rothman, J.A.; Saghir, A.; Zimmer-Faust, A.G.; Langlois, K.; Raygoza, K.; Steele, J.A.; Griffith, J.F.; Whiteson, K.L. Longitudinal Sequencing and Variant Detection of SARS-CoV-2 across Southern California Wastewater. Appl. Microbiol. 2024, 4, 635–649. [Google Scholar] [CrossRef]
- Ahmed, W.; Bivins, A.; Simpson, S.L.; Bertsch, P.M.; Ehret, J.; Hosegood, I.; Metcalfe, S.S.; Smith, W.J.; Thomas, K.V.; Tynan, J.; et al. Wastewater surveillance demonstrates high predictive value for COVID-19 infection on board repatriation flights to Australia. Environ. Int. 2022, 158, 106938–106938. [Google Scholar] [CrossRef]
- Barbé, L.; Schaeffer, J.; Besnard, A.; Jousse, S.; Wurtzer, S.; Moulin, L.; OBEPINE Consortium; Le Guyader, F. S.; Desdouits, M. SARS-CoV-2 Whole-Genome Sequencing Using Oxford Nanopore Technology for Variant Monitoring in Wastewaters. Front. Microbiol. 2022, 13, 889811. [Google Scholar] [CrossRef]
- Bar-Or, I.; Weil, M.; Indenbaum, V.; Bucris, E.; Bar-Ilan, D.; Elul, M.; Levi, N.; Aguvaev, I.; Cohen, Z.; Shirazi, R.; et al. Detection of SARS-CoV-2 variants by genomic analysis of wastewater samples in Israel. Sci. Total. Environ. 2021, 789, 148002. [Google Scholar] [CrossRef]
- Cancela, F.; Ramos, N.; Smyth, D.S.; Etchebehere, C.; Berois, M.; Rodríguez, J.; Rufo, C.; Alemán, A.; Borzacconi, L.; López, J.; et al. Wastewater surveillance of SARS-CoV-2 genomic populations on a country-wide scale through targeted sequencing. PLOS ONE 2023, 18, e0284483. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Kantor, R.S.; Olm, M.R.; Whitney, O.N.; Al-Shayeb, B.; Lou, Y.C.; Flamholz, A.; Kennedy, L.C.; Greenwald, H.; Hinkle, A.; et al. Genome Sequencing of Sewage Detects Regionally Prevalent SARS-CoV-2 Variants. mBio 2021, 12. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC Multiplex PCR Method for SARS-CoV-2 Genome Sequencing Using Nanopore. Biorxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Izquierdo-Lara, R.; Elsinga, G.; Heijnen, L.; Munnink, B.B.O.; Schapendonk, C.M.; Nieuwenhuijse, D.; Kon, M.; Lu, L.; Aarestrup, F.M.; Lycett, S.; et al. Monitoring SARS-CoV-2 Circulation and Diversity through Community Wastewater Sequencing, the Netherlands and Belgium. Emerg. Infect. Dis. 2021, 27, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Surya, K.; Buyukyoruk, M.; Cicha, C.; Vanderwood, K.K.; Wilkinson, R.; Wiedenheft, B. Temporal Detection and Phylogenetic Assessment of SARS-CoV-2 in Municipal Wastewater. Cell Rep. Med. 2020, 1, 100098–100098. [Google Scholar] [CrossRef]
- Vigil, K.; D'Souza, N.; Bazner, J.; Cedraz, F.M.-A.; Fisch, S.; Rose, J.B.; Aw, T.G. Long-term monitoring of SARS-CoV-2 variants in wastewater using a coordinated workflow of droplet digital PCR and nanopore sequencing. Water Res. 2024, 254, 121338. [Google Scholar] [CrossRef] [PubMed]
- Medema, G.; Heijnen, L.; Elsinga, G.; Italiaander, R.; Brouwer, A. Presence of SARS-Coronavirus-2 RNA in Sewage and Correlation with Reported COVID-19 Prevalence in the Early Stage of the Epidemic in The Netherlands. Environ. Sci. Technol. Lett. 2020, 7, 511–516. [Google Scholar] [CrossRef]
- World Health Organization. (2022). Methods-for-the-detection-char-SARS-CoV-2-variants_2nd update_final.pdf (pp. 1–14) [Technical Report]. https://www.ecdc.europa.eu/sites/default/files/documents/Methods-for-the-detection-char-SARS-CoV-2-variants_2nd%20update_final.pdf.
- Child, H.T.; Airey, G.; Maloney, D.M.; Parker, A.; Wild, J.; McGinley, S.; Evens, N.; Porter, J.; Templeton, K.; Paterson, S.; et al. Comparison of metagenomic and targeted methods for sequencing human pathogenic viruses from wastewater. mBio 2023, 14, e0146823. [Google Scholar] [CrossRef]
- Joshi, M.; Kumar, M.; Srivastava, V.; Kumar, D.; Rathore, D.S.; Pandit, R.; Graham, D.W.; Joshi, C.G. Genetic sequencing detected the SARS-CoV-2 delta variant in wastewater a month prior to the first COVID-19 case in Ahmedabad (India). Environ. Pollut. 2022, 310, 119757. [Google Scholar] [CrossRef]
- Tiwari, A.; Adhikari, S.; Zhang, S.; Solomon, T.B.; Lipponen, A.; Islam, A.; Thakali, O.; Sangkham, S.; Shaheen, M.N.F.; Jiang, G.; et al. Tracing COVID-19 Trails in Wastewater: A Systematic Review of SARS-CoV-2 Surveillance with Viral Variants. Water 2023, 15, 1018. [Google Scholar] [CrossRef]
- Lind, A.; Barlinn, R.; Landaas, E.T.; Andresen, L.L.; Jakobsen, K.; Fladeby, C.; Nilsen, M.; Bjørnstad, P.M.; Sundaram, A.Y.; Ribarska, T.; et al. Rapid SARS-CoV-2 variant monitoring using PCR confirmed by whole genome sequencing in a high-volume diagnostic laboratory. J. Clin. Virol. 2021, 141, 104906–104906. [Google Scholar] [CrossRef]
- Umunnakwe, C. , Makatini, Z., Maphanga, M., Mdunyelwa, A., Mlambo, K. M., Manyaka, P., Nijhuis, M., Wensing, A., & Tempelman, H. (2022). Evaluation of a commercial SARS-CoV-2 multiplex PCR genotyping assay for variant identification in resource-scarce settings. PLOS ONE, 17, e0269071. https://doi.org/10.1371/journal.pone.0269071Lind, A., Barlinn, R., Landaas, E. T., Andresen, L. L., Jakobsen, K., Fladeby, C., Nilsen, M., Bjørnstad, P. M., Sundaram, A. Y. M., Ribarska, T., Müller, F., Gilfillan, G. D., & Holberg-Petersen, M. (2021). Rapid SARS-CoV-2 variant monitoring using PCR confirmed by whole genome sequencing in a high-volume diagnostic laboratory. Journal of Clinical Virology, 141, 104906. https://doi.org/10.1016/j.jcv.2021.104906.
- D'Agostino, Y.; Rocco, T.; Ferravante, C.; Porta, A.; Tosco, A.; Cappa, V.M.; Lamberti, J.; Alexandrova, E.; Memoli, D.; Terenzi, I.; et al. Rapid and sensitive detection of SARS-CoV-2 variants in nasopharyngeal swabs and wastewaters. Diagn. Microbiol. Infect. Dis. 2022, 102, 115632–115632. [Google Scholar] [CrossRef]
- Tangwangvivat, R.; Wacharapluesadee, S.; Pinyopornpanish, P.; Petcharat, S.; Hearn, S.M.; Thippamom, N.; Phiancharoen, C.; Hirunpatrawong, P.; Duangkaewkart, P.; Supataragul, A.; et al. SARS-CoV-2 Variants Detection Strategies in Wastewater Samples Collected in the Bangkok Metropolitan Region. Viruses 2023, 15, 876. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.; Sashittal, P.; Zhou, A.; Wang, L.; El-Kebir, M.; Nguyen, T.H. Design of SARS-CoV-2 Variant-Specific PCR Assays Considering Regional and Temporal Characteristics. Appl. Environ. Microbiol. 2022, 88, e0228921. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.D.; Richter, S.R.; Midgley, S.E.; Franck, K.T. Detecting SARS-CoV-2 Omicron B.1.1.529 Variant in Wastewater Samples by Using Nanopore Sequencing. Emerg. Infect. Dis. 2022, 28, 1296–1298. [Google Scholar] [CrossRef]
- Gregory, D.A.; Wieberg, C.G.; Wenzel, J.; Lin, C.-H.; Johnson, M.C. Monitoring SARS-CoV-2 Populations in Wastewater by Amplicon Sequencing and Using the Novel Program SAM Refiner. Viruses 2021, 13, 1647. [Google Scholar] [CrossRef]
- Focosi, D.; Quiroga, R.; McConnell, S.; Johnson, M.C.; Casadevall, A. Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. Int. J. Mol. Sci. 2023, 24, 2264. [Google Scholar] [CrossRef] [PubMed]
- Pastorio, C.; Noettger, S.; Nchioua, R.; Zech, F.; Sparrer, K.M.; Kirchhoff, F. Impact of mutations defining SARS-CoV-2 Omicron subvariants BA.2.12.1 and BA.4/5 on Spike function and neutralization. iScience 2023, 26, 108299. [Google Scholar] [CrossRef]
- Biobot Analytics. (n.d.). COVID-19, Influenza, and RSV wastewater monitoring in the U.S. Retrieved May 1, 2023, from https://biobot.io/data/.
- Gupta, S.; Kumar, A.; Gupta, N.; Bharti, D.R.; Aggarwal, N.; Ravi, V. A two-step process for in silico screening to assess the performance of qRTPCR kits against variant strains of SARS-CoV-2. BMC Genom. 2022, 23, 755. [Google Scholar] [CrossRef]
- Sharma, S.; Shrivastava, S.; Kausley, S.B.; Rai, B.; Pandit, A.B. Coronavirus: a comparative analysis of detection technologies in the wake of emerging variants. Infection 2022, 51, 1–19. [Google Scholar] [CrossRef]
- Dip, S.D.; Sarkar, S.L.; Setu, A.A.; Das, P.K.; Pramanik, H.A.; Alam, A.S.M.R.U.; Al-Emran, H.M.; Hossain, M.A.; Jahid, I.K. Evaluation of RT-PCR assays for detection of SARS-CoV-2 variants of concern. Sci. Rep. 2023, 13, 2342. [Google Scholar] [CrossRef]
- Nextstrain. (2024). Nextstrain Annual Update March 2024. https://nextstrain.org/blog/2024-03-27-annual-update-march-2024.
- Fontenele, R.S.; Kraberger, S.; Hadfield, J.; Driver, E.M.; Bowes, D.; Holland, L.A.; Faleye, T.O.; Adhikari, S.; Kumar, R.; Inchausti, R.; et al. High-throughput sequencing of SARS-CoV-2 in wastewater provides insights into circulating variants. Water Res. 2021, 205, 117710. [Google Scholar] [CrossRef]
- Rader, B.; Gertz, A.; Iuliano, A.D.; Gilmer, M.; Wronski, L.; Astley, C.M.; Sewalk, K.; Varrelman, T.J.; Cohen, J.; Parikh, R.; et al. Use of At-Home COVID-19 Tests — United States, August 23, 2021–March 12, 2022. Mmwr. Morb. Mortal. Wkly. Rep. 2022, 71, 489–494. [Google Scholar] [CrossRef]
- Usher, A.D. FIND documents dramatic reduction in COVID-19 testing. Lancet Infect. Dis. 2022, 22, 949. [Google Scholar] [CrossRef] [PubMed]
- Flood, M.T.; Sharp, J.; Bruggink, J.; Cormier, M.; Gomes, B.; Oldani, I.; Zimmy, L.; Rose, J.B. Understanding the efficacy of wastewater surveillance for SARS-CoV-2 in two diverse communities. PLOS ONE 2023, 18, e0289343. [Google Scholar] [CrossRef] [PubMed]
- Rabe, A.; Ravuri, S.; Burnor, E.; Steele, J.A.; Kantor, R.S.; Choi, S.; Forman, S.; Batjiaka, R.; Jain, S.; León, T.M.; et al. Correlation between wastewater and COVID-19 case incidence rates in major California sewersheds across three variant periods. J. Water Heal. 2023, 21, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Sampling site locations (marked in red) in the eastern Upper Peninsula of Michigan.
Figure 2.
Normalized N1+N2 gene copies per 100 mL by site during the study period week of January 9, 2023 through the week of April 24, 2023.
Figure 2.
Normalized N1+N2 gene copies per 100 mL by site during the study period week of January 9, 2023 through the week of April 24, 2023.
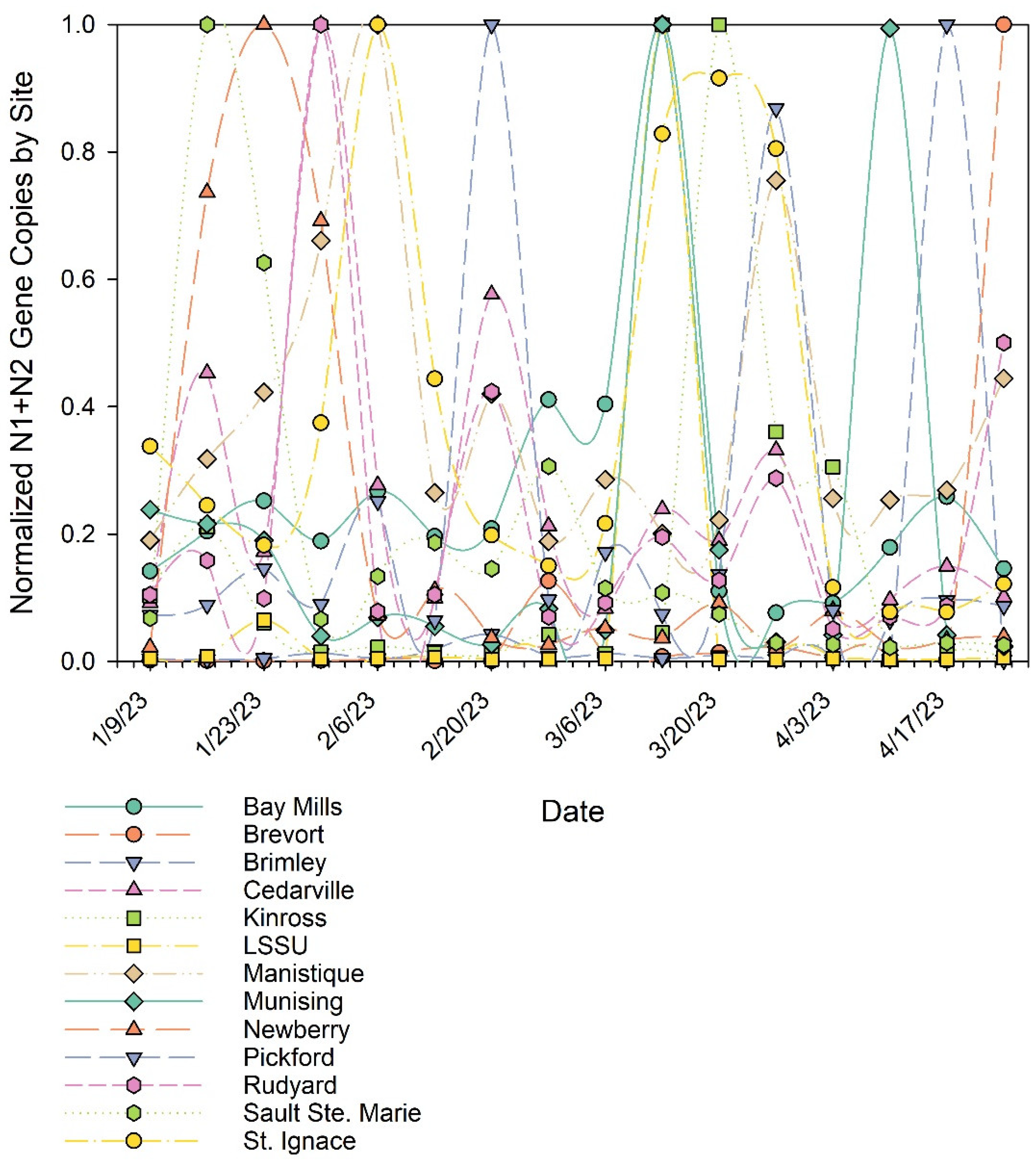
Figure 3.
Comparison of temporal subvariant occurrence between PCR (A) and ONT (B) methods for study period January 9, 2023 through April 27, 2023.
Figure 3.
Comparison of temporal subvariant occurrence between PCR (A) and ONT (B) methods for study period January 9, 2023 through April 27, 2023.
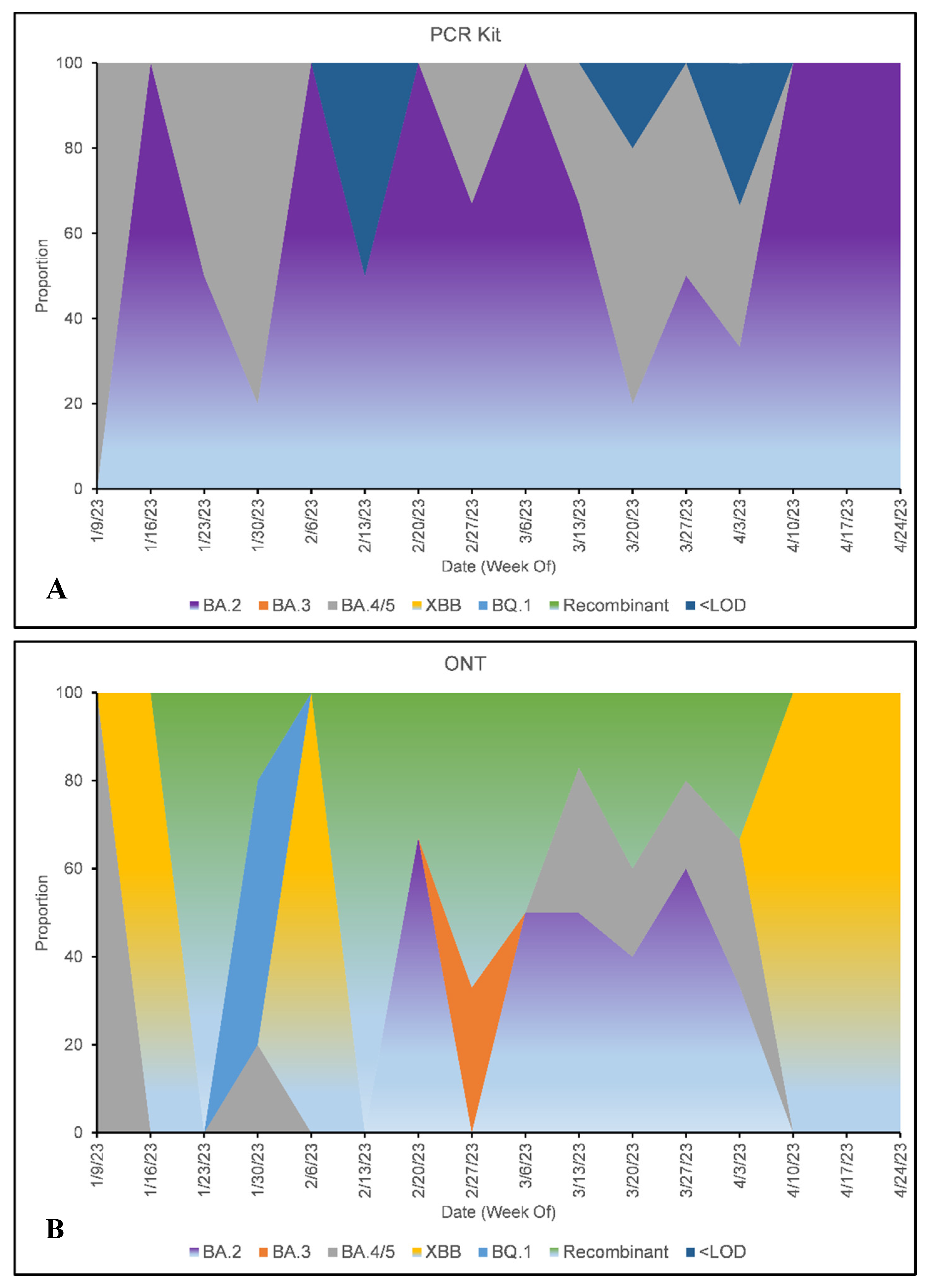

Table 1.
Comparison of subvariants identified using Oxford Nanopore Technologies sequencing and GT Molecular ddPCR BA.1/BA.2 discrimination kit over the study period January 9, 2023 - April 27, 2023. The number of sequencing instances was dependent upon meeting the 9000 N1+N2 gene copies per 100 mL minimum.
Table 1.
Comparison of subvariants identified using Oxford Nanopore Technologies sequencing and GT Molecular ddPCR BA.1/BA.2 discrimination kit over the study period January 9, 2023 - April 27, 2023. The number of sequencing instances was dependent upon meeting the 9000 N1+N2 gene copies per 100 mL minimum.
| DATE | SITE | ONT | PCR KIT |
| 3/14/2023 | Bay Mills Community | BA.5 | BA.1 and BA.2 (4/5) |
| 3/1/2023 | Brevort | BA.3 | BA.2 |
| 3/8/2023 | Brevort | BA.2 | BA.2 |
| 3/15/2023 | Brevort | BA.5 | BA.1 and BA.2 (4/5) |
| 3/22/2023 | Brevort | BA.5 | BA.1 and BA.2 (4/5) |
| 3/29/2023 | Brevort | BA.2 | BA.1 and BA.2 (4/5) |
| 4/5/2023 | Brevort | BA.5 | BA.1 and BA.2 (4/5) |
| 4/26/2023 | Brevort | XBB | BA.2 |
| 1/31/2023 | Brimley | Recombinant | BA.2 |
| 3/7/2023 | Brimley | Recombinant | BA.2 |
| 4/18/2023 | Brimley | XBB | BA.2 |
| 2/2/2023 | Cedarville | BQ.1 | BA.1 and BA.2 (4/5) |
| 2/23/2023 | Cedarville | BA.2.10 | BA.2 |
| 1/10/2023 | Kinross | BA.5 | BA.1 and BA.2 (4/5) |
| 2/28/2023 | Kinross | Recombinant | BA.1 and BA.2 (4/5) |
| 3/14/2023 | Kinross | Recombinant | BA.2 |
| 3/21/2023 | Kinross | BA.2.10 | BA.2 |
| 3/28/2023 | Kinross | BA.2.10 | BA.2 |
| 4/4/2023 | Kinross | BA.2 | BA.2 |
| 3/15/2023 | LSSU | BA.2.10 | BA.2 |
| 1/30/2023 | Manistique | BQ.1 | BA.1 and BA.2 (4/5) |
| 3/20/2023 | Manistique | BA.2 | <LOD |
| 1/23/2023 | Munising | Recombinant | BA.2 |
| 3/13/2023 | Munising | BA.2 | BA.2 |
| 3/20/2023 | Munising | Recombinant | BA.1 and BA.2 (4/5) |
| 4/10/2023 | Munising | XBB | BA.2 |
| 1/31/2023 | Newberry | BA.5 | BA.1 and BA.2 (4/5) |
| 2/14/2023 | Newberry | Recombinant | <LOD |
| 4/4/2023 | Newberry | Recombinant | <LOD |
| 2/22/2023 | Pickford | BA.2 | BA.2 |
| 3/29/2023 | Pickford | BA.5 | BA.1 and BA.2 (4/5) |
| 2/2/2023 | Rudyard | BQ.1 | BA.1 and BA.2 (4/5) |
| 2/23/2023 | Rudyard | Recombinant | BA.2 |
| 3/30/2023 | Rudyard | Recombinant | BA.2 |
| 4/27/2023 | Rudyard | XBB | BA.2 |
| 1/18/2023 | Sault Ste. Marie | XBB | BA.2 |
| 1/24/2023 | Sault Ste. Marie | Recombinant | BA.1 and BA.2 (4/5) |
| 2/14/2023 | Sault Ste. Marie | Recombinant | BA.2 |
| 2/28/2023 | Sault Ste. Marie | Recombinant | BA.2 |
| 2/8/2023 | St. Ignace | XBB | BA.2 |
| 3/15/2023 | St. Ignace | BA.2 | BA.2 |
| 3/22/2023 | St. Ignace | Recombinant | BA.1 and BA.2 (4/5) |
| 3/29/2023 | St. Ignace | BA.2.10 | BA.2 |
Table 2.
Percent of samples (n=43) identified as each subvariant using ONT sequencing and GT Molecular ddPCR kit.
Table 2.
Percent of samples (n=43) identified as each subvariant using ONT sequencing and GT Molecular ddPCR kit.
| SUBVARIANT | ONT | PCR KIT |
|---|---|---|
| BA.2 | 28% | 58% |
| BA.3 | 2% | 0% |
| BA.4 or BA.5 | 14% | 25% |
| XBB | 14% | 0% |
| BQ.1 | 7% | 0% |
| Recombinant | 35% | 0% |
| <LOD | N/A | 7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



