
Submitted:
04 September 2024
Posted:
05 September 2024
You are already at the latest version
Abstract
Helicobacter pylori infection constitutes a silent pandemic of global concern. In the last decades, the alarming increase of multidrug resistance evolved by this pathogen has led to a marked drop in the eradication rates of traditional therapies worldwide, encouraging scientific community to discover and develop new antimicrobial strategies. In the present work, we identified a battery of novel drug-like HsrA inhibitors with MIC values ranging from 0.031 to 4 mg/L against antibiotic-resistant strains of H. pylori. Most of these novel antimicrobials exhibited minor effects against both Gram-negative and Gram-positive species of human microbiota. The most potent anti-H. pylori candidate demonstrated a high therapeutic index, additive effect in combination with metronidazole and clarithromycin, as well as a strong antimicrobial action against Campylobacter jejuni, another clinically relevant pathogen of phylum Campylobacterota. Transcriptomic analysis suggested that in vivo inhibition of HsrA triggered lethal global disturbances in H. pylori physiology, including the arrest of protein biosynthesis, malfunction of respiratory chain, detriment in ATP generation, and oxidative stress. The novel drug-like HsrA inhibitors described here constitute valuable candidates to a new family of narrow-spectrum antibiotics that allow overcoming the current resistome, protecting from dysbiosis, and increasing therapeutic options for novel personalized treatments against H. pylori.
Keywords:
Helicobacter pylori
; antimicrobial resistance
; narrow-spectrum antibiotic
; antibacterial target
; response regulator
; HsrA
; Campylobacter jejuni
1. Introduction
Helicobacter pylori, a pathogenic member of phylum Campylobacterota (formerly Epsilonproteobacteria) [1], is recognized as the leading cause of several human gastric pathologies, including acute and chronic gastritis, peptic ulcer disease, gastric adenocarcinoma, and gastric mucosa-associated lymphoid tissue (MALT) lymphoma [2,3]. In addition, this microaerophilic Gram-negative bacterium has been associated to some extragastric disorders such as idiopathic thrombocytopenic purpura, idiopathic iron deficiency anaemia, vitamin B12 deficiency, cardiovascular and neurodegenerative diseases, among others [4]. H. pylori is presumably colonizing the gastric mucosa of more than half of the world population [5], causing chronic long-lasting inflammation that may progress to gastric cancer in about 1-3% of untreated or mistreated cases [6,7]. Despite H. pylori constitutes the only bacterial pathogen recognized as class I carcinogen by the International Agency for Research on Cancer [8], neither prophylactic nor therapeutic vaccines are currently available.
Effective treatment of H. pylori infection remains a challenge for clinicians and sanitary authorities worldwide [9,10,11]. Only a handful of antibiotics are currently used in clinical practice for H. pylori eradication, including metronidazole, clarithromycin, amoxicillin, tetracycline, and levofloxacin [12,13]. Due to the poor in vivo efficacy of antimicrobial monotherapies against this pathogen [14], current antibiotics are administrated as part of several aggressive and prolonged combinatory therapies which are commonly prescribed empirically, resulting frequently in H. pylori eradication failures, emergence of secondary antibiotic resistances, and refractory infections [15,16,17,18]. In the last two decades, the alarming increase of the antibiotic resistance levels to first-line and even “rescue” antibiotics, especially clarithromycin, metronidazole and levofloxacin [19,20,21], has led to a marked decrease of the eradication rates of traditional therapies [22,23]. This fact prompted the World Health Organization (WHO) to define H. pylori as a high priority pathogen in the current global efforts to R&D of novel antimicrobials [24].
On the other hand, even when current antimicrobial therapies against H. pylori infections are generally safe, the prolonged administration of high doses of broad-spectrum antibiotics into combinatory therapies frequently result in dysbiosis [25,26,27,28]. In some cases, H. pylori eradication regimens might cause major disruption of normal gut microbiome, promoting severe complications such as pseudomembranous colitis due to Clostridioides difficile proliferation [29,30,31,32].
The discovery and development of narrow-spectrum antibiotics, also known as precision antimicrobials, exemplify a current accurate strategy for reducing or slowing down the appearance and propagation of antimicrobial resistance as well as minimizing undesirable side effects on normal microbiome [33,34,35,36,37]. Ideally, a precision antimicrobial exerts its action on molecular targets shared by only a reduced number of closely related pathogenic species, but not expressed by most representative commensal microbes [38,39].
In previous works, we have validated the essential protein HsrA as an effective therapeutic target for H. pylori infection [40,41,42]. HsrA is an OmpR/PhoB-type orphan response regulator [43,44], unique and highly conserved in members of phylum Campylobacterota [45], which appears involved in a variety of crucial physiological processes including transcription, translation, redox homeostasis, chemotaxis and energy metabolism [46,47,48,49], and thereby resulting indispensable for cell viability [50,51]. In the present study, we have identified several novel low-molecular weight inhibitors of HsrA, which fit the Lipinski´s rule-of-five for oral drug-likeness and exhibited potent bactericidal activities against H. pylori, with minor antimicrobial effects on both Gram-negative and Gram-positive species of commensal bacteria. At least one of these novel precision antimicrobials exerted also a strong antimicrobial action against Campylobacter jejuni, another clinically relevant pathogen of phylum Campylobacterota.
2. Results
2.1. High-Throughput Screening of the Maybridge HitFinderTM Chemical Library Identified Novel HsrA Ligands
The Maybridge HitFinderTM chemical collection, comprising 12,000 compounds that fit the Lipinski´s rule-of-five for oral drug-likeness [52,53], was screened for the identification of low-molecular weight ligands of the H. pylori essential response regulator HsrA. The high-throughput screening (HTS) of the chemical library was carried out by using a previously described fluorescent thermal shift-based method [40,54]. With this approach, those compounds from the library that induced a thermostabilization of the protein in ≥ 5 ºC compared to reference controls with dimethyl sulfoxide (DMSO, vehicle) were considered as HsrA ligands (Figure 1). According to this selection criterion, our HTS led to identify 31 drug-like small molecules from the Maybridge collection than acted as ligands of HsrA (Supplementary Table S1). For practical purposes, the HsrA ligands were identified by consecutive Roman numerals according to the magnitude of the melting temperature (Tm) upshifts that they induced after binding to the native state of the protein. Almost a third part of the HsrA ligands identified by the HTS induced Tm upshifts above 10 ºC. Notably, two HsrA-ligand complexes increased protein thermal stability in 20 ºC or more. Chemical structures and some physicochemical properties of most relevant HsrA ligands identified and evaluated in this work are showed in Table 1.
2.2. Several Drug-like Ligands of HsrA Inhibited Its DNA Binding Activity In Vitro
The inhibitory capabilities of all HsrA ligands identified by the HTS on the in vitro DNA binding activity of the response regulator were evaluated by electrophoretic mobility shift assays (EMSA). Previous works have demonstrated an in vitro high affinity of HsrA for its target promoter PporGDAB in a concentration-dependent manner [40,41]. In the present work, titration of the HsrA activity confirmed that 120 ng of target DNA were completely and specifically complexed with at least 6 μM of freshly purified recombinant protein under the experimental conditions used in EMSA (data not shown). Accordingly, mixtures of these amounts of protein and target DNA were incubated in the presence of increasing concentrations of each HsrA ligands, from 100 μM to 2 mM. Twelve of the 31 low-molecular weight ligands of HsrA caused a noticeable inhibition of the in vitro DNA binding activity of this essential response regulator according to EMSA (Figure 2). Despite 2 mM of any of these compounds was sufficient to completely inhibit the in vitro biological activity of 6 μM of recombinant HsrA protein, the in vitro inhibitory effects of some ligands like IV and VIII appeared appreciably stronger than those exerted by other ligands such as XI and XXVI.
2.3. Novel HsrA Inhibitors Exhibited Potent Bactericidal Effects against H. pylori
The new twelve drug-like HsrA inhibitors identified by HTS and EMSA were evaluated for their antimicrobial properties against four different strains of H. pylori, including strains resistant to clarithromycin (ATCC 700684), metronidazole (ATCC 43504), and levofloxacin (strain Donostia 2). As shown in Table 2, at least six HsrA inhibitors (I, IV, V, VIII, XI, and XII) exhibited strong bactericidal activities against all the H. pylori strains tested, showing minimal inhibitory concentration (MIC) values ranging from 0.031 to 4 mg/L. Notably, the HsrA ligand V appeared specially effective against H. pylori viability, exhibiting a bactericidal potency even stronger than those demonstrated by first-line antibiotics like metronidazole and levofloxacin against the same H. pylori strains. Other HsrA inhibitors, such as IX, X, XVII, and XXVI demonstrated moderate anti-H. pylori action (MIC = 8 - 16 mg/L), while inhibitors XXVIII and XXXI appeared poorly effective as antimicrobials (MIC ≥ 32 mg/L). No relevant differences were observed in the antimicrobial activities of these molecules with respect to the antibiotic-resistance pattern of the H. pylori strains used in the assays.
To further characterize the bactericidal activities of most effective HsrA inhibitors, time-kill kinetic assays were carried out by exposing H. pylori strain ATCC 700392 to 4 × MIC of compounds I, IV, V, VIII, XI, and XII (Figure 3). Despite all compounds resulted completely lethal after 8 h of exposure, significant differences (p < 0.05) were noticed in the decline of bacterial counts produced by each HsrA inhibitor from 2 h of treatment. Thus, at the concentration used in this experiment, compounds V and VIII completely killed 104 H. pylori cells after 2 h of exposure. The same total bactericidal effect was observed after 4 h for compounds IV, XI, and XII. No live bacteria could be detected after 8 h of exposure with compound I.
Putative combinatory effects in the antimicrobial action of these bactericidal HsrA inhibitors with first-line antibiotics including clarithromycin, metronidazole and levofloxacin against H. pylori were assessed by checkerboard assays. As described in Table 3, only compound XII exhibited synergistic effect in combination with metronidazole, while compound IV did not demonstrate interaction with any of the antibiotics analysed. The highly bactericidal compounds V, VIII, and XI interacted additively with both clarithromycin and metronidazole; however, all HsrA inhibitors resulted neutral in combination with levofloxacin.
2.4. An HsrA Bactericidal Inhibitor Bound the HsrA Orthologue Protein CosR and Exhibited Strong Bactericidal Effect against C. jejuni
Since HsrA is an essential response regulator unique, but also highly conserved among members of phylum Campylobacterota, those HsrA inhibitors that previously demonstrated strong bactericidal activities against H. pylori (MIC ≤ 4 mg/L) were additionally evaluated for their antimicrobial properties against other Campylobacterota pathogenic species, C. jejuni. Notably, the most effective HsrA inhibitor against H. pylori, ligand V, exhibited also a strong antimicrobial activity against C. jejuni, with MIC and MBC values ≤ 0.5 mg/L (Table 4). However, the antimicrobial effect of other HsrA ligands against C. jejuni appeared moderate (16 – 32 mg/L) or quite low (≥ 64 mg/L) for the rest of compounds.
Considering that the essential response regulator CosR constitutes the orthologue of HsrA in C. jejuni [44,45], we analysed the effect of ligand V on the in vitro DNA binding activity of CosR by EMSA. As depicted in Figure 4, the HsrA inhibitor V also caused a noticeable inhibition of the in vitro affinity of CosR by its target promoter PsodB, suggesting that this low-molecular weight compound acts as an inhibitory ligand of both orthologues, HsrA and CosR.
2.5. HsrA Bactericidal Inhibitors Exhibited Low Antimicrobial Actions against Gram-Positive and Gram-Negative Species of Human Microbiota
Furthermore, we evaluated the antimicrobial activities of most potent bactericidal anti-H. pylori inhibitors against several Gram-negative and Gram-positive representative species of the human normal microbiota. MIC and MBC values of HsrA ligands I, IV, V, VIII, XI, and XII against control strains of E. coli, K. pneumoniae, E. faecalis, S. aureus, S. epidermidis, and S. agalactiae were determined according to the EUCAST guidelines. As shown in Table 4, most of the HsrA inhibitors exhibited a narrow spectrum of action against H. pylori, and did not exerted relevant antimicrobial effects against all the microbiota species tested. Unexpectedly, compounds VIII and XI appeared particularly effective against Staphylococcus sp. and Streptococcus sp. strains, which could suggest cross-inhibition of specific molecular targets expressed by these bacterial species.
2.6. Cytotoxicity and Therapeutic Index
As a preliminary study of toxicity, cytotoxicity effects of highly bactericidal anti-H. pylori inhibitors I, IV, V, VIII, XI, and XII were evaluated on HeLa cells. Mammalian cells were exposed during 24 h to increasing concentrations of each compound, from 0.125 to 128 mg/L, and cell viability was measured by the PrestoBlueTM method. None of the six HsrA inhibitors resulted cytotoxic for Hela cells under 2 mg/L, while compounds I, IV, V, and XI seemed completely safe even at 8 mg/L (Figure 5A). Compound VIII proved to be the most cytotoxic among the highly bactericidal HsrA ligands, causing 68% of cell death at 8 mg/L. Notably, the 50% cytotoxic concentrations (CC50) of compounds I, IV and V were 30 times or higher their values of MIC (Figure 5B), which supposes wide therapeutic windows of these novel anti-H. pylori antimicrobial candidates. Surprisingly, the most potent bactericidal HsrA ligand was also the safest candidate.
2.7. Thermodynamics and Predicted Models of HsrA-Inhibitor Interactions
Isothermal titration calorimetry (ITC) experiments and molecular docking analyses were combined in order to achieve a better understanding of thermodynamic, stoichiometry and putative structures of HsrA-inhibitor complexes. As shown in Table 5, all highly bactericidal inhibitors of HsrA appeared to interact with amino acid residues involved in the helix-turn-helix (HTH) DNA binding motif of the regulator and their immediate environments, following a 1:1 stoichiometry. Thus, each HsrA monomer binds one molecule of inhibitor with dissociation constants in the micromolar range. Ligands XI and XII exhibited significant lower binding affinities to HsrA according to their Kd values (Table 5, Supplementary Figure S1). ITC experiments also confirmed the molecular interaction of highly bactericidal ligand V with the orthologue of HsrA in C. jejuni, the orphan response regulator CosR (Supplementary Figure S2). This low-molecular weight inhibitor of HsrA bond to CosR with moderate affinity, showing a dissociation constant in the micromolar range, and following a 1:1 stoichiometry.
As previously observed with other HsrA low-molecular weight ligands [42], the novel drug-like inhibitors described here presumably interact with the C-terminal DNA binding domain of HsrA at a putative binding pocked, predominantly shaped by the nonpolar residues I121, I123, I126, I135, V142, V144, G146, P148, F149, L152, M195, P198, and L199, with few polar amino acids such as Y137, K145, and K194. Inside this predicted binding pocked, the best-ranked pose of the most effective HsrA bactericidal inhibitor against H. pylori, ligand V, appears to establish several non-covalent interactions with neighbouring amino acids that boosts the stabilization of the ligand-protein complex (Figure 6). Thus, the phenyl ring of ligand V establishes CH/π interactions with residues Leu126, Thr203, Leu199 and Leu152 (Figure 6C). In addition, the hydroxyl group in the side chain of Thr203 interacts through hydrogen bonding with the carbonyl group of ligand V, and through halogen bonding with the chloride substituent located in para-position of the ligand phenyl group. A series of hydrophobic interactions are formed between the heterocyclic thiophene group of ligand V and residues Lys194, Met195, Leu152, and Ileu135. Furthermore, the NH2 group of ligand V establishes a hydrogen bond with carbonyl group of Lys194, while the aromatic ring of Pro198 could interact with thiophene through perpendicular π/π stacking at 4.5 Å of distance. In the best ranked-pose of ligand V into the pocket of the C-terminal DNA binding domain of HsrA, the nitro group at C2 position in the thiophene of the ligand appears located very outside the binding pocket and therefore too far away from the amino groups of Leu152 and Lys194 to may stablish any interaction (Figure 6A,C).
2.8. In Vivo Inhibition of HsrA Uncovered New Insights into the Essential Role of this Orphan Response Regulator
The inhibitory effect of the highly bactericidal inhibitor V on the essential transcriptional regulatory role of HsrA in vivo was assessed by RNA-seq. For this purpose, ~3×107 CFU/mL freshly grown H. pylori 26695 cells were exposed to lethal concentrations of compound V (4 × MIC) until log10 CFU/mL resulted diminished in one unit (~2 h of exposure). As depicted in Figure 7, the transcriptomic analysis suggested that the in vivo inhibition of HsrA induced a global misregulation of H. pylori gene expression, which appears conducive to cell death as a result of several serious and possibly synergistic physiological alterations.
Treatment with the bactericidal HsrA inhibitor V significantly changed the transcript levels of 367 ORF (absolute log2 fold change > 1, p-value < 0.05), of which 212 genes appeared upregulated and 155 genes resulted in downregulation, as compared with control samples (Figure 7A,B, Supplementary Table S2). Thus, in vivo HsrA inhibition seems to influence, directly or indirectly, the expression of 23% of ORFs encoded by the H. pylori 26696 genome [55,56]. Among the 268 differentially expressed genes (DEGs) with defined functions, two functional categories were highly enriched with downregulated genes involved in essential physiological processes: (1) ribosome biogenesis, and (2) electron transfer and oxidative phosphorylation (Figure 7C, Supplementary Table S3).
At least 29 ribosomal structural proteins and 7 enzymes involved in ribosome biogenesis resulted strongly downregulated upon exposure of H. pylori cells to lethal concentrations of HsrA inhibitor V. In fact, some of these proteins corresponded with those DEGs with major decrease in transcript abundance, including the 50S ribosomal proteins L21, L35, and L27, but also the 30S ribosomal protein S10, all of which exhibited > 5.8-fold depletion in gene expression (Figure 7B,C, Supplementary Table S3). Notably, depletion in ribosomal protein abundances was accompanied by a noticeable increase in ribosomal RNAs transcription (Figure 7B,C, Supplementary Table S3). Taking into account that rRNA genes have been traditionally used as housekeeping controls in H. pylori due to their relatively constant rates of expression under very different environmental conditions, we hypothesize that the marked increase in rRNAs observed here could be a pleiotropic adaptive response of cell in an attempt to overcome the lethal disruption of ribosome biogenesis. In addition, the arrest of translation as a consequence of ribosome deficit could also explain the significant increase in tRNAs biosynthesis as well as in a variety of enzymes involved in amino acids metabolism. However, these hypotheses do not rule the possibility that some of these DEGs actually constitute unrecognized or little studied direct targets of HsrA transcriptional regulation [49].
In vivo inhibition of HsrA led also to decrease of transcript amounts of its previously recognized target cyt553 [49]. Moreover, other components of the respiratory chain and coupled oxidative phosphorylation, including subunit NuoN of NADH-quinone oxidoreductase, cytochrome b subunit of fumarate reductase (FrdC), and subunits A, B, B´, and D of ATP synthase also diminished their transcription (Supplementary Table S3).
Exposure of H. pylori to the bactericidal HsrA inhibitor V induced the expression of antioxidant enzymes and chaperones including katA, sodB, ccP, dnaK, groEL, clpB, grpE, and htpG, among other oxidative stress response proteins (Supplementary Table S3). Notably, the H. pylori nonhaem iron-containing ferritin (pfr, hp0653) and the ferritin-like iron-binding protein NapA appeared 4.7- and 2.3-fold upregulated, respectively, while the high-affinity ferrous iron transporter FeoB [57] was ~2.5-fold downregulated. Ferritin is required to sequester cytoplasmic free Fe2+ in order to minimize the formation of highly reactive hydroxyl radicals by the Fenton reaction, playing a major role in protection against oxidative stress in Campylobacterota [58,59]. A similar free-iron removal role has been described for the neutrophil-activating protein NapA in H. pylori [60,61].
Unexpectedly, transcriptomic analysis revealed that at least 42 genes related to H. pylori pathogenicity significantly changed their expression upon in vivo inhibition of HsrA, including cag pathogenicity island genes, flagellar proteins, adhesins, toxins and enzymes, among others (Figure 7C, Supplementary Table S3). In addition, a plethora of genes involved in other functional categories such as carbon and lipid metabolisms, biosynthesis of vitamins and cofactors, nucleotide metabolism, transcription and translation factors, DNA replication and repair, outer membrane proteins and membrane transporters, as well as metal-resistance, exhibited significant changes in their transcript abundances.
To validate the transcriptome analysis, we evaluated the transcription levels of 10 selected genes, including both upregulated and downregulated DEGs according to RNA-seq. As shown in Figure 7D and Supplementary Table S3, all the genes evaluated by qPCR showed the same variation trend observed in RNA-seq upon in vivo inhibition of HsrA.
3. Discussion
Infection with H. pylori constitutes a silent pandemic of global concern. About 50% of the world´s population is estimated to be infected with this carcinogenic bacterial pathogen [5,62], although this prevalence reaches 80% or more in several countries of Americas, Eastern Mediterranean, and Eastern Europe [63]. H. pylori is considered the most important risk factor for development of gastric cancer [7], the fifth most common malignancy and the fourth leading cause of all cancer-related deaths worldwide [64]. Despite eradication of H. pylori infection significantly reduces both incidence and mortality of gastric cancer [65,66,67], the increasing resistome accumulated by this pathogen in the last years has led to a considerable drop in the efficacy of current eradication therapies [10,68,69]. To face this concerning trend, efforts have being made to discover novel treatment strategies.
We previously validated the essential OmpR-like orphan response regulator HsrA as an effective therapeutic target against H. pylori [40,41,42]. HsrA affinity-based high-throughput screening (HTS) of 1,200 clinically approved drugs from Prestwick Chemical Library® revealed that some highly prescribed 1,4-dihydropyridine (DHP) calcium channel blockers, as well as several natural flavonoids, acted as HsrA low-molecular weight ligands leading to inhibition of the in vitro DNA-binding activity of the regulator. Some of these potentially repurposable drugs exhibited noticeable bactericidal effects against different antibiotic-resistant strains of H. pylori with MIC ranging 4 - 16 mg/L [40,41], and significantly decreased gastric colonization of H. pylori in mice [41]. Notably, flavonoids such as chrysin and hesperetin demonstrated strong synergistic bactericidal action against H. pylori in combination with clarithromycin or metronidazole, supporting the potential inclusion of these natural phytochemicals as valuable adjuvants in novel eradication therapies [40,70].
In the present work, we have identified novel low-molecular weight inhibitors of HsrA through the screening of 12,000 drug-like compounds from the Maybridge HitFinderTM chemical library. The collection is highly compliant with the Lipinski's rule-of-five, a rule of thumb to evaluate drug-likeness of novel potential medicines [52,53]. According to the Lipinski's rule-of-five, those compounds with molecular weight < 500 Da, calculated logarithm of the octanol-water partition coefficient (clog P) < 5, number of hydrogen bond acceptors ≤ 10, and number of hydrogen bond donors ≤ 5, will have better oral bioavailability and pharmacokinetics. Thus, molecules that violate more than one of these physicochemical parameters may show problems in terms of bioavailability after oral administration.
At least six of the novel drug-like HsrA ligands identified by affinity-based HTS of the HitFinderTM collection, denoted as I, IV, V, VIII, XI and XII, noticeably inhibited the DNA binding activity of the response regulator in vitro and demonstrated strong bactericidal activities against antibiotic-resistant strains of H. pylori. With MIC values ranging from 0.031 to 4 mg/L, some of these novel antimicrobial candidates constituted the most potent bactericidal HsrA inhibitors discovered to date. In addition, at least four of these highly bactericidal HsrA ligands exhibited narrow-spectrum antimicrobial activities against H. pylori, with MBC values ≥ 64 mg/L against several representative Gram-positive and Gram-negative members of normal human microbiota. Since protein HsrA is unique among members of phylum Campylobacterota (former class Epsilonproteobacteria) [45], the results suggest that certain low-molecular weight inhibitors of HsrA could specifically bind this response regulator without affecting biological activities of essential proteins from other bacterial species. These evidences push up the value of HsrA as an effective and selective therapeutic target for development of novel precision antibiotics against H. pylori, moving away the risk of dysbiosis observed with current eradication therapies [28,31].
Notably, one of the novel drug-like HsrA inhibitor described here, denoted as ligand V and named N'-{[(4-chloroanilino)carbonyl]}oxy-5-nitrothiophene-3-carboximidamide at the ChemSpider database (https://www.chemspider.com), showed significantly higher antimicrobial activities than the rest of compounds against all H. pylori strains used in the study. With MIC values ranging from 0.031 to 0.125 mg/L, the antimicrobial potency of ligand V was at least 4-fold higher than the second most active bactericidal HsrA inhibitor, the ligand XII, and more than 10-fold stronger than metronidazole against the same strains. In addition, ligand V demonstrated the highest therapeutic index among all highly bactericidal HsrA inhibitors described here, and showed additive antimicrobial action in combination with clarithromycin and metronidazole.
Several factors could be contributing to the higher antimicrobial potency exhibited by ligand V on H. pylori cells. For instance, a favourable balance among the physicochemical properties of this compound could make a difference in the microbial membrane translocation and intracellular concentration compared with other HsrA bactericidal ligands. In this context, the lipophilicity/hydrophilicity ratio, the molecular weight, the structural flexibility (rotatable bonds), the number of H-bond donors and acceptors, but also the polar surface area of small molecules may greatly influence their permeability through bacterial membranes as well as their possible efflux outside the pathogen [71,72,73,74]. Despite the contribution of each physicochemical property to membrane permeation and efflux varies among different bacteria species [74], it is well recognized that the polar nature of the outer membrane of Gram-negative pathogens hinders passive translocation of highly hydrophobic molecules [71,75]. Thus, decreasing the lipophilicity/hydrophilicity ratio of a low-molecular weight inhibitor of E.coli DNA gyrase by reducing logD from 2.59 to 1.75 led to 4-fold increase in its antibacterial potency [72]. Likewise, fine-tuning the physicochemical properties of a topoisomerase inhibitor in Pseudomonas aeruginosa by lowering logD from 2.0 to 0.9 resulted in 8-fold increase of its antibacterial activity [75]. On the other hand, Gram-negative pathogens express a plethora of broad-specific efflux pumps that actively export a variety of foreign molecules out of the cell, thereby decreasing the effective antibiotic concentration in the cytoplasm [76]. Some evidences suggested that highly polar and small compounds or very large and zwitterionic molecules are less susceptible to efflux [71].
Herein, time-kill kinetic studies demonstrated that ligand V and VIII produced a significant faster decline in bacterial counts compared to the rest of HsrA bactericidal inhibitors, suggesting similar permeability through H. pylori membrane. However, the polar surface area (TPSA) of ligand V almost duplicates this property in ligand VIII, which could be linked with a higher propensity of the last compound to be expelled by H. pylori efflux pumps. In fact, almost 30 genes related to different efflux pumps have been identified in the H. pylori genome [77].
The antimicrobial potency of HsrA ligands could also be influenced by the binding affinity of each molecule to its target protein. According to our ITC studies, the interaction of ligand V with HsrA occurs with > 3-fold higher affinity than the interaction of ligand XII, the second most potent bactericidal compound. Despite ligand IV bound HsrA with the highest affinity, its larger molecular weight and lipophilicity could negatively affect membrane permeation.
Besides its potent antimicrobial activity against H. pylori, ligand V stood up for its strong bactericidal effect against C. jejuni, another clinically relevant pathogen of phylum Campylobacterota. C. jejuni is considered the leading cause of food-borne bacterial gastroenteritis worldwide [78]. Infection in humans usually occurs by consumption of contaminated foods (mainly poultry) or direct contact with animal hosts (including pets) and environmental reservoirs [79,80]. Despite most cases of human campylobacteriosis produces mild and self-limiting diarrhoea and may not require antimicrobial therapy [78,81], the use of antibiotics could be necessary to shorten the duration of illness and to prevent severe complications in more susceptible populations, including very young children, elderly people and immunocompromised patients [78,82]. In addition, antibiotics must be used in cases of invasive or extra-gastrointestinal manifestations such as meningitis, bacteraemia and endocarditis, among others [81,83,84].
When antibiotics are needed, the recommended first-line treatment of campylobacteriosis consists of macrolides such as azithromycin and erythromycin [85]. Fluoroquinolones like ciprofloxacin can be used as alternative therapy, though intravenous aminoglycosides or carbapenems are recommended in severe systemic cases. Tetracyclines and chloramphenicol are also alternative therapies, but they are usually avoided in young children due to their adverse effects [82,85]. As in the case of H. pylori, the increasing rates of antibiotic resistance in Campylobacter strains isolated from humans and animals worldwide, especially to fluoroquinolones but also to macrolides [86,87,88], prompted the WHO to include C. jejuni as a high priority pathogen in the search of novel antimicrobials [24].
Among all the novel drug-like HsrA bactericidal inhibitors described in this work, only the ligand V exhibited a strong antimicrobial effect on C. jejuni, with MIC = 0.25 mg/L. This antimicrobial potency is similar to that observed with ciprofloxacin, and stronger than those observed with doxycycline, erythromycin and gentamicin against the same C. jejuni strain used herein [89]. Molecular interaction of ligand V with CosR was demonstrated by ITC analysis, while this compound noticeable inhibited the in vitro DNA binding activity of CosR according to EMSA experiments. Hence, the strong antimicrobial activity of ligand V on C. jejuni appears to rely on the inhibition of the essential biological activity of CosR [90,91,92,93], thereby validating this protein as an effective therapeutic target against C. jejuni [44]. The dual inhibition triggered by the same low-molecular weight ligand on the biological activities of HsrA and CosR could be expected given the high sequence identity of both orthologue proteins, especially in their C-terminal DNA binding domains (60% overall identity, 85% identity in the effector domain) [45].
Transcriptomic analysis was carried out in order to discern the global effects of lethal concentrations of ligand V on the H. pylori physiology. The results observed here appeared in correspondence with an effective in vivo inhibition of HsrA, and confirmed previous experimental evidences of Pelliciari and colleagues [49] about the key role of this essential response regulator in the control of protein biosynthesis and energy metabolism. Exposure of H. pylori to the highly bactericidal HsrA inhibitor V particularly disturbed transcription of genes involved in ribosome biogenesis [94]. The strong detriment observed in the expression of a plethora of ribosomal proteins could certainly arrest translation and protein biosynthesis leading to cell death [95]. In addition, the in vivo inhibition of HsrA led to a significant decrease in the transcription of several components of the respiratory chain and ATP synthase. Proteome imbalance in the electron transport chain could impair redox reactions and proton translocation across cytoplasmic membrane, shrinking the transmembrane electrochemical proton gradient that drives the synthesis of ATP [96,97]. Furthermore, disruption of ATP biosynthesis could be additionally exacerbated by an altered expression of ATP synthase subunits. On the other hand, imbalance in the respiratory chain may increase the production of reactive oxygen species leading to oxidative stress [98]. This deleterious condition could induce the expression of antioxidant enzymes, heat shock proteins, and other molecular chaperones [99,100], a transcriptional response observed in this work upon exposure of H. pylori to the HsrA inhibitor V. Taking together, the bactericidal effect triggered by the in vivo inhibition of the essential regulatory role of HsrA seems to be a consequence of synergistic deleterious disturbances in the H. pylori physiology, including the arrest of protein biosynthesis, malfunction of respiratory chain, detriment in ATP generation, and oxidative stress.
Due to the unsuccessful attempts to manipulate the expression of the essential hsrA gene in vivo [45,47], the elucidation of the HsrA regulon has represented a challenge. Previous studies have identified several dozens of putative target genes and a handful of well-characterized targets [46,47,48,49]. Our transcriptomic data suggest that HsrA acts as a global transcriptional regulator, modulating directly or indirectly the expression of up to 23% of the ORFs encoded by the H. pylori 26696 genome. Further analyses must be conducted in order to completely unravel the global regulatory role of HsrA and its crucial contribution to viability and pathogenicity of H. pylori.
In conclusion, the results presented here strongly support the use of HsrA and its orthologue proteins as effective and selective therapeutic targets for development of novel antimicrobial strategies against clinically relevant pathogenic members of phylum Campylobacterota. The novel drug-like highly bactericidal inhibitors of HsrA described here constitute valuable candidates to a new family of narrow-spectrum antibiotics that allow overcoming the current resistome, protecting from dysbiosis, and enhancing the battery of therapeutic options for novel personalized treatments against H. pylori.
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
H. pylori reference strains ATCC 700392 (aka 26695), ATCC 43504 (metronidazole-resistant), and ATCC 700684 (clarithromycin-resistant), as well as Campylobacter jejuni ATCC 33560 were purchased from the American Type Culture Collection (Rockville, MA, USA). The H. pylori strain Donostia 2 (levofloxacin-resistant) was isolated from gastroduodenal biopsies and kindly donated by Dr. Milagrosa Montes from Donostia University Hospital (San Sebastian, Spain). Helicobacter sp. and Campylobacter sp. strains were routinely grown in Blood Agar Base No. 2 (OXOID, Basingstoke, UK) supplemented with 8% defibrinated horse blood (OXOID) at 37 °C for 48-72 h, under microaerobic conditions (85% N2, 10% CO2, 5% O2). In some determinations, bacteria were cultured in brain heart infusion broth (OXOID) supplemented with 4% foetal bovine serum (Gibco, Carlsbad, CA, USA) at 37 °C for 48-72 h, under microaerobiosis.
Reference strains Escherichia coli ATCC 25922, Klebsiella pneumonia ATCC 700603, Enterococcus faecalis ATCC 29212, Staphylococcus aureus ATCC 29213, Staphylococcus epidermidis ATCC 12228, and Streptococcus agalactiae ATCC 12386 were kindly donated by the Microbiology Service of University Clinic Hospital Lozano Blesa (Zaragoza, Spain). These strains were routinely grown in Mueller-Hilton agar/broth (PanReac AppliChem, Barcelona, Spain), overnight at 37°C.
4.2. Chemicals
The Maybridge HitFinderTM chemical library was purchased from Thermo Fisher Scientific (Waltham, MA, USA). All compounds of the chemical library were provided as 10 mM solutions dissolved in 100% dimethyl sulfoxide (DMSO). Selected compounds of the library were purchased in additional amounts from Thermo Fisher Scientific, dissolved in 100% DMSO, and stored at -20ºC until use. Conventional antibiotics including clarithromycin (CLR), metronidazole (MTZ), levofloxacin (LVX), and ampicillin (AMP) were purchased from Merck (Rahway, NJ, USA). Stock solutions of these drugs at 10.24 g/L in 100% DMSO were stored at -20 °C for up to 30 days.
4.3. Recombinant Expression and Purification of Response Regulators
The H. pylori 26695 HsrA protein was overexpressed in E. coli BL21(DE3) (EMD Biosciences) and purified by immobilized metal-affinity chromatography (IMAC) as previously described [40]. The complete coding sequence of the cosR gene from C. jejuni ATCC 33560 was amplified by PCR and cloned into the expression vector pET-28a (Novagen). Sequences of primers used for cloning hsrA and cosR are described in Supplementary Table S4. Recombinant His-tagged CosR was overexpressed in E. coli BL21(DE3) and purified by IMAC-Ni2+, using an imidazole gradient for elution. Purified protein was dialyzed in 50 mM Tris–HCl (pH 8), 300 mM NaCl, and 10% glycerol. Concentration of purified recombinant proteins were estimated using the BCA™ Protein Assay kit (Thermo Fisher Scientific, Bothell, WA, United States). DNA binding activities of purified recombinant response regulators HsrA and CosR were assessed by EMSA, as described below.
4.4. High-Throughput Screening
High-throughput screening (HTS) of the Maybridge HitFinderTM chemical library for H. pylori HsrA ligands was assessed by a fluorescence-based thermal shift assay, according to previous described procedures [40,54]. Briefly, a reaction mixture containing 50 mM Tris-HCl (pH 8), 150 mM NaCl, 10% glycerol, 5 mM DTT, SYPRO® Orange ready-to-use fluorescent stain (Thermo Fisher Scientific) at a final concentration of 10 ×, and 10 µM HsrA was freshly prepared. Next, 90 μL was dispensed in all wells of V-shape 96-well plates ABgeneTM Thermo-FastTM 96 (Thermo Fisher Scientific). Wells of columns 1 and 12 received 10 μL of DMSO (vehicle) and were used as reference controls. The rest of wells (columns 2 to 11) received 2.5 μL of four different compounds from the HitFinder library (250 μM as final concentration each). The unfolding curve corresponding to each well was registered from 25°C to 75°C in 1°C steps using an Mx3005P™ qPCR System (Agilent Technologies, Inc., CA, USA), and analysed by Origin 7.0 (OriginLab, Northampton, MA, USA) using a homemade script that determines the midpoint temperature of unfolding (Tm). Those compounds that clearly increased the thermal stability of HsrA observed in reference controls (Tm ≥ 5ºC) were considered as HsrA ligands.
4.5. Electrophoretic Mobility Shift Assays
DNA binding activities of recombinant HsrA and CosR were assessed in vitro by electrophoretic mobility shift assays (EMSAs). A 300-bp promoter region of the H. pylori 26695 operon porGDAB was used as target sequence of HsrA in all EMSA experiments [48]. Likewise, a 420-bp sequence of the C. jejuni sodB promoter (ATCC 33560) was used as target DNA of CosR [90]. The oligonucleotides used for the synthesis of these promoter regions are described in Supplementary Table S4. Response regulators HsrA (6 μM) or CosR (700 nM) were mixed with 120 ng of their target promoters in reaction buffer 10 mM bis-Tris (pH 7.5), 40 mM KCl, 100 mg/L BSA, 1 mM DTT, and 5% glycerol. For the in vitro inhibition analyses, mixtures of target DNA and response regulator were exposed to 2, 1, 0.5 and 0.1 mM of each compound of interest, incubated at room temperature for 20 min, and then separated on a 6% non-denaturing electrophoresis. DMSO instead of inhibitors was included as vehicle controls, while an internal sequence of the Anabaena sp. pkn22 gene was used as non-specific competitor in all assays. For DNA-protein binding visualization, gels were stained with SYBR Safe DNA gel stain (Invitrogen) and analysed with a Gel Doc 2000 Image Analyser (Bio-Rad).
4.6. Minimal Inhibitory and Bactericidal Concentrations
The antimicrobial activity of HsrA inhibitors was evaluated by determination of minimal inhibitory concentration (MIC) against four different strains of H. pylori: ATCC 700392 (aka 26695), ATCC 43504 (MTZ-resistant reference strain), ATCC 700684 (CLR-resistant reference strain), and Donostia 2 (LVX-resistant clinical isolate). MIC were determined by the broth microdilution method, as previously described [42]. Briefly, H. pylori strains were grown in blood agar at 37 °C for 72 h under microaerobic conditions. Fresh inoculums adjusted at OD600 = 0.01 [~106 colony forming units (CFU) per mL] in BHI broth supplemented with 4% foetal bovine serum (BHI + FBS) were prepared and immediately exposed to final concentrations between 64 to 0.031 mg/L of each compound of interest. Compound solvent (DMSO) as well as conventional antibiotics (MTZ, CLR, LVX) were included as controls. MIC values were determined colourimetrically after 48 h at 37ºC by using resazurin. For minimal bactericidal concentration (MBC) determinations, aliquots from MIC plates were aseptically seeded on blood agar and incubated for 72 h at 37 ºC under microaerobic conditions. Each experiment was performed twice in triplicate. MIC and MBC values of selected compounds against C. jejuni ATCC 33560 were determined by using the same method and culture conditions described for H. pylori strains.
Antimicrobial activities of selected compounds against several Gram-negative and Gran-positive representative species of normal human microbiota were determined according to the EUCAST Guidelines [101]. Briefly, standardized inoculums (0.5 McFarland) were freshly prepared from overnight Mueller–Hinton agar plates of reference strains E. coli ATCC 25922, K. pneumonia ATCC 700603, E. faecalis ATCC 29212, S. aureus ATCC 29213, S. epidermidis ATCC 12228, and S. agalactiae ATCC 12386. Next, bacterial suspensions adjusted to 5 × 105 CFU/mL in Mueller–Hinton broth were exposed to a range of concentrations from 64 to 0.031 mg/L of selected compounds. DMSO and conventional antibiotics LVX and AMP were included as controls. MBC were determined by seeding aliquots from dilutions around the MIC values on Mueller-Hinton agar plates, which were incubated for 24 - 48 h at 37 ºC. Experiments were performed twice in triplicate.
4.7. Time-Kill Kinetic Assays
Time-kill kinetics of selected HsrA inhibitors were carried out as previously described [42], with slight modifications. Briefly, fresh grown cells of H. pylori strain ATCC 700392 were resuspended in BHI + FBS at 1.0 × 104 CFU/mL and exposed to 4 × MIC of each compound. Bacteria exposed to DMSO (vehicle) instead HsrA inhibitors was included as control. All cultures were incubated at 37 ºC under microaerobic conditions, and aliquots were taken after 0, 2, 4, 8, and 24 h of exposure for CFU determination. Experiments were performed twice in triplicate. Results were presented as log10 CFU/mL versus hours of exposure. Differences in H. pylori survival were subjected to statistical analysis by using the Mann-Whitney U test.
4.8. Checkerboard Assays
Potential synergisms between selected HsrA inhibitors and first-line antibiotics were evaluated by using checkerboard assays, as previous described [40,42]. Briefly, the antimicrobials to be tested were two-fold serially diluted in BHI + FBS using independent sterile microtiter plates. Then, concentration gradients from two antimicrobials were aseptically mixed at equal volumes in a new microtiter plate resulting in a matrix of 77 unique combinations of both compounds. Next, freshly prepared inoculum of H. pylori ATCC 700392 adjusted at 2×106 CFU/mL in BHI+ FBS were added to all wells, and plates were incubated for 72 h at 37 ºC under microaerophilic conditions. Microbial growth was revealed by adding filter-sterilized resazurin up to 0.01 mg/mL and further incubation for 6 hours. Fractional inhibitory concentration index (FICI) for each antimicrobial combination was calculated as previous described. Values of FICI ≤ 0.5, 0.5 < FICI ≤ 1, 1 < FICI ≤ 4, and FICI > 4 were considered as indicatives of synergistic, additive, neutral or antagonist interactions, respectively [40,42].
4.9. In Vivo Inhibition of HsrA
Bacterial growth from three blood agar plates of H. pylori ATCC 700392 (aka 26695) incubated for 48 h at 37 °C under microaerobic conditions was aseptically resuspended in BHI + FBS and adjusted to 107 CFU/mL. BHI broth cultures were incubated microaerobically at 37 °C for 24 h. Next, aliquots of 12 mL were exposed in triplicate to 4 × MIC of HsrA inhibitor V and further incubated in the same above conditions until log10 CFU/mL resulted diminished in one unit. At this time, bacterial growth was stopped by adding 1.5 mL ice-cold RNA stop solution (95% absolute ethanol, 5% acid-buffered phenol), and subsequently incubated on ice. Control samples were exposed to the same amount of DMSO instead inhibitor. Helicobacter cells from all samples were harvested by centrifugation (4,000 rpm at 4 ºC for 10 min), pellets were quickly frozen by immersion in liquid nitrogen and stored at -80 ºC.
4.10. RNA Sequencing
RNA-seq analysis was performed by GENEWIZ (Azenta Life Sciences, Leipzig, Germany). Frozen cell pellets from two biological replicates of each treatment condition were properly submitted to GENEWIZ in dry ice. RNA-seq libraries were prepared according to standard protocols via rRNA depletion, RNA fragmentation and random priming, reverse transcription, 5´-phosphorylation and dA-tailing of cDNAs, adaptor ligation and PCR enrichment. Once the quality of each library was confirmed, they were sequenced on Illumina NovaSeq 6000 (Illumina, San Diego, CA, USA) with 150-bp paired-ends. Raw sequence reads were filtered to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic v.0.36 [102]. The trimmed reads were subsequently mapped to the H. pylori strain 26695 reference genome NC_000915.1 available on NCBI, by using the Bowtie2 aligner v.2.2.6 [103]. Unique gene hit counts were calculated by using featureCounts [104] from the Subread package v.1.5.2. Only unique reads that fell within gene regions were counted. Using DESeq2 [105], a comparison of gene expression between control and treated groups of samples was performed. The Wald test was used to generate p-values and log2 fold changes. Genes with an adjusted p-value < 0.05 and absolute log2 fold change > 1 were considered as differentially expressed genes (DEG) for each comparison. The RNA-seq data generated in this study have been deposited in ArrayExpress with the accession number E-MTAB-14255.
4.11. Quantitative Real-Time PCR
For RNA extraction to qPCR analyses, frozen pellets were quickly resuspended in 850 μL lysis buffer containing 20 mM sodium acetate (pH 5.2), 0.5% SDS, 1 mM Na2-EDTA. Then, 850 μL of acid phenol preheated at 65ºC were immediately added to cell suspensions. Samples were vigorously mixed by vortex and incubated at 65ºC for 10 min. Cell extracts were centrifuged at 13,000 rpm during 5 min at room temperature and aqueous phase was transferred to clean RNase-free tubes. Next, samples were treated with 1 mL of TRIzolTM Reagent (Thermo Fisher Scientific) for 5 min, after which 200 μL of chloroform were added and vigorously mixed. Colourless upper aqueous phase was sequentially washed three times with 1V of chloroform, and total RNA was precipitated overnight at -80ºC with 2.5 V of ice-cold absolute ethanol. RNA samples were resuspended in 50 μL RNase-free water and genomic DNA was subsequently removed by using the TURBO DNA-free™ Kit (Thermo Fisher Scientific). Purity and integrity of RNA samples were checked by both NanoDrop spectrophotometer (Thermo Fisher Scientific) and electrophoresis. Absence of residual DNA was determined by qPCR.
Reverse transcription was carried out with SuperScript retrotranscriptase (Invitrogen) in reaction buffer containing 150 ng of random primers (Invitrogen), 1 mM dNTP mix (GE Healthcare), and 10 mM DTT. Quantitative PCRs were performed by using a QuantStudio™ 5 Real-Time PCR System (Applied Biosystems) in 30 μL of reaction mixtures containing 10 ng of cDNA, 12.5 μL of SYBR Green PCR Master Mix (Thermo Fisher Scientific), and 0.4 μL of primers at 25 μM in water. The sequences of primers used for each gene are described in Supplementary Table S4. Relative quantification was performed according to the ΔΔCt method [106]. Expression levels were normalized using the H. pylori 26695 glnA gene as housekeeping [107].
4.12. Cytotoxicity and Therapeutic Index
The in vitro toxicity of selected HsrA inhibitors was determined on HeLa cells by the PrestoBlueTM assay, as previously described [42]. Briefly, cells were cultured at 37 °C with 5% CO2 in Dulbecco’s modified Eagle’s medium containing 10% foetal bovine serum, 1% L-GlutaMAXTM solution (Thermo Fisher Scientific) and 1% penicillin/streptomycin. When 80% confluence was achieved, cells were detached, counted, seeded in 96-well microplates at a density of 10,000 cells per well, and allowed to adhere for 24 h. Next, the mammalian cells were exposed to selected compounds at a range of 0.125 to 128 mg/L. After 24 h of exposure, cell viability was measured with PrestoBlueTM cell viability reagent (Thermo Fisher Scientific), according to the manufacturer´s instructions. Experiments were performed twice in triplicate. The 50 % cytotoxic concentration (CC50) of each compound was calculated by regression analysis using Microsoft Excel. Therapeutic index (TI) values were calculated as the ratio of the CC50 to the higher MIC observed with each compound [108].
4.13. Isothermal Titration Calorimetry
Molecular interactions of selected ligands with proteins HsrA or CosR were studied by isothermal titration calorimetry (ITC), as previously described [42]. Titrations were assessed in a high-sensitivity MicroCal Auto-iTC200 calorimeter (Malvern Panalytical, Malvern, UK). Experiments were carried out at 25 °C in freshly prepared buffer containing 50 mM Tris-HCl [pH 8], 150 mM NaCl, 10% glycerol, and 1% DMSO. A 20 µM solution of protein was located in the calorimetric cell. Then, a sequence of 19 injections of 2 µL volume each with 200 µM solution of the corresponding ligand was programmed with a time spacing of 150 s, a stirring speed of 750 rpm, and a reference power of 10 µcal/s in the sample cell. Thermodynamic properties such as binding stoichiometry, dissociation constants and binding enthalpies were calculated by nonlinear least-squares regression data analysis using the Origin 7.0 software.
4.14. Molecular Docking
Predicted models of interaction between the response regulator HsrA and selected compounds were determined by molecular docking analyses using AutoDock Vina [109]. The 3D structures of compounds were built in Corina Classic (www.mn-am.com) and energy was minimized using the web server AMMOS2 [110]. 3D structure of HsrA (2HQR, model 1, chain A) was obtained from Protein Data Bank. The protein structure was considered rigid while ligands were treated as flexible molecules with free rotatable bonds. Interaction energy was estimated by AutoGrid4 [111] and top-ranked pose for each ligand was considered as the predicted model of interaction. Protein-ligand complexes were visualized by PyMOL (www.pymol.org).
5. Patents
The authors declare that a patent has been filed concerning the use of compounds for the treatment and/or prevention of an infection or disease caused by Helicobacter or Campylobacter.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Isothermal titration calorimetry (ITC) analyses of the interaction between the H. pylori response regulator HsrA and its low-molecular weight ligands I, IV, V, VIII, XI and XII.; Figure S2: ITC analysis of the interaction between the C. jejuni response regulator CosR and compound V.; Table S1: Compounds from the Maybridge HitFinderTM chemical collection identified as HsrA ligands according the fluorescent thermal shift-based HTS.; Table S2: Differential expression analysis of H.pylori 26695 exposed to lethal concentrations of the highly bactericidal HsrA ligand V ; Table S3: Functional classification of DEGs.; Table S4: List of oligonucleotides used in this study.
Author Contributions
Conceptualization, A.G.; methodology, A.G., J.C., I.O.-M., S.A., E.C., S.S. and A.V.-C.; software, A.G., J.C., I.O-M., and A.V.-C.; validation, A.G., J.C., I.O.-M., S.S., A.V.-C., E.P., M.F. and J.S.; formal analysis, A.G., I.O.-M., A.V.-C., E.P., M.F. and J.S ; investigation, A.G., J.C., I.O.-M., S.A., E.C., S.S. and A.V.-C.; resources, S.A., M.F., and A.L.; data curation, A.G., J.C., I.O.-M., S.A., E.C., S.S. and A.V.-C.; writing—original draft preparation, A.G.; writing—review and editing, A.G., J.C., I.O.-M., M.F., J.S. and A.L.; visualization, A.G., J.C., I.O.-M. and A.V.-C.; supervision, A.G., E.P., M.F., J.S., A.L.; project administration, A.G. and A.L.; funding acquisition, M.F. and A.L. All authors have read and agreed to the published version of the manuscript..
Funding
This research was funded by the Government of Aragon (Spain), grant numbers B25_20R, B25_23R, and E35_20R).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
RNA-seq data have been deposited in ArrayExpress under accession number E-MTAB-14255, and are available for reviewers through this link: https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-14255?key=5f27c51b-0a47-402f-a663-8fee6c188cc7. The rest of data supporting the findings of this study are available within the paper and its Supplementary Information files.
Acknowledgments
Authors greatly appreciate the logistic and technical support of Dr. José Antonio Aínsa, from Department of Microbiology, Faculty of Medicine, University of Zaragoza. Authors specially thank Dr. Milagrosa Montes from Donostia University Hospital (San Sebastian, Spain) for the kind donation of H. pylori levofloxacin-resistant strain Donostia 2. Financial support by the National Institute of Health Carlos III (ISCIII) to Javier Casado through the PFIS predoctoral fellow programme (FI21/00098) is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int J Syst Evol Microbiol 2021, 71, 005056. [Google Scholar] [CrossRef]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol 2010, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, F.; Covino, M.; Roubaud Baudron, C. Review: Helicobacter pylori and extragastric diseases. Helicobacter 2019, 24 Suppl 1, e12636. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Moss, S.F. The clinical evidence linking Helicobacter pylori to gastric cancer. Cell Mol Gastroenterol Hepatol 2017, 3, 183–191. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum 1994, 61, 1–241. [Google Scholar]
- Hu, Y.; Zhang, M.; Lu, B.; Dai, J. Helicobacter pylori and antibiotic resistance, a continuing and intractable problem. Helicobacter 2016, 21, 349–363. [Google Scholar] [CrossRef]
- Tshibangu-Kabamba, E.; Yamaoka, Y. Helicobacter pylori infection and antibiotic resistance - from biology to clinical implications. Nat Rev Gastroenterol Hepatol 2021, 18, 613–629. [Google Scholar] [CrossRef]
- Shah, S.C.; Iyer, P.G.; Moss, S.F. AGA clinical practice update on the management of refractory Helicobacter pylori infection: expert review. Gastroenterology 2021, 160, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner, P.; Megraud, F.; Rokkas, T.; Gisbert, J.P.; Liou, J.M.; Schulz, C.; Gasbarrini, A.; Hunt, R.H.; Leja, M.; O'Morain, C.; et al. Management of Helicobacter pylori infection: the Maastricht VI/Florence consensus report. Gut 2022, 71, 1724–1762. [Google Scholar] [CrossRef]
- Gisbert, J.P.; Alcedo, J.; Amador, J.; Bujanda, L.; Calvet, X.; Castro-Fernandez, M.; Fernandez-Salazar, L.; Gene, E.; Lanas, A.; Lucendo, A.J.; et al. V Spanish Consensus Conference on Helicobacter pylori infection treatment. Gastroenterol. Hepatol. 2022, 45, 392–417. [Google Scholar] [CrossRef]
- Salcedo, J.A.; Al-Kawas, F. Treatment of Helicobacter pylori infection. Arch. Intern. Med. 1998, 158, 842–851. [Google Scholar] [CrossRef]
- Arslan, N.; Yilmaz, O.; Demiray-Gurbuz, E. Importance of antimicrobial susceptibility testing for the management of eradication in Helicobacter pylori infection. World J Gastroenterol 2017, 23, 2854–2869. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, T.; Tang, H.; Tang, X.; Shen, Y.; Benghezal, M.; Tay, A.; Marshall, B. Helicobacter pylori infection is an infectious disease and the empiric therapy paradigm should be changed. Precis Clin Med 2019, 2, 77–80. [Google Scholar] [CrossRef]
- Megraud, F.; Bruyndonckx, R.; Coenen, S.; Wittkop, L.; Huang, T.D.; Hoebeke, M.; Benejat, L.; Lehours, P.; Goossens, H.; Glupczynski, Y.; et al. Helicobacter pylori resistance to antibiotics in Europe in 2018 and its relationship to antibiotic consumption in the community. Gut 2021, 70, 1815–1822. [Google Scholar] [CrossRef]
- De Francesco, V.; Zullo, A.; Manta, R.; Gatta, L.; Fiorini, G.; Saracino, I.M.; Vaira, D. Helicobacter pylori eradication following first-line treatment failure in Europe: What, how and when chose among different standard regimens? A systematic review. Eur. J. Gastroenterol. Hepatol. 2021, 33, e66–e70. [Google Scholar] [CrossRef]
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of antibiotic resistance in Helicobacter pylori: a systematic review and meta-analysis in World Health Organization regions. Gastroenterology 2018, 155, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Sukri, A.; Hanafiah, A.; Yusoff, H.; Shamsul Nizam, N.A.; Nameyrra, Z.; Wong, Z.; Raja Ali, R.A. Multidrug-resistant Helicobacter pylori strains: a five-year surveillance study and its genome characteristics. Antibiotics (Basel) 2022, 11, 1391. [Google Scholar] [CrossRef]
- Ho, J.J.C.; Navarro, M.; Sawyer, K.; Elfanagely, Y.; Moss, S.F. Helicobacter pylori antibiotic resistance in the United States between 2011 and 2021: a systematic review and meta-analysis. Am. J. Gastroenterol. 2022, 117, 1221–1230. [Google Scholar] [CrossRef]
- Kim, S.E.; Park, M.I.; Park, S.J.; Moon, W.; Choi, Y.J.; Cheon, J.H.; Kwon, H.J.; Ku, K.H.; Yoo, C.H.; Kim, J.H.; et al. Trends in Helicobacter pylori eradication rates by first-line triple therapy and related factors in eradication therapy. Korean J. Intern. Med. 2015, 30, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Boyanova, L.; Evstatiev, I.; Yordanov, D.; Markovska, R.; Mitov, I. Three unsuccessful treatments of Helicobacter pylori infection by a highly virulent strain with quadruple antibiotic resistance. Folia Microbiol. (Praha) 2016, 61, 307–310. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Hsu, P.I.; Pan, C.Y.; Kao, J.Y.; Tsay, F.W.; Peng, N.J.; Kao, S.S.; Wang, H.M.; Tsai, T.J.; Wu, D.C.; Chen, C.L.; et al. Helicobacter pylori eradication with bismuth quadruple therapy leads to dysbiosis of gut microbiota with an increased relative abundance of Proteobacteria and decreased relative abundances of Bacteroidetes and Actinobacteria. Helicobacter 2018, 23, e12498. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.M.; Chen, C.C.; Chang, C.M.; Fang, Y.J.; Bair, M.J.; Chen, P.Y.; Chang, C.Y.; Hsu, Y.C.; Chen, M.J.; Chen, C.C.; et al. Long-term changes of gut microbiota, antibiotic resistance, and metabolic parameters after Helicobacter pylori eradication: a multicentre, open-label, randomised trial. Lancet Infect Dis 2019, 19, 1109–1120. [Google Scholar] [CrossRef]
- Ye, Q.; Shao, X.; Shen, R.; Chen, D.; Shen, J. Changes in the human gut microbiota composition caused by Helicobacter pylori eradication therapy: a systematic review and meta-analysis. Helicobacter 2020, 25, e12713. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ye, Z.; Wang, Y.; Huang, Z.; Zheng, C.; Shi, J.; Tang, W.; Zhang, P.; Wang, S.; Huang, Y. Long-term changes in the gut microbiota after triple therapy, sequential therapy, bismuth quadruple therapy and concomitant therapy for Helicobacter pylori eradication in Chinese children. Helicobacter 2021, 26, e12809. [Google Scholar] [CrossRef]
- Trifan, A.; Girleanu, I.; Cojocariu, C.; Sfarti, C.; Singeap, A.M.; Dorobat, C.; Grigore, L.; Stanciu, C. Pseudomembranous colitis associated with a triple therapy for Helicobacter pylori eradication. World J Gastroenterol 2013, 19, 7476–7479. [Google Scholar] [CrossRef]
- Sato, S.; Chinda, D.; Yamai, K.; Satake, R.; Soma, Y.; Shimoyama, T.; Fukuda, S. A case of severe pseudomembranous colitis diagnosed by colonoscopy after Helicobacter pylori eradication. Clin J Gastroenterol 2014, 7, 247–250. [Google Scholar] [CrossRef]
- Nei, T.; Hagiwara, J.; Takiguchi, T.; Yokobori, S.; Shiei, K.; Yokota, H.; Senoh, M.; Kato, H. Fatal fulminant Clostridioides difficile colitis caused by Helicobacter pylori eradication therapy; a case report. J Infect Chemother 2020, 26, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, M.; Suzuki, R.; Tanaka, N.; Fukunaga, H.; Kinoshita, Y.; Kimura, H.; Tsutsui, S.; Murata, M.; Morita, S. Community-acquired fulminant Clostridioides (Clostridium) difficile infection by ribotype 027 isolate in Japan: a case report. Surg Case Rep 2021, 7, 137. [Google Scholar] [CrossRef]
- Melander, R.J.; Zurawski, D.V.; Melander, C. Narrow-spectrum antibacterial agents. Medchemcomm 2018, 9, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, C.N.; Klein, R.D.; Schreiber, H.L.t.; Janetka, J.W.; Hultgren, S.J. Precision antimicrobial therapeutics: the path of least resistance? NPJ Biofilms Microbiomes 2018, 4, 4. [Google Scholar] [CrossRef]
- Alm, R.A.; Lahiri, S.D. Narrow-spectrum antibacterial agents-benefits and challenges. Antibiotics (Basel) 2020, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Landick, R.; Campbell, E.A. A roadmap for designing narrow-spectrum antibiotics targeting bacterial pathogens. Microb Cell 2022, 9, 136–138. [Google Scholar] [CrossRef]
- Diamantis, S.; Retur, N.; Bertrand, B.; Lieutier-Colas, F.; Carenco, P.; Mondain, V.; On Behalf Of Promise Professional Community Network On Antimicrobial, R. The production of antibiotics must be reoriented: repositioning old narrow-spectrum antibiotics, developing new microbiome-sparing antibiotics. Antibiotics (Basel) 2022, 11, 924. [Google Scholar] [CrossRef]
- De la Fuente-Nunez, C.; Torres, M.D.; Mojica, F.J.; Lu, T.K. Next-generation precision antimicrobials: towards personalized treatment of infectious diseases. Curr. Opin. Microbiol. 2017, 37, 95–102. [Google Scholar] [CrossRef]
- Paharik, A.E.; Schreiber, H.L.t.; Spaulding, C.N.; Dodson, K.W.; Hultgren, S.J. Narrowing the spectrum: the new frontier of precision antimicrobials. Genome Med 2017, 9, 110. [Google Scholar] [CrossRef]
- González, A.; Salillas, S.; Velázquez-Campoy, A.; Espinosa Angarica, V.; Fillat, M.F.; Sancho, J.; Lanas, Á. Identifying potential novel drugs against Helicobacter pylori by targeting the essential response regulator HsrA. Sci Rep 2019, 9, 11294. [Google Scholar] [CrossRef]
- González, A.; Casado, J.; Chueca, E.; Salillas, S.; Velázquez-Campoy, A.; Espinosa Angarica, V.; Benejat, L.; Guignard, J.; Giese, A.; Sancho, J.; et al. Repurposing dihydropyridines for treatment of Helicobacter pylori infection. Pharmaceutics 2019, 11, 681. [Google Scholar] [CrossRef] [PubMed]
- González, A.; Casado, J.; Gündüz, M.G.; Santos, B.; Velázquez-Campoy, A.; Sarasa-Buisan, C.; Fillat, M.F.; Montes, M.; Piazuelo, E.; Lanas, Á. 1,4-Dihydropyridine as a promising scaffold for novel antimicrobials against Helicobacter pylori. Front Microbiol 2022, 13, 874709. [Google Scholar] [CrossRef]
- Lee, H.M.; Hong, E.; Jeon, B.Y.; Kim, D.U.; Byun, J.S.; Lee, W.; Cho, H.S. Crystallization and preliminary X-ray crystallographic study of HP1043, a Helicobacter pylori orphan response regulator. Biochim. Biophys. Acta 2006, 1764, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Casado, J.; Lanas, A.; González, A. Two-component regulatory systems in Helicobacter pylori and Campylobacter jejuni: Attractive targets for novel antibacterial drugs. Front Cell Infect Microbiol 2022, 12, 977944. [Google Scholar] [CrossRef]
- Muller, S.; Pflock, M.; Schar, J.; Kennard, S.; Beier, D. Regulation of expression of atypical orphan response regulators of Helicobacter pylori. Microbiol. Res. 2007, 162, 1–14. [Google Scholar] [CrossRef]
- Delany, I.; Spohn, G.; Rappuoli, R.; Scarlato, V. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 2002, 184, 4800–4810. [Google Scholar] [CrossRef]
- Olekhnovich, I.N.; Vitko, S.; Chertihin, O.; Hontecillas, R.; Viladomiu, M.; Bassaganya-Riera, J.; Hoffman, P.S. Mutations to essential orphan response regulator HP1043 of Helicobacter pylori result in growth-stage regulatory defects. Infect. Immun. 2013, 81, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Olekhnovich, I.N.; Vitko, S.; Valliere, M.; Hoffman, P.S. Response to metronidazole and oxidative stress is mediated through homeostatic regulator HsrA (HP1043) in Helicobacter pylori. J. Bacteriol. 2014, 196, 729–739. [Google Scholar] [CrossRef]
- Pelliciari, S.; Pinatel, E.; Vannini, A.; Peano, C.; Puccio, S.; De Bellis, G.; Danielli, A.; Scarlato, V.; Roncarati, D. Insight into the essential role of the Helicobacter pylori HP1043 orphan response regulator: genome-wide identification and characterization of the DNA-binding sites. Sci. Rep. 2017, 7, 41063. [Google Scholar] [CrossRef]
- Beier, D.; Frank, R. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 2000, 182, 2068–2076. [Google Scholar] [CrossRef]
- McDaniel, T.K.; Dewalt, K.C.; Salama, N.R.; Falkow, S. New approaches for validation of lethal phenotypes and genetic reversion in Helicobacter pylori. Helicobacter 2001, 6, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Cremades, N.; Velázquez-Campoy, A.; Martínez-Júlvez, M.; Neira, J.L.; Pérez-Dorado, I.; Hermoso, J.; Jiménez, P.; Lanas, A.; Hoffman, P.S.; Sancho, J. Discovery of specific flavodoxin inhibitors as potential therapeutic agents against Helicobacter pylori infection. ACS Chem Biol 2009, 4, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef]
- Wuchty, S.; Muller, S.A.; Caufield, J.H.; Hauser, R.; Aloy, P.; Kalkhof, S.; Uetz, P. Proteome data improves protein function prediction in the interactome of Helicobacter pylori. Mol Cell Proteomics 2018, 17, 961–973. [Google Scholar] [CrossRef]
- Velayudhan, J.; Hughes, N.J.; McColm, A.A.; Bagshaw, J.; Clayton, C.L.; Andrews, S.C.; Kelly, D.J. Iron acquisition and virulence in Helicobacter pylori: a major role for FeoB, a high-affinity ferrous iron transporter. Mol. Microbiol. 2000, 37, 274–286. [Google Scholar] [CrossRef]
- Wai, S.N.; Nakayama, K.; Umene, K.; Moriya, T.; Amako, K. Construction of a ferritin-deficient mutant of Campylobacter jejuni: contribution of ferritin to iron storage and protection against oxidative stress. Mol. Microbiol. 1996, 20, 1127–1134. [Google Scholar] [CrossRef]
- Waidner, B.; Greiner, S.; Odenbreit, S.; Kavermann, H.; Velayudhan, J.; Stahler, F.; Guhl, J.; Bisse, E.; van Vliet, A.H.; Andrews, S.C.; et al. Essential role of ferritin Pfr in Helicobacter pylori iron metabolism and gastric colonization. Infect. Immun. 2002, 70, 3923–3929. [Google Scholar] [CrossRef]
- Olczak, A.A.; Olson, J.W.; Maier, R.J. Oxidative-stress resistance mutants of Helicobacter pylori. J. Bacteriol. 2002, 184, 3186–3193. [Google Scholar] [CrossRef]
- Wang, G.; Hong, Y.; Olczak, A.; Maier, S.E.; Maier, R.J. Dual roles of Helicobacter pylori NapA in inducing and combating oxidative stress. Infect. Immun. 2006, 74, 6839–6846. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Choi, H.; Leung, K.; Jiang, F.; Graham, D.Y.; Leung, W.K. Global prevalence of Helicobacter pylori infection between 1980 and 2022: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol 2023, 8, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Malfertheiner, P.; Yu, H.T.; Kuo, C.L.; Chang, Y.Y.; Meng, F.T.; Wu, Y.X.; Hsiao, J.L.; Chen, M.J.; Lin, K.P.; et al. Global prevalence of Helicobacter pylori infection and incidence of gastric cancer between 1980 and 2022. Gastroenterology 2024, 166, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020-40: A population-based modelling study. EClinicalMedicine 2022, 47, 101404. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.H.; Chang, W.J.; Chen, S.L.; Yen, A.M.; Fann, J.C.; Chiu, S.Y.; Chen, Y.R.; Chuang, S.L.; Shieh, C.F.; Liu, C.Y.; et al. Mass eradication of Helicobacter pylori to reduce gastric cancer incidence and mortality: a long-term cohort study on Matsu Islands. Gut 2021, 70, 243–250. [Google Scholar] [CrossRef]
- Yan, L.; Chen, Y.; Chen, F.; Tao, T.; Hu, Z.; Wang, J.; You, J.; Wong, B.C.Y.; Chen, J.; Ye, W. Effect of Helicobacter pylori eradication on gastric cancer prevention: updated report from a randomized controlled trial with 26.5 years of follow-up. Gastroenterology 2022, 163, 154–162. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chiang, T.H.; Chou, C.K.; Tu, Y.K.; Liao, W.C.; Wu, M.S.; Graham, D.Y. Association between Helicobacter pylori eradication and gastric cancer incidence: a systematic review and meta-analysis. Gastroenterology 2016, 150, 1113–1124. [Google Scholar] [CrossRef]
- Boyanova, L.; Hadzhiyski, P.; Gergova, R.; Markovska, R. Evolution of Helicobacter pylori resistance to antibiotics: a topic of increasing concern. Antibiotics (Basel) 2023, 12, 332. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Q.; Yuan, Y.; Gong, Y. The antibiotic resistance of Helicobacter pylori to five antibiotics and influencing factors in an area of China with a high risk of gastric cancer. BMC Microbiol 2019, 19, 152. [Google Scholar] [CrossRef]
- González, A.; Casado, J.; Lanas, A. Fighting the antibiotic crisis: flavonoids as promising antibacterial drugs against Helicobacter pylori infection. Front Cell Infect Microbiol 2021, 11, 709749. [Google Scholar] [CrossRef]
- Brown, D.G.; May-Dracka, T.L.; Gagnon, M.M.; Tommasi, R. Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem. 2014, 57, 10144–10161. [Google Scholar] [CrossRef]
- Kolaric, A.; Kokot, M.; Hrast, M.; Weiss, M.; Zdovc, I.; Trontelj, J.; Zakelj, S.; Anderluh, M.; Minovski, N. A Fine-tuned lipophilicity/hydrophilicity ratio governs antibacterial potency and selectivity of bifurcated halogen bond-forming NBTIs. Antibiotics (Basel) 2021, 10, 862. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Weiss, M.; Zdovc, I.; Hrast, M.; Anderluh, M.; Minovski, N. Structurally optimized potent dual-targeting NBTI antibacterials with an enhanced bifurcated halogen-bonding propensity. ACS Med Chem Lett 2021, 12, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Leus, I.V.; Adamiak, J.; Chandar, B.; Bonifay, V.; Zhao, S.; Walker, S.S.; Squadroni, B.; Balibar, C.J.; Kinarivala, N.; Standke, L.C.; et al. Functional diversity of Gram-negative permeability barriers reflected in antibacterial activities and intracellular accumulation of antibiotics. Antimicrob. Agents Chemother. 2023, 67, e0137722. [Google Scholar] [CrossRef] [PubMed]
- Surivet, J.P.; Zumbrunn, C.; Bruyere, T.; Bur, D.; Kohl, C.; Locher, H.H.; Seiler, P.; Ertel, E.A.; Hess, P.; Enderlin-Paput, M.; et al. Synthesis and characterization of tetrahydropyran-based bacterial topoisomerase inhibitors with antibacterial activity against Gram-negative bacteria. J. Med. Chem. 2017, 60, 3776–3794. [Google Scholar] [CrossRef]
- Blair, J.M.; Richmond, G.E.; Piddock, L.J. Multidrug efflux pumps in Gram-negative bacteria and their role in antibiotic resistance. Future Microbiol 2014, 9, 1165–1177. [Google Scholar] [CrossRef]
- Alfaray, R.I.; Saruuljavkhlan, B.; Fauzia, K.A.; Torres, R.C.; Thorell, K.; Dewi, S.R.; Kryukov, K.A.; Matsumoto, T.; Akada, J.; Vilaichone, R.K.; et al. Global antimicrobial resistance gene study of Helicobacter pylori: comparison of detection tools, ARG and efflux pump gene analysis, worldwide epidemiological distribution, and information related to the antimicrobial-resistant phenotype. Antibiotics (Basel) 2023, 12, 1118. [Google Scholar] [CrossRef]
- Igwaran, A.; Okoh, A.I. Human campylobacteriosis: A public health concern of global importance. Heliyon 2019, 5, e02814. [Google Scholar] [CrossRef]
- Facciola, A.; Riso, R.; Avventuroso, E.; Visalli, G.; Delia, S.A.; Lagana, P. Campylobacter: from microbiology to prevention. J Prev Med Hyg 2017, 58, E79–E92. [Google Scholar]
- Thepault, A.; Rose, V.; Queguiner, M.; Chemaly, M.; Rivoal, K. Dogs and cats: reservoirs for highly diverse Campylobacter jejuni and a potential source of human exposure. Animals (Basel) 2020, 10, 838. [Google Scholar] [CrossRef]
- Allos, B.M. Campylobacter jejuni infections: update on emerging issues and trends. Clin. Infect. Dis. 2001, 32, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Same, R.G.; Tamma, P.D. Campylobacter infections in children. Pediatr. Rev. 2018, 39, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Yokoyama, K.; Nukada, T.; Ikeda, Y.; Hara, S. Campylobacter jejuni bacteremia in the term infant a rare cause of neonatal hematochezia. Pediatr. Infect. Dis. J. 2022, 41, e156–e157. [Google Scholar] [CrossRef] [PubMed]
- Mearelli, F.; Casarsa, C.; Breglia, A.; Biolo, G. Septic shock with multi organ failure due to fluoroquinolones resistant Campylobacter jejuni. Am J Case Rep 2017, 18, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Eiland, L.S.; Jenkins, L.S. Optimal treatment of Campylobacter dysentery. J Pediatr Pharmacol Ther 2008, 13, 170–174. [Google Scholar] [CrossRef]
- Luangtongkum, T.; Jeon, B.; Han, J.; Plummer, P.; Logue, C.M.; Zhang, Q. Antibiotic resistance in Campylobacter: emergence, transmission and persistence. Future Microbiol 2009, 4, 189–200. [Google Scholar] [CrossRef]
- Bolinger, H.; Kathariou, S. The current state of macrolide resistance in Campylobacter spp.: trends and impacts of resistance mechanisms. Appl. Environ. Microbiol. 2017, 83, e00416–17. [Google Scholar] [CrossRef]
- Portes, A.B.; Panzenhagen, P.; Pereira Dos Santos, A.M.; Junior, C.A.C. Antibiotic resistance in Campylobacter: a systematic review of South American isolates. Antibiotics (Basel) 2023, 12, 548. [Google Scholar] [CrossRef]
- McDermott, P.F.; Bodeis, S.M.; Aarestrup, F.M.; Brown, S.; Traczewski, M.; Fedorka-Cray, P.; Wallace, M.; Critchley, I.A.; Thornsberry, C.; Graff, S.; et al. Development of a standardized susceptibility test for Campylobacter with quality-control ranges for ciprofloxacin, doxycycline, erythromycin, gentamicin, and meropenem. Microb Drug Resist 2004, 10, 124–131. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, M.; Ryu, S.; Jeon, B. Regulation of oxidative stress response by CosR, an essential response regulator in Campylobacter jejuni. PLoS One 2011, 6, e22300. [Google Scholar] [CrossRef]
- Hwang, S.; Zhang, Q.; Ryu, S.; Jeon, B. Transcriptional regulation of the CmeABC multidrug efflux pump and the KatA catalase by CosR in Campylobacter jejuni. J. Bacteriol. 2012, 194, 6883–6891. [Google Scholar] [CrossRef] [PubMed]
- Grinnage-Pulley, T.; Mu, Y.; Dai, L.; Zhang, Q. Dual repression of the multidrug efflux pump CmeABC by CosR and CmeR in Campylobacter jejuni. Front Microbiol 2016, 7, 1097. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Hwang, S.; Ryu, S.; Jeon, B. CosR regulation of perR transcription for the control of oxidative stress defense in Campylobacter jejuni. Microorganisms 2021, 9, 1281. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowska, M.; Ryden-Aulin, M. Ribosome biogenesis and the translation process in Escherichia coli. Microbiol. Mol. Biol. Rev. 2007, 71, 477–494. [Google Scholar] [CrossRef] [PubMed]
- Stokes, J.M.; Selin, C.; Cardona, S.T.; Brown, E.D. Chemical inhibition of bacterial ribosome biogenesis shows efficacy in a worm infection model. Antimicrob. Agents Chemother. 2015, 59, 2918–2920. [Google Scholar] [CrossRef]
- Farha, M.A.; Verschoor, C.P.; Bowdish, D.; Brown, E.D. Collapsing the proton motive force to identify synergistic combinations against Staphylococcus aureus. Chem. Biol. 2013, 20, 1168–1178. [Google Scholar] [CrossRef]
- Berube, B.J.; Parish, T. Combinations of respiratory chain inhibitors have enhanced bactericidal activity against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e01677–17. [Google Scholar] [CrossRef]
- Kashyap, D.R.; Kuzma, M.; Kowalczyk, D.A.; Gupta, D.; Dziarski, R. Bactericidal peptidoglycan recognition protein induces oxidative stress in Escherichia coli through a block in respiratory chain and increase in central carbon catabolism. Mol. Microbiol. 2017, 105, 755–776. [Google Scholar] [CrossRef]
- Stohl, E.A.; Criss, A.K.; Seifert, H.S. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol. Microbiol. 2005, 58, 520–532. [Google Scholar] [CrossRef]
- Wang, S.; Deng, K.; Zaremba, S.; Deng, X.; Lin, C.; Wang, Q.; Tortorello, M.L.; Zhang, W. Transcriptomic response of Escherichia coli O157:H7 to oxidative stress. Appl. Environ. Microbiol. 2009, 75, 6110–6123. [Google Scholar] [CrossRef]
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society for Clinical Microbiology and Infectious Diseases (ESCMID). EUCAST Discussion Document E. Dis 5.1: Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin Microbiol Infec 2003, 9, 1–7. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- van Amsterdam, K.; van Vliet, A.H.; Kusters, J.G.; Feller, M.; Dankert, J.; van der Ende, A. Induced Helicobacter pylori vacuolating cytotoxin VacA expression after initial colonisation of human gastric epithelial cells. FEMS Immunol. Med. Microbiol. 2003, 39, 251–256. [Google Scholar] [CrossRef]
- Pitucha, M.; Wos, M.; Miazga-Karska, M.; Klimek, K.; Miroslaw, B.; Pachuta-Stec, A.; Gladysz, A.; Ginalska, G. Synthesis, antibacterial and antiproliferative potential of some new 1-pyridinecarbonyl-4-substituted thiosemicarbazide derivatives. Med Chem Res 2016, 25, 1666–1677. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Labbe, C.M.; Pencheva, T.; Jereva, D.; Desvillechabrol, D.; Becot, J.; Villoutreix, B.O.; Pajeva, I.; Miteva, M.A. AMMOS2: a web server for protein-ligand-water complexes refinement via molecular mechanics. Nucleic Acids Res 2017, 45, W350–W355. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Upshifts in the thermal unfolding curve of response regulator HsrA triggered by ligands IV and XII. The high-throughput screening (HTS) of the Maybridge HitFinderTM chemical library for HsrA ligands was assessed by a fluorescence-based thermal shift assay. Mixtures of DMSO (vehicle) instead of compounds were used as reference controls. Any compound that preferentially binds to the native state of HsrA would increase protein stability and cause an increase of the midpoint temperature of unfolding (Tm). Those compounds that clearly increased the thermal stability of HsrA observed in reference controls (Tm ≥ 5ºC) were considered as HsrA ligands.
Figure 1.
Upshifts in the thermal unfolding curve of response regulator HsrA triggered by ligands IV and XII. The high-throughput screening (HTS) of the Maybridge HitFinderTM chemical library for HsrA ligands was assessed by a fluorescence-based thermal shift assay. Mixtures of DMSO (vehicle) instead of compounds were used as reference controls. Any compound that preferentially binds to the native state of HsrA would increase protein stability and cause an increase of the midpoint temperature of unfolding (Tm). Those compounds that clearly increased the thermal stability of HsrA observed in reference controls (Tm ≥ 5ºC) were considered as HsrA ligands.
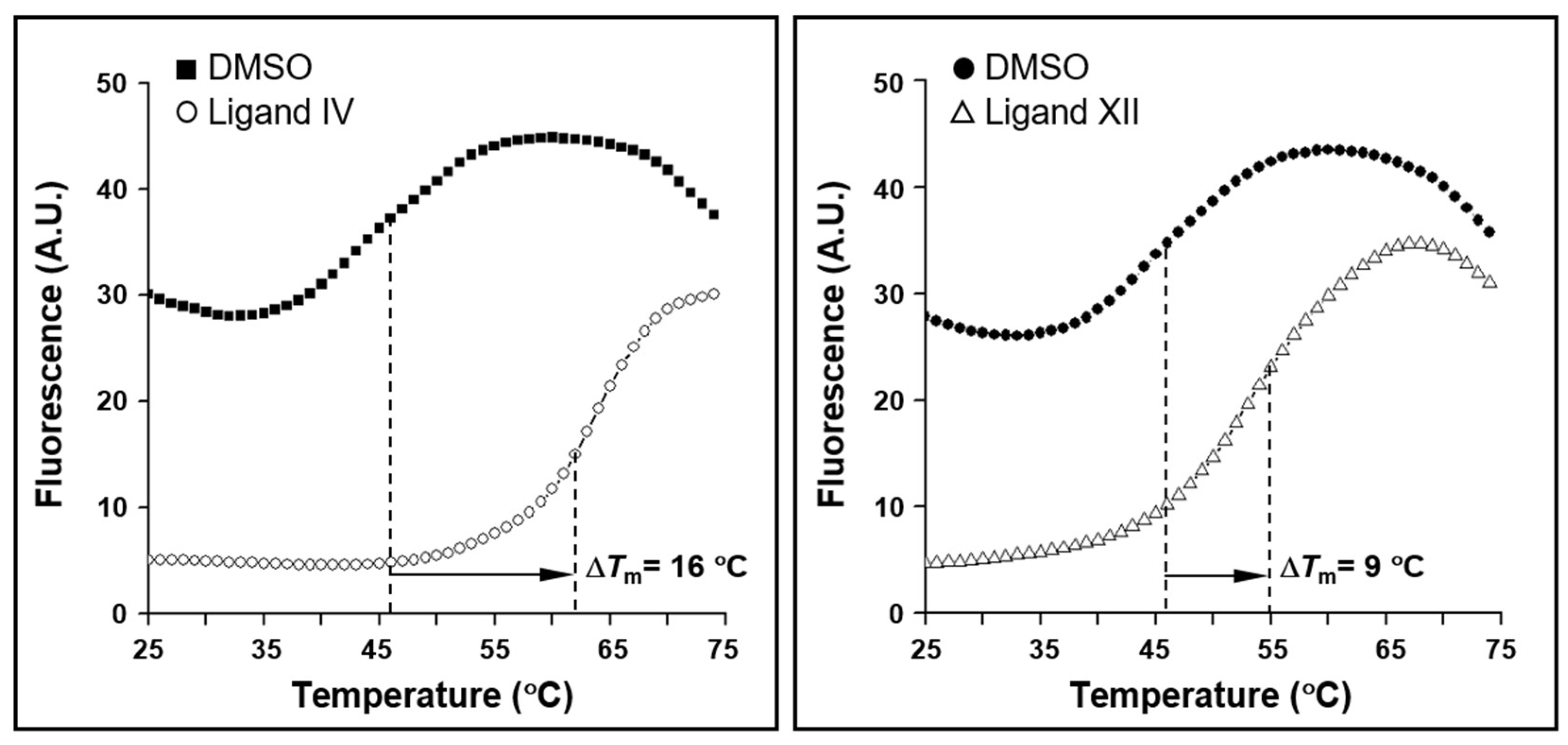
Figure 2.
Specific in vitro inhibition of the DNA binding activity of response regulator HsrA by selected ligands, according to EMSA. Recombinant HsrA was mixed with its target promoter PporG in the presence of 2, 1, 0.5, and 0.1 mM of ligands. DMSO instead of inhibitors was included as vehicle controls, while an internal sequence of the Anabaena sp. pkn22 gene was used as non-specific competitor in all assays. Addition of protein in the reaction mixtures are indicate by +. Protein-DNA interactions were analysed by 6% non-denaturing electrophoresis.
Figure 2.
Specific in vitro inhibition of the DNA binding activity of response regulator HsrA by selected ligands, according to EMSA. Recombinant HsrA was mixed with its target promoter PporG in the presence of 2, 1, 0.5, and 0.1 mM of ligands. DMSO instead of inhibitors was included as vehicle controls, while an internal sequence of the Anabaena sp. pkn22 gene was used as non-specific competitor in all assays. Addition of protein in the reaction mixtures are indicate by +. Protein-DNA interactions were analysed by 6% non-denaturing electrophoresis.
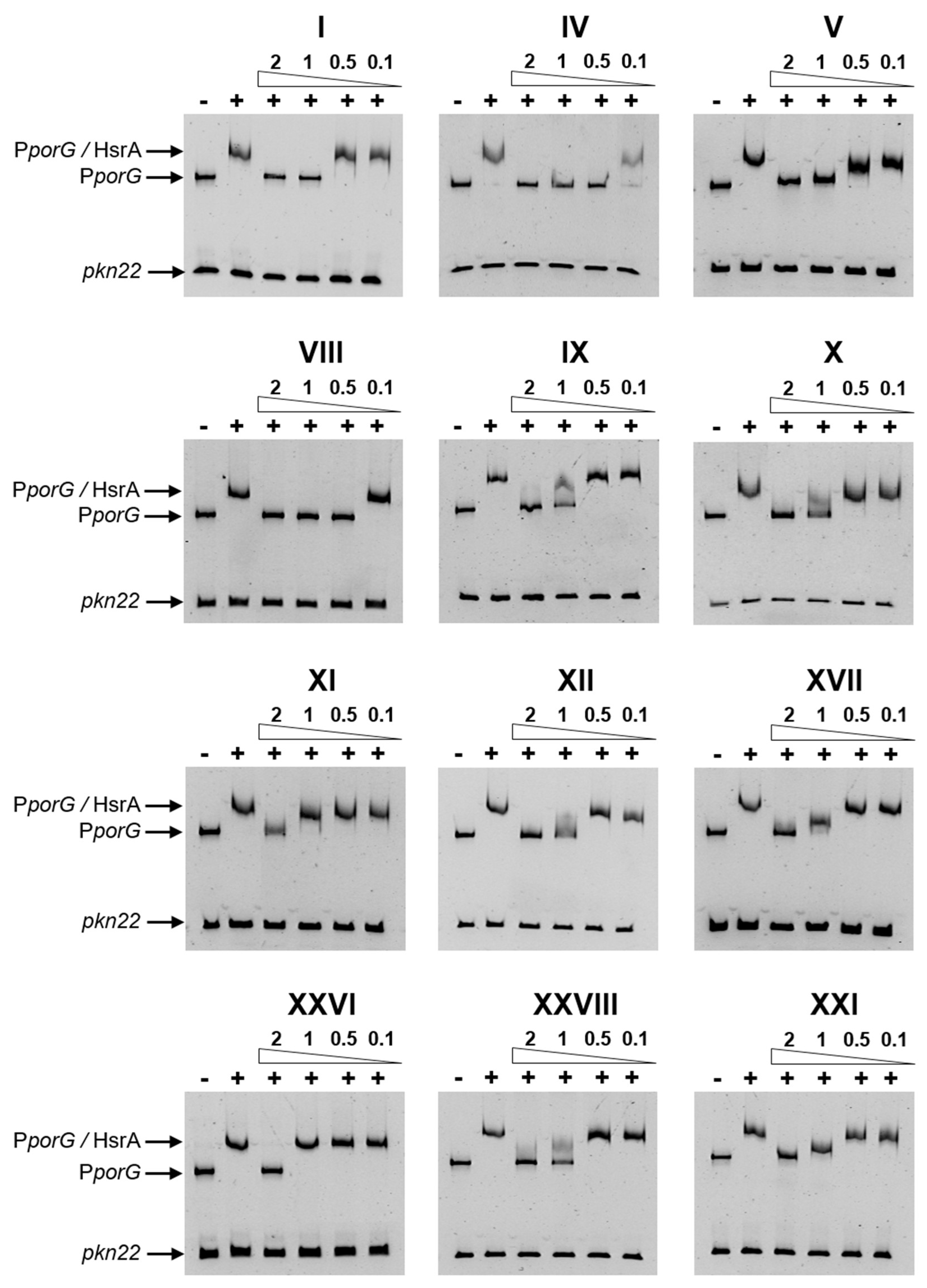
Figure 3.
Time-kill kinetics of selected HsrA inhibitors against Helicobacter pylori strain ATCC 700392. Fresh grown cells of H. pylori (104 CFU/mL) were exposed to 4 × MIC of each compound. Mixtures of bacteria with DMSO (vehicle) instead of inhibitors were used as controls. CFU were determined at time zero and after 2, 4, 8, and 24 h of exposure. Values are the means of three independent determinations; standard deviations are represented by vertical bars.
Figure 3.
Time-kill kinetics of selected HsrA inhibitors against Helicobacter pylori strain ATCC 700392. Fresh grown cells of H. pylori (104 CFU/mL) were exposed to 4 × MIC of each compound. Mixtures of bacteria with DMSO (vehicle) instead of inhibitors were used as controls. CFU were determined at time zero and after 2, 4, 8, and 24 h of exposure. Values are the means of three independent determinations; standard deviations are represented by vertical bars.
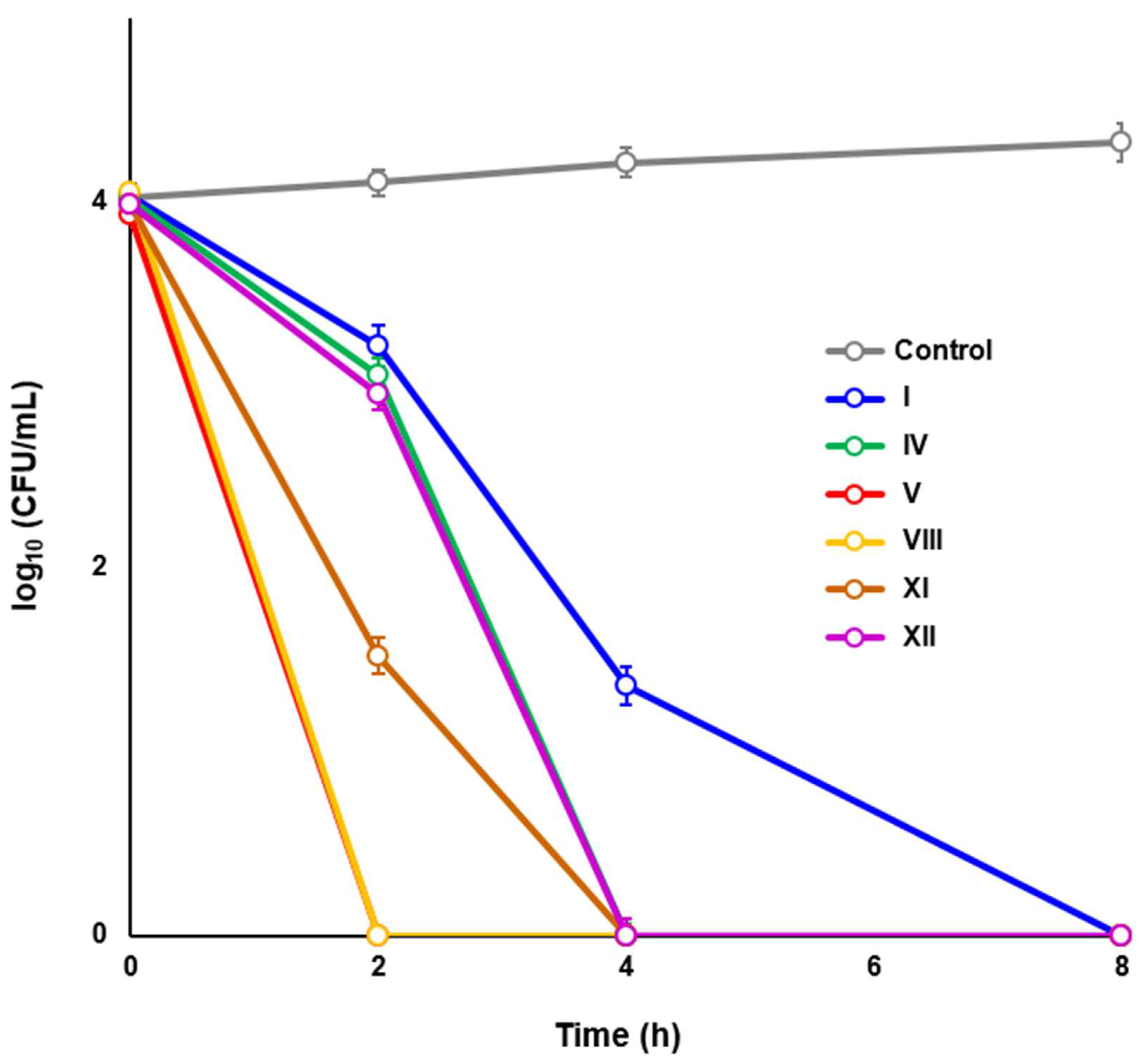
Figure 4.
The HsrA ligand V noticeably inhibited the in vitro DNA binding activity of the C. jejuni response regulator CosR, an orthologue protein of HsrA. Recombinant CosR was mixed with its target promoter PsodB in the presence of 2, 1, 0.5, and 0.1 mM of ligand V. DMSO instead of inhibitors was included as vehicle controls, while an internal sequence of the Anabaena sp. pkn22 gene was used as non-specific competitor in all assays. Addition of protein in the reaction mixtures are indicate by +. Protein-DNA interactions were analysed by 6% non-denaturing electrophoresis.
Figure 4.
The HsrA ligand V noticeably inhibited the in vitro DNA binding activity of the C. jejuni response regulator CosR, an orthologue protein of HsrA. Recombinant CosR was mixed with its target promoter PsodB in the presence of 2, 1, 0.5, and 0.1 mM of ligand V. DMSO instead of inhibitors was included as vehicle controls, while an internal sequence of the Anabaena sp. pkn22 gene was used as non-specific competitor in all assays. Addition of protein in the reaction mixtures are indicate by +. Protein-DNA interactions were analysed by 6% non-denaturing electrophoresis.
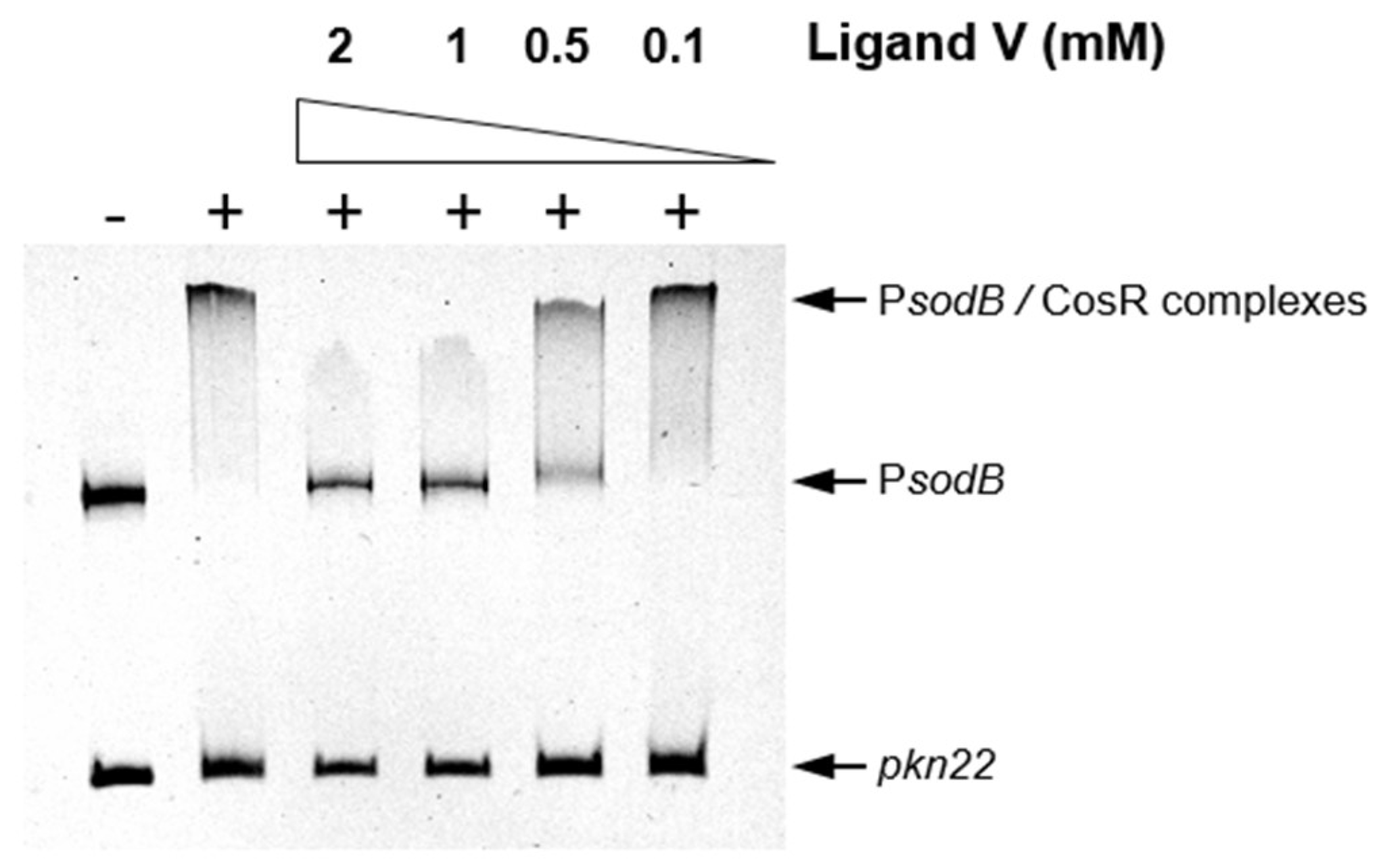
Figure 5.
Cytotoxicity and therapeutic index of selected highly bactericidal HsrA inhibitors. (A) Cytotoxicity of HsrA inhibitors I, IV, V, VIII, XI, and XII toward HeLa cells was assessed at 24 h of exposure through the PrestoBlueTM method. Experiments were performed twice in triplicate, vertical bars represent standard deviations. (B) The 50% cytotoxic concentration (CC50) was defined as the ligand concentration that reduced the viability of DMSO (vehicle) - treated cell cultures by 50%. Therapeutic index (TI) of each compound was calculated as the ratio between CC50 and MIC.
Figure 5.
Cytotoxicity and therapeutic index of selected highly bactericidal HsrA inhibitors. (A) Cytotoxicity of HsrA inhibitors I, IV, V, VIII, XI, and XII toward HeLa cells was assessed at 24 h of exposure through the PrestoBlueTM method. Experiments were performed twice in triplicate, vertical bars represent standard deviations. (B) The 50% cytotoxic concentration (CC50) was defined as the ligand concentration that reduced the viability of DMSO (vehicle) - treated cell cultures by 50%. Therapeutic index (TI) of each compound was calculated as the ratio between CC50 and MIC.
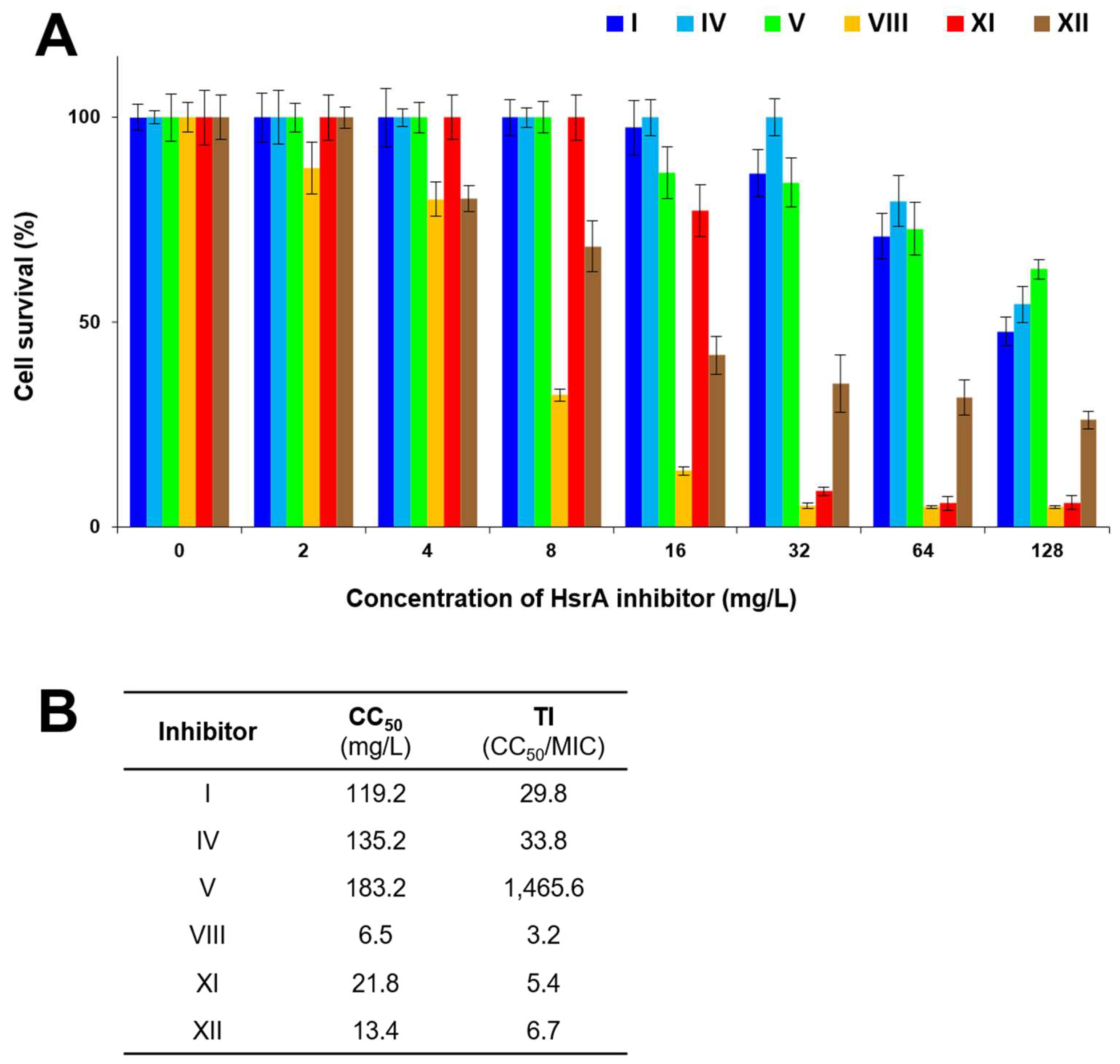
Figure 6.
Predicted model of molecular interaction between HsrA and ligand V. (A) 3D view (molecular surface) and (B) ribbon diagram of the HsrA effector domain and the best-ranked docking pose of ligand V. The helix-turn-helix (HTH) DNA binding motif has been highlighted in blue. (C) Detailed view of the amino acid residues directly involved in the interaction of HsrA with ligand V. Hydrophobic interactions are indicated with yellow dashed lines, H-bonds and halogen bonds are indicated with orange dashed lines.
Figure 6.
Predicted model of molecular interaction between HsrA and ligand V. (A) 3D view (molecular surface) and (B) ribbon diagram of the HsrA effector domain and the best-ranked docking pose of ligand V. The helix-turn-helix (HTH) DNA binding motif has been highlighted in blue. (C) Detailed view of the amino acid residues directly involved in the interaction of HsrA with ligand V. Hydrophobic interactions are indicated with yellow dashed lines, H-bonds and halogen bonds are indicated with orange dashed lines.
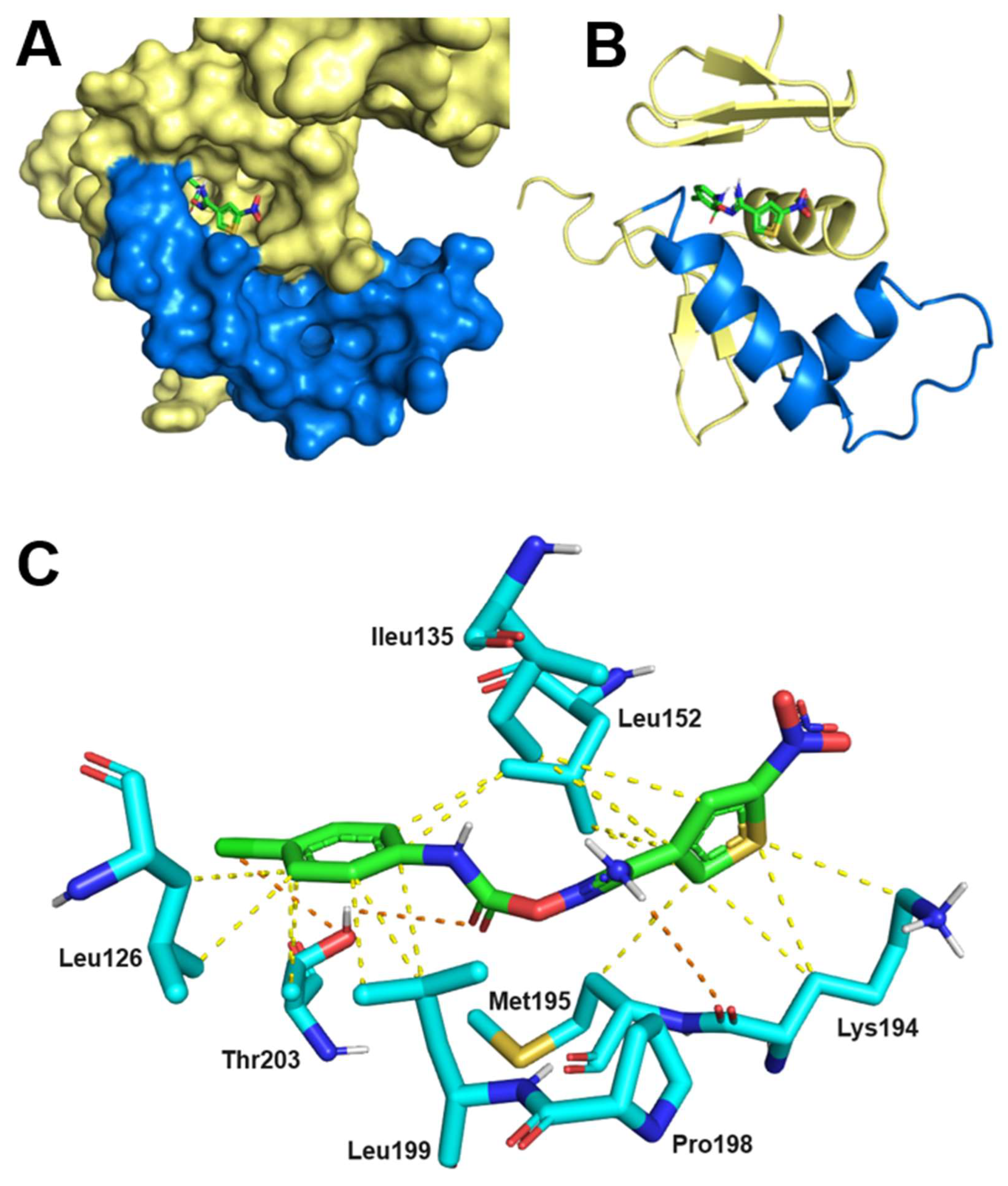
Figure 7.
Transcriptomic analysis of H. pylori cells exposed to lethal concentrations of HsrA bactericidal inhibitor V. Fresh cultures of H. pylori 26695 adjusted to 107 CFU/mL were exposed in triplicate to 4 × MIC of HsrA inhibitor V and incubated under controlled conditions (microaerobiosis, 37 °C) until log10 CFU/mL resulted diminished in one unit. (A) Heat map showing hierarchical cluster analysis of selected differentially expressed genes (DEG, absolute log2 fold change > 1 and p-value < 0.05). Each row represents a gene, and each column represents a sample. Red and green indicate up-regulated and down-regulated genes, respectively. The gradient colour barcode at the top right indicates the log10 normalized hit counts. (B) Volcano plot indicating distribution of DEGs. (C) Distribution of up-regulated (column in red) and down-regulated (column in blue) known-function genes by functional categories. (D) qPCR analysis of selected genes. Dashed grey lines indicate fold change cut-off values of +/- 1.5.
Figure 7.
Transcriptomic analysis of H. pylori cells exposed to lethal concentrations of HsrA bactericidal inhibitor V. Fresh cultures of H. pylori 26695 adjusted to 107 CFU/mL were exposed in triplicate to 4 × MIC of HsrA inhibitor V and incubated under controlled conditions (microaerobiosis, 37 °C) until log10 CFU/mL resulted diminished in one unit. (A) Heat map showing hierarchical cluster analysis of selected differentially expressed genes (DEG, absolute log2 fold change > 1 and p-value < 0.05). Each row represents a gene, and each column represents a sample. Red and green indicate up-regulated and down-regulated genes, respectively. The gradient colour barcode at the top right indicates the log10 normalized hit counts. (B) Volcano plot indicating distribution of DEGs. (C) Distribution of up-regulated (column in red) and down-regulated (column in blue) known-function genes by functional categories. (D) qPCR analysis of selected genes. Dashed grey lines indicate fold change cut-off values of +/- 1.5.
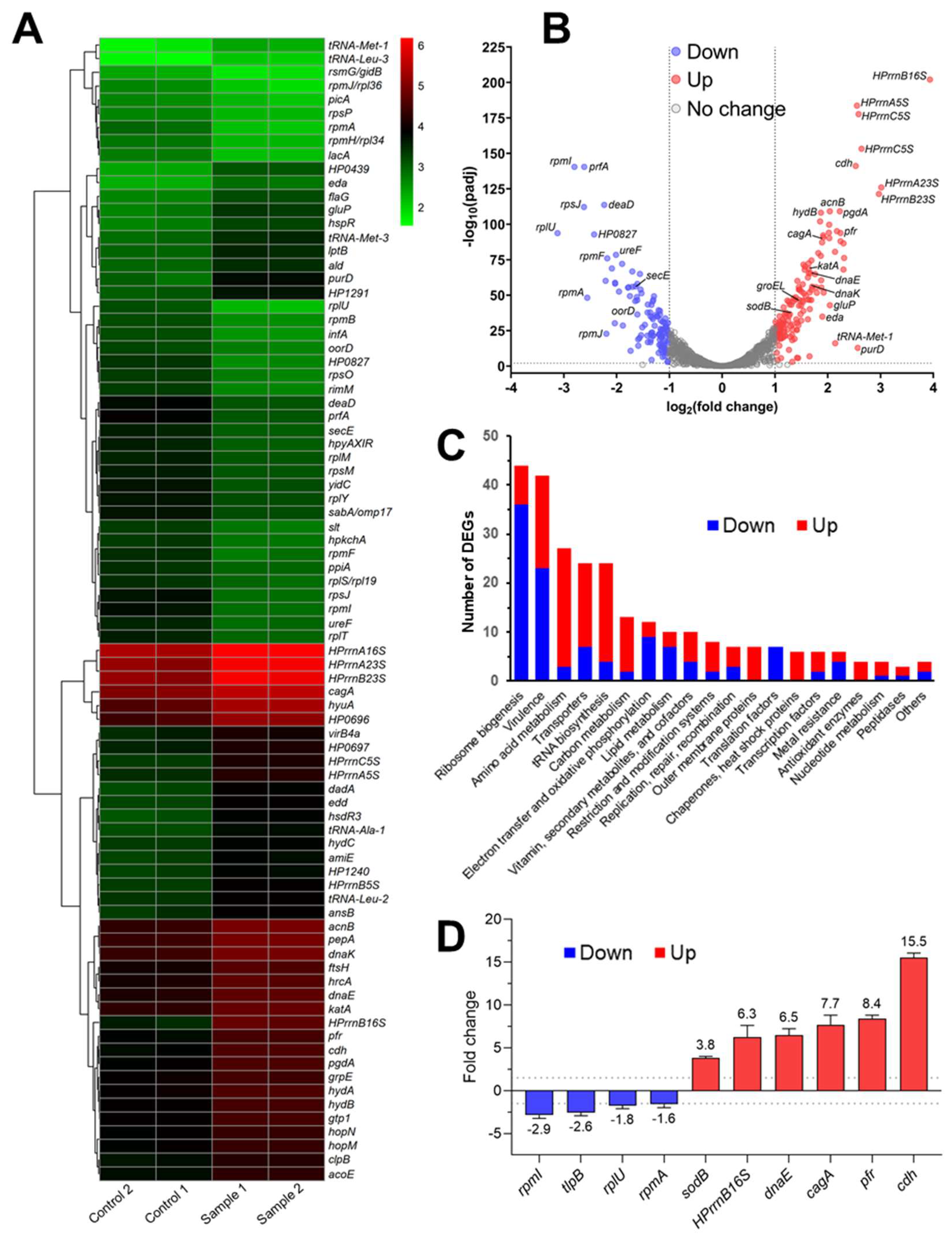

Table 1.
Chemical structure and physicochemical properties of relevant HsrA ligands identified by a fluorescent thermal shift-based HTS of the Maybridge HitFinderTM chemical library.
Table 1.
Chemical structure and physicochemical properties of relevant HsrA ligands identified by a fluorescent thermal shift-based HTS of the Maybridge HitFinderTM chemical library.
| Ligand | Chemical structure | Formula | Molecular Weight (Da) | log P1 | H-bond donors | H-bond acceptors | Lipinski violations | TPSA (Å)2 | Rotatable bonds | Log D3 |
| I | 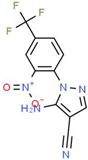 |
C11H6F3N5O2 | 297.19 | 1.56 | 1 | 7 | 0 | 113.45 | 3 | 2.26 |
| IV | 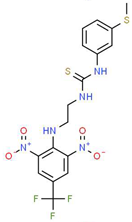 |
C17H16F3N5O4S2 | 475.47 | 3.08 | 3 | 7 | 0 | 185.12 | 11 | 4.97 |
| V | 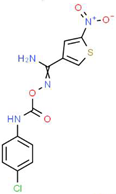 |
C12H9ClN4O4S | 340.74 | 1.99 | 2 | 5 | 0 | 150.77 | 6 | 2.92 |
| VIII | 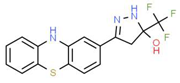 |
C16H12F3N3OS | 351.35 | 3.50 | 3 | 5 | 0 | 81.95 | 2 | 3.60 |
| XI | 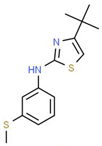 |
C14H18N2S2 | 278.44 | 4.18 | 1 | 1 | 0 | 78.46 | 4 | 4.77 |
| XII | 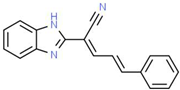 |
C18H13N3 | 271.32 | 3.53 | 1 | 2 | 0 | 52.47 | 3 | 3.79 |
Molecular properties obtained from the SwissADME web tool (http://www.swissadme.ch/) and the ChemSpider database (www.chemspider.com). 1Log P values correspond to consensus Log Po/w from SwissADME. 2TPSA, Topological Polar Surface Area. 3Log D values correspond to ACD/LogD (pH 7.4) from ChemSpider. .
Table 2.
Minimal inhibitory and bactericidal concentrations of HsrA inhibitors against different strains of H. pylori.
Table 2.
Minimal inhibitory and bactericidal concentrations of HsrA inhibitors against different strains of H. pylori.
| Inhibitor or drug | MIC (MBC), mg/L | |||
|---|---|---|---|---|
|
H. pylori ATCC 700392 (26695) |
H. pylori
ATCC 43504 (MTZ-R) |
H. pylori
ATCC 700684 (CLR-R) |
H. pylori
Donostia 2 (LVX-R) |
|
| I | 4 (4) | 1 (1) | 4 (4) | 2 (2) |
| IV | 2 (4) | 2 (4) | 4 (4) | 4 (4) |
| V | 0.063 (0.125) | 0.125 (0.125) | 0.031 (0.031) | 0.125 (0.125) |
| VIII | 1 (2) | 1 (2) | 1 (2) | 2 (2) |
| IX | 8 (16) | 4 (8) | 4 (8) | 8 (16) |
| X | 8 (16) | 8 (8) | 8 (8) | 8 (16) |
| XI | 4 (4) | 2 (4) | 2 (4) | 2 (4) |
| XII | 2 (2) | 0.5 (0.5) | 0.5 (0.5) | 1 (1) |
| XVII | 16 (16) | 4 (8) | 8 (8) | 8 (8) |
| XXVI | 16 (16) | 8 (8) | 8 (8) | 8 (16) |
| XXVIII | 32 (64) | 16 (16) | 32 (32) | 32 (32) |
| XXXI | > 64 (> 64) | 64 (64) | > 64 (> 64) | > 64 (> 64) |
| MTZ | 1 (2) | 64 (128) | 1 (2) | 8 (8) |
| CLR | < 0.03 (< 0.03) | < 0.03 (< 0.03) | 16 (32) | < 0.03 (< 0.03) |
| LVX | 0.12 (0.12) | 0.5 (0.5) | 0.12 (0.12) | 16 (32) |
MTZ-R, metronidazole-resistant strain. CLR-R, clarithromycin-resistant strain. LVX-R, levofloxacin-resistant strain.
Table 3.
Interaction of selected bactericidal HsrA inhibitors with first-line antibiotics against H. pylori ATCC 700392.
Table 3.
Interaction of selected bactericidal HsrA inhibitors with first-line antibiotics against H. pylori ATCC 700392.
| Antibiotic1 | HsrA inhibitor | FICantibiotic | FICinhibitor | FICI2 | Interaction3 |
| CLR | I | 1 | 1 | 2 | neutral |
| IV | 1 | 1 | 2 | neutral | |
| V | 0.5 | 0.06 | 0.56 | additive | |
| VIII | 0.5 | 0.03 | 0.53 | additive | |
| XI | 0.25 | 0.5 | 0.75 | additive | |
| XII | 0.25 | 0.5 | 0.75 | additive | |
| MTZ | I | 0.125 | 0.5 | 0.625 | additive |
| IV | 1 | 1 | 2 | neutral | |
| V | 0.125 | 0.5 | 0.625 | additive | |
| VIII | 0.5 | 0.125 | 0.625 | additive | |
| XI | 0.5 | 0.063 | 0.563 | additive | |
| XII | 0.25 | 0.25 | 0.5 | synergism | |
| LVX | I | 1 | 1 | 2 | neutral |
| IV | 1 | 1 | 2 | neutral | |
| V | 1 | 1 | 2 | neutral | |
| VIII | 1 | 1 | 2 | neutral | |
| XI | 1 | 1 | 2 | neutral | |
| XII | 0.5 | 0.5 | 1 | neutral |
1CLR, clarithromycin. MTZ, metronidazole. LVX, levofloxacin. 2Fractional inhibitory concentration index (FICI) is calculated as: FICA (MICA in the presence of B/MICA alone) + FICB (MICB in the presence of A/MICB alone). 3According to the FICI value, the interaction between two compounds against a particular bacterial strain can be classified as synergism (FICI ≤ 0.5), additive (FICI > 0.5 to ≤1), no interaction or neutral (FICI > 1 to ≤4), and antagonism (FICI > 4).
Table 4.
Antimicrobial activities of relevant HsrA inhibitors against C. jejuni and several representative members of normal human microbiota.
Table 4.
Antimicrobial activities of relevant HsrA inhibitors against C. jejuni and several representative members of normal human microbiota.
| Inhibitor or drug | MIC (MBC), mg/L | ||||||
|---|---|---|---|---|---|---|---|
|
C. jejuni
ATCC 33560 |
E. coli
ATCC 25922 |
K. pneumoniae ATCC 700603 |
E. faecalis
ATCC 29212 |
S. aureus
ATCC 29213 |
S. epidermidis
ATCC 12228 |
S. agalactiae
ATCC 12386 |
|
| I | 32 (32) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | 32 (64) |
| IV | 64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) |
| V | 0.25 (0.5) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) |
| VIII | 16 (32) | >64 (>64) | >64 (>64) | 64 (64) | 16 (32) | 4 (4) | 16 (32) |
| XI | >64 (>64) | >64 (>64) | >64 (>64) | 64 (64) | 16 (32) | 16 (64) | 8 (8) |
| XII | 64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) | >64 (>64) |
| LVX | < 0.12 (< 0.12) | N.D. | 0.5 (0.5) | N.D. | N.D. | N.D. | N.D. |
| AMP | N.D. | 4 (4) | N.D. | 4 (4) | 2 (4) | 4 (4) | 0.25 (0.5) |
LVX, levofloxacin. AMP, ampicillin.
Table 5.
Thermodynamic parameters and interacting amino acid residues of selected HsrA-inhibitor complexes, according to ITC and molecular docking analyses.
Table 5.
Thermodynamic parameters and interacting amino acid residues of selected HsrA-inhibitor complexes, according to ITC and molecular docking analyses.
| Inhibitor | ITC1 | Molecular Docking2 | |||
|---|---|---|---|---|---|
| n |
Kd (μM) |
ΔH (kcal/mol) |
ΔG (kcal/mol) |
Interacting Residues | |
| I | 1.0 | 13 | –1.2 | –6.7 | I121, I123, L126, I135, L152, L155, A156, R159, M195, L199, T203 |
| IV | 0.9 | 3.9 | –1.6 | –7.4 | I135, V142, V144, K145, G146, P148, L152, K194, P198 |
| V | 1.0 | 13 | –1.9 | –6.7 | L126, I135, L152, K194, M195, P198, L199, T203 |
| VIII | 0.9 | 10 | 0.8 | –6.8 | V144, K145, G146, F149, L152, I191, K194, M195 |
| XI | 1.1 | 49 | –3.1 | –5.9 | I123, I128, L152, R159, D160, M195, L199, T203, C215, Y216 |
| XII | 1.0 | 43 | 0.8 | –6.0 | I123, L126, I128, I135, L152, M195, P198, L199, T203 |
1Relative error in Kd is 15%, absolute error in ΔH is 0.4 kcal/mol, absolute error in ΔG is 0.1 kcal/mol. 2Amino acid residues directly involved in forming the helix-turn-helix (HTH) DNA binding motif of HsrA are highlighted in bold fonts.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



