
Submitted:
04 September 2024
Posted:
06 September 2024
You are already at the latest version
Abstract
Background: Patients with mutations in the monocarboxylate transporter 8 (MCT8) suffer from Allan-Herndon-Dudley syndrome (AHDS), characterized by developmental delay and a highly disabling movement disorder. Despite the potential of thyroid hormone derivatives to overcome the transporter defect, current trials did not achieve patient-oriented therapeutic goals. Objectives: Since most neurological symptoms are related to the dopaminergic system, we investigated the role of dopamine and its metabolites in MCT8 deficiency with regard to pathophysiology and potential therapeutic strategies in an observational cohort study. Methods: We present longitudinal data from the DEEPTYPE registry of ten patients with video-documentation, standardized phenotyping, cerebrospinal fluid analysis, treatment response to levodopa/carbidopa supplementation, and neuroimaging data. To establish a cell-based model for pathophysiological studies, we differentiated healthy human induced pluripotent stem cells (hiPSCs) into dopaminergic neurons. Results: Children presented with signs of parkinsonism in childhood, including hypokinesia, hypomimia, inability to sit or stand, rigidity, dystonia, and autonomic dysfunction along the classification of Leuzzi and colleagues. Cerebrospinal fluid homovanillic acid concentrations were decreased (n=11), suggesting isolated dopamine pathway impairment. Six out of seven patients responded favorably to levodopa/carbidopa supplementation and we did not see any adverse drug reactions. Our cell-based studies showed that hiPSC-derived dopaminergic neurons expressed MCT8 and produced quantifiable levels of biogenic amines. Conclusions: Parkinsonism is part of AHDS's clinical presentation and may be amenable to treatment. The precise impact of MCT8 deficiency on the dopamine metabolism needs to be further elucidated, e.g. by using patient iPSC-derived dopaminergic neurons in future studies.
Keywords:
MCT8
; SLC16A2
; movement disorder
; parkinsonism
; dopamine
; iPSC cell model
Introduction
Allan-Herndon-Dudley syndrome (AHDS) was described in 1944 as one of the first X-linked intellectual disability syndromes [1]. In addition to cognitive impairment, neurological symptoms such as “dysarthria”, “athetoid movements”, “extrapyramidal signs”, “muscular hypotonia”, and “severe motor developmental delay” were described as characteristic features of the disease [1]. Sixty years later, in 2004, researchers discovered that variants in the SLC16A2 gene on the X chromosome, which encodes the thyroid hormone transporter monocarboxylate transporter 8 (MCT8), are the cause of AHDS [2,3]. MCT8 deficiency does not affect thyroid hormone synthesis. Therefore, in contrast to congenital hypothyroidism, patients have normal to elevated triiodothyronine (T3), and thyroid-stimulating hormone (TSH), as well as normal to decreased thyroxine (T4) serum concentrations (Figure 1). However, in some tissues, mainly in the central nervous system, thyroid hormone is unable to reach its intracellular targets and receptors due to the MCT8 transporter defect [4]. Since intracellular thyroid hormone receptors act as transcription factors, the lack of intracellular T3 results in a state of local (central) hypothyroidism [5]. In contrast, other organs such as the heart are not solely dependent on the MCT8 transporter and settle in a hyperthyroid state with symptoms such as tachycardia. The severity of symptoms in patients with MCT8 deficiency varies from severe phenotypes functionally similar to children with dyskinetic cerebral palsy Gross Motor Function Classification System Grade V (GMFCS V) to milder phenotypes with varying developmental delay. Patients have recently been diagnosed by routine diagnostic exome sequencing and functional analysis such as determination of the fT3/fT4 ratio (http://www.thyroid-hormone-ratio.org) [6].
While the initial phenotypic description of AHDS as a novel X-linked disorder was mainly in the realm of geneticists, the subsequent clinical phenotyping was performed with a strong endocrinologic focus. Only recently, the neurological symptoms, especially the movement disorder, have received more attention [7,8,9]. These neurological symptoms represent the main disease burden for patients and their caregivers [10]. Based on the pathogenetic concept of reduced central thyroid hormone action, researchers have developed therapeutic strategies. These include supplementation of patients with thyroid hormone analogues such as tiratricol (Triac) and diiodothyropropionic acid (DITPA), which can bypass the transport by MCT8 in vitro [11,12]. Unfortunately, clinical trials with Triac and DITPA did not substantially improve clinical outcomes in patients. While reducing elevated peripheral T3 concentrations, the study drugs failed to achieve patient-oriented therapeutic goals such as improvement of gross, fine motor and verbal skills, and a reduction of “muscle cramps/stiffness” [10]. Given that most of the neurological symptoms are related to the patients’ movement disorder, established therapies such as levodopa/carbidopa supplementation could be an alternative strategy, until suitable gene therapy becomes available [13,14]. Here we present a retrospective longitudinal study of a larger group of patients with this ultra-rare disease, documented by videos and motor symptom scoring. In addition, we measured cerebrospinal fluid (CSF) concentrations of neurotransmitters in n=11 patients and followed n=6 patients with regular scoring before and after treatment with levodopa/carbidopa for up to 12 months. Thereby, we show that parkinsonism as part of AHDS may indeed be treatable.
Methods
Patient Enrollment and Follow-Up
Patients and their families were recruited into the DEEPTYPE registry study (https://clinicaltrials.gov/study/NCT06566066) for patients with thyroid hormone resistance between July 2021 and July 2024 from different centers in Germany. All patients carried a hemizygous mutation in SLC16A2. Patients’ caregivers provided written informed consent according to the Declaration of Helsinki for participation in the study and for publication of results and patient images. The deep phenotyping study was observational only and did not influence treatment decisions. Data were collected in a REDCap registry. Ethical approval for the study was granted by the Institutional Review Board of Charité (EA2/026/20).
During regular visits of our patients, we recorded videos according to the protocol of the Dyskinesia Impairment Scale [15] and phenotyped the patients at regular intervals applying the following scales: Burke-Fahn-Marsden Dystonia Rating Scale [16], Infantile Parkinsonism-Dystonia Rating Scale (IPDRS) [17] (Supplementary methods 1), Bayley Scales of Infant and Toddler Development III [18], Cerebral Palsy Child Health Index of Life with Disabilities [19], which are relevant for the evaluation of disease progression and treatment response. Patients were evaluated by a multidisciplinary team of pediatric neurologists, psychologists, physical, occupational, and speech therapists. The Burke-Fahn-Marsden Dystonia Rating Scale was modified by excluding items assessing swallowing function because of its inability to differentiate dysphagia of different etiologies, as described elsewhere [7]. The presence of dystonia and parkinsonism required a lumbar puncture to test for dopamine deficiency. CSF was flash frozen in liquid nitrogen and analyzed for biogenic amine, 5-methyltetrahydrofolate (5-MTHF), and pterin concentrations with minor modifications as described [20]. Six patients received levodopa/carbidopa 4:1 suspension orally or via gastric tube. Medication was started at 1.00/0.25 mg/kg/d in four doses per day under inpatient supervision for at least 4 days. Subsequently, the dose was increased by 1.00/0.25 mg/kg/d per week depending on the clinical response up to a maximum dose of 10.0/2.5 mg/kg/d in all children.
Neuroimaging
Neuroimaging data were also collected retrospectively, the details of which are listed in Supplementary methods 2. Briefly, for volumetric neuroimaging, MRI images were segmented according to the Desikan-Killiany atlas [21] using Freesurfer v6.0 [22]. 18F-DOPA positron emission tomography (PET) data were recorded from patients 7 and 9 and compared to normal values [23].
Differentiation of Human Induced Pluripotent Stem Cells (hiPSCs) into Dopaminergic Neurons
To generate dopaminergic neurons from iPSCs, we obtained the NCRM-1 iPSC line (https://hpscreg.eu/cell-line/CRMi003-A) commercially from RUCDR Infinite Biologics (RUCDR). The protocols for differentiation of iPSCs into neuronal progenitor cells (NPCs) and then into dopaminergic neurons [24,25], the measurement of intracellular biogenic amines [26], and the image acquisition protocol [27] are deceived in detail in the Supplementary methods 3.
Statistical Analysis
Statistical differences and significance levels between groups were determined using GraphPad Prism 10 (GraphPad Software) with various statistical tests indicated in the respective figure legends. A p-value of <0.05 was considered statistically significant. Results are presented as scatter plots, with bars representing the mean and standard deviation (SD). For natural history data, we attempted to contact the corresponding authors of each study or digitized data from published figures using ImageJ.
Results
Patients Present with Signs of Parkinsonism in Childhood
We collected retrospective data from n=10 male patients with AHDS (age range: 1.0-25.5 years, mean: 7.4 years) (Figure 1D). All had fT3/fT4 ratios above the 97th percentile [6] and carried pathogenic SLC16A2 mutations, inherited from an unaffected mother in 63% (5/8) or arising de novo in 37% (3/8) of the patients.
Consistent with published phenotypic data [7,28], the AHDS phenotype varied in severity, with most patients (80%, 8/10) presenting with severe global developmental delay and movement disorder, functionally similar to dyskinetic cerebral palsy with GMFCS V. According to the classification of Leuzzi and colleagues [29], these severely affected patients showed signs and symptoms of parkinsonism in childhood with infantile onset of symptoms. In all cases, head control did not develop during the first months of life. Further symptoms comprised hypokinesia (hypomimia, reduced spontaneous and voluntary movements), bradykinesia, rigidity (increasing with age, independent of a task or posture), dystonia, severe axial hypotonia (head lag and trunk hypotonia), autonomic dysfunction (excessive diaphoresis, obstipation, audible obstructive breathing, sleep disturbance), and motor developmental delay (Video 1, for a detailed list see Figure 1D). On glabellar reflex testing, the patients were unable to resist blinking (positive Myerson’s sign) and their startle response was strikingly pronounced. Oculogyric crises were neither described by the parents nor observed during clinical examination.
A 2.1-year-old patient (patient 3) was classified as moderately affected with global developmental delay (head control at 10 months, free sitting at 12 months, pulling to a standing position at 18 months, and walking along objects at 24 months), suspected intellectual disability and functionally disabling dystonia and hypokinesia (GMFCS II). Another 7.4-year-old patient (patient 8) was classified as mildly affected with mild intellectual disability and infrequent action-specific dystonia of the limbs (GMFCS I) (videos of the last two cases are available at [6].
Altered Biogenic Amine Concentrations Indicate an Isolated Dopamine Pathway Impairment
Biogenic amines, 5-MTHF and pterins were measured in n=11 CSF samples from n=10 patients (Figure 2). Patient 9 underwent lumbar puncture at the age of 3 years during the initial clinical evaluation and later at the age of 13.7 years because of suspected parkinsonism in childhood. While 5-MTHF and pterin levels were normal, biogenic amine analyses revealed significantly lower CSF homovanillic acid (HVA) concentrations in AHDS patients with a mean difference of -164.8 nmol/l compared to normal age-matched controls (p<0.001) (Figure 2A). However, CSF HVA concentrations were reduced to an average of 65% of normal HVA concentrations, not as markedly reduced as would be expected for a primary dopamine synthesis deficiency as seen in tyrosine hydroxylase deficiency [30]. The 5-hydroxyindoleacetic acid (5-HIAA) concentrations were normal (Figure 2B) and consequently the HVA/5-HIAA ratio was significantly reduced in the CSF of AHDS patients with a median difference of -0.9 (p<0.001) (Figure 2C). We did not find any changes in 3-O-methyldopa, levodopa, or 5-hydroxytryptophan concentrations. In conclusion, the constellation of biogenic amine alterations in the CSF of AHDS patients strongly suggests an isolated impairment of the dopamine pathway, consistent with the symptoms of parkinsonism in childhood [29].
Patients Persistently Respond to Levodopa/Carbidopa Treatment
We initiated levodopa/carbidopa treatment in n=6 patients at Charité - Universitätsmedizin Berlin because of suspected parkinsonism in childhood. Only two patients were not treated, one (patient 8) with a rare and functionally irrelevant dystonia and one patient (patient 6) who was lost to follow-up. The levodopa/carbidopa dosage was slowly increased to 10.0/2.5 mg/kg/d over 10 weeks in all patients. All parents reported improvement in patient-oriented outcomes [10]: 100% (6/6) improvement in expressive language (syllables), 83% (5/6) improvement in gross motor skills (head control, first steps) and improved targeted movements (reaching for objects, selective opening and closing of hands), 67% (4/6) had less muscle “cramps/stiffness”, and 50% (3/6) had improved dysphagia (less hypersalivation and choking on food intake) (data not shown, Video 2).
Parental impressions were objectively confirmed using the Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS), the Infantile Parkinsonism-Dystonia Rating Scale (IPDS), and the Bayley Scales of Infant and Toddler Development III (BSID-III). The dystonia movement subscale of the BFMDRS improved significantly in all patients with a mean difference of -16.2 points (p<0.01) at six months and in two patients with sustained improvement after 12 months of treatment (Figure 3A). The disability subscale of the BFMDRS improved less, but also significantly, with a mean difference of -1.8 points (p<0.05) (Figure 3B). In addition, the total score of the IPDRS improved significantly by +9.5 points (p<0.001) (Figure 3C). Within the subcategories, patients showed significant improvement in parent-reported non-motor signs (autonomic, mood dysfunction) (p<0.05), and investigator-rated motor signs of parkinsonism and dystonia (hypokinesia, rigidity, dystonia, axial hypotonia, motor developmental delay) (p<0.001) (Figure 3D). No oculogyric crisis was ever observed. Dyskinesia (myoclonus) was present in 50% (3/6) of the patients but did not improve with therapy.
Consistent with the parents' perception of improvement in goal-directed movements, patients had significantly higher age equivalents by 1.2 months on the BSID-III fine motor subscale (p<0.05) (Supplementary figure 1B). We did not observe any significant improvement in gross motor skills, expressive/receptive language, or cognition (Supplementary figure 2A, C-E). However, none of the patients showed developmental regression, but rather a trend toward improvement. The Cerebral Palsy Child Health Index of Life with Disabilities (CPCHILD) did not improve. One family (patient 4) even reported a drastic decrease in quality of life due to multiple surgeries on their child (placement of a gastric tube and orthopedic hip surgery) (Supplementary figure 1F). Under levodopa/carbidopa therapy, the patients did not experience any adverse drug reactions or laboratory abnormalities (Supplementary figure 2B-F). The z-score of the body weight (compared to healthy children) decreased significantly by 0.2 points over the 6 months (p<0.05), which was, however, within the body weight trajectory of the entire cohort of MCT8 deficient patients (Supplemental figure 2A). The z-score of the body mass index, however, did not change significantly in the levodopa/carbidopa treated patients. Patient 10 received levodopa/carbidopa at the Georg-August-University Göttingen up to 9.5/2.4 mg/kg/d for only one week and showed no signs of improvement.
Patients with MCT8 Deficiency Have a Reduced Subcortical Gray Matter Volume
We compared the segmented MRI brain volumes of patients 7 and 9 with previously published brain maps of 58,836 MRI scans from across the human lifespan [31] (Figure 4A). The results for both individuals were strikingly consistent. Total intracranial volume was reduced by more than two standard deviations (SD), with the most pronounced reduction in subcortical gray matter (-6.4 and -6.5 SD). In particular, (i) the Globus pallidus, as a major output structure of the basal ganglia and site of dopamine action, and (ii) the ventral diencephalon, including the Substantia nigra with the dopaminergic neurons were most affected by volume reduction (-2.4 to -4.1 SD). To differentiate between primary brain volume loss and secondary atrophy, we collected head circumference measurements in the first postnatal week and before initiation of levodopa/carbidopa treatment. 4/6 patients had crossed percentiles with a reduction in head circumference and the oldest patient 9 had developed secondary microcephaly at the age of 14.2 years (Figure 4B). More data are needed to draw conclusions. In agreement with previously published data [9], we did not observe signal changes in the basal ganglia on MRI in 7/7 patients (data not shown). F18-DOPA PET MRI, which assesses the functional integrity of nigrostriatal dopaminergic networks, appeared normal in patients 7 and 9 compared to adult controls (n=44) [23], as would be expected in monoamine biogenesis disorders other than aromatic l-amino acid decarboxylase deficiency (Figure 4C-D) [32].
Establishment of a Cell-Based Model to Analyze the Role of MCT8 Deficiency on Dopamine Metabolism
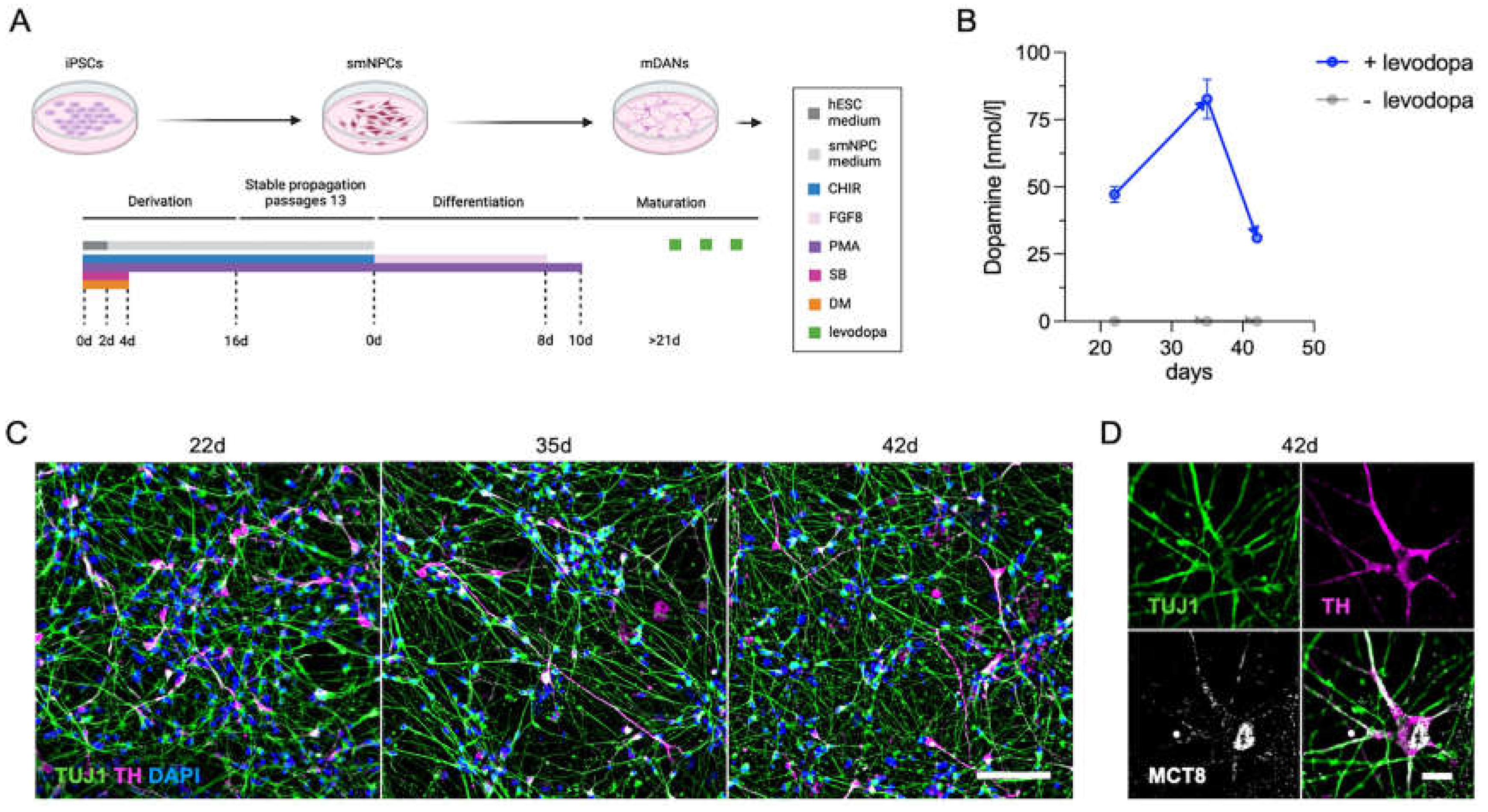
Neuronal cell cultures derived from hiPSCs provide a patient-derived model to test mechanisms of human diseases directly in human material, thereby eliminating any species bias. To this end, we used established protocols [24,25] for the differentiation of dopaminergic neurons from iPSCs (here the standard NCRM-1 line) using small molecules (Figure 5A). After 22 days, neuronal networks (TUJ1+, green) with dopaminergic cells (TH+, pink) had formed and remained consistent over an observation period of 42 days (Figure 5C). As described by Mahajani and colleagues [26], intracellular dopamine synthesis could only be measured after the addition of the precursor levodopa to the media at 28 h and 4 h before cell extraction (Figure 5B). The dopaminergic cells (TH+) expressed MCT8 (white) around the nuclear and plasma membranes of the soma, as well as in axons and dendrites (Figure 5D). Dopaminergic neurons derived from patients [33] versus control iPSCs may therefore be a promising model to study the interaction between MCT8 and dopamine synthesis.
Discussion
With this retrospective study, we describe Allan-Herndon-Dudly syndrome as a further rare disease that can be classified as “childhood parkinsonism”. In addition to the clinical signs of parkinsonism, the distinct constellation of biogenic amines with decreased HVA and HVA/5-HIAA ratios in the cerebrospinal fluid suggests an isolated dopamine dyshomeostasis [34]. This is supported by reduced subcortical gray matter volumes, especially of the Globus pallidus as a site of dopamine action and of the ventral diencephalon (including the Substantia nigra) as the site of dopamine synthesis. According to this hypothesis, substitution with levodopa/carbidopa led to a significant and long-lasting improvement in patient-oriented outcomes (improved axial tone and fine motor skills, reduced hypokinesia, rigidity and dystonia). Only one patient did not benefit from therapy, but this was only tested for a (too) short period of one week. Underdosing (<10.0/2.5 mg/kg/d) or too short a treatment period may account for the lack of response in single case reports of levodopa/carbidopa supplementation [8,9,28].
Infantile-onset parkinsonism is an exceedingly rare and underdiagnosed condition. It has distinct features from Parkinson’s disease (PD), one of the most common neurological disorders in the adult population. Whereas in PD neurodegeneration of dopaminergic neurons in the Pars compacta of the Substantia nigra [35] leads to the cardinal signs of bradykinesia plus tremor or rigidity [36], the dysfunction of dopamine action in the immature brain causes a more complex clinical picture. Because consensus clinical diagnostic criteria of pediatric-onset parkinsonism have long been lacking, Leuzzi and colleagues recently proposed an initial classification for parkinsonism in childhood [29]. These included (i) degenerative conditions with a monogenetic variant of PD and very early onset (juvenile parkinsonism and dystonia-parkinsonism) [37,38], (ii) acquired conditions leading to parkinsonism, e.g. due to asphyxia, intoxication, or infection [39], (iii) multisystem brain disorders with neurodegenerative or metabolic etiology (e.g. Wilson’s disease, Pantothenate kinase-associated neurodegeneration), and (iv) genetic disorders with generalized disruption of neurodevelopment (developmental parkinsonism, infantile degenerative parkinsonism, and parkinsonism in the setting of neurodevelopmental disorders). While (i) juvenile parkinsonism and dystonia-parkinsonism mimic PD with an earlier onset, children with conditions of the latter category (iv) certainly present with different symptoms [29]. Patients with developmental parkinsonism present with hypo-, bradykinesia, rigidity, dystonia, inability to sit or stand, and global developmental delay [29]. Additional clinical signs may include oculogyric crises, abnormal fetal movement patterns, oscillatory jerks, and periodic fluctuations [29]. The pathophysiology is a selective, non-degenerative derangement of dopaminergic connectivity, as seen in several neurotransmitter disorders (e.g., tyrosine hydroxylase or aromatic L-amino acid decarboxylase deficiency). If treatment is initiated early, patients can respond dramatically to neurotransmitter substitution [29]. Regarding AHDS, we postulate that it can be classified as developmental parkinsonism (given the findings of reduced HVA in CSF and the favorable response to levodopa/carbidopa treatment) with some overlap to parkinsonism in the setting of neurodevelopmental disorders, where cognitive disability is the key finding while parkinsonism emerges later in life. Patients with AHDS showed only a partial response to levodopa/carbidopa substitution while the global developmental disorder persisted. This partial response to therapy may be due to the fact that, in addition to the functional impairment of dopamine synthesis, the patient's structural defect (reduced dopaminergic neurons and basal ganglia volume) may not be treatable by neurotransmitter substitution alone.
The observed structural abnormalities may be a consequence of prenatal MCT8 deficiency. This is supported by the fact that patients with the rather "historical" disease of “neurological type of cretinism”, who are affected by hypothyroidism in utero due to severe maternal iodine deficiency during pregnancy, present a clinical picture that resembles most features of childhood parkinsonism, including dystonia [40,41,42]. In contrast, children with untreated congenital hypothyroidism due to agenesis of the thyroid gland show global developmental delay but no evidence of movement disorder [43]. While children with congenital hypothyroidism are hypothyroid only in the later stages of pregnancy and after birth due to their own impaired thyroid hormone synthesis, fetuses of iodine-deficient mothers are exposed to hypothyroidism throughout gestation due to inadequate transplacental thyroid hormone supply. Thus, it seems likely that thyroid hormones play a critical role during the early period of neurodevelopment, including the formation of dopaminergic circuits, which may not be fully rescued by levodopa/carbidopa substitution after birth.
At the molecular level, we hypothesize that MCT8 deficiency disrupts thyroid hormone transport to neuronal target cells, resulting in altered gene transcription patterns via thyroid hormone receptors that act as transcription factors. In mice, over 1,000 genes are regulated by T3, and T3-dependent genes (e.g., sonic hedgehog, orthodenticle homeobox 2) play important roles in neurodevelopmental processes such as cell proliferation, cell fate decision, axonogenesis, synaptogenesis, and myelinogenesis [44]. The consequences of thyroid hormone deficiency on the developing human brain are not yet known and are currently being investigated in human disease models such as human forebrain organoids [45]. These emerging data already suggest a critical role for MCT8 in cerebral cortex development and regulation of multiple genes beyond the dopamine pathways, which may explain the only partial response to therapy in our patients.
Regarding the effect of MCT8 deficiency on dopamine action, Hassan and colleagues have shown in vitro that hypothyroid condition inhibit the tyrosine hydroxylase as the rate-limiting enzyme of dopamine synthesis [46], whereas, inversely, hyperthyroid states lead to an increase of dopamine metabolism [47]. Whether this is also true for MCT8 deficiency remains to be determined in future studies. Given the species differences and the numerous limitations of the Mct8-deficient mouse model, which does not resemble the human phenotype of patients, we would suggest the use of human iPSC-derived dopaminergic midbrain neurons expressing MCT8 [48] as a future in vitro experimental system to address these important questions of the pathogenesis of parkinsonism in AHDS [33].
Taken together, our data establish AHDS as another syndrome that fulfills the clinical criteria of parkinsonism in childhood. While there is no definitive treatment for the underlying MCT8 deficiency while gene therapies are still under development [13,14], patients appear to benefit from symptomatic treatment with levodopa/carbidopa.
Limitations: This study is an initial observation in a small cohort of patients that had been entered into a dedicated patient registry. The initial observation of the positive effect of levodopa/carbidopa treatment has to be confirmed by a formal prospective, controlled blinded, cross-over phase II/III trial.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Authors' roles
NMW, ALH, MTH, AT, HK, TO, and MS contributed to the design and NMW, ALH, MTH, ADO, MB, CG, SM, CR, CL, CF, MW, KB, SJK, SC, AT, TO, and MS to the conduct of the study. NMW, MTH, ADO, MB, SM, AZ, AP, CL, SC, HK, TO, and MS analyzed the data. NMW and MS provided funding, NMW, HK, TO, and MS wrote the first draft of the manuscript, and all authors revised and approved the final version of the manuscript.
Funding sources
NMW was supported by the Deutsche Forschungsgemeinschaft (DFG) Research Unit 2841 “Beyond the Exome”, and she is participant in the BIH Charité Junior Clinician Scientist Program for Rare funded by the Charité - Universitätsmedizin Berlin, the Berlin Institute of Health at the Charité (BIH), the Alliance4Rare, and the Berliner Sparkassenstiftung Medizin. MS was funded by the DFG under the German Excellence Strategy (EXC-2049-390688087) through the NeuroCure Consortium at Charité - Universitätsmedizin Berlin. AMK was supported by the Einstein Stiftung Fellowship through the Günter Endres Fond. AMK and MS were supported by the Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) as part of the German Center for Child and Adolescent Health (DZKJ).
Institutional Review Board Statement
Ethical approval for the study was obtained from the Institutional Review Board of Charité - Universitätsmedizin Berlin, Campus Virchow Klinikum in the year 2020 (EA2/026/20). The study was conducted in accordance with the tenets of the Declaration of Helsinki.
Acknowledgements
We thank the patients and their families. We thank Sylvia Richter for excellent technical assistance in the measurement of biogenic amine metabolites.
Conflicts of interest
NMW and CR participated in a paid consultancy with Primus Consulting Group GmbH. NMW consulted for the film production company Hellinger-Doll, and was paid by Biogen for a congress presentation. The funders had no role in the design of the study, in the collection, analysis, or interpretation of the data, in the writing of the manuscript, or in the decision to publish the results.
References
- Allan W, Herndon, C. Nash, Dudley, Forence C. Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microencephaly. Am. J. Ment. Defic. 1944, 48, 325–334. [Google Scholar]
- Dumitrescu, A.M.; Liao, X.H.; Best, T.B.; Brockmann, K.; Refetoff, S. A Novel Syndrome Combining Thyroid and Neurological Abnormalities Is Associated with Mutations in a Monocarboxylate Transporter Gene. Am. J. Hum. Genet. 2004, 74, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.; Grueters, A.; Biebermann, H.; Krude, H.; von Moers, A.; Reeser, M.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. The Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Vatine, G.D.; Al-Ahmad, A.; Barriga, B.K.; Svendsen, S.; Salim, A.; Garcia, L.; et al. Modeling Psychomotor Retardation using iPSCs from MCT8-Deficient Patients Indicates a Prominent Role for the Blood-Brain Barrier. Cell Stem Cell 2017, 20, 831–843. [Google Scholar] [CrossRef]
- Groeneweg, S.; van Geest, F.S.; Abacı, A.; Alcantud, A.; Ambegaonkar, G.P.; Armour, C.M.; et al. Disease characteristics of MCT8 deficiency: an international, retrospective, multicentre cohort study. Lancet Diabetes Endocrinol. 2020, 8, 594–605. [Google Scholar] [CrossRef]
- Wilpert, N.M.; Thamm, R.; Thamm, M.; Kratzsch, J.; Seelow, D.; Vogel, M.; et al. Normal Values for the fT3/fT4 Ratio: Centile Charts (0–29 Years) and Their Application for the Differential Diagnosis of Children with Developmental Delay. Int. J. Mol. Sci. 2024, 25, 8585. [Google Scholar] [CrossRef]
- Masnada, S.; Sarret, C.; Antonello, C.E.; Fadilah, A.; Krude, H.; Mura, E.; et al. Movement disorders in MCT8 deficiency/Allan-Herndon-Dudley Syndrome. Mol. Genet. Metab. 2022, 135, 109–113. [Google Scholar] [CrossRef]
- Ramon-Gomez, J.L.; Cortés-Rojas, M.C.; Polania-Puentes, M.J.; Guerrero-Ruiz, G.D.P. Movement Disorder Perspectives on Monocarboxylate 8 Deficiency: A Case Series of 3 Colombian Patients with Allan-Herndon-Dudley Syndrome. Mov. Disord. Clin. Pract. 2024, 11, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Tonduti, D.; Vanderver, A.; Berardinelli, A.; Schmidt, J.L.; Collins, C.D.; Novara, F.; et al. MCT8 Deficiency: Extrapyramidal Symptoms and Delayed Myelination as Prominent Features. J. Child Neurol. 2013, 28, 795–800. [Google Scholar] [CrossRef]
- Wilpert, N.M.; Tonduti, D.; Vaia, Y.; Krude, H.; Sarret, C.; Schuelke, M. Establishing Patient-Centered Outcomes for MCT8 Deficiency: Stakeholder Engagement and Systematic Literature Review. Neuropsychiatr. Dis. Treat. 2023, 19, 2195–2216. [Google Scholar] [CrossRef]
- Groeneweg, S.; Peeters, R.P.; Moran, C.; Stoupa, A.; Auriol, F.; Tonduti, D.; et al. Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: an international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Verge, C.F.; Konrad, D.; Cohen, M.; Di Cosmo, C.; Dumitrescu, A.M.; Marcinkowski, T.; et al. Diiodothyropropionic Acid (DITPA) in the Treatment of MCT8 Deficiency. J. Clin. Endocrinol. Metab. 2012, 97, 4515–4523. [Google Scholar] [CrossRef]
- Iwayama, H.; Liao, X.H.; Braun, L.; Bárez-López, S.; Kaspar, B.; Weiss, R.E.; et al. Adeno Associated Virus 9–Based Gene Therapy Delivers a Functional Monocarboxylate Transporter 8, Improving Thyroid Hormone Availability to the Brain of Mct8-Deficient Mice. Thyroid 2016, 26, 1311–1319. [Google Scholar] [CrossRef]
- Sundaram, S.M.; Arrulo Pereira, A.; Müller-Fielitz, H.; Köpke, H.; De Angelis, M.; Müller, T.D.; et al. Gene therapy targeting the blood–brain barrier improves neurological symptoms in a model of genetic MCT8 deficiency. Brain 2022, 145, 4264–4274. [Google Scholar] [CrossRef]
- Vanmechelen, I.; Danielsson, A.; Lidbeck, C.; Tedroff, K.; Monbaliu, E.; Krumlinde-Sundholm, L. The Dyskinesia Impairment Scale, Second Edition: Development, construct validity, and reliability. Dev. Med. Child Neurol. 2023, 65, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; Fahn, S.; Marsden, C.D.; Bressman, S.B.; Moskowitz, C.; Friedman, J. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology 1985, 35, 73–77. [Google Scholar] [CrossRef]
- Pons, R.; Pearson, T.; Perez-Dueñas, B.; Garcia-Cazorla, A.; Kurian, M.; Dalibigka, Z.; et al. Validation of a Parkinsonism-dystonia scale for infants and young children. 7th International Symposium on Paediatric Movement Disorders, Barcelona, February 09-11: 2022.
- Bayley, N. Bayley Scales of Infant and Toddler Development. 3rd ed. APA Psyc Tests; 2005.
- Narayanan, U.G.; Fehlings, D.; Weir, S.; Knights, S.; Kiran, S.; Campbell, K. Initial development and validation of the Caregiver Priorities and Child Health Index of Life with Disabilities (CPCHILD). Dev. Med. Child Neurol. 2006, 48, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Hyland, K.; Surtees, R.A.H.; Heales, S.J.R.; Bowron, A.; Howells, D.W.; Smith, I. Cerebrospinal Fluid Concentrations of Pterins and Metabolites of Serotonin and Dopamine in a Pediatric Reference Population. Pediatr. Res. 1993, 34, 10–14. [Google Scholar] [CrossRef]
- Desikan, R.S.; Ségonne, F.; Fischl, B.; Quinn, B.T.; Dickerson, B.C.; Blacker, D.; et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 2006, 31, 968–980. [Google Scholar] [CrossRef]
- Fischl, B. FreeSurfer. NeuroImage 2012, 62, 774–781. [Google Scholar] [CrossRef]
- Lorenz, R.C.; Gleich, T.; Buchert, R.; Schlagenhauf, F.; Kühn, S.; Gallinat, J. Interactions between glutamate, dopamine, and the neuronal signature of response inhibition in the human striatum. Hum. Brain Mapp. 2015, 36, 4031–4040. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PloS One 2013, 8, e59252. [Google Scholar] [CrossRef]
- Zink, A.; Lisowski, P.; Prigione, A. Generation of Human iPSC-derived Neural Progenitor Cells (NPCs) as Drug Discovery Model for Neurological and Mitochondrial Disorders. Bio-Protoc. 2021, 11, e3939. [Google Scholar] [CrossRef] [PubMed]
- Mahajani, S.; Raina, A.; Fokken, C.; Kügler, S.; Bähr, M. Homogenous generation of dopaminergic neurons from multiple hiPSC lines by transient expression of transcription factors. Cell Death Dis. 2019, 10, 898. [Google Scholar] [CrossRef]
- Wilpert, N.M.; Krueger, M.; Opitz, R.; Sebinger, D.; Paisdzior, S.; Mages, B.; et al. Spatiotemporal Changes of Cerebral Monocarboxylate Transporter 8 Expression. Thyroid 2020, 30, 1366–1383. [Google Scholar] [CrossRef]
- Remerand, G.; Boespflug-Tanguy, O.; Tonduti, D.; Touraine, R.; Rodriguez, D.; Curie, A.; et al. Expanding the phenotypic spectrum of Allan–Herndon–Dudley syndrome in patients with SLC16A2 mutations. Dev. Med. Child Neurol. 2019, 61, 1439–1447. [Google Scholar] [CrossRef]
- Leuzzi, V.; Nardecchia, F.; Pons, R.; Galosi, S. Parkinsonism in children: Clinical classification and etiological spectrum. Parkinsonism Relat. Disord. 2021, 82, 150–157. [Google Scholar] [CrossRef]
- Willemsen, M.A.; Verbeek, M.M.; Kamsteeg, E.J.; de Rijk-van Andel, J.F.; Aeby, A.; Blau, N.; et al. Tyrosine hydroxylase deficiency: a treatable disorder of brain catecholamine biosynthesis Brain. J. Neurol. 2010, 133, 1810–1822. [Google Scholar] [CrossRef]
- Rutherford, S.; Fraza, C.; Dinga, R.; Kia, S.M.; Wolfers, T.; Zabihi, M.; et al. Charting brain growth and aging at high spatial precision. eLife 2022, 11, e72904. [Google Scholar] [CrossRef]
- Lee, W.T.; Weng, W.C.; Peng, S.F.; Tzen, K.Y. Neuroimaging findings in children with paediatric neurotransmitter diseases. J. Inherit. Metab. Dis. 2009, 32, 361–370. [Google Scholar] [CrossRef]
- Ludwik, K.A.; Opitz, R.; Jyrch, S.; Megges, M.; Weiner, J.; Beule, D.; et al. Generation of iPSC lines with SLC16A2:G401R or SLC16A2 knock out. Stem Cell Res. 2023, 73, 103256. [Google Scholar] [CrossRef]
- Brennenstuhl, H.; Jung-Klawitter, S.; Assmann, B.; Opladen, T. Inherited Disorders of Neurotransmitters: Classification and Practical Approaches for Diagnosis and Treatment. Neuropediatrics 2019, 50, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet Lond. Engl. 2009, 373, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Poewe, W.; Litvan, I.; Lewis, S.; Lang, A.E.; Halliday, G.; et al. Validation of the MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord Off. J. Mov. Disord. Soc. 2018, 33, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Morales-Briceño, H.; Mohammad, S.S.; Post, B.; Fois, A.F.; Dale, R.C.; Tchan, M.; et al. Clinical and neuroimaging phenotypes of genetic parkinsonism from infancy to adolescence. Brain. J. Neurol. 2020, 143, 751–770. [Google Scholar] [CrossRef]
- Niemann, N.; Jankovic, J. Juvenile parkinsonism: Differential diagnosis, genetics, and treatment. Parkinsonism Relat. Disord. 2019, 67, 74–89. [Google Scholar] [CrossRef]
- Pranzatelli, M.R.; Mott, S.H.; Pavlakis, S.G.; Conry, J.A.; Tate, E.D. Clinical spectrum of secondary parkinsonism in childhood: a reversible disorder. Pediatr. Neurol. 1994, 10, 131–140. [Google Scholar] [CrossRef]
- Cao, X.Y.; Jiang, X.M.; Dou, Z.H.; Rakeman, M.A.; Zhang, M.L.; O’Donnell, K.; et al. Timing of Vulnerability of the Brain to Iodine Deficiency in Endemic Cretinism. N. Engl. J. Med. 1994, 331, 1739–1744. [Google Scholar] [CrossRef]
- Korevaar, T.I.M.; Muetzel, R.; Medici, M.; Chaker, L.; Jaddoe, V.W.V.; de Rijke, Y.B.; et al. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: a population-based prospective cohort study. Lancet Diabetes Endocrinol. 2016, 4, 35–43. [Google Scholar] [CrossRef]
- Zimmermann, M.B. Iodine Deficiency. Endocr. Rev. 2009, 30, 376–408. [Google Scholar] [CrossRef]
- Grüters, A.; Krude, H. Detection and treatment of congenital hypothyroidism. Nat. Rev. Endocrinol. 2011, 8, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J. Thyroid hormone regulated genes in cerebral cortex development. J. Endocrinol. 2017, 232, R83–R97. [Google Scholar] [CrossRef] [PubMed]
- Salas-Lucia, F.; Escamilla, S.; Bianco, A.C.; Dumitrescu, A.; Refetoff, S. Impaired T3 uptake and action in MCT8-deficient cerebral organoids underlie Allan-Herndon-Dudley syndrome. JCI Insight 2024, 9, e174645. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.A.; Aly, M.S.; Rahman, T.A.; Shahat, A.S. Impact of experimental hypothyroidism on monoamines level in discrete brain regions and other peripheral tissues of young and adult male rats. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2013, 31, 225–233. [Google Scholar] [CrossRef]
- Gaum, P.M.; Gube, M.; Esser, A.; Schettgen, T.; Quinete, N.; Bertram, J.; et al. Depressive Symptoms After PCB Exposure: Hypotheses for Underlying Pathomechanisms via the Thyroid and Dopamine System. Int. J. Environ. Res. Public. Health 2019, 16, 950. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Y.; Montero-Pedrazuela, A.; Prensa, L.; Guadaño-Ferraz, A.; Rausell, E. Thyroid Hormone Transporters MCT8 and OATP1C1 Are Expressed in Projection Neurons and Interneurons of Basal Ganglia and Motor Thalamus in the Adult Human and Macaque Brains. Int. J. Mol. Sci. 2023, 24, 9643. [Google Scholar] [CrossRef]
Figure 1.
Schematic and phenotypic representation of MCT8 deficiency. (A, B) In MCT8 deficiency, thyroid hormones are released from the thyroid gland into the bloodstream with increased triiodothyronine (T3) and normal to decreased thyroxine (T4) concentrations. However, due to inactivating mutations of the SLC16A2 gene, which encodes the thyroid hormone transporter MCT8 (depicted in blue), T3 and T4 cannot be transported across blood-organ barriers that depend on MCT8, such as the (C) neurovascular unit (endothelial cells, red; pericytes, orange; astrocytes, purple; neurons, green). The brain is subsequently hypothyroid in the prenatal and in the postnatal phase. In other organs that express alternative thyroid hormone transporters, increased T3 concentrations can reach their intracellular thyroid hormone receptors and lead to hyperthyroid states, such as tachycardia. TRH, thyrotropin releasing hormone; TSH, thyroid stimulating hormone. The figure was created with BioRender.com. (D) Table depicting the patients' geno- and phenotypes. Age, current age; +, positive; (+), slightly positive; -, negative; na, not assessed.
Figure 1.
Schematic and phenotypic representation of MCT8 deficiency. (A, B) In MCT8 deficiency, thyroid hormones are released from the thyroid gland into the bloodstream with increased triiodothyronine (T3) and normal to decreased thyroxine (T4) concentrations. However, due to inactivating mutations of the SLC16A2 gene, which encodes the thyroid hormone transporter MCT8 (depicted in blue), T3 and T4 cannot be transported across blood-organ barriers that depend on MCT8, such as the (C) neurovascular unit (endothelial cells, red; pericytes, orange; astrocytes, purple; neurons, green). The brain is subsequently hypothyroid in the prenatal and in the postnatal phase. In other organs that express alternative thyroid hormone transporters, increased T3 concentrations can reach their intracellular thyroid hormone receptors and lead to hyperthyroid states, such as tachycardia. TRH, thyrotropin releasing hormone; TSH, thyroid stimulating hormone. The figure was created with BioRender.com. (D) Table depicting the patients' geno- and phenotypes. Age, current age; +, positive; (+), slightly positive; -, negative; na, not assessed.
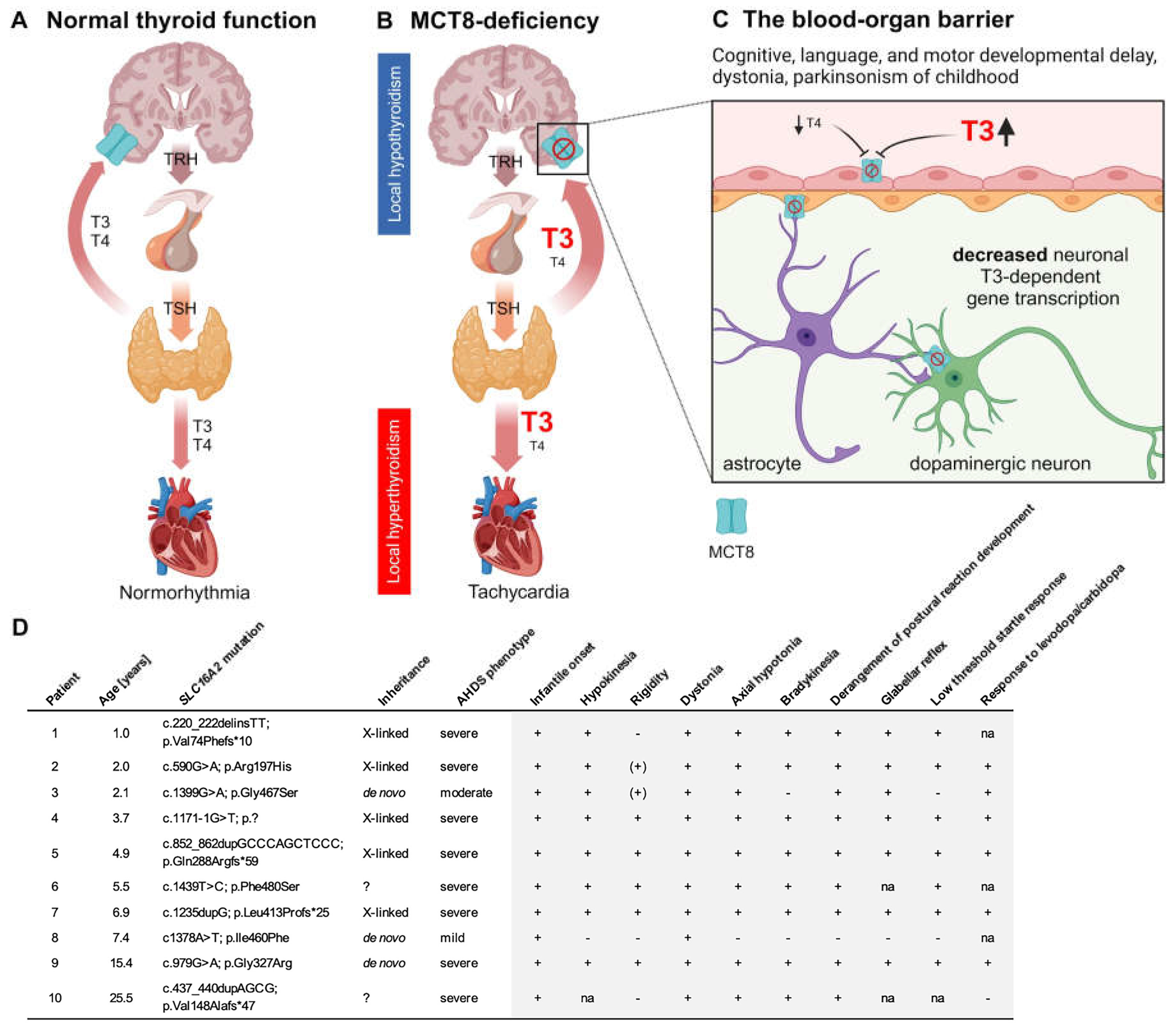
Figure 2.
Biogenic amines in the cerebrospinal fluid (CSF). (A) The homovanillic acid (HVA) concentrations in the CSF were below the reference ranges (depicted in gray) in 36% (4/11) of patients (depicted in blue). In the remaining 64% (7/11) of cases, HVA concentrations were observed to fall within the two lower quartiles of the normal range. In general, the HVA concentrations of patients were markedly lower than the mean of the reference data. Patient 1 (depicted in pink) had undergone treatment with Triac (97 µg/kg/d) for a period of six months prior to the lumbar puncture. However, the biogenic amine concentrations in the CSF of this patient were comparable to those of untreated children. (B) The 5-hydroxyindoleacetic acid (5-HIAA) concentrations were within the reference range, and (C) the HVA/5-HIAA ratios were significantly reduced in patients. This constellation of biogenic amine alterations indicates an isolated defect of dopamine deficiency, as observed in tyrosine hydroxylase deficiency, albeit with less pronounced HVA decreases. (A, B) The normality of the distribution was tested with the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was analyzed by applying a paired t-test. (C) If the data were not normally distributed, the statistical significance of the differences between the groups was tested by applying the Wilcoxon test. Ns, not significant; ***, p ≤ 0.001; NR, normal range; P, patients.
Figure 2.
Biogenic amines in the cerebrospinal fluid (CSF). (A) The homovanillic acid (HVA) concentrations in the CSF were below the reference ranges (depicted in gray) in 36% (4/11) of patients (depicted in blue). In the remaining 64% (7/11) of cases, HVA concentrations were observed to fall within the two lower quartiles of the normal range. In general, the HVA concentrations of patients were markedly lower than the mean of the reference data. Patient 1 (depicted in pink) had undergone treatment with Triac (97 µg/kg/d) for a period of six months prior to the lumbar puncture. However, the biogenic amine concentrations in the CSF of this patient were comparable to those of untreated children. (B) The 5-hydroxyindoleacetic acid (5-HIAA) concentrations were within the reference range, and (C) the HVA/5-HIAA ratios were significantly reduced in patients. This constellation of biogenic amine alterations indicates an isolated defect of dopamine deficiency, as observed in tyrosine hydroxylase deficiency, albeit with less pronounced HVA decreases. (A, B) The normality of the distribution was tested with the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was analyzed by applying a paired t-test. (C) If the data were not normally distributed, the statistical significance of the differences between the groups was tested by applying the Wilcoxon test. Ns, not significant; ***, p ≤ 0.001; NR, normal range; P, patients.
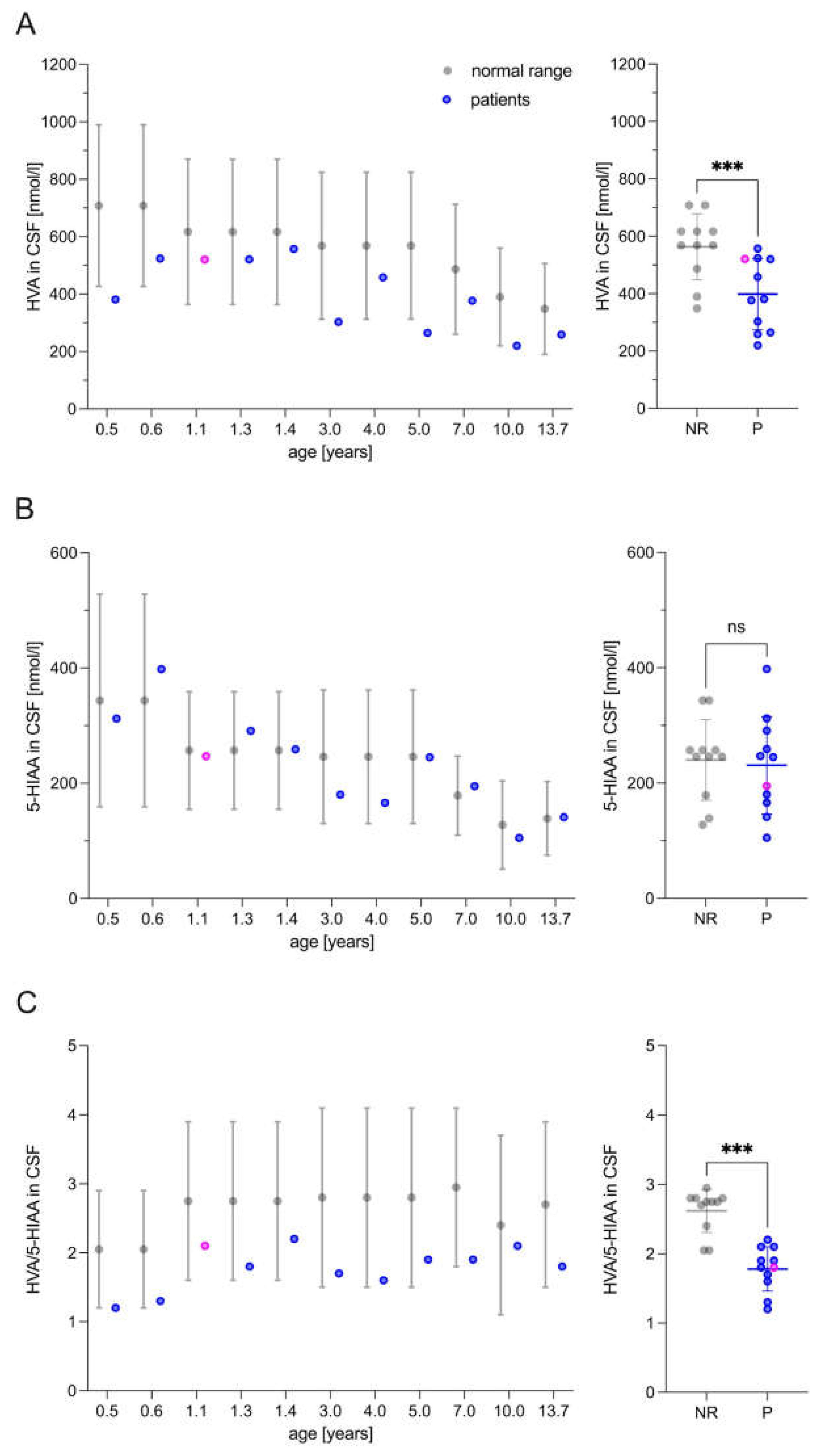
Figure 3.
Patients’ responses to levodopa/carbidopa therapy. (A) Patients (depicted in blue) exhibited a notable improvement in dystonic movement and (B) overall disability, as evaluated by the Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS). The natural history data (depicted in gray) were taken from previously published work and the BFMDRS was modified by excluding items pertaining to swallowing abilities in accordance with the requisite modifications [7]. (C) Additionally, the patients exhibited significantly lower ratings on the Infantile Parkinsonism-Dystonia Rating Scale (IPDRS) following six months of levodopa/carbidopa treatment. This was particularly evident in the reduction of non-motor symptoms, including autonomic dysfunction and mood dysregulation, as well as motor signs of parkinsonism and dystonia, such as hypokinesia, rigidity, dystonia, axial hypotonia, and motor developmental delay. (A-D) The normality of the distributions was assessed using the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was evaluated through the application of a paired t-test. The following symbols were used to indicate the level of significance: Ns, not significant; *, p≤0.05; **, p≤0.01; ***, p≤0.001. T0 represents the baseline measurement, while T6 denotes the assessment conducted after 6 months of levodopa/carbidopa therapy.
Figure 3.
Patients’ responses to levodopa/carbidopa therapy. (A) Patients (depicted in blue) exhibited a notable improvement in dystonic movement and (B) overall disability, as evaluated by the Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS). The natural history data (depicted in gray) were taken from previously published work and the BFMDRS was modified by excluding items pertaining to swallowing abilities in accordance with the requisite modifications [7]. (C) Additionally, the patients exhibited significantly lower ratings on the Infantile Parkinsonism-Dystonia Rating Scale (IPDRS) following six months of levodopa/carbidopa treatment. This was particularly evident in the reduction of non-motor symptoms, including autonomic dysfunction and mood dysregulation, as well as motor signs of parkinsonism and dystonia, such as hypokinesia, rigidity, dystonia, axial hypotonia, and motor developmental delay. (A-D) The normality of the distributions was assessed using the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was evaluated through the application of a paired t-test. The following symbols were used to indicate the level of significance: Ns, not significant; *, p≤0.05; **, p≤0.01; ***, p≤0.001. T0 represents the baseline measurement, while T6 denotes the assessment conducted after 6 months of levodopa/carbidopa therapy.
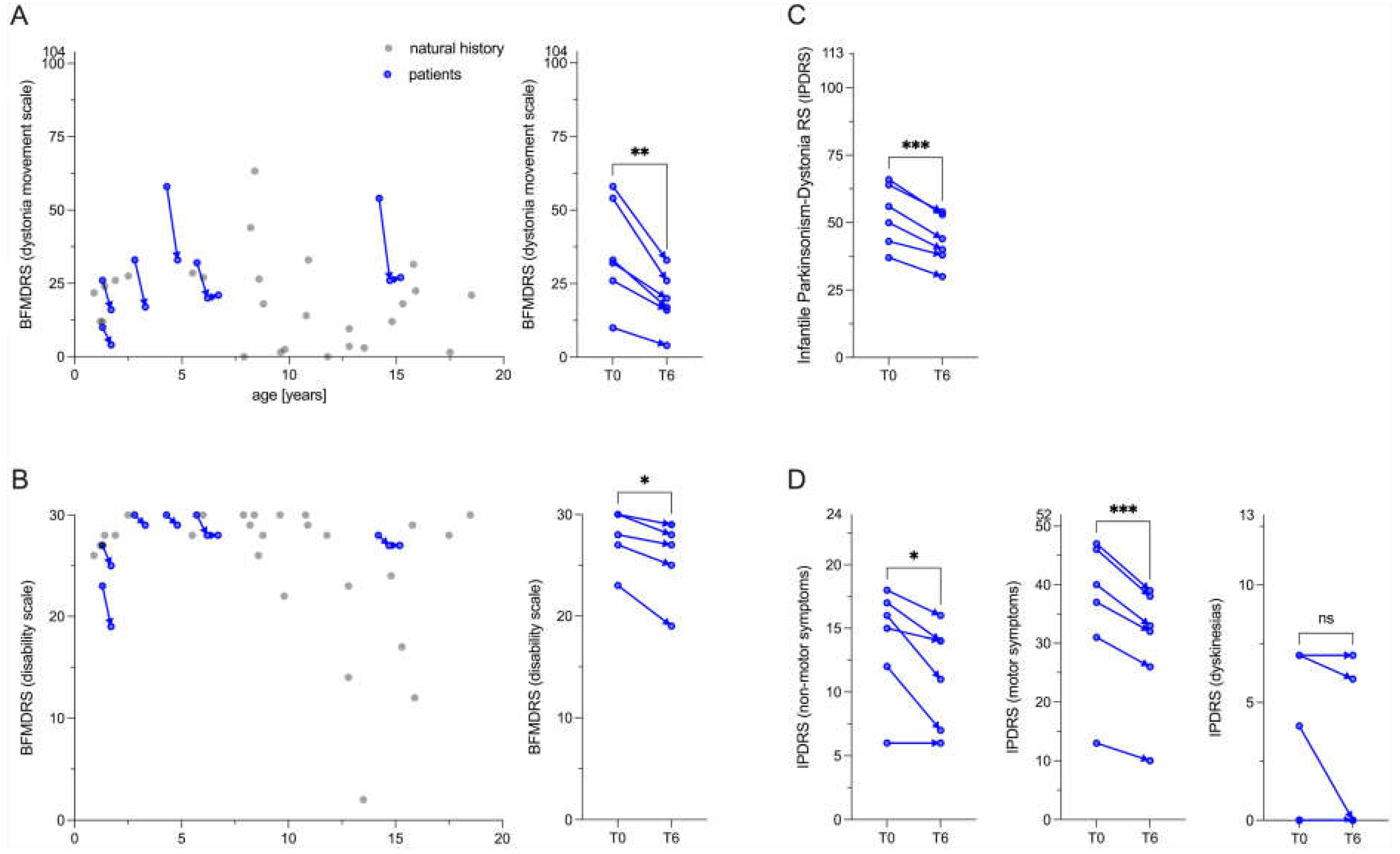
Figure 4.
Neuroimaging findings. (A) Patients with MCT8 deficiency exhibited a reduction in total intracranial brain volume below two standard deviations (z-scores, < -2.0z). This was particularly evident in the subcortical gray matter with a notable reduction in the size of the Globus pallidus (the site of dopamine action) and the ventral diencephalon, including the Substantia nigra (the site of dopamine synthesis). le, left; ri right. (B) Left panel: the z-scores of the head circumferences (two-point measurements) changed into both directions, n=4 patients with decreasing z-scores, n=2 patients with increasing z-scores. In terms of z-scores, we did not see an overall significant change with progressing age. Additional data are required to distinguish between primary reduced brain volume and secondary brain atrophy in patients with MCT8 deficiency. The patients identified as 7 and 9, for which volumetric analyses were conducted, are indicated in red. The normality of the distribution was evaluated using the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was evaluated through the application of a paired t-test. Right panel: B, measurement of head circumference within the first week after birth; T0, measurement of head circumference before levodopa/carbidopa therapy. (C) For illustrative purposes, a non-smoothed representative transversal image of the voxelwise modeled 18F-DOPA rate constant (Ki) is presented. (D) The 18F-DOPA uptake in the left caudate nucleus was found to be comparable to that observed in a previously published cohort of n=44 healthy adults [23], indicating that this parameter may serve as a reliable indicator of nigrostriatal dopaminergic network integrity. The normality of the distribution was evaluated using the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was evaluated using an ordinary one-way ANOVA test. The result was not statistically significant (ns).
Figure 4.
Neuroimaging findings. (A) Patients with MCT8 deficiency exhibited a reduction in total intracranial brain volume below two standard deviations (z-scores, < -2.0z). This was particularly evident in the subcortical gray matter with a notable reduction in the size of the Globus pallidus (the site of dopamine action) and the ventral diencephalon, including the Substantia nigra (the site of dopamine synthesis). le, left; ri right. (B) Left panel: the z-scores of the head circumferences (two-point measurements) changed into both directions, n=4 patients with decreasing z-scores, n=2 patients with increasing z-scores. In terms of z-scores, we did not see an overall significant change with progressing age. Additional data are required to distinguish between primary reduced brain volume and secondary brain atrophy in patients with MCT8 deficiency. The patients identified as 7 and 9, for which volumetric analyses were conducted, are indicated in red. The normality of the distribution was evaluated using the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was evaluated through the application of a paired t-test. Right panel: B, measurement of head circumference within the first week after birth; T0, measurement of head circumference before levodopa/carbidopa therapy. (C) For illustrative purposes, a non-smoothed representative transversal image of the voxelwise modeled 18F-DOPA rate constant (Ki) is presented. (D) The 18F-DOPA uptake in the left caudate nucleus was found to be comparable to that observed in a previously published cohort of n=44 healthy adults [23], indicating that this parameter may serve as a reliable indicator of nigrostriatal dopaminergic network integrity. The normality of the distribution was evaluated using the Shapiro-Wilk test (α = 0.05). The statistical significance of the differences between the groups was evaluated using an ordinary one-way ANOVA test. The result was not statistically significant (ns).
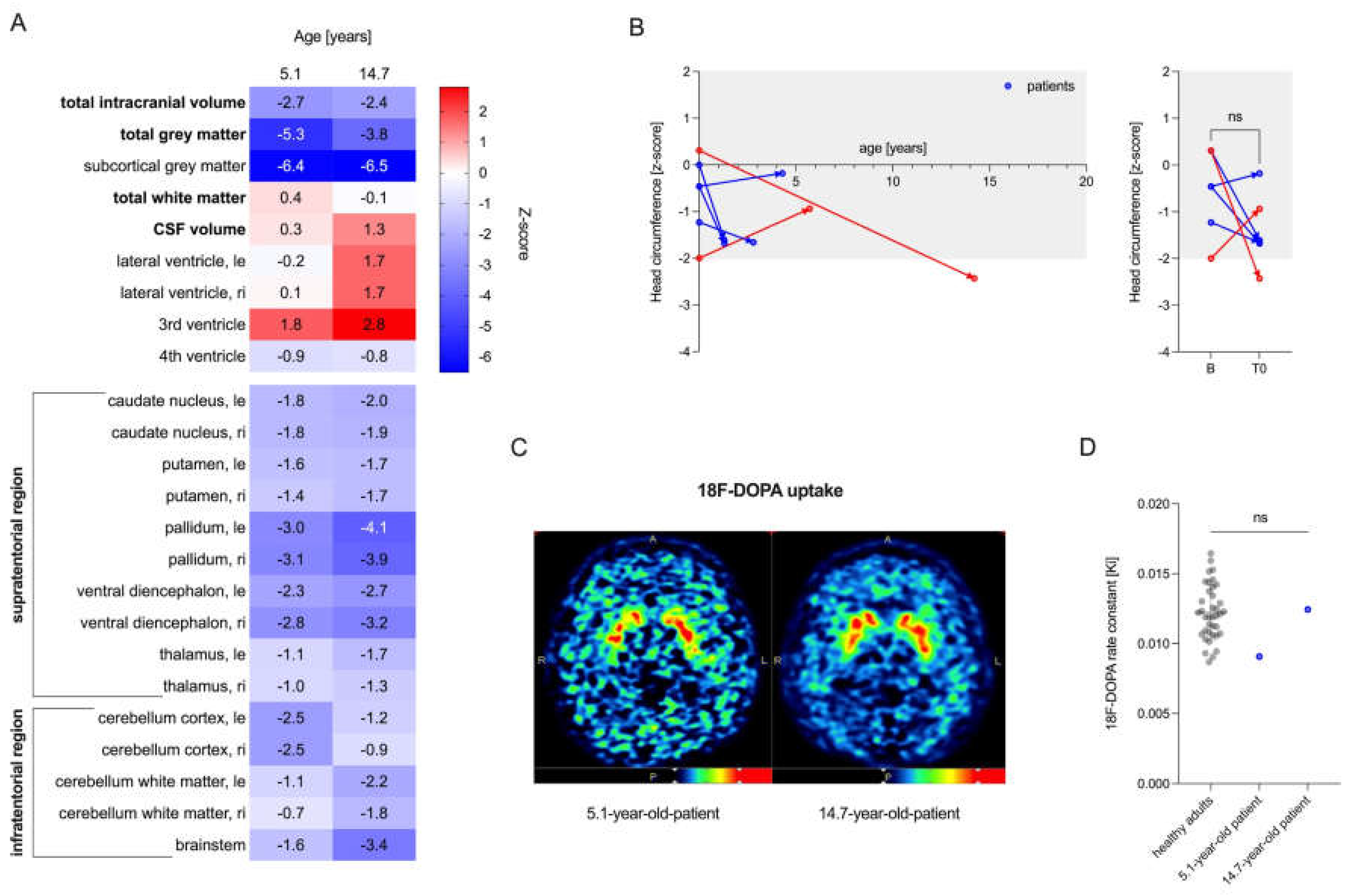
Figure 5.
iPSC-derived dopaminergic neurons for analyzing the interaction of MCT8 and dopamine pathways. (A) Schematic representation of the protocol for the differentiation of small molecule neural precursor cells (smNPCs) to midbrain dopaminergic neurons (mDANs) from induced pluripotent stem cells (iPSCs). The figure was created with BioRender.com and adapted from previously published work [24,25]. hESC, human embryonic stem cells; CHIR, CHIR99021; DM, dorsomorphin; PMA, purmorphamine; SB, SB43152; FGF8, fibroblast growth factor 8; d, days. (B) In accordance with previously published data [26], intracellular dopamine synthesis could only be detected and quantified following levodopa substitution (depicted in blue) in the growth media 28 and 4 hours prior to cell extraction. (C) After 22, 35, and 42 days, neuronal networks (TUJ1+, green) had developed, including populations of dopaminergic neurons (TH+, purple). Scale bar, 100 µm. (D) Dopaminergic neurons express MCT8 (depicted in white) in the nuclear and cell membrane of the soma, dendrites, and axon. Scale bar, 10 µm; TUJ1, tubulin beta III isoform; TH, tyrosine hydroxylase; DAPI, 4,6-diamidino-2-phenylindole; d, number of days.
Figure 5.
iPSC-derived dopaminergic neurons for analyzing the interaction of MCT8 and dopamine pathways. (A) Schematic representation of the protocol for the differentiation of small molecule neural precursor cells (smNPCs) to midbrain dopaminergic neurons (mDANs) from induced pluripotent stem cells (iPSCs). The figure was created with BioRender.com and adapted from previously published work [24,25]. hESC, human embryonic stem cells; CHIR, CHIR99021; DM, dorsomorphin; PMA, purmorphamine; SB, SB43152; FGF8, fibroblast growth factor 8; d, days. (B) In accordance with previously published data [26], intracellular dopamine synthesis could only be detected and quantified following levodopa substitution (depicted in blue) in the growth media 28 and 4 hours prior to cell extraction. (C) After 22, 35, and 42 days, neuronal networks (TUJ1+, green) had developed, including populations of dopaminergic neurons (TH+, purple). Scale bar, 100 µm. (D) Dopaminergic neurons express MCT8 (depicted in white) in the nuclear and cell membrane of the soma, dendrites, and axon. Scale bar, 10 µm; TUJ1, tubulin beta III isoform; TH, tyrosine hydroxylase; DAPI, 4,6-diamidino-2-phenylindole; d, number of days.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



