
Submitted:
05 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
Background/Objectives: The Perilla frutescens plant has historically been used to protect against inflammation and redox stress. This has been attributed in part to the high content of various polyphenols. Despite this, the complement of polyphenolic components in Perilla extract is not yet fully defined. The aim of this study was to characterise the polyphenolic composition in Perilla extract, and its effect on the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), which during inflammation and oxidative stress binds to the antioxidant response element (ARE) found on the promoter of phase II antioxidant enzymes.
Methods: Following extraction using four solvents of different properties, the total phenolic, flavonoid and ortho-diphenolic content of Perilla extract were determined. Individual phenolic compounds were identified and quantified by RP-HPLC. Dual Luciferase assay using a reporter plasmid containing a human NQO1 ARE-luciferase fusion was employed to determine the potential protective effect of the Perilla fractions against redox stress.
Results: HPLC analysis revealed that the highest phenolic content was present in the polar extracts. A total of 35 distinct polyphenolic compounds were identified, with rosmarinic acid being the major constituent. On cells in vitro, the methanol Perilla fraction showed protection against redox stress, with the luciferase assay displaying up to 1.5 fold increase in the induction of the human NQO1 ARE-luciferase reporter.
Conclusions: While Perilla extract seems to be mainly protecting against inflammation and redox stress through the action of rosmarinic acid, further work is required on the synergystic effects between different polyphenols.
Keywords:
Perilla frutescens
; polyphenols
; redox stress
; anti-inflammatory response
; antioxidant response element
1. Introduction
The Perilla frutescens is a mint family (Lamiaceae) member common across South East Asia [1]. The leaves of P. frutescens have been used in traditional medicine to treat various ailments [2,3] and ample research has been performed presenting the impact on biological processes such as inflammation, redox stress, allergic reaction and even carcinogenesis [4,5,6,7,8,9,10,11,12,13,14]
The active constituents in Perilla extracts have been sub-divided into a number of classes, with the ones of greatest relevance to antioxidant activity being: alkaloids, phenylpropane analogues, terpenoids, and polyphenols (phenolic acids, flavonoids including anthocyanins, tannins, stilbenes, and lignans) [14].
Polyphenols are secondary metabolites widespread throughout the plant kingdom that are able to act as antioxidants mainly by neutralising reactive oxygen (such as hydroxyl radical and superoxide ion) and nitrogen (such as nitric oxide, peroxynitrite) radicals via hydrogen donation from their hydroxyl groups [15,16,17]. They can also chelate transition metal ions such as Fe3+ Cu2+ involved in the production of free radicals [17]. Synergistic effects have also been observed between various pairs of polyphenols [18], which in the context of natural extracts can have significance.
They present strong interactions with proteins due to the hydrophobicity of their rings and hydrogen-bonding potential of the phenolic hydroxyl groups [19], and have the potential to inhibit some enzymes involved in radical generation, including xanthine oxidase, myeloperoxidase and lipoxygenase [20], as well as regulation of intracellular glutathione levels [21]. Over and above their antioxidant action, polyphenols also display anti-inflammatory, anti-allergenic, anti-viral, anti-microbial, anti-mutagenic, anti-cancer and cardio-protective properties [22].
Specifically looking at flavonoids, their free radical scavenging capacity depends on their structure, on the number of hydroxyl groups in the structure, on the position of these hydroxyl groups, and on the ability of the hydroxyl groups to donate hydrogen atoms to radicals in order to stabilise them [23]. Flavonoids have also been reported to present interesting anti-cancer properties by modulating several biological signalling pathways including redox metabolism (detoxification, oxidation and reduction), inflammation, suppression of oncogenes and tumour formation, cell growth and proliferation, cell cycle checkpoints, DNA repair, senescence, intrinsic and extrinsic apoptosis, autophagy, stimulation of the immune system [24,25].
Similarly, phenolic acids have varying levels of antioxidant activity based on the number and position of hydroxyl groups within these molecules, with higher antioxidant activity upon introduction of a second ortho- or para-hydroxyl group or methoxyl groups [26]. Additionally, they also present anti-cancer properties such as their ability to inhibit cell proliferation, angiogenic factors, oncogenic signaling cascades, growth and differentiation, preventing cellular migration and metastasis, whilst inducing apoptosis [27].
Inflammation and oxidative stress are known to play a role in a variety of human conditions including auto-immune disorders and cancer [28,29,30,31]. A major transcription factor involved in protecting cells against oxidative stress is nuclear factor erythroid 2-related factor 2 (Nrf2). It functions by binding to a motif called the antioxidant response element (ARE) found on the promoter of phase II antioxidant enzymes [32].
In order to better understand the administration dosing, timing and molecular targeting in the therapeutic use of Perilla leaf extract, it is important to understand the overall contribution of the polyphenolic, flavonoid and ortho-diphenolic components to the bioactive functions. However, there has so far been only limited investigation of the extent of antioxidant activity by these compounds [33,34,35,36].
The overall aim of this study was first of all to define the polyphenolic composition of Perilla leaf extract following fractionation using solvents of varying polarity. To achieve this the biological activity of these components was quantified through the determination of the metal cation reduction activity and their scavenging power. The fractions of the Perilla leaf extract were then incubated with cells cultured in vitro to quantify the induction of Nrf2.
2. Materials and Methods
Sample Preparation
The Perilla leaf extract used in this study was provided by Amino Up (Sapporo, Japan). Unlike the Perilla extract products sold by Amino Up Co., Ltd., the powder used in this study was 100% Perilla-water-soluble components. Portions of 0.2g Perilla leaf extract were first treated using different pH conditions (pH 2, 4, 7, 9) at 70°C, after which the solution underwent exhaustive maceration extraction using a 1:5 dry weight to solvent ratio in butanol, ethyl acetate and ether or direct extraction from the powder using methanol. The fractions were dried at 30°C under vacuum, followed by a reconstitution in 1ml of methanol for all the subsequent testing.
Determination of Total Phenolic content
The Folin-Ciocalteu colourimetric method [37] was used in order to determine the total phenolic content present in the hydroalcoholic extracts derived from the Perilla leaf extract against a standard calibration curve made with caffeic acid (Sigma Aldrich). The concentrated extracts derived from liquid–liquid extraction were diluted by a factor of 10 using 1:1 methanol to acetonitrile (v/v). Then, 20 µL of the resulting solution were oxidised with 100 µL of 5-fold diluted Folin-Ciocalteu reagent followed by neutralisation by the addition of Na2CO3 (80 µL, 7.5%) in a 96-well microtiter plate. After incubation at room temperature for 2 hours in the dark, the absorbance at 600 nm was recorded using a microtiter plate reader. The total phenolic content was expressed as mg caffeic acid equivalents (mg/ml CAE).
Determination of Total Flavonoid content
The total flavonoid content assay was determined according to Mabry et al. [38] with minor modifications. For this analysis 25 µL of the diluted extract were mixed with 7.5 µL of 10 % aluminium chloride, 7.5 µL of 7 % w/v sodium nitrite and 80 µL of distilled water, and left at room temperature for 30 min, after which 100 µL of 1M NaOH solution was added. The plate was shaken vigorously, and the absorbance of the reaction was recorded at 415 nm. The calibration curve was prepared by using catechin. The total flavonoid content was expressed as mg catechin equivalents (mg/ml CE).
Determination of ortho-diphenolic content
The Arnow’s colourimetric method [39] was used for the determination of o-diphenolic compounds present in the extracts against a standard calibration curve made with pyrocatechol (Sigma Aldrich). To perform this determination, 20 µL of 5-fold diluted phenolic extract were added to 20 µL of 1M HCl in a 96-well microtiter plate. The resulting mixture was briefly mixed followed by the addition of 20 µL of Arnow’s reagent, previously prepared by dissolving 10g of sodium nitrite and 10 g of sodium molybdate dihydrate in 100 ml 1:1 ethanol to water (v/v). The plate was shaken vigorously and after 15 min, 80 µL of water and 40 µL of 1M NaOH were added and the absorbance was measured at 370 nm using a microtiter plate reader. The ortho-diphenolic content was expressed as mg pyrocatechol equivalents (mg/ml PyCE).
Determination of 2 2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging activity
The radical scavenging activity of the phenolic extracts was measured using the DPPH assay. In order to determine the radical scavenging activity of phenolic compounds derived from Perilla extract, a stock solution of 60 µM DPPH was prepared in methanol on the day and the solution was kept in the dark at 4 °C. Initially, 25 µl of phenolic stock solution was added into the well and it was further down diluted to the lowest concentration (7.8 µg/ml) by performing a serial two-fold dilution in a 96-well microtiter plate. A row of negative DPPH control was added in the same 96-well microtiter plates by adding 100 µl of MeOH into each well. Then, 150 µl of methanolic DPPH were added into each well and the reaction was allowed to proceed for 30 minutes in the dark. The absorbance was measured at 517 nm by a microplate reader. The % of DPPH radicals scavenged by the phenolic extracts was calculated using the equation:
Determination of 2,2-azinobis-(3-ethylbenzothiazoline-6-sulfonate) (ABTS) radical cation stabilisation
The ABTS•+ was produced by the reaction of 7 mM stock solution of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) with 2.45 mM potassium persulfate and allowing the mixture to stand in the dark at room temperature for 12 h before use. The concentration of the blue-green ABTS radical solution was adjusted with methanol to an absorbance of 0.700 (0.020 ± mean ± SD) at 734 nm. To 280 µL of this solution was added 20 µL of sample or solvent in a 96-well plate. For phenolic extracts, the stock solution of 500 µg/ml was added into the well and two-fold serially diluted down to the lowest concentration of 7.8 µg/ml. The mixture was incubated for 5 min at 30°C, and the absorbance at 734 nm was measured with a microplate reader. The percentage inhibition of ABTS•+ was calculated as follows:
Determination of Cupric Reducing Antioxidant Power
The copper ion reducing antioxidant capacity assay employs the use of copper (II) neocuproine reagent as the chromogenic oxidising agent. The method was adjusted for microtiter plates whereby 20 µL of diluted extract were added 100 µL of 10 mM CuCl2 solution followed by 100 µL of 1M ammonium acetate buffer at pH 7.0. To the resulting solution, 100 µL of 7.5 mM neocuproine ethanolic solution was added and the reaction was allowed to proceed for 30 minutes, after which the absorbance at 450 nm was recorded.
Determination of Ferric Reducing Antioxidant Power
Ferric-reducing antioxidant power (FRAP) was determined by the spectrophotometric method previously described by Benzie and Strain [40], using the reduction of a ferric tripyridyl triazine (Fe(III)(TPTZ)2) complex at low pH. The FRAP reagent was prepared by mixing 25 ml of 300.0 mmol/L acetate buffer, 2.5 ml of 10 mmol/L ferric tripyridyl triazine (TPTZ) solution, and 2.5 ml of 20 mmol/L FeCl3 solution in a 10:1:1 ratio. 10 μL of sample were mixed with 200 μL of FRAP reagent. The contents were mixed vigorously. The reduction of ferric tripyridyl triazine (Fe(III)-TPTZ) complex to ferrous tripyridyltriazine (FeII-TPTZ) in the presence of antioxidants was measured at 593 nm. The results were calculated from an ascorbic acid calibration curve and expressed as mg/ml ascorbic acid.
Determination of Nitrous oxide radical scavenging activity
Nitric oxide (NO) was generated from sodium nitroprusside and was measured by the Griess reagent (naphthylethylenediamine). 50 µL of 10 mM sodium nitroprusside in phosphate buffer saline at pH 7 was incubated with 10 µL of the test compounds at room temperature for 180 minutes. After 180 minutes, 100 µL of freshly prepared Griess reagent were added and the absorbance was measured at 546 nm [41]. Griess reagent was prepared by mixing equal amounts of 1% sulphanilamide in 2.5% phosphoric acid and 0.1% naphthylethylene diamine dihydrochloride in 2.5% phosphoric acid immediately before use. Control samples without the extracts but with an equal volume of buffer were prepared in a similar manner as was done for the test samples. For coloured extracts absorbing at 540 nm the reaction was carried out with no sodium nitroprusside and the absorbance was subtracted. The absorbance was read at 540 nm and the percentage nitric oxide inhibition by the extracts was calculated using the following equation:
Determination of hydrogen peroxide scavenging activity
This assay is based on the reaction of ferrous ions (Fe+2) with 1,10-phenanthroline. The hydrogen peroxide scavenging assay was performed in accordance with the method described by Zhang et al., [42], with slight modifications. The scavenging activity for hydroxyl radicals was measured using the reaction mixture contained 3.6 μl of 1.0 mM FeCl3, 5.4 μl of 1mM 1,10-phenanthroline, 100 µl of 0.2 M phosphate buffer (pH 7.8), 9 μl of 0.17 M H2O2, and 90 µL of extract. Adding H2O2 started the reaction. After incubation at room temperature for 5 minutes, the absorbance of the mixture at 510 nm was measured with a spectrophotometer.
High performance liquid chromatography (HPLC)
Identification and quantification of phenolic compounds was carried out by a Shimadzu LC-20 AB (Kyoto, Japan) HPLC system equipped with a binary pump, autosampler (SIL-20AC), and UV/vis detector (SPD-20AV) monitoring at 280 nm and 320 nm. Samples were centrifuged and syringe filtered through a 0.45 micron PVDF filter. The analysis was carried out using 20 μL injection volume on an ACE® C18 analytical column (250 × 4.6 mm i.d.) with a particle size of 5 μm (Aberdeen, Scotland). The mobile phases were degassed and consisted of (A) water: Acetic acid (95:5, v/v) and (B) methanol: Acetonitrile (1:1, v/v) at a constant flow rate of 1 mL/min. A gradient elution was performed using the following solvent program: 95% (A): 5% (B) 0–30 min; 70% (A): 30% (B) 30–35 min; 50% (A): 50% (B) 35–40 min; 100% (B) 40-50 min and then 95% (A): 5% (B) for the final 2 min as a post-equilibration step. The column temperature was set to 35 °C. The column was maintained at 35 °C using a Shimadzu CTO-10AC thermostatically controlled column compartment, whilst the sample chamber was set to 4°C to prevent phenolic degradation. Compounds were identified by peak identification using standards and relative retention time.
Cell Culture
Human embryonic kidney, Hek293, cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% foetal bovine serum and 1% penicillin/streptomycin and kept at 37°C, 5% CO2 and > 95% humidity.
Cell Viability
Hek293 cells were seeded at 5,000 cells/well in a 96 well plate in complete medium and allowed to adhere for 24 hours. The cells were then treated with the different Perilla extracts (aqueous, methanol, butanol, ethyl acetate and ether) at 1 or 5mg/ml. The plates were incubated at 37 °C, 5% CO2 and >95% humidity for 72 hours. After 72 hours incubation with the various extracts, the cell proliferation assay was performed by adding 20 µl of 3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS; CellTiter 96®Aqueous One Solution Cell ProliferationAssay; Promega) to each well. Absorbance readings were taken after 1h incubation using the Mithras LB940 microplate reader at 490 nm.
Construction of the human NQO1 ARE-luciferase reporter
The human NQO1 ARE (GCAGTCACAGTGACTCAGCAGAATCT) was amplified from HepG2 (liver cancer cell line) cDNA using the primers hNQO1-ARE-Forward: CCTGAGCTCGCTAGCCTCGACAGGGGTGGTGCAGTGGCAT; hNQO1-ARE-Reverse: CCAGATCTTGATATCCTCGAGGCTCTGGTGCAGTCCGGGG. The PCR involved 5 minutes initial denaturation at 95 °C, 30 s denaturation at 95 °C, 30 s annealing at 55 °C, 1 minute extension at 72 °C repeated 35 cycles and a final 1 minute extension at 72 °C. The pGL4.1 luciferase reporter vector (Promega) was digested using the Xho1 restriction enzyme. The 723 bp PCR product was ligated into the linearised plasmid using the In-Fusion HD Cloning Mix (Takara Clontech) following the manufacturer instructions. A number of colonies that successfully grew on the ampicillin plates were picked and sequenced in order to ensure that the human NQO1 ARE construct did not present any mutations.
Transfection
Once the Hek293 cells transfered to the wells reached 80% confluency, they were transfected with 100 ng pGL3 reporter plasmid containing the human NQO1 ARE-luciferase fusion and 5 ng pRL-SV40, using a magnetofection procedure (OZ Biosciences), with a DNA : NeuroMag ratio of 100 ng : 0.1 µl per well. After mixing the DNA and NeuroMag, the complex was incubated at RT for 20 minutes, before being added to the cells and incubated on the magnet for 30 minutes.
Dual Luciferase Reporter Assay
The effect of the different solvent extractions of the Perilla leaf extract (0.5 mg/ml) on redox stress was determined by using the human NQO1 ARE-luciferase fusion in a pGL3-Enhancer vector (Promega) as the reporter. Following a 24, 48 or 72 h incubation, the Hek293 cells in each well were lysed using 20 µl Passive Lysis Buffer. To 5 µl of lysate, 100 µl of LAR II were added and firefly luciferase activity was measured. Following that, 100 µl of Stop & Glo Reagent were added and Renilla luciferase activity was measured. The firefly luciferase values were normalised using the co-transfected Renilla luciferase for triplicate experiments. Relative fold changes of the averages for each of the extract fractions acting upon the firefly reporter were then generated compared to untreated, transfected cells.
Western blotting
Hek293 cells treated with 0.5 mg/ml of the various extracts for 24, 48 or 72 hours were harvested and lysed using urea lysis buffer (8 M urea, 1.5 M thiourea, 0.5 M NaCl). The protein concentration was determined by Bradford Assay using the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad) following the manufacturer’s instructions and measured on a Bio Photometer Plus UV/Vis spectrophotometer (Eppendorf). SDS-PAGE was performed to separate 20 μg of protein using an AE-6450 Dual Mini Slab Electrophoresis Kit (Atto) at 20 mA constant current and 300 V for 60 minutes.
Semi-dry electroblotting was carried out using the Trans-Blot SD Semi-Dry Transfer Cell (BioRad) set at 120 mA and 100 V for 60 minutes. Following electroblotting, transfer success was evaluated using 0.1% Ponceau S solution and the membrane was then blocked with 5% skimmed milk in Tris Buffer Saline (TBS) for 1 hour. The membrane was then incubated overnight at 4°C with primary antibodies against HSP27 (NBP2-32972, Novus Bio; 1:1000), HSP70 (NB110-61582, Novus Bio; 1:1000), HSP90 (NB110-61640, Novus Bio; 1:1000), Actin (NB100-74340, Novus Bio; 1:1000), mono-methyl lysine (14679, Cell Signaling Technology: 1:1000) and tri-methyl lysine (14680, Cell Signaling Technology; 1:1000), diluted using a 5% Bovine Serum Albumin (BSA) solution in TBS. The membrane was then incubated with IRDye 800CW goat anti-mouse secondary IgG antibody (926-32210, Li-Cor; 1:5000) or IRDye 800CW goat anti-rabbit secondary IgG antibody (926-32211, Li-Cor; 1:10000) diluted using 5% BSA in TBS. The bands were visualised using the Odyssey (Li-Cor) imaging system.
3. Results
Determination of total phenolic, flavonoid and ortho-diphenolic content
The data (Table 1) showed that the highest phenolic, flavonoid and ortho-diphenolic content was present in the polar solvent extracts using methanol and butanol. More specifically, the methanol extracts presented the highest phenolic content compared to all the butanol extracts at the different pHs. Total flavonoid content was actually the highest for the butanol extract at pH 7. There was no observable difference between the flavonoid content in the methanol extracts and the butanol extracts at pH 2 and pH 4, whilst the butanol extract at pH 9 presented the lowest flavonoid content. Similarly for the ortho-diphenolic content, the butanol extract at pH 7 presented the highest value, with no observable difference between the ortho-diphenolic content in the methanol extracts and the butanol extracts at pH 2 and pH 4, whilst the butanol extract at pH 9 presented the lowest ortho-diphenolic content. On the other hand, the extraction of phenolic, flavonoid and ortho-diphenolic compounds was extremely inefficient using either ethyl acetate or ether.
Determination of the DPPH radical scavenging activity and ABTS radical cation stabilisation
The data (Table 2 and Figure 1) showed that the highest percentage inhibition of DPPH radicals was for the methanol extract, whilst the lowest by far was for the ether extract. The butanol extracts all clustered together, but at double the extract content. Similarly, the data (Table 2 and Figure 2) showed the highest percentage inhibition of ABTS radicals or the methanol extract and the lowest for the ether extract, with a relatively close clustering of the methanol and butanol extracts.
Determination of cupric and ferric reducing antioxidant power
The data (Table 3) showed that the highest cupric and ferric reducing antioxidant power was present in the polar solvent extracts using methanol and butanol. More specifically, the methanol extracts presented the highest cupric and ferric reducing antioxidant power compared to all the butanol extracts at the different pHs. Of all the butanol extracts, the extract at pH 7 presented slightly higher cupric reducing antioxidant power, while the extracts at pH 7 and pH 9 presented higher ferric reducing antioxidant power. The butanol extracts at pH 2 and pH 4 presented comparable cupric but low ferric reducing antioxidant power particularly pH 4. On the other hand, the cupric and ferric reducing antioxidant power was extremely low for both the ethyl acetate and ether extracts.
Determination of nitrous oxide radical and hydrogen peroxide scavenging activity
The data (Table 4) showed that the highest nitrous oxide radical and hydrogen peroxide scavenging activity was present in the polar solvent extracts using methanol and butanol. More specifically, the butanol extract at pH 9 presented the highest nitrous oxide radical scavenging activity, with all other butanol extracts as well as the methanol extracts being comparable in their nitrous oxide radical scavenging activity. Hydrogen peroxide scavenging activity was highest for the methanol extracts. The ethyl acetate and ether extracts showed comparable nitrous oxide radical scavenging activity to the polar solvent extracts, however they showed poor hydrogen peroxide scavenging activity.
Qualitative and quantitative assessment of extracts using HPLC
HPLC (Figure 3 and Figure 4) identified a total of 35 distinct compounds, of which 18 were quantified for each extract.
Table 5 indicates the regression formula obtained and the regression coefficient for each compound based on triplicate injections and 6-point calibration. The table highlights the limits of detection and limits of quantification for each compound.
The data comparing the hydrophilic and lipophilic fractions of the Perilla extract (Table 6) shows that the 18 polyphenols quantifed (out of the 35 identified) are significantly higher in the methanol extract than in the ether extract, with rosmarinic acid being by far the major constituent.
The butanol extract was subjected to 4 different pHs (Table 7). The extracts subjected to acidic pH values had a higher concentration of most phenolic acids, with some acids including rosmarinic acid being more abundant at neutral pH, whilst the concentrations of flavonoids and their derivatives were not affected by pH.
Cell Viability
The microscopy data (Figure 5) as well as the cell viability assay data (Figure 6) performed in parallel showed that the various fractions of the Perilla extract presented no toxic effects on the cells at 1 mg/mL but had varying levels of cytotoxicity at 5 mg/mL.
Dual Luciferase Reporter Assay
The dual luciferase assay data (Figure 7) showed that the most effective components for the induction of Nrf2 found in the crude extract are extracted by methanol as evident by the high activity of the methanol extract, only second to that of the crude extract. Butanol seems to be less effective in extracting these components as the observed effect is somewhat lower compared to the methanol extract. Ethyl ethanoate also presents a similarly low activity, which can be considered to be practically negligible. Interestingly the aqueous leftover fraction initially presents a detrimental effect, which is reduced over time.
Western Blotting
The Western blotting data for HSP70 and HSP27 (Figure 8) show little variation between the different conditions over the 72 hour period of testing. Interestingly, the blots for mono-methyl and tri-methyl lysine (Figure 9) show that different doses of the crude Perilla extract bring about changes in the band intensities of methylated proteins throughout the 10-250 kDa range, although the band intensity is not always apparent due to the low abundances of such modifications.
4. Discussion
The identity of the main polyphenolic, flavonoid and ortho-diphenolic components in Perilla leaf extract, and their bioactive functions, was investigated so as to obtain a better understanding of the dose, action time and molecular targets for therapeutic applications.
To maximise the polyphenolic extraction from the crude Perilla leaf extract, this was fractionated using solvents of varying polarity, namely methanol, n-butanol, ethyl ethanoate and ether. The highest phenolic, flavonoid and ortho-diphenolic content was obtained using the polar solvents methanol and n-butanol, with very low recoveries obtained using either ethyl acetate or ether (Table 1). Considering different pHs of n-butanol, phenolic extraction was not pH dependent, whilst flavonoid and ortho-diphenolic extraction were best at neutral pH (pH 7). Acidic pHs (pH 2 and pH 4) presented only slightly lower extarction efficiency, whilst the n-butanol extract at alkaline pH (pH 9) presented the lowest flavonoid and ortho-diphenolic recovery. Upon performing HPLC analysis on the different solvent fractions, a total of 35 distinct polyphenolic compounds were identified (Figure 3 and Figure 4), and 18 of these were further quantified (Table 5). The comparative quantification of the polyphenolic composition of the hydrophilic (methanol) and lipophilic (ether) fractions identified rosmarinic acid as being the major constituent, 20-fold more abundant than any other compenent (Table 6). The n-butanol extract subjected to acidic pH values had a higher concentration of most of the quantified phenolic acids, with some acids including rosmarinic acid having a higher abundant in the neutral pH extract. On the other hand, flavonoids and their derivatives showed similar concentrations at all pHs (Table 7).
Other studies that investigated the composition of Perilla extract. For example, Ueda et al. [43] identified caffeic acid, rosmarinic acid and 3′,4′,5,7-tetrahydroxyflavone (luteolin) as the main constituents of Perilla extract. Izumi et al. [34] identified 2′,3′-dihydroxy-4′,6′-dimethoxychalcone from an ether extract. Kwon et al. [33] identified protocatechuic acid, chlorogenic acid, caffeic acid, 4-methyoxycinnamic acid, oleanolic acid, kaempferol-3-O-rutinoside, rosmarinic acid, luteolin, methyl-rosmarinic acid, apigenin and 4′,5,7-trimethoxyflavone but according to them the EtOAc fraction had the highest activity. Tantipaiboonwong et al. [9] identified rosmarinic acid, chlorogenic acid, caffeic acid, ferulic acid and luteolin as the major constituents, with rosmarinic acid being the predominant compound. Adam et al. [36] identified 18 compounds, with the major components being caffeic acid, syringic acid, cinnamic acid, rosmarinic acid, quercitin, kaemferol and pinostrobin. The concentrations extracted vary greatly between the studies mainly due to the different procedure and solvents employed.
Rosmarinic acid has been known to be present in Perilla for 30 years [44] and was later proven to be one of the main polyphenolic constituent found in P. frutescens leaf extracts [45]. It has also been identified in numerous other medicinal plant species of the Lamiaceae family including basil, sage, rosemary, and mint [46,47]. Rosmarinic acid is produced from the aromatic amino acids l-phenylalanine and l-tyrosine through the intermediary precursors 4-coumaroyl-CoA and 4-hydroxyphenyllactic acid respectively, and formed as an ester of caffeic acid and 3,4-dihydroxyphenyllactic [48]. It is well known to suppress inflammation reactions and protect against redox stress [49,50,51,52,53,54,55,56,57].
Of note is that the yields of the various polyphenolics extracted from Perilla frutescens is influenced by several factors. The primary factor is the existence of multiple cultivars of Perilla frutescens [36,58]. Additionally, geographical location [59] and growing conditions, including soil type, play a significant role. Consequently the antioxidant effect exhibited is greatly impacted by both the solvent used and the process used for the extraction. This variability arises from the compatibility between the chemical characteristics of the polyphenolic compounds and the polarity of the solvent used, which determines how much of each compound is recovered by a specific solvent [60].
Further to this, there has been only limited investigation of the extent of antioxidant activity by these compounds as distributed following various solvent extractions from the same starting material. Moreover, when this has been investigated it was done in very different ways, making comparison difficult. The closest extraction procedure to the one used in this study was that of Hong et al. [52], which reported much lower total phenolic and flavonoid content than the current study.
The biological activity of these components has been generally quantified through the determination of the metal cation reduction activity and their scavenging power. Adam et al. [36] determined that the ethanolic extracts had the highest hydroxyl radical scavenging capacity and superoxide anion radical neutralisation potential, similar to the methanolic extract of the current study. However, Hong et al. [52] reported that the DPPH radical scavenging ability was similar between ethyl acetate and n-butanol fraction, while in this study the DPPH radical scavenging ability for the ethyl acetate fraction was found to be lower than that of the n-butanol fraction. Furthermore, Hong et al. [52] reported that the reducing power (determined by potassium ferricyanide reduction method) was higher for the ethyl acetate fraction than the butanol fraction. However in the current study the reducing power of the butanol fraction was twice as high as the ethyl acetate fraction. Yet different conclusions were obtained by Kwon et al. [33] that reported the aqueous fraction had the highest hydroxyl radical scavenging activity, while the ethylethanoate fraction had the highest hydrogen peroxide scavenging activity, nitric oxide scavenging activity, ferric reducing/antioxidant power, ABTS scavenging activity and DPPH scavenging activity, whilst n-butanol had the highest ferrous ion chelating activity of the various fractions prepared.
To quantify the biological significance of the Perilla leaf extract fractions, Nrf2 induction was used as an indicator. Firstly, the cytotoxicity of the various fractions of the Perilla extract on Hek293 cells was determined to be negligible up to 1mg/ml but presented varying levels of cytotoxicity at 5 mg/mL (Figure 5). This is in line with a number of other studies that described cytotoxicity at concentrations within the same range. Similar to the current study, Kim et al. [61] found no toxic effects of the aqueous extract on rat hepatocytes up to a concentration of 1 mg/mL. Adam et al. [36] reported cytotoxicity from 0.5mg/ml on Mg-63 (osteosarcoma) cells and A431 (squamous cell carcinoma) cells.
In this study Nrf2 induction was highest with the crude extract, followed by the methanol extract. Butanol and ethyl ethanoate were less effective at extracting the active components (Figure 7). Interestingly, the induction effect seems to be time-dependent, with most extracts showing the highest activation within the first 24 hours, followed by a decline. Conversely, the aqueous leftover fraction initially exhibited a cytotoxic or inhibitory effect, which diminished by the 72 hour mark. Similarly, Adam et al. [36] demonstrated that different extracts exerted their cytotoxic effects at varying time points over the 72 hour experimental period.
The cytoprotective and antioxidant nature of Perilla extract has been shown to be through inhibition of enzymes including cytochrome P450 isoforms, lipoxygenases, cyclooxygenase, and xanthine oxidase involved in radical generation [62]. This inhibition is achieved by precipitation or the formation of various complexes that hinder enzyme activity [63]. It has been shown that both Perilla extracts and rosmarinic acid reduce liver injury induced by d-galactosamine and lipopolysaccharide (LPS) in mice by scavenging superoxide radicals produced by Kupffer cells as well as inhibiting peroxynitrite formation induced by inducible nitric oxide synthase (iNOS) [64]. Kwon et al. [33] described the major antioxidant constituents of ethanolic Perilla extracts as being protocatechuic acid, chlorogenic acid, caffeic acid, rosmarinic acid, luteolin, methyl-rosmarinic acid, apigenin and 4′,5,7-trimethoxyflavone.
The Western blotting data for both HSP70 and HSP27 (Figure 8) did not show any markable difference in expression of the 72 hours of incubation with the extract fractions. This could have been due to the absence of a redox insult or the low-dose induction produced by the single dose used. That being said, both mono-methyl and tri-methyl lysine (Figure 9) presented changes in the band intensities of methylated proteins throughout the 10-250 kDa range upon incubation with different doses of the crude Perilla extract. Low abundances hinder quantification and the large number of overlapping proteins makes identification of the modified proteins impossible. That being said, using shotgun mass spectrometry or Western blotting for specific targets could make identification of some of these proteins possible, which opens up a whole new avenue for antioxidant research.
Since inflammation is closely associated with elevated levels of reactive species [65], further work could potentially focus on the effect of the extract fractions on controlling redox modifications of inflammatory mediators, particularly investigating post-translational modifications such as phosphorylation and methylation. Understanding which components best reduce inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) and suppress the production of inflammatory cytokines (tumour necrosis factor alpha (TNF-a), interleukin (IL)-1, and IL-6) would be of great clinical benefit. Furthermore, investigating the roles played by Perilla extract biomolecules on the activity of NF-κB signaling, regulation of AMP-activated protein kinase (AMPK) and HIF-1α expression could provide mechanistic insight into the processes involved in controlling inflammation by polyphenolics.
5. Conclusions
This study has shown that Perilla extract contains a large variety of polyphenolic, flavonoid and ortho-diphenolic components. Using four solvents these could be fractionated and yields differed based on the polarity of the solvent employed. The antioxidant properties were evident both through chemical analysis as well as through induction of the human NQO1 ARE luciferase reporter. This information provides a strong basis for further investigation of the proteins and pathways targetted by these components as well as a better understanding of the synergistic effect of multiple polyphenolics.
Author Contributions
Conceptualization, F.L. and B.B.; methodology, F.L. and B.B.; validation, F.L. and B.B.; formal analysis, F.L. and B.B.; investigation, F.L. and B.B.; resources, F.L. and B.B.; writing—original draft preparation, B.B.; writing—review and editing, F.L.; visualization, F.L. and B.B.; project administration, B.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Amino Up Co. Ltd. for producing and providing custom grade Perilla leaf extract for this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ahmed, H.M. Ethnomedicinal, phytochemical and pharmacological investigations of Perilla frutescens (L.) Britt. Molecules, 2018, 24(1), 102.
- Yu, H.; Qiu, J.F.; Ma, L.J.; Hu, Y.J.; Li, P.; Wan, J.B. Phytochemical and phytopharmacological review of Perilla frutescens L.(Labiatae), a traditional edible-medicinal herb in China. Food and Chemical Toxicology, 2017, 108, 375-391. [CrossRef]
- Igarashi, M.; Miyazaki, Y. A review on bioactivities of perilla: progress in research on the functions of perilla as medicine and food. Evidence-based complementary and alternative medicine, 2013. [CrossRef]
- Kwak, Y.; Ju, J. Inhibitory activities of Perilla frutescens britton leaf extract against the growth, migration, and adhesion of human cancer cells. Nutrition research and practice, 2015, 9(1), 11.
- Pintha, K.; Tantipaiboonwong, P.; Yodkeeree, S.; Chaiwangyen, W.; Chumphukam, O.; Khantamat, O.; Khanaree, C.; Kangwan, N.; Thongchuai, B.; Suttajit, M. Thai perilla (Perilla frutescens) leaf extract inhibits human breast cancer invasion and migration. Maejo International Journal of Science and Technology, 2018, 12(2), pp.112-123.
- Hou, T.; Netala, V.R.; Zhang, H.; Xing, Y.; Li, H.; Zhang, Z.. Perilla frutescens: A rich source of pharmacological active compounds. Molecules, 2022, 27(11), p.3578. [CrossRef]
- Vanita, K.; Megh, T.; Shivam, D. Perilla Frutescens – A review on pharmacological activities, extraction procedure and applications. Asian J Pharm Clin Res, 2022, 15(8), pp.34-40.
- Youn, I.; Han, S.; Jung, H.J.; Noh, S.G.; Chung, H.Y.; Koo, Y.K.; Shin, S.; Seo, E.K. Anti-Inflammatory Activity of the Constituents from the Leaves of Perilla frutescens var. acuta. Pharmaceuticals, 2023, 16(12), p.1655.
- Tantipaiboonwong, P.; Pintha, K.; Chaiwangyen, W.; Suttajit, M.; Khanaree, C.; Khantamat, O. Bioefficacy of Nga-Mon (Perilla frutescens) fresh and dry leaf: Assessment of antioxidant, antimutagenicity, and anti-inflammatory potential. Plants, 2023, 12(11), p.2210.
- Adam, G.; Robu, S.; Flutur, M.M.; Cioanca, O.; Vasilache, I.A.; Adam, A.M.; Mircea, C.; Nechita, A.; Harabor, V.; Harabor, A.; Hancianu, M. Applications of Perilla frutescens extracts in clinical practice. Antioxidants, 2023, 12(3), p.727. [CrossRef]
- Takemoto, K.; Ganlin, T.; Iji, M.; Narukawa, T.; Koyama, T.; Hao, L.; Watanabe, H. Vegetable Extracts as Therapeutic Agents: A Comprehensive Exploration of Anti-Allergic Effects. Nutrients, 2024, 16(5), p.693. [CrossRef]
- Khanaree, C.; Punfa, W.; Tantipaiboonwong, P.; Nuntaboon, P.; Suttajit, M.; Topanurak, S.; Dukaew, N.; Mon, M.T.; Hu, R.; Pintha, K. In vitro anti-metastasis of Perilla frutescens leaf water extract on aggressive human breast cancer cells. Journal of Associated Medical Sciences, 2022, 55(3), pp.51-59. [CrossRef]
- Jeong, J.H.; Park, H.J.; Chi, G.Y.; Choi, Y.H.; Park, S.H. An Ethanol Extract of Perilla frutescens Leaves Suppresses Adrenergic Agonist-Induced Metastatic Ability of Cancer Cells by Inhibiting Src-Mediated EMT. Molecules, 2023, 28(8), p.3414. [CrossRef]
- Huang, S.; Nan, Y.; Chen, G.; Ning, N.; Du, Y.; Lu, D.; Yang, Y.; Meng, F.; Yuan, L. The role and mechanism of Perilla frutescens in Cancer treatment. Molecules, 2023, 28(15), p.5883. [CrossRef]
- Kaurinovic, B.; Vastag, D. Flavonoids and phenolic acids as potential natural antioxidants. 2019, (pp. 1-20). London, UK: IntechOpen.
- Makhaik, M.S.; Shakya, A.K.; Kale, R. Dietary phytochemicals: As a natural source of antioxidants. Antioxidants-benefits, sources, mechanisms of action, 2021, p.646.
- Chaudhary, P.; Janmeda, P.; Docea, A.O.; Yeskaliyeva, B.; Abdull Razis, A.F.; Modu, B.; Calina, D.; Sharifi-Rad, J. Oxidative stress, free radicals and antioxidants: Potential crosstalk in the pathophysiology of human diseases. Frontiers in chemistry, 2023, 11, p.1158198. [CrossRef]
- Mitra, S.; Tareq, A.M.; Das, R.; Emran, T.B.; Nainu, F.; Chakraborty, A.J.; Ahmad, I.; Tallei, T.E.; Idris, A.M.; Simal-Gandara, J. Polyphenols: A first evidence in the synergism and bioactivities. Food Reviews International, 2023, 39(7), pp.4419-4441. [CrossRef]
- Sun, X.; Sarteshnizi, R.A.; Udenigwe, C.C. Recent advances in protein–polyphenol interactions focusing on structural properties related to antioxidant activities. Current Opinion in Food Science, 2022, 45, p.100840. [CrossRef]
- Filipsky, T.; Riha, M.; Macakova, K.; Anzenbacherová, E.; Karlickova, J.; Mladenka, P. Antioxidant effects of coumarins include direct radical scavenging, metal chelation and inhibition of ROS-producing enzymes. Current topics in medicinal chemistry, 2015, 15(5), pp.415-431. [CrossRef]
- Cianfruglia, L.; Morresi, C.; Bacchetti, T.; Armeni, T.; Ferretti, G. Protection of polyphenols against glyco-oxidative stress: Involvement of glyoxalase pathway. Antioxidants, 2020, 9(10), p.1006. [CrossRef]
- Grgić, J.; Šelo, G.; Planinić, M.; Tišma, M.; Bucić-Kojić, A. Role of the encapsulation in bioavailability of phenolic compounds. Antioxidants 2020, 9, 923. [CrossRef]
- Treml, J.; Šmejkal, K. Flavonoids as potent scavengers of hydroxyl radicals. Comprehensive reviews in food science and food safety, 2016, 15(4), pp.720-738.
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients, 2020, 12(2), p.457. [CrossRef]
- Ponte, L.G.S.; Pavan, I.C.B.; Mancini, M.C.S.; da Silva, L.G.S.; Morelli, A.P.; Severino, M.B.; Bezerra, R.M.N.; Simabuco, F.M. The hallmarks of flavonoids in cancer. Molecules, 2021, 26(7), p.2029. [CrossRef]
- Chen, J.; Yang, J.; Ma, L.; Li, J.; Shahzad, N.; Kim, C.K. Structure-antioxidant activity relationship of methoxy, phenolic hydroxyl, and carboxylic acid groups of phenolic acids. Scientific reports, 2020, 10(1), p.2611.
- Abotaleb, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Therapeutic potential of plant phenolic acids in the treatment of cancer. Biomolecules, 2020, 10(2), p.221. [CrossRef]
- El Menyiy, N.; El Allam, A.; Aboulaghras, S.; Jaouadi, I.; Bakrim, S.; El Omari, N.; Shariati, M.A.; Miftakhutdinov, A.; Wilairatana, P.; Mubarak, M.S.; Bouyahya, A. Inflammatory auto-immune diseases of the intestine and their management by natural bioactive compounds. Biomedicine & Pharmacotherapy, 2022, 151, p.113158. [CrossRef]
- Vaidya, F.U.; Chhipa, A.S.; Sagar, N.; Pathak, C. Oxidative stress and inflammation can fuel cancer. Role of oxidative stress in pathophysiology of diseases, 2020, pp.229-258.
- Neganova, M.; Liu, J.; Aleksandrova, Y.; Klochkov, S.; Fan, R. Therapeutic influence on important targets associated with chronic inflammation and oxidative stress in cancer treatment. Cancers, 2021, 13(23), p.6062. [CrossRef]
- Bardelčíková, A.; Šoltys, J.; Mojžiš, J. Oxidative stress, inflammation and colorectal cancer: an overview. Antioxidants, 2023, 12(4), p.901. [CrossRef]
- Zhao, C.R.; Qu, X.J. Nrf2–ARE signaling pathway and natural products for cancer chemoprevention. Cancer epidemiology, 2010, 34(5), 523-533. [CrossRef]
- Kwon, S.H.; Wang, Z.; Hwang, S.H.; Kang, Y.H.; Lee, J.Y.; Lim, S.S. Comprehensive evaluation of the antioxidant capacity of Perilla frutescens leaves extract and isolation of free radical scavengers using step-wise HSCCC guided by DPPH-HPLC. International journal of food properties, 2017, 20(sup1), 921-934. [CrossRef]
- Izumi, Y.; Matsumura, A.; Wakita, S.; Akagi, K.I.; Fukuda, H.; Kume, T.; Irie, K.; Takada-Takatori, Y.; Sugimoto, H.; Hashimoto, T.; Akaike, A. Isolation, identification, and biological evaluation of Nrf2-ARE activator from the leaves of green perilla (Perilla frutescens var. crispa f. Viridis). Free Radical Biology and Medicine, 2012, 53(4), pp.669-679. [CrossRef]
- Lee, A.Y.; Wu, T.T.; Hwang, B.R.; Lee, J.; Lee, M.H.; Lee, S.; Cho, E.J. The neuro-protective effect of the methanolic extract of Perilla frutescens var. japonica and rosmarinic acid against H2O2-induced oxidative stress in C6 glial cells. Biomolecules & therapeutics, 2016, 24(3), 338.
- Adam, G.; Cojocaru, F.D.; Verestiuc, L.; Cioanca, O.; Vasilache, I.A.; Adam, A.M.; Mircea, C.; Nechita, A.; Harabor, V.; Huzum, B.; Harabor, A. Assessing the Antioxidant Properties, In Vitro Cytotoxicity and Antitumoral Effects of Polyphenol-Rich Perilla leaves Extracts. Antioxidants, 2023, 13(1), p.58. https://www.mdpi.com/2076-3921/13/1/58.
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. Methods in Enzymology, 1999, 299, 152-178. [CrossRef]
- Mabry, T.J.; Markham, K.R.; Thomas, M.B.; Mabry, T.J.; Markham, K.R.; Thomas, M.B. The ultraviolet spectra of flavones and flavonols, 1970.
- Mateos, R.; Espartero, J.L.; Trujillo, M.; Ríos, J.J.; León-Camacho, M.; Alcudia, F.; Cert, A. Determination of phenols, flavones, and lignans in virgin olive oils by solid-phase extraction and high-performance liquid chromatography with diode array ultraviolet detection. Journal of Agricultural and Food Chemistry, 2001, 49(5), pp.2185-2192. [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Analytical biochemistry, 1996, 239(1), pp.70-76.
- Polshettiwar, S.A.; Ganjiwale, R.O.; Wadher, S.J.; Yeole, P.G. Spectrophotometric estimation of total tannins in some ayurvedic eye drops. Indian Journal of Pharmaceutical Sciences, 2007, 69(4).
- Zhang, G.; Zong, R.; Tseng, H.W.; Thummel, R.P. Ru (II) Complexes of Tetradentate Ligands Related to 2, 9-Di (pyrid-2 ‘-yl)-1, 10-phenanthroline. Inorganic chemistry, 2008, 47(3), pp.990-998.
- Ueda, H.; Yamazaki, C.; Yamazaki, M. Luteolin as an anti-inflammatory and anti-allergic constituent of Perilla frutescens. Biological and Pharmaceutical Bulletin, 2002, 25(9), pp.1197-1202. [CrossRef]
- Tada, M.; Matsumoto, R.; Yanmaguchi, H.; Chiba, K. Novel antioxidants isolated from Perilla flutescens britton var. crispa (Thunb). Bioscience, biotechnology, and biochemistry, 1996, 60(7), pp.1093-1095.
- Zhu, F.; Asada, T.; Sato, A.; Koi, Y.; Nishiwaki, H.; Tamura, H. Rosmarinic acid extract for antioxidant, antiallergic, and α-glucosidase inhibitory activities, isolated by supramolecular technique and solvent extraction from Perilla leaves. Journal of agricultural and food chemistry, 2014, 62(4), 885-892.
- Alagawany, M.; Abd El-Hack, M.E.; Farag, M.R.; Gopi, M.; Karthik, K.; Malik, Y.S.; Dhama, K. Rosmarinic acid: modes of action, medicinal values and health benefits. Animal health research reviews, 2017, 18(2), 167. [CrossRef]
- Noor, S.; Mohammad, T.; Rub, M.A.; Raza, A.; Azum, N.; Yadav, D.K.; Hassan, M.I.; Asiri, A.M. Biomedical features and therapeutic potential of rosmarinic acid. Archives of Pharmacal Research, 2022, 45(4), pp.205-228. [CrossRef]
- Petersen, M.; Abdullah, Y.; Benner, J.; Eberle, D.; Gehlen, K.; Hücherig, S.; Janiak, V.; Kim, K.H.; Sander, M.; Weitzel, C.; Wolters, S. Evolution of rosmarinic acid biosynthesis. Phytochemistry, 2009, 70(15-16), pp.1663-1679. [CrossRef]
- Lim, H.J.; Woo, K.W.; Lee, K.R.; Lee, S.K.; Kim, H.P. Inhibition of proinflammatory cytokine generation in lung inflammation by the leaves of Perilla frutescens and its constituents. Biomolecules & therapeutics, 2014, 22(1), 62. [CrossRef]
- Stansbury, J. Rosmarinic acid as a novel agent in the treatment of allergies and asthma. Journal of restorative medicine, 2014, 3(1), 121-126. [CrossRef]
- Jin, B.R.; Chung, K.S.; Cheon, S.Y.; Lee, M.; Hwang, S.; Noh Hwang, S.; Rhee, K.J.; An, H.J. Rosmarinic acid suppresses colonic inflammation in dextran sulphate sodium (DSS)-induced mice via dual inhibition of NF-κB and STAT3 activation. Scientific reports, 2017, 7(1), p.46252.
- Hong, E.; Park, K.H.; Kim, G.H. Phenolic-enriched fractions from perilla frutescens var. acuta: Determinating rosmarinic acid and antioxidant activity.Journal of Food Biochemistry, 2011, 35(6), 1637-1645. [CrossRef]
- Han, J.; Wang, D.; Ye, L.; Li, P.; Hao, W.; Chen, X.; Ma, J.; Wang, B.; Shang, J.; Li, D.; Zheng, Q. Rosmarinic acid protects against inflammation and cardiomyocyte apoptosis during myocardial ischemia/reperfusion injury by activating peroxisome proliferator-activated receptor gamma. Frontiers in pharmacology, 2017, 8, p.456. [CrossRef]
- Mushtaq, N.; Schmatz, R.; Ahmed, M.; Pereira, L.B.; da Costa, P.; Reichert, K.P.; Dalenogare, D.; Pelinson, L.P.; Vieira, J.M.; Stefanello, N.; de Oliveira, L.S. Protective effect of rosmarinic acid against oxidative stress biomarkers in liver and kidney of strepotozotocin-induced diabetic rats. Journal of physiology and biochemistry, 2015, 71, pp.743-751. [CrossRef]
- Luo, C.; Zou, L.; Sun, H.; Peng, J.; Gao, C.; Bao, L.; Ji, R.; Jin, Y.; Sun, S. A review of the anti-inflammatory effects of rosmarinic acid on inflammatory diseases. Frontiers in pharmacology, 2020, 11, p.153. [CrossRef]
- Gui, H.; Jin, Y.; Lin, A.; Wang, P.; Wang, Y.; Zhu, H. Rosmarinic acid relieves cisplatin-induced ovary toxicity in female mice via suppression of oxidative stress and inflammation. Journal of Biochemical and Molecular Toxicology, 2021, 35(9), p.e22839. [CrossRef]
- Lu, Y.H.; Hong, Y.; Zhang, T.Y.; Chen, Y.X.; Wei, Z.J.; Gao, C.Y. Rosmarinic acid exerts anti-inflammatory effect and relieves oxidative stress via Nrf2 activation in carbon tetrachloride-induced liver damage. Food & Nutrition Research, 2022, 66. [CrossRef]
- Deguchi, Y.; Ito, M. Caffeic acid and rosmarinic acid contents in genus Perilla. J. Nat. Med. 2020, 74, 834–839. [CrossRef]
- Li, Y.; Zhang, Y.; Wang, Y.; Li, X.; Zhou, L.; Yang, J.; Guo, L. Metabolites and chemometric study of Perilla (Perilla frutescens) from different varieties and geographical origins. J. Food Sci. 2022, 87, 5240–5251. [CrossRef]
- Sultana, B.; Anwar, F.; Ashraf, M. Effect of extraction solvent/technique on the antioxidant activity of selected medicinal plant extracts. Molecules, 2009, 14, 2167–2180. [CrossRef]
- Kim, M.K.; Lee, H.S.; Kim, E.J.; Won, N.H.; Chi, Y.M.; Kim, B.C.; Lee, K.W. Protective effect of aqueous extract of Perilla frutescens on tert-butyl hydroperoxide-induced oxidative hepatotoxicity in rats. Food and Chemical Toxicology, 2007, 45(9), pp.1738-1744. [CrossRef]
- Parr, A.; Bolwell, G.P. Phenols in the plant and in man. The potential for possible nutritional enhancement of the diet by modifying the phenols content or profile. J. Sci. Food Agric. 2000, 80, 985–1012.
- Le Bourvellec, C.; Renard, C.M. Interactions between polyphenols and macromolecules: Quantification methods and mechanisms. Critical reviews in food science and nutrition, 2012, 52(3), pp.213-248. [CrossRef]
- Osakabe, N.; Yasuda, A.; Natsume, M.; Sanbongi, C.; Kato, Y.; Osawa, T.; Yoshikawa, T. Rosmarinic acid, a major polyphenolic component of Perilla frutescens, reduces lipopolysaccharide (LPS)-induced liver injury in D-galactosamine (D-GalN)-sensitized mice. Free Radical Biology and Medicine, 2002, 33(6), pp.798-806.
- Lei, Y.; Wang, K.; Deng, L.; Chen, Y.; Nice, E.C.; Huang, C. Redox regulation of inflammation: old elements, a new story. Medicinal research reviews, 2015, 35(2), pp.306-340. [CrossRef]
Figure 1.
Dose response curve of various extracts and the % inhibition of DPPH radicals. The solid black line represents the effective concentration at which 50% of the DPPH radical are stabilised.
Figure 1.
Dose response curve of various extracts and the % inhibition of DPPH radicals. The solid black line represents the effective concentration at which 50% of the DPPH radical are stabilised.
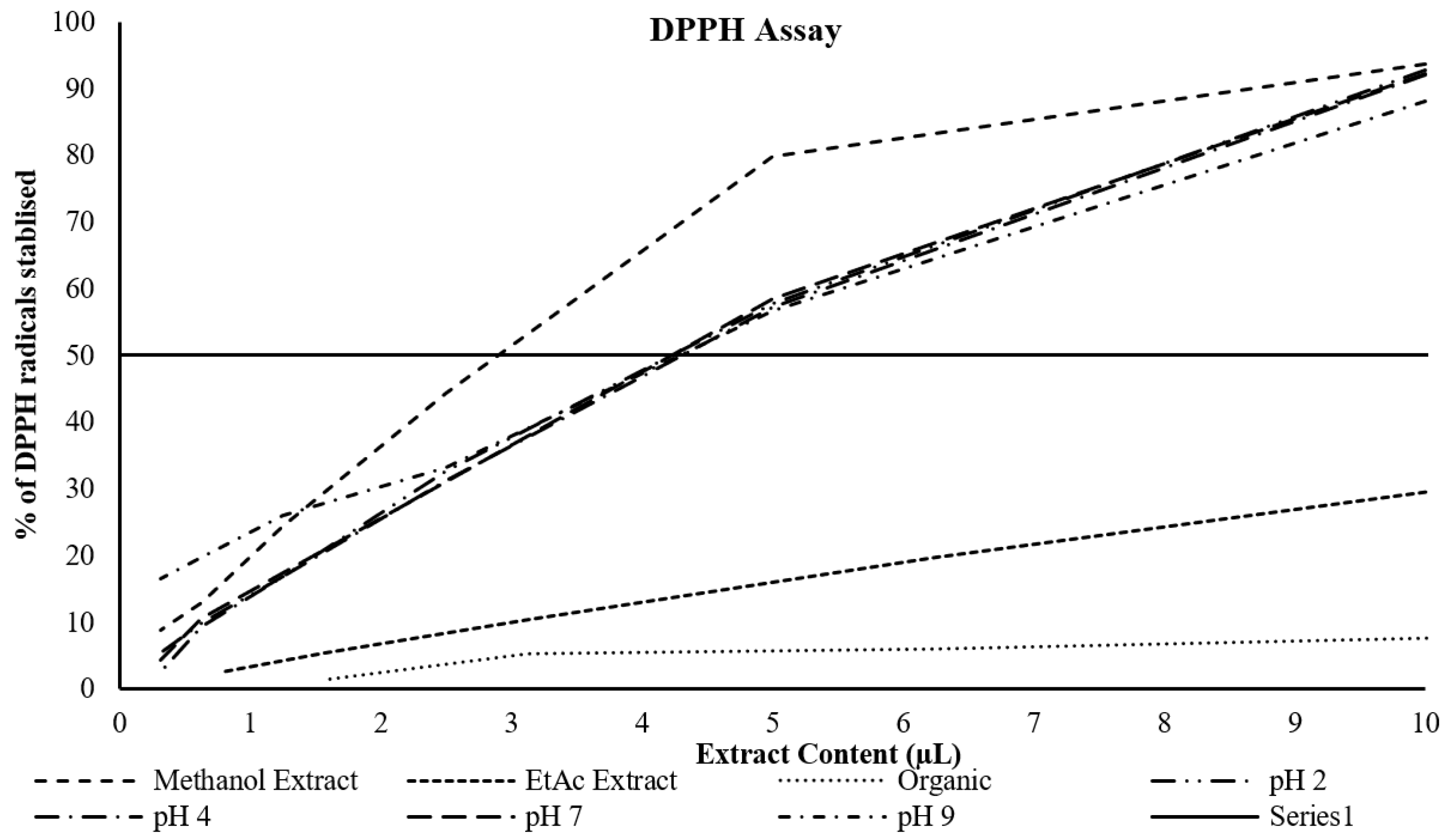
Figure 2.
Dose response curve of various extracts and the % inhibition of ABTS radical cations. The solid black line represents the effective concentration at which 50% of the ABTS radical cations are stabilised.
Figure 2.
Dose response curve of various extracts and the % inhibition of ABTS radical cations. The solid black line represents the effective concentration at which 50% of the ABTS radical cations are stabilised.
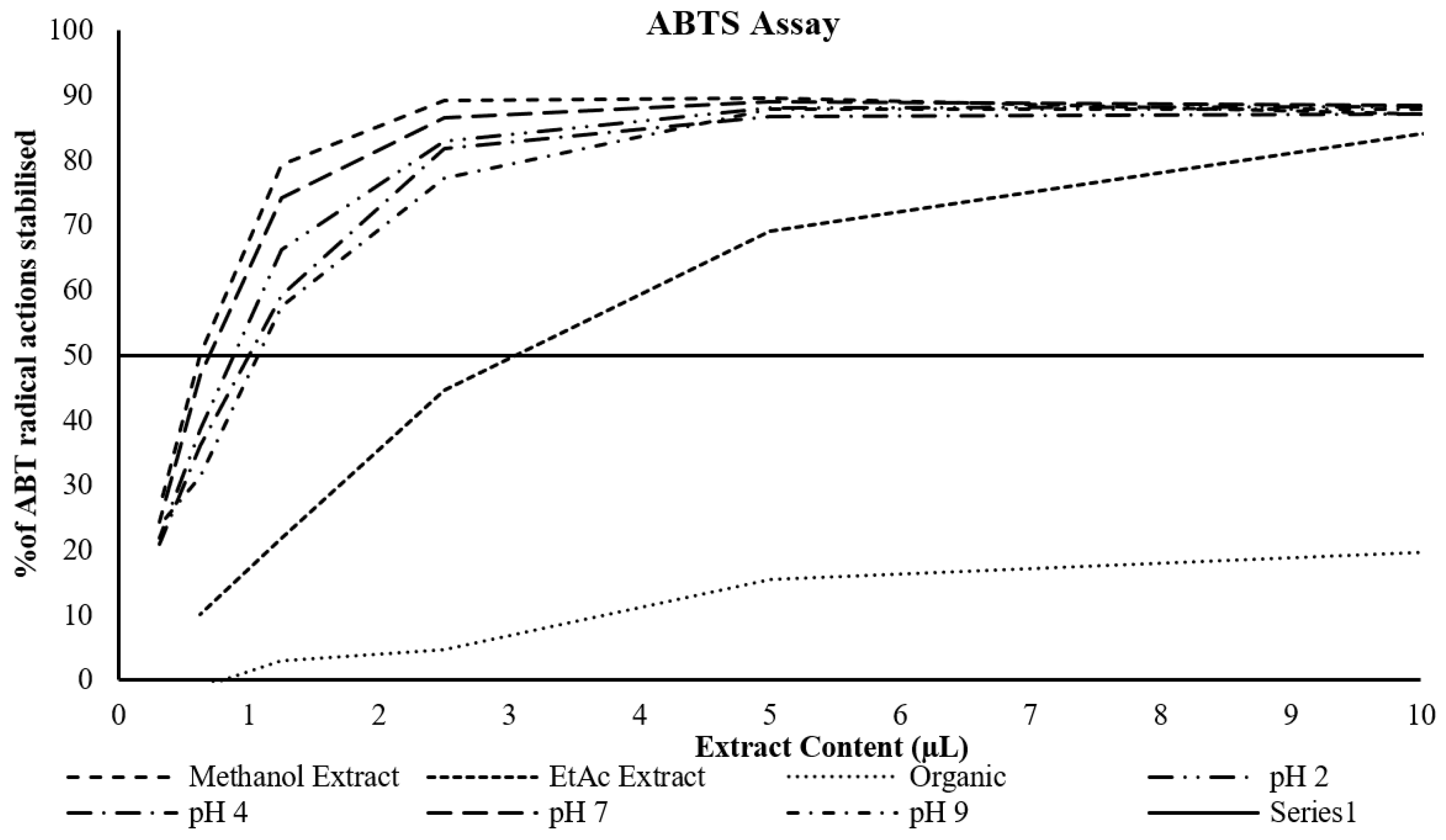
Figure 3.
Chromatogram of Perilla phenolic extract obtained using methanol and different hydrolysis conditions observed at 280 nm.
Figure 3.
Chromatogram of Perilla phenolic extract obtained using methanol and different hydrolysis conditions observed at 280 nm.
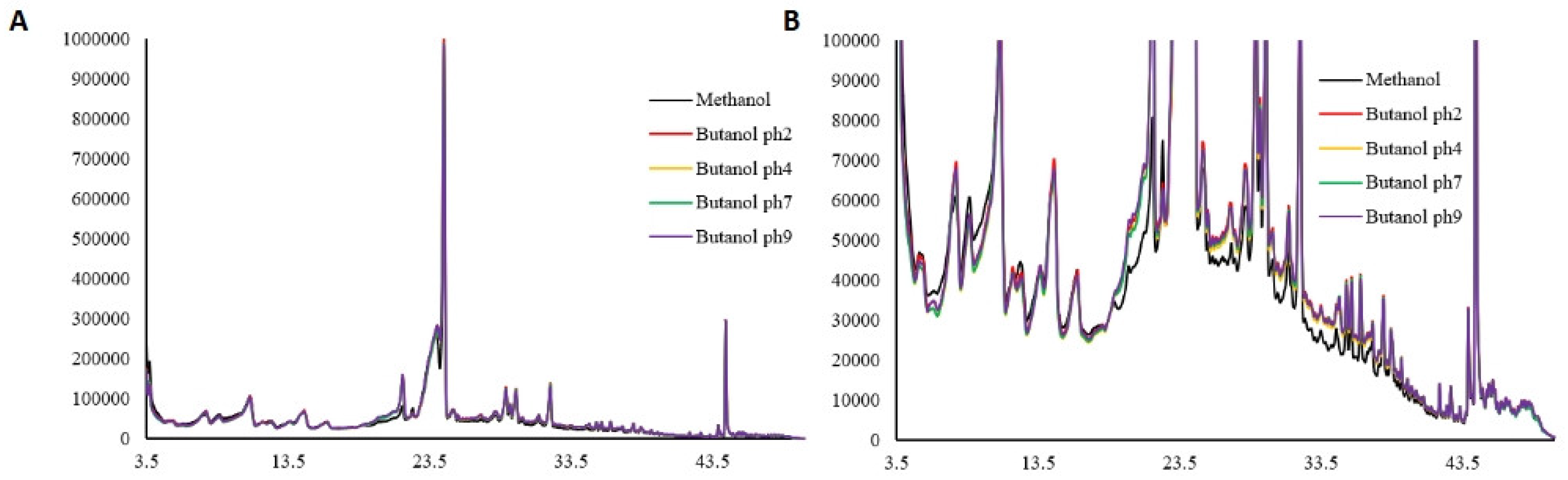
Figure 4.
Chromatogram of Perilla phenolic extract obtained using methanol and different hydrolysis conditions observed at 320 nm.
Figure 4.
Chromatogram of Perilla phenolic extract obtained using methanol and different hydrolysis conditions observed at 320 nm.
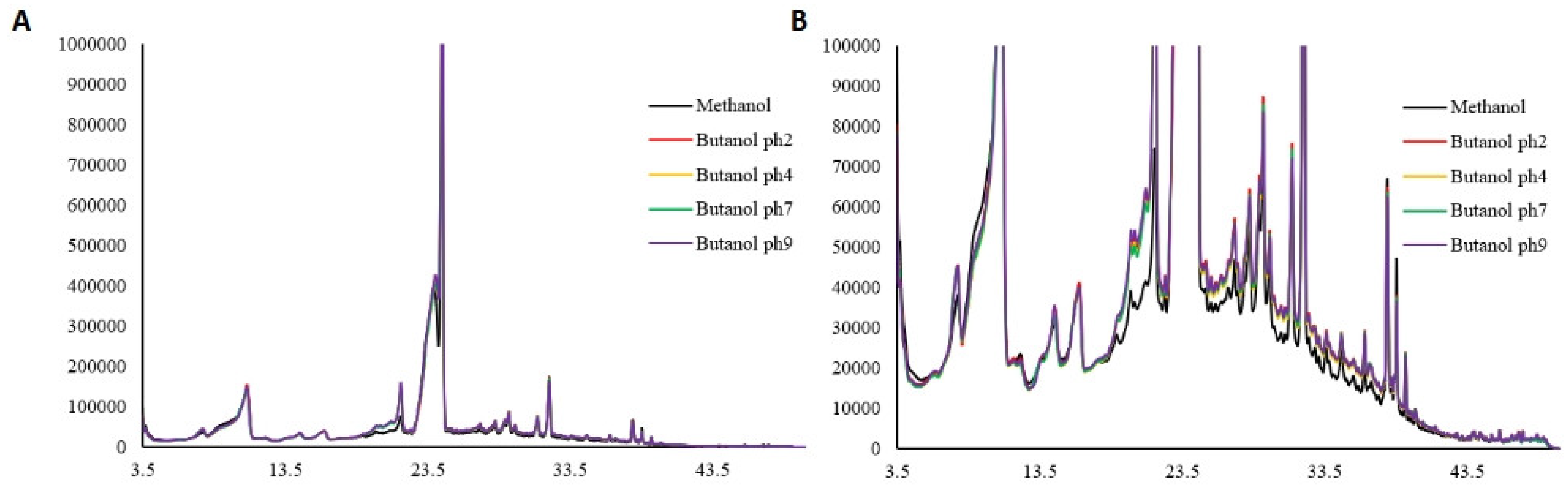
Figure 5.
Cell imaging following incubation with the different Perilla extracts (Mag x100; scale bar 50 μm).
Figure 5.
Cell imaging following incubation with the different Perilla extracts (Mag x100; scale bar 50 μm).
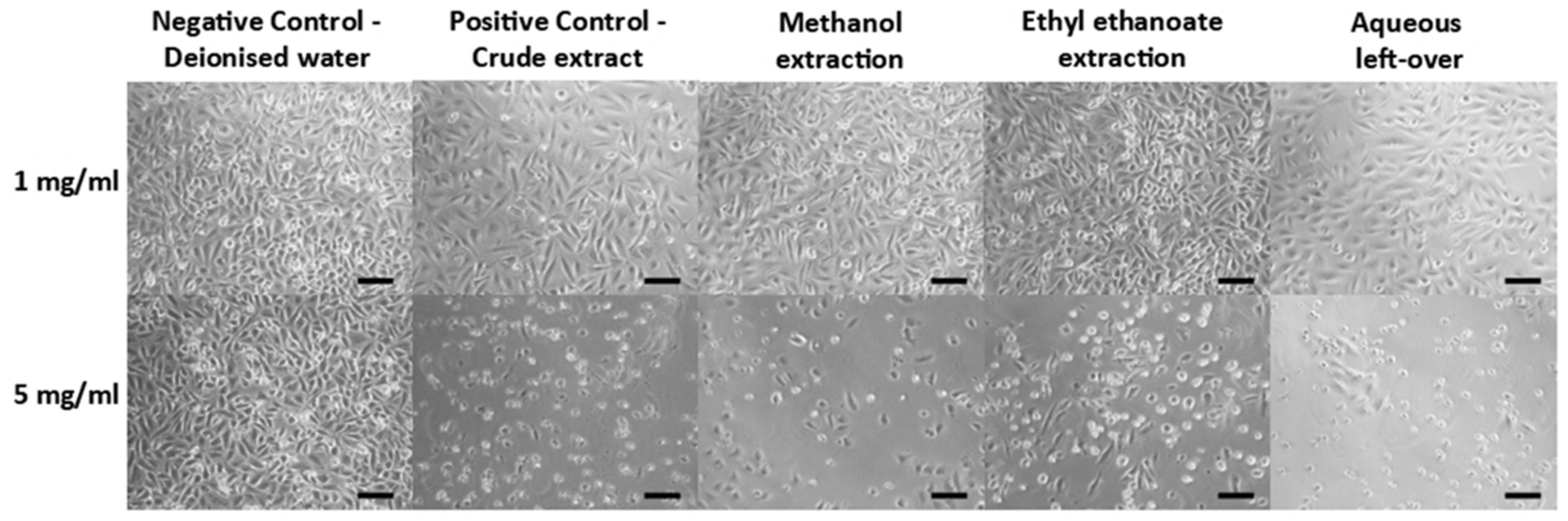
Figure 6.
Cell viability assay following incubation with the different Perilla extracts.
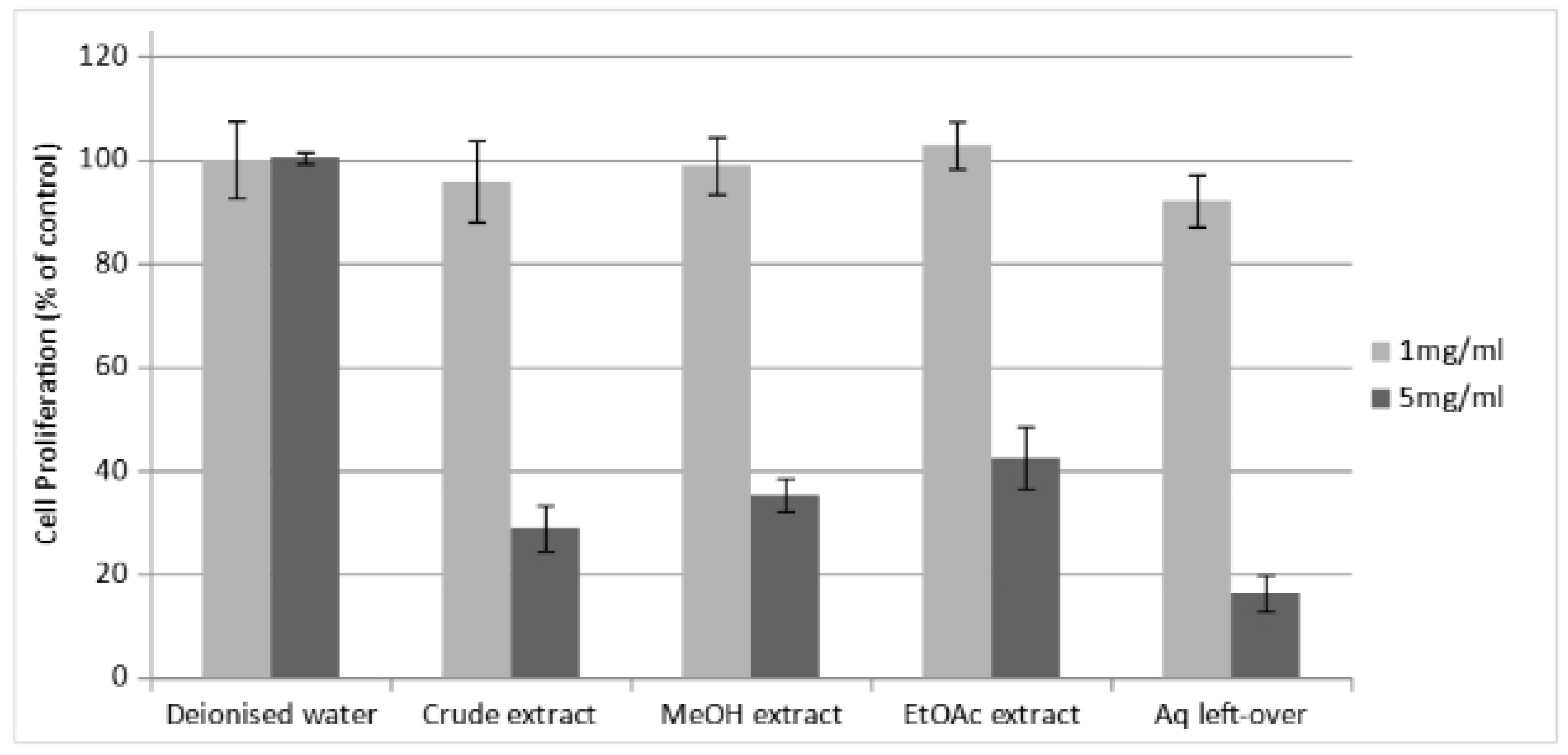
Figure 7.
Dual luciferase assay for the different Perilla extracts using the human NQO1 ARE, performed at 24, 48 and 72 hours.
Figure 7.
Dual luciferase assay for the different Perilla extracts using the human NQO1 ARE, performed at 24, 48 and 72 hours.
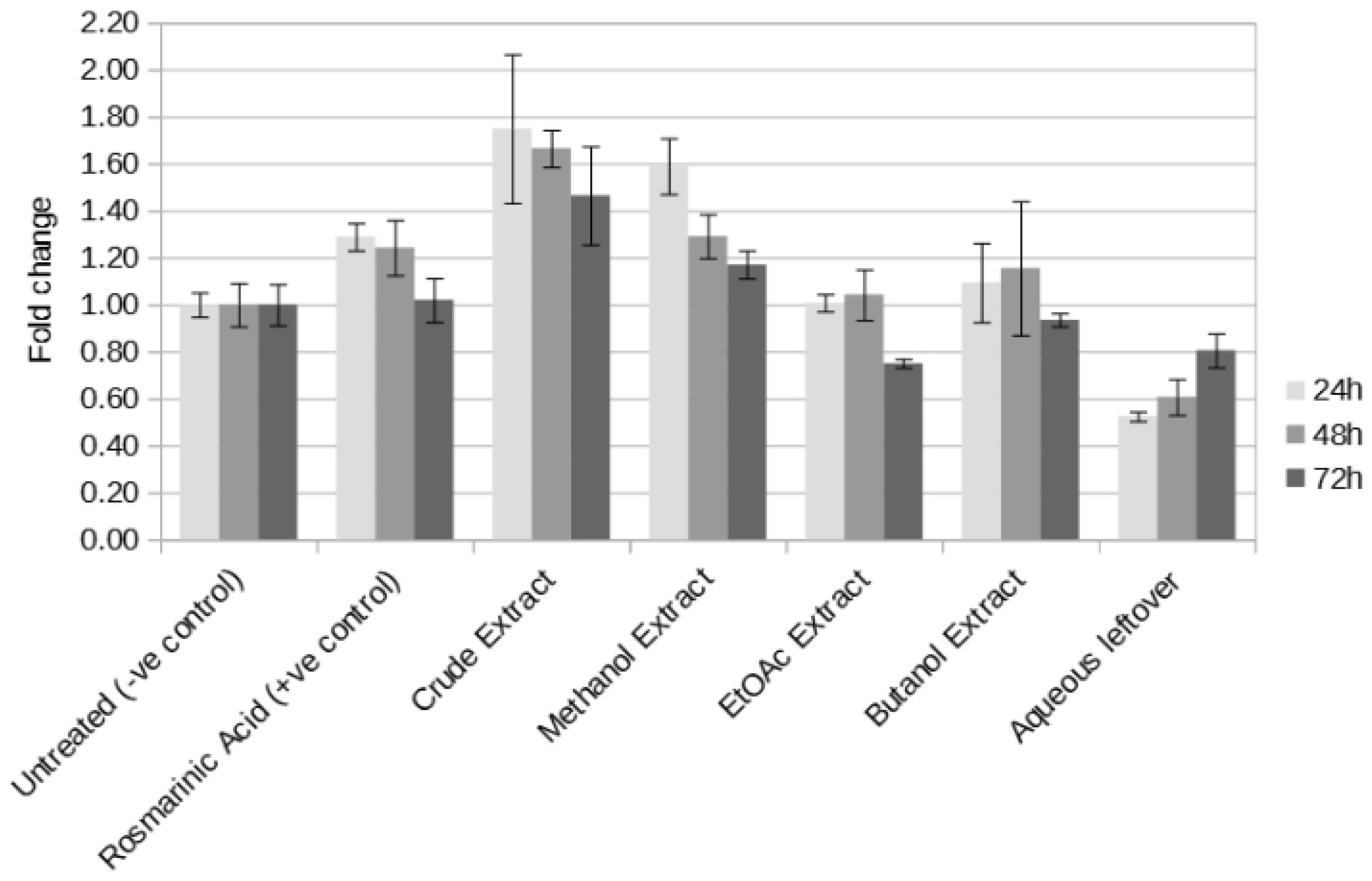
Figure 8.
Western blots for HSP70, HSP27 and Actin (as loading control) at 0, 24, 48 and 72 h for the various Perilla extract fractions, where: Control = untreated, Crude = whole extract, MeOH = methanol fraction, EtOAc = ethyl-ethanoate fraction, BuOH = n-butanol fraction, Leftover = water-soluble remnant.
Figure 8.
Western blots for HSP70, HSP27 and Actin (as loading control) at 0, 24, 48 and 72 h for the various Perilla extract fractions, where: Control = untreated, Crude = whole extract, MeOH = methanol fraction, EtOAc = ethyl-ethanoate fraction, BuOH = n-butanol fraction, Leftover = water-soluble remnant.
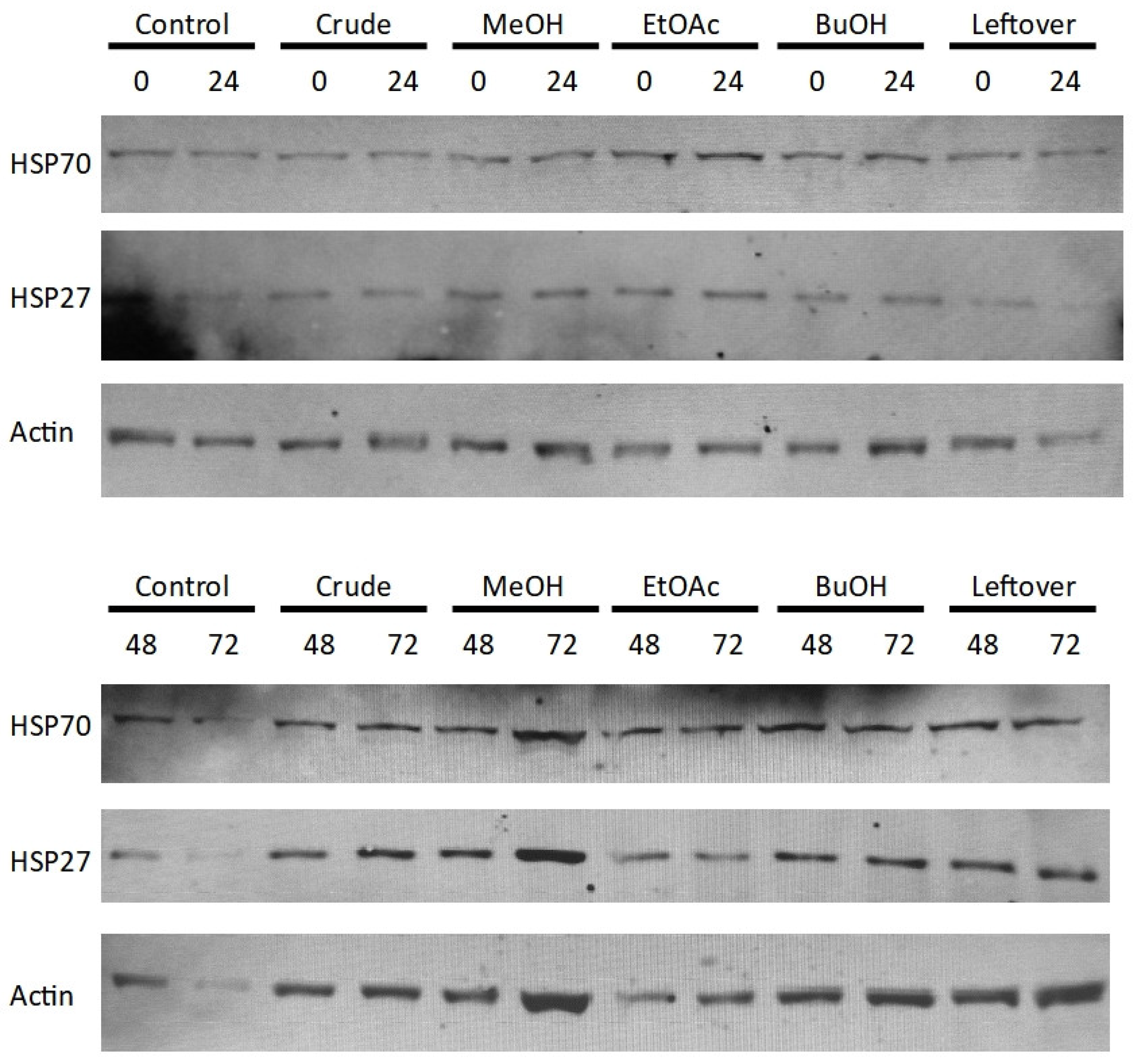
Figure 9.
Western blotting for mono-methyl lysine, tri-methyl lysine and HSP90 (as loading control) after 24 hours exposure to increasing concentrations (25, 50, 100 and 1,000 μg/ml) of the crude Perilla extract.
Figure 9.
Western blotting for mono-methyl lysine, tri-methyl lysine and HSP90 (as loading control) after 24 hours exposure to increasing concentrations (25, 50, 100 and 1,000 μg/ml) of the crude Perilla extract.


Table 1.
Total phenolic content expressed mg/L of caffeic acid equivalents, total flavonoid content expressed as mg/L of catechin equivalents and total orto-diphenolic content expressed as mg/L of pyrocatechol equivalents.
Table 1.
Total phenolic content expressed mg/L of caffeic acid equivalents, total flavonoid content expressed as mg/L of catechin equivalents and total orto-diphenolic content expressed as mg/L of pyrocatechol equivalents.
| |
TPC (mg/L CAE) | TFC (mg/L CatE) | TdPC (mg/L PyE) |
|---|---|---|---|
| Methanol Extract 1 | 1705.43 ± 144.9 | 1199.43 ±26.5 | 1401.95 ± 33.2 |
| Methanol Extract 2 | 1865.75 ± 266.7 | 1240.86 ± 13.8 | 1580.15 ± 76.1 |
| Ethyl Acetate Extract 1 | 321.83 ± 37.1 | 208.00 ± 5.2 | 247.33 ± 9.9 |
| Ethyl Acetate Extract 2 | 371.44 ± 6.1 | 304.19 ± 14.5 | 340.41 ± 4.9 |
| Ether Extract 1 | 46.36 ± 8.7 | 77.52 ± 6.4 | 6.31 ± 5.4 |
| Ether Extract 2 | 55.96 ± 0.5 | 87.05 ± 22.4 | 17.85 ± 2.0 |
| Butanol Extract pH 2 | 1267.33 ± 224.3 | 1253.24 ± 15.9 | 1496.05 ± 134.9 |
| Butanol Extract pH 4 | 1302.89 ± 42.6 | 1278.48 ± 52.5 | 1525.15 ± 45.6 |
| Butanol Extract pH 7 | 1472.10 ± 94.2 | 1414.67 ± 38.3 | 1747.85 ± 63.7 |
| Butanol Extract pH 9 | 1289.27 ± 61.9 | 987.52 ± 51.0 | 1356.82 ± 106.4 |
Table 2.
Calculated effective concentration of different extracts against DPPH and ABTS radicals.
| |
EC50 DPPH· | EC50 ABTS·+ |
|---|---|---|
| Methanol Extract 1 | 2.98 ± 0.0 | 0.75 ± 0.0 |
| Ethyl Acetate Extract 1 | > 10 | 3.24 ± 0.2 |
| Ether Extract 1 | >10 | >10 |
| Butanol Extract pH2 | 4.23 ± 0.1 | 1.08 ± 0.1 |
| Butanol Extract pH4 | 4.28 ± 0.1 | 1.19 ± 0.1 |
| Butanol Extract pH7 | 4.22 ± 0.1 | 0.86 ± 0.1 |
| Butanol Extract pH9 | 4.25 ± 0.8 | 1.27 ± 0.2 |
Table 3.
Metal cation Cu2+ and Fe3+ reduction activity of the various extracts using different solvents expressed as mg/L of caffeic acid equivalents for CUPRAC and mg/L of ascorbic acid equivalents for FRAP.
Table 3.
Metal cation Cu2+ and Fe3+ reduction activity of the various extracts using different solvents expressed as mg/L of caffeic acid equivalents for CUPRAC and mg/L of ascorbic acid equivalents for FRAP.
| |
CUPRAC (mg/L CAE) | FRAP (mg/L AcE) |
|---|---|---|
| Methanol Extract 1 | 557.96 ± 59.4 | 1892.22 ± 117.1 |
| Methanol Extract 2 | 514.44 ± 49.4 | 2147.78 ± 19.2 |
| Ethyl Acetate Extract 1 | 109.63 ± 15.2 | 747.78 ± 96.2 |
| Ethyl Acetate Extract 2 | 193.15 ± 7.3 | 847.78 ± 50.9 |
| Organic Extract 1 | 54.63 ± 6.1 | 292.22 ± 69.4 |
| Organic Extract 2 | 77.96 ± 4.5 | 281.11 ± 38.5 |
| Butanol Extract pH 2 | 438.33 ± 24.6 | 814.44 ± 245.7 |
| Butanol Extract pH 4 | 475.19 ± 60.0 | 381.11 ± 279.5 |
| Butanol Extract pH 7 | 512.59 ± 18.1 | 1770.00 ± 100.0 |
| Butanol Extract pH 9 | 487.96 ± 54.8 | 1792.22 ± 77.0 |
Table 4.
The % of Nitric oxide and hydroxyl radicals scavenging activity of the various extracts using different solvents.
Table 4.
The % of Nitric oxide and hydroxyl radicals scavenging activity of the various extracts using different solvents.
| |
% NOS | % OH |
|---|---|---|
| Methanol Extract 1 | 76.18 ± 5.1 | 72.95 ± 1.5 |
| Methanol Extract 2 | 71.18 ± 3.2 | 67.32 ± 2.0 |
| Ethyl Acetate Extract 1 | 75.00 ± 0.9 | 27.83 ± 1.0 |
| Ethyl Acetate Extract 2 | 72.65 ± 1.2 | 34.23 ± 2.1 |
| Organic Extract 1 | 59.47 ± 0.3 | 12.87 ± 3.7 |
| Organic Extract 2 | 58.74 ± 0.5 | 12.33 ± 0.4 |
| Butanol Extract pH 2 | 80.19 ± 0.0 | 55.00 ± 0.9 |
| Butanol Extract pH 4 | 78.62 ± 1.0 | 56.73 ± 0.5 |
| Butanol Extract pH 7 | 71.91 ± 12.1 | 56.79 ± 1.7 |
| Butanol Extract pH 9 | 84.80 ± 0.7 | 50.75 ± 0.6 |
Table 5.
Quantification of different phenolic compounds identified in the Perilla extract.
| Chemical Compound | Rt | Formula | R2 | LOD | LOQ |
|---|---|---|---|---|---|
| Caffeic Acid | 10.81 | y = 106920x + 179396 | 0.9846 | 0.088 | 0.29 |
| Syringic | 12.36 | y = 67460x - 355785 | 0.9919 | 0.050 | 0.17 |
| Vanillin | 14.59 | y = 108318x + 38533 | 0.9957 | 0.054 | 0.18 |
| p-coumaric acid | 16.24 | y = 118736x - 174419 | 0.9997 | 0.034 | 0.11 |
| Ferulic acid | 18.90 | y = 73150x - 84158 | 0.9999 | 0.048 | 0.16 |
| Ellagic Acid | 20.62 | y = 48178x + 25528 | 0.9968 | 0.041 | 0.14 |
| Rosmarinic Acid | 23.81 | y = 29517x + 164232 | 0.9946 | 0.055 | 0.18 |
| Trans-Cinnamic | 28.55 | y = 50153x + 36860 | 0.9998 | 0.091 | 0.30 |
| 3,4,5,7 tetrahydroxyflavone | 29.17 | y = 57138x + 68835 | 0.9954 | 0.041 | 0.14 |
| Quercetin | 29.51 | y = 102718x + 116121 | 0.9994 | 0.050 | 0.17 |
| 4′,5,7-trihydroxyisoflavone | 31.03 | y = 77186x + 43243 | 0.9998 | 0.077 | 0.26 |
| 5,7-dihydroxy flavone | 37.57 | y = 170069x - 714585 | 0.9833 | 0.172 | 0.57 |
| Rutin | 38.55 | y = 194800x + 49409 | 0.9999 | 0.077 | 0.26 |
Table 6.
Chemical comparison of hydrophilic and lipophilic fractions of the Perilla extract.
| Methanol Extract | Ether Extract |
|
|---|---|---|
| Caffeic Acid | 39.45±2.61 | 0.20±0.01 |
| Syringic | 7.13±0.59 | ND |
| Vanillin | 14.85±0.32 | 0.17±0.00 |
| p-coumaric acid | 4.87±0.18 | 0.03±0.00 |
| Ferulic acid | 3.07±0.04 | ND |
| Ellagic Acid | 23.87±0.27 | 0.33±0.01 |
| Rosmarinic Acid | 786.88±1.51 | 4.60±0.04 |
| Trans-Cinnamic | 34.02±0.29 | 0.58±0.17 |
| 3,4,5,7-tetrahydroxyflavone | 10.20±0.20 | 0.10±0.04 |
| Quercetin | 7.84±0.06 | 2.69±4.50 |
| 4′,5,7-trihydroxyisoflavone | 8.69±0.08 | 0.09±0.01 |
| 5,7-dihydroxyflavone | 1.09±0.06 | 1.17±0.02 |
| Rutin | 1.29±0.03 | 0.89±0.00 |
| Methyl 3,4,5-trihydroxybenzoate1 | 8.04±0.49 | ND |
| Tryptophanol1 | 11.13±0.17 | 0.02±0.00 |
| 4′,5,7-trihydroxyflavone2 | 18.03±0.02 | 0.26±0.20 |
| Kaempferol2 | 0.40±0.02 | ND |
| Pinocembrin2 | 0.56±0.02 | 0.06±0.00 |
1 Quantified and expressed in terms of caffeic acid equivalent. 2 Quantified and expressed in terms of 5,7-dihydroxyflavone equivalent.
Table 7.
Chemical comparison of butanol extracts of Perilla subjected to different hydrolysis conditions.
Table 7.
Chemical comparison of butanol extracts of Perilla subjected to different hydrolysis conditions.
| Compound | Butanol Extract pH 2 | Butanol Extract pH 4 | Butanol Extract pH 7 | Butanol Extract pH 9 |
|---|---|---|---|---|
| Caffeic acid | 38.61±0.39 | 37.85±0.31 | 39.70±0.69 | 38.62±0.00 |
| Syringic acid | 5.75±0.01 | 5.81±0.02 | 6.30±0.35 | 6.26±0.02 |
| Vanillin | 14.90±0.16 | 14.65±0.14 | 15.55±0.28 | 14.97±0.10 |
| p-coumaric acid | 5.23±0.11 | 5.04±0.07 | 5.37±0.12 | 5.03±0.06 |
| Ferulic acid | 5.32±0.07 | 3.92±1.70 | 1.72±0.11 | 1.29±0.81 |
| Ellagic acid | 20.71±15.92 | 30.03±6.28 | 17.47±1.67 | 19.55±0.07 |
| Rosmarinic acid | 905.51±3.21 | 859.48±51.51 | 523.96±7.18 | 482.18±2.17 |
| Trans-Cinnamic acid | 40.32±1.33 | 33.29±10. 47 | 46.59±0.49 | 26.46±0.12 |
| 3,4,5,7- tetrahydroxyflavone | 12.89±0.59 | 10.35±3.65 | 14.90±0.05 | 7.64±0.02 |
| Quercetin | 8.51±0.35 | 7.12±1.97 | 9.64±0.03 | 5.80±0.03 |
| 4′,5,7-trihydroxyisoflavone | 14.57±1.96 | 9.83±7.98 | 19.01±0.12 | 4.09±0.07 |
| 5,7di hydroxy flavone | 1.01±0.01 | 0.98±0.04 | 1.06±0.01 | 1.00±0.01 |
| Rutin | 1.07±0.00 | 1.06±0.01 | 1.09±0.01 | 1.06±0.11 |
| Methyl 3,4,5-trihydroxybenzoate1 | 8.29±0.01 | 8.14±0.00 | 8.35±0.18 | 8.32±0.01 |
| Tryptophanol1 | 9.36±1.03 | 7.31±3.30 | 1.24±0.59 | 1.06±0.13 |
| 4′,5,7-trihydroxyflavone2 | 20.66±1.16 | 18.20±3.88 | 23.39±0.15 | 15.54±0.09 |
| Kaempferol2 | 1.98±0.66 | 1.27±1.63 | 3.18±0.05 | 0.12±0.00 |
| Pinocembrin2 | 0.48±0.00 | 0.47±0.00 | 0.49±0.00 | 0.47±0.01 |
|
1 Quantified and expressed in terms of caffeic acid equivalent 2 Quantified and expressed in terms of 5,7-dihydroxyflavone equivalent | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



