
Submitted:
05 September 2024
Posted:
06 September 2024
You are already at the latest version
Abstract
Dual-fraction proteomics reveals a novel class of proteins impacted by nanoparticle exposure. Background: Nanoparticles (NPs) interact with cellular proteomes, altering biological processes. Understanding these interactions requires comprehensive analyses beyond solely characterizing the NP corona. Methods: We utilized a dual-fraction mass spectrometry (MS) approach analyzing both NP-bound and unbound proteins in Saccharomyces cerevisiae sp. protein extracts exposed to silica nanoparticles (SiNPs). We identified unique protein signatures for each fraction and quantified protein abundance changes using spectral counts. Results: Strong correlations were observed between protein profiles in each fraction and non-exposed controls, while minimal correlation existed between the fractions themselves. Linear models demonstrated equal contributions from both fractions in predicting control sample abundance. Combining both fractions revealed a larger proteomic response to SiNP exposure compared to single-fraction analysis. We identified 302/56 proteins bound/unbound to SiNPs and an additional 196 "impacted" proteins demonstrably affected by SiNPs. Conclusion: This dual-fraction MS approach provides a more comprehensive understanding of nanoparticle interactions with cellular proteomes. It reveals a novel class of "impacted" proteins, potentially undergoing conformational changes or aggregation due to NP exposure. Further research is needed to elucidate their biological functions and the mechanisms underlying their impact.
Keywords:
Silica Nanoparticles
; Protein Extracts
; Proteomics
; Mass Spectrometry
; Protein-Nanoparticle Interactions
; Corona
1. Introduction
Nanomaterials (NMs) have become ubiquitous in manufactured products across a diverse range of industries, including food processing, pharmaceuticals, cosmetics, and electronics. Among these NMs, silica-based nanoparticles (SiNPs) exhibit the highest prevalence due to their versatile properties and wide applicability [1]. SiNPs find use in various sectors, such as: food packaging and additives, materials production (glass, cement, fiberglass, concrete), optics and fiber technologies, textile manufacturing, agricultural fungicides, and biomedical/pharmaceutical applications (biological carriers, excipients). This widespread utilization has sparked growing societal concern regarding potential health and environmental consequences [2,3,4], prompting extensive research into the toxicity of NMs. Investigation at the molecular level reveals crucial insights into SiNP interactions with biomolecules. Analyzing protein-SiNP interactions allows for elucidation of affected biological mechanisms. For instance, studies have demonstrated that SiNPs can specifically target RNA binding proteins [5]. Furthermore, nanoscale analysis elucidates the influence of SiNP size on corona formation and protein conformational changes [6,7], highlighting the importance of considering both size and surface properties when assessing potential risks or beneficial effects associated with SiNPs.
Understanding the intricate interactions between proteins and nanoparticles (NPs) is crucial for assessing their potential impact on biological systems and guiding the development of safe and effective nanotechnology applications. Numerous methods have been established to elucidate these interactions, with techniques such as fluorescence flow cytometry offering insights into binding events with purified proteins. However, complex protein samples derived from protein cellular extracts, such as those obtained from Saccharomyces cerevisiae sp., pose a greater challenge.
In this study, we employ a dedicated protocol detailed in Mathé C. et al. [8] to identify protein-NP interactions within these complex matrices. Briefly, the protocol involves incubating protein extracts with NPs of interest, followed by centrifugation and drying steps to retrieve aggregates of proteins and NPs (hereafter denote “pellet”). Proteins are subsequently desorbed from these complexes for identification using high-throughput label-free nano-liquid chromatography coupled with tandem mass spectrometry (nano-LC MS/MS). Bioinformatics analysis and database harvesting enable protein identification based on spectral matching.
Differential analysis, comparing the identified proteins in exposed samples to those in control samples lacking NP exposure [5], allows us to pinpoint significantly adsorbed proteins. Notably, most existing studies focusing on NP toxicity primarily analyze the pellet fraction, which represents proteins physically bound to NPs. However, this approach overlooks potential impacts on unbound proteins that may undergo structural alterations, aggregation, or other functional changes upon NP exposure. Besides, certain NP types, such as carbon-based NPs, exhibit exceptionally strong binding affinities for proteins [9], rendering traditional desorption methods ineffective. Similarly, smaller plastic particles can aggregate with proteins in solution, preventing their retrieval through centrifugation. In these instances, the fraction containing unbound proteins (hereafter denote “supernatant”), becomes crucial for a comprehensive understanding of NP-induced effects [10]. Noteworthy, in most large-scale proteins NM interactions studies usually only one fraction, the pellet or the supernatant, is analyzed.
To our knowledge, no previous studies have implemented a dual analysis approach encompassing both pellet and supernatant fractions within the context of protein-NP interactions. This study aims to shed light on this novel approach by addressing key questions regarding the correlation and complementarity of these fractions. We hypothesize that analyzing both fractions provides a more complete picture of NP-induced protein alterations, encompassing both direct binding events and indirect effects on the proteome. High-throughput mass spectrometry is a sampling procedure and the number of distinct proteins mapped in a solution is correlated with: the proteins extract concentration, dynamic and complexity; the acquisition duration. Thus, proteins identified in the dual fractions are representative samples of the respective population of proteins: bounded or not bounded to the NP. This sampling effect rises many interrogations. In particular, how these fractions correlate and complement each other? Does the combined pellet and supernatant fractions are representative of the control population (i.e., not exposed to the NP)? What can we learn on the pellet (resp. supernatant) fraction composition by analyzing the dual supernatant (resp. pellet) fraction.
To investigate this hypothesis, we exposed Saccharomyces cerevisiae sp. protein extracts to SiNPs. We identified adsorbed proteins (pellet) and unbound proteins (supernatant) using nano-LC MS/MS, with spectral count (SC) serving as a proxy for relative protein concentration. Furthermore, we included control samples devoid of NP exposure for differential analysis. In this research, we present a comparison between biological protein concentrations and their corresponding SC ratios obtained from mass spectrometry. Subsequently, we employ various linear models to demonstrate the complementarity of pellet and supernatant fractions. Finally, we utilize differential analysis to highlight the benefits of our dual approach in identifying novel protein subsets impacted by SiNP exposure.
2. Materials and Methods
Sample Preparation
Yeast protein extracts (YPE) were prepared from the Saccharomyces cerevisiae sp. strain S288C (Matα SUC2 mal mel gal2 CUP1), as described in [10]. Silica NP (13 nm radius, LUDOX® TM-50, Sigma-Aldrich) were dialyzed using a membrane with a 3.5 kDa cut-off against Milli-Q water at 4 °C. YPE (0.6 g.L-1) was incubated with silica NP (1 g.L-1) in phosphate buffer (100 mM, pH7) using a ThermoMixer ® (Eppendorf, Hamburg, Germany) at 20 °C during 3 h (cycles of 15 sec at 800 rpm followed by 285 sec at rest). The YPE concentration was chosen at the start of the adsorption isotherm plateau (see Figure 1), to have the minimum protein quantity in saturation conditions.
A centrifugation (20 °C, 20,000 rpm, 10 min) allowed the separation of free proteins in the supernatant and adsorbed proteins in the pellet. 500 µL of supernatant were recovered for the proteomic analysis. Then, for the pellet, two washings were realized as following: the pellet (40 µL) was resuspended in 1.5 mL of phosphate buffer, centrifuged (20 °C, 20,000 rpm, 5 min) and the supernatant (1.46 mL) was removed. After the two washings, a desorption protocol was performed: the 40 µL of pellet were resuspended in phosphate buffer and sodium dodecyl sulfate (SDS UltraPure™ 10 %, Invitrogen). The 750 µL final solution contains 1% of SDS, concentration often used to desorb proteins [6]. Then, mixing during 1 h at 20 °C was realized using a Thermomixer ® (cycles of 15 sec at 800 rpm followed by 105 sec at rest). A last centrifugation (20 °C, 20,000 rpm, 10 min) was performed and the 100 µL of pellet were recovered for the proteomic analysis. The protein concentration was determined using the peptide bond absorbance at 205 nm with an absorption coefficient of 31 L.g-1.cm-1 [11].
Since SDS impacts the determination of protein concentration, a calibration curve was performed to correct raw concentration values (see Figure 2).
Proteomics Analysis
Tubes containing 60% of their maximum volume of YPE in a phosphate buffer (100 mM) were mixed during 24 hours at 3 rpm, 6 °C. Samples concentrations were within the recommended range for optimal detection and quantification. Proteomic experiments were performed at the Proteomic Analysis Platform of Paris Sud-Ouest (PAPPSO). YPE samples were deposited on SDS-PAGE gels and proteins separated using short migration time. A classic protein digestion protocol was applied (described in Henry C. & al. [12]). Samples were analyzed by LC–MS/MS on an Orbitrap Fusion Lumos Tibrid (Thermo Fisher Scientific, MA, United-States) mass spectrometer. The protein identification was performed using the Saccharomyces cerevisiae sp. strain S288c protein database (41,6750 entries, version 2020).
Spectral Count Normalisation
Spectral counts (SC) were calculated for each protein detected by MS. Then these raw SC values are normalized in two steps. Firstly, for each fraction we adjust SC of the three replicates to a constant average ratio of 40 SC per mg.L-1. Secondly, we perform a global normalization to equals the total SC of pellet and supernatant sum to the control total SC. Normalized SC for each replicate and averaged are shown on Table 1.
Additionally, proteins identify with less than three SC were filtered out from the final datasets.
Statistical Analysis
All statistical analysis were performed using the R language [13] and the standard packages for the correlation analysis, the statistical tests (Pearson and Spearman) and the fitting of linear models. Linear mixed effects models were adjusted using the “lme4” R package [14]. Generalized linear models used for the bayesian protein differential analysis (detailed in Marichal L. & al. [6]) were calculated using the following packages: “rstanarm” [15] to fit generalized linear models using a gaussian link function; “bridgesampling” [16] to compute the log marginal likelihoods; the “BayesFactor” [17] to calculate the Bayes Factors and posterior probabilities from marginal likelihoods.
3. Results
Biological Fractions Complementarity
Protein concentration was determined for each technical replicate in all samples using optical density measurements. Table 2 presents the average protein concentrations for each fraction. As the pellet and supernatant fractions are derived from the control sample, we anticipate their combined protein concentration to closely approximate that of the control. Our data demonstrate a slightly higher total protein concentration for the combined pellet and supernatant fractions (0.73 g.L-1) compared to the control (0.63 g.L-1). Remarkably, both the supernatant and pellet fractions exhibited protein concentrations approximately equal to (50%) or close to (65%) half of that observed in the control sample.
Spectral-Counts Distributions Complementarity
Following protein identification via mass spectrometry, raw spectral count (SC) for each protein was determined as the sum of its corresponding spectra. To account for experimental and technical biases (e.g., sample preparation and mass spectrometry acquisition variability), raw SC values were normalized using a two-step process described in detail within the Methods section. Indeed, raw SC data exhibited variations in average spectral counts per mg.L-1 between fractions: ~40 SC per mg.L-1 in the pellet, ~23 SC per mg.L-1 in the supernatant, and ~20 SC per mg.L-1 in the control.
At the protein level, high correlations between technical replicates were observed across all three fractions using both Pearson (range [0.993-1.000]) and the non-parametric Spearman (range [0.902-1.000]) correlation coefficients.
Figure 3 presents a visualization of Pearson correlation coefficient values (represented by a red to green color scale). In particular, SC in the control exhibited stronger correlations with the supernatant (ρ = 0.878) compared to the pellet (ρ = 0.606). Conversely, a weak correlation was observed between the pellet and supernatant (ρ = 0.196). These findings underscore the complementary nature of information derived from both the pellet and supernatant fractions relative to the control.
Regression Analysis
To evaluate the complementary nature of the three fractions, control, pellet, and supernatant, linear regression models were employed. The independent variable was defined as control spectral counts (SC), while the dependent variables were pellet and supernatant SC. The model equation is represented as:
Table 3 presents the fitted coefficients and regression performance metrics. The model demonstrates that both the pellet and supernatant contribute equally to the estimation of control SC, as indicated by the coefficients β1 = 1.07 and β2 = 1.05, respectively. The model exhibits strong adjustment, with an adjusted R2 of 0.98 and a Root Mean Square Error (RMSE) of 8.29. Student t-tests yielded highly significant p-values (<0.001) for both the pellet and supernatant coefficients, falling within narrow confidence intervals (see Table 3). Noteworthy, the intercept coefficient is nearly zero and deemed irrelevant to the model based on a Student t-test p-value of 0.83.
To directly compare the “reconstructed” protein subset derived from the combined pellet and supernatant data with the original control set, we summed the averaged spectral counts for each identified protein across the respective fractions. A linear regression model was then fitted to this new dataset, excluding the intercept term (α = 0) as previously determined to be irrelevant. The model is defined as:
The fitted linear coefficient (β) for this model is 1.05 ± 0.01, with an adjusted R2 of 0.98. Figure 4 visually represents the data, plotting proteins as dots colored according to their majority fraction and overlaying the fitted linear regression line. The strong collinearity between the detected and control sets is evident, with a similar dispersion across each fraction observed for proteins exhibiting the highest SC values.
To assess potential replicate effects on spectral count relationships, linear mixed effects models were employed. The model structure is defined as:
where “(1 | replica)” in equation 3 indicates a random intercept for each replicate. Both pellet and supernatant fitted coefficients remained similar to the initial model, with β1 = 1.07 and β2 = 1.01, respectively. The model exhibited strong adequacy (conditional R2 = 0.93 and RMSE = 15.43). Furthermore, an analysis of variance comparing the models with and without replicate mixed effects revealed no significant influence of this parameter (p-value < 10-16). Based on these findings, we conclude that random effects attributable to sample replicates are absent.
Differential Analysis between Fractions
To identify enriched or depleted proteins between the pellet and supernatant fractions relative to the control, a differential analysis was conducted (see Methods section). Prior to this analysis, normalization was performed to equalize the average total spectral counts (SC) across each fraction. Proteins with an average SC below five were subsequently filtered, mitigating potential biases arising from differences in overall protein concentration between samples. Figure 5 presents a Venn diagram illustrating the overlap of proteins identified by MS across the three fractions. Notably, 98% of proteins identified in the control were also detected in either the supernatant or pellet fraction. The pellet exhibited the highest number of identified proteins (1,077), including 301 unique to this fraction.
To determine if differences in spectral counts (SC) between proteins across different conditions are statistically significant, a Bayesian approach was employed (see Methods section). The Bayes factor (BF) served as a threshold for identifying enriched or depleted proteins. This approach effectively distinguished proteins adsorbed onto nanoparticle (NM) surfaces (enriched in the pellet fraction) from those unaffected by the NMs (enriched in the supernatant fraction), as detailed in Table 4.
Table 5 presents a cross analysis between proteins enriched in the pellet and those depleted in the supernatant for the threshold BF ≥ 3.
Table 5 highlights distinct protein partitioning patterns based on Bayes Factor (BF) thresholds. At a substantial threshold BF ≥ 3, over 69% (i.e., 124/179) of pellet-enriched proteins and detected in the supernatant are also depleted in this fraction, indicating robust adsorption onto nanoparticles (NPs). Conversely, 59% (i.e., (302-179)/302) of pellet-enriched proteins at this threshold are absent from the supernatant, suggesting highly specific NP binding that results in undetectable levels within the supernatant due to limited sample sensitivity. Notably, no proteins exhibited simultaneous depletion in both fractions (data not shown), indicating a low rate of false positives.
The analysis extends to non-adsorbed proteins present in the supernatant and their relationship with pellet-depleted proteins. Table 6 presents this cross analysis, demonstrating that at threshold BF ≥ 3, nearly all (over 88.9%) supernatant-enriched proteins and detected in the pellet are also depleted in this fraction, reinforcing a clear distinction between adsorbed and non-adsorbed populations. However, a significant disparity exists: 56 non-adsorbed proteins were detected in the supernatant at BF ≥ 3 compared to 302 adsorbed proteins in the pellet. This observation underscores the substantial difference in abundance between these protein groups.
To encompass the full spectrum of protein responses to nanoparticle exposure, we introduce a novel category “impacted proteins” encompassing those significantly altered in abundance compared to the control. This category extends beyond simple adsorption/non-adsorption distinctions. We define a protein P as “directly impacted” if it meets criteria i) P is detected in either the pellet or supernatant fraction; and ii) P is enriched in the pellet and detected but not depleted in the supernatant. Conversely, a protein P is stated “indirectly impacted” if it fulfills criteria i) and iii) P is depleted in the supernatant but not enriched in the pellet. The less specific set of “impacted” proteins is the union of the “directly impacted” and “indirectly impacted” proteins subsets.
Applying a BF ≥ 3 threshold (see Table 5), there is 179-124=55 directly impacted proteins (condition ii) and 265-124=141 indirectly impacted proteins (condition iii). This analysis reveals a total of 196 proteins impacted by the SiNPs exposure at this threshold. Furthermore, 178 (resp. 24) proteins were enriched and detected only in the pellet (resp. supernatant). These subsets identify proteins with a “very high” bound/unbound affinity for the SiNP surface. Table 7 summarizes the distribution of proteins across each category.
4. Discussion
Nanotoxicological investigations strive to elucidate the composition of the nanoparticle (NP) corona, providing crucial insights into the biological mechanisms affected by nanomaterials. Utilizing complex protein extracts enables a broader scale investigation of molecular mechanisms perturbed by nanomaterials, such as silica nanoparticles (SiNPs) in this study. While conventional molecular biology techniques are effective for analyzing individual or small sets of peptides, deciphering protein-NP interactions within complex matrices necessitates technologies capable of sampling larger set of proteins. Mass spectrometry (MS) has emerged as one of the preferred methods for high-throughput and large-scale proteomics analyses, proving particularly valuable in characterizing protein-NP interactions [8]. Most studies employing MS have primarily focused on identifying the subset of proteins shaping the NP corona [18,19]. Characterizing the corona offers valuable information for diverse applications, for example: Identifying specific proteins within the corona allows for targeted NP functionalization strategies aimed at modulating desired biological interactions. Analyzing the composition of the protein corona can help to decipher the specific molecular pathways impacted by NP exposure, shedding light on the mechanisms underlying potential toxicity or beneficial effects. Identifying adsorbed proteins on the NP surface requires the following steps: isolation of NP aggregates followed by desorption of bound proteins from the surface, identification and relative quantification (e.g., spectral counts) using MS. While this protocol effectively identifies adsorbed proteins, it overlooks unbound proteins remaining in solution which may exhibit altered fates, such as conformational changes or aggregation, upon NP exposure. This study demonstrates that a dual fractions approach, encompassing both NP aggregate-bound proteins and unbound proteins in solution, provides a more comprehensive understanding of the proteomic alterations induced by NP exposure in protein extracts.
Utilizing SiNPs and Saccharomyces cerevisiae sp. protein extracts, we observed strong correlations between protein abundance profiles in both fractions with non-exposed controls (Pearson correlation coefficient ρ ranging from 0.6 to 0.9). Notably, minimal correlation was observed between the two fractions themselves (ρ = 0.196), indicating distinct proteomic signatures for bound and unbound proteins. Linear models fitting both fractions demonstrated equal contributions to predicting protein abundance in the non-exposed control sample (linear coefficients ranging from 1.05 to 1.07, adjusted R2 = 0.98). This suggests that each fraction independently captures unique aspects of the proteomic response to SiNPs exposure. Due to inherent limitations of MS sampling, which can only detect a subset of the total expressed proteome, a dual-fraction approach provides a more comprehensive understanding of SiNPs-induced effects than single-fraction analysis (e.g., solely focusing on NP aggregates). We identified 1078 unique proteins in the NP aggregates fraction and 1322 (+22.6%) by analyzing both fractions combined. These results show that both fractions complement each other and are representative of the control population. Furthermore, these findings also demonstrates that in conditions where proteins cannot be desorbed from NP (e.g., carbon nanotubes [9]) or extracted in solution (e.g., plastic particles), analyzing the subset of proteins in solution is relevant to study the effects of exposure to these particular NPs [10].
Our results also highlight the utility of the opposite fraction (unbound to SiNPs) for identifying a novel proteins subset, those unbound but potentially impacted by SiNPs. Indeed, proteins enriched in one fraction are expected to be depleted in the other. Differential analysis between the supernatant (i.e., proteins in solution) and control fractions at a substantial Bayesian factor evidence level (BF ≥ 3) identified 265 depleted proteins (see Table S1), with only 124 also showing enrichment in the pellet fraction (i.e., NP aggregates). This discrepancy underscores the value of considering both bound and unbound protein populations for a comprehensive understanding of protein-nanoparticle interactions. Based on these observations, we propose a formal definition for the subset of “impacted” proteins in the context of dual-fraction proteomic analysis: a protein P, detected at least in one fraction, enriched in the pellet (resp. depleted in the supernatant) and detected but not depleted in the supernatant (resp. not detected or enriched in the pellet) is directly (resp. indirectly) impacted by the NP. This dual-fraction approach expands upon conventional analyses focusing solely on the pellet fraction, which identified 302 proteins (BF ≥ 3) adsorbed onto the SiNP surface. Notably, our dual-fraction analysis further reveals an additional 196 proteins impacted by SiNPs exposure and 56 proteins non-adsorbed onto the SiNP surface, highlighting the value of considering both bound and unbound protein populations for a comprehensive understanding of protein-nanoparticle interactions.
To the best of our knowledge, this is the first MS-based proteomic study, relying on a dual fractions approach, to analyze both bound and unbound protein populations from YPE exposed to SiNPs. Defining a novel category of “impacted” proteins based on their differential relative abundance in bound and unbound fractions offers a promising avenue for enriching our understanding of the biochemical and biophysical interactions involved. The “impacted” proteins subset warrants further investigation to elucidate its characteristics, still several hypotheses can be proposed: Directly interacting proteins may arise from transient binding events, exemplified by the Vroman effect [20], wherein proteins bind either directly to the NP surface (i.e., hard corona) or indirectly via the protein corona (i.e., soft corona) [8,21]; Indirectly impacted proteins could result from protein denaturation or aggregation, facilitated by interactions with plastic surfaces (e.g., tubes) and mechanical agitation [10]. To elucidate the fate of these impacted proteins, some key questions to work on include: Do they undergo conformational modifications? Are they aggregating within solution? While our results show that this approach is more sensitive and generate a more in-depth analysis of NP exposure, it does require a doubling of the quantities of biological material, sample preparation and MS acquisition times, and most probably total costs.
To enhance comparability between samples within our dual-fraction mass spectrometry approach, two key modifications were implemented in the MS analysis protocol: i) Maintaining non-normalized and unequal sample concentrations was critical. This allowed for a direct relationship between spectral count (SC) values and protein abundance within each sample; ii) Identical acquisition times were enforced for all samples, ensuring that each sample underwent the same level of sampling depth. These modifications aimed to achieve comparable SC per unit concentration across all samples. However, when sample concentrations fall below detection limits, a correction factor can be applied, followed by normalization, as exemplified with SiNPs (detailed in the Methods section). This approach introduces potential sampling biases. Oversampling may inflate SC for identified proteins while overlooking low-abundance proteins. Conversely, under-sampling risks suppressing true protein identifications. The implementation of semi-absolute quantification MS techniques [22], employing external standards such as UPS2, could potentially mitigate these biases and optimize the quantitation process.
This article demonstrates that a dual-fraction mass spectrometry approach facilitates a more comprehensive understanding of nanoparticle exposure at the molecular level. We introduce a novel protein subset, characterized by the absence of corona binding but demonstrable NP impact. In Saccharomyces cerevisiae sp. protein cellular extracts exposed to SiNPs, 196 proteins exhibit this characteristic. While a detailed investigation into the biological mechanisms and altered metabolic pathways associated with these impacted proteins is beyond the scope of this work, such an analysis would significantly enhance our understanding of SiNP exposure effects. Future research should explore the applicability of this method to other proteomes, particularly those characterized by larger size or greater protein translation dynamic range [23]. Additionally, optimization of the MS protocol may mitigate current sampling limitations and further improve method efficiency.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: Cross analysis of adsorbed proteins in the pellet and supernatant fractions; Table S2: Cross Analysis of non-adsorbed proteins in the pellet and supernatant fractions. Supplementary File ‘Yeast_SiO2_raw.xlsx’: Excel file containing raw spectral counts for the three replicates of the pellet, supernatant and control; Supplementary File ‘Yeast_SiO2_raw.RData’: R datafile containing a tibble data frame (tidyr R package) with raw spectral counts for the three replicates of the pellet, supernatant and control.
Author Contributions
Conceptualization, methodology and validation, Y.B, J.-P.R., S.C., S.P. and J.-C.A.; software and formal analysis, J.-C.A.; investigation, M.S., F.S. and J.-C.A.; proteomics resources, C.H.; data curation, M.S., F.S., S.C. and J.-C.A.; writing—original draft preparation, M.S. and J.-C.A.; writing—review and editing, M.S., Y.B, J.-P.R., S.C., S.P. and J.-C.A.; visualization, M.S. and J.-C.A.; supervision, S.P. and J.-C.A.; funding acquisition, Y.B., J.-P.R. and S.P. All authors have read and agreed to the published version of the manuscript.
Funding
Proteomics analyses were performed on the PAPPSO platform (http://pappso.inrae.fr/en) which is supported by INRAE, the Ile-de-France regional council, IBiSA and CNRS. This project was funded by the CNRS MITI, under the call “Plastiques et micro-plastiques en milieux aquatiques”. Schvartz M. was supported by a PhD grant overseen by the CEA and the Région Pays de la Loire and Saudrais F. by a CEA PhD grant.
Data Availability Statement
Data are provided as supplementary material.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nayl, A.A.; Abd-Elhamid, A.I.; Aly, A.A.; Bräse, S. Recent Progress in the Applications of Silica-Based Nanoparticles. RSC Adv. 2022, 12, 13706–13726. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yu, D.; Feng, J.; You, H.; Bai, Y.; He, J.; Cao, H.; Che, Q.; Guo, J.; Su, Z. Toxicity Evaluation of Silica Nanoparticles for Delivery Applications. Drug Deliv. Transl. Res. 2023, 13, 2213–2238. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, P.; Zhao, R.; Zhao, L.; Liu, J.; Peng, S.; Fu, X.; Wang, X.; Luo, R.; Wang, R.; et al. Silica Nanoparticles: Biomedical Applications and Toxicity. Biomed. Pharmacother. Biomedecine Pharmacother. 2022, 151, 113053. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, J.; Zhang, Y.; Zhang, G.; Kang, Y.; Chen, A.; Feng, X.; Shao, L. The Toxicity of Silica Nanoparticles to the Immune System. Nanomed. 2018, 13, 1939–1962. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Mathé, C.; Biola-Clier, M.; Devineau, S.; Drouineau, E.; Hatem, E.; Marichal, L.; Alonso, B.; Gaillard, J.-C.; Lagniel, G.; et al. RNA Binding Proteins Are a Major Target of Silica Nanoparticles in Cell Extracts. Nanotoxicology 2016, 10, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Marichal, L.; Klein, G.; Armengaud, J.; Boulard, Y.; Chédin, S.; Labarre, J.; Pin, S.; Renault, J.P.; Aude, J.C. Protein Corona Composition of Silica Nanoparticles in Complex Media: Nanoparticle Size Does Not Matter. Nanomaterials 2020, 10, 240. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J. Protein–Nanoparticle Interaction: Corona Formation and Conformational Changes in Proteins on Nanoparticles. Int. J. Nanomedicine 2020, 15, 5783–5802. [Google Scholar] [CrossRef] [PubMed]
- Mathé, C.; Devineau, S.; Aude, J.-C.; Lagniel, G.; Chédin, S.; Legros, V.; Mathon, M.-H.; Renault, J.-P.; Pin, S.; Boulard, Y.; et al. Structural Determinants for Protein Adsorption/Non-Adsorption to Silica Surface. PloS One 2013, 8, e81346. [Google Scholar] [CrossRef] [PubMed]
- Teeguarden, J.G.; Webb-Robertson, B.-J.; Waters, K.M.; Murray, A.R.; Kisin, E.R.; Varnum, S.M.; Jacobs, J.M.; Pounds, J.G.; Zanger, R.C.; Shvedova, A.A. Comparative Proteomics and Pulmonary Toxicity of Instilled Single-Walled Carbon Nanotubes, Crocidolite Asbestos, and Ultrafine Carbon Black in Mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2011, 120, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Schvartz, M.; Saudrais, F.; Devineau, S.; Aude, J.-C.; Chédin, S.; Henry, C.; Millán-Oropeza, A.; Perrault, T.; Pieri, L.; Pin, S.; et al. A Proteome Scale Study Reveals How Plastic Surfaces and Agitation Promote Protein Aggregation. Sci. Rep. 2023, 13, 1227. [Google Scholar] [CrossRef] [PubMed]
- Scopes, R.K. Measurement of Protein by Spectrophotometry at 205 Nm. Anal. Biochem. 1974, 59, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.; Haller, L.; Blein-Nicolas, M.; Zivy, M.; Canette, A.; Verbrugghe, M.; Mézange, C.; Boulay, M.; Gardan, R.; Samson, S.; et al. Identification of Hanks-Type Kinase PknB-Specific Targets in the Streptococcus Thermophilus Phosphoproteome. Front. Microbiol. 2019, 10, 1329. [Google Scholar] [CrossRef] [PubMed]
- R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022.
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Goodrich B., A.I., Gabry J.; S, B. Rstanarm: Bayesian Applied Regression Modeling via Stan; 2023.
- Gronau, Q.F.; Singmann, H.; Wagenmakers, E.-J. Bridgesampling: An R Package for Estimating Normalizing Constants. J. Stat. Softw. 2020, 92, 1–29. [Google Scholar] [CrossRef]
- Morey, R.D.; Rouder, J.N. BayesFactor: Computation of Bayes Factors for Common Designs; 2022.
- Partikel, K.; Korte, R.; Stein, N.C.; Mulac, D.; Herrmann, F.C.; Humpf, H.-U.; Langer, K. Effect of Nanoparticle Size and PEGylation on the Protein Corona of PLGA Nanoparticles. Eur. J. Pharm. Biopharm. 2019, 141, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Biola-Clier, M.; Gaillard, J.-C.; Rabilloud, T.; Armengaud, J.; Carriere, M. Titanium Dioxide Nanoparticles Alter the Cellular Phosphoproteome in A549 Cells. Nanomaterials 2020, 10, 185. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, S.L.; McKenzie, D.R.; Nosworthy, N.J.; Denman, J.A.; Sezerman, O.U.; Bilek, M.M.M. The Vroman Effect: Competitive Protein Exchange with Dynamic Multilayer Protein Aggregates. Colloids Surf. B Biointerfaces 2013, 103, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Schvartz, M.; Saudrais, F.; Devineau, S.; Chédin, S.; Jamme, F.; Leroy, J.; Rakotozandriny, K.; Taché, O.; Brotons, G.; Pin, S.; et al. Role of the Protein Corona in the Colloidal Behavior of Microplastics. Langmuir 2023, 39, 4291–4303. [Google Scholar] [CrossRef] [PubMed]
- Millán-Oropeza, A.; Blein-Nicolas, M.; Monnet, V.; Zivy, M.; Henry, C. Comparison of Different Label-Free Techniques for the Semi-Absolute Quantification of Protein Abundance. Proteomes 2022, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A. The Challenge of the Proteome Dynamic Range and Its Implications for In-Depth Proteomics. PROTEOMICS 2013, 13, 723–726. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Adsorption isotherm of YPE on silica NP (1 g.L-1) in phosphate buffer (100 mM, pH7) realized by depletion. Ten samples at YPE concentration from 2.5x10-2 to 2 g.L-1 were incubated with silica NP using a Thermomixer ® at 20 °C during 3 h (cycles of 15 sec at 800 rpm followed by 285 sec at rest). Samples were centrifuged at 20 °C, 20,000 rpm during 10 min and the supernatant concentration (unbound proteins) was determined using the absorbance at 205 nm with an absorption coefficient of 31 L.g-1.cm-1. Horizontal and vertical error bars represent standard error of the mean.
Figure 1.
Adsorption isotherm of YPE on silica NP (1 g.L-1) in phosphate buffer (100 mM, pH7) realized by depletion. Ten samples at YPE concentration from 2.5x10-2 to 2 g.L-1 were incubated with silica NP using a Thermomixer ® at 20 °C during 3 h (cycles of 15 sec at 800 rpm followed by 285 sec at rest). Samples were centrifuged at 20 °C, 20,000 rpm during 10 min and the supernatant concentration (unbound proteins) was determined using the absorbance at 205 nm with an absorption coefficient of 31 L.g-1.cm-1. Horizontal and vertical error bars represent standard error of the mean.
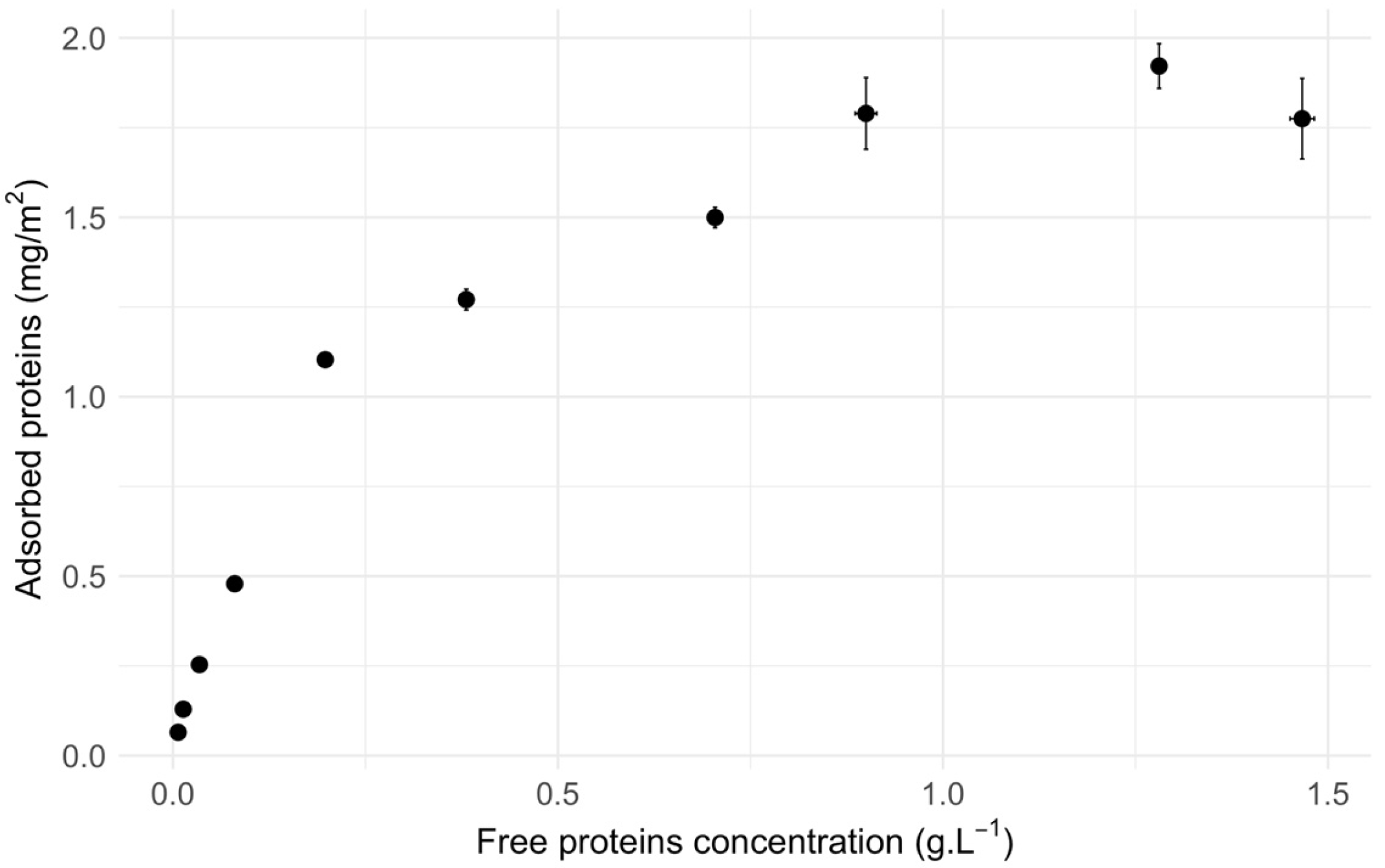
Figure 2.
Calibration curve for the YPE concentration with SDS 1%. Concentrations levels are determined using the absorbance at 205 nm with an absorption coefficient of 31 L.g-1.cm-1. The blue curve depicts the linear regression model fitted to the data points.
Figure 2.
Calibration curve for the YPE concentration with SDS 1%. Concentrations levels are determined using the absorbance at 205 nm with an absorption coefficient of 31 L.g-1.cm-1. The blue curve depicts the linear regression model fitted to the data points.
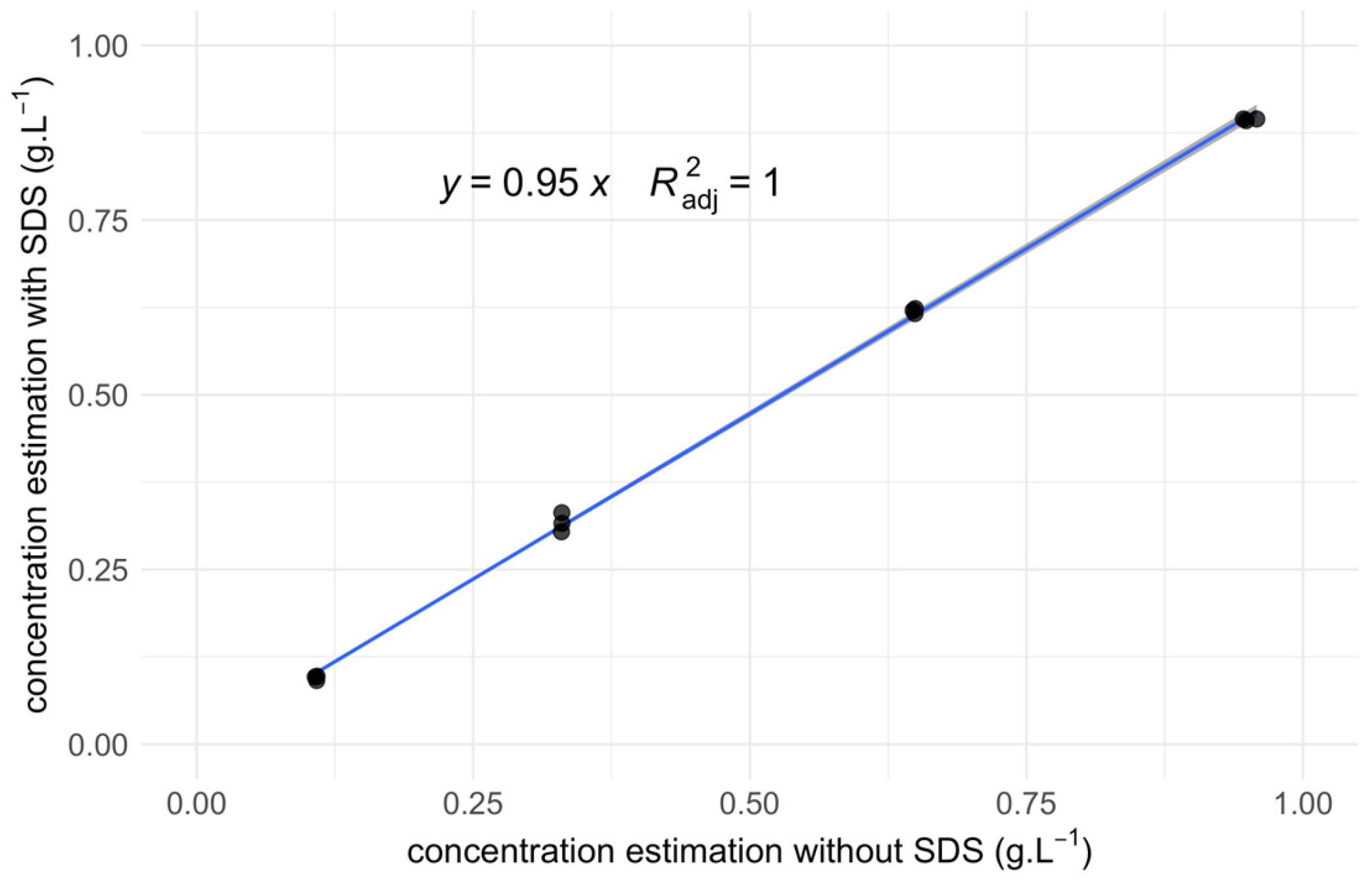
Figure 3.
Spectral counts correlation plot between the pellet, supernatant and control fractions. Pearson correlation coefficients between replicates are calculated and depicted as squares which size and color (scale given on the right of the plot) depends on its value. Average correlation coefficients values between fraction pairs are indicated on the lower triangular matrix.
Figure 3.
Spectral counts correlation plot between the pellet, supernatant and control fractions. Pearson correlation coefficients between replicates are calculated and depicted as squares which size and color (scale given on the right of the plot) depends on its value. Average correlation coefficients values between fraction pairs are indicated on the lower triangular matrix.
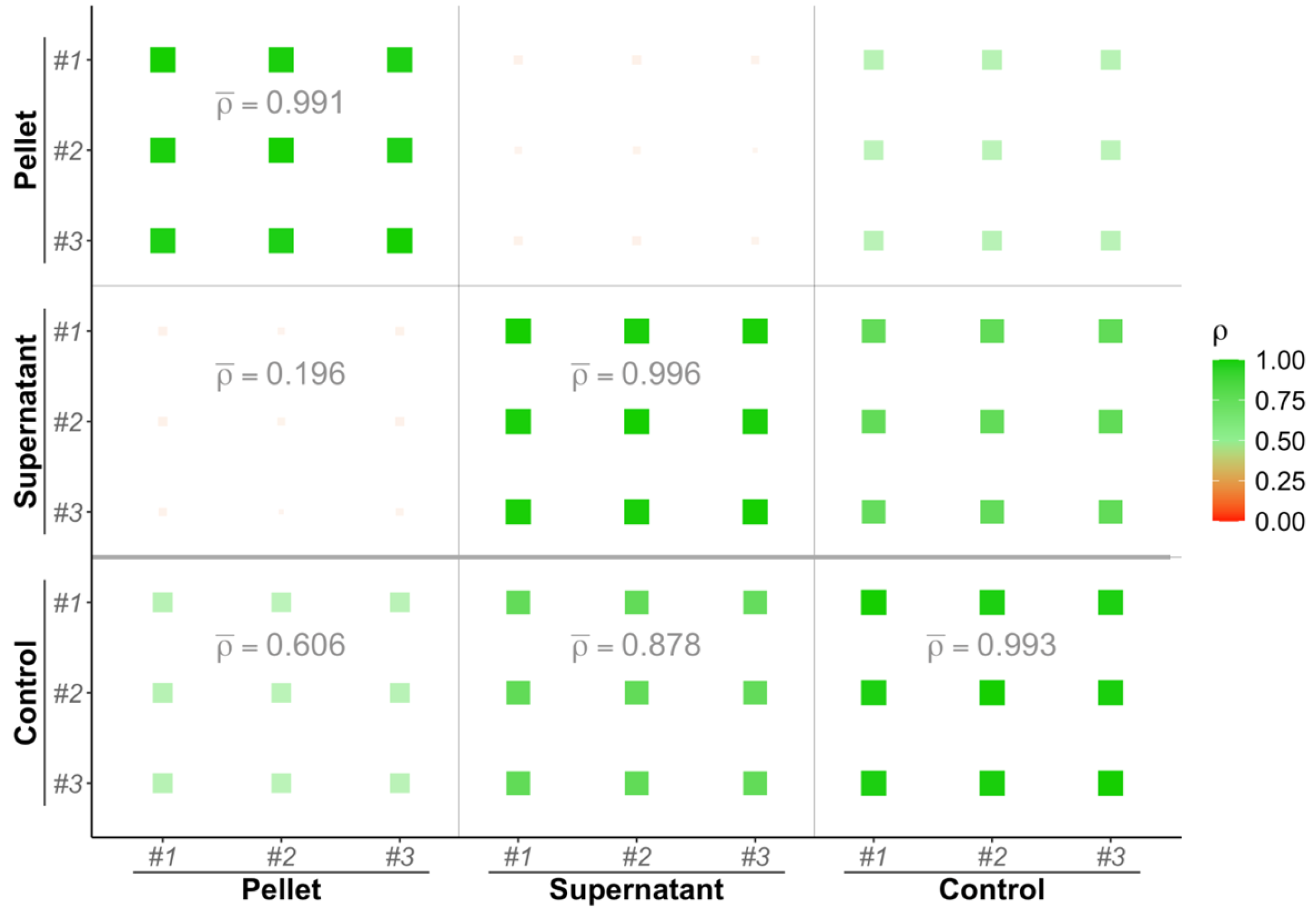
Figure 4.
Spectral Counts (SC) linear regression model. This plot depicts the linear regression, as a blue line, between the cumulated SC of each protein in the pellet and supernatant, with the SC of these proteins in the control (see Equation 2). Besides the regression line, each dot depicts the related SC in the control (Y axis) and in the cumulated pellet and supernatant (X axis). The color of the dot is red when the protein is more abundant in the pellet than in the supernatant and blue otherwise.
Figure 4.
Spectral Counts (SC) linear regression model. This plot depicts the linear regression, as a blue line, between the cumulated SC of each protein in the pellet and supernatant, with the SC of these proteins in the control (see Equation 2). Besides the regression line, each dot depicts the related SC in the control (Y axis) and in the cumulated pellet and supernatant (X axis). The color of the dot is red when the protein is more abundant in the pellet than in the supernatant and blue otherwise.
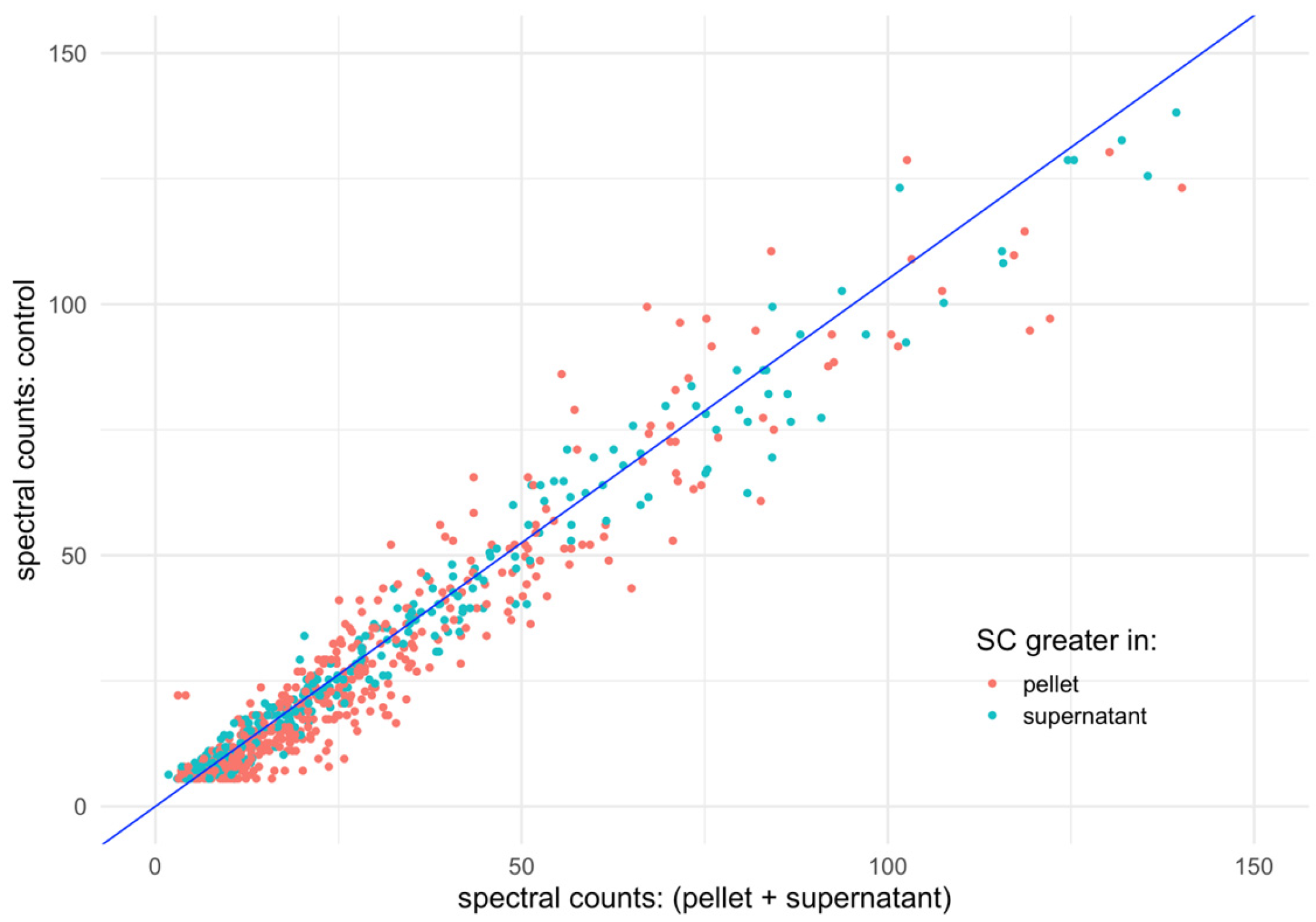
Figure 5.
Venn diagram of the number of detected proteins in the pellet, supernatant and control fractions. The relative percentage is indicated within parenthesis.
Figure 5.
Venn diagram of the number of detected proteins in the pellet, supernatant and control fractions. The relative percentage is indicated within parenthesis.
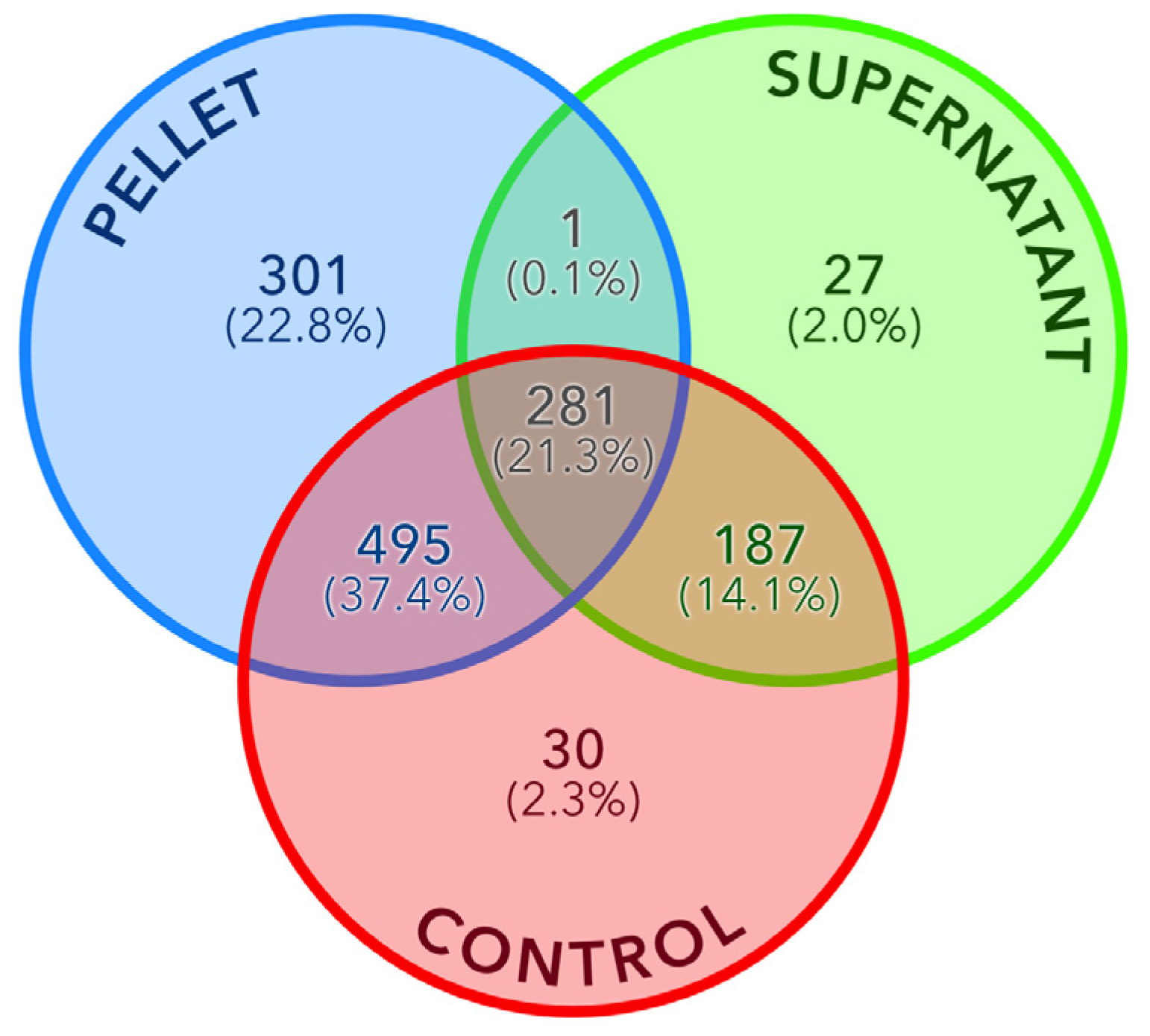

Table 1.
Spectral counts summary. Fractions are indicated in the first column. Columns 2 to 4 contains the total spectral count for each replica. Column 5 indicates the average total SC. Column 6 shows the percentage of the average SC value, for each fraction, compared to the control average SC.
Table 1.
Spectral counts summary. Fractions are indicated in the first column. Columns 2 to 4 contains the total spectral count for each replica. Column 5 indicates the average total SC. Column 6 shows the percentage of the average SC value, for each fraction, compared to the control average SC.
| Fraction | Replica 1 | Replica 2 | Replica 3 | Average | % of control |
|---|---|---|---|---|---|
| Control | 20,346 | 33,223 | 33,791 | 29,120 | 100.0% |
| Pellet | 15,629 | 17,503 | 16,068 | 16,400 | 56.3% |
| Supernatant | 12,453 | 9,768 | 15,939 | 12,720 | 43.7% |
Table 2.
Average proteins concentration in each fraction.
| Fraction | Pellet | Supernatant | Control |
|---|---|---|---|
| Average proteins concentration | 0.41 g.L-1 | 0.32 g.L-1 | 0.63 g.L-1 |
Table 3.
Coefficients of the linear model, Equation (1). For each parameter of the model (first column), the coefficient value and its deviation are indicated in the second column. The third column contains the confidence interval. The Student t-test statistic and the associated p-values are provided in the last two columns.
Table 3.
Coefficients of the linear model, Equation (1). For each parameter of the model (first column), the coefficient value and its deviation are indicated in the second column. The third column contains the confidence interval. The Student t-test statistic and the associated p-values are provided in the last two columns.
| Parameter | Coefficient | Confidence Interval | t-student | p-value |
|---|---|---|---|---|
| Intercept (α) | -0.09 ±0.40 | [-0.88, 0.70] | 29,120 | 0.83 |
| SC Pellet (β1) | 1.07 ±0.01 | [1.04, 1.09] | 16,400 | <0.001 |
| SC Supernatant (β2) | 1.05 ±0.01 | [1.03, 1.06] | 12,720 | <0.001 |
Table 4.
Pellet and supernatant Bayesian differential analysis. For Bayesian factor thresholds 3,10 and 30 we indicate the number of proteins with a significant difference in spectral counts between fractions: pellet and control (columns 2 and 3); supernatant and control (columns 4 and 5).
Table 4.
Pellet and supernatant Bayesian differential analysis. For Bayesian factor thresholds 3,10 and 30 we indicate the number of proteins with a significant difference in spectral counts between fractions: pellet and control (columns 2 and 3); supernatant and control (columns 4 and 5).
| Bayes Factor (BF) evidence |
 Pellet fraction Pellet fraction
|
 Supernatant fraction Supernatant fraction
|
||
|---|---|---|---|---|
| enriched | depleted | enriched | depleted | |
| BF ≥ 3: substantial | 302 | 83 | 56 | 265 |
| BF ≥ 10: strong | 139 | 55 | 11 | 159 |
| BF ≥ 30: very strong | 54 | 27 | 2 | 88 |
Table 5.
Cross Analysis of adsorbed proteins in the pellet and supernatant fractions. This table displays the overlap between proteins enriched in the pellet (indicating nanoparticle adsorption) and those depleted in the supernatant (poorly detected in solution) for a Bayes Factor (BF) threshold 3 (see additional Table S1 for thresholds 3, 10, and 30). The first column indicates the number of supernatant-depleted proteins. Blue number along the top row represent the number of pellet-enriched proteins and the green number indicate the subset of pellet-enriched proteins also detected in the supernatant fraction. Black numbers represent the number of proteins concurrently enriched in the pellet and depleted in the supernatant. Blue (resp. green) percentage represent the ratio of overlapped proteins to total pellet-enriched proteins (resp. and detected in the supernatant).
Table 5.
Cross Analysis of adsorbed proteins in the pellet and supernatant fractions. This table displays the overlap between proteins enriched in the pellet (indicating nanoparticle adsorption) and those depleted in the supernatant (poorly detected in solution) for a Bayes Factor (BF) threshold 3 (see additional Table S1 for thresholds 3, 10, and 30). The first column indicates the number of supernatant-depleted proteins. Blue number along the top row represent the number of pellet-enriched proteins and the green number indicate the subset of pellet-enriched proteins also detected in the supernatant fraction. Black numbers represent the number of proteins concurrently enriched in the pellet and depleted in the supernatant. Blue (resp. green) percentage represent the ratio of overlapped proteins to total pellet-enriched proteins (resp. and detected in the supernatant).
| depleted in the supernatant |
Enriched in the pellet AND detected in the supernatant 
|
|||
|---|---|---|---|---|
| 302 | 179 | |||
| 265 | 124 | 41.1% | 124 | 69.3% |
Table 6.
Cross Analysis of non-adsorbed proteins in the pellet and supernatant fractions. This table displays the overlap between proteins enriched in the supernatant (indicating unbound proteins) and those depleted in the pellet (poorly adsorbed) for a Bayes Factor (BF) threshold 3 (see additional table S2 for thresholds 3, 10, and 30). The first column indicates the number of pellet-depleted proteins. Green number along the top row represent the number of supernatant-enriched proteins and the blue number indicate the subset of supernatant-enriched proteins also detected in the pellet fraction. Black numbers represent the number of proteins concurrently enriched in the supernatant and depleted in the pellet. Green (resp. blue) percentage represent the ratio of overlapped proteins to total supernatant-enriched proteins (resp. and detected in the pellet).
Table 6.
Cross Analysis of non-adsorbed proteins in the pellet and supernatant fractions. This table displays the overlap between proteins enriched in the supernatant (indicating unbound proteins) and those depleted in the pellet (poorly adsorbed) for a Bayes Factor (BF) threshold 3 (see additional table S2 for thresholds 3, 10, and 30). The first column indicates the number of pellet-depleted proteins. Green number along the top row represent the number of supernatant-enriched proteins and the blue number indicate the subset of supernatant-enriched proteins also detected in the pellet fraction. Black numbers represent the number of proteins concurrently enriched in the supernatant and depleted in the pellet. Green (resp. blue) percentage represent the ratio of overlapped proteins to total supernatant-enriched proteins (resp. and detected in the pellet).
| depleted in the pellet |
Enriched in the supernatant / AND detected in the pellet
|
|||
|---|---|---|---|---|
| 56 | 36 | |||
| 83 | 32 | 57.1% | 32 | 88.9% |
Table 7.
Proteins were categorized based on their interaction with silica nanoparticles (SiNPs) using a Bayes Factor threshold ≥ 3. Proteins are classified as: Unbound (blue background), proteins not associated with SiNPs; Impacted (grey background), proteins showing altered abundance despite not being directly bound to SiNPs; Bound (red background), proteins directly interacting with the SiNP surface. Within each category, proteins are further subcategorized: Very high, exclusively detected in either the pellet (bound) or supernatant (unbound) fraction; High, detected in both the pellet and supernatant fraction; Direct, enriched in the pellet and detected but not depleted in the supernatant fraction; Indirect, depleted in the supernatant but not enriched in the pellet fraction. Protein counts and relative percentages (%) are indicated in parentheses for each category and subcategory.
Table 7.
Proteins were categorized based on their interaction with silica nanoparticles (SiNPs) using a Bayes Factor threshold ≥ 3. Proteins are classified as: Unbound (blue background), proteins not associated with SiNPs; Impacted (grey background), proteins showing altered abundance despite not being directly bound to SiNPs; Bound (red background), proteins directly interacting with the SiNP surface. Within each category, proteins are further subcategorized: Very high, exclusively detected in either the pellet (bound) or supernatant (unbound) fraction; High, detected in both the pellet and supernatant fraction; Direct, enriched in the pellet and detected but not depleted in the supernatant fraction; Indirect, depleted in the supernatant but not enriched in the pellet fraction. Protein counts and relative percentages (%) are indicated in parentheses for each category and subcategory.
| Unbound | Impacted | Bound | |||
|---|---|---|---|---|---|
| very high | high | indirect | direct | high | very high |
| 24 (4.3%) | 32 (5.8%) | 141 (25.5%) | 55 (9.9%) | 124 (22.4%) | 178 (32.1%) |
| 56 (10.1%) | 196 (35.4%) | 302 (54.5%) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



