
Submitted:
05 September 2024
Posted:
06 September 2024
You are already at the latest version
Abstract
The human brain is highly dependent on oxygen, utilizing approximately 20% of the body’s oxygen at rest. Oxygen deprivation to the brain can lead to loss of consciousness within seconds and death within minutes. Recent studies have identified regions of the brain with spontaneous episodic hypoxia, referred to as "hypoxic pockets." Hypoxia can also result from impaired blood flow due to conditions such as heart disease, blood clots, stroke, or hemorrhage, as well as from reduced oxygen intake or excessive oxygen consumption caused by factors like low ambient oxygen, pulmonary diseases, infections, inflammation, and cancer. Severe hypoxia in the brain can manifest symptoms similar to Parkinson’s disease (PD), including cerebral edema, mood disturbances, and cognitive impairments. Additionally, the development of PD appears to be closely associated with hypoxia and hypoxic pathways. This review seeks to investigate the molecular interactions between hypoxia and PD, emphasizing the pathological role of hypoxic pathways in PD and exploring their potential as therapeutic targets.
Keywords:
Parkinson’s disease
; hypoxia
; hypoxia pathways
; neurodegenerative diseases
; transmembrane protein 175
; DJ-1
; LRRK2
1. Introduction
Hypoxia, or reduced oxygen supply, is a significant factor in the development of various neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and other age-related disorders[1,2,3,4,5]. The neurotoxic effects of hypoxia are linked to brain injury and the onset of neurodegenerative conditions such as AD, vascular dementia, and PD[6]. Prolonged hypoxia can lead to irreversible neuronal loss, thereby exacerbating these neurodegenerative pathologies[7].
Parkinson’s disease is primarily characterized by both motor and non-motor symptoms, along with the gradual loss of dopaminergic neurons in various brain structures, particularly the substantia nigra pars compacta (SNpc). Dysfunction in the SNpc results in motor symptoms such as bradykinesia (slow movements), rigidity, resting tremor, impaired postural control, and gait deficits[8,9]. Additionally, non-motor symptoms, including fatigue, sleep disturbances, anxiety, depression, and impaired autonomic regulation, significantly affect the quality of life in PD patients[10]. Addressing these concerns, it is crucial to identify new targets or pathways for developing therapies that can alleviate symptoms or potentially combat the disease.
Research has shown that hypoxia is associated with cell death mechanisms relevant to neurodegenerative diseases, particularly through the accumulation of reactive oxygen species, which are implicated in conditions such as Huntington's disease, PD, and AD[11]. Abnormal misfolding and aggregation of alpha-synuclein in the brain are pathological features of Parkinson's disease. Hypoxia is closely associated with the activation of alpha-synuclein, suggesting that low oxygen levels may influence the progression of PD[12].
This review article aims to explore the complex and multifaceted role of hypoxia in the pathogenesis of PD and to highlight hypoxia as a potential therapeutic target.
2. Hypoxia Pathways
Before discussing hypoxia pathways, it is crucial to differentiate between hypoxia training and hypoxia insult[13]. Hypoxia training involves mild, intermittent, and controlled hypoxia stimuli designed to enhance an individual’s resistance and cellular adaptation to hypoxia insult. Examples include intermittent hypoxia cycles and frequent physical exercise, which may alleviate PD symptoms by improving hypoxia pathways[12,14,15]. In contrast, hypoxia insult is typically caused by pathological processes, toxins, chronic hypoxia, or a severe decrease in ambient oxygen levels, leading to cellular injury and exacerbation of PD symptoms[1,16]. Different treatments, animal models, and in vitro models reflect various hypoxia conditions, resulting in paradoxical effects within the same pathway. Understanding the nuanced role of hypoxia can help clarify the complex evidence surrounding hypoxia pathways.
At the molecular level, low oxygen levels reduce the availability of electron acceptors in the mitochondrial electron transport chain, leading to increased reactive oxygen species (ROS) production and diminished ATP synthesis. These changes can directly cause neuronal damage and, in rare cases, contribute to PD symptoms[17,18,19]. Hypoxia-inducible factors (HIFs) and nuclear factor erythroid 2-related factor 2 (Nrf2) are central regulators of cellular adaptation to hypoxia, and their neuroprotective functions are well established [20,21,22]. Hypoxic exposure has been shown to induce α-synuclein(α-syn) overexpression and oligomer formation, leading to cell injury in HEK293 cells[23]. Given that hypoxia can influence the pathogenesis of PD, regulating HIF-1 and Nrf2 activity may be a crucial neuroprotective strategy for promoting the survival of dopaminergic neurons.
2.1. HIF Pathway
HIFs are transcription factors that respond to low oxygen levels by regulating molecular adaptations to maintain oxygen supply and energy metabolism[24]. HIFs are part of the PER-ARNT-SIM (PAS) subfamily within the basic helix-loop-helix (bHLH) family of heterodimeric transcription factors[25]. Currently, three HIF members have been identified: HIF-1, HIF-2 (also known as endothelial PAS domain-containing protein 1), and HIF-3[26,27,28]. Northern blot analysis reveals minimal expression of HIF-3 in brain tissue, suggesting a limited connection between HIF-3 and PD[29].
HIF-1 and HIF-2 are both heterodimeric transcription factors composed of α- and β-subunits. HIF-1α and HIF-2α share 48% amino acid sequence similarity, with 83% identity in their basic bHLH domains and approximately 70% homology in their PER-ARNT-SIM regions[30]. Additionally, the oxygen-dependent degradation domains of these HIF-α subunits, including the two critical proline residues that interact with prolyl hydroxylase (PHD), also exhibit a high degree of homology[31]. This significant sequence and structural similarity suggest that HIF-1 and HIF-2 have similar regulatory mechanisms involving PHD-von Hippel-Lindau (pVHL) complex and target genes.
Both HIF-1 and HIF-2 share the same β-subunit, HIF-1β, which is stably expressed and located in the nucleus, binding to hypoxia response elements (HREs), upstream of hypoxia-inducible genes[32,33]. Under normoxic conditions, HIF-α subunits are continuously degraded through hydroxylation, which is catalyzed by PHD and factor inhibiting HIF (FIH). Prolyl hydroxylation promotes interaction with the pVHL E3 ubiquitin ligase complex, leading to degradation by the 26S proteasome[34]. During moderate hypoxia, PHD activity is impaired, but FIH can still perform hydroxylation. This allows HIF-α to enter the nucleus, where it binds with HIF-1β to form the functional HIF complex, which then binds to HREs to promote the transcription of target genes involved in PHD regulation.
Under severe hypoxia, reduced oxygen levels inhibit PHD activity, preventing the degradation of HIF-α subunits (both HIF-1α and HIF-2α)[35]. Consequently, HIF-α escapes the ubiquitin-proteasome system (UPS), binds to the coactivator p300/CBP, and is transported into the nucleus to activate transcription with HIF-1β[36]. Typically, these genes enhance oxygen supply to tissues and facilitate metabolic adaptation to hypoxia. Despite their considerable similarities, HIF-1 and HIF-2 exhibit distinct functions and spatio-temporal regulations[37,38].
HIF-1α is rapidly activated during acute, severe hypoxia (1–2% O2). A higher prevalence of HIF-1α polymorphisms has been observed in PD patients, with some single nucleotide variants associated with an increased risk of developing PD (Figure 1)[39]. Hypoxia has been reported to upregulate the transcription and expression of tyrosine hydroxylase, the rate-limiting enzyme for dopamine synthesis, and the dopamine transporter (DAT), both crucial for dopaminergic neuronal function, via HIF-1α regulation[1,40]. Conditional knockout mice lacking HIF-1α exhibit reduced levels of vascular endothelial growth factor (VEGF) and TH in midbrain-derived neural precursor cells, leading to decreased differentiation of dopaminergic neurons[41,42,43]. This evidence indicates that HIF-1α plays a crucial role in the survival and functioning of dopaminergic neurons in the SNpc region.
Additionally, activation of HIF-1α is associated with suppressed mitochondrial function. HIF-1α negatively regulates mitochondrial function by repressing the transcriptional activity and inhibiting the activity of cellular myelocytomatosis viral oncogene in VHL-deficient renal cell carcinoma, which may contribute to mitochondrial dysfunction in PD[44]. However, the function of HIF-1α appears to be diminished in PD patients. Gene expression profiling analysis reveals decreased levels of HIF-1α and its target genes (including VEGF and hexokinase) in PD patients, with an upregulation of PHD2 in the SNpc homogenate of PD patients compared to age-matched controls.
The evidence suggests that HIF-1α exhibits promising neuroprotective functions against PD. However, paradoxical findings indicate that during severe hypoxia in the brain, HIF-1α may also facilitate inflammasome formation, mitochondrial dysfunction, and cell death[45]. For instance, in neuron-specific conditional knockout mice, deficiency of neuronal HIF-1α and HIF-2α improves neuronal survival in the early acute phase following ischemic stroke. Conversely, similar experimental designs have yielded different outcomes, with some studies showing that neuron-specific HIF-1α knockdown increases tissue damage and reduces survival rates in response to transient focal cerebral ischemia. These discrepancies might be due to variations in murine stroke models used[46]. Additionally, HIF-1α regulates genes involved in autophagy and apoptosis, such as BNIP3 and Noxa (or PMAIP1, phorbol-12-myristate-13-acetate-induced protein 1)[47,48]. This adds an extra layer of complexity to its role in cellular regulation, making HIF-1α a potential double-edged sword in the context of hypoxia, oxidative stress, or ischemia. While moderate stress levels might activate the neuroprotective aspects of HIF-1α, even slight deviations could trigger detrimental effects[49]. These paradoxes highlight the intricate and sophisticated role of HIF-1α, which must be carefully considered in PD therapy development.
In contrast, HIF-2α gradually accumulates during prolonged, moderate hypoxia (<5% O2) and regulates similar genes[50,51]. Although research on the correlation between HIF-2 and PD is limited, HIF-2 likely has functions that overlap significantly with those of HIF-1. This is suggested by the comparable upregulation of HIF-1-regulated genes observed in response to short hypoxic episodes in brain tissue from HIF-1 knockout mice[52]. In post-mortem PD brains, accumulation of HIF-2α is observed as a marker of chronic hypoxia[53]. Additionally, data suggest that HIF-2α promotes α-syn hyperphosphorylation at the serine 129 site (pS129-αSyn) and abnormal aggregation of pS129-αSyn by upregulating alkaline ceramidase 2 (Acer2), which leads to cognitive impairment in mice. This finding is consistent with elevated levels of pS129-αSyn in the plasma of patients with obstructive sleep apnea, a condition associated with chronic intermittent hypoxia[54]. Thus, like HIF-1α, HIF-2α also exhibits paradoxical effects, potentially being either neuroprotective or detrimental to neurons.
2.2. Nrf2/HO-1 Pathway
As a central regulator of cellular redox states, Nrf2 plays a crucial role in protecting cells against oxidative stress by activating the transcription of a wide range of antioxidant and anti-inflammatory genes. This regulation impacts metabolic pathways and initiates the production of NADPH and ATP[55]. Nrf2 is regulated by Kelch-like ECH-associated protein 1 (KEAP1), a substrate adaptor that forms a complex with Cullin-3 (CUL3) to target specific proteins for degradation. Under normal conditions, KEAP1 and CUL3 form a ubiquitin E3 ligase complex that polyubiquitinates Nrf2, leading to its rapid degradation via the proteasome system. Whether under normoxia or hypoxia, Nrf2 degradation through the UPS depends on direct interaction with KEAP1, but this degradation is more rapid in normoxia than in hypoxia[56], aligning with Nrf2’s role as a hypoxia regulator [57].
Under oxidative stress or specific conditions, Nrf2 translocates from the cytoplasm to the nucleus, where it forms a heterodimer with small MAF (sMAF) proteins and binds to the antioxidant response element (ARE) to activate a range of phase II detoxification genes, thus functioning as a major regulatory transcription factor (Figure 2). The beneficial role of Nrf2 in neurodegenerative conditions and the potential of specific Nrf2 activators as therapeutic agents are well-documented[58]. While the antioxidant functions of Nrf2 have been a primary focus, in vitro studies also show that Nrf2 accelerates the clearance of α-syn and mutant leucine-rich repeat kinase 2 (LRRK2) through the UPS[59]. As a transcription factor, Nrf2 exerts its neuroprotective effects by activating multiple protective genes involved in various molecular events of PD[60]. Among these genes, one notable candidate is heme oxygenase-1 (HO-1).
HO-1 is a potent antioxidant enzyme that degrades heme into carbon monoxide, free iron, and biliverdin[61]. The promoter region of the HO-1 gene contains various binding sites for transcription factors such as Nrf2 and HIF-1[62]. Significantly elevated levels of HO-1 have been observed in the plasma of early-stage PD patients and in the cerebrospinal fluid of children following severe traumatic brain injury[63]. These observations suggest an underlying connection between HO-1 and PD.
Consistent with findings on Nrf2, in vitro studies have shown that overexpression of HO-1 enhances the degradation of α-syn through the UPS. The α-syn mutant A30P exhibits resistance to HO-1-dependent degradation, leading to increased toxic intracellular aggregation. This finding suggests that Nrf2 may facilitate α-syn clearance through the Nrf2/HO-1 pathway[64]. Furthermore, HO-1 helps prevent dopaminergic neuronal death by promoting the expression of neurotrophic factors and enhancing the antioxidant response in both in vivo and in vitro PD models[65]. Chemicals that activate the Nrf2/HO-1 pathway have demonstrated therapeutic effects against PD models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 1-Methyl-4-phenylpyridinium (MPP+), and other agents[66,67]. Nevertheless, the neuroprotective function of HO-1 appears to be closely associated with both the Nrf2 and HIF-1 pathways, highlighting the potential of targeting the Nrf2/HO-1 and HIF-1/HO-1 pathways for PD therapy.
3. The Correlation between Hypoxia and PD Pathogenesis
Hypoxia significantly affects neuronal health, particularly impacting dopaminergic neurons in the substantia nigra, which are crucial for motor control[68,69,70]. Hypoxia regulators and pathways play a key role in neuroprotection and in mitigating the progression of PD[71]. PD patients show a marked reduction in antioxidant defenses within the SNpc and lower levels of antioxidant proteins in peripheral blood[72]. Drugs targeting hypoxia pathways have also shown great potential in alleviating PD symptoms[73,74]. Research on the relationship between hypoxia and PD has been conducted in the Han Chinese population[75]. Therefore, elucidating how PD development and symptoms interact with hypoxia pathways is crucial for understanding and potentially improving therapeutic strategies (Figure 3).
3.1. Dopaminergic Neurons’ Susceptibility to Hypoxia
Dopaminergic neurons in the substantia nigra, which innervate the striatum through long branching axons, are highly susceptible to hypoxia[76]. These neurons have high oxygen and ATP demands due to their extensive axonal projections, making them particularly vulnerable to oxidative stress and hypoxia[77,78]. The oxygen dependence of dopamine biosynthesis and metabolism exacerbates this vulnerability[79]. Additionally, dopamine metabolism is prone to generating oxidative stress, necessitating a robust antioxidant system[80]. The susceptibility of dopaminergic neurons to oxidative damage is further highlighted by their sensitivity to hypoxia and energy deficiency. Studies in rat brains have demonstrated that hypoxia or glucose deprivation can inhibit neuronal firing and induce hyperpolarization in dopaminergic neurons[81], indicating reduced neuronal excitability and activity.
3.2. Evidence of Hypoxia in PD
Evidence of hypoxia-related events in PD brains suggests that hypoxia may contribute to the pathogenesis of PD. In PD patients, the chemo sensitive receptors that control respiration by sensing blood pH fluctuations are impaired, leading to a diminished sensation of dyspnea and a reduced hypoxic response[82]. The inspiratory muscle strength is compromised even in very early-stage PD patients[83], and detrimental changes in lung volumes are observed, such as airflow limitations[84], restrictive patterns[85], or mixed patterns[86]. Acute levodopa administration has been found to improve inspiratory muscle function in both anesthetized dogs[87] and patients, and dopamine has been shown to enhance diaphragm function during acute respiratory failure in patients with chronic obstructive pulmonary disease[88]. These findings suggest that respiratory symptoms in PD may be related to the loss of dopaminergic neurons.
Furthermore, reduced respiratory functions, declines in autonomic regulation of the circulatory system, and inadequate cerebral perfusion could exacerbate the hypoxic environment in the brains of PD patients. At the molecular level, HIF-2α, a marker of chronic hypoxia, is elevated in the substantia nigra of post-mortem PD patients. Additionally, higher polymorphisms of HIF-1α have been observed in PD patients. Reduced levels of brain-derived neurotrophic factor (BDNF), which is important for synaptic development and plasticity[89], have also been noted in PD, schizophrenia, and Alzheimer's disease[90]. The promoter region of the BDNF gene contains a bHLH-PAS transcription factor response element that interacts with HIF-1β to activate transcription by HIF[91]. Thus, decreased BDNF levels may indicate impaired hypoxia resistance in PD patients.
3.3. The Role of Hypoxia in PD Pathogenesis
Hypoxia disrupts normal mitochondrial function at the cellular level, leading to increased oxidative stress and decreased energy production. This disruption causes an accumulation of ROS, which further damages brain tissue and accelerates neurodegeneration. Dopaminergic neurons are particularly vulnerable to hypoxia due to their close association with mitochondrial function. Notably, the accumulation of ROS can result in two distinct outcomes: extreme hypoxia leads to significant oxidative stress, while moderate oxygen deprivation acts as a signaling mechanism that enhances cellular resilience[92].
4. The Mechanism of Hypoxia in Causing PD
Several theories regarding the pathogenesis of PD emphasize the roles of α-syn accumulation, mitochondrial dysfunction, lysosomal damage, and alterations in receptors/neurotransmitters in both sporadic and familial cases of PD. Hypoxia, a significant environmental factor, may influence the development of PD by affecting these pathways through various mechanisms. The interaction between these mechanisms forms a complex network, where they mutually influence and exacerbate each other throughout the pathological process of PD (Figure 4).
4.1. α-Syn Accumulation
The main pathological feature of PD is the degeneration and death of dopaminergic and other pigmented neurons in the substantia nigra pars compacta. The link between α-syn and PD was first identified in 1997. In familial PD, six missense point mutations in the SNCA gene (also known as PARK1) promote the formation of α-syn aggregates and fibrils, thereby increasing the risk of PD[93].
α-syn is a soluble protein composed of 140 amino acid residues encoded by the SNCA gene, primarily found in the presynaptic nerve endings of the central nervous system. The protein consists of three regions: an amphiphilic helical structure at the N-terminal, a central hydrophobic region known as the NAC, which is critical for detecting α-syn aggregation, and a highly negatively charged C-terminal that forms an acidic, irregular curl structure[94,95]. α-syn has several physiological functions, including maintaining synaptic function[96], regulating dopamine and other neurotransmitter release[97], preserving cell membrane homeostasis[98], and influencing the production and function of microglia[99]. It also affects mitochondrial defects, autophagy, and lysosomal dysfunction[100].
Studies indicate that α-syn transmission relies on the release, internalization, and misfolding of α-syn[101]. Under physiological conditions, α-syn stabilizes into a tetramer of about 58 kDa[102]. However, in pathological states, α-syn misfolds, leading to abnormal aggregate formation[103]. The pathogenic amyloid aggregates formed by phosphorylated α-syn at tyrosine 39 (pY39α-syn) are highly cytotoxic to neurons and can induce more endogenous α-syn to undergo liquid-solid phase transformation, resulting in pathological protein aggregates[104].
The abnormal aggregation and transmission of α-syn play a crucial role in PD pathogenesis and are exacerbated under hypoxic conditions[14]. Research has shown that patients with obstructive sleep apnea syndrome have increased levels of total α-syn and phosphorylated α-syn (p-α-syn) in their plasma, which correlates positively with the oxygen desaturation index, suggesting that chronic intermittent hypoxia elevates α-syn levels[7]. During chronic hypoxia, hyperphosphorylation of α-syn occurs, enhancing ceramide catabolism through HIF-2α and HIF-2α-dependent transcriptional activation of Acer2, which is implicated in α-syn pathology and cognitive impairment in mice[54]. Additionally, exogenous α-syn oligomers promote HIF-1α accumulation in normoxic primary microglia via TLR7/8 mediation, thereby enhancing their migratory capacity[105]. The α-syn/HIF-1α axis induces neuroinflammatory injury through the IL6ST-AS/STAT3/HIF-1α axis. Inhibition of this axis in microglia, induced by α-syn stimulation, reduces HIF-1α expression and increases oxidative stress injury in SH-SY5Y cells[106]. Hypoxia/ischemia-related increases in α-syn levels may contribute to protein aggregation, and hypoxia leads to toxic α-syn oligomer formation within cells[23,107]. Moreover, abnormal α-syn aggregation further impairs the function of the ubiquitin-proteasome system, which mediates HIF-1α degradation[2,108].
This connection between hypoxia/ischemia and PD is further supported by other evidence. The intestine is considered a possible origin of α-syn pathology, with hypoxia and hypoxia signaling pathways playing significant roles in intestinal diseases. Chronic intestinal inflammation can also drive PD progression[109], with a clear mutual promotion between hypoxia and inflammation, the latter being a critical factor in promoting α-syn aggregation and dissemination[56]. However, specific studies linking hypoxia to inflammation-driven α-syn aggregation and dissemination are lacking.
Calcium ions are important signaling molecules in cells. Studies have shown that in human cell lines expressing α-syn, transient intracellular calcium increases lead to higher α-syn aggregate levels[110]. Calcium-binding protein calmodulin alters its conformation and binds to α-syn, thereby inducing α-syn fibrillation[111]. Additionally, when intracellular calcium concentration rises, the Ca2+/calmodulin complex can stabilize HIF-1α, enhancing its transcriptional activity[112]. Preventing iron accumulation is associated with α-syn aggregation and dopaminergic neuron vulnerability[113,114]. HIF promotes iron transport in the blood and iron absorption by hematopoietic cells, providing a theoretical basis for the positive effect of HIF-1α stabilization in PD treatment[115].
Hypoxia may trigger, promote, and exacerbate the abnormal pathology of α-syn through these mechanisms, though the underlying processes remain unclear. Additionally, there is currently no research indicating that hypoxia has a more profound impact on PD, highlighting the need for further investigation.
4.2. Mitochondrion Dysfunction
Mitochondria are critical organelles within cells. The introduction of toxins like MPTP inhibits mitochondrial complex I and disrupts the mitochondrial electron transport chain (ETC), leading to severe degeneration of dopaminergic neurons in the substantia nigra and striatum. This process induces an acute and irreversible PD-like syndrome, highlighting the importance of mitochondrial function in PD research[69]. Increasing evidence suggests that PD is characterized by mitochondrial defects, including reduced mitochondrial numbers, structural damage, and decreased activity of respiratory chain complex I. Sporadic Parkinson’s disease, which accounts for over 90% of PD cases, is associated with disruptions in pathways that regulate mitochondrial energy production in nerve cells[116].
Although there is no direct evidence that PD-related genes cause hypoxia, mitochondrial dysfunction is a key mechanism through which these genes contribute to the disease, leading to oxygen utilization disorders. Hypoxic conditions can alter mitochondrial membrane structure and interactions with α-syn, potentially exacerbating Lewy body pathology and mitochondrial dysfunction. Therefore, hypoxia-induced mitochondrial dysfunction may interact with α-synuclein pathology[2]. Overexpression of α-syn can cause mitochondrial rupture, changes in mitochondrial membrane permeability, and decreased respiratory function, ultimately leading to neuronal death[117,118]. Additionally, α-syn can impair autophagy, including mitophagy, preventing the clearance of dysfunctional mitochondria and further exacerbating damage. The heterozygous GBA1 mutation, a common genetic risk factor for PD, leads to α-syn aggregation and mitochondrial dysfunction, increasing PD risk by more than 20 times[119].
Reactive oxygen species plays a crucial role in the interplay between PD and hypoxia. Mitochondria are a primary source of intracellular ROS, and elevated ROS levels can cause oxidative stress, leading to severe cellular damage[120]. This imbalance can result in the oxidative modification of α-syn[121], as well as lipid peroxidation, protein oxidation, and DNA damage, contributing to the loss of dopaminergic cells in the substantia nigra, a hallmark of PD[122].
Hypoxia can increase mitochondrial ROS production by limiting oxygen availability to complex III and regulating antioxidant gene expression[1]. Activation of the HIF-1 signaling pathway inhibits the expression of key mitochondrial ribosome components and ETC proteins COX2 and CYTB, resulting in abnormal mitochondrial membrane potential[123]. ROS can increase HIF-1α stability, while HIF-1α helps mitigate excessive ROS production by modulating mitochondrial oxygen consumption and inhibiting mtDNA-encoded mRNA expression[1]. Compared with wild-type cells, HIF-1α-knockout cells produce more ROS due to increased ATP production and impaired mitophagy[124]. Data show that arsenide stabilizes HIF-1α in a ROS-dependent manner by consuming ascorbic acid and Fe (II), which inhibits PHD activity and contributes to HIF-1α stability[125]. ROS-induced attenuation of NO-mediated HIF-1α regulation is mediated through the active proteasome degradation pathway[126]. Inhibition of mitochondrial ROS can eliminate Ang II-induced HIF-1 protein stability, HIF-1-dependent transcription, and VSMC migration[127]. ROS also influences the regulation of HIF-1-activated VEGF[128] and up-regulates HIF-1α expression through the PI3K/AKT signaling pathway[129].
Notably, ROS activates Nrf2, another key hypoxia regulator. Knockdown of Nrf2 results in decreased antioxidant gene expression and increased ROS levels[130]. The Keap1-Nrf2 pathway regulates mitochondrial and cytoplasmic ROS production through NADPH oxidase[131]. Interestingly, while hypoxia-induced mitochondrial ROS formation stabilizes HIF-1α[132], HIF-1α is also stabilized in cells lacking mitochondrial respiration (U0 cells)[133]. Further research is needed to explore how factors such as hypoxia duration and oxygen concentration may influence these interactions.
4.3. Lysosome Damage
Mitochondrial dysfunction can lead to lysosomal damage[134,135,136]. Lysosomes play a crucial role in supporting mitochondrial metabolism by regulating iron homeostasis[137,138]. Dysfunctional lysosomes can impair mitochondrial function, leading to the accumulation of damaged mitochondria[139]. The physical and functional communication between mitochondria and lysosomes underscores this interaction.
Under hypoxic conditions, mitochondrial-lysosomal contact is enhanced, with some lysosomes being engulfed by enlarged mitochondria, a process termed giant mitochondrial engulfing lysosomes (MMEL). Following MMEL, the lysosomal membrane ruptures, releasing lysosomal proteases into the enlarged mitochondria. This process, combined with mitochondrial proteases, facilitates mitochondrial self-digestion under hypoxia and promotes mitochondrial ROS production[140].
Lysosomes are central to cellular homeostasis, managing the turnover of proteins, lipids, and other macromolecules through endocytosis, phagocytosis, and autophagy[141]. In neurodegenerative diseases like PD, frontotemporal dementia, and ALS, numerous risk variants in lysosomal genes have been identified[142,143]. Dysfunction in lysosomes of glial and neuronal cells can lead to the spread of pathological features, such as amyloid-β and tau in AD and α-syn in PD[144,145,146].
Hypoxia can alter the distribution of lysosomal proteins, resulting in lysosomal membrane permeability [147]. The ATPase H+ transporter V1 subunit A (ATP6V1A) is vital for maintaining lysosomal homeostasis. Hypoxia impairs lysosomal function in head and neck squamous cell carcinoma by binding HIF-1α to the E-box motif in the ATP6V1A promoter, leading to down-regulation of ATP6V1A expression[148]. This impairment reduces the fusion of multivesicular bodies (MVBs) with lysosomes and decreases the secretion of intracavitary vesicles (ILVs) into extracellular vesicles (EVs). Hypoxia disrupts lysosomal function by affecting lysosomal acidification rather than its biosynthesis. Re-expressing ATP6V1A under hypoxic conditions can restore lysosomal function and EVs release levels.
Furthermore, overexpression of HIF-1α and inhibition of HIF-1α ubiquitination impair lysosomal function and enhance EV secretion. Several studies have documented the impact of HIF-1α on EV release[149,150]. For instance, King et al. reported that increased expression of plasma membrane receptors regulated by HIF-1α can lead to self-activation, promoting endocytosis and EV release. Studies have also shown that lysosomal-associated membrane proteins (LAMPs) and the key lysosomal protease cathepsin D (CTSD) are down-regulated in hypoxic-exposed trophoblast cells, eventually leading to lysosomal dysfunction[151]. In addition, hypoxia and the resulting acidosis in tumors can cause lysosomal movement, abnormal expression of LAMPs, lysosomal compartment expansion, and lysosomal deacidification, all associated with impaired lysosomal activity[152,153,154,155].
Under hypoxic conditions, chondrocytes exhibit increased levels of the autophagy marker LC3, the lysosomal marker LAMP1, and CTSD. This results in an increase in the number of autophagosomes, lysosomes, and defective lysosomes. Transcription factor EB, a key regulator of lysosomal biosynthesis, has transcription binding sites in its promoter region regulated by HIF-1α and HIF-2α, indicating that HIF-α is involved in regulating lysosomal biosynthesis and function. Additionally, hypoxia increases endoplasmic reticulum (ER) stress in chondrocytes via the PERK signaling pathway, which is upstream of the autophagy-lysosomal signaling pathway[156]. Hypoxia can also enhance ROS elimination by activating autophagy, thereby increasing cellular resistance to radiation[157].
4.4. PD and Receptors/Neurotransmitters
Hypoxia and neurotoxicity are potential contributors to PD, with δ-opioid receptors (DOR) showing neuroprotective effects against hypoxia and ischemic injury. In the brain, MPTP is metabolized by monoamine oxidase B (MAO-B) in glial cells into an intermediate called MPDP+. This intermediate is further oxidized to form the toxic MPP+. MPP+ is a neurotoxin that mimics PD by disrupting dopaminergic neurons in the substantia nigra both in vivo and in vitro [158,159]. In SH-SY5Y PD model cells, inhibition of complex I by rotenone or MPP+ also results in decreased levels of HIF-1α[160].
Both hypoxia and MPP+ stress have been identified as potential pathogenic factors leading to PD[81,161]. Prolonged hypoxia or high concentrations of MPP+ reduce cell viability, decrease PINK1 protein levels, and increase caspase 3 cleavage. DOR activation protects cells from hypoxia and MPP+ injury by up-regulating PINK1 and down-regulating cleaved caspase 3. In contrast, inhibition of DOR exacerbates these effects.
Previous studies have demonstrated that they induce apoptosis by disrupting mitochondrial stability[162,163,164]. Additionally, apoptosis has been found to be caspase-dependent[165]. Activation of DOR by UFP-512 significantly reduces hypoxia-induced cell damage and α-syn overexpression. DOR antagonists like naltrindole or DOR siRNA knockdown reduce hypoxia-induced cell damage and α-syn overexpression. These findings suggest that DOR activation mitigates MPP+ or severe hypoxia-induced aggregation of α-syn through the CREB pathway[23]. However, unlike MPP+, hypoxia-induced mitophagy is not dependent on DOR-PINK1 signaling pathway[166]. Additionally, in PD models, changes in the serotonin system, beyond the dopamine pathway, have been noted[167,168,169].
5-Hydroxytryptamine (5-HT), a monoamine neurotransmitter, regulates hypoxic ventilatory responses. The 5-HT 2 receptor is involved in both normoxic and hypoxic respiration in PD model rats. Unilateral injection of 6-hydroxydopamine (6-OHDA) into the rat medial forebrain bundle (MFB) affects the hypoxic ventilation response via 5-HT 2 receptor stimulation[170]. However, some studies suggest that the raphe in the medulla oblongata and its 5-HT neurons do not mediate the respiratory response to hypoxia, and 5-HT absence in transgenic mice does not affect long-term ventilation after intermittent hypoxia[171]. Patients with mild PD exhibit weakened hyperventilation responses to hypoxic stimulation. In the rat MFB PD model, dopamine depletion primarily leads to enhanced ventilation responses to hypoxia and the disappearance of central and peripheral D2 dopamine receptor antagonism on respiration under both hypoxic and normoxic conditions[170].
DJ-1 plays a crucial role in the expression of rearrangement (RET), a receptor for neurotrophic factors and a neuroprotective molecule for dopaminergic neurons. Its ligand, glial cell line-derived neurotrophic factor, promotes the survival and differentiation of midbrain DA cells, making RET a key target in PD degeneration[172]. Inducible loss of DJ-1 triggers hypoxia and ROS production, stabilizing HIF-1α. HIF-1α expression is essential for RET downregulation[173].
The synthesis of neurotransmitters and modulators is regulated by oxygen-demand rate-limiting enzymes, meaning hypoxia, which disrupts O2 homeostasis, can affect neuronal function by altering neurotransmitter synthesis. Hypoxia can be classified into persistent hypoxia and intermittent hypoxia [174]. Dysfunction of the nicotinic acetylcholine receptor (nAChR) is associated with PD. In addition, HIF-1α also interacts with mitochondrial nAChRs. Nicotine stimulates HIF-1α expression via activation of α7 nAChRs, which interact with HIF-1α to regulate its translocation to the nucleus and mitochondria[175]. Nitric oxide (NO) is involved in neurotransmitter-mediated activation and inhibition of the inflammatory cascade. Excessive NO production by activated microglia is associated with neuroinflammation and neurodegenerative diseases. Short-term inhibition of neuronal nitric oxide synthase (nNOS) by CH is due to reduced oxygen supply[176].
As mentioned, molecular events specific to PD pathogenesis can interfere with the expression and stability of HIF-1α. These disruptions can impair cellular antioxidant capacity, mitochondrial function, and metabolic processes. HIF-1α activates several transcriptional processes targeting oxidative stress, including autophagy and mitochondrial function, which influence PD development[1]. It is important to note that the pathogenesis of PD remains unclear and may involve various molecular events, each affecting hypoxic pathways and treatment responses differently.
5. The Role of PD-Related Genes in Hypoxia
To date, over 20 genetic variations associated with PD have been identified, collectively accounting for about 5-10% of PD cases. Among these, mutations in genes encoding the proteins Parkin, PINK1, DJ-1, and TMEM175 can lead to autosomal recessive PD. These genetic mutations are closely related to PD risk and provide significant insights into its pathogenesis. Notably, several of these genes are linked to mitochondrial dysfunction and hypoxic responses (Figure 5)[113].
5.1. Hypoxia Promotes Parkin/PINK1 Pathway
Parkin (PARK2) is a RING-in-between-RING (RBR) type E3 ubiquitin ligase. The PARK2 mutation is the most common cause of recessive PD, particularly in familial early-onset cases, accounting for over 70%[177]. Mutations in Parkin increase oxidative stress, promote mitochondrial swelling and fission, and affect mitochondrial phagocytosis, leading to mitochondrial defects and ultimately resulting in the loss of dopamine neurons[178,179]. Parkin is involved in ubiquitination, and mutations disrupt its E3 ligase activity, including its binding to α-syn, which contributes to the formation of Lewy bodies[180]. Interestingly, recent studies using quantitative proteomics have identified HIF-1α as a potential ubiquitination substrate for Parkin[181]. Parkin has been shown to reduce HIF-1α levels in glioblastoma cells. It binds to HIF-1α and promotes HIF-1α degradation through ubiquitination, thereby inhibiting breast cancer cell metastasis. This effect is independent of oxygen levels[182].
HIF-1α, in turn, promotes Parkin/PINK1-mediated mitophagy[183] and can upregulate mitophagy in the spinal cord of mice with diabetic neuropathic pain through the Parkin signaling pathway[184]. Additionally, FSH activates mitophagy via the HIF-1α-PINK1-Parkin pathway, reducing hypoxia-induced apoptosis in porcine ovarian granulosa cells[185]. Parkin regulates the expression of HIF-1α and HIF-3α in glioblastoma-derived cell lines in vitro. Under normoxic conditions, Parkin silencing leads to HIF-1α accumulation in the nucleus, while under hypoxic conditions, Parkin affects HIF expression. Parkin knockdown decreases HIF-3α immunoreactivity in both the cytoplasm and nucleus, suggesting a positive role for Parkin in regulating HIF-3α[186]. Conversely, the inhibitory PAS domain protein (IPAS), a splicing variant of HIF-3α, generally inhibits HIF-1, and reduced PINK1-Parkin activity stabilizes IPAS, increasing its mitochondrial level and affecting HIF-1α expression[187]. The relationship between Parkin and hypoxia pathways warrants further investigation.
PTEN-induced putative kinase 1 (PINK1) is a serine/threonine kinase located in mitochondria and cytoplasm, crucial for regulating mitochondrial function. By phosphorylating mitochondrial proteins, PINK1 helps protect against mitochondrial dysfunction during cell stress[188]. PINK1 gene knockout leads to mitochondrial dysfunction and potential dopaminergic neuron death. Mutations in PINK1 account for about 9% of familial early-onset PD cases.
PINK1 is essential for the translocation of Parkin to depolarized mitochondria, promoting the clearance of Parkin through mitophagy[189]. PINK1 recruits Parkin from the cytoplasm to mitochondria, where Parkin facilitates mitochondrial phagocytosis[190]. Studies have demonstrated a strong correlation between PINK1 and HIF-1α. PINK1 knockout alters the expression of many genes in the HIF-1α pathway[191]. DOR protects neurons from hypoxia and MPP+ by upregulating PINK1[192].
Using hypoxic stress models, researchers have found that PINK1 deficiency attenuates the induction of HIF-1α across three different cell types. PINK1 mediates the HRE gene response required for handling hypoxic stress response through HIF-1α translation regulation[193]. This suggests that changes in PINK1 expression may be an adaptive response to hypoxic environments. Under hypoxic conditions, PINK1 knockout mice show significantly reduced HIF-1α protein accumulation, transcriptional activity, and expression of hypoxia-responsive genes[194]. HIF-1α mitigates oxidative stress through the HEY1/PINK1 pathway, and PINK1 expression can be modulated by changes in oxygen levels, impacting mitochondrial development in tumor cell lines[195]. Additionally, the JCYSTL formula activates PINK1/Parkin-mediated mitophagy by stabilizing HIF-1α[196]. Given the close interaction between Parkin and PINK1 in mitochondrial regulation, the links between PINK1 and HIF-1α are also likely involved in Parkin’s function.
5.2. DJ-1 Reduces Oxidative Stress
DJ-1 (PARK7) has over 20 mutations associated with early-onset PD[197]. It mitigates oxidative stress through various signaling pathways, serving as a cellular protective mechanism[198]. In the human brain, DJ-1 is highly expressed in reactive astrocytes but less so in neurons[199]. DJ-1 is crucial for mitochondrial function and acts as a redox sensor within mitochondria. Abnormal DJ-1 expression leads to mitochondrial defects and increased oxidative stress, affecting mitochondrial phagocytosis[200]. DJ-1 is involved in the hypoxia pathway and can be induced by oxidative stress, mediating hypoxia-induced cell responses.
As early as 1997, researchers identified DJ-1's role in protecting neurons from hypoxia-induced cell death[201]. DJ-1 is essential for HIF-1α target gene expression. In U2OS cells, DJ-1 deficiency alters HIF-1α-induced gene expression and impairs transcription of many HIF-1α target genes. Additionally, DJ-1 is crucial for maintaining HIF-1α-stable Akt and mTOR activity[202]. DJ-1 deficiency reduces HIF-1α expression in neuroblastoma cells, and its regulation of the DJ-1/PI3K/AKT pathway impacts HIF-1α protein expression and transcriptional activity[203]. DJ-1 inhibits the VHL ubiquitination activity of HIF-1α subunit by inhibiting HIF-1α-VHL interaction. The decreased expression of VEGF mRNA after DJ-1 deletion suggests that DJ-1 mediates neuronal survival through the VHL-HIF-1α pathway[204].
5.3. LRRK2 can Be Activated by HIF-1α
LRRK2, also known as PARK8 is widely expressed throughout the brain and surrounding tissues, where it influences neurite growth and autophagic protein degradation[205]. LRRK2 plays a role in mitochondrial function and cytoskeleton dynamics[206], which are critical for various cellular processes. Mutations in LRRK2 are a major cause of autosomal dominant PD and are associated with either mitochondrial phagocytosis and other normal function or α-syn pathology and other neurodegenerative proteins[207]. After traumatic brain injury, HIF-1α directly binds to the LRRK2 proximal promoter, leading to increased LRRK2 transcription and exacerbating neuronal cell death[208]. In cases of anemia or hypoxia, HIF binds to the erythropoietin (EPO) 5' hypoxia response element, resulting in increased EPO gene transcription[209]. In the liver of LRRK2 knockout mice with hemangiomas, there is an increase in HIF-2α protein expression and significant reactivation of HIF-2α target gene EPO, reflecting the role of the HIF-2α pathway in angiogenesis[210]. Additionally, in human cancers, LRRK2 phosphorylates HIF-1α at Ser797, which enhances HIF-1α's binding affinity to its cofactor p300, thereby increasing the production of HIF-1α target genes.
5.4. TMEM175 May Influence HIF-1α.
Lysosomal K+ conductance, mediated by the lysosomal membrane protein Transmembrane Protein 175 (TMEM175), is essential for maintaining lysosomal membrane potential, pH stability, and regulating lysosomal-autophagosome fusion[211,212]. TMEM175 acts as a proton-permeable, cation-selective channel, helping to stabilize intracellular pH by facilitating lysosomal hydrogen ion efflux. Compared to wild-type neurons, TMEM175 knockout neurons exhibit increased aggregation of phosphorylated α-syn, while TMEM175 overexpression reduces these inclusions, suggesting that TMEM175 enhances the lysosomal degradation of α-syn aggregates.
TMEM175 knockdown decreases Cathepsin B and Glucosylceramidase Beta (GBA) enzyme activity and promotes α-syn accumulation[213]. In mice, the loss of TMEM175 function results in dopaminergic neuron loss and progressive dyskinesia[214]. Dysfunction in mitochondrial autophagy caused by TMEM175 loss-of-function mutations is linked to several central nervous system diseases, including Alzheimer's[215] and Parkinson's diseases[216]. Additionally, TMEM175 protein levels decreased after cerebral ischemia-reperfusion (I/R) injury, and its overexpression can mitigate brain cell death and neurobehavioral deficits caused by cerebral artery occlusion/reperfusion in vivo [217]. Excessive nitric oxide (NO) production during cerebral I/R injury leads to upregulation of HIF-1α, triggering an inflammatory response and ultimately neuronal apoptosis, which may explain TMEM175's effect on HIF-1α.
The pathogenic genes associated with PD discussed above are predominantly linked to hypoxic pathways and HIF-1α. They primarily influence PD progression by modulating cellular antioxidant capacity, mitochondrial function, autophagy pathways, and the aggregation of α-syn proteins.
6. Hypoxia-Based Therapeutic Strategies for PD
Currently, there is no cure for PD, but symptoms can be managed with medication. Approved treatments include DOPA decarboxylase inhibitors/ replenishing lost dopamine (DA) precursors, COMT inhibitors, DA agonists, MAO-B inhibitors, and other drugs with mixed mechanism[218]. These medications primarily address PD symptoms by dopamine and alleviating DA deficiency. Since hypoxia signaling is intrinsically involved in the pathogenesis of PD, targeting the HIF and Nrf2 pathways has been proposed as a strategy to ameliorate or prevent PD[219,220]. Hypoxia pathways, particularly HIF-1α and Nrf2, are considered promising targets due to their connection with PD-related molecular events, including protein degradation, mitochondrial function, ROS generation, and genetic mutations[1]. Consequently, an increasing number of studies are investigating drugs that target HIF-1α and Nrf2, as well as treatments aimed at improving hypoxia, as novel approaches to modulate aberrant pathways and treat PD[221].
6.1. Drugs Targeting Hypoxia Pathways
Deferoxamine mesylate (DFO) is an iron chelator that also functions as a hypoxia-mimetic agent, often referred to as a HIF-1α activator [167]. DFO stabilizes HIF-1α by inhibiting PHD activity[222]. Additionally, DFO alleviates iron imbalances induced by cypermethrin in rats and reduces α-syn A53T aggregation in PC12 cells[223,224]. In SH-SY5Y cells, DFO treatment decreases apoptosis and ROS generation caused by H2O2 or 6-OHDA, a compound that selectively destroys dopaminergic neurons[225].
Furthermore, DFO enhances autophagy in SH-SY5Y cells treated with rotenone or MPP+ through the HIF-1α and Beclin-1 pathways[172]. Notably, DFO also promotes AMPK-mediated Nrf2 pathway activation by inhibiting ferroptosis, which leads to reduced ROS generation[226,227]. Other iron chelators, including M30, lactoferrin, clioquinol, and FG-0041, also block PHD activity through iron chelation and subsequently activate HIF-1α and potentially Nrf2, thereby enhancing neuronal function and viability[228,229].
Albendazole (ABZ), a benzimidazole anthelmintic, enhances the expression of HIF-1α and its downstream target, VEGF, while inhibiting PHD activity[230]. It significantly reduces histological alterations in the SNpc, restores DA levels and motor functions, and decreases α-syn expression in a rat model of PD induced by rotenone[231]. Additionally, ABZ increases the expression of nuclear receptor-related 1 (Nurr1) in the SNpc and enhances the transcriptional activation of Nurr1-controlled genes, which are crucial for regulating DA synthesis[231].
Agmatine, a natural metabolite of the amino acid arginine, activates HIF-1α and thus prevents ROS production, mitochondrial membrane potential loss, and increases in caspase 3 and cytochrome c in rotenone-treated differentiated SH-SY5Y cells[232]. Consequently, agmatine alleviates motor symptoms in PD rats, inhibits inflammatory and oxidative stress pathways, and increases striatal dopamine levels through the activation of Nrf2 and other pathways[233,234].
6.2. Behavioral Modulation Improving Hypoxia
Preclinical and clinical evidence suggests that regular exposure to intermittent hypoxia may benefit PD patients both in the short and long term, highlighting potential treatment targets[9,14,235]. One study involving a 7-day walk in the Swedish mountains demonstrated significant improvements in motor performance in PD patients, with effects observed 4 days post-intervention and lasting for 3 months, although not beyond 6 months[236]. Quality-of-life assessments showed improvements 1 week after the intervention, but these benefits did not persist for 18 weeks[237,238]. Additionally, negative feelings related to daily life appeared to be alleviated following the intervention[239].
Another study used a 14-day hypoxia protocol involving controlled oxygen levels of 8-10% for 5-7 minutes, three times per day, to investigate the interaction between DA metabolism and hypoxia-induced ventilatory responses (HVRs)[240]. This intervention significantly improved HVRs in the PD group, and DA metabolism also showed improvement compared to healthy controls[241]. These findings suggest that hypoxic training can enhance dopamine metabolism, HVRs, motor symptoms, and overall quality of life in PD patients. Consistent with these results, intensive exercise has been shown to improve motor and cognitive symptoms in rat models of PD and to restore striatal synaptic plasticity damaged by α-syn aggregation[242].
7. Conclusions and Perspectives
As the second most common neurodegenerative disease, PD imposes a significant burden on society, which is increasing due to the rising life expectancy of the population. Understanding the pathogenesis and progression of PD is therefore of great concern. Hypoxia, which occurs both as a cause and a consequence of PD symptoms, exacerbates the disease. Hypoxic insults can directly induce PD-like features, including motor disorders, mood changes, and brain lesions in susceptible individuals. Conversely, PD patients exhibit impaired responses to hypoxic stimuli, reduced ventilatory function, and diminished recovery following cumulative hypoxia.
These observations suggest that hypoxia pathways, particularly the HIF-1α and Nrf2 pathways, are intrinsically linked to PD-related molecular events. Various molecular changes specific to PD influence HIF-1α and Nrf2 in different ways: some induce ROS and other stresses, leading to the activation of HIF-1α and Nrf2, while others accelerate their degradation or inhibit their functions, impairing the neurons' ability to adapt to hypoxia. Over-activation of HIF-1α and Nrf2 pathways can lead to apoptosis, whereas reduced activity of these pathways increases vulnerability to oxidative stress, iron dysregulation, and protein misfolding in neurons. The connection between hypoxia and PD is strongly supported by numerous studies. As a result, targeting the hypoxia pathways presents a promising strategy for developing new PD therapies.
However, it is crucial to recognize that hypoxia plays a dual role in PD, influencing various aspects of the disease's etiology. On one hand, hypoxia-induced neurotoxicity highlights the detrimental effects of low oxygen levels on neuronal health, contributing to brain damage and the onset of neurodegenerative disorders like PD[243]. As previously noted, hypoxia is a common feature in many neurological disorders, including PD[244]. For PD patients, particularly those with obstructive sleep apnea syndrome, hypoxia is associated with an increased risk of disease progression[7]. On the other hand, HIF-1α plays a unique neuroprotective role in PD. Insufficient levels of HIF-1α have been linked to the development of PD, underscoring the importance of this transcription factor in promoting cell survival, especially in dopaminergic neurons affected by PD. Additionally, δ-opioid receptor activation, as a HIf-1α stabilizer, has been shown to prevent hypoxia-related mitochondrial dysfunction associated with PD, suggesting a potential therapeutic target for mitigating the disease's pathology under low oxygen conditions[6].
Admittedly, the research on the relationship between hypoxia and PD still faces several debates and unanswered questions due to the complex and multifaceted effects of hypoxia on the disease. As mentioned before, the relationship between hypoxia and the onset and progression of neurodegenerative disorders continues to be debated[6]. Additionally, while mitochondria play a crucial role in regulating HIF-1α expression under hypoxic conditions, the mechanisms governing this regulation remain controversial[245]. Furthermore, studies indicate that activated hypoxia response pathways may exert varying effects across the spectrum of PD. There is ongoing debate about the optimal doses of hypoxic adaptation therapies and their potential risks[14]. Thus, there are unresolved issues regarding the effects of hypoxia on PD and other neurodegenerative conditions, as well as the diverse impacts of hypoxia response pathways and the regulatory mechanisms of HIF-1α in hypoxic environments.
To gain a clearer understanding of the connection between hypoxia and PD and to develop effective treatment strategies, further research is essential. The reciprocal relationship between hypoxia and PD underscores the need to balance the harmful effects of low oxygen on neuronal health with the potential neuroprotective mechanisms that could mitigate the disease's pathology. Future studies on the role of hypoxia in PD should focus on the effectiveness and underlying molecular mechanisms of hypoxia-based neuroprotective strategies[246]. Investigating the connections between hypoxia and PD pathophysiology—such as its effects on α-syn pathology, mitochondrial dysfunction, and the vulnerability of dopaminergic neurons—could provide crucial insights for developing novel treatment approaches. Additionally, research should explore the potential benefits of hypoxia-based therapy protocols, including continuous and intermittent hypoxia, with a focus on specific treatment plans and outcomes for PD patients[247]. Addressing these research gaps will enhance our understanding of hypoxia's role in PD progression and may lead to the development of innovative therapeutic strategies.
Author Contributions
Y.G. and J.Z., writing-original draft and editing; T.T., writing-review and editing; Z.L., writing-review, Open Access fee acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
The article processing charge (APC) for this manuscript was supported by the Natural Science Foundation of China, grant number 32200782 and the Science and Technology Development Plan Project of Jilin Province, China, grant number 20240404014ZP
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
The authors thank Boshuo Jian for organizing the original draft and all lab members to discuss this review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lestón Pinilla, L.; Ugun-Klusek, A.; Rutella, S.; De Girolamo, L.A. Hypoxia signaling in parkinson’s disease: There is use in asking “What HIF?”. Biology 2021, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.V.; Vedunova, M.V. The Role of Oxygen Homeostasis and the HIF-1 Factor in the Development of Neurodegeneration. International Journal of Molecular Sciences 2024, 25, 4581. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, Q.-Y.; Chao, D.-M.; Deng, Y.-D.; Liu, Y.-H.; Cai, B.-C.; Zhao, J.-N.; Wen, G.-Q.; Xia, Y. Interactive Responses of Enkephalin, δ-opioid Receptor and Dopamine Receptor D1/D2 to Hypoxia and/or MPP+ Stress in PC12 Cells. 2021.
- Auti, A.; Alessio, N.; Ballini, A.; Dioguardi, M.; Cantore, S.; Scacco, S.; Vitiello, A.; Quagliuolo, L.; Rinaldi, B.; Santacroce, L. Protective effect of resveratrol against hypoxia-induced neural oxidative stress. Journal of Personalized Medicine 2022, 12, 1202. [Google Scholar] [CrossRef] [PubMed]
- Fiskum, V.; Sandvig, A.; Sandvig, I. Silencing of activity during hypoxia improves functional outcomes in motor neuron networks in vitro. Frontiers in Integrative Neuroscience 2021, 15, 792863. [Google Scholar] [CrossRef]
- C Correia, S.; Carvalho, C.; Cardoso, S.; X Santos, R.; I Plácido, A.; Candeias, E.; I Duarte, A.; I Moreira, P. Defective HIF signaling pathway and brain response to hypoxia in neurodegenerative diseases: not an “iffy” question! Current pharmaceutical design 2013, 19, 6809–6822. [Google Scholar] [CrossRef]
- Sun, H.L.; Sun, B.L.; Chen, D.W.; Chen, Y.; Li, W.W.; Xu, M.Y.; Shen, Y.Y.; Xu, Z.Q.; Wang, Y.J.; Bu, X.L. Plasma α-synuclein levels are increased in patients with obstructive sleep apnea syndrome. Annals of clinical and translational neurology 2019, 6, 788–794. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E. MDS clinical diagnostic criteria for Parkinson's disease. Movement disorders 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Mirelman, A.; Bonato, P.; Camicioli, R.; Ellis, T.D.; Giladi, N.; Hamilton, J.L.; Hass, C.J.; Hausdorff, J.M.; Pelosin, E.; Almeida, Q.J. Gait impairments in Parkinson's disease. The Lancet Neurology 2019, 18, 697–708. [Google Scholar] [CrossRef]
- Sanchez-Luengos, I.; Lucas-Jiménez, O.; Ojeda, N.; Peña, J.; Gómez-Esteban, J.C.; Gómez-Beldarrain, M.Á.; Vázquez-Picón, R.; Foncea-Beti, N.; Ibarretxe-Bilbao, N. Predictors of health-related quality of life in Parkinson’s disease: the impact of overlap between health-related quality of life and clinical measures. Quality of Life Research 2022, 31, 3241–3252. [Google Scholar] [CrossRef]
- Brimson, J.M.; Tencomnao, T. Rhinacanthus nasutus protects cultured neuronal cells against hypoxia induced cell death. Molecules 2011, 16, 6322–6338. [Google Scholar] [CrossRef]
- Burtscher, J.; Syed, M.M.K.; Lashuel, H.A.; Millet, G.P. Hypoxia conditioning as a promising therapeutic target in Parkinson's disease? Movement Disorders 2021, 36, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Opazo, A.; Mitchell, G.S. Therapeutic potential of intermittent hypoxia: a matter of dose. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2014, 307, R1181–R1197. [Google Scholar] [CrossRef] [PubMed]
- Janssen Daalen, J.M.; Koopman, W.J.; Saris, C.G.; Meinders, M.J.; Thijssen, D.H.; Bloem, B.R. The Hypoxia Response Pathway: A Potential Intervention Target in Parkinson's Disease? Movement Disorders 2024, 39, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson's disease: preclinical and clinical outcomes. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2014, 1842, 1282–1294. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Sharma, R.; Kumar, D.; Ambasta, R.K.; Kumar, P. Hypoxia-induced signaling activation in neurodegenerative diseases: targets for new therapeutic strategies. Journal of Alzheimer's Disease 2018, 62, 15–38. [Google Scholar] [CrossRef]
- Jagečić, D.; Petrović, D.J.; Šimunić, I.; Isaković, J.; Mitrečić, D. The Oxygen and Glucose Deprivation of Immature Cells of the Nervous System Exerts Distinct Effects on Mitochondria, Mitophagy, and Autophagy, Depending on the Cells’ Differentiation Stage. Brain Sciences 2023, 13, 910. [Google Scholar] [CrossRef]
- Tian, H.-Y.; Huang, B.-Y.; Nie, H.-F.; Chen, X.-Y.; Zhou, Y.; Yang, T.; Cheng, S.-W.; Mei, Z.-G.; Ge, J.-W. The interplay between mitochondrial dysfunction and ferroptosis during ischemia-associated central nervous system diseases. Brain Sciences 2023, 13, 1367. [Google Scholar] [CrossRef]
- Diptendu, S.; Dev, M.G.; Basha, K.A.; Kumar, R.T.; Jahirhussain, G.; Sarasa, D. International Journal of Zoological Investigations.
- Baranich, T.; Voronkova, A.; Anufriev, P.; Brydun, A.; Turygina, S.; Glinkina, V.; Sukhorukov, V. Hypoxia-inducible Factors—Their Regulation and Function in Neural Tissue. Human Physiology 2020, 46, 895–899. [Google Scholar] [CrossRef]
- Lewczuk, A.; Zablocka, B.; Beresewicz-Haller, M. Is Nrf2 Behind Endogenous Neuroprotection of the Hippocampal CA2-4, DG Region? Molecular Neurobiology 2023, 60, 1645–1658. [Google Scholar] [CrossRef]
- Potteti, H.R.; Noone, P.M.; Tamatam, C.R.; Ankireddy, A.; Noel, S.; Rabb, H.; Reddy, S.P. Nrf2 mediates hypoxia-inducible HIF1α activation in kidney tubular epithelial cells. American Journal of Physiology-Renal Physiology 2021, 320, F464–F474. [Google Scholar] [CrossRef]
- Chen, T.; Li, J.; Chao, D.; Sandhu, H.K.; Liao, X.; Zhao, J.; Wen, G.; Xia, Y. δ-Opioid receptor activation reduces α-synuclein overexpression and oligomer formation induced by MPP+ and/or hypoxia. Experimental neurology 2014, 255, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, Z.; Fan, Y.; Fang, Y. An overview of biological research on hypoxia-inducible factors (HIFs). Endokrynologia Polska 2020, 71, 432–440. [Google Scholar] [CrossRef]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nature Reviews Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. The FASEB journal 2002, 16, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Cowman, S.J.; Koh, M.Y. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends in cancer 2022, 8, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef]
- Pasanen, A.; Heikkilä, M.; Rautavuoma, K.; Hirsilä, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3α is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. The international journal of biochemistry & cell biology 2010, 42, 1189–1200. [Google Scholar]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proceedings of the National Academy of Sciences 1997, 94, 4273–4278. [Google Scholar] [CrossRef]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; Kriegsheim, A. v.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin Jr, W.G. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Paltoglou, S.; Roberts, B. HIF-1α and EPAS ubiquitination mediated by the VHL tumour suppressor involves (flexibility in the ubiquitination mechanism, similar to other RING E3 ligases. Oncogene 2007, 26, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Camagni, G.F.; Minervini, G.; Tosatto, S.C. Structural Characterization of Hypoxia Inducible Factor α—Prolyl Hydroxylase Domain 2 Interaction through MD Simulations. International Journal of Molecular Sciences 2023, 24, 4710. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Discher, D.J.; Hu, J.; Bishopric, N.H.; Webster, K.A. Molecular regulation of the endothelin-1 gene by hypoxia: contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, and p300/CBP. Journal of Biological Chemistry 2001, 276, 12645–12653. [Google Scholar] [CrossRef]
- Lyubimov, A.V.; Cherkashin, D.V.; Efimov, S.V.; Alanichev, A.E.; Ivanov, V.S.; Kutelev, G.G. Mechanisms and triggers of adaptation to hypoxia. Reviews on Clinical Pharmacology and Drug Therapy 2021, 19, 269–280. [Google Scholar] [CrossRef]
- Ivy, C.M.; Velotta, J.P.; Cheviron, Z.A.; Scott, G.R. Genetic variation in HIF-2α attenuates ventilatory sensitivity and carotid body growth in chronic hypoxia in high-altitude deer mice. The Journal of physiology 2022, 600, 4207–4225. [Google Scholar] [CrossRef]
- Chen, H.; Ma, D.; Yue, F.; Qi, Y.; Dou, M.; Cui, L.; Xing, Y. The potential role of hypoxia-inducible factor-1 in the progression and therapy of central nervous system diseases. Current Neuropharmacology 2022, 20, 1651. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Savyuk, M.O.; Ponimaskin, E.; Vedunova, M.V. Hypoxia-inducible factor (HIF) in ischemic stroke and neurodegenerative disease. Frontiers in cell and developmental biology 2021, 9, 703084. [Google Scholar] [CrossRef]
- Sun, H.-y.; Wu, J.; Wang, R.; Zhang, S.; Xu, H.; Kaznacheyeva, Е.; Lu, X.-j.; Ren, H.-g.; Wang, G.-h. Pazopanib alleviates neuroinflammation and protects dopaminergic neurons in LPS-stimulated mouse model by inhibiting MEK4-JNK-AP-1 pathway. Acta Pharmacologica Sinica 2023, 44, 1135–1148. [Google Scholar] [CrossRef]
- Sheikh, M.A.; Malik, Y.S.; Xing, Z.; Guo, Z.; Tian, H.; Zhu, X.; Chen, X. Polylysine-modified polyethylenimine (PEI-PLL) mediated VEGF gene delivery protects dopaminergic neurons in cell culture and in rat models of Parkinson's Disease (PD). Acta biomaterialia 2017, 54, 58–68. [Google Scholar] [CrossRef]
- Yasuhara, T.; Shingo, T.; Muraoka, K.; Kameda, M.; Takeuchi, A.; Yano, A.; Nishio, S.; Matsui, T.; Miyoshi, Y.; Hamada, H. The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson's disease model. Brain research 2005, 1038, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer cell 2007, 11, 407–420. [Google Scholar] [CrossRef]
- Choi, Y.K. Detrimental Roles of Hypoxia-Inducible Factor-1α in Severe Hypoxic Brain Diseases. International Journal of Molecular Sciences 2024, 25, 4465. [Google Scholar] [CrossRef] [PubMed]
- Barteczek, P.; Li, L.; Ernst, A.-S.; Böhler, L.-I.; Marti, H.H.; Kunze, R. Neuronal HIF-1α and HIF-2α deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. Journal of Cerebral Blood Flow & Metabolism 2017, 37, 291–306. [Google Scholar]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proceedings of the National Academy of Sciences 2000, 97, 9082–9087. [Google Scholar] [CrossRef]
- Fei, P.; Wang, W.; Kim, S.-h.; Wang, S.; Burns, T.F.; Sax, J.K.; Buzzai, M.; Dicker, D.T.; McKenna, W.G.; Bernhard, E.J. Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer cell 2004, 6, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Vatte, S.; Ugale, R. HIF-1, an important regulator in potential new therapeutic approaches to ischemic stroke. Neurochemistry International 2023, 105605. [Google Scholar] [CrossRef]
- Koh, M.Y.; Powis, G. Passing the baton: the HIF switch. Trends in biochemical sciences 2012, 37, 364–372. [Google Scholar] [CrossRef]
- Moszyńska, A.; Jaśkiewicz, M.; Serocki, M.; Cabaj, A.; Crossman, D.K.; Bartoszewska, S.; Gebert, M.; Dąbrowski, M.; Collawn, J.F.; Bartoszewski, R. The hypoxia induced changes in miRNA-mRNA in RNA-induced silencing complexes and HIF-2 induced miRNAs in human endothelial cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 2022, 36, e22412. [Google Scholar] [CrossRef]
- Albadari, N.; Deng, S.; Li, W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert opinion on drug discovery 2019, 14, 667–682. [Google Scholar] [CrossRef]
- Formica, V.; Riondino, S.; Morelli, C.; Guerriero, S.; D’Amore, F.; Di Grazia, A.; Del Vecchio Blanco, G.; Sica, G.; Arkenau, H.-T.; Monteleone, G. HIF2α, Hepcidin and their crosstalk as tumour-promoting signalling. British Journal of Cancer 2023, 129, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, J.; Guo, M.; Gu, Y.; Guan, Y.; Shao, Q.; Ma, W.; Ji, X. Chronic hypoxia leads to cognitive impairment by promoting HIF-2α-mediated ceramide catabolism and alpha-synuclein hyperphosphorylation. Cell Death Discovery 2022, 8, 473. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ru, X.; Wen, T. NRF2, a transcription factor for stress response and beyond. International journal of molecular sciences 2020, 21, 4777. [Google Scholar] [CrossRef]
- La Vitola, P.; Balducci, C.; Baroni, M.; Artioli, L.; Santamaria, G.; Castiglioni, M.; Cerovic, M.; Colombo, L.; Caldinelli, L.; Pollegioni, L. Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson's models. Neuropathology and applied neurobiology 2021, 47, 43–60. [Google Scholar] [CrossRef]
- Yu, C.; Xiao, J.-H. The Keap1-Nrf2 system: a mediator between oxidative stress and aging. Oxidative Medicine and Cellular Longevity 2021, 2021, 6635460. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a therapeutic target in neurodegenerative diseases. ASN neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Skibinski, G.; Hwang, V.; Ando, D.M.; Daub, A.; Lee, A.K.; Ravisankar, A.; Modan, S.; Finucane, M.M.; Shaby, B.A.; Finkbeiner, S. Nrf2 mitigates LRRK2-and α-synuclein–induced neurodegeneration by modulating proteostasis. Proceedings of the National Academy of Sciences 2017, 114, 1165–1170. [Google Scholar] [CrossRef]
- Cuadrado, A. Brain-protective mechanisms of transcription factor NRF2: toward a common strategy for neurodegenerative diseases. Annual Review of Pharmacology and Toxicology 2022, 62, 255–277. [Google Scholar] [CrossRef] [PubMed]
- Padda, I.; Sethi, Y.; Das, M.; Fabian, D.; Ralhan, T.; Aziz, D.; Sexton, J.; Johal, G. Heme Oxygenase-1, Cardiac Senescence, and Myocardial Infarction: A Critical Review of the Triptych. Cardiovascular Drugs and Therapy 2024, 1–12. [Google Scholar] [CrossRef]
- Medina, M.V.; Sapochnik, D.; Garcia Solá, M.; Coso, O. Regulation of the expression of heme oxygenase-1: Signal transduction, gene promoter activation, and beyond. Antioxidants & Redox Signaling 2020, 32, 1033–1044. [Google Scholar]
- Sun, W.; Zheng, J.; Ma, J.; Wang, Z.; Shi, X.; Li, M.; Huang, S.; Hu, S.; Zhao, Z.; Li, D. Increased plasma heme oxygenase-1 levels in patients with early-stage Parkinson’s disease. Frontiers in Aging Neuroscience 2021, 13, 621508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. Journal of advanced research 2021, 34, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-H.; Hsieh, H.-L. Roles of heme oxygenase-1 in neuroinflammation and brain disorders. Antioxidants 2022, 11, 923. [Google Scholar] [CrossRef]
- Wang, H.; Dou, S.; Zhu, J.; Shao, Z.; Wang, C.; Cheng, B. Ghrelin mitigates MPP+-induced cytotoxicity: involvement of ERK1/2-mediated Nrf2/HO-1 and endoplasmic reticulum stress PERK signaling pathway. Peptides 2020, 133, 170374. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.-Y.; Hwang, H.; Rhim, H.; Kim, M.O. Gintonin mitigates MPTP-induced loss of nigrostriatal dopaminergic neurons and accumulation of α-synuclein via the Nrf2/HO-1 pathway. Molecular Neurobiology 2019, 56, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Mallet, R.T.; Burtscher, J.; Pialoux, V.; Pasha, Q.; Ahmad, Y.; Millet, G.P.; Burtscher, M. Molecular mechanisms of high-altitude acclimatization. International journal of molecular sciences 2023, 24, 1698. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.-Y.; Ho, P.W.-L.; Liu, H.-F.; Leung, C.-T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.-L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Translational Neurodegeneration 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Burtscher, J.; Mallet, R.T.; Burtscher, M.; Millet, G.P. Hypoxia and brain aging: Neurodegeneration or neuroprotection? Ageing research reviews 2021, 68, 101343. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative stress in Parkinson's disease: a systematic review and meta-analysis. Frontiers in molecular neuroscience 2018, 11, 236. [Google Scholar] [CrossRef]
- Lee, D.W.; Rajagopalan, S.; Siddiq, A.; Gwiazda, R.; Yang, L.; Beal, M.F.; Ratan, R.R.; Andersen, J.K. Inhibition of prolyl hydroxylase protects against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced neurotoxicity: model for the potential involvement of the hypoxia-inducible factor pathway in Parkinson disease. Journal of Biological Chemistry 2009, 284, 29065–29076. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Rajagopalan, S.; Ganesan, A.; Andersen, J.K. A Possible Novel Anti-Inflammatory Mechanism for the Pharmacological Prolyl Hydroxylase Inhibitor 3, 4-Dihydroxybenzoate: Implications for Use as a Therapeutic for Parkinson’s Disease. Parkinson’s disease 2012, 2012, 364684. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Shu, L.; Zhong, J.; Pan, H.; Guo, J.; Sun, Q.; Yan, X.; Tang, B.; Xu, Q. Association of HIF1A and Parkinson’s disease in a Han Chinese population demonstrated by molecular inversion probe analysis. Neurological Sciences 2019, 40, 1927–1931. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Duderstadt, Y.; Gatterer, H.; Burtscher, M.; Vozdek, R.; Millet, G.P.; Hicks, A.A.; Ehrenreich, H.; Kopp, M. Hypoxia Sensing and Responses in Parkinson’s Disease. International Journal of Molecular Sciences 2024, 25, 1759. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nature Reviews Neuroscience 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.-É. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Current Biology 2015, 25, 2349–2360. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Communication and Signaling 2013, 11, 1–18. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson's disease. Journal of neurochemistry 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Karunasinghe, R.N.; Lipski, J. Oxygen and glucose deprivation (OGD)-induced spreading depression in the Substantia Nigra. Brain research 2013, 1527, 209–221. [Google Scholar] [CrossRef]
- Onodera, H.; Okabe, S.; Kikuchi, Y.; Tsuda, T.; Itoyama, Y. Impaired chemosensitivity and perception of dyspnoea in Parkinson's disease. The Lancet 2000, 356, 739–740. [Google Scholar] [CrossRef]
- Baille, G.; Perez, T.; Devos, D.; Deken, V.; Defebvre, L.; Moreau, C. Early occurrence of inspiratory muscle weakness in Parkinson’s disease. PLoS One 2018, 13, e0190400. [Google Scholar] [CrossRef] [PubMed]
- Herer, B.; Arnulf, I.; Housset, B. Effects of levodopa on pulmonary function in Parkinson's disease. Chest 2001, 119, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Pal, P.K.; Sathyaprabha, T.N.; Tuhina, P.; Thennarasu, K. Pattern of subclinical pulmonary dysfunctions in Parkinson's disease and the effect of levodopa. Movement disorders: official journal of the Movement Disorder Society 2007, 22, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Sabaté, M.; Gonzalez, I.; Ruperez, F.; Rodríguez, M. Obstructive and restrictive pulmonary dysfunctions in Parkinson's disease. Journal of the neurological sciences, 1996; 138, 114–119. [Google Scholar]
- Fujii, Y. Olprinone/dopamine combination for improving diaphragmatic fatigue in pentobarbital-anesthetized dogs. Current therapeutic research 2006, 67, 204–213. [Google Scholar] [CrossRef]
- Aubier, M.; Murciano, D.; Menu, Y.; Boczkowski, J.; Mal, H.; Pariente, R. Dopamine effects on diaphragmatic strength during acute respiratory failure in chronic obstructive pulmonary disease. Annals of internal medicine 1989, 110, 17–23. [Google Scholar] [CrossRef]
- Binder, D.K. Neurotrophins in the dentate gyrus. Progress in brain research 2007, 163, 371–397. [Google Scholar]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. Journal of neurochemistry 2005, 93, 1412–1421. [Google Scholar] [CrossRef]
- Hartman, W.; Helan, M.; Smelter, D.; Sathish, V.; Thompson, M.; Pabelick, C.M.; Johnson, B.; Prakash, Y. Role of hypoxia-induced brain derived neurotrophic factor in human pulmonary artery smooth muscle. PloS one 2015, 10, e0129489. [Google Scholar] [CrossRef]
- Burtscher, J.; Citherlet, T.; Camacho-Cardenosa, A.; Camacho-Cardenosa, M.; Raberin, A.; Krumm, B.; Hohenauer, E.; Egg, M.; Lichtblau, M.; Müller, J. Mechanisms underlying the health benefits of intermittent hypoxia conditioning. The Journal of physiology 2023. [Google Scholar] [CrossRef]
- Hatano, T.; Okuzumi, A.; Matsumoto, G.; Tsunemi, T.; Hattori, N. α-Synuclein: A Promising Biomarker for Parkinson’s Disease and Related Disorders. Journal of Movement Disorders 2024, 17, 127. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. Journal of Biological Chemistry 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Longhena, F.; Faustini, G.; Spillantini, M.G.; Bellucci, A. Living in promiscuity: the multiple partners of alpha-synuclein at the synapse in physiology and pathology. International journal of molecular sciences 2019, 20, 141. [Google Scholar] [CrossRef] [PubMed]
- Salmina, A.B.; Kapkaeva, M.R.; Vetchinova, A.S.; Illarioshkin, S.N. Novel approaches used to examine and control neurogenesis in Parkinson′ s disease. International Journal of Molecular Sciences 2021, 22, 9608. [Google Scholar] [CrossRef]
- Fusco, G.; Sanz-Hernandez, M.; De Simone, A. Order and disorder in the physiological membrane binding of α-synuclein. Current opinion in structural biology 2018, 48, 49–57. [Google Scholar] [CrossRef]
- Booms, A.; Coetzee, G.A. Functions of intracellular alpha-synuclein in microglia: implications for Parkinson’s disease risk. Frontiers in Cellular Neuroscience 2021, 15, 759571. [Google Scholar] [CrossRef]
- Tripathi, T.; Chattopadhyay, K. Interaction of α-Synuclein with ATP Synthase: switching Role from Physiological to Pathological. ACS Chemical Neuroscience 2018, 10, 16–17. [Google Scholar] [CrossRef]
- Han, D.; Zheng, W.; Wang, X.; Chen, Z. Proteostasis of α-synuclein and its role in the pathogenesis of Parkinson’s disease. Frontiers in cellular neuroscience 2020, 14, 45. [Google Scholar] [CrossRef]
- Irwin, D.J.; Abrams, J.Y.; Schonberger, L.B.; Leschek, E.W.; Mills, J.L.; Lee, V.M.-Y.; Trojanowski, J.Q. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA neurology 2013, 70, 462–468. [Google Scholar] [CrossRef]
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-like mechanisms in Parkinson’s disease. Frontiers in neuroscience 2019, 13, 552. [Google Scholar] [CrossRef]
- Zhao, K.; Lim, Y.-J.; Liu, Z.; Long, H.; Sun, Y.; Hu, J.-J.; Zhao, C.; Tao, Y.; Zhang, X.; Li, D. Parkinson’s disease-related phosphorylation at Tyr39 rearranges α-synuclein amyloid fibril structure revealed by cryo-EM. Proceedings of the National Academy of Sciences 2020, 117, 20305–20315. [Google Scholar] [CrossRef] [PubMed]
- Qiao, H.; He, X.; Zhang, Q.; Zhang, N.; Li, L.; Hui, Y.; Li, W.; Wang, D.; Wu, Z. A-synuclein induces microglial cell migration through stimulating HIF-1α accumulation. Journal of Neuroscience Research 2017, 95, 1809–1817. [Google Scholar] [CrossRef]
- Lin, D.; Zhang, H.; Zhang, J.; Huang, K.; Chen, Y.; Jing, X.; Tao, E. α-Synuclein induces neuroinflammation injury through the IL6ST-AS/STAT3/HIF-1α axis. International Journal of Molecular Sciences 2023, 24, 1436. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke induction of α-synuclein mediates ischemic brain damage. Journal of Neuroscience 2016, 36, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Suthar, S.K.; Lee, S.-Y. Truncation or proteolysis of α-synuclein in Parkinsonism. Ageing Research Reviews 2023, 90, 101978. [Google Scholar] [CrossRef] [PubMed]
- Weimers, P.; Halfvarson, J.; Sachs, M.C.; Saunders-Pullman, R.; Ludvigsson, J.F.; Peter, I.; Burisch, J.; Olén, O. Inflammatory bowel disease and Parkinson’s disease: a nationwide Swedish cohort study. Inflammatory bowel diseases 2019, 25, 111–123. [Google Scholar] [CrossRef]
- Nath, S.; Goodwin, J.; Engelborghs, Y.; Pountney, D. Raised calcium promotes α-synuclein aggregate formation. Molecular and Cellular Neuroscience 2011, 46, 516–526. [Google Scholar] [CrossRef]
- Martinez, J.; Moeller, I.; Erdjument-Bromage, H.; Tempst, P.; Lauring, B. Parkinson's disease-associated α-synuclein is a calmodulin substrate. Journal of Biological Chemistry 2003, 278, 17379–17387. [Google Scholar] [CrossRef]
- Yuan, G.; Nanduri, J.; Bhasker, C.R.; Semenza, G.L.; Prabhakar, N.R. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. Journal of Biological Chemistry 2005, 280, 4321–4328. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, J.; Chang, Y.; ShiDu Yan, S.; Shi, H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Current medicinal chemistry 2011, 18, 4335–4343. [Google Scholar] [CrossRef]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and α-synuclein pathology in Parkinson's disease. Free Radical Biology and Medicine 2019, 141, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, C.; Tsuchiya, K.; Maeda, K. Hypoxia-inducible factor prolyl hydroxylase inhibitors and iron metabolism. International Journal of Molecular Sciences 2023, 24, 3037. [Google Scholar] [CrossRef]
- Magalhaes, J.; Tresse, E.; Ejlerskov, P.; Hu, E.; Liu, Y.; Marin, A.; Montalant, A.; Satriano, L.; Rundsten, C.F.; Carlsen, E.M.M. PIAS2-mediated blockade of IFN-β signaling: a basis for sporadic Parkinson disease dementia. Molecular psychiatry 2021, 26, 6083–6099. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J. Direct membrane association drives mitochondrial fission by the parkinson disease-associated protein α-Synuclein*. Journal of Biological Chemistry 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Du, T.; Wang, X.; Duan, C.; Gao, G.; Zhang, J.; Lu, L.; Yang, H. α-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain research 2014, 1591, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Kong, Q.; Lin, C.-l. G. Oxidative damage to RNA: mechanisms, consequences, and diseases. Cellular and Molecular Life Sciences 2010, 67, 1817–1829. [Google Scholar] [CrossRef]
- Burmann, B.M.; Gerez, J.A.; Matečko-Burmann, I.; Campioni, S.; Kumari, P.; Ghosh, D.; Mazur, A.; Aspholm, E.E.; Šulskis, D.; Wawrzyniuk, M. Regulation of α-synuclein by chaperones in mammalian cells. Nature 2020, 577, 127–132. [Google Scholar] [CrossRef]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.-L.; Zhang, C.-w.; Li, L. The sources of reactive oxygen species and its possible role in the pathogenesis of Parkinson’s disease. Parkinson’s disease 2018, 2018, 9163040. [Google Scholar] [CrossRef]
- Peng, Y.; He, J.; Xiang, H.; Xie, L.; She, J.; Cheng, D.; Liu, B.; Hu, J.; Qian, H. Potential Impact of Hypoxic Astrocytes on the Aggravation of Depressive Symptoms in Parkinson’s Disease. Journal of Molecular Neuroscience 2024, 74, 28. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. Journal of Biological Chemistry 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-N.; Xi, M.-M.; Guo, Y.; Hai, C.-X.; Yang, W.-L.; Qin, X.-J. NADPH oxidase-mitochondria axis-derived ROS mediate arsenite-induced HIF-1α stabilization by inhibiting prolyl hydroxylases activity. Toxicology letters 2014, 224, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Köhl, R.; Zhou, J.; Brüne, B. Reactive oxygen species attenuate nitric-oxide-mediated hypoxia-inducible factor-1α stabilization. Free Radical Biology and Medicine 2006, 40, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: the essential role of mitochondrial-derived reactive oxygen species. Molecular biology of the cell 2010, 21, 3247–3257. [Google Scholar] [CrossRef]
- Hu, Z.; Dong, N.; Lu, D.; Jiang, X.; Xu, J.; Wu, Z.; Zheng, D.; Wechsler, D.S. A positive feedback loop between ROS and Mxi1-0 promotes hypoxia-induced VEGF expression in human hepatocellular carcinoma cells. Cellular Signalling 2017, 31, 79–86. [Google Scholar] [CrossRef]
- Chen, C.; Guo, Z.; Shi, X.; Guo, Y.; Ma, G.; Ma, J.; Yu, Q. H2O2-induced oxidative stress improves meat tenderness by accelerating glycolysis via hypoxia-inducible factor-1α signaling pathway in postmortem bovine muscle. Food Chemistry: X 2022, 16, 100466. [Google Scholar] [CrossRef]
- Gremmels, H.; De Jong, O.G.; Hazenbrink, D.H.; Fledderus, J.O.; Verhaar, M.C. The transcription factor Nrf2 protects angiogenic capacity of endothelial colony-forming cells in high-oxygen radical stress conditions. Stem cells international 2017, 2017, 4680612. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochimica et Biophysica Acta (BBA)-General Subjects 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. Journal of Biological Chemistry 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Srinivas, V.; Leshchinsky, I.; Sang, N.; King, M.P.; Minchenko, A.; Caro, J. Oxygen sensing and HIF-1 activation does not require an active mitochondrial respiratory chain electron-transfer pathway. Journal of Biological Chemistry 2001, 276, 21995–21998. [Google Scholar] [CrossRef]
- Baixauli, F.; Acín-Pérez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabandé-Rodriguez, E.; Ledesma, M.D.; Blázquez, A.; Martin, M.A.; Falcón-Pérez, J.M. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell metabolism 2015, 22, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Mosquera, L.; Yambire, K.F.; Couto, R.; Pereyra, L.; Pabis, K.; Ponsford, A.H.; Diogo, C.V.; Stagi, M.; Milosevic, I.; Raimundo, N. Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy 2019, 15, 1572–1591. [Google Scholar] [CrossRef] [PubMed]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of mitochondrial function impairs lysosomes. Journal of biological chemistry 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Coody, T.K.; Jeong, M.-Y.; Berg, J.A.; Winge, D.R.; Hughes, A.L. Cysteine toxicity drives age-related mitochondrial decline by altering iron homeostasis. Cell 2020, 180, 296–310.e18. [Google Scholar] [CrossRef]
- Weber, R.A.; Yen, F.S.; Nicholson, S.P.; Alwaseem, H.; Bayraktar, E.C.; Alam, M.; Timson, R.C.; La, K.; Abu-Remaileh, M.; Molina, H. Maintaining iron homeostasis is the key role of lysosomal acidity for cell proliferation. Molecular cell 2020, 77, 645–655.e7. [Google Scholar] [CrossRef]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–lysosome crosstalk: from physiology to neurodegeneration. Trends in molecular medicine 2020, 26, 71–88. [Google Scholar] [CrossRef]
- Hao, T.; Yu, J.; Wu, Z.; Jiang, J.; Gong, L.; Wang, B.; Guo, H.; Zhao, H.; Lu, B.; Engelender, S. Hypoxia-reprogramed megamitochondrion contacts and engulfs lysosome to mediate mitochondrial self-digestion. Nature Communications 2023, 14, 4105. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nature reviews Molecular cell biology 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Wallings, R.L.; Humble, S.W.; Ward, M.E.; Wade-Martins, R. Lysosomal dysfunction at the centre of Parkinson’s disease and frontotemporal dementia/amyotrophic lateral sclerosis. Trends in neurosciences 2019, 42, 899–912. [Google Scholar] [CrossRef]
- Baker, M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome. Nature, LETTERS 2006, 17, 1–4. [Google Scholar] [CrossRef]
- Xu, H.; O’Reilly, M.; Gibbons, G.S.; Changolkar, L.; McBride, J.D.; Riddle, D.M.; Zhang, B.; Stieber, A.; Nirschl, J.; Kim, S.-J. In vitro amplification of pathogenic tau conserves disease-specific bioactive characteristics. Acta neuropathologica 2021, 141, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nature communications 2020, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.-C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiology of disease 2014, 65, 82–92. [Google Scholar] [CrossRef]
- Troncoso, M.; Bannoud, N.; Carvelli, L.; Asensio, J.; Seltzer, A.; Sosa, M. Hypoxia-ischemia alters distribution of lysosomal proteins in rat cortex and hippocampus. Biology Open 2018, 7, bio036723. [Google Scholar] [CrossRef]
- Wang, X.; Wu, R.; Zhai, P.; Liu, Z.; Xia, R.; Zhang, Z.; Qin, X.; Li, C.; Chen, W.; Li, J. Hypoxia promotes EV secretion by impairing lysosomal homeostasis in HNSCC through negative regulation of ATP6V1A by HIF-1α. Journal of Extracellular Vesicles 2023, 12, e12310. [Google Scholar] [CrossRef]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC cancer 2012, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muñiz-García, A.; Romero, M.; Falcόn-Perez, J.M.; Murray, P.; Zorzano, A.; Mora, S. Hypoxia-induced HIF1α activation regulates small extracellular vesicle release in human embryonic kidney cells. Scientific Reports 2022, 12, 1443. [Google Scholar] [CrossRef]
- Nakashima, A.; Cheng, S.-B.; Ikawa, M.; Yoshimori, T.; Huber, W.J.; Menon, R.; Huang, Z.; Fierce, J.; Padbury, J.F.; Sadovsky, Y. Evidence for lysosomal biogenesis proteome defect and impaired autophagy in preeclampsia. Autophagy 2020, 16, 1771–1785. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Tafreshi, N.K.; Lloyd, M.C.; Sprung, R.; Estrella, V.; Wojtkowiak, J.W.; Morse, D.L.; Koomen, J.M.; Bui, M.M.; Gatenby, R.A. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nature communications 2015, 6, 8752. [Google Scholar] [CrossRef]
- Glunde, K.; Guggino, S.E.; Solaiyappan, M.; Pathak, A.P.; Ichikawa, Y.; Bhujwalla, Z.M. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia 2003, 5, 533–545. [Google Scholar] [CrossRef]
- Hong, J.; Wuest, T.R.; Min, Y.; Lin, P.C. Oxygen tension regulates lysosomal activation and receptor tyrosine kinase degradation. Cancers 2019, 11, 1653. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Palmfeldt, J.; Lin, L.; Colaço, A.; Clemmensen, K.K.; Huang, J.; Xu, F.; Liu, X.; Maeda, K.; Luo, Y. STAT3 associates with vacuolar H+-ATPase and regulates cytosolic and lysosomal pH. Cell Research 2018, 28, 996–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Hypoxia Regulates Type II Collagen Secretion by Chondrocytes through the Autophagy-lysosomal Pathway. The Chinese University of Hong Kong (Hong Kong): 2019.
- Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. Journal of bone oncology 2016, 5, 67–73. [Google Scholar] [CrossRef]
- Monti, C.; Bondi, H.; Urbani, A.; Fasano, M.; Alberio, T. Systems biology analysis of the proteomic alterations induced by MPP+, a Parkinson's disease-related mitochondrial toxin. Frontiers in Cellular Neuroscience 2015, 9, 14. [Google Scholar] [CrossRef]
- Chen, T.; Hou, R.; Li, C.; Wu, C.; Xu, S. MPTP/MPP+ suppresses activation of protein C in Parkinson's disease. Journal of Alzheimer's Disease 2015, 43, 133–142. [Google Scholar] [CrossRef]
- Dong, S.-Y.; Guo, Y.-J.; Feng, Y.; Cui, X.-X.; Kuo, S.-H.; Liu, T.; Wu, Y.-C. The epigenetic regulation of HIF-1α by SIRT1 in MPP+ treated SH-SY5Y cells. Biochemical and biophysical research communications 2016, 470, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Grande, B.; Blackabey, V.; Gittens, B.; Pinteaux, E.; Denes, A. Loss of substance P and inflammation precede delayed neurodegeneration in the substantia nigra after cerebral ischemia. Brain, Behavior, and Immunity 2013, 29, 51–61. [Google Scholar] [CrossRef]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; De Rosa, P.; Cilia, E.; Jonas, E.; Valente, E.M. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death & Differentiation 2013, 20, 920–930. [Google Scholar]
- Gautier, C.; Corti, O.; Brice, A. Mitochondrial dysfunctions in Parkinson's disease. Revue neurologique 2014, 170, 339–343. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Progress in neurobiology 2013, 106, 17–32. [Google Scholar] [CrossRef]
- Liu, X.-R.; Li, Y.-Q.; Hua, C.; Li, S.-J.; Zhao, G.; Song, H.-M.; Yu, M.-X.; Huang, Q. Oxidative stress inhibits growth and induces apoptotic cell death in human U251 glioma cells via the caspase-3-dependent pathway. European Review for Medical & Pharmacological Sciences, 2015; 19. [Google Scholar]
- Xu, Y.; Zhi, F.; Mao, J.; Peng, Y.; Shao, N.; Balboni, G.; Yang, Y.; Xia, Y. δ-opioid receptor activation protects against Parkinson’s disease-related mitochondrial dysfunction by enhancing PINK1/Parkin-dependent mitophagy. Aging (Albany NY) 2020, 12, 25035. [Google Scholar] [CrossRef] [PubMed]
- Hornykiewicz, O. Brain monoamines and parkinsonism. Natl Inst Drug Abuse Res Monogr Ser 1975, 3, 13–21. [Google Scholar]
- Reader, T.; Dewar, K. Review article Effects of denervation and hyperinnervation on dopamine and serotonin systems in the rat neostriatum: Implications for human Parkinsons disease. Neurochemistry international 1999, 34, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Scholtissen, B.; Verhey, F.; Steinbusch, H.; Leentjens, A. Serotonergic mechanisms in Parkinson’s disease: opposing results from preclinical and clinical data. Journal of neural transmission 2006, 113, 59–73. [Google Scholar] [CrossRef]
- Andrzejewski, K.; Kaczyńska, K.; Zaremba, M. Serotonergic system in hypoxic ventilatory response in unilateral rat model of Parkinson’s disease. Journal of Biomedical Science 2017, 24, 1–10. [Google Scholar] [CrossRef]
- Hickner, S.; Hussain, N.; Angoa-Perez, M.; Francescutti, D.M.; Kuhn, D.M.; Mateika, J.H. Ventilatory long-term facilitation is evident after initial and repeated exposure to intermittent hypoxia in mice genetically depleted of brain serotonin. Journal of Applied Physiology 2014, 116, 240–250. [Google Scholar] [CrossRef]
- Lin, L.-F. H.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Foti, R.; Zucchelli, S.; Biagioli, M.; Roncaglia, P.; Vilotti, S.; Calligaris, R.; Krmac, H.; Girardini, J.E.; Del Sal, G.; Gustincich, S. Parkinson disease-associated DJ-1 is required for the expression of the glial cell line-derived neurotrophic factor receptor RET in human neuroblastoma cells. Journal of Biological Chemistry 2010, 285, 18565–18574. [Google Scholar] [CrossRef]
- Kumar, G.K. Hypoxia. 3. Hypoxia and neurotransmitter synthesis. American Journal of Physiology-Cell Physiology 2011, 300, C743–C751. [Google Scholar] [CrossRef]
- Kalashnyk, O.; Lykhmus, O.; Koval, L.; Uspenska, K.; Obolenskaya, M.; Chernyshov, V.; Komisarenko, S.; Skok, M. α7 Nicotinic acetylcholine receptors regulate translocation of HIF-1α to the cell nucleus and mitochondria upon hypoxia. Biochemical and Biophysical Research Communications 2023, 657, 35–42. [Google Scholar] [CrossRef]
- Rengasamy, A.; Johns, R. Characterization of endothelium-derived relaxing factor/nitric oxide synthase from bovine cerebellum and mechanism of modulation by high and low oxygen tensions. Journal of Pharmacology and Experimental Therapeutics 1991, 259, 310–316. [Google Scholar] [PubMed]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain research bulletin 2017, 133, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harbor perspectives in medicine 2012, 2, a008888. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, L.; Liu, X.; Hou, Z.; Yu, B. PINK1 alleviates myocardial hypoxia-reoxygenation injury by ameliorating mitochondrial dysfunction. Biochemical and biophysical research communications 2017, 484, 118–124. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Frontiers in aging neuroscience 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nature communications 2017, 8, 1823. [Google Scholar] [CrossRef]
- Li, C.; Zhou, J.; Liu, Z.; Zhou, J.; Yao, W.; Tao, J.; Shen, M.; Liu, H. FSH prevents porcine granulosa cells from hypoxia-induced apoptosis via activating mitophagy through the HIF-1α-PINK1-Parkin pathway. The FASEB Journal 2020, 34, 3631–3645. [Google Scholar] [CrossRef]
- He, J.; Qin, Z.; Chen, X.; He, W.; Li, D.; Zhang, L.; Le, Y.; Xiong, Q.; Zhang, B.; Wang, H. HIF-1α Ameliorates Diabetic Neuropathic Pain via Parkin-Mediated Mitophagy in a Mouse Model. BioMed Research International 2022, 2022, 5274375. [Google Scholar] [CrossRef]
- Yu, L.; Wang, Y.; Guo, Y.H.; Wang, L.; Yang, Z.; Zhai, Z.H.; Tang, L. HIF-1α alleviates high-glucose-induced renal tubular cell injury by promoting Parkin/PINK1-mediated mitophagy. Frontiers in Medicine 2022, 8, 803874. [Google Scholar] [CrossRef]
- Maugeri, G.; D’Amico, A.G.; Reitano, R.; Saccone, S.; Federico, C.; Cavallaro, S.; D’Agata, V. Parkin modulates expression of HIF-1α and HIF-3α during hypoxia in gliobastoma-derived cell lines in vitro. Cell and tissue research 2016, 364, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Torii, S.; Kakita, A.; Sogawa, K. Inhibitory PAS domain protein is a substrate of PINK1 and Parkin and mediates cell death in a Parkinson's disease model. Cell Death & Disease 2015, 6, e1886. [Google Scholar]
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. Journal of neurochemistry 2008, 106, 464–474. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Priyadarshini, M.; Tuimala, J.; Chen, Y.; Panula, P. A zebrafish model of PINK1 deficiency reveals key pathway dysfunction including HIF signaling. Neurobiology of disease 2013, 54, 127–138. [Google Scholar] [CrossRef]
- Xu, Y.; Zhi, F.; Peng, Y.; Shao, N.; Khiati, D.; Balboni, G.; Yang, Y.; Xia, Y. δ-Opioid receptor activation attenuates hypoxia/MPP+-Induced downregulation of PINK1: a novel mechanism of neuroprotection against parkinsonian injury. Molecular Neurobiology 2019, 56, 252–266. [Google Scholar] [CrossRef]
- Wadlington, N.L. The Role of PINK1 in the Hypoxic Stress Response Through the Regulation of Hif-1alpha Translation. University of Chicago, Division of the Biological Sciences, and The Pritzker …: 2013.
- Lin, W.; Wadlington, N.L.; Chen, L.; Zhuang, X.; Brorson, J.R.; Kang, U.J. Loss of PINK1 attenuates HIF-1α induction by preventing 4E-BP1-dependent switch in protein translation under hypoxia. Journal of Neuroscience 2014, 34, 3079–3089. [Google Scholar] [CrossRef]
- Kung-Chun Chiu, D.; Pui-Wah Tse, A.; Law, C.-T.; Ming-Jing Xu, I.; Lee, D.; Chen, M.; Kit-Ho Lai, R.; Wai-Hin Yuen, V.; Wing-Sum Cheu, J.; Wai-Hung Ho, D. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell death & disease 2019, 10, 934. [Google Scholar]
- Zhang, X.; Guo, J.; Wang, Y.; Jiang, Y.; Li, S.; Liu, Y.N.; Liu, W.J. JinChan YiShen TongLuo Formula ameliorate mitochondrial dysfunction and apoptosis in diabetic nephropathy through the HIF-1α-PINK1-Parkin pathway. Journal of Ethnopharmacology 2024, 328, 117863. [Google Scholar] [CrossRef]
- Mencke, P.; Boussaad, I.; Romano, C.D.; Kitami, T.; Linster, C.L.; Krüger, R. The role of DJ-1 in cellular metabolism and pathophysiological implications for Parkinson’s disease. Cells 2021, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Sandrelli, F.; Beltramini, M.; Greggio, E.; Bubacco, L.; Bisaglia, M. Recent findings on the physiological function of DJ-1: Beyond Parkinson's disease. Neurobiology of disease 2017, 108, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Rizzu, P.; Hinkle, D.A.; Zhukareva, V.; Bonifati, V.; Severijnen, L.A.; Martinez, D.; Ravid, R.; Kamphorst, W.; Eberwine, J.H.; Lee, V.M.Y. DJ-1 colocalizes with tau inclusions: a link between parkinsonism and dementia. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 2004, 55, 113–118. [Google Scholar] [CrossRef]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. Journal of neurochemistry 2012, 121, 830–839. [Google Scholar] [CrossRef]
- Aleyasin, H.; Rousseaux, M.W.; Phillips, M.; Kim, R.H.; Bland, R.J.; Callaghan, S.; Slack, R.S.; During, M.J.; Mak, T.W.; Park, D.S. The Parkinson's disease gene DJ-1 is also a key regulator of stroke-induced damage. Proceedings of the National Academy of Sciences 2007, 104, 18748–18753. [Google Scholar] [CrossRef]
- Vasseur, S.; Afzal, S.; Tardivel-Lacombe, J.; Park, D.S.; Iovanna, J.L.; Mak, T.W. DJ-1/PARK7 is an important mediator of hypoxia-induced cellular responses. Proceedings of the National Academy of Sciences 2009, 106, 1111–1116. [Google Scholar] [CrossRef]
- Zheng, H.; Zhou, C.; Lu, X.; Liu, Q.; Liu, M.; Chen, G.; Chen, W.; Wang, S.; Qiu, Y. DJ-1 promotes survival of human colon cancer cells under hypoxia by modulating HIF-1α expression through the PI3K-AKT pathway. Cancer management and research 2018, 4615–4629. [Google Scholar] [CrossRef]
- Parsanejad, M.; Zhang, Y.; Qu, D.; Irrcher, I.; Rousseaux, M.W.; Aleyasin, H.; Kamkar, F.; Callaghan, S.; Slack, R.S.; Mak, T.W. Regulation of the VHL/HIF-1 pathway by DJ-1. Journal of Neuroscience 2014, 34, 8043–8050. [Google Scholar] [CrossRef]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A. Interplay of LRRK2 with chaperone-mediated autophagy. Nature neuroscience 2013, 16, 394–406. [Google Scholar] [CrossRef]
- Häbig, K.; Gellhaar, S.; Heim, B.; Djuric, V.; Giesert, F.; Wurst, W.; Walter, C.; Hentrich, T.; Riess, O.; Bonin, M. LRRK2 guides the actin cytoskeleton at growth cones together with ARHGEF7 and Tropomyosin 4. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2013, 1832, 2352–2367. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.; Niso-Santano, M.; Rodríguez-Arribas, M.; Gómez-Sánchez, R.; Martínez-Chacón, G.; Uribe-Carretero, E.; Navarro-García, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M. Impaired mitophagy and protein acetylation levels in fibroblasts from Parkinson’s disease patients. Molecular neurobiology 2019, 56, 2466–2481. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.-H.; Joo, H.; Bae, J.; Hyeon, S.J.; Her, S.; Ko, E.; Choi, H.G.; Ryu, H.; Hur, E.-M.; Bu, Y. Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell death & disease 2018, 9, 1125. [Google Scholar]
- Shih, H.-M.; Wu, C.-J.; Lin, S.-L. Physiology and pathophysiology of renal erythropoietin-producing cells. Journal of the Formosan Medical Association 2018, 117, 955–963. [Google Scholar] [CrossRef]
- Wu, B.; Xiao, K.; Zhang, Z.; Ma, L. Altered expression of EPO might underlie hepatic hemangiomas in LRRK2 knockout mice. BioMed Research International 2016, 2016, 7681259. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Aranda, K.; Seo, Y.-j.; Gasnier, B.; Ren, D. TMEM175 is an organelle K+ channel regulating lysosomal function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef]
- Tang, T.; Jian, B.; Liu, Z. Transmembrane protein 175, a lysosomal ion channel related to Parkinson’s disease. Biomolecules 2023, 13, 802. [Google Scholar] [CrossRef]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.-K.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proceedings of the National Academy of Sciences 2017, 114, 2389–2394. [Google Scholar] [CrossRef]
- Wie, J.; Liu, Z.; Song, H.; Tropea, T.F.; Yang, L.; Wang, H.; Liang, Y.; Cang, C.; Aranda, K.; Lohmann, J. A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 2021, 591, 431–437. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer's disease. Journal of Alzheimer's Disease, 2010; 20, S401–S412. [Google Scholar]
- Vives-Bauza, C.; Przedborski, S. Mitophagy: the latest problem for Parkinson's disease. Trends in molecular medicine 2011, 17, 158–165. [Google Scholar] [CrossRef]
- Zhang, M.; Lu, H.; Xie, X.; Shen, H.; Li, X.; Zhang, Y.; Wu, J.; Ni, J.; Li, H.; Chen, G. TMEM175 mediates Lysosomal function and participates in neuronal injury induced by cerebral ischemia-reperfusion. Molecular Brain 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Pardo-Moreno, T.; García-Morales, V.; Suleiman-Martos, S.; Rivas-Domínguez, A.; Mohamed-Mohamed, H.; Ramos-Rodríguez, J.J.; Melguizo-Rodríguez, L.; González-Acedo, A. Current treatments and new, tentative therapies for Parkinson’s disease. Pharmaceutics 2023, 15, 770. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, L.; Chen, J.; Li, Q.; Huo, L.; Wang, Y.; Wang, H.; Du, J. Pharmacological modulation of Nrf2/HO-1 signaling pathway as a therapeutic target of Parkinson’s disease. Frontiers in pharmacology 2021, 12, 757161. [Google Scholar] [CrossRef]
- Yang, X.-x.; Yang, R.; Zhang, F. Role of Nrf2 in Parkinson’s disease: toward new perspectives. Frontiers in pharmacology 2022, 13, 919233. [Google Scholar] [CrossRef]
- Guo, M.; Ji, X.; Liu, J. Hypoxia and alpha-Synuclein: inextricable link underlying the pathologic progression of Parkinson's disease. Frontiers in Aging Neuroscience 2022, 14, 919343. [Google Scholar] [CrossRef]
- You, H.; Wang, D.; Wei, L.; Chen, J.; Li, H.; Liu, Y. Deferoxamine Inhibits Acute Lymphoblastic Leukemia Progression through Repression of ROS/HIF-1α, Wnt/β-Catenin, and p38MAPK/ERK Pathways. Journal of Oncology 2022, 2022, 8281267. [Google Scholar] [CrossRef]
- Guo, C.; Hao, L.-J.; Yang, Z.-H.; Chai, R.; Zhang, S.; Gu, Y.; Gao, H.-L.; Zhong, M.-L.; Wang, T.; Li, J.-Y. Deferoxamine-mediated up-regulation of HIF-1α prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Experimental neurology 2016, 280, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Sachan, N.; Srikrishna, S.; Patel, D.K.; Singh, M.P. Deferoxamine Ameliorates Cypermethrin-Induced Iron Accumulation and Associated Alterations. Molecular Neurobiology 2024, 61, 4178–4187. [Google Scholar] [CrossRef]
- Pieńkowska, N.; Bartosz, G.; Sadowska-Bartosz, I. Effect of 6-hydroxydopamine increase the glutathione level in SH-SY5Y human neuroblastoma cells. Acta Biochimica Polonica 2023, 70, 457–464. [Google Scholar] [CrossRef]
- Chen, G.-H.; Song, C.-C.; Pantopoulos, K.; Wei, X.-L.; Zheng, H.; Luo, Z. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radical Biology and Medicine 2022, 180, 95–107. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, Y.; Chen, D.; Reddy, M.B. The Protection of EGCG Against 6-OHDA-Induced Oxidative Damage by Regulating PPARγ and Nrf2/HO-1 Signaling. Nutrition and Metabolic Insights 2024, 17, 11786388241253436. [Google Scholar] [CrossRef]
- Yong, S.J.; Veerakumarasivam, A.; Lim, W.L.; Chew, J. Neuroprotective effects of lactoferrin in Alzheimer’s and Parkinson’s diseases: A narrative review. ACS Chemical Neuroscience 2023, 14, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.P.; Pandey, A.; Vishwakarma, S.; Modi, G. A review on iron chelators as potential therapeutic agents for the treatment of Alzheimer’s and Parkinson’s diseases. Molecular diversity 2019, 23, 509–526. [Google Scholar] [CrossRef]
- Zhou, F.; Du, J.; Wang, J. Albendazole inhibits HIF-1α-dependent glycolysis and VEGF expression in non-small cell lung cancer cells. Molecular and cellular biochemistry 2017, 428, 171–178. [Google Scholar] [CrossRef]
- Kandil, E.A.; Sayed, R.H.; Ahmed, L.A.; Abd El Fattah, M.A.; El-Sayeh, B.M. Hypoxia-inducible factor 1 alpha and nuclear-related receptor 1 as targets for neuroprotection by albendazole in a rat rotenone model of Parkinson's disease. Clinical and Experimental Pharmacology and Physiology 2019, 46, 1141–1150. [Google Scholar] [CrossRef]
- Ferlazzo, N.; Currò, M.; Giunta, M.L.; Longo, D.; Rizzo, V.; Caccamo, D.; Ientile, R. Up-regulation of HIF-1α is associated with neuroprotective effects of agmatine against rotenone-induced toxicity in differentiated SH-SY5Y cells. Amino Acids 2020, 52, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Azar, Y.O.; Badawi, G.A.; Zaki, H.F.; Ibrahim, S.M. Agmatine-mediated inhibition of NMDA receptor expression and amelioration of dyskinesia via activation of Nrf2 and suppression of HMGB1/RAGE/TLR4/MYD88/NF-κB signaling cascade in rotenone lesioned rats. Life Sciences 2022, 311, 121049. [Google Scholar] [CrossRef]
- El-Sayed, E.; Ahmed, A.; Morsy, E.E.; Nofal, S. Neuroprotective effect of agmatine (decarboxylated l-arginine) against oxidative stress and neuroinflammation in rotenone model of Parkinson’s disease. Human & experimental toxicology 2019, 38, 173–184. [Google Scholar]
- Janssen Daalen, J.M.; Meinders, M.J.; Mathur, S.; van Hees, H.W.; Ainslie, P.N.; Thijssen, D.H.; Bloem, B.R. Randomized controlled trial of intermittent hypoxia in Parkinson’s disease: study rationale and protocol. BMC Neurology 2024, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Berking, S.; Astalosch, M.; Grünheid, R.; Kühn, A.A. Motor and non-motor improvements following short-term multidisciplinary day-clinic care in Parkinson´ s disease. Journal of Neural Transmission 2022, 129, 1419–1426. [Google Scholar] [CrossRef]
- Papastergiou, D.; Kokaridas, D.; Bonotis, K.; Digelidis, N.; Patsiaouras, A. Intervention effect of supportive group therapy and physical exercise on the quality of life of cancer patients. Central European Journal of Sport Sciences and Medicine, 2019; 25. [Google Scholar]
- Aimée, N.R.; Damé-Teixeira, N.; Alves, L.S.; Borges, G.Á.; Foster Page, L.; Mestrinho, H.D.; Carvalho, J.C. Responsiveness of oral health-related quality of life questionnaires to dental caries interventions: systematic review and meta-analysis. Caries research 2019, 53, 585–598. [Google Scholar] [CrossRef]
- Ford, B.Q.; Lam, P.; John, O.P.; Mauss, I.B. The psychological health benefits of accepting negative emotions and thoughts: Laboratory, diary, and longitudinal evidence. Journal of personality and social psychology 2018, 115, 1075. [Google Scholar] [CrossRef] [PubMed]
- Bialkowska, M.; Zajac, D.; Mazzatenta, A.; Di Giulio, C.; Pokorski, M. Inhibition of peripheral dopamine metabolism and the ventilatory response to hypoxia in the rat. Neurotransmitter Interactions and Cognitive Function 2015, 9–17. [Google Scholar]
- Kaczyńska, K.; Orłowska, M.E.; Andrzejewski, K. Respiratory abnormalities in Parkinson’s disease: What do we know from studies in humans and animal models? International Journal of Molecular Sciences 2022, 23, 3499. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Campanelli, F.; Natale, G.; De Carluccio, M.; Servillo, F.; Ferrari, E.; Gardoni, F.; Caristo, M.E.; Picconi, B.; Cardinale, A. Intensive exercise ameliorates motor and cognitive symptoms in experimental Parkinson’s disease restoring striatal synaptic plasticity. Science Advances 2023, 9, eadh1403. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.N.; Kim, S.-H.; Lee, B.; Yoo, H.S.; Lee, J.H.; Jo, M.G.; Seong, H.M.; Song, C.; Kim, S.J.; Park, S.W. A2-astrocyte activation by short term hypoxia rescue α-synuclein preformed fibril induced neuronal cell death. 2024.
- Ogunshola, O.; Antoniou, X. Contribution of hypoxia to Alzheimer’s disease: is HIF-1α a mediator of neurodegeneration? Cellular and Molecular Life Sciences 2009, 66, 3555–3563. [Google Scholar] [CrossRef]
- Agani, F.H.; Pichiule, P.; Chavez, J.C.; LaManna, J.C. The role of mitochondria in the regulation of hypoxia-inducible factor 1 expression during hypoxia. Journal of Biological Chemistry 2000, 275, 35863–35867. [Google Scholar] [CrossRef]
- Kalva-Filho, C.A.; Faria, M.H.; Papoti, M.; Barbieri, F.A. Acute and cumulative effects of hypoxia exposure in people with Parkinson’s disease: A scoping review and evidence map. Parkinsonism & Related Disorders, 2023; 105885. [Google Scholar]
- Janssen Daalen, J.M.; Meinders, M.J.; Giardina, F.; Roes, K.C.; Stunnenberg, B.C.; Mathur, S.; Ainslie, P.N.; Thijssen, D.H.; Bloem, B.R. Multiple N-of-1 trials to investigate hypoxia therapy in Parkinson’s disease: study rationale and protocol. BMC neurology 2022, 22, 262. [Google Scholar] [CrossRef]
Figure 1.
The HIF pathway in hypoxia. Under conditions of sufficient oxygen, PHD and FIH hydroxylate HIF-1α. Hydroxylated HIF-1α is subsequently recognized by VHL, leading to its ubiquitination and degradation. As a result, the concentration of HIF-1α remains very low. In moderate hypoxia, only FIH hydroxylates HIF-1α, allowing HIF-1α to translocate into the nucleus, where it binds with HIF-1β to form the HIF complex. This complex then binds to HREs in the genome, initiating transcription of target genes. In severe hypoxia, unhydroxylated HIF-1α not only enters the nucleus and binds to HIF-1β, but also interacts with the coactivator p300. This interaction enhances the transcription of downstream regulatory genes. Abbreviations: PD: Parkinson's disease; HIF-1α: hypoxia inducing factor-1α; FIH: Factor Inhibiting HIF; PHD: hypoxia-inducible factor prolyl hydroxylase; pVHL: von Hippel-Lindau protein; Ub: ubiquitin; Proteasome: 26S proteasome; HIF-1β: hypoxia inducing factor-1β; CBP/P300: the coactivator CBP/P300; HER: hypoxia response elements; TH: tyrosine hydroxylase; DAT: dopamine transporter; O2: oxygen.
Figure 1.
The HIF pathway in hypoxia. Under conditions of sufficient oxygen, PHD and FIH hydroxylate HIF-1α. Hydroxylated HIF-1α is subsequently recognized by VHL, leading to its ubiquitination and degradation. As a result, the concentration of HIF-1α remains very low. In moderate hypoxia, only FIH hydroxylates HIF-1α, allowing HIF-1α to translocate into the nucleus, where it binds with HIF-1β to form the HIF complex. This complex then binds to HREs in the genome, initiating transcription of target genes. In severe hypoxia, unhydroxylated HIF-1α not only enters the nucleus and binds to HIF-1β, but also interacts with the coactivator p300. This interaction enhances the transcription of downstream regulatory genes. Abbreviations: PD: Parkinson's disease; HIF-1α: hypoxia inducing factor-1α; FIH: Factor Inhibiting HIF; PHD: hypoxia-inducible factor prolyl hydroxylase; pVHL: von Hippel-Lindau protein; Ub: ubiquitin; Proteasome: 26S proteasome; HIF-1β: hypoxia inducing factor-1β; CBP/P300: the coactivator CBP/P300; HER: hypoxia response elements; TH: tyrosine hydroxylase; DAT: dopamine transporter; O2: oxygen.
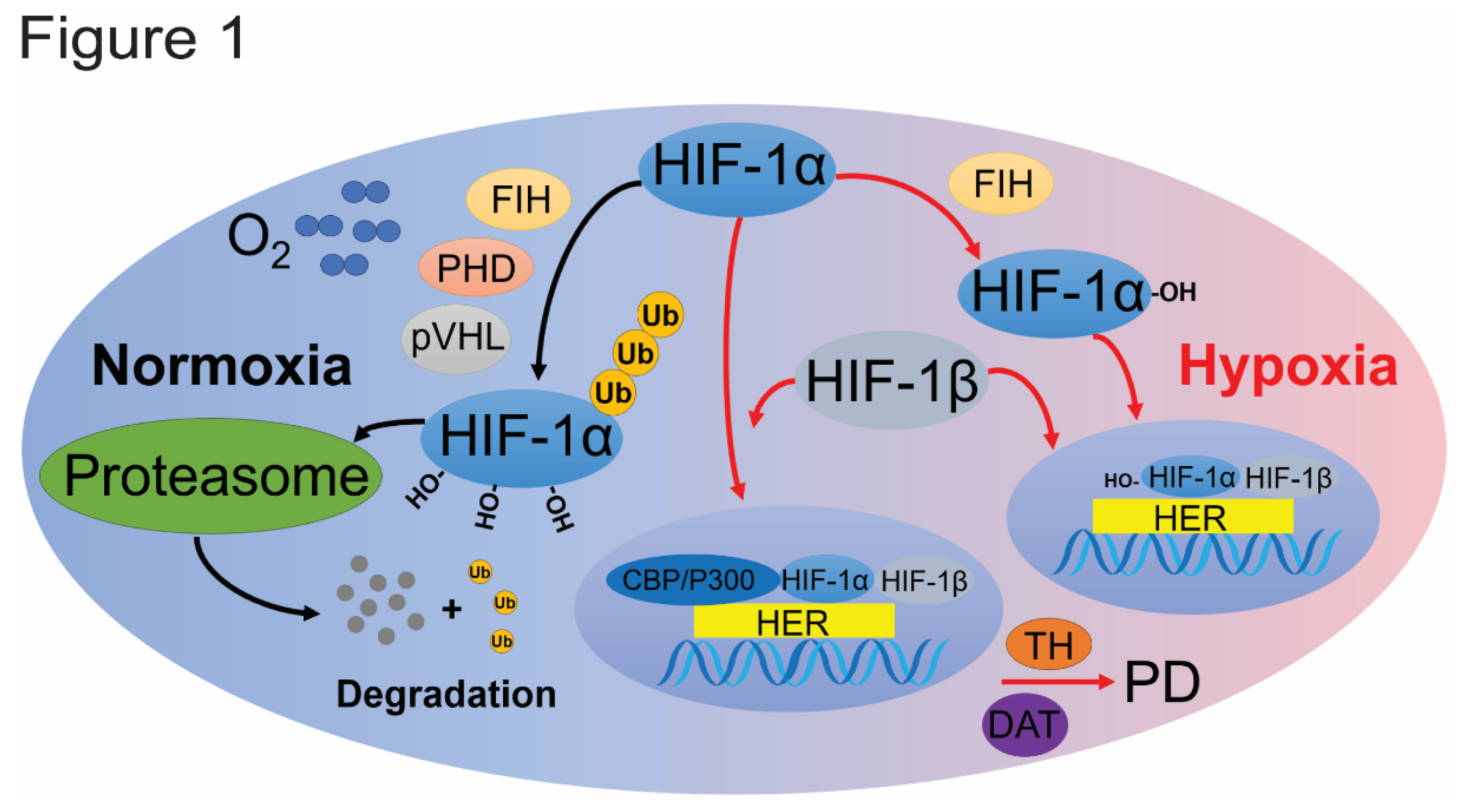
Figure 2.
The Nrf2/HO-1 pathway in hypoxia. In the cytoplasm, KEAP1 forms a ubiquitin E3 ligase complex with CUL3 to polyubiquitinate NRF2, leading to its rapid degradation by the proteasomal system. Under oxidative stress, NRF2 is released from KEAP1-mediated inhibition. NRF2 translocates to the nucleus, where it heterodimerizes with the small Maf protein. Then, this NRF2-Maf complex binds to the ARE in the genome, allowing the expression of a series of downstream protective phase II detoxification enzyme and antioxidant enzyme genes and proteins, such as HO-1. Abbreviations: Nrf2: nuclear factor erythroid 2-related factor 2; sMAF: Myeloma-associated factors; ARE: antioxidant response element; HO-1: heme oxygenase-1; UPS: ubiquitin proteasome system; MPTP/MPP+: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; α-syn: α-synuclein; Lrrk2: leucine-rich repeat kinase 2; Keap1: Kelch-like ECH-associated protein 1; Cul3: Cullin-3; Ub: ubiquitin; Proteasome: 26S proteasome.
Figure 2.
The Nrf2/HO-1 pathway in hypoxia. In the cytoplasm, KEAP1 forms a ubiquitin E3 ligase complex with CUL3 to polyubiquitinate NRF2, leading to its rapid degradation by the proteasomal system. Under oxidative stress, NRF2 is released from KEAP1-mediated inhibition. NRF2 translocates to the nucleus, where it heterodimerizes with the small Maf protein. Then, this NRF2-Maf complex binds to the ARE in the genome, allowing the expression of a series of downstream protective phase II detoxification enzyme and antioxidant enzyme genes and proteins, such as HO-1. Abbreviations: Nrf2: nuclear factor erythroid 2-related factor 2; sMAF: Myeloma-associated factors; ARE: antioxidant response element; HO-1: heme oxygenase-1; UPS: ubiquitin proteasome system; MPTP/MPP+: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; α-syn: α-synuclein; Lrrk2: leucine-rich repeat kinase 2; Keap1: Kelch-like ECH-associated protein 1; Cul3: Cullin-3; Ub: ubiquitin; Proteasome: 26S proteasome.
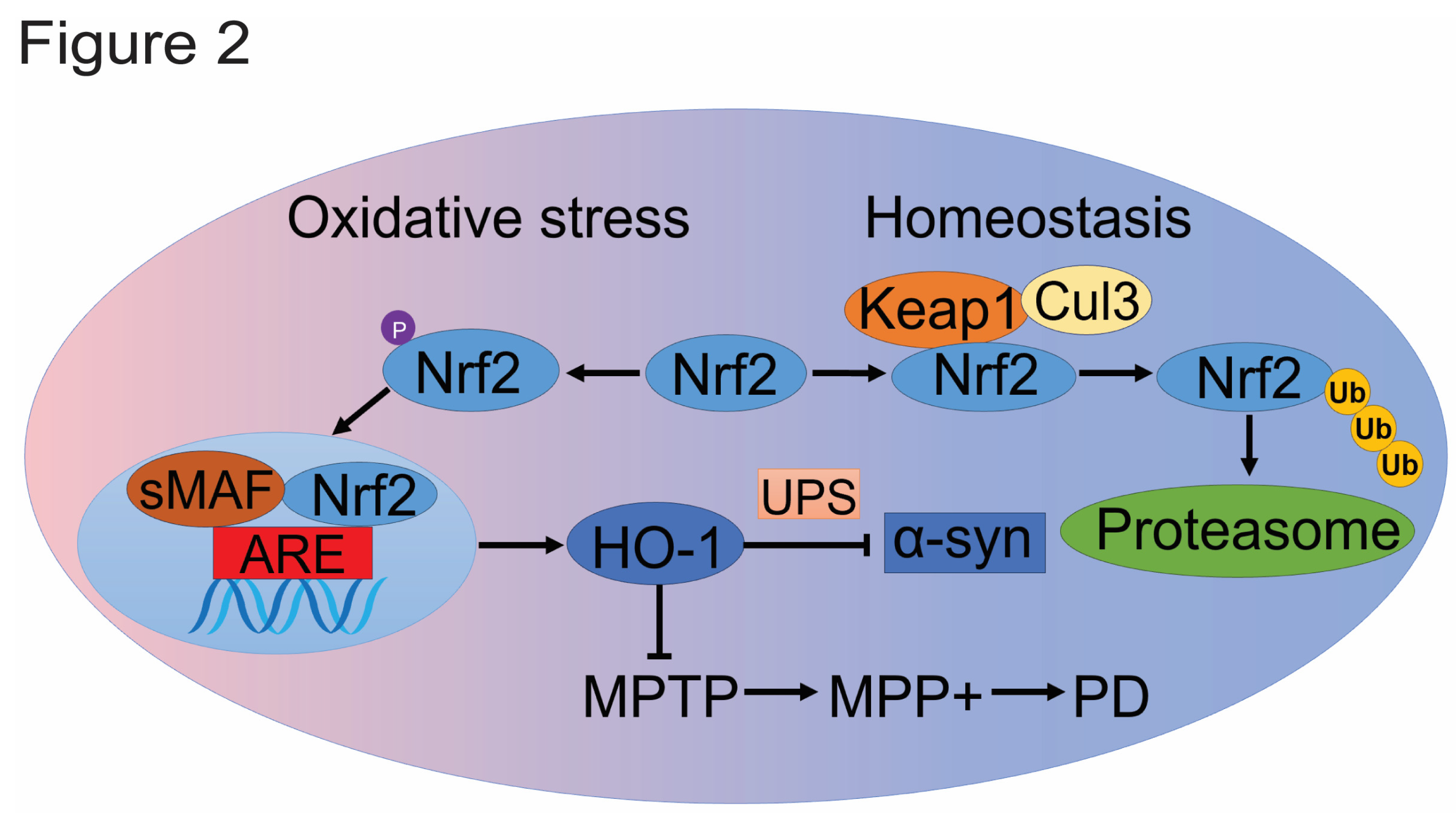
Figure 3.
Hypoxia-induced neuronal damage and PD. Three evidence of hypoxia-induced neuronal damage and Parkinson’s disease: (1) the susceptibility of dopaminergic neurons to hypoxia, (2) clinical evidence of hypoxia in PD patients, and (3) the impact of cellular oxygen deficit on PD progression. Abbreviations: ROS: reactive oxygen species; L-DOPA: L-3,4-dihydroxyphenylalanine; Ub: ubiquitin; HIF-2α: hypoxia inducing factor-2α; SNpc: substantia nigra pars compacta; BDNF: brain-derived neurotrophic factor.
Figure 3.
Hypoxia-induced neuronal damage and PD. Three evidence of hypoxia-induced neuronal damage and Parkinson’s disease: (1) the susceptibility of dopaminergic neurons to hypoxia, (2) clinical evidence of hypoxia in PD patients, and (3) the impact of cellular oxygen deficit on PD progression. Abbreviations: ROS: reactive oxygen species; L-DOPA: L-3,4-dihydroxyphenylalanine; Ub: ubiquitin; HIF-2α: hypoxia inducing factor-2α; SNpc: substantia nigra pars compacta; BDNF: brain-derived neurotrophic factor.
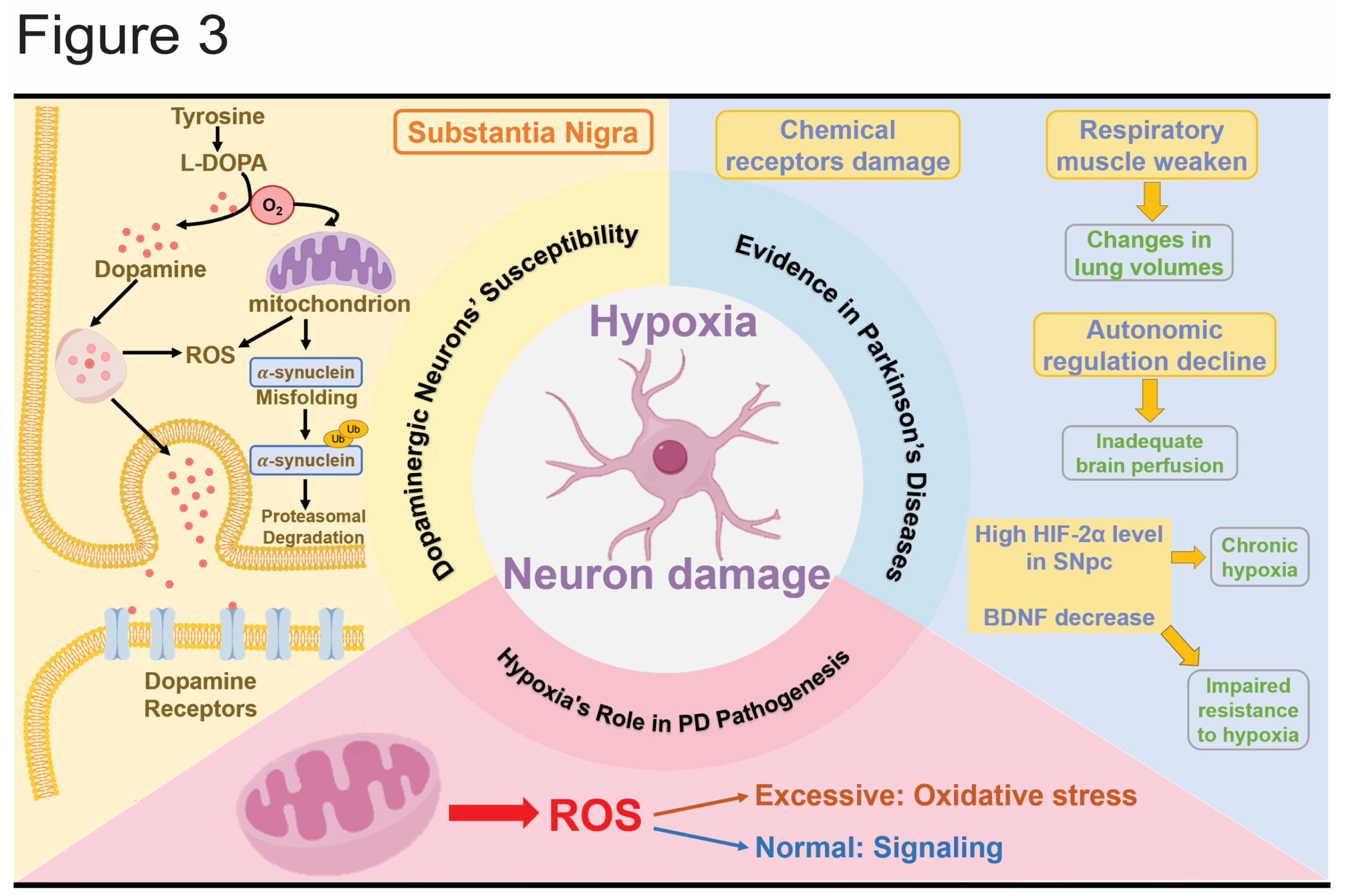
Figure 4.
Hypoxia contributes to PD. HIF-1α influences the development of PD through multiple pathways, including the accumulation of α-syn, mitochondrial dysfunction, lysosomal damage, and alterations in receptor and neurotransmitter systems. Abbreviations: HIF-1α: hypoxia inducing factor-1α; EV: extracellular vesicle; ILVs: intracavitary vesicles; ATP6V1A: ATPase H+ transporting V1 subunit A.
Figure 4.
Hypoxia contributes to PD. HIF-1α influences the development of PD through multiple pathways, including the accumulation of α-syn, mitochondrial dysfunction, lysosomal damage, and alterations in receptor and neurotransmitter systems. Abbreviations: HIF-1α: hypoxia inducing factor-1α; EV: extracellular vesicle; ILVs: intracavitary vesicles; ATP6V1A: ATPase H+ transporting V1 subunit A.
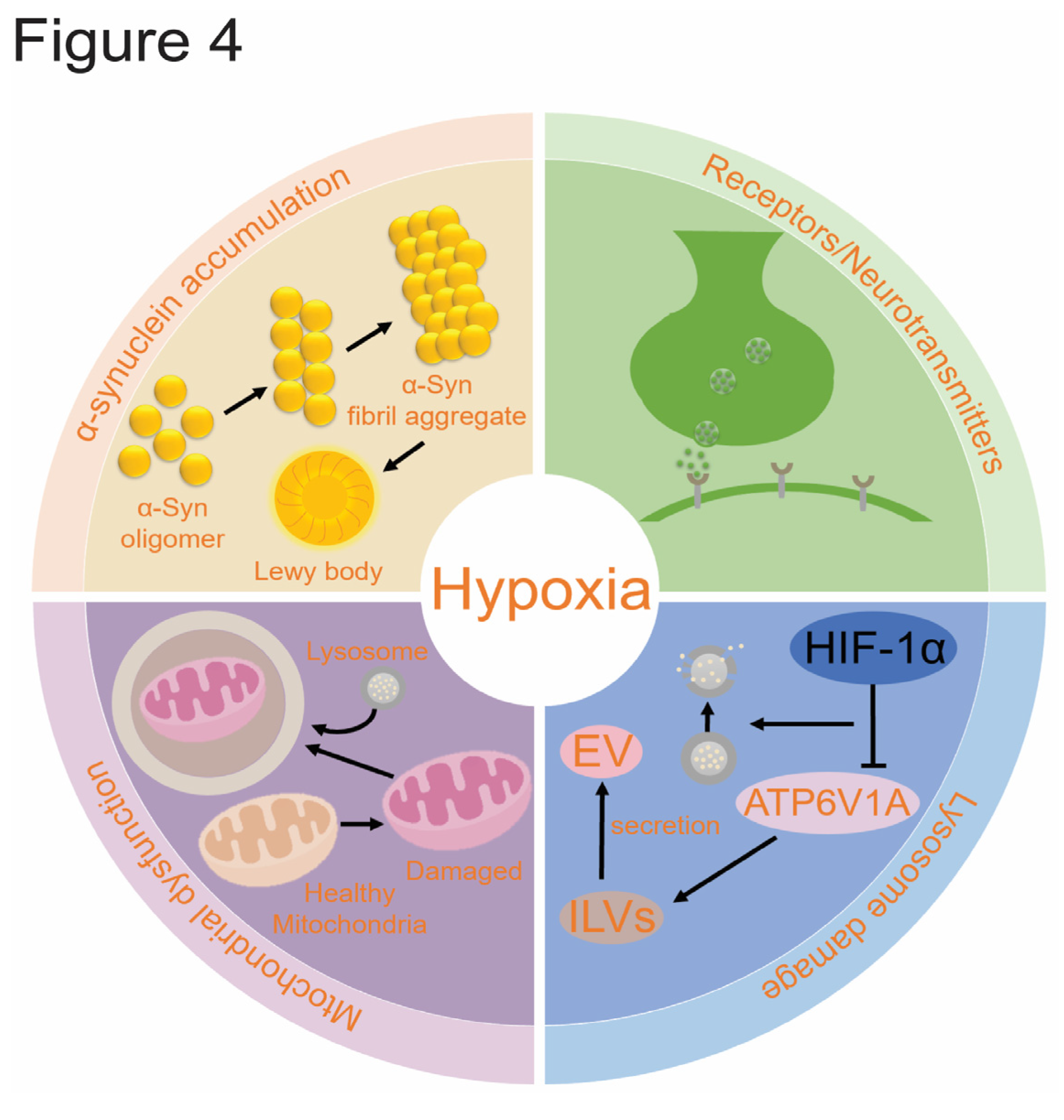
Figure 5.
The role of five PD-related genes in hypoxia. The PRKN, PINK1, LRRK2, DJ-1, and TMEM175 genes play significant roles in the onset and progression of PD. Under certain conditions, these genes interact with HIF-1α, influencing the disease’s development. Abbreviations: PRKN: Parkin RBR E3 ubiquitin protein ligase; LRRK2: Leucine-rich repeat kinase 2; PINK1: PTEN-induced putative kinase 1; DJ-1: PARK7, Parkinson protein 7; TMEM175: Transmembrane Protein 175; HER:hypoxia response elements; HIF: hypoxia inducing factor; pVHL: von Hippel-Lindau; PI3K: phosphatidylin-ositol-3-kinase; AKT: Protein Kinase B, PKB; TBI: traumatic brain injury; p-α-syn: phosphorylated α-Syn; I/R: ischemia-reperfusion; NO: Nitric Oxide; Ub: ubiquitin; Proteasome: 26S proteasome.
Figure 5.
The role of five PD-related genes in hypoxia. The PRKN, PINK1, LRRK2, DJ-1, and TMEM175 genes play significant roles in the onset and progression of PD. Under certain conditions, these genes interact with HIF-1α, influencing the disease’s development. Abbreviations: PRKN: Parkin RBR E3 ubiquitin protein ligase; LRRK2: Leucine-rich repeat kinase 2; PINK1: PTEN-induced putative kinase 1; DJ-1: PARK7, Parkinson protein 7; TMEM175: Transmembrane Protein 175; HER:hypoxia response elements; HIF: hypoxia inducing factor; pVHL: von Hippel-Lindau; PI3K: phosphatidylin-ositol-3-kinase; AKT: Protein Kinase B, PKB; TBI: traumatic brain injury; p-α-syn: phosphorylated α-Syn; I/R: ischemia-reperfusion; NO: Nitric Oxide; Ub: ubiquitin; Proteasome: 26S proteasome.
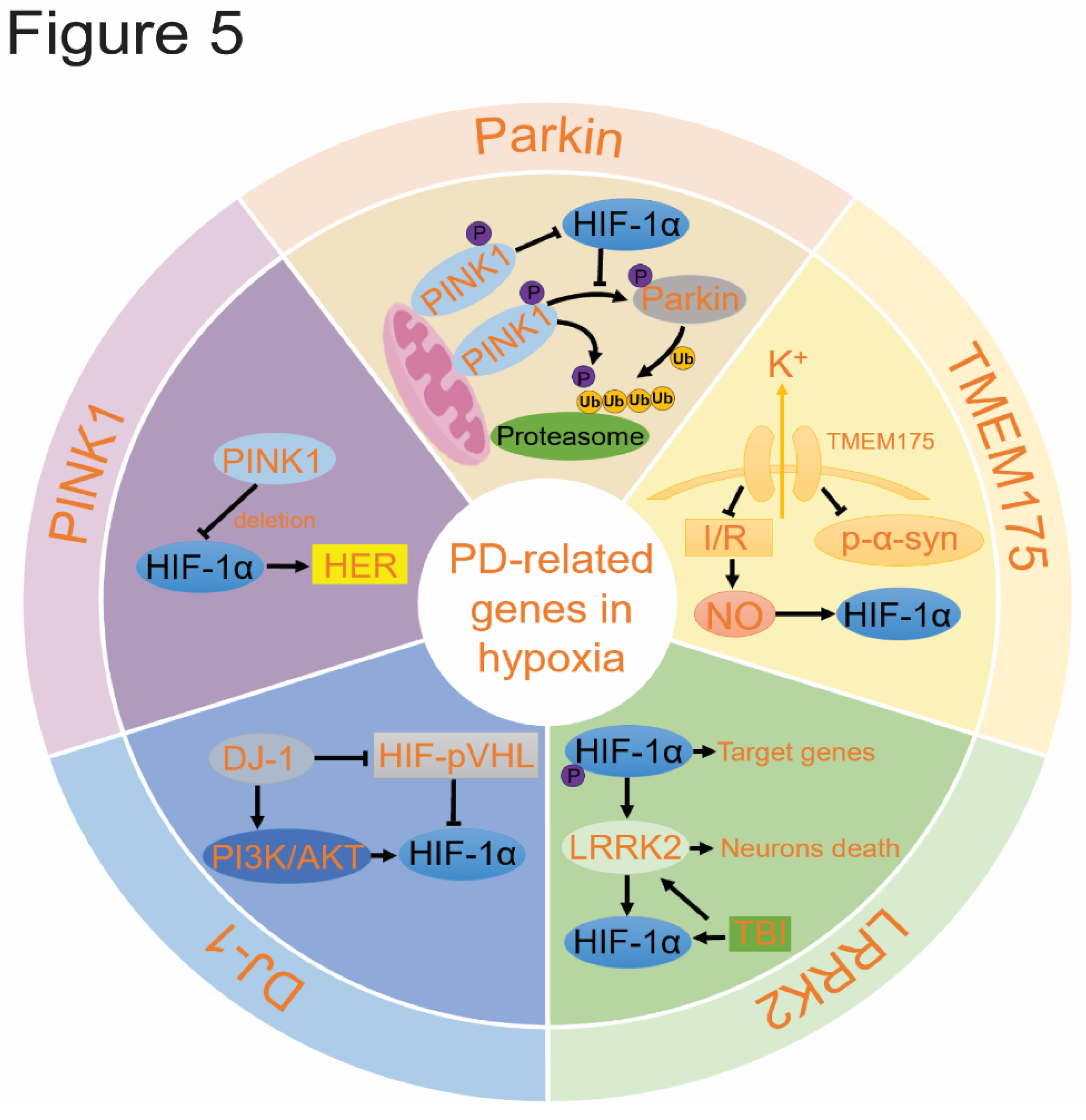
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



