
Submitted:
05 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
Inherited retinal diseases (IRDs) are clinically complex and genetically heterogeneous visual impairment disorders with varying penetrance and severity. Disease-causing variants in at least 289 nuclear and mitochondrial genes have been implicated in their pathogenesis. In the current study, we performed exome sequencing on a 51 years-old Ashkenazi Jewish patient with non-syndromic retinitis pigmentosa (RP) and identified compound heterozygous variants in the CLRN1 gene: a known pathogenic missense [p.(N48K)] and a novel deep intronic variant c.254-643G>T. A minigene splicing assay that was performed aiming to study the effect of the c.254-643G>T variant on CLRN1 pre mRNA splicing revealed the inclusion of a pseudo-exon that was also reported to be included in the transcript due to an adjacent variant, c.254-649T>G. However, unlike the reported c.254-649T>G variant, c.254-643G>T showed aberrant splicing in a leaky manner, implying that the identified variant is not totally penetrant. The non-syndromic phenotype observed in this index case may be attributed to the leaky nature of this variant, which is causing some normal transcripts to be produced. To conclude, we report on a novel deep intronic variant in CLRN1 causing non-syndromic RP.
Keywords:
CLRN1
; deep intronic
; inherited retinal diseases
; pseudo-exon
; retinitis pigmentosa
; splicing
1. Introduction
Inherited retinal diseases (IRDs) encompass a large group of retinal phenotypes characterized by extensive clinical variability and large genetic heterogeneity [1, 2]. Retinitis pigmentosa (RP) is the most prevalent and heterogeneous IRD and can be inherited in autosomal dominant (AD), autosomal recessive (AR), digenic, mitochondrial and X-linked (XL) modes [3].
Although it is acceptable that most IRD-associated genes have been identified [4], next-generation sequencing (NGS) used as gene panels, whole exome sequencing (WES) or whole genome sequencing (WGS), fails to identify the causative genes in about 30% of IRD cases [5,6]. Although genes currently not associated with IRDs, and therefore not well-covered in such analyses, might cause a fraction of these cases, elusive variants might play an important role in such cases. One example might be deep intronic variants that affect splicing, usually by introducing pseudo exons into the transcripts [7,8,9]. A second possibility might be novel variants in genes that are associated with a similar, but not identical, phenotype. For example, genes in which all, or the vast majority, of pathogenic variants cause a syndromic multiorgan phenotype, might not always serve as candidates for the isolated non-syndromic phenotypes. Usher syndrome (RP and sensorineural hearing loss- SNHL), for example, is caused by disease-causing variants in 15 genes (https://retnet.org/), while variants in some of these genes can cause either nonsyndromic RP or nonsyndromic SNHL [10,11,12]. For some of these genes, this phenomenon is well-established with a high number of variants causing either the syndromic or the non-syndromic phenotypes (e.g., USH2A), while for other genes, e.g., USH1C, all variants but one cause the USH1 while a single variant in an alternative exon causes RP with late-onset hearing loss [13].
The combination of elusive variants and genes causing multiple phenotypes is therefore challenging and requires in some cases the development of functional assays to support the pathogenicity of such variants. In the current study, we report such an elusive, deep intronic variant in the CLRN1 gene, variants which are known to cause mainly USH type 3. This variant was found in compound heterozygosity with a well-established CLRN1 pathogenic variant that causes USH3. The intronic variant introduces a pseudo-exon in a leaky fashion, allowing normal hearing in a 51-year-old (YO) case who suffers from non-syndromic retinitis pigmentosa.
2. Materials and Methods
Patient Recruitment
Blood samples were collected from the index cases and unaffected family members. The sample collection process adhered to ethical standards, receiving approval from the Hadassah Hospital Institutional Review Board. Informed consent, obtained in writing, ensured compliance with all relevant ethical regulations and safeguarded the rights and privacy of participants. Genomic DNA extraction was carried out from blood samples using the Promega kit and Promega Maxwell (Madison, WI, USA) DNA extraction system.
Sanger and Next-Generation Sequencing (NGS)
WES was performed by Pronto Diagnostic (Tel-Aviv, Israel), and an IRD gene panel was performed using the smMIPs platform [14]. Fastq files were uploaded to the Genoox pipeline and analyzed using Franklin (https://franklin.genoox.com/). For segregation analysis primers were designed using primer3 web (https://primer3.ut.ee/), primer sequence F: TCTAATGGTCTGTCTTCTCCCA; R:AGCCTTTAATGACCTTTCTCGG.
In Silico Splicing Analysis
The effect of the deep intronic c.254-643G>T CLRN1 (NM_174878.2) variant on its pre-mRNA splicing was predicted using Alternative Splice Site Predictor (ASSP), Berkeley Drosophila Genome Project (https://www.fruitfly.org/seq_tools/splice.html) and the SpliceAI prediction tool.
Cloning
A minigene exontrap plasmid (pET01) was purchased from MoBiTec. A 792 bp CLRN1 intron 1 sequence flanking the c.254-643G>T variant was PCR amplified from genomic DNA of the patient and a control individual using (PCR Taq Mix Red, PB045625-071-0, London). These PCR products and the pET01plasmid were digested with BamHI and XhoI restriction enzymes for 2 h at 37 oc. Digested products were gel purified using a Qiagen gel extraction kit (Cat. no: 28706X4), and ligated using T4 DNA ligase (NEB, Cat. no: M0202S) for 2 h at room temperature. Ligated plasmids were transformed into DH5 alpha competent cells (NEB, Cat. no: C2987H) and plated on LB agar plates with ampicillin for selectivity of transfected cells. Positive clones were identified using colony PCR and confirmed by Sanger sequencing. Primer sequence used for cloning F-BamHI-CTCGAG-AGAGTAGCCTGGACTTCTTGG; XhoI-R-GGATCC-TCTGGGGATATCTGGGGAGT.
Transfection, RNA Isolation and cDNA Synthesis
For splicing analysis, HeLa cells were seeded into a 24-well plate and cultured till they reached 70% confluence. Cells were transfected separately with empty pET01 plasmid, wild-type and mutant CLRN1 plasmids using (TransfeX™ Transfection Reagent, ACS-4005 ™, ATCC, USA) transfection reagent. Cells were harvested 48 h post-transfection and the total RNA was isolated using (Quick-RNA MiniPrep, R1055, ZYMO RESEARCH, California) by following the manufacturer’s protocol. cDNA synthesis was carried out using qScript cDNA Synthesis Kit (Cat. No: 733-1178).
Automated Electrophoresis - TapeStation
Quality control of the RT-PCR products was assessed using the Agilent Technologies D1000 ScreenTape system kit and Agilent Technologies 4200 TapeStation instrument (Agilent Technologies, Waldron, Germany). After automated electrophoresis, the results were analyzed on the TapeStation Controller Software to detect product size and quantity with a sensitivity of 0.1 ng/µL and sizing accuracy of ±10%. Primer sequence used for PCR amplification and Sanger sequencing of CLRN1 cDNA analysis F-GGATTCTTCTACACACCC; R-AGGTGGGTCGAGGTCAAC.
3. Results
3.1. Clinical analysis
MOL0377-1 is a 51 YO male descending from a non-consanguineous Ashkenazi family (Figure1A).
At age 37 YO, he was referred to our retina clinic due to complaints of blurred vision and constricted visual fields since the age of 16 years, which led to a diagnosis of RP at the age of 25 years. The best-corrected visual acuity (BCVA) at presentation was 0.80 in the right eye (RE) and 0.40 in the left eye (LE), quiet anterior segments with mild central posterior subcapsular cataract and in fundoscopy diffuse retinal atrophy and coalescing bone specula-like pigmentations (BSPs) encroaching the macula along with narrow vessels and waxy-appearance optic discs were seen. Optical coherence tomography (OCT) showed an atrophic and dry macula in the RE while cystoid macular edema (CME) was present in the LE (Figure 2).
Full-field electroretinography (ffERG) at age of 25 years showed extinguished rod responses and reduced cone responses, compatible with RP, and electrooculography (EOG) responses were moderately reduced. On Goldmann kinetic perimetry testing, 4IIIE target, preservation of the central 70 degrees in BE was seen. Farnsworth-Munsell D-15 and Ishihara tests were normal and he was treated for the CME in the LE (PO Diamox 125mgx3). He was also treated in BE with Brominidine Tartrate (Alphagan 1x3/d) following his previous participation in a study examining the neuroprotective characteristics of alpha agonist inhibitors. He has been followed up for 15 years and in the last follow-up, his BCVA was 0.32 in the BE. Of note, he underwent cataract surgery at the age of 50 years in BE including implantation of an artificial intraocular lens. On fundoscopy, classic RP findings were observed including retinal atrophy mixed with dense BSPs, attenuated retinal vessels, and pale optic discs in BE (Figure 2). Throughout the follow-up period, he was treated with different regimens with Dorzolamide (Trusopt 2%) and Nepafenac (Nevanac 0.1%) drops for alternating CME in BE. Fundus autofluorescence images demonstrated a hyperfluorescent retina encroaching on a hyperfluorescent ring surrounding the fovea. Repeated Farnsworth-Munsell D-15 tests revealed a tendency toward the tritanopic axis. Recent Goldmann kinetic perimetry testing, 4VE target, showed preservation of the central 15-20 degrees in BE. Of note, following the genetic results (see below), the patient underwent audiometry which revealed slightly reduced responses in both ears at high frequencies which were attributed to aging and environmental exposure rather than sensorineural causes.
3.2. Variants Identification
WES analysis of MOL0377-1 revealed 124,202 variants, 10,935 of which had MAF of less than 1%. Only two clear-cut reported pathogenic variants were identified in this case, both were heterozygous identified in AR genes: FTCD- c.1366dupG has been reported to cause Formiminoglutamic Aciduria and is non-relevant to the case phenotype, and CLRN1- c.144T>G [p.(N48K); Figure 1] which is well-described in the literature as a founder mutation in the Ashkenazi population known to cause Usher type 3 [15,16]. Three additional CLRN1 heterozygous variants were identified by WES and smMIPs in MOL0377-1, none of them within the open reading frame, and two of the variants showed high MAF of over 20%, excluding them as possible pathogenic variants. The remaining variant was deep intronic, c.254-643G>T (Figure 1B), identified in intron 1. This variant is extremely rare and was not found in gnomAD or other genetic databases. The variant shows a moderate score of 0.49 as splice-altering by the SpliceAI tool. Interestingly, a nearby variant, c.254-649T>G (Figure 3), with a strong SpliceAI score of 0.85 as splice-altering, was previously reported to generate a pseudo-exon [17] in a homozygous state in patients with Usher syndrome type 1. We therefore suspected that the variant we identified, c.254-643G>T, might also affect splicing. Both CLRN1 variants identified in MOL0377-1 were verified by Sanger sequencing (Figure 1C-D). In addition, two unaffected brothers of the index case were found by Sanger sequencing to carry heterozygous missense variant, c.144T>G [p.(N48K)], but not the deep intronic variant, indicating a compound heterozygous state in the index case (Figure 1). In addition, we used Sanger sequencing to screen for the deep intronic variants in 143 ethnicity-matched index cases with IRDs, none of whom was found to carry it.
3.3. Splicing Assay
To investigate the impact of the c.254-643G>T deep-intronic variant on the pre-mRNA splicing, we designed an in-vitro minigene splicing assay (Figure 3A). To this end, we cloned a 792 bp fragment of intron 1 flanking the c.254-643G>T variant. Three clones were generated including the wild-type (WT) sequence, c.254-643G>T, or c.254-649T>G bearing fragments inserted into the pET01 exontrap plasmid. All three clones were validated through Sanger sequencing. The plasmids were transfected separately into HeLa cells followed by RNA isolation 48 hours post-transfection. RT-PCR analysis in combination with Sanger sequencing (Figure 4) and TapeStation (Figure 5) analysis revealed a single 260 bp transcript (TS1) in cells transfected with an empty plasmid (Figure 3B), indicating the normal splicing of the pET01 plasmid minigene.
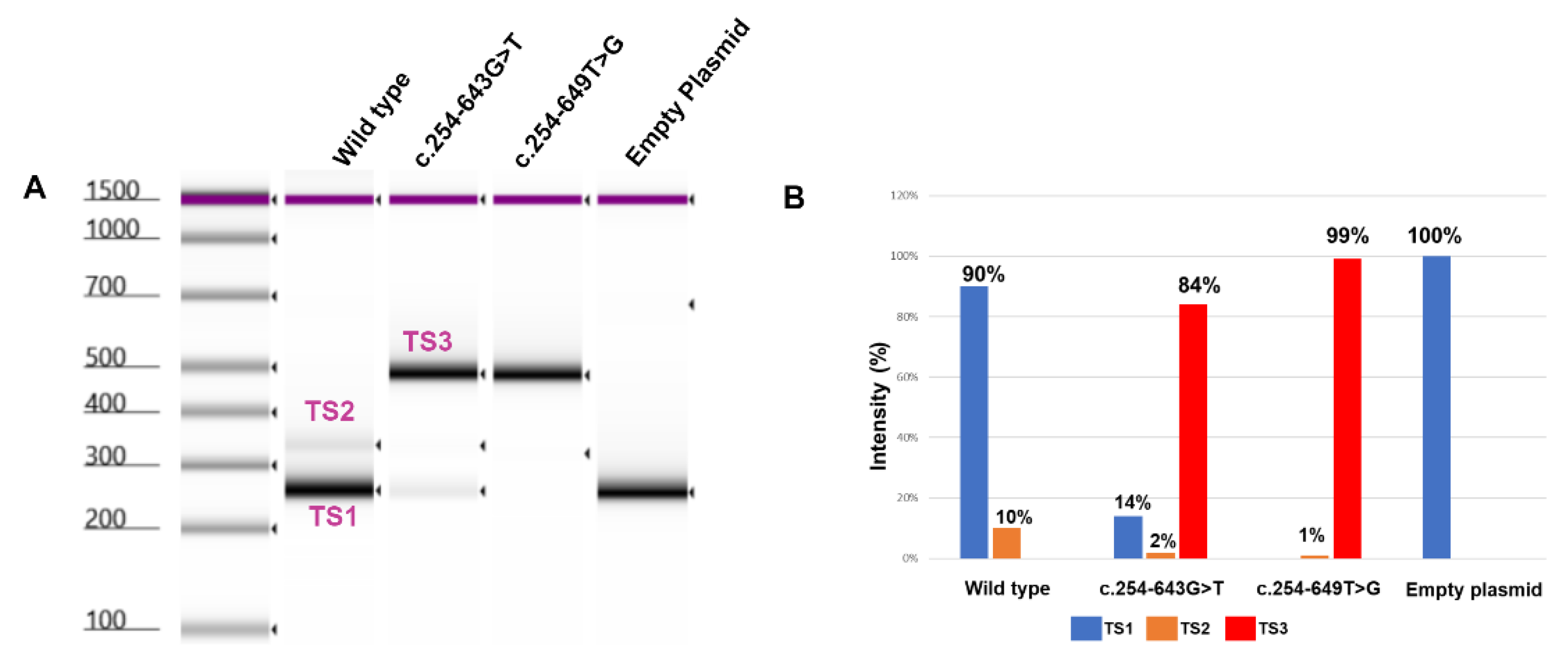
In the WT cDNA we observed two different bands (Figure 3B): a higher intensity TS1 band and a lower intensity 346 bp (TS2) band indicating the inclusion of an 86 bp pseudo-exon in TS2 (Figure 4). On the other hand, in mutant (c.254-643G>T) cDNA we observed three different transcripts: a high-intensity 490 bp (TS3) band, and faint TS1 and TS2 bands. The 490 bp band (Figure 3) represents an abnormal transcript (TS3) that includes pseudoexon 1 (an addition of 230 bp). However, the presence of the other two splice variants (TS1 and TS2) suggests that c.254-643G>T is leaky and not fully penetrant and some normal transcripts were also produced. On the other hand, the reported c.254-649T>G variant resulted in the production of the same pseudoexon (#1) in a fully penetrant way, with no production of the WT transcript (Figure 3, 5). A more detailed analysis of the PCR products using TapeStation (Figure 5A) revealed similar results. We observed a high-intensity (99%) TS3 transcript in c.254-649T>G cDNA with very low to undetectable levels (1%) of TS2 and complete absence of TS1, similarly, we observed a high-intensity TS3 (84%) and a 14% of TS1 in 254-643G>T cDNA (Figure 5B).
4. Discussion
Following the identification of CLRN1 as the causative USH3A gene in 2001 [18,19], many CLRN1 disease-causing variants have been reported. CLRN1 can produce 11 transcripts through various mechanisms [19,20] and therefore exon numbering might vary between studies. The LOVD-CLRN1 database (https://databases.lovd.nl/shared/genes/CLRN1) includes 34 LP/P variants with c.528T>G [p.(Y176*)] being the most common one identified on 44% of the mutated alleles, followed by the c.144T>G [p.(N48K)] missense variants identified on 23% of alleles. The former is relatively common in Europe (MAF of 0.005% in the gnomAD database) and especially among Finnish (MAF of 0.6%) [21]. The latter is a founder mutation in the Ashkenazi Jewish population with a MAF of 0.6%. The vast majority of CLRN1 variants cause USH3, with a variable age of hearing loss onset and a progressive nature [22]. The average age of hearing loss diagnosis in USH3A patients is 8–10 years. However, in rare cases, patients who harbor relatively mild missense variants might not be aware of their hearing conditions even until the fourth decade of life or might still have normal hearing function. For example, two cases who were homozygous for c.606T>G [p.(N202K)] were aware of their hearing loss condition only at the age of 40 [23]. In addition, two missense variants [p.(P31L) and p.(L154W)] were reported to cause non-syndromic RP with normal audiometric assessment in two Pakistani families [24].
The deep intronic variant we identified is situated within intron 1 downstream to an intronic sequence that can potentially serve as a splice acceptor site (c.254-879_880). A previous study identified a homozygous founder mutation in Saudi Arabia patients, c.254-649T>G, that creates an intronic splice donor site, generating a 230 bp pseudoexon within intron 1 in 87% of PCR products, with the variant located at the last nucleotide of this pseudoexon [17]. The variant we identified introduces the same pseudogene, but based on our splicing assay, studying both variants in the same experimental setup, the variant we identified is leaky, resulting in the production of normal protein, with levels that might be sufficient for normal hearing but are not enough to protect the retina from the RP phenotype. It should be noted here that all the described-above splice assays were performed in cell lines that might not fully reflect the actual splicing events in photoreceptor cells.
While patients who were homozygous for the c.254-649T>G variant were reported to suffer from a severe USH type1 phenotype with early-onset hearing loss, the index case we are reporting here, who is a compound heterozygous for p.(N48K) (known to cause USH3 in homozygous and compound heterozygous states) and c.254-643G>T, suffers from non-syndromic RP at the age of 51 years. Our results therefore support those reported by others that relatively mild CLRN1 variants can cause non-syndromic RP or RP with late-onset hearing loss. Therefore, both USH and non-syndromic RP cases should be screened for CLRN1 variants, not only within the ORF but also in intronic regions that might result in the inclusion of pseudoexons, when mutated. Such deep intronic variants might be corrected using either CRISPR/Cas9 or antisense oligonucleotides that were found to be efficient for correcting the showed positive results [25]. Both approaches can potentially rescue the c.254-643G>T we are reporting here.
Supplementary Materials
TThe following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.B., D.S. and A.S.S.; methodology, M.A.E., S.K, D.M.P.; validation, M.A.E., and A.S.S.; formal analysis, M.A.E., S.K., and A.S.S.; investigation, M.A.E., S.K., and A.S.S.; resources, S.R., E.B., D.S. and A.S.S.; data curation, M.A.E., and D.S.; writing—original draft preparation, M.A.E., S.K., D.S., and A.S.S.; writing—review and editing, D.M.P., S.R., S.K., and E.B.; supervision, S.R., E.B., and D.S.; funding acquisition, S.R., E.B., and D.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Israel Science Foundation, grant number 1778/20 (DS), the European Union’s Horizon 2020 Research and Innovation Programme under the EJP RD COFUND-EJP N° 825575 (to SR), the Foundation Fighting Blindness Career Development Award CDGE-0621-0809-RAD (to SR), the Foundation Fighting Blindness Project Program Award PPA-0123-0841-UCL (to SR), and by the HRCI HRB Joint Funding Scheme 2020-007 (to FPMC and SR). This study also received funding from Novartis. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Institutional Review Board Statement
Ethical approval was obtained from Hadassah University Medical Center Institutional Review Board (approval code 21-03.08.07, approved on 3 August 2007). The tenets of the Declaration of Helsinki were followed. Participants provided written informed consent after receiving an explanation about the study and its possible consequences and before donating blood samples.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data of this study are presented within the article and its Supplementary Materials. The authors are willing to share materials, data sets, and protocols used in the acquisition of data presented in this publication with other researchers upon request (contact Dror Sharon, E-mail: dror.sharon1@mail.huji.ac.il).
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
The authors would like to thank the individuals who participated in the study for their role and Prof. Tamar Ben-Yosef for sharing research ideas and information.
References
- Schneider, N.; Sundaresan, Y.; Gopalakrishnan, P.; Beryozkin, A.; Hanany, M.; Levanon, E.Y.; Banin, E.; Ben-Aroya, S.; Sharon, D. Inherited Retinal Diseases: Linking Genes, Disease-Causing Variants, and Relevant Therapeutic Modalities. Prog Retin Eye Res 2021, 101029. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-Syndromic Retinitis Pigmentosa. Prog Retin Eye Res 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis Pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yosef, T. Inherited Retinal Diseases. Int J Mol Sci 2022, 23, 13467. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am J Hum Genet 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Hayman, T.; Millo, T.; Hendler, K.; Chowers, I.; Gross, M.; Banin, E.; Sharon, D. Whole Exome Sequencing of 491 Individuals with Inherited Retinal Diseases Reveals a Large Spectrum of Variants and Identification of Novel Candidate Genes. J Med Genet 2023, 61, 224–231. [Google Scholar] [CrossRef]
- Weisschuh, N.; Sturm, M.; Baumann, B.; Audo, I.; Ayuso, C.; Bocquet, B.; Branham, K.; Brooks, B.P.; Catalá-Mora, J.; Giorda, R.; et al. Deep-Intronic Variants in CNGB3 Cause Achromatopsia by Pseudoexon Activation. Hum Mutat 2020, 41, 255–264. [Google Scholar] [CrossRef]
- Runhart, E.H.; Valkenburg, D.; Cornelis, S.S.; Khan, M.; Sangermano, R.; Albert, S.; Bax, N.M.; Astuti, G.D.N.; Gilissen, C.; Pott, J.W.R.; et al. Late-Onset Stargardt Disease Due to Mild, Deep-Intronic ABCA4 Alleles. Invest Ophthalmol Vis Sci 2019. [Google Scholar] [CrossRef]
- Fadaie, Z.; Khan, M.; Del Pozo-Valero, M.; Cornelis, S.S.; Ayuso, C.; Cremers, F.P.M.; Roosing, S.; The, A. Identification of Splice Defects Due to Noncanonical Splice Site or Deep-Intronic Variants in ABCA4. Hum Mutat 2019, 40, 2365–2376. [Google Scholar] [CrossRef]
- Hufnagel, R.B.; Liang, W.; Duncan, J.L.; Brewer, C.C.; Audo, I.; Ayala, A.R.; Branham, K.; Cheetham, J.K.; Daiger, S.P.; Durham, T.A.; et al. Tissue-Specific Genotype–Phenotype Correlations among USH2A-Related Disorders in the RUSH2A Study. Hum Mutat 2022, 43, 613–624. [Google Scholar] [CrossRef]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense Mutation in the USH2A Gene: Association with Recessive Retinitis Pigmentosa without Hearing Loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Huang, M.; Wei, X.; Sun, J.; Zhang, F. Case Report: Compound Heterozygous Nonsense PCDH15 Variant and a Novel Deep-Intronic Variant in a Chinese Child with Profound Hearing Loss. Mol Genet Genomic Med 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Zelinger, L.; Ben-Yosef, T.; Merin, S.; Crystal-Shalit, O.; Gross, M.; Banin, E.; Sharon, D. Exome Sequencing Identifies a Founder Frameshift Mutation in an Alternative Exon of USH1C as the Cause of Autosomal Recessive Retinitis Pigmentosa with Late-Onset Hearing Loss. PLoS One 2012, 7, e51566. [Google Scholar] [CrossRef] [PubMed]
- Panneman, D.M.; Hitti-Malin, R.J.; Holtes, L.K.; de Bruijn, S.E.; Reurink, J.; Boonen, E.G.M.; Khan, M.I.; Ali, M.; Andréasson, S.; De Baere, E.; et al. Cost-Effective Sequence Analysis of 113 Genes in 1,192 Probands with Retinitis Pigmentosa and Leber Congenital Amaurosis. Front Cell Dev Biol 2023, 11, 1112270. [Google Scholar] [CrossRef]
- Herrera, W.; Aleman, T.S.; Cideciyan, A. V.; Roman, A.J.; Banin, E.; Ben-Yosef, T.; Gardner, L.M.; Sumaroka, A.; Windsor, E.A.M.; Schwartz, S.B.; et al. Retinal Disease in Usher Syndrome III Caused by Mutations in the Clarin-1 Gene. Invest Ophthalmol Vis Sci 2008, 49, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Ness, S.L.; Ben-Yosef, T.; Bar-Lev, A.; Madeo, A.C.; Brewer, C.C.; Avraham, K.B.; Kornreich, R.; Desnick, R.J.; Willner, J.P.; Friedman, T.B.; et al. Genetic Homogeneity and Phenotypic Variability among Ashkenazi Jews with Usher Syndrome Type III. J Med Genet 2003, 40, 767–772. [Google Scholar] [CrossRef]
- Khan, A.O.; Becirovic, E.; Betz, C.; Neuhaus, C.; Altmüller, J.; Maria Riedmayr, L.; Motameny, S.; Nürnberg, G.; Nürnberg, P.; Bolz, H.J. A Deep Intronic CLRN1 (USH3A) Founder Mutation Generates an Aberrant Exon and Underlies Severe Usher Syndrome on the Arabian Peninsula. Sci Rep 2017, 7. [Google Scholar] [CrossRef]
- Joensuu, T.; Hamalainen, R.; Yuan, B.; Johnson, C.; Tegelberg, S.; Gasparini, P.; Zelante, L.; Pirvola, U.; Pakarinen, L.; Lehesjoki, A.E.; et al. Mutations in a Novel Gene with Transmembrane Domains Underlie Usher Syndrome Type 3. Am.J.Hum.Genet. 2001, 69, 673–684. [Google Scholar] [CrossRef]
- Adato, A.; Vreugde, S.; Joensuu, T.; Avidan, N.; Hamalainen, R.; Belenkiy, O.; Olender, T.; Bonne-Tamir, B.; Ben-Asher, E.; Espinos, C.; et al. USH3A Transcripts Encode Clarin-1, a Four-Transmembrane-Domain Protein with a Possible Role in Sensory Synapses. Eur J Hum Genet 2002, 10, 339–350. [Google Scholar] [CrossRef]
- Fields, R.R.; Zhou, G.; Huang, D.; Davis, J.R.; Möller, C.; Jacobson, S.G.; Kimberling, W.J.; Sumegi, J. Usher Syndrome Type III: Revised Genomic Structure of the USH3 Gene and Identification of Novel Mutations. The American Journal of Human Genetics 2002, 71, 607–617. [Google Scholar] [CrossRef]
- Pakarinen, L.; Tuppurainen, K.; Laippala, P.; Mäntyjärvi, M.; Puhakka, H. The Ophthalmological Course of Usher Syndrome Type III. Int Ophthalmol 19 307–311. [CrossRef] [PubMed]
- Marouf, A.; Johnson, B.; Alagramam, K.N. Usher Syndrome IIIA: A Review of the Disorder and Preclinical Research Advances in Therapeutic Approaches. Hum Genet 2022, 141, 759–783. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, H.; Oshikawa, C.; Nakayama, J.; Moteki, H.; Usami, S.I. Identification of a Novel CLRN1 Gene Mutation in Usher Syndrome Type 3: Two Case Reports. Annals of Otology, Rhinology and Laryngology 2015, 124, 94S–99S. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Kersten, F.F.J.; Azam, M.; Collin, R.W.J.; Hussain, A.; Shah, S.T.A.; Keunen, J.E.E.; Kremer, H.; Cremers, F.P.M.; Qamar, R.; et al. CLRN1 Mutations Cause Nonsyndromic Retinitis Pigmentosa. Ophthalmology 2011, 118, 1444–1448. [Google Scholar] [CrossRef]
- Panagiotopoulos, A.L.; Karguth, N.; Pavlou, M.; Böhm, S.; Gasparoni, G.; Walter, J.; Graf, A.; Blum, H.; Biel, M.; Riedmayr, L.M.; et al. Antisense Oligonucleotide- and CRISPR-Cas9-Mediated Rescue of MRNA Splicing for a Deep Intronic CLRN1 Mutation. Mol Ther Nucleic Acids 2020, 21, 1050–1061. [Google Scholar] [CrossRef]
Figure 1.
Variant detection and familial segregation analysis. (A) Two generation family pedigree; (B) BAM files showing the two CLRN1 variants: c.144T>G and c.254-643G>T; (C-D) Familial segregation analysis in proband (C) and one of his unaffected siblings (D).
Figure 1.
Variant detection and familial segregation analysis. (A) Two generation family pedigree; (B) BAM files showing the two CLRN1 variants: c.144T>G and c.254-643G>T; (C-D) Familial segregation analysis in proband (C) and one of his unaffected siblings (D).
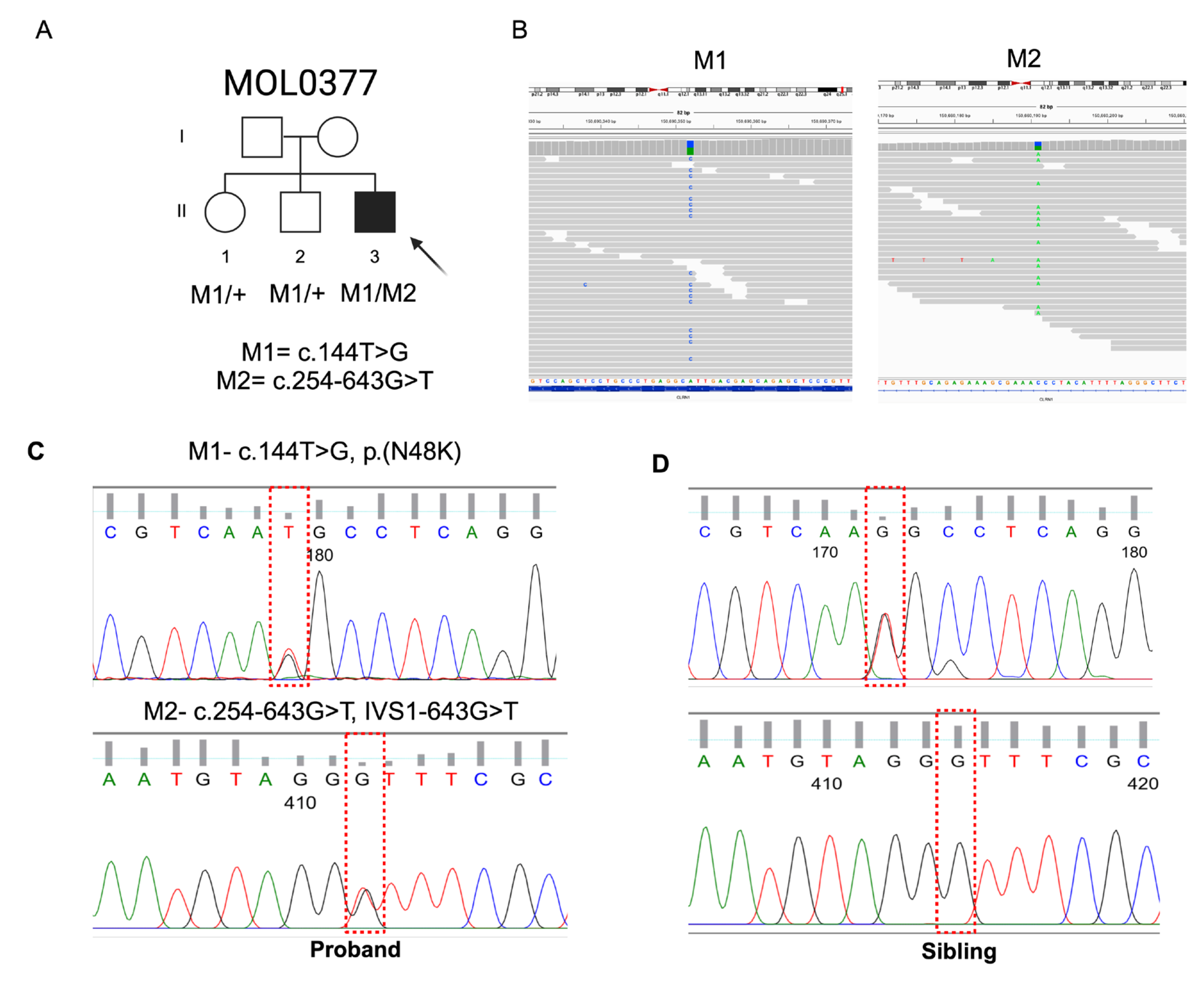
Figure 2.
Retinal imaging of MOL377-1 at the age of 40 years (A-F) and 50 (G-L). A-B and G-H represent ultra-wide field pseudocolor and autofluorescence (FAF) fundus photos, respectively, taken using the Optos Panoramic 200 Optomap Fundus Camera. Characteristic peripheral dense BSPs mixed with retinal atrophy encroaching the temporal vascular arcades can be seen. C-D and I-J Show heterogeneous autofluorescence compatible with the atrophic retina along with hyperfluorescent ring surrounding the fovea. E-F and K-L are Horizontal optical coherence tomography (OCT) sections showing preserved foveal islands surrounded by retinal thinning and loss of the outer retinal layers in the macular area. Cystoid macular edema (CME) was observed in the LE and RE in the first and the last follow-up visits, respectively.
Figure 2.
Retinal imaging of MOL377-1 at the age of 40 years (A-F) and 50 (G-L). A-B and G-H represent ultra-wide field pseudocolor and autofluorescence (FAF) fundus photos, respectively, taken using the Optos Panoramic 200 Optomap Fundus Camera. Characteristic peripheral dense BSPs mixed with retinal atrophy encroaching the temporal vascular arcades can be seen. C-D and I-J Show heterogeneous autofluorescence compatible with the atrophic retina along with hyperfluorescent ring surrounding the fovea. E-F and K-L are Horizontal optical coherence tomography (OCT) sections showing preserved foveal islands surrounded by retinal thinning and loss of the outer retinal layers in the macular area. Cystoid macular edema (CME) was observed in the LE and RE in the first and the last follow-up visits, respectively.
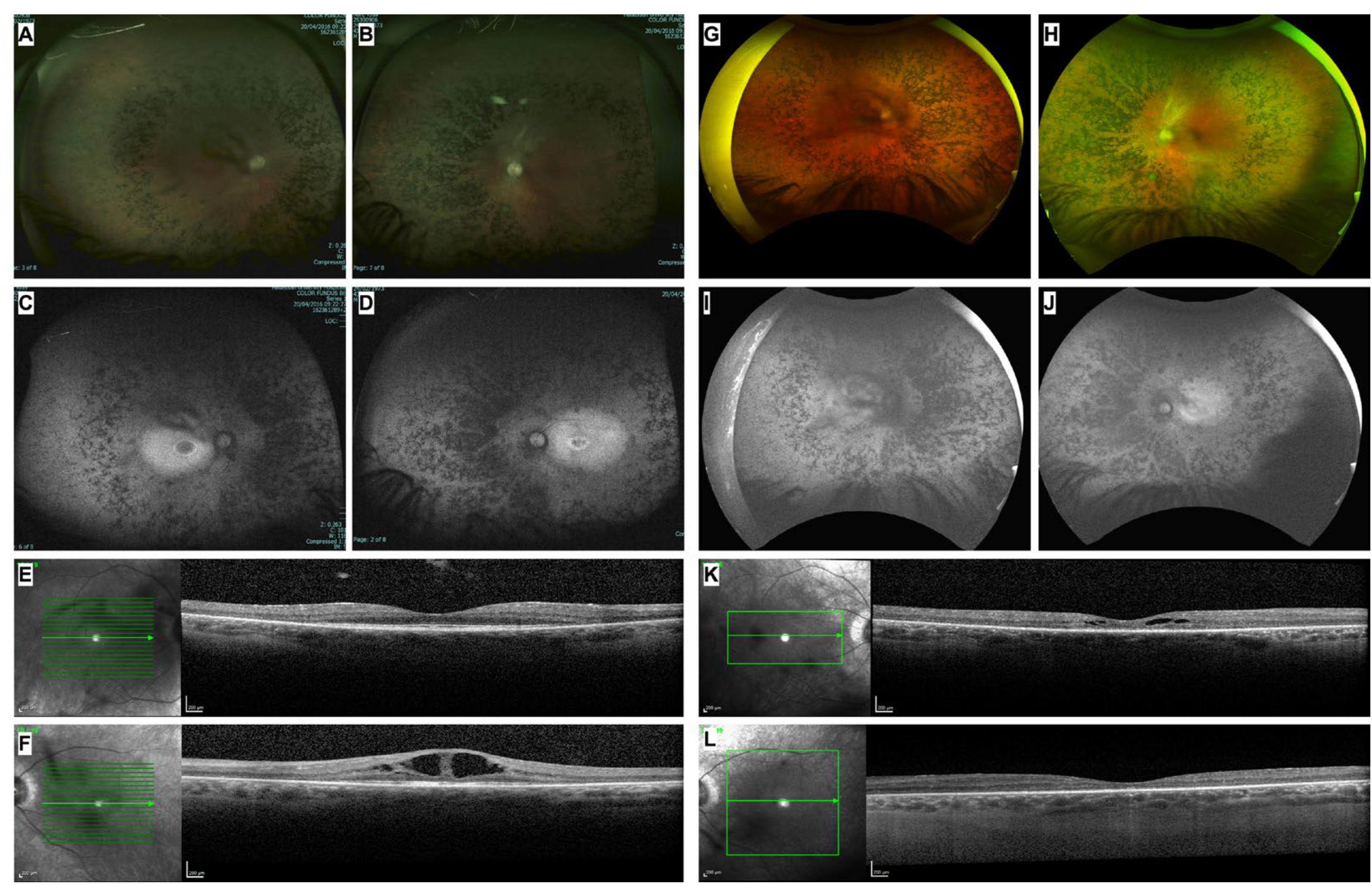
Figure 3.
Analyzing the effect of two CLRN1 variants (c.254-643G>T and c.254-649T>G) on its pre-mRNA splicing. (A) Graphical representation of pET01 minigene plasmid with a 792 bp CLRN1 intron 1 insert; (B) A representative agarose gel image of cDNA analysis from HeLa cells transfected with different plasmid constructs. A 1.5% agarose gel was used to separate the PCR products; the experiment was performed in biological triplicates. Bands labeled as TS1-3 represent the different transcripts shown in panel C; (C) Graphical representation of the different splicing patterns in the pET01-CLRN1 minigene due to the two CLRN1 variants c.254-643G>T and c.254-649T>G. (D) A graphical representation of the human CLRN1 gene (top panel) and the insertion of a pseudo-exon due to the c.254-643G>T and c.254-649T>G variants (bottom panel). Int- intron, TS- transcript, Ex- exon, WT- wild-type, DSS- donor splice-site, BP- branch point, ASS- acceptor splice-site.
Figure 3.
Analyzing the effect of two CLRN1 variants (c.254-643G>T and c.254-649T>G) on its pre-mRNA splicing. (A) Graphical representation of pET01 minigene plasmid with a 792 bp CLRN1 intron 1 insert; (B) A representative agarose gel image of cDNA analysis from HeLa cells transfected with different plasmid constructs. A 1.5% agarose gel was used to separate the PCR products; the experiment was performed in biological triplicates. Bands labeled as TS1-3 represent the different transcripts shown in panel C; (C) Graphical representation of the different splicing patterns in the pET01-CLRN1 minigene due to the two CLRN1 variants c.254-643G>T and c.254-649T>G. (D) A graphical representation of the human CLRN1 gene (top panel) and the insertion of a pseudo-exon due to the c.254-643G>T and c.254-649T>G variants (bottom panel). Int- intron, TS- transcript, Ex- exon, WT- wild-type, DSS- donor splice-site, BP- branch point, ASS- acceptor splice-site.
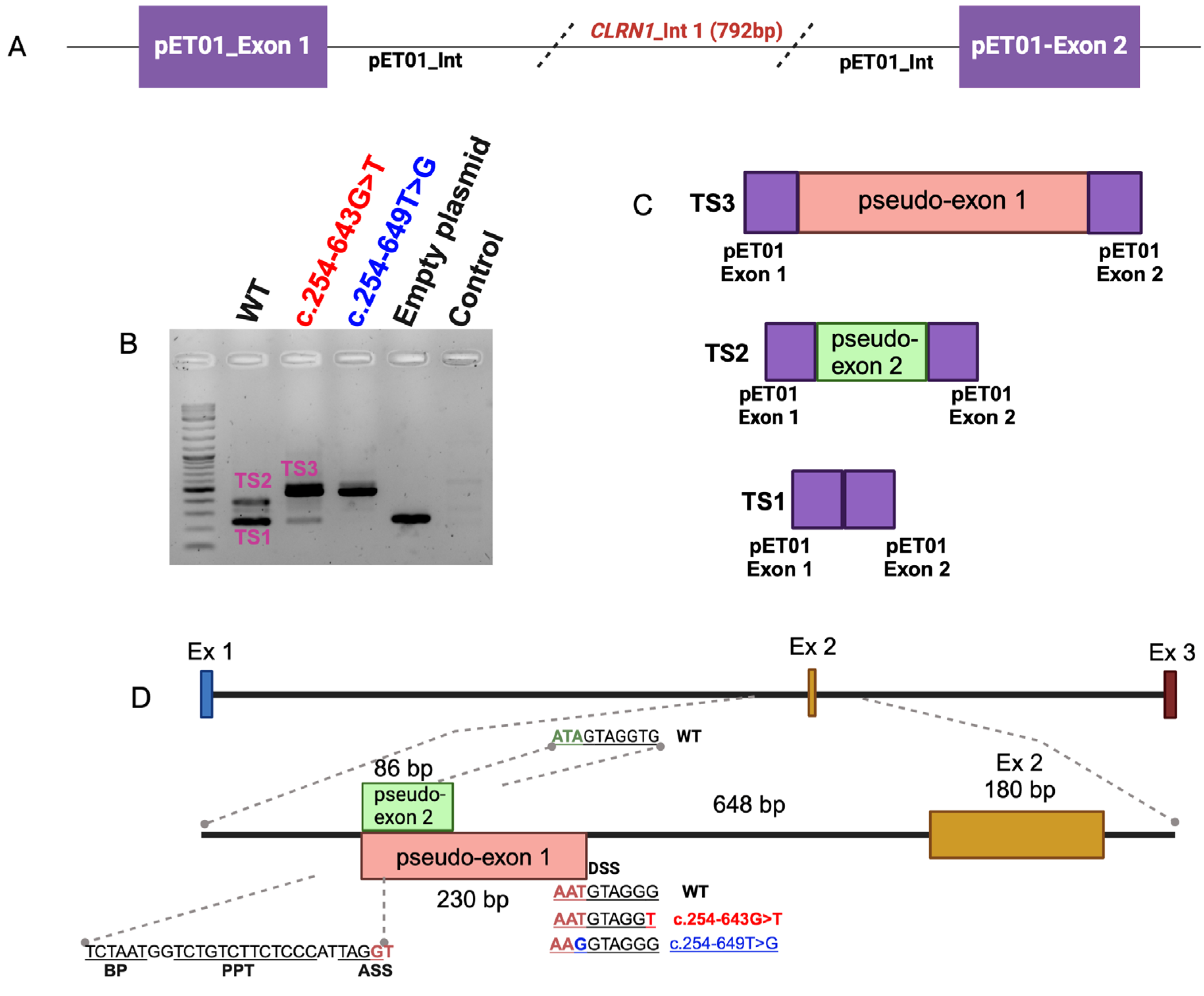
Figure 4.
Sanger sequencing of the wild type and mutant CLRN1 transcripts. (A) Sanger sequencing of transcript TS3 containing pseudo exon 1; (B) Sanger sequencing of transcript TS2 containing pseudo-exon 2; (C) Sanger sequencing of transcript TS1 (wild type transcript).
Figure 4.
Sanger sequencing of the wild type and mutant CLRN1 transcripts. (A) Sanger sequencing of transcript TS3 containing pseudo exon 1; (B) Sanger sequencing of transcript TS2 containing pseudo-exon 2; (C) Sanger sequencing of transcript TS1 (wild type transcript).
Figure 5.
Detailed analysis of CLRN1 transcripts using TapeStation. (A) Automated gel electrophoresis of the PCR products using Agilent Technologies 4200 TapeStation. (B) Bar graph showing the intensity of each band in WT compared to the two studied variants.
Figure 5.
Detailed analysis of CLRN1 transcripts using TapeStation. (A) Automated gel electrophoresis of the PCR products using Agilent Technologies 4200 TapeStation. (B) Bar graph showing the intensity of each band in WT compared to the two studied variants.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



