
Submitted:
07 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
Control of oxidation/antioxidation homeostasis is important for cellular protective functions, and disruption of the antioxidation balance by exogenous and endogenous ligands can lead to profound pathological consequences of cancerous commitment within cells. Although cancers are sensitive to antioxidation drugs, these drugs are sometimes associated with problems including tumor resistance or dose-limiting toxicity in host animals and patients. These problems are often caused by the imbalance between the levels of oxidative stress induced reactive oxygen species (ROS) and the redox efficacy of antioxidants. Increased ROS levels, because of abnormal function, including metabolic abnormality and signaling aberrations, can promote tumorigenesis and the progression of malignancy, which are generated by genome mutations and activation of pro-oncogene signaling. This hypothesis is supported by various experiments to show that the balance of oxidative stress and redox control is important for cancer therapy. Although many antioxidant drugs exhibit therapeutic potential, there is a heterogeneity of antioxidation functions, including cell growth, cell survival, invasion abilities, and tumor formation, as well as the expression of marker genes including tumor suppressor proteins, cell cycle regulators, nuclear factor erythroid 2-related factor 2, and Jun dimerization protein 2; and their effectiveness in cancer remains unproven. Here, we summarize the rationale for the use of antioxidative drugs in preclinical and clinical antioxidant therapy of cancer, and recent advances in this area using cancer cells and their organoids, including the targeting of ROS homeostasis.
Keywords:
Antioxidation
; Heterogeneity
; Reactive oxygen species
; Redox homeostasis
; Cancer therapy
; Tumor suppressor p53
1. Introduction
It has been known that the control of balance between reactive oxygen species (ROS) and redox status is an important way to address the occurrence and development of tumor cells without compromising normal cells [1]. This idea depends upon the theory that cancers have a homeostatic balance of oxidation, which is produced by phase I enzyme ligands, and antioxidation, which is mediated by phase II enzyme ligands and is arranged to favor the hallmark actions of normal cells or cancer cells, such as proliferation, survival, migration, and metastasis (Figure 1) [2]. Thus, the ROS balance is critical for regulating oxidative stress and antioxidation: mediated by the transcription factors such as to aryl hydrocarbon receptor (AHR) and nuclear factor erythroid 2-related factor 2 (NRF2), respectively. The extensive redox conditions damage biocomponents, and cancers are known to compensate for this damage by enhancing the expression of antioxidants. Thus, the development of cancers depends on these antioxidant proteins to regulate the oxidation and antioxidation status of macromolecules within cells to maintain the ROS balance. Thus, a switch in cancer redox activity produced by antioxidation or increased ROS production causes the threshold of the ROS balance of cancer cells to be altered, which causes cell cycle arrest or cell death [3]. Indeed, redox-modulating drugs that cause apoptosis are important in defining ROS-induced cell death, which is important for anticancer therapy [3]. Although drugs that control ROS are expected to have therapeutic applications in cancer, drugs targeting ROS have shown only limited success in preclinical trials compared with other anticancer drugs [4]. These results raise the possibility that tumor cells may not be more sensitive to ROS than normal cells. Thus, these trials offered less success than anticipated. The lack of biomarkers to measure endogenous redox levels might also prevent success [5]. Furthermore, the complexity and redundancy of drugs targeting ROS/redox are not well characterized. Recent reports have shown the heterogeneity of phase II enzyme reagents against cancer organoids [6]. Thus, the use of antioxidants for cancer treatment should be applied with caution. In this review, the heterogeneity of phase II enzyme agents is discussed in the context of cancer treatment and how to avoid these difficulties in preclinical trials of phase II drugs for cancer treatment.
2. ROS in Cancer Cells
Assuming a crucial role for ROS in cancer cells, controlling endogenous ROS by altering the function of mitochondrial constituents is a possible anticancer strategy. These strategies may inhibit ROS-mediated cancer occurrence and progression by causing oxidative damage, such as ROS-mediated apoptosis [7,8]. Thus, preclinical research on antioxidant production and weak pro-oxidants was conducted to understand their merits. Compared with normal cells, cancer cells generate excessive ROS, increasing their sensitivity to further increases in ROS-related cell damage and committing them to apoptosis. Pro-oxidants may thus have antitumor functions. Therapeutic trials of antioxidants targeting ROS control have included nonenzymatic drugs, including NRF2 agonists [9] and vitamins [10,11], or targeting ROS via the enzymatic production of antioxidants, such as nitrogen oxide inhibitors [12,13], superoxide dismutase mimetics [14], N-acetylcysteine, and glutathione (GSH) esters [15,16]. Many such clinical trials have been conducted (Table 1), and many such agents against ROS production have been developed, but their actions involve ROS homeostasis and antioxidation, which are not specific to cancer.
For example, the repression of glutathione peroxidase 2 (GPx2) in breast cancer may enhance cancer progression due to hypoxic signals, aberrant vascularization, and a metabolic switch to the aerobic glycolysis/oxidative phosphorylation (OXPHOS) axis. The complex expression of GPx2 depends on metabolic specific drives and microenvironments, such as hyperproliferation-derived hypoxia.
Another consideration is that ROS has a dual function in cancer occurrence, which might depend on endogenous ROS (Figure 1) [17]. This dichotomy further complicates the antioxidation response to the decomposition of H2O2 through a change in the glutathione-SH (GSH)–glutathione sulfide (GSSG) axis. Hypoxia-inducible factor-1β which was named differently as AHR nuclear translocator (ARNT), is a component of the complex of phase I enzymes with AHR. Thus, crosstalk between AHR signals and hypoxia signals might occur, leading to overlap. In addition, the development of cancers also involves contrasting levels of GPx2 expression, which are correlated with the progression of cancer and poor prognosis. Decreased GPx2 expression was detected in colon cancer, pancreatic cancer, cancers of the bladder and urinary tract, and esophageal colon carcinomas, whereas increased GPx2 expression was detected in several carcinoma [18]. This apparent contradiction of GPx2 levels might be due to differences in the levels of the intact endogenous ROS production machinery [18].
3. NRF2 Has Dual Roles in Tumorigenesis
NRF2 has dual and contradictory roles in cancer [19]. Abnormal increased expression of NRF2 can be related with a poor prognosis. The constitutive level of NRF2 in many cancer cells might induce the expression of pro-survival genes and promote the proliferation of tumor cells via metabolic alterations, inhibit cancer specific apoptosis, and increase the self-renewal function of cancer stem cells. Moreover, NRF2 contributed to chemoresistance, radio-resistance, and inflammation, inducing carcinogenesis. Many NRF2 inhibitors have been reported to treat cancers [20]. These trials targeted NRF2 might provide a new tool in cancer treatment.
3.1. Tumor Suppressive Actions of NRF2
The NRF2 protein is activated by tumor suppressor gene products such as breast cancer susceptibility gene I and p21Cip1 by inhibiting KEAP1 (Kelch like ECH associated protein 1/NRF2 complex formation in microenvironmental cells in tumors [21,22]. The degradation of the oncogene tyrosine kinase Fyn blocks its activation [23]. Nrf2-deficient mice reduced the production of antioxidants and repressed the expression of markers specific for phase II enzyme encoded genes. The quinone induced the severe oxidative stress rendered Nrf2-deficient mice more prone to skin cancer, while the Nrf2-medited expressions of NAD(P)H quinone dehydrogenase 1 (Nqo1) and Gst were inhibited [24]. Similarly, Nrf2-deficient mice exposed to carcinogens developed more cancers included stomach [25], liver [26], and urinary bladder [27] as compared with wild type mice, suggesting that Nrf2 is the critical factor controlling inflammation.
Many compounds isolated from plants, including carnosol, curcumin, resveratrol, and sulforaphane, and synthetic chemicals like oltipraz and oleanane triterpenoids, exhibited the chemo-preventive functions by activating the Nrf2-antioxidant response element (ARE)-regulated genes. These Nrf2 activators mainly controlled the antioxidation reaction by changing the intermolecular disulfide (S–S) bonds between two cystine residues of Keap1 proteins at Cys273 and Cys288 to commit the accumulation of Nrf2 [28]. Sulforaphane (SFN) is committed to inducing the transcription of phase II enzyme-encoding genes while inhibiting phase I enzyme-encoding gene and facilitating the cell death of cancer cells through the TP53 mechanism [29]. In addition, SFN targeted nuclear factor kappa B (NF-κB) [30] and Activation protein-1 (AP-1) complexes with an anti-inflammatory effect [31]. Other synthetic oleanane triterpenoids inhibited tumorigenesis by inhibiting the transcription of oncogenes, including k-RAS, TP53, BRCA1, and Erb-B2 Receptor Tyrosine Kinase 2, in some cancers [32,33].
In fact, a lot of therapeutics that controlled the NRF2 activity have been currently applied in clinical trials; these drugs played a significant role in repressing the cancers and could be augmented further by chemo-preventive compounds. Paradoxically, NRF2-deficient cancer cells were more prone to death of cancerous cells by oxidative stress but more resistant to chemo-preventive compounds. Thus, the effective drugs targeting NRF2 pathways represented a crucial strategy to develop the useful chemo-preventive medications. However, we need accumulate the further pre-clinical data for convincing the efficiency of these drugs against cancer patients.
Nrf2-knockout mice showed that the level of nitric oxide synthase activated by cyclooxygenase 2 and the level of tumor necrosis factor (Tnf) are greater than those in normal mice, indicating that Nrf2 is able to suppress the proinflammatory molecules [34]. In addition, Nrf2-dependent activation of Nqo1 decreased the levels of Tnf and Interleukin 1 after incubation with lipopolysaccharide. Although ROS are the molecular targets of NRF2-mediated anti-inflammatory effects, NRF2 might function as an anti-inflammation mediator without inducing ROS, which were reported by regulating the expressions of axis encoding the macrophage receptor with collagenous structure and CD36, which are not concerned with the oxidative response [35]. Furthermore, NRF2 protected against cell damages induced by H2O2 exposure via the p38/MAPK pathway [36,37]. Similarly, NRF2 inhibited the NF-κB pathway by stabilizing the inhibitor of nuclear factor κB kinase subunit-α (IKK-α) to inhibit the degradation of IKK-β [38]. By contrast, the NF-κB p65 subunit was reported to compete with NRF2 through the CH-KIK domain of the cyclic AMP response element binding protein (CREB) binding protein CBP coactivator [39]. Thus, these different mechanistic inputs of NRF2 should be clarified to determine how it suppressed the oncogenicity by controlling the level of ROS within cells.
3.2. Oncogenic Functions of NRF2
NRF2 is usually constitutively activated in tumorigenic cells. For example, somatic mutations in KEAP1 and/or NRF2 genes, exon skipping in the NRF2 genome, methylation/demethylation of the KEAP1 promoter, accumulation of p62/sequestosome-1, and mutation of fumarate hydrolase were reported [40,41] Constitutively activated NRF2 promoted cell proliferation through metabolic changes; inhibiting apoptosis; promoting angiogenesis, and invasion/ metastasis; and promoting drug resistance in various cancers [40,41]. Forced expression of NRF2 promoted the transcriptions of the oncogenes like MYC, KRAS, and BRAF [42]. Moreover, NRF2 activated oncogenes by suppressing the activity of tumor suppressor gene products like phosphatase tensin homolog/glycogen synthetase kinase 3/β-transduction repeat-containing E3 ubiquitin-protein ligase [43]. Indeed, NRF2 increased the proliferation of tumor cells by increasing the expression of glycolytic enzymes included glucose-6-phosphate dehydrogenase, phosphoglucomutase dehydrogenase, transketolase and trans-aldolase [44], which controlled genes involved in fatty acid-lipid metabolism [45], growth-associated genes [46], and cell cycle regulators [47]. The activation of NRF2 via the glucose-regulated protein 78/phosphorylated protein kinase RNA-like ER kinase/NRF2 signaling pathway enhanced the transcriptions of glycolytic genes and simultaneously to induced the inhibition of the TCA cycle related genes, which enhanced the Warburg effect [48].
Increased levels of NRF2 suppressed the endogenous ROS level in cancer stem cells (CSCs) compared with noncancer stem cells, suggesting the enrichment of stemness phenotypes [49,50,51,52,53]. For example, the CSCs with reduced mitochondrial-derived ROS exhibited the cancer stemness-associated properties, such as epithelial-to-mesenchymal transition (EMT) [48,61]. Similarly, persistent activation of NRF2 improved the self-renew ability of CSCs, by maintaining the cell quiescence and reducing the intracellular levels of ROS [54,55]. Mesenchymal stem cells (MSCs) in the microenvironment cells promoted the spread of cancer cells by enhancing their motility and invasion ability [56,57]. Thus, NRF2 was required to maintain the stemness of MSCs and caused their cell deaths under the oxidative stress condition [58]. Furthermore, because the continuous expression of NRF2 significantly benefited the cancer cells, these cells frequently developed to be the NRF2 dependency [59,60].
In addition, NRF2 derived oncogenic activity also stimulated the angiogenesis capacity, mainly by activating heme oxygenase 1 (HO-1) [61], which in turn stimulated the vascular endothelial growth factor to enhance the angiogenesis [62]. siRNA against NRF2 downregulated HO-1 and sensitized the acute myeloid leukemia cells to TNF-induced cell death. This effect indicates that NRF2 inhibits the apoptosis of cancer cells by regulating HO-1 [63]. In addition, NRF2 upregulated the antiapoptotic protein B-cell lymphoma 2 (Bcl2), while it downregulated the activity of BAX and caspase 3 to protect against the etoposide/radiation-mediated cell death following drug resistance [64]. Furthermore, NRF2 inhibited the activation of proapoptotic JNKs [65] and induced selective autophagy reactions via Keap1 degradation [66,67]. However, the forced expression of NRF2 allowed the autophagy-dependent cancer cells to overcome the loss of autophagy function [68].
Increased accumulation of NRF2 in the nucleus was associated with proliferation capacity. For instance, phosphoinositide 3-kinase (PI3K)-Akt activation coupled with depletion of Keap1 resulted in Nrf2-dependent proliferation of hepatocytes and cholangiocytes [69,70]. However, because the ability of stable Nrf2 was not sufficient to alter from a cancer defender to a cancer driver, the additional mutations of oncogenes and tumor suppressor genes were absolutely required [71,72,73]. Keap1 mutations, such as Kras/Hras mutations and Trp53 loss of function, are required to produce the NRF2-dependent cancer models [74]. Furthermore, NRF2-dependent malignancies with somatic mutations of KEAP1 and NRF2 differed depending on the specific organs. Thus, tissue-specific variation is another issue for the study of NRF2-dependent cancer, which might be discussed further.
In resistance to medical therapy, NRF2-controled drug efflux transporters are predictors of resistance for drug inoculations. For example, multidrug resistance protein 1 (MDR1), multidrug resistance-associated protein 1-5 (MRP1-5), and breast cancer-resistant protein (BCRP) are critical for controlling issues to regulate the therapy because they induced the abnormal NRF2 activation to induce the widespread chemoresistance [75,76,77,78]. These issues might be the complementary trials for drug delivery to the cancer cells.
4. NRF2-Tragetted Drugs in Cancer Prevention
In the initiation stage of cancer, NRF2 activation can suppress carcinogenesis. Under normal conditions, NRF2 maintains antioxidation homeostasis and exerts anti-inflammatory effects and antitumorigenic effects, thus supporting cell survival.
Heterogeneity of Antioxidation Drugs against Cancer
Wu et al. (2023) recently reported the heterogeneity of redox ligands controlling the growth and progression of human gastric cancer organoids [6]. They performed experiments to compare the efficiency of three drugs, perillaldehyde (PEA) and cinnamaldehyde (CE), and the regular antioxidative drug sulforaphane (1-isothiocyanato-4- (methane sulfinyl) butane; SFN), and measured cancer occurrence by assessing xenotransplantation in SCID mice. PEA extracted from Perilla frutescens [79] showed the antifungal, and antioxidant activities in addition to other biological functions [80,81,82]. PEA activates NRF2 and represses oxidative stress-induced innate immunity in human keratinocytes [83]. CIN is a β-unsaturated aldehyde from cinnamon [84]. In addition, the antioxidant activity of CIN was demonstrated in mice [85]. Exposure to CIN induced autophagy-dependent cell death through epigenetic alteration by the histone methylating enzyme G9a in the endoplasmic reticulum [86]. CIN inhibited the AHR axis and induced an NRF2-mediated antioxidation response [87]. Thus, PEA and CIN might function in NRF2-dependent antioxidation to inhibit ROS generation without affecting AHR-mediated oxidative stress pathway. SFN, as a dietary isothiocyanate synthesized from a precursor in Brassica, was also examined. It is one of the most typical ligands of phase II enzymes pathway including NQO1, GST α-1, and HO-1, which are required to eliminate chemically damaged DNA. SFN induced cell cycle arrest at S-phase and apoptosis in a TP53-dependent manner in gastric cancer (GC) cells [88]. In addition, SFN inhibited the metastasis of breast cancer [89].
Another issue is the differential outputs on function of AHR- and NRF2-mediated phase I and II pathways. Both PEA and CIN induced an NRF2-mediated phase II enzyme response, which was mediated by ARE-containing genes [86] but was not involved in the phase I pathway [87]. By contrast, the effects of SFN were reported to be involved in both enzymatic pathways; SFN produced NRF2-dependent phase II promoter activation and AHR-dependent activation of phase II promoters such as NQO1 [90,91]. This activation provides unambiguous evidence of the different actions of PEA/CIN and SFN in preventing cancer-inducing activity. It is surprising to observe the contrasting actions of PEA/CIN and SFN in inhibiting gastric cancer development, ROS generation, and cellular apoptosis. The TP53 mutation might explain these differential effects because 3-D organoids exhibited TP53 mutations, which might affect the alterations of phase I and phase II enzyme reactions (Figure 2).
5. Phase I Drugs in Clinical Trials
Phase I and II enzyme pathways are known to overlap and are regulated by the “ARF–NRF2” gene battery [92]. In addition, the AP1/ATF transcription factor and histone chaperone Jun dimerization protein 2 (Jdp2) played a role in regulating AHR promoter activity via the NRF2 complex [93]. This mechanism was called as the AHR–NRF2–JDP2 gene battery.
In the case of AHR therapeutics, the present application of AHR antagonists in cancer disorders is a reasonable strategy for preclinical trials. AHR antagonists inhibit immune suppression, and AHR agonists overcome chronic inflammation and autoimmune disorders in cancer patients. The AHR antagonist IK-175 is well known to be effective in an anticancer therapeutic trial when combined with anti-PD1 antibodies. A phase Ia/b open-label study of OK175, alone or in combining with nivolumab, which suppressed locally advanced solid tumors and urothelial carcinoma, has been reported (NCT04200963) [94]. The microbial metabolite indole-3-aldehyde (3-IAld) exhibited strong anti-inflammatory activity via AHR [95,96]. Compelling results with 3-IAld were obtained in a dextran sodium sulfate (DSS)-mediated inflammatory bowel disease of mouse model. 3-IAld repaired colon damage and improved epithelial barrier integrity through AHR, and IL22 cured the immune checkpoint inhibitor (ICI)-induced colitis but did not block the antitumor effect of ICI [97]. These data indicate that the AHR ligand of 3-IAld could be developed as an agent for treating human disease.
BAY 2416964 was a novel oral AhR inhibitor that prevent the AHR ligand-induced immunosuppressive effects and enhanced the proinflammatory activity of antigen-in-human phase I clinical trials [98]. Studies in vitro showed that BAY 2416964 restored immunological activity in human and mouse cells, stimulated antigen-specific cytotoxic T-cell responses, and killed tumors. Studies in vivo showed that its oral application was well tolerated and demonstrated antitumor activity in a syngeneic mouse cancer model.
6. Phase II Drugs in Clinical Trials
Clinical trials using phase II enzyme drugs have been reported. For example, SFN has achieved only limited success in prostate cancer patients [99]. However, SFN is not effective for treating breast cancer patients [100]. SFN inhibited the progression of GC [88,101,102,103,104]. Thus, further studies are required to determine the usefulness of SFN.
In addition, the mutation status of oncogenes and antioncogenes is also crucial for the efficacy of treatment with phase II enzyme drugs. The expressions of TP53, NRF2, and JDP2 were tremendously repressed in the growth of organoids exposed with PEA and CIN as compared with those in control normal organoids. However, a 1.5- to 1.75-fold increase in NRF2 and TP53 expression was found in SFN-treated organoids compared with in control organoids. Exposure to PEA or CIN repressed the expression of proteins of the TP53–NRF2 axis and a third factor histone chaperoneJDP2, but SFN increased the expression of TP53 and NRF2 proteins. In the case of a tumor, the p53 mutation itself did not induce the tumor formation, but tumors developing from areas with p53 mutation and loss of heterozygosity were larger and demonstrated extensive chromosomal instability compared with lesions arising in normal epithelium [105]. Most TP53 mutations in cancers are found as the missense mutations rather than truncations or deletions. Both dominant-negative effects and gain-of-function activities were observed in the case of mutant p53 [106]. Mutant TP53 interacts with NRF2, but positive or negative effects of NRF2 have been reported [107,108,109]. Non-small cell lung cancers carrying mutant TP53 but not mutant NRF2 or KEAPl displayed higher levels of NRF2 mRNA than wild-type TP53 tumors [110]. In similar, the oncogenes KRAS, BRAF, and MYC promoted the increased transcription of NRF2 gene and its target genes, which might induce a greater decrease in cellular molecules [42]. Mutant p53 prolonged TNFα-induced NF-κB signaling [111], and mutation of p53 can upregulate Nrf2 via the NF-κB axis. However, in 3-D gastric cancer organoids, PEA and CIN treatment reduced the protein expression of TP53 and Nrf2 and reduced ROS and caspase 3 activity [6].
By contrast, SFN exposure induced NRF2 and other redox functions to be dominant. Some reports have indicated that the levels of phase II enzymes, such as NQO1, are increased in cancer tissues compared with healthy tissues and that NQO1 stabilized the wild-type TP53, especially under oxidative stress [112]. Compared with those of wild-type TP53, the TP53 mutants exhibited increased binding to NQO1 [113]. By contrast, the function of NRF2 as an antioxidation response factor was blocked in the case of R273H in p53-expressing cancer cells upon oxidative stress, and the NRF2 antioxidant response was impaired because of the decreased expression of NQO1 and HO-1 [114].
Mutant p53 enhanced the Warburg effect by activating the glucose transporters GLUT5/6 and GLUT3 via NF-κB axis and GLUT1 by stimulating its transferring to the plasma membrane. Various glycolytic enzymes are also stimulated by mutant p53, which caused increased glucose uptake and glycolysis [115]. The effect of the microenvironment with p53 mutations can also affect cancer generation.
One TP53 mutant, APR-246, has been tested in clinical applications [116]. The APR-246 mutant inhibited thioredoxin reductase 1 (TrxR1) [117], thioredoxin (Trx), glutaredoxin, and ribonucleotide reductase [118] and depleted cellular GSH [116,119,120]. Recent studies revealed that APR-246 was not specific to cancer cells, but its effects might be cell-type specific [121].
In general, the expression of p53 counteracted the expression of ARE-containing antioxidant genes such as cystine/glutamate transporter (SLC7a11; x-CT), NQO1, and GST-α1, which are Nrf2 targets [122]. p53 deletion increased ROS, DNA oxidation, and mutations. The introduction of the dietary supplementation with the antioxidant N-acetylcysteine subsequently improved karyotype stability and prevented the early onset of mutations for tumorigenesis. Three p53 mutations, K117R, K161R, and K162R, were generated and resulted in impaired p53-mediated cell cycle arrest, senescence, and apoptosis. Unlike p53-knockout mice, this p53 mutant mice did not develop early-stage lymphoma [123,124]. KRAS depletion and the depletion of RalB and IκB-related TANK-binding kinase 2 (TBK1) induced to activate p53 in a ROS- and NRF2-dependent manner. Similarly, the IκB kinase inhibitor BAY 11-7085 and dominant-negative mutant IκB-αM inhibited the NF-κB activity and increased the levels of phosphorylated p53, p53, and p21Cip1 in a ROS-dependent manner [125]. The p53–Mdm2 protooncogene (MDM2)–ARF network can lead to unconventional and unique innovative approaches for developing a new generation of genetically informed and clinically effective cancer therapies [126].
Wild-type TP53 repressed Jdp2 promoter activity, but TP53-knockout mutant cells did not, which was demonstrated using HCT116 p53−/− cells [127]. JDP2 enhances the NRF2-dependent antioxidant response [128]. JDP2 functions not only as a transcription factor but also as a histone chaperone for targeting specific acetylated histones. In addition, it has inhibitory effects on p300-mediated histone acetylation [129]. Therefore, the expression of TP53 and JDP2 was reported to be coregulated during transcription and chromatin regulation. These alterations might influence the function of NRF2 in TP53-mutant cancer cells or organoids. Thus, the molecular interactions of mutated TP53, JDP2, and NRF2 should be clarified.
Therefore, the contradictory results with PEA or CIN and SFN should be studied at the molecular level to identify the distinct pathways involved in human GC.
7. Discussion
This review highlights the role of the transcriptional balance of key factors between cellular antioxidation and oxidation in cancer development. The dedicated interplay to control NRF2 and AHR master regulators of ROS homeostasis should be defined as the context-dependent nature of NRF2/AHR actions in each cancer case. Ingenuity pathway analysis (IPA) of the signal networks between AHR and NRF2 revealed that they connected to shared target genes, such as genes related to the cell cycle, and chromatin regulators, including cyclin-dependent kinase inhibitor 1 (CDKN1A; p21CIP1) and EP300 (histone acetylase p300) (Figure 3). JDP2, such as CDKN1A, has been reported to play a role in cell cycle regulation by controlling cell growth via cyclin A2 [130] and p300/CBP acetyltransferase to function as a HAT on histones [129].
Given the complexity of antioxidation mechanisms in different cancer patients, the cancer types, and stages of cancer progression, manipulating redox status may be inappropriate. Thus, the precise regulation of redox balance is important and has become a key target in searching the next-generation of redox-regulating agents for cancer therapy. A further understanding of redox mechanisms in cancer initiation and progression is still challenging by repeating the examples to see the experimental validation.
A variety of responses to NRF2-mediated cancer development, which are obtained by 2-D cell culture, should be reexamined using 3-D organoids. These responses might be due to the intratumor heterogeneity of CSCs and to committed subpopulations with distinct microenvironments. The following questions should be addressed: (i) Do antioxidant exposures, as cancer treatments, demonstrate the positive and negative dual effects in cancer types, caner stages or the endogenous ROS dependent manner? (ii) Are there specific treatments with different doses in different tumor tissues that need treatment with antioxidant therapies for required clinical outcomes? (iii) Is JDP2–NRF2-induced metabolic reprogramming in response to phase II enzyme ligands are required for CSCs or niches which are dependent upon the status of TP53 mutation? Such questions are currently ongoing. The techniques used to measure endogenous ROS levels in vivo and in 3-D organoid tips might be the next targets for applying redox drug therapy for cancer treatment, in addition to further identifying markers for CSCs and microenvironments. These technologies might validate antioxidant therapy for tumors. Thus, we need more experiments of mechanistic analysis of phase II drugs against cancer development.
Funding
This work was supported by grants from the Ministry of Science and Technology (MOST111-2314-B-037-009), the National Health Research Institutes (NHRI-EX109-10720SI), Kaohsiung Medical University Hospital (KMUH111-1R77; KMUH-DK(A)112002), the National Sun-Yat-sen University-Kaohsiung Medical University Joint Research Project (NYCUKMU-113-I006), and Kaohsiung Medical University (KMU-TC113A02).
CRediT authorship contribution statement
YCL wrote, reviewed the original manuscript, and generated the figures. CC-K performed the conception and design the work and wrote the original draft, and KW reviewed the references in Table 1 and drew the Figueres as well as the revising the draft. DCW performed the investigation and reviewed the manuscript. KKY wrote the original and revised manuscript. All the authors have read and approved the manuscript.
Declaration of Competing Interest
The authors declare no potential conflicts of interest with respect to the research, authorship, or publication of this article.
Data Availability
No data were used for the research described in the article.
Acknowledgments
This article was supported by grants from the Ministry of Science and Technology (MOST111-2314-B-037-009), the National Health Research Institutes (NHRI-EX109-10720SI), Kaohsiung Medical University Hospital (KMUH111-1R77; KMUH-DK(A)112002), the National Sun-Yat-sen University-Kaohsiung Medical University Joint Research Project (NYCUKMU-113-I006) and Kaohsiung Medical University (KMU-TC113A02).
Abbreviations
| AhR | aryl hydrocarbon receptor |
| AP-1 | Activation protein-1 |
| ARE | antioxidant response element |
| BAX | Bcl-2-associated X protein |
| BCL2 | B-cell lymphoma 2 |
| BCRP | breast cancer-resistant protein |
| BRCA1 | breast cancer susceptibility gene 1 |
| CBP | CREB binding protein |
| CDKN1A | cyclin-dependent kinase inhibitor 1 |
| CE | cinnamaldehyde |
| COX2 | cyclooxygenase 2 |
| CRC | colorectal cancer |
| CREB | cyclic AMP response element binding protein |
| CSCs | cancer stem cells |
| DPx2 | glutathione peroxidase 2 |
| DSS | dextran sodium sulfate |
| EMT | epithelial-to-mesenchymal transition |
| ERBB2 | Erb-B2 Receptor Tyrosine Kinase 2 |
| GC | gastric cancer |
| GLUTs | glucose transporters |
| G6PD | glucose-6-phosphate dehydrogenase |
| GPR78 | glucose-regulated protein 78 |
| GSH | glutathione-SH |
| GSK3 | glycogen synthetase kinase 3 |
| GSSG | glutathione sulfide |
| H2O2 | hydrogen peroxide |
| HIF-1β | hypoxia inducible factor beta |
| HNSCC | head and neck squamous cell carcinoma |
| HO-1 | heme oxygenase 1 |
| 3-IAld | indole-3-aldehyde |
| ICI | immune checkpoint inhibitor |
| IKK-α:-β | inhibitor of nuclear factor κB kinase subunit-α,-β |
| IPA | Ingenuity pathway analysis |
| iNOS | nitric oxide synthase |
| JDP2 | Jun dimerization protein 2 |
| JNKs | c-Jun N-terminal kinases |
| Keap1 | Kelch like ECH associated protein 1 |
| LPS | lipopolysaccharide |
| MARCO | macrophage receptor with collagenous structure |
| MDR1 | multidrug resistance protein 1 |
| MRP1-5 | multidrug resistance-associated protein 1-5 |
| MSCs | mesenchymal stem cells |
| NAC | N-acetylcysteine |
| NFkB | nuclear factor kappa B |
| NO | nitrogen oxide |
| NQO1 | NAD(P)H:quinone dehydrogenase 1 |
| NRF2 | nuclear factor erythroid 2-related factor 2 |
| NSCLC | non-small cell lung cance |
| OXPHOS | oxidative phosphorylation |
| PEA | perillaldehyde |
| PGD | phosphoglucomutase dehydrogenase |
| PI3K | phosphoinositide 3-kinase |
| p-PERK | phosphorylated protein kinase RNA-like ER kinase |
| PSMA2 | proteasome 20S alpha 2 |
| PSMC4 | 26S proteasome regulatory subunit 4 |
| ROS | reactive oxygen species |
| SFN | sulforaphane |
| SOD | superoxide dismutase |
| SQSRM1 | sequestosome-1 |
| TALDO1 | trans-aldolase |
| TBK1 | TANK-binding kinase 2 |
| TKT | transketolase |
| TNF | tumor necrosis factor |
| β-TrCP | β-transduction repeat-containing E3 ubiquitin-protein ligase |
| Trx | thioredoxin |
| TrxR1 | thioredoxin reductase 1 |
| VEGF | vascular endothelial growth factor |
| x-CT = SLC7a11 cystine/glutamate transporter | |
References
- C. Gorrini, I.S.; Harris, T.W.; Mak, Modulation of oxidative stress as an anticancer strategy, Nat. Rev. Drug Discov. 12 (12) (2013) 931-947. [CrossRef]
- F.; Xing, Q.; Hu, Y.; Qin, J.; Xu, B. Zhang, et al., The Relationship of Redox With Hallmarks of Cancer: The Importance of Homeostasis and Context, Front. Oncol. 12 (2022) 862743. [CrossRef]
- B.; Perillo, M. Di Donato, A.; Pezone, E. Di Zazzo, P. Giovannelli, et al., ROS in cancer therapy: the bright side of the moon, Exp. Mol. Med. 52 (2) (2020) 192-203. [CrossRef]
- D.L.; Kirkpatrick, G.; Powis, Clinically Evaluated Cancer Drugs Inhibiting Redox Signaling, Antioxid. Redox Signal. 26 (6) (2017) 262-273. [CrossRef]
- X.; Wu, Z.; Zhou, K.; Li, S.; Liu, Nanomaterials-Induced Redox Imbalance: Challenged and Opportunities for Nanomaterials in Cancer Therapy, Adv. Sci. (Weinh) 11 (16) (2024) e2308632. [CrossRef]
- D.C.; Wu, C.C.; Ku, J.B.; Pan, K.; Wuputra, Y.H. Yang, et al., Heterogeneity of Phase II Enzyme Ligands on Controlling the Progression of Human Gastric Cancer Organoids as Stem Cell Therapy Model, Int. J. Mol. Sci. 24 (21) (2023) 15911. [CrossRef]
- P.; Poprac, K.; Jomova, M.; Simunkova, V.; Kollar, C.J. Rhodes, et al., Targeting Free Radicals in Oxidative Stress-Related Human Diseases, Trends Pharmacol. Sci. 38 (7) (2017) 592-607. [CrossRef]
- S.; Liu, M.; Yue, Y.; Lu, Y.; Wang, S. Luo, et al., Advancing the frontiers of colorectal cancer treatment: harnessing ferroptosis regulation, Apoptosis 29 (1-2) (2024) 86-102. [CrossRef]
- C.J.; Schmidlin, A.; Shakya, M.; Dodson, E.; Chapman, D.D.; Zhang, The intricacies of NRF2 regulation in cancer, Semin. Cancer Biol. 76 (2021) 110-119. [CrossRef]
- M.R.I.; Young, Y.; Xiong, Influence of vitamin D on cancer risk and treatment: Why the variability?, Trends Cancer Res. 13 (2018) 43-53.
- R.; Bakalova, Z.; Zhelev, T.; Miller, I.; Aoki, T.; Higashi, New potential biomarker for stratification of patients for pharmacological vitamin C in adjuvant settings of cancer therapy, Redox Biol. 28 (2020) 101357. [CrossRef]
- F.; Augsburger, A.; Filippova, D.; Rasti, T.; Seredenina, M. Lam, et al., Pharmacological characterization of the seven human NOX isoforms and their inhibitors, Redox Biol. 26 (2019) 101272. [CrossRef]
- S.; Liang, H.Y.; Ma, Z.; Zhong, D.; Dhar, X. Liu, et al., NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice, Gastroenterology 156 (4) (2019) 1156-1172.e6. [CrossRef]
- ; Spasojevic, Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue Against Oxidative Injury, Antioxid. Redox Signal. 29 (16) (2018) 1691-1724. [CrossRef]
- Z.Y.; Wu, H.J.; Kim, J.W.; Lee, I.Y.; Chung, J.S. Kim, et al., Breast Cancer Recurrence in the Nipple-Areola Complex After Nipple-Sparing Mastectomy With Immediate Breast Reconstruction for Invasive Breast Cancer, JAMA Surg. 154 (11) (2019) 1030-1037. [CrossRef]
- J.H.; Wu, G.; Batist, Glutathione and glutathione analogues; therapeutic potentials, Biochim. Biophys. Acta. 1830 (5) (2013) 3350-3353. [CrossRef]
- X.; Luo, C.; Cheng, Z.; Tan, N.; Li, M. Tang, et al., Emerging roles of lipid metabolism in cancer metastasis, Mol. Cancer 16 (1) (2017) 76. [CrossRef]
- H.S. Selistre-de-Araujo, B.C.; Pachane, W.F.; Altei, Tumor heterogeneity and the dilemma of antioxidant therapies in cancer, Ann. Transl. Med. 10 (19) (2022) 1074. [CrossRef]
- S.; Wu, H.; Lu, Y.; Bai, Nrf2 in cancers: A double-edged sword, Cancer Med. 8 (5) (2019) 2252-2267. [CrossRef]
- H.; Jiang, J.; Zuo, B.; Li, R.; Chen, K. Luo, et al., Drug-induced oxidative stress in cancer treatments: Angel or devil?, Redox Biol. 63 (2023) 102754. [CrossRef]
- C.; Gorrini, P.S.; Baniasadi, I.S.; Harris, J.; Silvester, S. Inoue, et al., BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival, J. Exp. Med. 210 (8) (2013) 1529-1544. [CrossRef]
- W.; Chen, Z.; Sun, X.J.; Wang, T.; Jiang, Z. Huang, et al., Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response, Mol. Cell 34 (6) (2009) 663-673. [CrossRef]
- S.K.; Niture, R.; Khatri, A.K.; Jaiswal, Regulation of Nrf2-an update, Free Radic. Biol. Med. 66 (2014) 36-44. [CrossRef]
- D.J. Long, 2nd, R.L.; Waikel, X.J.; Wang, L.; Perlaky, D.R. Roop, et al., NAD(P)H:quinone oxidoreductase 1 deficiency increases susceptibility to benzo(a)pyrene-induced mouse skin carcinogenesis, Cancer Res. 60 (21) (2000) 5913-5915.
- M. Ramos-Gomez, M.K.; Kwak, P.M.; Dolan, K.; Itoh, M. Yamamoto, et al., Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice, Proc. Natl. Acad. Sci. USA 98 (6) (2001) 3410-3415. [CrossRef]
- Y.; Kitamura, T.; Umemura, K.; Kanki, Y.; Kodama, S. Kitamoto, et al., Increased susceptibility to hepatocarcinogenicity of Nrf2-deficient mice exposed to 2-amino-3-methylimidazo[4,5-f]quinoline, Cancer Sci. 98 (1) (2007) 19-24. [CrossRef]
- K.; Iida, K.; Itoh, J.M.; Maher, Y.; Kumagai, R. Oyasu, et al., Nrf2 and p53 cooperatively protect against BBN-induced urinary bladder carcinogenesis, Carcinogenesis 28 (11) (2007) 2398-2403. [CrossRef]
- N.; Wakabayashi, A.T. Dinkova-Kostova, W.D.; Holtzclaw, M.I.; Kang, A. Kobayashi, et al., Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers, Proc. Natl. Acad. Sci. USA 101 (7) (2004) 2040-2045. [CrossRef]
- L. Gamet-Payrastre, P.; Li, S.; Lumeau, G.; Cassar, M.A. Dupont, et al., Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells, Cancer Res. 60 (5) (2000) 1426-1433.
- E.; Heiss, C.; Herhaus, K.; Klimo, H.; Bartsch, C.; Gerhauser, Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms, J. Biol. Chem. 276 (34) (2001) 32008-32015. [CrossRef]
- S.E.; Dickinson, T.F.; Melton, E.R.; Olson, J.; Zhang, K. Saboda, et al., Inhibition of activator protein-1 by sulforaphane involves interaction with cysteine in the cFos DNA-binding domain: implications for chemoprevention of UVB-induced skin cancer, Cancer Res. 69 (17) (2009) 7103-7110. [CrossRef]
- K.T.; Liby, D.B.; Royce, R.; Risingsong, C.R.; Williams, A. Maitra, et al., Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer, Cancer Prev. Res. (Phila) 3 (11) (2010) 1427-1434. [CrossRef]
- E.H.; Kim, C.; Deng, M.B.; Sporn, D.B.; Royce, R. Risingsong, et al., CDDO-methyl ester delays breast cancer development in BRCA1-mutated mice, Cancer Prev. Res. (Phila) 5 (1) (2012) 89-97. [CrossRef]
- S.S.; Boyanapalli, X. Paredes-Gonzalez, F.; Fuentes, C.; Zhang, Y. Guo, et al., Nrf2 knockout attenuates the anti-inflammatory effects of phenethyl isothiocyanate and curcumin, Chem. Res. Toxicol. 27 (12) (2014) 2036-2043. [CrossRef]
- E.H.; Kobayashi, T.; Suzuki, R.; Funayama, T.; Nagashima, M. Hayashi, et al., Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription, Nat. Commun. 7 (2016) 11624. [CrossRef]
- X.; Kong, R.; Thimmulappa, F.; Craciun, C.; Harvey, A. Singh, et al., Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis, Am. J. Respir. Crit. Care Med. 184 (8) (2011) 928-938. [CrossRef]
- X.L.; Chen, G.; Dodd, S.; Thomas, X.; Zhang, M.A. Wasserman, et al., Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression, Am. J. Physiol. Heart Circ. Physiol. 290 (5) (2006) H1862-1870. [CrossRef]
- J.E.; Kim, D.J.; You, C.; Lee, C.; Ahn, J.Y. Seong, et al., Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation, Cell Signal. 22 (11) (2010) 1645-1654. [CrossRef]
- W.; Gao, L.; Guo, Y.; Yang, Y.; Wang, S. Xia, et al., Dissecting the Crosstalk Between Nrf2 and NF-kappaB Response Pathways in Drug-Induced Toxicity, Front. Cell Dev. Biol. 9 (2021) 809952. [CrossRef]
- Y.C.; Kim, H.; Masutani, Y.; Yamaguchi, K.; Itoh, M. Yamamoto, et al., Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors, J. Biol. Chem. 276 (21) (2001) 18399-18406. [CrossRef]
- N.; Hanada, T.; Takahata, Q.; Zhou, X.; Ye, R. Sun, et al., Methylation of the KEAP1 gene promoter region in human colorectal cancer, BMC Cancer 12 (2012) 66. [CrossRef]
- G.M. DeNicola, F.A.; Karreth, T.J.; Humpton, A.; Gopinathan, C. Wei, et al., Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis, Nature 475 (7354) (2011) 106-109. [CrossRef]
- A.I.; Rojo, P.; Rada, M.; Mendiola, A. Ortega-Molina, K. Wojdyla, et al., The PTEN/NRF2 axis promotes human carcinogenesis, Antioxid. Redox Signal. 21 (18) (2014) 2498-2514. [CrossRef]
- Y.; Mitsuishi, K.; Taguchi, Y.; Kawatani, T.; Shibata, T. Nukiwa, et al., Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming, Cancer Cell 22 (1) (2012) 66-79. [CrossRef]
- N.R.; Kitteringham, A.; Abdullah, J.; Walsh, L.; Randle, R.E. Jenkins, et al., Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver, J. Proteomics 73 (8) (2010) 1612-1631. [CrossRef]
- D.; Malhotra, E. Portales-Casamar, A.; Singh, S.; Srivastava, D. Arenillas, et al., Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis, Nucleic Acids Res. 38 (17) (2010) 5718-5734. [CrossRef]
- N.M.; Reddy, S.R.; Kleeberger, J.H.; Bream, P.G.; Fallon, T.W. Kensler, et al., Genetic disruption of the Nrf2 compromises cell-cycle progression by impairing GSH-induced redox signaling, Oncogene 27 (44) (2008) 5821-5832. [CrossRef]
- C.W.; Chang, Y.S.; Chen, Y.G.; Tsay, C.L.; Han, Y.J. Chen, et al., ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells, Cell Death Dis. 9 (2) (2018) 194. [CrossRef]
- C.W.; Chang, Y.S.; Chen, S.H.; Chou, C.L.; Han, Y.J. Chen, et al., Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity, Cancer Res. 74 (21) (2014) 6291-6305. [CrossRef]
- H.; Kumar, R.M.; Kumar, D.; Bhattacharjee, P.; Somanna, V.; Jain, Role of Nrf2 Signaling Cascade in Breast Cancer: Strategies and Treatment, Front. Pharmacol. 13 (2022) 720076. [CrossRef]
- I.G.; Ryoo, S.H.; Lee, M.K.; Kwak, Redox Modulating NRF2: A Potential Mediator of Cancer Stem Cell Resistance, Oxid. Med. Cell Longev. 2016 (2016) 2428153. [CrossRef]
- A.; Singh, S. Boldin-Adamsky, R.K.; Thimmulappa, S.K.; Rath, H. Ashush, et al., RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy, Cancer Res. 68 (19) (2008) 7975-7984. [CrossRef]
- T.; Yasuda, T.; Ishimoto, H.; Baba, Conflicting metabolic alterations in cancer stem cells and regulation by the stromal niche, Regen. Ther. 17 (2021) 8-12. [CrossRef]
- L.; Gao, Y.; Morine, S.; Yamada, Y.; Saito, T. Ikemoto, et al., Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells, PLoS One 16 (9) (2021) e0256755. [CrossRef]
- H.; Kahroba, M.; Shirmohamadi, M.S.; Hejazi, N.; Samadi, The Role of Nrf2 signaling in cancer stem cells: From stemness and self-renewal to tumorigenesis and chemoresistance, Life Sci. 239 (2019) 116986. [CrossRef]
- S.M.; Ridge, F.J.; Sullivan, S.A.; Glynn, Mesenchymal stem cells: key players in cancer progression, Mol. Cancer 16 (1) (2017) 31. [CrossRef]
- M. Mohammadzadeh-Vardin, M. Habibi Roudkenar, A. Jahanian-Najafabadi, Adenovirus-Mediated Over-Expression of Nrf2 Within Mesenchymal Stem Cells (MSCs) Protected Rats Against Acute Kidney Injury, Adv. Pharm. Bull 5 (2) (2015) 201-208. [CrossRef]
- Z.; Yuan, J.; Zhang, Y.; Huang, Y.; Zhang, W. Liu, et al., NRF2 overexpression in mesenchymal stem cells induces stem-cell marker expression and enhances osteoblastic differentiation, Biochem. Biophys. Res. Commun. 491 (1) (2017) 228-235. [CrossRef]
- H.; Kitamura, H.; Motohashi, NRF2 addiction in cancer cells, Cancer Sci. 109 (4) (2018) 900-911. [CrossRef]
- K.; Okazaki, T.; Papagiannakopoulos, H.; Motohashi, Metabolic features of cancer cells in NRF2 addiction status, Biophys. Rev. 12 (2) (2020) 435-441. [CrossRef]
- S.; Zhou, W.; Ye, M.; Zhang, J.; Liang, The effects of nrf2 on tumor angiogenesis: a review of the possible mechanisms of action, Crit. Rev. Eukaryot. Gene Expr. 22 (2) (2012) 149-160. [CrossRef]
- B.; Bussolati, J.C.; Mason, Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis, Antioxid. Redox Signal. 8 (7-8) (2006) 1153-1163. [CrossRef]
- S.A.; Rushworth, D.J. MacEwan, HO-1 underlies resistance of AML cells to TNF-induced apoptosis, Blood 111 (7) (2008) 3793-3801. [CrossRef]
- S.K.; Niture, A.K.; Jaiswal, Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis, J. Biol. Chem. 287 (13) (2012) 9873-9886. [CrossRef]
- R.; Elsby, N.R.; Kitteringham, C.E.; Goldring, C.A.; Lovatt, M. Chamberlain, et al., Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi, J. Biol. Chem. 278 (25) (2003) 22243-22249. [CrossRef]
- A.; Jain, T.; Lamark, E.; Sjottem, K.B.; Larsen, J.A. Awuh, et al., p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription, J. Biol. Chem 285 (29) (2010) 22576-22591. [CrossRef]
- M.; Komatsu, H.; Kurokawa, S.; Waguri, K.; Taguchi, A. Kobayashi, et al., The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1, Nat. Cell Biol. 12 (3) (2010) 213-223. [CrossRef]
- C.G.; Towers, B.E.; Fitzwalter, D.; Regan, A.; Goodspeed, M.J. Morgan, et al., Cancer Cells Upregulate NRF2 Signaling to Adapt to Autophagy Inhibition, Dev. Cell 50 (6) (2019) 690-703 e696. [CrossRef]
- K.; Taguchi, I.; Hirano, T.; Itoh, M.; Tanaka, A. Miyajima, et al., Nrf2 enhances cholangiocyte expansion in Pten-deficient livers, Mol. Cell Biol. 34 (5) (2014) 900-913. [CrossRef]
- K.; Shirasaki, K.; Taguchi, M.; Unno, H.; Motohashi, M.; Yamamoto, NF-E2-related factor 2 promotes compensatory liver hypertrophy after portal vein branch ligation in mice, Hepatology 59 (6) (2014) 2371-2382. [CrossRef]
- T.; Suzuki, S.; Seki, K.; Hiramoto, E.; Naganuma, E.H. Kobayashi, et al., Hyperactivation of Nrf2 in early tubular development induces nephrogenic diabetes insipidus, Nat. Commun. 8 (2017) 14577. [CrossRef]
- S.; Murakami, T.; Suzuki, H.; Harigae, P.H.; Romeo, M. Yamamoto, et al., NRF2 Activation Impairs Quiescence and Bone Marrow Reconstitution Capacity of Hematopoietic Stem Cells, Mol. Cell Biol. 37 (19) (2017) e00086-17. [CrossRef]
- K.; Taguchi, J.M.; Maher, T.; Suzuki, Y.; Kawatani, H. Motohashi, et al., Genetic analysis of cytoprotective functions supported by graded expression of Keap1, Mol. Cell Biol. 30 (12) (2010) 3016-3026. [CrossRef]
- Y.; Jeong, N.T.; Hoang, A.; Lovejoy, H.; Stehr, A.M. Newman, et al., Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance, Cancer Discov. 7 (1) (2017) 86-101. [CrossRef]
- X.; Bai, Y.; Chen, X.; Hou, M.; Huang, J.; Jin, Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters, Drug Metab. Rev. 48 (4) (2016) 541-567. [CrossRef]
- I.G.; Ryoo, G.; Kim, B.H.; Choi, S.H.; Lee, M.K.; Kwak, Involvement of NRF2 Signaling in Doxorubicin Resistance of Cancer Stem Cell-Enriched Colonospheres, Biomol. Ther. (Seoul) 24 (5) (2016) 482-488. [CrossRef]
- H.; Sasaki, M.; Shitara, K.; Yokota, Y.; Hikosaka, S. Moriyama, et al., MRP3 gene expression correlates with NRF2 mutations in lung squamous cell carcinomas, Mol. Med. Rep. 6 (4) (2012) 705-708. [CrossRef]
- A.M.; Gao, Z.P.; Ke, J.N.; Wang, J.Y.; Yang, S.Y. Chen, et al., Apigenin sensitizes doxorubicin-resistant hepatocellular carcinoma BEL-7402/ADM cells to doxorubicin via inhibiting PI3K/Akt/Nrf2 pathway, Carcinogenesis 34 (8) (2013) 1806-1814. [CrossRef]
- H.M.; Ahmed, Ethnomedicinal, Phytochemical and Pharmacological Investigations of Perilla frutescens (L.) Britt, Molecules 24 (1) (2018). [CrossRef]
- W.W.; Ji, S.Y.; Wang, Z.Q.; Ma, R.P.; Li, S.S. Li, et al., Effects of perillaldehyde on alternations in serum cytokines and depressive-like behavior in mice after lipopolysaccharide administration, Pharmacol. Biochem. Behav. 116 (2014) 1-8. [CrossRef]
- Y.; Song, R.; Sun, Z.; Ji, X.; Li, Q. Fu, et al., Perilla aldehyde attenuates CUMS-induced depressive-like behaviors via regulating TXNIP/TRX/NLRP3 pathway in rats, Life Sci. 206 (2018) 117-124. [CrossRef]
- T.; Uemura, T.; Yashiro, R.; Oda, N.; Shioya, T. Nakajima, et al., Intestinal Anti-Inflammatory Activity of Perillaldehyde, J. Agric. Food Chem. 66 (13) (2018) 3443-3448. [CrossRef]
- Y.; Fuyuno, H.; Uchi, M.; Yasumatsu, S. Morino-Koga, Y. Tanaka, et al., Perillaldehyde Inhibits AHR Signaling and Activates NRF2 Antioxidant Pathway in Human Keratinocytes, Oxid. Med. Cell Longev. 2018 (2018) 9524657. [CrossRef]
- C.M.; Cabello, W.B. Bair, 3rd, S.D.; Lamore, S.; Ley, A.S. Bause, et al., The cinnamon-derived Michael acceptor cinnamic aldehyde impairs melanoma cell proliferation, invasiveness, and tumor growth, Free Radic. Biol. Med. 46 (2) (2009) 220-231. [CrossRef]
- J.; Zhao, X.; Zhang, L.; Dong, Y.; Wen, X. Zheng, et al., Cinnamaldehyde inhibits inflammation and brain damage in a mouse model of permanent cerebral ischaemia, Br. J. Pharmacol. 172 (20) (2015) 5009-5023. [CrossRef]
- T.W.; Kim, Cinnamaldehyde induces autophagy-mediated cell death through ER stress and epigenetic modification in gastric cancer cells, Acta Pharmacol. Sin. 43 (3) (2022) 712-723. [CrossRef]
- H.; Uchi, M.; Yasumatsu, S. Morino-Koga, C.; Mitoma, M.; Furue, Inhibition of aryl hydrocarbon receptor signaling and induction of NRF2-mediated antioxidant activity by cinnamaldehyde in human keratinocytes, J Dermatol. Sci. 85 (1) (2017) 36-43. [CrossRef]
- Y.; Wang, H.; Wu, N.; Dong, X.; Su, M. Duan, et al., Sulforaphane induces S-phase arrest and apoptosis via p53-dependent manner in gastric cancer cells, Sci. Rep. 11 (1) (2021) 2504. [CrossRef]
- Y.; Zhang, Q.; Lu, N.; Li, M.; Xu, T. Miyamoto, et al., Sulforaphane suppresses metastasis of triple-negative breast cancer cells by targeting the RAF/MEK/ERK pathway, NPJ Breast Cancer 8 (1) (2022) 40. [CrossRef]
- Ostolga-Chavarria, C. Sanchez-Garibay, P. Rojas-Morales, S. Galvan-Arzate, et al., Sulforaphane protects from myocardial ischemia-reperfusion damage through the balanced activation of Nrf2/AhR, Free Radic. Biol. Med. 143 (2019) 331-340. [CrossRef]
- S.; Cao, S.; Hu, P.; Jiang, Z.; Zhang, L. Li, et al., Effects of sulforaphane on breast cancer based on metabolome and microbiome, Food Sci. Nutr. 11 (5) (2023) 2277-2287. [CrossRef]
- R.L.; Yeager, S.A.; Reisman, L.M.; Aleksunes, C.D.; Klaassen, Introducing the "TCDD-inducible AhR-Nrf2 gene battery", Toxicol. Sci. 111 (2) (2009) 238-246. [CrossRef]
- K.; Wuputra, M.H.; Tsai, K.; Kato, C.C.; Ku, J.B. Pan, et al., Jdp2 is a spatiotemporal transcriptional activator of the AhR via the Nrf2 gene battery, Inflamm. Regen. 43 (1) (2023) 42. [CrossRef]
- K. McGovern, A.C.; Castro, J.; Cavanaugh, S.; Coma, M. Walsh, et al., Discovery and Characterization of a Novel Aryl Hydrocarbon Receptor Inhibitor, IK-175, and Its Inhibitory Activity on Tumor Immune Suppression, Mol. Cancer Ther. 21 (8) (2022) 1261-1272. [CrossRef]
- F. D'Onofrio, G.; Renga, M.; Puccetti, M.; Pariano, M.M. Bellet, et al., Indole-3-Carboxaldehyde Restores Gut Mucosal Integrity and Protects from Liver Fibrosis in Murine Sclerosing Cholangitis, Cells 10 (7) (2021). [CrossRef]
- M.; Puccetti, G.; Paolicelli, V.; Oikonomou, A. De Luca, G. Renga, et al., Towards Targeting the Aryl Hydrocarbon Receptor in Cystic Fibrosis, Mediators Inflamm. 2018 (2018) 1601486. [CrossRef]
- G.; Renga, E.; Nunzi, M.; Pariano, M.; Puccetti, M.M. Bellet, et al., Optimizing therapeutic outcomes of immune checkpoint blockade by a microbial tryptophan metabolite, J. Immunother. Cancer 10 (3) (2022). [CrossRef]
- C.; Kober, J.; Roewe, N.; Schmees, L.; Roese, U. Roehn, et al., Targeting the aryl hydrocarbon receptor (AhR) with BAY 2416964: a selective small molecule inhibitor for cancer immunotherapy, J. Immunother. Cancer 11 (11) (2023). [CrossRef]
- S.V.; Singh, S.K.; Srivastava, S.; Choi, K.L.; Lew, J. Antosiewicz, et al., Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species, J. Biol. Chem. 280 (20) (2005) 19911-19924. [CrossRef]
- L.L.; Atwell, Z.; Zhang, M.; Mori, P.; Farris, J.T. Vetto, et al., Sulforaphane Bioavailability and Chemopreventive Activity in Women Scheduled for Breast Biopsy, Cancer Prev. Res. (Phila) 8 (12) (2015) 1184-1191. [CrossRef]
- M.; Ge, L.; Zhang, L.; Cao, C.; Xie, X. Li, et al., Sulforaphane inhibits gastric cancer stem cells via suppressing sonic hedgehog pathway, Int. J. Food Sci. Nutr. 70 (5) (2019) 570-578. [CrossRef]
- S.; Li, P.N.; Khoi, H.; Yin, D.K.; Sah, N.H. Kim, et al., Sulforaphane Suppresses the Nicotine-Induced Expression of the Matrix Metalloproteinase-9 via Inhibiting ROS-Mediated AP-1 and NF-kappaB Signaling in Human Gastric Cancer Cells, Int. J. Mol. Sci. 23 (9) (2022) 5172. [CrossRef]
- M.; Honma, M.; Yamada, M.; Yasui, K.; Horibata, K.I. Sugiyama, et al., In vivo and in vitro mutagenicity of perillaldehyde and cinnamaldehyde, Genes Environ. 43 (1) (2021) 30. [CrossRef]
- J.; Puschhof, C. Pleguezuelos-Manzano, A. Martinez-Silgado, N.; Akkerman, A. Saftien, et al., Intestinal organoid cocultures with microbes, Nat. Protoc. 16 (10) (2021) 4633-4649. [CrossRef]
- K.; Murai, S.; Dentro, S.H.; Ong, R.; Sood, D. Fernandez-Antoran, et al., p53 mutation in normal esophagus promotes multiple stages of carcinogenesis but is constrained by clonal competition, Nat. Commun. 13 (1) (2022) 6206. [CrossRef]
- K.; Sabapathy, D.P.; Lane, Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others, Nat. Rev. Clin. Oncol. 15 (1) (2018) 13-30. [CrossRef]
- K.; Liu, W.; Chen, S.; Lei, L.; Xiong, H. Zhao, et al., Wild-type and mutant p53 differentially modulate miR-124/iASPP feedback following pohotodynamic therapy in human colon cancer cell line, Cell Death Dis. 8 (10) (2017) e3096. [CrossRef]
- K.; Lisek, E.; Campaner, Y.; Ciani, D.; Walerych, G. Del Sal, Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells, Oncotarget 9 (29) (2018) 20508-20523. [CrossRef]
- D.; Walerych, K.; Lisek, R.; Sommaggio, S.; Piazza, Y. Ciani, et al., Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer, Nat. Cell Biol. 18 (8) (2016) 897-909. [CrossRef]
- M.C.; Tung, P.L.; Lin, Y.C.; Wang, T.Y.; He, M.C. Lee, et al., Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2, Oncotarget 6 (39) (2015) 41692-41705. [CrossRef]
- S.A.; Rushworth, L.; Zaitseva, M.Y.; Murray, N.M.; Shah, K.M. Bowles, et al., The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance, Blood 120 (26) (2012) 5188-5198. [CrossRef]
- G.; Asher, J.; Lotem, R.; Kama, L.; Sachs, Y.; Shaul, NQO1 stabilizes p53 through a distinct pathway, Proc. Natl. Acad. Sci. USA 99 (5) (2002) 3099-3104. [CrossRef]
- G.; Asher, J.; Lotem, P.; Tsvetkov, V.; Reiss, L. Sachs, et al., P53 hot-spot mutants are resistant to ubiquitin-independent degradation by increased binding to NAD(P)H:quinone oxidoreductase 1, Proc. Natl. Acad. Sci. USA 100 (25) (2003) 15065-15070. [CrossRef]
- E.; Kalo, I. Kogan-Sakin, H.; Solomon, E. Bar-Nathan, M. Shay, et al., Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species, J. Cell Sci. 125 (Pt 22) (2012) 5578-5586. [CrossRef]
- A.S.; Gomes, H.; Ramos, J.; Soares, L. Saraiva, p53 and glucose metabolism: an orchestra to be directed in cancer therapy, Pharmacol. Res. 131 (2018) 75-86. [CrossRef]
- V.J.N.; Bykov, S.E.; Eriksson, J.; Bianchi, K.G.; Wiman, Targeting mutant p53 for efficient cancer therapy, Nat. Rev. Cancer 18 (2) (2018) 89-102. [CrossRef]
- X.; Peng, M.Q.; Zhang, F.; Conserva, G.; Hosny, G. Selivanova, et al., APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase, Cell Death Dis. 4 (10) (2013) e881. [CrossRef]
- L.; Haffo, J.; Lu, V.J.N.; Bykov, S.S.; Martin, X. Ren, et al., Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246, Sci. Rep. 8 (1) (2018) 12671. [CrossRef]
- B.; Tessoulin, G.; Descamps, P.; Moreau, S.; Maiga, L. Lode, et al., PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance, Blood 124 (10) (2014) 1626-1636. [CrossRef]
- N.; Mohell, J.; Alfredsson, A.; Fransson, M.; Uustalu, S. Bystrom, et al., APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells, Cell Death Dis. 6 (6) (2015) e1794. [CrossRef]
- Z.; Wang, H.; Hu, L.; Heitink, K.; Rogers, Y. You, et al., The anti-cancer agent APR-246 can activate several programmed cell death processes to kill malignant cells, Cell Death Differ. 30 (4) (2023) 1033-1046. [CrossRef]
- R.; Faraonio, P.; Vergara, D. Di Marzo, M.G.; Pierantoni, M. Napolitano, et al., p53 suppresses the Nrf2-dependent transcription of antioxidant response genes, J. Biol. Chem. 281 (52) (2006) 39776-39784. [CrossRef]
- Y.; Liang, J.; Liu, Z.; Feng, The regulation of cellular metabolism by tumor suppressor p53, Cell Biosci. 3 (1) (2013) 9. [CrossRef]
- T.; Li, N.; Kon, L.; Jiang, M.; Tan, T. Ludwig, et al., Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence, Cell 149 (6) (2012) 1269-1283. [CrossRef]
- H.; Yang, S.; Xiang, A.; Kazi, S.M.; Sebti, The GTPase KRAS suppresses the p53 tumor suppressor by activating the NRF2-regulated antioxidant defense system in cancer cells, J. Biol. Chem. 295 (10) (2020) 3055-3063. [CrossRef]
- C.P.; Kung, J.D.; Weber, It's Getting Complicated-A Fresh Look at p53-MDM2-ARF Triangle in Tumorigenesis and Cancer Therapy, Front. Cell Dev. Biol. 10 (2022) 818744. [CrossRef]
- Y.; Xu, C.; Jin, Z.; Liu, J.; Pan, H. Li, et al., Cloning and characterization of the mouse JDP2 gene promoter reveal negative regulation by p53, Biochem. Biophys. Res. Commun. 450 (4) (2014) 1531-1536. [CrossRef]
- S.; Tanigawa, C.H.; Lee, C.S.; Lin, C.C.; Ku, H. Hasegawa, et al., Jun dimerization protein 2 is a critical component of the Nrf2/MafK complex regulating the response to ROS homeostasis, Cell Death Dis. 4 (11) (2013) e921. [CrossRef]
- C.; Jin, K.; Kato, T.; Chimura, T.; Yamasaki, K. Nakade, et al., Regulation of histone acetylation and nucleosome assembly by transcription factor JDP2, Nat. Struct. Mol. Biol. 13 (4) (2006) 331-338. [CrossRef]
- J.; Pan, K. Nakade, Y.C. Huang, Z.W. Zhu, S. Masuzaki, et al., Suppression of cell-cycle progression by Jun dimerization protein-2 (JDP2) involves downregulation of cyclin-A2, Oncogene 29 (47) (2010) 6245-6256. [CrossRef]
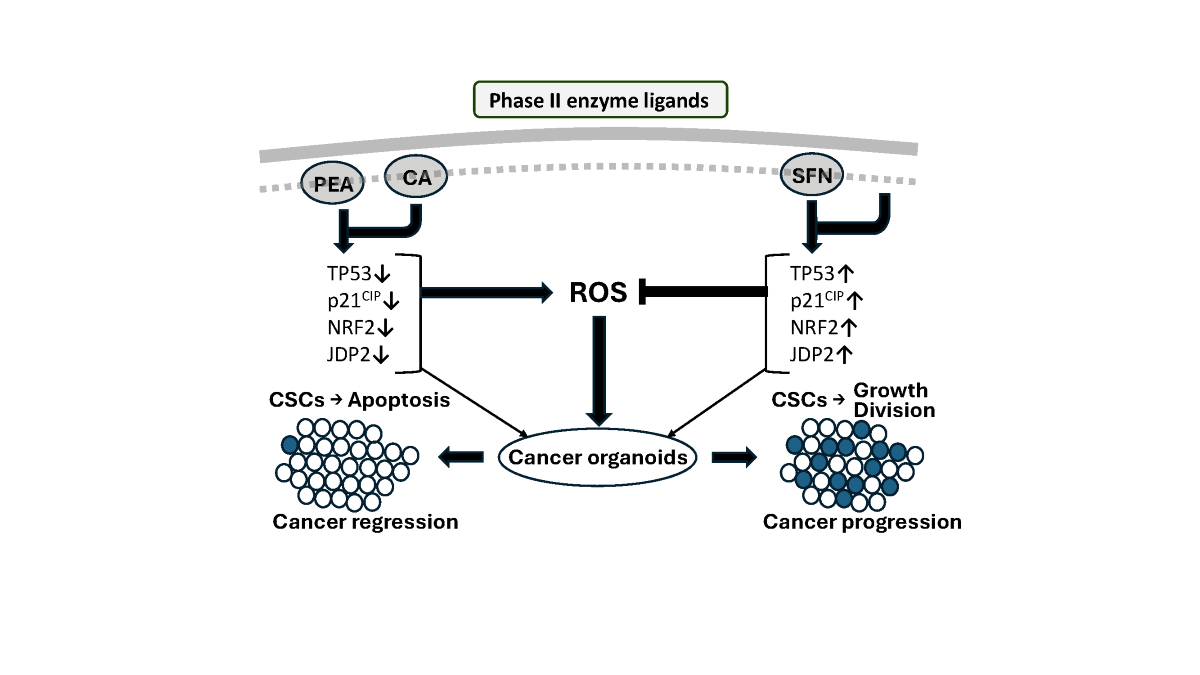
Figure 1.
Schematic model of ROS balance induced by phase I enzyme ligands and phase II enzyme ligands. Phase I enzyme ligands or phase II enzyme metabolites are able to induce the AhR activation to generate ROS production. By contrast, the phase II enzyme ligands induced Nrf2 activation to decrease the level of AhR and ROS production, which resulted in the phase II enzyme metabolites. Thus, ROS balance is controlled by AhR and Nrf2. Cyp1A1 (p450 family); cytochrome p450 family Cyp1A1, NOX; nitrogen oxides, COX; cyclooxygenase, AKR; Aldo-keto reductase, NQO1; NAD(P)H dehydrogenase (quinone) family, GSTA1/2; Glutathione S-Transferase Alpha 1 and 2, UGT1A; UDP Glucuronosyltransferase Family 1 Member A Complex Locus.
Figure 1.
Schematic model of ROS balance induced by phase I enzyme ligands and phase II enzyme ligands. Phase I enzyme ligands or phase II enzyme metabolites are able to induce the AhR activation to generate ROS production. By contrast, the phase II enzyme ligands induced Nrf2 activation to decrease the level of AhR and ROS production, which resulted in the phase II enzyme metabolites. Thus, ROS balance is controlled by AhR and Nrf2. Cyp1A1 (p450 family); cytochrome p450 family Cyp1A1, NOX; nitrogen oxides, COX; cyclooxygenase, AKR; Aldo-keto reductase, NQO1; NAD(P)H dehydrogenase (quinone) family, GSTA1/2; Glutathione S-Transferase Alpha 1 and 2, UGT1A; UDP Glucuronosyltransferase Family 1 Member A Complex Locus.
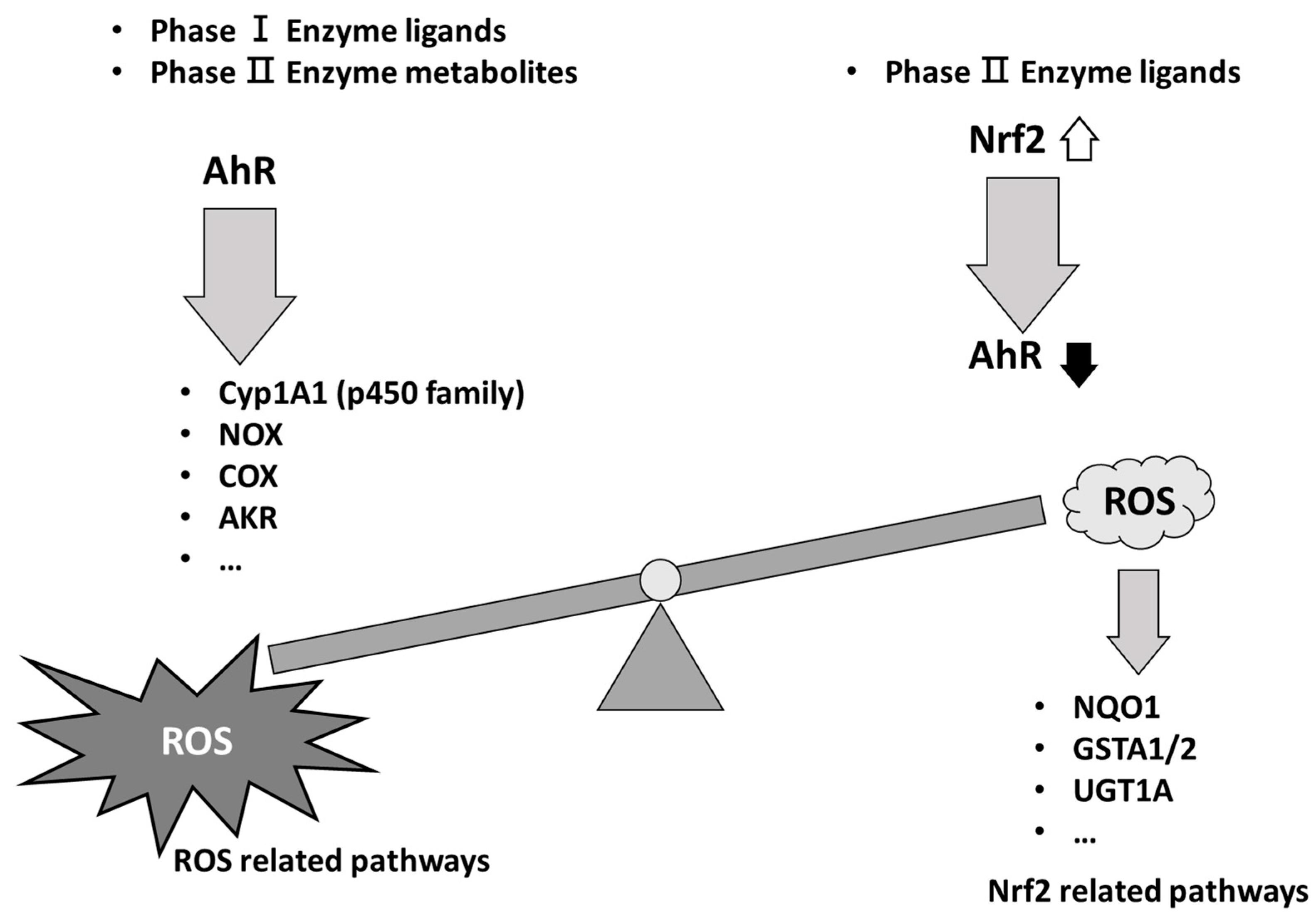
Figure 2.
Divergent functions of PER, CIN, and SNF to control gene expressions of AHR and NRF2. The heterogeneous function of phase II enzyme ligands to affect the ROS level and Nrf2 gene expression is illustrated. The p53 mutation status and the normal and the cancer cells context are critical for the bifunctional effects of antioxidation drugs to control the balance of ROS. Thus, the endogenous ROS in the cells we targeted should be examined. Higher ROS levels decreased the expression levels of Nrf2, p53, and Jdp2, and lower ROS decreased the generation of ROS and increased p53 and Jdp2 levels. The ROS level was controlled by the AhR expression and antioxidation was controlled by Nrf2 mediated responses. PER; perillaldehyde, CIN; cinnamon aldehyde, SFN; Sulforaphane, ARE; antioxidative response element.
Figure 2.
Divergent functions of PER, CIN, and SNF to control gene expressions of AHR and NRF2. The heterogeneous function of phase II enzyme ligands to affect the ROS level and Nrf2 gene expression is illustrated. The p53 mutation status and the normal and the cancer cells context are critical for the bifunctional effects of antioxidation drugs to control the balance of ROS. Thus, the endogenous ROS in the cells we targeted should be examined. Higher ROS levels decreased the expression levels of Nrf2, p53, and Jdp2, and lower ROS decreased the generation of ROS and increased p53 and Jdp2 levels. The ROS level was controlled by the AhR expression and antioxidation was controlled by Nrf2 mediated responses. PER; perillaldehyde, CIN; cinnamon aldehyde, SFN; Sulforaphane, ARE; antioxidative response element.
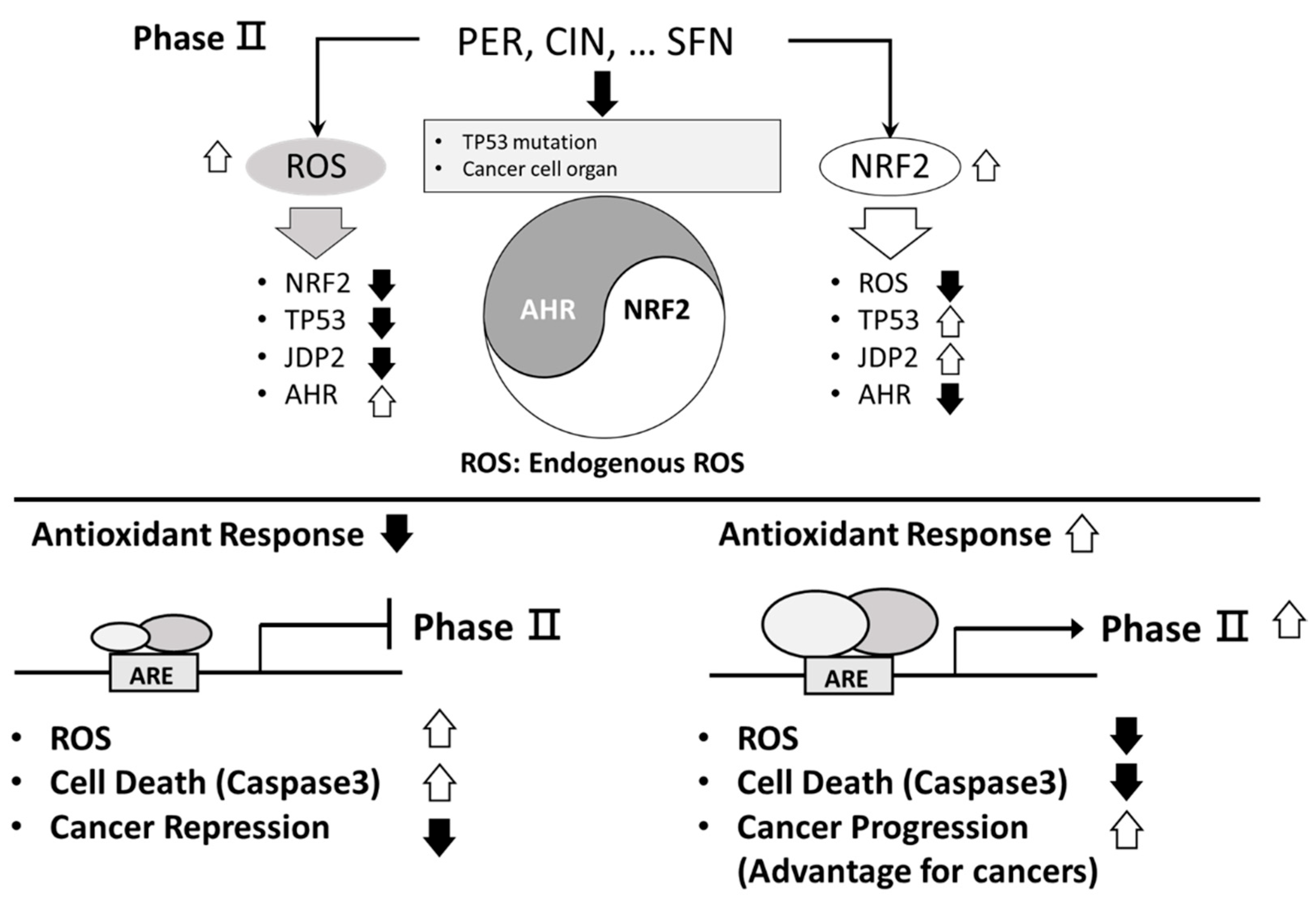
Figure 3.
IPA analysis of the signaling networks between AhR signaling and NRF2 signaling. The canonical pathways of Aryl Hydrocarbon Receptor (AhR) Signaling, NRF2-mediated Oxidative Stress Response Pathway, and KEAP1-NEF2L2 (NRF2) Pathway are selected from Ingenuity Pathway Analysis (IPA) System (https://www.nihlibrary.nih.gov/resources/tools/ingenuity-pathways-analysis-ipa). For the construction of signaling networks, AhR signaling is considered as the major axis, the other two pathways are indicated with “CP: KEAP1-NEF2L2 Pathway” and “CP: NRF2-mediated Oxidative Stress Response”, respectively. The web-based IPA bioinformatic software is applied to overlay with each other signaling pathway and to indicate the crosstalk molecules by the cross line between two signaling pathways.
Figure 3.
IPA analysis of the signaling networks between AhR signaling and NRF2 signaling. The canonical pathways of Aryl Hydrocarbon Receptor (AhR) Signaling, NRF2-mediated Oxidative Stress Response Pathway, and KEAP1-NEF2L2 (NRF2) Pathway are selected from Ingenuity Pathway Analysis (IPA) System (https://www.nihlibrary.nih.gov/resources/tools/ingenuity-pathways-analysis-ipa). For the construction of signaling networks, AhR signaling is considered as the major axis, the other two pathways are indicated with “CP: KEAP1-NEF2L2 Pathway” and “CP: NRF2-mediated Oxidative Stress Response”, respectively. The web-based IPA bioinformatic software is applied to overlay with each other signaling pathway and to indicate the crosstalk molecules by the cross line between two signaling pathways.
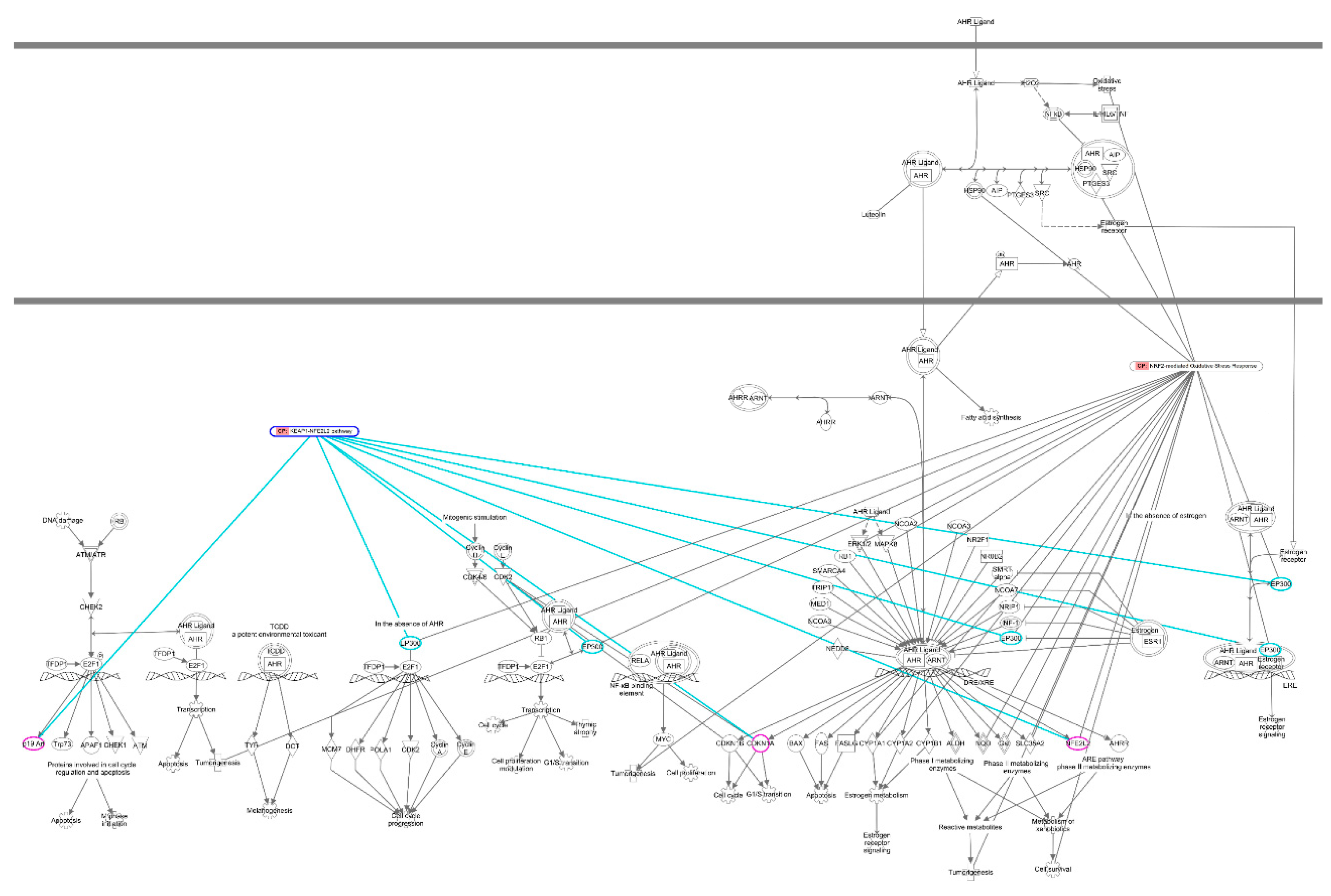

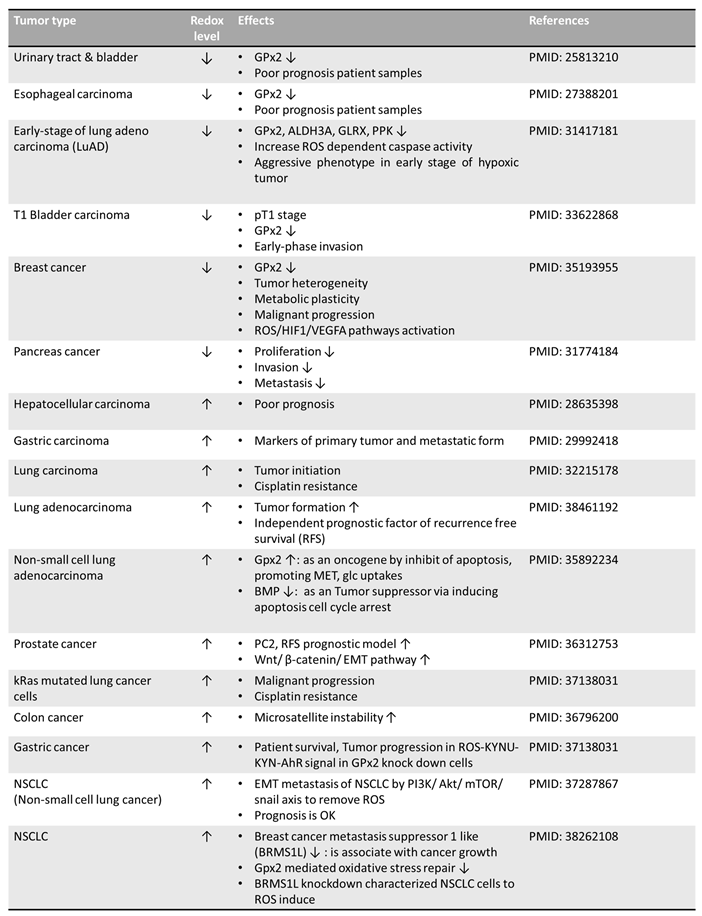
Table 1.
The variation of redox level in different cancer cells. Various effects of antioxidant functions are also shown.
Table 1.
The variation of redox level in different cancer cells. Various effects of antioxidant functions are also shown.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



