
Submitted:
05 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
This review explores the crucial role of glycosphingolipids (GSLs) in the context of cardiovascular diseases (CVD), focusing on their biosynthesis, metabolic pathways, and implications for clinical outcomes. GSLs are pivotal in regulating a myriad of cellular functions that are essential for heart health and disease progression. Highlighting findings from both human cohorts and animal models, this review emphasizes the potential of GSLs as biomarkers and therapeutic targets. We advocate for more detailed mechanistic studies to deepen our understanding of GSL functions in cardiovascular health, which could lead to innovative strategies for diagnosis, treatment, and personalized medicine in cardiovascular care.
Keywords:
glycosphingolipids
; heart failure
; HFrEF
; HFpEF
; ceramide
1. Introduction
Glycosphingolipids (GSLs) are a diverse group of bioactive molecules essential for cell membrane structure and function across various organisms, from bacteria to humans [1]. Each GSL comprises a hydrophobic ceramide backbone—made up of a fatty acid and a sphingoid base—and a hydrophilic carbohydrate headgroup, which ranges from a simple sugar to complex oligosaccharides. The structural variety is significant, featuring over 400 glycan structures in vertebrates, varying in length from one to twenty sugar residues and incorporating up to twelve different sugar types [2]. Additionally, the ceramide backbone introduces further diversity with more than 200 distinct species identified in mammalian cells, differentiated by variations in chain length, double bonds, and hydroxyl groups [2].
GSLs are broadly categorized into seven main types based on their glycan components—ganglio-, globo-, isoglobo-, lacto-, and neolacto-series prevalent in vertebrates, and mollu- and arthro-series primarily found in mollusks and arthropods (Table 1) [1]. These GSLs are also classified by charge into neutral, sialylated, or sulfated forms, where neutral GSLs contain uncharged sugars, and sialylated GSLs include negatively charged sialic acid residues [3].
Glycosphingolipids (GSLs) are integral in mediating a variety of biological processes, including cell division, differentiation, and signaling [4,5]. Positioned strategically within plasma membranes, GSLs facilitate critical interactions that regulate cell adhesion, motility, and communication. These interactions are central to signaling pathways that control cell proliferation, migration, autophagy, apoptosis, and mitochondrial function [6].
In the bloodstream, GSLs notably associate with low-density lipoproteins (LDL), playing a pivotal role in lipid transport and metabolism [7]. This association is crucial for receptor-dependent endocytic pathways that are fundamental to lipid homeostasis and cellular signaling. The dynamic interactions of GSLs with various cellular components significantly impact angiogenesis and inflammation, which are vital in both disease progression and the development of therapeutic strategies [8].
At the cellular level, GSLs influence essential membrane proteins such as growth factor receptors (GFRs) and integrins. For example, the GM3 ganglioside can inhibit the autophosphorylation of the epidermal growth factor receptor (EGFR), thereby disrupting signaling pathways crucial for cancer progression [9]. GSLs also play roles in cell adhesion through interactions with galectin-3 and E-selectin, which are crucial for cell tethering and migration. Furthermore, they regulate cell motility by interacting with tetraspanins and integrins within glycosynapses, specialized domains that contain clusters of GSLs and modulate cellular responses to external stimuli [10].
Alterations in GSL metabolism are implicated in various diseases, including cancer, where they promote tumor growth and invasiveness [11], as well as in neurodegenerative diseases like Alzheimer’s and Parkinson’s, where they influence protein aggregation [12]. Additionally, in cardiovascular disorders, GSLs are linked to atherosclerosis and heart failure [5,13,14]. The therapeutic targeting of GSL biosynthesis pathways highlights their potential as universal targets in oncology, neurology, and cardiovascular medicine.
2. Biosynthesis and Metabolism of Glycoshingolipids
2.1. GSL Synthesis
GSL synthesis initiates in the endoplasmic reticulum (ER), where serine palmitoyltransferase (SPT), a key enzyme located in the ER membrane, catalyzes the condensation of L-serine and palmitoyl-CoA. This reaction forms 3-ketosphingosine, which is subsequently reduced to sphinganine by 3-ketodihydrosphingosine reductase (KDHR), a reaction dependent on nicotinamide adenine dinucleotide phosphate (NADPH). Following sphinganine formation, it is acylated by ceramide synthase (CerS) with fatty acyl-CoA to form dihydroceramide. This step is pivotal as different CerS isoforms (CerS1-6), with specificity for various acyl chain lengths, direct the synthesis of distinct ceramides, influencing diverse cellular functions from apoptosis to cell survival [15,16]. These dihydroceramides are then desaturated by dihydroceramide desaturase to form ceramides, integral components of complex GSLs [17].
In parallel, the salvage or catabolic pathway plays a crucial role in GSL metabolism, in which sphingolipids are recycled through hydrolysis at the plasma membrane or in lysosomes. Here, complex GSLs are first reverted to ceramide, which serves as a substrate for the generation of new sphingolipids or for catabolism into sphingosine and subsequently into sphingosine-1-phosphate (S1P), a potent signaling molecule [18].
2.2. Transport and Glycoslyation
Once synthesized, ceramides are transported to the Golgi apparatus. This transfer is facilitated by the ceramide transfer protein (CERT), which shuttles ceramide from the ER to the trans-Golgi network for further processing into GSLs like glucosylceramide (GlcCer) and lactosylceramide (LacCer). These molecules are foundational for the synthesis of higher order GSLs such as gangliosides and globosides [12,19]. Glycosylation reactions in the Golgi extend the ceramide backbone into diverse GSL structures, which are then transported to the plasma membrane, contributing to the lipid raft architecture and affecting signal transduction processes (Figure 1).
2.3. GSL Degradation
GSL degradation involves endocytosis of membrane-bound GSLs and transport to endosomal vesicles, followed by the sequential removal of sugar residues from the non-reducing end, a process which is catalyzed by glycosyl hydrolases (GHs). GH activity is often facilitated by sphingolipid activator proteins, or saposins (SAPs), which provide the water-soluble hydrolases access to their membrane-embedded substrates. The precursor protein prosaposin, upon cleavage in the lysosome, gives rise to four SAP variants (SapA, B, C, and D), which each have affinities to specific GSLs and function specifically with their associated hydrolase [20]. Following the degradation of the glycan chains, ceramidases hydrolyze ceramide, resulting in sphingosine and fatty acid [21]. There are 3 types of ceramidases: acid, neutral, and alkaline ceramidase. Acid ceramidase localizes to the lysosomes and hydrolyzes ceramides from the endosomal membrane system from the plasma membrane. Neutral ceramidase localizes to the plasma membrane and regulates sphingosine and S1P production across the plasma membrane. Finally, there are 3 alkaline ceramidases that localize to the ER (ACER1), Golgi (ACER2), or both (ACER3, which catalyzes the hydrolysis of ceramides with unsaturated long acyl chains) [16].
3. Spatial Dynamics of Glycosphingolipid Signaling in Cardiac Cellular Compartments
3.1. GSLs in Plasma Membranes and Lipid Microdomains
GSLs play vital roles in the structure of plasma membranes, with varying concentrations and compositions of different GSLs leading to changes in various membrane properties. For example, an increase of GlcCer within fluid POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, a commonly used model lipid for biophysical experiments) membranes was shown to drive the formation of gel domains, and even higher levels lead to the formation of crystal-like structures. In vesicles, high concentrations of GlcCer led to the protrusion of tubule-like structures from the vesicles, which may be driven by changes in membrane curvature and lipid phase changes [22]. These results point to the functions of GSLs in structuring and reorganizing membranes.
In most cells, GSLs are found primarily in lipid rafts, microdomains within the membrane that are enriched in sphingolipids, cholesterol, and specific proteins. The unique properties of GSLs, including their geometry, ability to form hydrogen bonds, and specific content of the saturated alkyl chains, allow them to form and stabilize lipid rafts [23]. These lipid rafts act as signaling centers, enhancing intracellular communication by bringing signaling molecules and their substrates together [3]. Indeed, GSLs are also enriched in extracellular vesicles (EVs), which package specific cargo to facilitate intracellular signaling. These lipid-enriched EVs can cause altered cell processes, including inducing apoptosis and activating immune responses [24].
In the cardiovascular system, lipid rafts regulate signaling pathways, controlling heart rate, vascular tone, and endothelial function [25]. GSLs within lipid rafts modulate the activity of ion channels and receptors, such as G-protein coupled receptors and integrins, crucial for the physiological responses of the heart and blood vessels. Alterations in GSL composition can disrupt raft integrity and functionality, impairing signal transduction and contributing to cardiovascular diseases such as hypertrophy, atherosclerosis, hypertension, and cardiac arrhythmias [3,5,13,14,23]. These structural and functional modifications by GSLs directly influence cardiac efficiency and health, with further implications for cardiovascular diseases discussed in subsequent sections of this paper.
Caveolae, invaginated lipid microdomains rich in GSLs, are significant in cardiovascular health. The loss of caveolin function, the major membrane proteins of caveolae, is linked to vascular disorders and hypertension, possibly due to its role in endothelial nitric oxide synthase (eNOS) signaling. eNOS, localized in caveolae, associates with caveolin-1 (Cav1), inhibiting eNOS activity and nitric oxide (NO) production [26]. Increased Cav1 expression is observed in patients with insulin resistance and type 2 diabetes [27], while Cav1 knockout mice exhibit increased systemic NO and dilated ventricles [28]. Reduced NO production is associated with atherosclerosis, the pathological basis of many cardiovascular disorders.
3.2. GSLs in Mitochondria-Associated ER Membranes (MAMs)
Another crucial membrane site is the mitochondria-associated ER membrane (MAM). MAMs are contact sites between the ER and mitochondria that play roles in Ca2+ dynamics, lipid synthesis and transport, apoptosis, and energy metabolism [29]. Gangliosides play an important role in MAMs, and variations in their concentration within these microdomains can lead to different cell fate decisions. For example, the ganglioside GD3 has been implicated in inducing autophagy through its interactions with calnexin, another resident protein of MAMs. Another important ganglioside is GM1, which binds Ca2+. The presence of GM1 in MAMs (especially when increased under disease conditions) can disturb the membrane Ca2+ buffering capacity. This results in mitochondrial dysfunction, including depolarization and membrane permeabilization, eventually leading to apoptosis [29]. In mice with a β-galactosidase (β-gal) mutation, defective lysosomal β-gal leads to impaired degradation of GM1. The increased concentration of GM1 leads to a buildup within MAMs. This buildup of GM1 may cause clustering of IP3R-1 (an intracellular calcium channel) and, in turn, leads to the formation of a Ca2+ mega pore. Furthermore, the Ca2+ mega pore induces an increase in grp75 and VDAC-1 levels within mitochondria, which leads to greater diffusion of Ca2+, disrupting the mitochondrial membrane potential [30] (Figure 2). The disruption of MAM functions, particularly in cardiac cells, is critical as it influences not only cellular health but also overall heart function, potentially exacerbating conditions like heart failure and arrhythmias [31].
3.3. GSLs in Mitochonrial Function and Dynamics
GSLs play critical roles in modulating mitochondrial dynamics and signaling, impacting cellular processes like energy production and apoptosis (Figure 3). One key area of regulation that GSLs are involved in is mitochondrial membrane permeabilization, a critical event in apoptosis. Ceramide, a fundamental GSL, forms channels within mitochondrial membranes, leading to mitochondrial outer membrane permeabilization [16]. The ganglioside GD3 has also been shown to suppress the proton pumping activity and/or the increase in small-conductance channels, inducing a gradual depolarization of the inner mitochondrial membrane [32]. This permeabilization and depolarization facilitates the release of cytochrome c, triggering caspase activation and cell death, mediated through interactions with B-cell lymphoma-2 (BCL-2) family proteins [33]. In the cardiac system, this relationship is especially important, considering the critical role of mitochondrial function to cardiomyocyte performance, and dysregulation of this pathway may lead to adverse cardiac outcomes.
Furthermore, GSLs are integral to the regulation of mitochondrial fission and fusion dynamics, processes vital for maintaining mitochondrial integrity and function. Mitochondrial dynamics control the transport, size, morphology, and turnover of mitochondria, directly affecting cell viability and ATP production, an important aspect for cells such as cardiomyocytes, which have high metabolic demand. Disruption of this fission/fusion balance can lead to alterations in mitochondrial function, which in turn is associated with several cardiac diseases [34]. GD3, a ganglioside present in MAMs, has also been shown to interact with proteins involved in initiating autophagy, such as WIPI1 and AMBRA1 [35]. Furthermore, following CD95/Fas triggering (activation of death receptors that trigger apoptosis), GD3 has been shown to localize to mitochondrial fission regions, along with other fission-involved proteins such as dynamin-like protein 1 (DLP1) and human Fis1 protein (hFis1) [36]. This relationship may also occur with less complex GSLs such as glucosylceramide and lactosylceramide, resulting in increased synthesis of mitochondrial fusion and fission proteins, indicating increased mitochondrial turnover [35]. Ceramide also influences the recruitment of proteins like Drp1 to mitochondrial raft-like domains, promoting mitochondrial fission [37].
Additionally, GSLs play a role in regulating mitochondrial calcium retention capacity and energy production. Mitochondrial Ca2+ uptake is important in regulating contractile activity through the modulation of cellular metabolism and energy. In the hearts of mice models with type-1 diabetes, lactosylceramide was shown to inhibit the electron transport chain and decrease calcium retention capacity. Specifically, LacCer suppressed state 3 respiration (characterized by maximal mitochondrial oxygen consumption after the addition of ADP) by targeting complex I, II, and IV in the ETC. In terms of calcium retention, addition of LacCer to mitochondria led to the release of Ca2+ from the mitochondria, causing mitochondrial dysfunction. A possible mechanism for this release could be a LacCer-induced increased Ca2+ sensitivity of the mitochondrial permeability transition pore (mPTP) [38]. GD3 also impacts the protein complexes of the ETC. Dysregulation of GD3 synthesis and metabolism leads to ROS production, activating the PI3K/Akt/mTOR pathway, which regulates mitochondrial biogenesis and oxidative phosphorylation [35].
3.4. GSLs in Cellular Signaling Pathways
Sphingolipids such as ceramide and sphingosine-1-phosphate (S1P) play pivotal roles in cardiac function and disease by modulating a vast array of cellular processes. Ceramide, for instance, is known to escalate cytosolic Ca2+ levels, thereby influencing cardiac hypertrophy or contractile function during ischemia-reperfusion (IR) events. It activates key signaling pathways such as p38 MAPK and PKC, which concurrently inhibit AKT and JNK pathways, thus triggering pro-apoptotic BAX signaling. This dual role of ceramide in promoting cellular adaptations to stress while inducing detrimental effects highlights its complex involvement in cardiomyocyte physiology [39].
In contrast, S1P offers cardioprotective effects through its intricate signaling mechanisms. It is produced under the influence of hypoxic transcription factors like HIF1α and HIF2α, or pro-inflammatory mediators such as TNF-α, activating a cascade of pro-survival pathways including AKT, PI3K, Pak1, and Rac. These pathways notably decrease infarct size and promote endothelial cell migration and angiogenesis, enhancing the viability of cardiomyocytes in both in vitro and in vivo IR models [40]. Additionally, S1P interactions with its specific receptors activate pathways like Pak1 and protein phosphatase 2A (PP2A), essential for cardiac protection in conditions such as ischemic and Takotsubo cardiomyopathy [41]. S1P also plays a critical systemic role by mediating renin release and stimulating aldosterone secretion within the renin-angiotensin-aldosterone system (RAAS), crucial for maintaining electrolyte and fluid balance and thus stabilizing blood pressure in conditions like hypertrophic cardiomyopathy [42].
Furthermore, GM1 ganglioside, through its oligosaccharide chain, independently interacts with plasma membrane receptors such as TrkA, the receptor for nerve growth factor (NGF). This interaction initiates the MAPK/ERK signaling pathway, promoting cellular differentiation and survival, crucial in cardiac stress responses and repair mechanisms [43]. This function of GM1 illustrates the non-structural, signaling-mediated roles of GSL oligosaccharides in cellular modulation, emphasizing their potential in therapeutic strategies targeting various cardiomyopathies and cardiovascular dysfunctions.
4. Glycosphingolipids in Cardiovascular Disease Pathogenesis
Heart disease remains the leading cause of mortality in the United States, accounting for 1 in every 5 deaths in 2021 and representing a substantial portion of healthcare expenditures [44,45]. Cardiovascular disorders, including hypertension, coronary artery disease, cardiomyopathy, atherosclerosis, and heart failure, are influenced by diabetes, obesity, poor diet, physical inactivity, and excessive alcohol use [45]. GSLs have been linked to the progression of these cardiovascular conditions [5,13,33,44,46,47].
4.1. Atherosclerosis
The role of GSLs in modulating the inflammatory response within the cardiovascular system contributes to atherosclerosis, a crucial factor in heart failure development (Figure 4). While the complete mechanisms of atherosclerosis progression remain to be fully elucidated, lipoprotein retention in the vascular intima is a known initiating factor. Accumulated lipoproteins, predominantly LDLs, prompt endothelial cells to recruit monocytes, which differentiate into macrophages and subsequently into foam cells [47]. GSLs have been observed accumulating in the aortic walls of patients with atherosclerosis: tissue samples from these patients, showing fatty streaks and atherosclerotic plaques, revealed increased levels of glucosylceramide, lactosylceramide, and GM3. Furthermore, an upsurge in gangliosides GD3 and GD1a was detected in the media beneath the atherosclerotic lesions [48]. The pro-atherosclerotic mechanism likely involves GSLs binding LDLs. Gangliosides have been demonstrated to bind LDLs, leading to alterations in LDL structure by inducing conformational changes in apoB molecules on the LDL surface, promoting LDL particle aggregation and enhanced uptake by macrophages [7]. Our previous studies have also shown that inhibiting glycosphingolipid synthesis reduces atherosclerosis and arterial stiffness in apolipoprotein E(-/-) mice and rabbits. Feeding these animals a Western diet significantly increased aortic pulse-wave velocity, intima-media thickening, levels of oxidized low-density lipoprotein, Ca2+ deposits, and glucosylceramide and lactosylceramide synthase activity. These effects were dose-dependently reduced by the GSL synthesis inhibitor D-PDMP. In the liver, D-PDMP lowered cholesterol and triglyceride levels by enhancing the expression of SREBP2, LDL receptor, HMGCo-A reductase, and cholesterol efflux genes such as ABCG5 and ABCG8. D-PDMP also affected VLDL catabolism by upregulating the expression of lipoprotein lipase and the VLDL receptor. Rabbits fed a Western diet for 90 days exhibited extensive atherosclerosis, accompanied by a 17.5-fold increase in total cholesterol levels and a 3-fold increase in lactosylceramide levels, all of which were completely prevented by D-PDMP administration [14]. However, a study by Glaros et al. (2008) indicated that inhibiting GSL synthesis through glucosylceramide synthesis inhibition did not impact the atherosclerotic lesion area in mice, suggesting a complex role for GSLs in atherosclerosis [49].
S1P, a GSL precursor, has been demonstrated to affect atherosclerosis both positively and negatively. By inhibiting macrophage apoptosis and endothelial inflammation via the PI3K/Akt pathway, S1P’s interaction with S1PR1 may have anti-atherosclerotic effects. Conversely, its interaction with S1PR2/3 may inhibit the proliferation and migration of smooth muscle cells by interacting with Rac protein, thus delaying or inhibiting neointima formation. However, S1P has also been shown to activate pro-inflammatory and endothelial factors, including ICAM-1, VCAM-1, and IL-6, potentially exacerbating atherosclerosis [50]. Ceramides also play a significant role in atherosclerosis development. They accumulate in atherosclerotic plaques and may initiate plaque formation. Inhibition of SPT reduces plasma sphingolipid concentrations, including ceramides, leading to smaller atherosclerotic lesions in the aortas of atherosclerotic mice [47]. Endothelial dysfunction, a precursor to atherosclerosis, can be induced by ROS generation and reduced nitric oxide (NO) availability. Ceramides have been linked to elevated ROS levels, and conversely, ROS can stimulate ceramide synthesis. Ceramides also activate ROS-generating enzymes such as NADPH oxidase, uncoupled eNOS, and xanthine oxidase following stimulation by pro-inflammatory cytokines such as TNF. Other critical processes in atherosclerotic lesion development, such as LDL aggregation and monocyte recruitment, have been associated with ceramide levels [51].
4.2. GSLs in Angiogenesis and Inflammation
GSLs may influence cardiac remodeling through their role in angiogenesis. Globo H ceramide (GHCer), a prevalent cancer-associated GSL, exhibits proangiogenic activity. GHCer induced migration, tube formation, and intracellular calcium mobilization in human umbilical vein endothelial cells (HUVEC). Additionally, subcutaneous injection of GHCer plugs into mice increased blood vessel formation. These effects are linked to the co-localization of GHCer with translin-associated factor X (TRAX), wherein GHCer competes with phospholipase C β1 (PLCb1) for binding to TRAX. GHCer effectively inhibits TRAX, activating PLCb1 and leading to calcium mobilization and angiogenesis [8].
GSL-enriched microdomains modulate immune receptor signaling across various immune cell types, affecting both innate and adaptive responses. Lactosylceramide (LacCer) is highly expressed in human neutrophils and binds various pathogenic microorganisms. LacCer-enriched lipid rafts contain the Src family tyrosine kinase Lyn and mediate the generation of proinflammatory cytokines and superoxide in neutrophils [52]. Changes in the expression of GSL metabolism genes are associated with fibroblast differentiation in keloids, pathological scars characterized by inflammation and excess collagen [53]. Specifically within the cardiovascular system, in patients with myocardial infarction, sphingomyelin, ceramide, and glucosylceramide have been shown to positively correlate with high-sensitivity C-reactive protein, a marker of acute inflammation [54]. Cer and sphingomyelin have also been shown to modulate cellular responses to cytokines. For example, C2 and C6 both potentiated the induction of C-reactive protein (CRP) and serum amyloid A (SAA) in Hep3B cells [55].
In macrophages, GM3-enriched microdomains interact with insulin receptors to inhibit insulin signaling, impacting metabolic responses [56]. Similarly, in adipocytes, these interactions suppress the autophosphorylation of EGF receptors, thereby modulating growth signaling pathways [57]. Furthermore, GSL microdomains regulate toll-like receptor (TLR) functions in dendritic cells and macrophages; for example, the interaction of glucosylceramide with TLR4 influences its orientation and signaling response to lipopolysaccharide, enhancing immune activation via MyD88 adapter proteins [58]. This mechanistic role extends to T-cell regulation, where GM1 microdomains interact with neurotrophin receptors like TrkA, influencing T-cell differentiation and survival [59]. On the other hand, proteins involved in GSL metabolism, such as S1PR1, may protect against myocardial injury by soothing inflammatory responses, stimulating the proliferation of repair macrophages, and inhibiting myocardial fibrosis through the Akt/eNOS dependent pathway [50]. These specific interactions across different immune cells underscore the sophisticated regulatory capabilities of GSL-enriched microdomains in orchestrating immune responses and maintaining cellular homeostasis.
4.3. Hypertrophy and Heart Failure
Cardiac hypertrophy, defined by an increase in cardiomyocyte size in response to ventricular wall stress and pressure overload, initially serves as a compensatory mechanism to enhance contractility by increasing the number of sarcomere units and reducing left ventricular wall stress, thus maintaining cardiac efficiency. However, this adaptive hypertrophy can progress to pathological hypertrophy, leading to heart failure. Heart failure, characterized by the heart’s inability to pump blood effectively, is divided into two major types: heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF) [44].
4.3.1. GSL Insights from HFrEF
HFrEF is characterized by dilated ventricles, decreased contractility, and reduced ejection fraction. This form of heart failure can arise from ischemic causes, such as ischemic injury, or non-ischemic causes. Disrupted GSL metabolism has been linked to the pathogenesis of heart failure. Specifically, serine palmitoyltransferase (SPT), which catalyzes the condensation of serine and palmitoyl-CoA, is crucial for cardiovascular health. In mice, the expression of SPTLC3, a subunit that broadens the substrate specificity of SPT, enables the generation of d16-derived sphingolipids from myristoyl-CoA, promoting cell death and cleavage of poly(ADP-ribose) in cardiomyocytes [60]. Furthermore, the ER membrane protein Nogo-A, which inhibits SPT, is upregulated in cardiomyocytes following pressure overload, thereby preventing ceramide accumulation and protecting the heart from failure [61]. In other mouse models of heart failure, altered lipid content within the heart is associated with the onset and progression of heart failure. In mice subjected to transverse aortic constriction, early sphingolipid shifts include an increase in dihydrosphingosine, with subsequent decreases in erythro-sphingosylphosphorylcholine and stearoyl sphingomyelin as heart failure progresses [62]. Additionally, in mice with cardiac-specific angiotensin II overexpression (TG1306/R1), studies highlight a distinct sphingolipid profile characterized by exacerbated heart size, systolic dysfunction, and cardiac fibrosis, accompanied by significant changes in ceramide species [63]. This finding correlates with emerging evidence that suggests a relationship between the fatty acyl chain length of ceramides and their impact on cardiac function. A higher ratio of C16:0 to C24:0 ceramides is associated with worse left ventricular dysfunction, decreased left atrial function, increased left atrial size, overall mortality, and increased incidence of heart failure [33].
Oxidative stress plays a critical role in cardiomyocyte death and fibrosis, leading to pathological hypertrophy and heart failure. Our studies have shown that GSLs such as lactosylceramide (LacCer) can generate reactive oxygen species (ROS) and activate ERK-1/p44 MAPK in cardiomyocytes, ultimately leading to hypertrophy [5]. Treatment with the GSL synthesis inhibitor D-PDMP prevented cardiac hypertrophy in mice fed a high-fat, high-cholesterol diet, and reduced the expression of BNP and ANP, biomarkers of cardiac hypertrophy. D-PDMP also decreased GSL mass in heart tissue by inhibiting glycosyltransferase activity [13].
Furthermore, UGCG (GlcCer synthase) and B4GalT5 (the enzyme converting glucosylceramides to lactosylceramides) may form a complex regulating mitochondrial oxidative stress. Inhibition of UGCG in cardiomyocytes led to reduced mitochondrial ROS levels, while its overexpression increased ROS levels. Inhibition of B4GalT5 improved mitochondrial ROS levels even after UGCG overexpression, suggesting an involvement mediated by ERK signaling: activation of the UGCG-B4GalT5 axis correlated with ERK activation, and downregulation of UGCG reduced ERK activation and myocardial hypertrophy [64]. Elevated levels of glucosylceramide (GlcCer) have been observed in heart tissue from patients with reduced ejection fraction (EF) following an ischemic event, compared to those with normal heart function. This trend is also evident in mice subjected to induced myocardial infarction, indicating a potential role for GSLs in the remodeling heart [65]. These findings are supported by studies in mouse models with pressure-overloaded hearts, where UGCG was upregulated. Downregulation of UGCG ameliorated heart hypertrophy, evidenced by reduced ventricular wall hypertrophy, lower heart weight to body weight ratios, decreased left ventricular weight to body weight ratios, reduced heart weight to tibial length ratios, and decreased cardiac fibrosis. Conversely, overexpression of UGCG exacerbated heart hypertrophy following transverse aortic constriction (TAC) modeling [64]. Another study found an inverse relationship between specific ceramides (glycosyl ceramide, glycosyl-N-nervonoyl-sphingosine, lactosyl-N-nervonoyl-sphingosine, and lactosyl-N-palmitoyl-sphingosine) and the left ventricular mass to volume ratio, a measure of cardiac concentric remodeling [66].
These findings underscore the significance of increased GSL levels in contributing to oxidative stress and cardiac hypertrophy, ultimately leading to heart failure (Figure 5).
4.3.2. GSL Insights from Cardiometabolic Diseases and HFpEF
Heart failure with preserved ejection fraction (HFpEF) is a growing concern due to its increasing prevalence and strong association with metabolic disturbances. Obesity and type 2 diabetes mellitus are major drivers of HFpEF pathophysiology, primarily due to their significant impact on metabolic regulation. GSLs are increasingly recognized for their role in metabolic dysregulation, including insulin resistance and lipid metabolism, which contribute to myocardial remodeling and vascular dysfunction characteristic of HFpEF [44].
A hallmark of cardiometabolic diseases is the excess lipid deposition in non-adipose tissues, including the heart. Normally, fatty acids are metabolized via beta-oxidation to produce ATP, with excess free fatty acids stored as inert triglycerides within cells. However, when these metabolic pathways become saturated, bioactive lipids such as complex sphingolipids and ceramides accumulate in non-adipose tissues, including blood vessels and cardiac tissue. This lipid accumulation, or lipotoxicity, contributes to the development of cardiometabolic disorders [5,13,47]. For example, studies in Dahl salt-sensitive (Dahl/SS rats), a model for HFpEF due to sodium sensitivity, show a pre-heart failure increase in N-palmitoyl-sphingosine (C16:0) and glycosyl-N-stearoyl-sphingosine (C18:0), pointing to their potential as early biomarkers of heart failure [67].
GSLs are particularly involved in lipotoxic cardiomyopathy, acting as second messengers in pathways that lead to disease phenotypes such as proliferation, adhesion, migration, autophagy, apoptosis, and mitochondrial dysfunction [35] (Figure 5). Specific GSLs participate in signaling pathways that regulate cellular energy utilization, lipid metabolism, and inflammatory responses, all crucial in HFpEF pathology. Overaccumulation of certain GSLs in cardiac cells disrupts normal metabolic processes, leading to inefficient energy use and increased oxidative stress, which are hallmarks of HFpEF. Additionally, GSLs influence the inflammatory environment within the myocardium, affecting fibrotic processes that contribute to heart muscle stiffening [46]. Modulating GSL pathways may mitigate metabolic disturbances and inflammatory responses, thereby improving cardiac function and patient outcomes.
Obesity is linked to an altered lipidome and is a common comorbidity in HFpEF. A study investigating the effects of weight loss in women with obesity and HFpEF demonstrated that gastric bypass surgery led to significant cardiac improvements, including reduced left ventricular mass, relative wall thickness, and enhanced left ventricular relaxation. Moreover, weight loss was associated with decreased plasma levels of several sphingolipids [68]. These findings underscore the pivotal role of GSLs in cardiometabolic diseases.
4.3.3. Diabetes
In diabetes, the dysregulation of sphingolipid metabolism, particularly ceramides, plays a pivotal role in modulating cellular signaling pathways that are crucial for maintaining metabolic homeostasis. Ceramides induce mitochondrial dysfunction and elevate oxidative stress, which are implicated in the disruption of insulin signaling pathways. Specifically, ceramides facilitate the inhibition of AKT translocation and activation of conventional protein kinase C (PKC) isoforms, mechanisms that critically attenuate insulin-mediated glucose uptake [69]. Furthermore, ceramides trigger the activation of the mitogen-activated protein kinase (MAPK) pathway via p38, which concurrently inhibits AKT and activates c-Jun N-terminal kinase (JNK). This sequence of events promotes the activation of the pro-apoptotic B-cell lymphoma 2 associated X protein (BAX), leading to apoptosis and contributing to myocardial hypertrophy in diabetic cardiomyopathy [70]. Diacylglycerol acyltransferase (DGAT1), the enzyme that catalyzes the final step in triglyceride synthesis, has been shown to affect insulin resistance [71], and inhibition of this enzyme may be beneficial in patients with type 2 diabetes. However, humans with severe heart failure have notably reduced levels of DGAT1 mRNA in the heart. Indeed, cardiomyocyte-specific DGAT1 knockout mice exhibit severe increases in ceramide and diacylglycerol, as well as cardiomyopathy, linking aberrant metabolism of these lipids to diabetes and heart failure [72]. Additionally, the glycosphingolipid glucosylceramide (GlcCer) exerts effects independent from ceramide, impairing insulin signaling through yet-to-be-elucidated mechanisms, thus exacerbating insulin resistance [73]. This detailed mechanistic insight underscores the complex interplay between sphingolipid metabolism and key regulatory pathways affecting cellular function in diabetes.
4.4. Fabry’s Disease
Fabry’s disease (FD) is an X-linked genetic disorder caused by mutations in the GLA gene, which encodes the enzyme α-galactosidase A (AGAL). This enzyme is crucial for the degradation of GSLs, and its deficiency leads to the accumulation of globotriaosylceramide (Gb3) in lysosomes. This accumulation affects multiple organs, including the kidneys, heart, and blood vessels, causing symptoms such as renal insufficiency, cardiomyopathy, strokes, and gastrointestinal pain. FD affects 1 in 40,000 to 117,000 people, but it is likely underdiagnosed [74]. Over 1,000 mutations in GLA have been identified, with disease severity influenced by environmental factors and blood type; individuals with AB or B blood types often experience more severe symptoms due to additional GSL accumulation in erythrocyte membranes [75]. FD presents differently in males and females, with females often developing symptoms later due to X-chromosome inactivation, which creates a mosaic of normal and mutant cells. Diagnosis in females often requires genetic analysis, as AGAL activity can appear normal [76].
Cardiac symptoms are reported in 40-60% of FD patients, especially those with late-onset FD. The accumulation of Gb3 in FD significantly affects various cardiac structures, including myocytes, intramyocardial vessels, endocardium, valvular fibroblasts, and conduction tissue. For example, endomyocardial GSL deposition causes enlarged myocytes, eventually making the ventricular walls more rigid and impeding ventricular filling. Ultimately, patients with FD suffer from left ventricular hypertrophy, which may progress to heart failure with preserved ejection fraction [77].
Impaired mitochondrial function and disrupted energy metabolism may also underlie the development of cardiac symptoms in patients with FD. Cultured fibroblasts obtained from FD patients showed significant impairment of mitochondrial function, leading to reduced energy metabolism. Additionally, compared to healthy controls, FD patients demonstrated reduced phosphocreatine and ATP concentrations. [78]. Gb3 accumulation has also been implicated in dysregulated autophagy, which can lead to cell death and disease progression. In a human podocyte model of FD, the accumulation of intracellular Gb3 was accompanied by an increased abundance of LC3-II, a marker for autophagosomes, and a loss of mTOR kinase activity, an inhibitor of autophagic activity [79]. A similar mechanism in cardiomyocytes could contribute to the onset of cardiovascular disease seen in FD patients. Electrocardiographic abnormalities have been shown in FD cardiomyocytes that have an overabundance of Gb3. Compared to control cardiomyocytes, these FD cardiomyocytes showed increased excitability and altered calcium handling [80].
Inflammation also plays a critical role in the progression of FD. Elevated Gb3 levels have been shown to increase apoptotic states in peripheral blood mononuclear cells from FD patients and elevate proinflammatory cytokine expression and production [81,82]. FD patients exhibit higher levels of inflammatory biomarkers such as TNF, IL-6, TNFR1, and TNFR2, which correlate with deteriorating cardiac function [83]. These findings suggest that glycosphingolipid-induced inflammatory responses significantly contribute to cardiomyopathy in FD.
Understanding the multifaceted impact of Gb3 accumulation on cardiac tissues and the role of inflammation in FD can provide valuable insights into the pathophysiology of cardiac complications and potential therapeutic targets for managing FD.
4.5. Gaucher’s Disease
Gaucher’s disease is another lysosomal storage disorder resulting from deficiency in lysosomal glucocerebrosidase (GCase, also called glucosylceramidase). There are three types of this disease: Type 1 is non-neuronopathic and the most common of the three, affecting about 1 in 40,000 people. Type 2 is acute neuropathic, and type 3 is subacute neuronopathic, and these occur in less than 1 in 100,000 people [84].
GCase catalyzes the cleavage of glucocerebroside into glucose and ceramide in the intralysosomal membrane. Mutations in the glucocerebrosidase gene (GBA) leads to misfolding of the protein in the ER, disrupting protein trafficking to the lysosomes, and ultimately inhibiting GCase activity. Deficiency in GCase activity leads to glucocerebroside accumulation, affecting many organs and organ systems, including the spleen and liver, as well as the skeletal, neurologic, immune, and hematologic systems. Since glucocerebroside is a precursor to many complex GSLs, the absence of functional GCase disrupts a crucial step of sphingolipid metabolism. In GD, glucocerebroside is deacetylated to form glucosylsphingosine (lyso-Gl-1), which accumulates in the lysosomes, causing pH increases and lysosomal destabilization. The dysfunctional lysosomes begin to amass the cells, interfering with cellular pathways [85]. The deposition of glucocerebroside within these cells, primarily macrophages, results in the appearance of Gaucher cells, which are abnormal cells with small, displaced nuclei and wrinkled or striated cytoplasm [86].
Cardiac involvement in Gaucher’s disease generally manifests in valvular calcification. The cardiovascular type of GD is GD Type 3c (GD3c), resulting from a rare homozygosity to the p.Asp448His (D409H) GBA mutation. It is primarily the ascending aorta and aortic and mitral valves that are affected, causing stenosis and, eventually, heart failure [87]. Pericarditis and intramyocardial infiltration by Gaucher’s cells can also occur, leading to constriction and cardiomyopathy [88].
4.6. Niemann-Pick Disease
Niemann-Pick disease refers to a group of lysosomal storage disorders that is separated into two subcategories. Niemann-Pick disease types A and B result from mutations in the SMPD1 gene leading to acid sphingomyelinase deficiency and consequent accumulation of sphingomyelin. Niemann-Pick disease type C results from mutations to either the NPC1 or, rarely, the NPC2 gene, leading to accumulation of cholesterol, as well as gangliosides and other GSLs, in endosomal compartments. Clinical manifestations of Niemann-Pick disease include liver failure, pulmonary disorder, neurological deficits, and psychiatric symptoms [89]. This review focuses on Niemann-Pick disease type C (NPC).
The full functions of NPC1 and NPC2 have yet to be elucidated. NPC1 is localized to vesicles involved in the recycling of unesterified cholesterol from late endosome/lysosome to the ER and Golgi, and may also function in GSL homeostasis [90]. Therefore, disruptions in NPC1 activity disrupt the transfer of cholesterol and other lipids, causing lower than normal amounts of these lipids to reach the plasma membrane and ER and an accumulation of lipids in multiple tissues. Indeed, tissue extracted from mice with an NPC1 knockout was highly enriched in GSLs, sphingosine, and cholesterol compared to normal mice. Furthermore, NPC cell culture models show altered endocytic trafficking and decreased fluid-phase uptake, which was reversed by GSL-lowering drugs and inhibition of GSL synthesis [91]. Similarly, murine and feline models of NPC treated with NB-DNJ, a GSL synthesis inhibitor, showed reduced ganglioside accumulation and ameliorated neurological disease symptoms [90]. These results suggest a pivotal role of GSLs and GSL synthesis in NPC disease.
In the cardiovascular system, NPC1 has been linked to atherosclerosis. Apoe-/- mice, the standard mouse model for atherosclerosis, were crossed with Npc1-/- mice to create a double mutant. These mice showed greater atherosclerotic lesion area compared to their Apoe-/- littermates and also had a greater risk for atherothrombosis and medial degradation, acute complications in patients with atherosclerosis [92]. On the other hand, NPC1 heterozygosity in mice conferred protection against lesional necrosis and subsequent atherosclerotic plaque instability compared to NPC1+/+ mice [93]. The role of NPC1 in the cardiovascular system is further supported by cohort studies linking common genetic variants associated with NPC with cardiovascular complications. For example, Afzali et al. (2013) showed that methylation of the NPC1 promoter was a risk factor for cardiovascular disease (CVD), and levels of total triglycerides, cholesterol, HDL-C and LDL-C vary with methylation level, with the unmethylated promoter correlating with lower levels compared to the methylated promoter [94]. Similarly, Ma et al. (2010) showed a correlation between NPC1 variants and coronary heart disease (CHD) [95]. Finally, Zhao et al. (2022) showed a relationship between an indel polymorphism downstream of NPC1 (rs150703258) with sudden cardiac death (SCD) [28]. However, these studies are limited by a relatively small cohort size and low ethnic diversity.
5. Clinical Studies on Glycosphingolipids in Cardiovascular Disorders
Clinical studies exploring glycosphingolipids (GSLs) in cardiovascular disorders primarily focus on biomarker identification for high cardiovascular risk. However, emerging research has begun to unravel the mechanistic roles of GSLs in acute cardiovascular events. For instance, a pivotal study by Knapp et al. (2013) investigated the alterations in sphingolipid plasma concentrations following acute ST-segment elevation myocardial infarction (STEMI) [96]. The study revealed that patients with STEMI exhibited significantly reduced plasma levels of sphingoid base-1-phosphates such as S1P and sphinganine-1-phosphate (SA1P) [96], suggesting a disruption in sphingolipid-mediated signaling pathways that are crucial for cardiac protection.
Further, erythrocytes from STEMI patients showed transient increases in sphingoid bases and sphingolipids [96], suggesting a compensatory response to the acute reduction in plasma levels. This response may involve altered sphingolipid metabolism within erythrocytes or a shift in their release or degradation dynamics, possibly mediated by vascular endothelium interactions. The persistent discrepancy in plasma and erythrocyte sphingolipid levels post-STEMI underscores the complex regulatory mechanisms governing cardiac sphingolipid homeostasis.
Moreover, the impact of standard antiplatelet therapies on sphingolipid profiles complicates the interpretation of these differential distributions of sphingolipids. Subsequent research by Knapp et al. (2013) evaluated the influence of aspirin on sphingolipid concentrations across different blood compartments [96]. Results indicated that aspirin administration could modulate sphingolipid metabolism, affecting the levels of cardioprotective sphingoid base-1-phosphates in the bloodstream [96].
These studies collectively highlight the critical role of GSLs and their metabolic pathways in modulating cardiac function and response to ischemic injury [96]. Understanding these pathways offers potential therapeutic avenues for enhancing cardioprotection and tailoring interventions to manage and prevent the progression of heart failure.
6. Glycosphingolipids as Biomarkers in Cardiovascular Diseases
The compound that has been most consistently studied as an indicator of cardiovascular disease is ceramide. Many studies have suggested that disturbances in the balance between ceramide and sphingosine-1-phosphate may move forward the induction of apoptosis [50], thereby underscoring why it is so important to examine them as biomarkers.
Spijkers et al. (2011) investigated plasma ceramide levels in patients diagnosed with Stage 2 and Stage 3 essential hypertension and found that ceramide levels were significantly higher as compared to normotensive controls. Even more interestingly, the ceramide levels correlated to severity of disease, with increases in C24:1 and C24:0 ceramides accounting for the majority of the increase in plasma ceramide concentrations. They did not find any significant changes in sphingomyelin or ceramide-1-phosphate (C1P) levels, though C1P did trend downwards for hypertensive patients [97].
Delving deeper into the different forms of ceramides at play in vascular health, a recent study evaluated the effect of a Mediterranean diet on various indices, including lipid and ceramide serum concentrations, through a randomized controlled trial. The intervention of the Mediterranean diet was intended to mitigate the effects of higher ceramide concentrations on cardiovascular disease risk. At six months, subjects in the intervention arm showed an increased serum concentration of Cer-24 and decreased serum concentrations of Cer-22 and Cer-24/Cer-16 ratio. At 12 months, they showed even higher serum concentrations of Cer-24/Cer-16 ratio and lower serum concentrations of C24:0 and C18. The study indicated that an increase in the Cer-24/Cer-16 ratio is inversely related to cardiovascular risk, suggesting a potential cardioprotective role [98]. Similarly, a meta-analysis investigated the association between specific ceramide species and cardiovascular disease, finding that major adverse cardiovascular events (MACE) were associated with elevated plasma concentrations of Cer(d18:1/16:0), Cer(d18:1/18:0), and Cer(d18:1/24:1), whereas levels of Cer(d18:1/22:0) and Cer(d18:1/24:0) were not similarly increased [99]. In the PREDIMED trial, patients were randomized to different variations of the Mediterranean diet. Researchers calculated a “ceramide score” for each participant, based on a weighted sum of plasma concentrations of C16:0, C22:0, C24:0, and C24:1. They discovered that a higher ceramide score was associated with more than double the risk of cardiovascular disease (CVD), including non-fatal acute myocardial infarction, non-fatal stroke, or cardiovascular death. This finding suggests that these specific plasma ceramide concentrations are independently correlated with an increased risk of CVD (non-fatal acute myocardial infarction, non-fatal stroke, or cardiovascular death) [100] and that the ratios of ceramide species were not more strongly associated with CVD risk than individual ceramides, which is notably different than the evidence put forth by Daidone et al. (2024).
The idea that total ceramide levels are more relevant than ceramide composition in predicting CVD risk has become less prominent over the years, as recent studies like that conducted by Yin et al. (2021) have demonstrated the utility of factoring in specific species ratios to evaluate risk. Yin et al. (2021) proposed that different populations may have different relevant combinations of ceramides to examine, and they chose to find a good fit for hypertensive patients at high CVD risk, as compared to ceramide score 1 (CERT-1), which is a conventional scoring system based on plasma concentrations of specific ceramides that groups patients into one of four risk groups. The novel ceramide score for hypertensive patients (CERT-HBP) significantly improved the predictive value of the measured plasma concentrations of ceramides. The CERT-HBP consisted of Cer(d18:1/16:0) and its ratio to Cer(d18:1/22:0), as well as Cer(d18:1/24:1) and its ratio to Cer(d18:1/24:0), as the species had previously been proven to be associated with CVD risk [101]. In contrast, the CERT-2 score employed by Hilvo et al. (2020) to detect residual risk in patients with stable CAD is comprised of three lipid ratios (Cer(d18:1/24:1)/Cer(d18:1/24:0), Cer(d18:1/18:0)/phosphatidylcholine 14:0/22:6, and Cer(d18:1/16:0)/phosphatidylcholine 16:0/22:5) and a single lipid (phosphatidylcholine 16:0/16:0) [102]. This particular metric proved useful in evaluating residual risk in stable CAD patients, just as the prediction of MACE in hypertensive patients based on the CERT-HBP metric was also significantly improved. Other sphingolipids of note for positive associations with CAD include sphingosine, dihydro-Cer(d18:0/16:0), dihydro-Cer(d18:0/18:1), dihydro-SM(d18:0/24:1), dihydro-SM(d18:0/22:0), SM(d18:1/18:0), Cer(d18:1/18:0), and Cer(d18:1/24:0), all of which were identified by unbiased machine-learning conducted on targeted serum lipidomics [103]. This underscores the suggestion that different combinations of ceramides may be necessary to assess a variety of CVD risk in different patient populations.
Interestingly, Saleem et al. (2013) examined the role of C22:0 and C24:0 in the pathological neurodegeneration associated with coronary artery disease (CAD) by assessing them as predictors for verbal memory performance in CAD patients. High concentrations of both ceramide species were found to be significantly predictive of less verbal memory performance improvement over one year, adding a new dimension to the use of ceramides as a biomarker of disease [104].
In addition to ceramide, sphingomyelin (SM) and secretory acid sphingomyelinase (S-SMase) have been investigated as potential biomarkers. When comparing plasma concentrations of all three compounds between healthy controls, patients with stable angina pectoris (SAP), patients with unstable angina pectoris (UAP), and patients with acute myocardial infarction (AMI), ceramide and S-SMase levels were highest in the AMI and UAP groups. Surprisingly, S-SMase activity was higher in the UAP group than in the AMI group [105].
Recent studies, including a significant analysis from the PREDIMED and EPIC-Potsdam cohorts, have shown that certain sphingolipid species, particularly Cer C16:0, are not only increased in individuals with heart failure but also serve as top lipid cluster networks associated with the condition [106]. These findings suggest a deeper interconnection between sphingolipid profiles and heart failure pathogenesis, expanding our understanding of how these biomarkers could potentially influence diagnostic and therapeutic strategies in cardiac care.
7. Therapeutic Potential of Targeting Glycosphingolipids
7.1. Enzyme Replacement Therapy (ERT)
In diseases caused by deficiency of enzyme activity, resulting in a build-up of GSLs, enzyme replacement therapy (ERT) may compensate for the lack of normal enzymatic activity and provide a viable treatment option. Gaucher’s disease, which is characterized by a deficiency in glucocerebrosidase (the enzyme that hydrolyzes the glucose moiety from glucosylceramide) is one such disease. ERT for GD was introduced in 1991. There are 3 available ERTs for GD: imiglucerase (a recombinant GCase produced from Chinese hamster ovaries), velaglucerase alfa (a gene-activated GCase produced from human fibroblasts), and taliglucerase alfa (produced from carrot cells). In long-term observational studies, patients treated with these ERTs have shown improvements that sustained for 20 years with continued treatment. These results were consistent regardless of pre-treatment disease severity and were accompanied by limited side effects [107,108,109]. However, due to the low stability of the enzyme in blood, ERT is a lifelong treatment requiring periodic intravenous administration, leading to extremely high costs. In addition, due to the impermeability of the blood-brain barrier, ERT is less successful in treating the neurologic symptoms of GD. These drawbacks have led to the use of nanotechnology to develop improved delivery of ERT [110,111].
Similarly, Fabry’s disease, which is characterized by a deficiency in α-galactosidase A activity, causing a buildup of globotriaosylceramide (Gb3), can be treated with ERT. Agalsidase beta and agalsidase alfa have been commercially available since 2003, and treated patients show improved outcomes [112,113]. Pegunigalsidase alfa, a novel, PEGylated, chemically modified α-Gal A enzyme with covalently crosslinked monomers, was also recently approved to treat FD [114]. It has a plasma half-life of about 80 hours, compared to about 2 hours for the other two ERTs, and shows reduced immunogenicity [114].
7.2. Substrate Reduction Therapy (SRT)
Another strategy for treating diseases caused by an accumulation of GSLs is substrate reduction therapy (SRT), which involves decreasing the biosynthesis of the accumulated GSL. Miglustat acts as a competitive inhibitor of glucosylceramide synthase, decreasing the synthesis and accumulation of glucosylceramide in GD. However, adverse events such as gastrointestinal disturbance have been reported in some patients [115]. Eligustat is also approved for GD patients and has proven to be equivalent to ERT and less toxic than miglustat, making it the frontline treatment for patients with GD1 [116]. For patients with FD, venglustat and lucerastat (the galactose form of miglustat) are two SRTs that are being tested [117]. SRTs may be preferable to ERTs due to the relative ease of administration (oral vs intravenous), ability of some to cross the BBB, and reduced likelihood of inducing anti-drug antibodies (ADAs) [117].
In addition, glucosylceramide synthase inhibitors (GCSi) have been studied in mice for the treatment of cardiac hypertrophy. As mentioned above, D-PDMP, an inhibitor of glucosylceramide synthase and lactosylceramide synthase activity, decreased GSL load and inhibited cardiac hypertrophy in a dose-dependent manner [13,14]. Similarly, Baccam et al. (2022) demonstrated that GZ667161, a small molecule GCSi, protects mice against isoproterenol- and chronic kidney disease-induced cardiac dysfunction in mice [118]. Other GCSi’s, like Genz-123346, have been studied for their efficacy in arresting the growth of tumors, in which glucosylceramide synthase is commonly overexpressed [119]. These studies, in combination with the proven safety and efficacy of GCSi’s in treating lysosomal storage disorders through SRT, suggest that a similar strategy can be utilized to treat cardiac hypertrophy.
7.3. Chaperone Mediated Therapy (CMT) or Pharmacological Chaperone Therapy (PCT)
Chaperone mediated therapy (CMT) uses pharmacologically active molecules to recover enzyme activity by correcting misfolding and enhancing the enzyme’s stability. In Gaucher’s disease, misfolded GCase is degraded in the ER, leading to decreased enzymatic activity. Several chaperones are used to treat GD, including ambroxol and N-Octyl-b-valienamine [120]. Similarly, for Fabry’s disease, migalastat is used to bind and stabilize mutant α-galactosidase A, increasing its activity and improving lysosomal trafficking [121]. These treatments result in reduced GSL load, leading to improved health outcomes. However, because CMT is mutation-specific, results can vary from patient to patient, illustrating the need for personalized medicine approaches in these therapies [120].
8. Conclusion and Future Directions
Our comprehension of glycosphingolipids (GSLs) within cardiovascular contexts is presently constricted, mainly deriving from explorations that emphasize GSLs’ roles in cellular signaling and structural support. The molecular complexity of GSLs has historically impeded detailed studies, a challenge further exacerbated by limitations in the analytical techniques available. Recent technological advancements in glycan analysis, such as MALDI-TOF MS and ESI-MS, have significantly enhanced our capacity to identify and quantify GSLs, facilitating more comprehensive investigations into their diverse roles in health and disease. While emerging data offer some insights into heart failure, extensive mechanistic studies are essential, considering much of the current literature predominantly addresses correlations without establishing causality. As research progresses, the integration of GSL metabolic pathways into the broader framework of cardiovascular disease management promises not only to refine therapeutic strategies but also to improve outcomes for patients with complex cardiovascular conditions. This continued exploration is poised to unveil pivotal insights into the systemic impacts of GSLs, potentially driving the development of personalized medicine approaches that tailor treatment efficacy based on specific GSL profiles.
Author Contributions
Writing – original draft, SH and SM; writing – review & editing, SH and SM; writing – review, KA; conceptualization, SM; supervision, SM; funding acquisition, SM. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the American Heart Association grant 938718 to SM.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors would like to thank Dr. Tiffany Draper for reading and formatting the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest. Additionally, the funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Jin, X. and Yang, G. Y. (2023) ‘Pathophysiological roles and applications of glycosphingolipids in the diagnosis and treatment of cancer diseases’, Prog Lipid Res, 91, pp. 101241. [CrossRef]
- D’Angelo, G., Capasso, S., Sticco, L. and Russo, D. (2013) ‘Glycosphingolipids: synthesis and functions’, FEBS J, 280(24), pp. 6338-53. [CrossRef]
- Varki, A., Cummings, R. D., Esko, J. D., Freeze, H. H., Stanley, P., Bertozzi, C. R., Hart, G. W. and Etzler, M. E. (2009) ‘Essentials of Glycobiology.
- Liang, Y. J. (2022) ‘Glycosphingolipids in human embryonic stem cells and breast cancer stem cells, and potential cancer therapy strategies based on their structures and functions’, Glycoconj J, 39(2), pp. 177-195. [CrossRef]
- Mishra, S. and Chatterjee, S. (2014) ‘Lactosylceramide promotes hypertrophy through ROS generation and activation of ERK1/2 in cardiomyocytes’, Glycobiology, 24(6), pp. 518-31. [CrossRef]
- Ando, H. and Komura, N. (2024) ‘Recent progress in the synthesis of glycosphingolipids’, Curr Opin Chem Biol, 78, pp. 102423. [CrossRef]
- Prokazova, N. V. and Bergelson, L. D. (1994) ‘Gangliosides and atherosclerosis’, Lipids, 29(1), pp. 1-5. [CrossRef]
- Wang, S. H., Wu, T. J., Lee, C. W. and Yu, J. (2020) ‘Dissecting the conformation of glycans and their interactions with proteins’, J Biomed Sci, 27(1), pp. 93. [CrossRef]
- Wang, X. Q., Sun, P. and Paller, A. S. (2003) ‘Ganglioside GM3 blocks the activation of epidermal growth factor receptor induced by integrin at specific tyrosine sites’, J Biol Chem, 278(49), pp. 48770-8. [CrossRef]
- Regina Todeschini, A. and Hakomori, S. I. (2008) ‘Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains’, Biochim Biophys Acta, 1780(3), pp. 421-33. [CrossRef]
- Cumin, C., Huang, Y. L., Everest-Dass, A. and Jacob, F. (2021) ‘Deciphering the Importance of Glycosphingolipids on Cellular and Molecular Mechanisms Associated with Epithelial-to-Mesenchymal Transition in Cancer’, Biomolecules, 11(1). [CrossRef]
- Pan, X., Dutta, D., Lu, S. and Bellen, H. J. (2023) ‘Sphingolipids in neurodegenerative diseases’, Front Neurosci, 17, pp. 1137893. [CrossRef]
- Mishra, S., Bedja, D., Amuzie, C., Avolio, A. and Chatterjee, S. (2015a) ‘Prevention of cardiac hypertrophy by the use of a glycosphingolipid synthesis inhibitor in ApoE-/- mice’, Biochem Biophys Res Commun, 465(1), pp. 159-64. [CrossRef]
- Mishra, S., Bedja, D., Amuzie, C., Foss, C. A., Pomper, M. G., Bhattacharya, R., Yarema, K. J. and Chatterjee, S. (2015b) ‘Improved intervention of atherosclerosis and cardiac hypertrophy through biodegradable polymer-encapsulated delivery of glycosphingolipid inhibitor’, Biomaterials, 64, pp. 125-135. [CrossRef]
- Levy, M. and Futerman, A. H. (2010) ‘Mammalian ceramide synthases’, IUBMB Life, 62(5), pp. 347-56. [CrossRef]
- Hernández-Corbacho, M. J., Salama, M. F., Canals, D., Senkal, C. E. and Obeid, L. M. (2017) ‘Sphingolipids in mitochondria’, Biochim Biophys Acta Mol Cell Biol Lipids, 1862(1), pp. 56-68. [CrossRef]
- Michel, C., van Echten-Deckert, G., Rother, J., Sandhoff, K., Wang, E. and Merrill, A. H. (1997) ‘Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide’, J Biol Chem, 272(36), pp. 22432-7. [CrossRef]
- Marchesini, N. and Hannun, Y. A. (2004) ‘Acid and neutral sphingomyelinases: roles and mechanisms of regulation’, Biochem Cell Biol, 82(1), pp. 27-44. [CrossRef]
- Hanada, K., Kumagai, K., Yasuda, S., Miura, Y., Kawano, M., Fukasawa, M. and Nishijima, M. (2003) ‘Molecular machinery for non-vesicular trafficking of ceramide’, Nature, 426(6968), pp. 803-9. [CrossRef]
- Hill, C. H., Cook, G. M., Spratley, S. J., Fawke, S., Graham, S. C. and Deane, J. E. (2018) ‘The mechanism of glycosphingolipid degradation revealed by a GALC-SapA complex structure’, Nat Commun, 9(1), pp. 151. [CrossRef]
- Ryckman, A. E., Brockhausen, I. and Walia, J. S. (2020) ‘Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders’, Int J Mol Sci, 21(18). [CrossRef]
- Varela, A. R., Gonçalves da Silva, A. M., Fedorov, A., Futerman, A. H., Prieto, M. and Silva, L. C. (2013) ‘Effect of glucosylceramide on the biophysical properties of fluid membranes’, Biochim Biophys Acta, 1828(3), pp. 1122-30. [CrossRef]
- Sonnino, S., Mauri, L., Chigorno, V. and Prinetti, A. (2007) ‘Gangliosides as components of lipid membrane domains’, Glycobiology, 17(1), pp. 1R-13R. [CrossRef]
- Horbay, R., Hamraghani, A., Ermini, L., Holcik, S., Beug, S. T. and Yeganeh, B. (2022) ‘Role of Ceramides and Lysosomes in Extracellular Vesicle Biogenesis, Cargo Sorting and Release’, Int J Mol Sci, 23(23). [CrossRef]
- Das, M. and Das, D. K. (2009) ‘Lipid raft in cardiac health and disease’, Curr Cardiol Rev, 5(2), pp. 105-11. [CrossRef]
- Sessa, W. C. (2004). eNOS at a glance. J Cell Sci, 117(Pt 12), 2427 – 2429. [CrossRef]
- Catalán, V., Gómez-Ambrosi, J., Rodríguez, A., Silva, C., Rotellar, F., Gil, M. J.,…Frühbeck, G. (2008). Expression of caveolin-1 in human adipose tissue is upregulated in obesity and obesity-associated type 2 diabetes mellitus and related to inflammation. Clin Endocrinol (Oxf), 68(2), 213-219. [CrossRef]
- Zhao, W., Zhang, Q., Wang, J., Yu, H., Zhen, X., Li, L., Qu, Y., He, Y., Zhang, J., Li, C., Zhang, S., Luo, B., Huang, J. and Gao, Y. (2022) ‘Novel Indel Variation of NPC1 Gene Associates With Risk of Sudden Cardiac Death’, Front Genet, 13, pp. 869859. [CrossRef]
- Annunziata, I., Sano, R. and d’Azzo, A. (2018) ‘Mitochondria-associated ER membranes (MAMs) and lysosomal storage diseases’, Cell Death Dis, 9(3), pp. 328. [CrossRef]
- Sano, R., Annunziata, I., Patterson, A., Moshiach, S., Gomero, E., Opferman, J., Forte, M. and d’Azzo, A. (2009) ‘GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis’, Mol Cell, 36(3), pp. 500-11. [CrossRef]
- Zhang, Y., Yao, J., Zhang, M., Wang, Y. and Shi, X. (2023) ‘Mitochondria-associated endoplasmic reticulum membranes (MAMs): Possible therapeutic targets in heart failure’, Front Cardiovasc Med, 10, pp. 1083935. [CrossRef]
- Higuchi, Y., Miura, T., Kajimoto, T. and Ohta, Y. (2005) ‘Effects of disialoganglioside GD3 on the mitochondrial membrane potential’, FEBS Lett, 579(14), pp. 3009-13. [CrossRef]
- Park, L. K., Garr Barry, V., Hong, J., Heebink, J., Sah, R. and Peterson, L. R. (2022) ‘Links between ceramides and cardiac function’, Curr Opin Lipidol, 33(1), pp. 47-56. [CrossRef]
- Scheffer, D. D. L., Garcia, A. A., Lee, L., Mochly-Rosen, D. and Ferreira, J. C. B. (2022) ‘Mitochondrial Fusion, Fission, and Mitophagy in Cardiac Diseases: Challenges and Therapeutic Opportunities’, Antioxid Redox Signal, 36(13-15), pp. 844-863. [CrossRef]
- Schömel, N., Geisslinger, G. and Wegner, M. S. (2020) ‘Influence of glycosphingolipids on cancer cell energy metabolism’, Prog Lipid Res, 79, pp. 101050. [CrossRef]
- Ciarlo, L., Manganelli, V., Garofalo, T., Matarrese, P., Tinari, A., Misasi, R., Malorni, W. and Sorice, M. (2010) ‘Association of fission proteins with mitochondrial raft-like domains’, Cell Death Differ, 17(6), pp. 1047-58. [CrossRef]
- Dany, M. and Ogretmen, B. (2015) ‘Ceramide induced mitophagy and tumor suppression’, Biochim Biophys Acta, 1853(10 Pt B), pp. 2834-45. [CrossRef]
- Novgorodov, S. A., Riley, C. L., Yu, J., Keffler, J. A., Clarke, C. J., Van Laer, A. O., Baicu, C. F., Zile, M. R. and Gudz, T. I. (2016) ‘Lactosylceramide contributes to mitochondrial dysfunction in diabetes’, J Lipid Res, 57(4), pp. 546-62. [CrossRef]
- He, X. and Schuchman, E. H. (2018) ‘Ceramide and Ischemia/Reperfusion Injury’, J Lipids, 2018, pp. 3646725. [CrossRef]
- Wang, N., Li, J. Y., Zeng, B. and Chen, G. L. (2023) ‘Sphingosine-1-Phosphate Signaling in Cardiovascular Diseases’, Biomolecules, 13(5). [CrossRef]
- Egom, E. E., Bae, J. S., Capel, R., Richards, M., Ke, Y., Pharithi, R. B., Maher, V., Kruzliak, P. and Lei, M. (2016) ‘Effect of sphingosine-1-phosphate on L-type calcium current and Ca(2+) transient in rat ventricular myocytes’, Mol Cell Biochem, 419(1-2), pp. 83-92. [CrossRef]
- Brizuela, L., Rábano, M., Peña, A., Gangoiti, P., Macarulla, J. M., Trueba, M. and Gómez-Muñoz, A. (2006) ‘Sphingosine 1-phosphate: a novel stimulator of aldosterone secretion’, J Lipid Res, 47(6), pp. 1238-49. [CrossRef]
- Mutoh, T., Tokuda, A., Miyadai, T., Hamaguchi, M. and Fujiki, N. (1995) ‘Ganglioside GM1 binds to the Trk protein and regulates receptor function’, Proc Natl Acad Sci U S A, 92(11), pp. 5087-91. [CrossRef]
- Mishra, S. and Kass, D. A. (2021) ‘Cellular and molecular pathobiology of heart failure with preserved ejection fraction’, Nat Rev Cardiol, 18(6), pp. 400-423. [CrossRef]
- Heart Disease Facts (2023). Heart Disease: CDC (Accessed: January 23 2024).
- Balram, A., Thapa, S. and Chatterjee, S. (2022) ‘Glycosphingolipids in Diabetes, Oxidative Stress, and Cardiovascular Disease: Prevention in Experimental Animal Models’, Int J Mol Sci, 23(23). [CrossRef]
- Choi, R. H., Tatum, S. M., Symons, J. D., Summers, S. A. and Holland, W. L. (2021) ‘Ceramides and other sphingolipids as drivers of cardiovascular disease’, Nat Rev Cardiol, 18(10), pp. 701-711. [CrossRef]
- Mukhin, D. N., Chao, F. F., & Kruth, H. S. (1995). Glycosphingolipid accumulation in the aortic wall is another feature of human atherosclerosis. Arterioscler Thromb Vasc Biol, 15(10), 1607-1615. [CrossRef]
- Glaros, E. N., Kim, W. S., Rye, K. A., Shayman, J. A., & Garner, B. (2008). Reduction of plasma glycosphingolipid levels has no impact on atherosclerosis in apolipoprotein E-null mice. J Lipid Res, 49(8), 1677-1681. [CrossRef]
- Borodzicz-Jażdżyk, S., Jażdżyk, P., Łysik, W., Cudnoch-Jȩdrzejewska, A. and Czarzasta, K. (2022) ‘Sphingolipid metabolism and signaling in cardiovascular diseases’, Front Cardiovasc Med, 9, pp. 915961. [CrossRef]
- Piccoli, M., Cirillo, F., Ghiroldi, A., Rota, P., Coviello, S., Tarantino, A.,…Anastasia, L. (2023). Sphingolipids and Atherosclerosis: The Dual Role of Ceramide and Sphingosine-1-Phosphate. Antioxidants (Basel), 12(1). [CrossRef]
- Iwabuchi, K. (2015) ‘Involvement of glycosphingolipid-enriched lipid rafts in inflammatory responses’, Front Biosci (Landmark Ed), 20(2), pp. 325-34. [CrossRef]
- Song, B., Zheng, Y., Chi, H., Zhu, Y., Cui, Z., Chen, L., Chen, G., Gao, B., Du, Y. and Yu, Z. (2023) ‘Revealing the roles of glycosphingolipid metabolism pathway in the development of keloid: a conjoint analysis of single-cell and machine learning’, Front Immunol, 14, pp. 1139775. [CrossRef]
- Park, J. Y., Lee, S. H., Shin, M. J. and Hwang, G. S. (2015) ‘Alteration in metabolic signature and lipid metabolism in patients with angina pectoris and myocardial infarction’, PLoS One, 10(8), pp. e0135228. [CrossRef]
- Lozanski, G., Berthier, F. and Kushner, I. (1997) ‘The sphingomyelin-ceramide pathway participates in cytokine regulation of C-reactive protein and serum amyloid A, but not alpha-fibrinogen’, Biochem J, 328( Pt 1)(Pt 1), pp. 271-5. [CrossRef]
- Yokoyama, N., Hanafusa, K., Hotta, T., Oshima, E., Iwabuchi, K. and Nakayama, H. (2021) ‘Multiplicity of Glycosphingolipid-Enriched Microdomain-Driven Immune Signaling’, Int J Mol Sci, 22(17). [CrossRef]
- Coskun, Ü., Grzybek, M., Drechsel, D. and Simons, K. (2011) ‘Regulation of human EGF receptor by lipids’, Proc Natl Acad Sci U S A, 108(22), pp. 9044-8. [CrossRef]
- Mobarak, E., Håversen, L., Manna, M., Rutberg, M., Levin, M., Perkins, R., Rog, T., Vattulainen, I. and Borén, J. (2018) ‘Glucosylceramide modifies the LPS-induced inflammatory response in macrophages and the orientation of the LPS/TLR4 complex in silico’, Sci Rep, 8(1), pp. 13600. [CrossRef]
- Chiricozzi, E., Di Biase, E., Lunghi, G., Fazzari, M., Loberto, N., Aureli, M., Mauri, L. and Sonnino, S. (2021) ‘Turning the spotlight on the oligosaccharide chain of GM1 ganglioside’, Glycoconj J, 38(1), pp. 101-117. [CrossRef]
- Russo, S. B., Tidhar, R., Futerman, A. H. and Cowart, L. A. (2013) ‘Myristate-derived d16:0 sphingolipids constitute a cardiac sphingolipid pool with distinct synthetic routes and functional properties’, J Biol Chem, 288(19), pp. 13397-409. [CrossRef]
- Sasset, L., Manzo, O. L., Zhang, Y., Marino, A., Rubinelli, L., Riemma, M. A., Chalasani, M. L. S., Dasoveanu, D. C., Roviezzo, F., Jankauskas, S. S., Santulli, G., Bucci, M. R., Lu, T. T. and Di Lorenzo, A. (2023) ‘Nogo-A reduces ceramide de novo biosynthesis to protect from heart failure’, Cardiovasc Res, 119(2), pp. 506-519. [CrossRef]
- Sansbury, B. E., DeMartino, A. M., Xie, Z., Brooks, A. C., Brainard, R. E., Watson, L. J., DeFilippis, A. P., Cummins, T. D., Harbeson, M. A., Brittian, K. R., Prabhu, S. D., Bhatnagar, A., Jones, S. P. and Hill, B. G. (2014) ‘Metabolomic analysis of pressure-overloaded and infarcted mouse hearts’, Circ Heart Fail, 7(4), pp. 634-42. [CrossRef]
- Pellieux, C., Montessuit, C., Papageorgiou, I., Pedrazzini, T. and Lerch, R. (2012) ‘Differential effects of high-fat diet on myocardial lipid metabolism in failing and nonfailing hearts with angiotensin II-mediated cardiac remodeling in mice’, Am J Physiol Heart Circ Physiol, 302(9), pp. H1795-805. [CrossRef]
- Cui, S., Zhang, X., Li, Y., Hu, S., Wu, B., Fang, Z., Gao, J., Li, M., Wu, H., Tao, B., Xia, H. and Xu, L. (2023) ‘UGCG modulates heart hypertrophy through B4GalT5-mediated mitochondrial oxidative stress and the ERK signaling pathway’, Cell Mol Biol Lett, 28(1), pp. 71. [CrossRef]
- Andersson, L., Cinato, M., Mardani, I., Miljanovic, A., Arif, M., Koh, A., Lindbom, M., Laudette, M., Bollano, E., Omerovic, E., Klevstig, M., Henricsson, M., Fogelstrand, P., Swärd, K., Ekstrand, M., Levin, M., Wikström, J., Doran, S., Hyötyläinen, T., Sinisalu, L., Orešič, M., Tivesten, Å., Adiels, M., Bergo, M. O., Proia, R., Mardinoglu, A., Jeppsson, A., Borén, J. and Levin, M. C. (2021) ‘Glucosylceramide synthase deficiency in the heart compromises β1-adrenergic receptor trafficking’, Eur Heart J, 42(43), pp. 4481-4492. [CrossRef]
- Brady, E. M., Cao, T. H., Moss, A. J., Athithan, L., Ayton, S. L., Redman, E., Argyridou, S., Graham-Brown, M. P. M., Maxwell, C. B., Jones, D. J. L., Ng, L., Yates, T., Davies, M. J., McCann, G. P. and Gulsin, G. S. (2024) ‘Circulating sphingolipids and relationship to cardiac remodelling before and following a low-energy diet in asymptomatic Type 2 Diabetes’, BMC Cardiovasc Disord, 24(1), pp. 25. [CrossRef]
- Li, J., Kemp, B. A., Howell, N. L., Massey, J., Mińczuk, K., Huang, Q., Chordia, M. D., Roy, R. J., Patrie, J. T., Davogustto, G. E., Kramer, C. M., Epstein, F. H., Carey, R. M., Taegtmeyer, H., Keller, S. R. and Kundu, B. K. (2019) ‘Metabolic Changes in Spontaneously Hypertensive Rat Hearts Precede Cardiac Dysfunction and Left Ventricular Hypertrophy’, J Am Heart Assoc, 8(4), pp. e010926. [CrossRef]
- Mikhalkova, D., Holman, S. R., Jiang, H., Saghir, M., Novak, E., Coggan, A. R., O’Connor, R., Bashir, A., Jamal, A., Ory, D. S., Schaffer, J. E., Eagon, J. C. and Peterson, L. R. (2018) ‘Bariatric Surgery-Induced Cardiac and Lipidomic Changes in Obesity-Related Heart Failure with Preserved Ejection Fraction’, Obesity (Silver Spring), 26(2), pp. 284-290. [CrossRef]
- Stratford, S., Hoehn, K. L., Liu, F. and Summers, S. A. (2004) ‘Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B’, J Biol Chem, 279(35), pp. 36608-15. [CrossRef]
- Chen, C. L., Lin, C. F., Chang, W. T., Huang, W. C., Teng, C. F. and Lin, Y. S. (2008) ‘Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway’, Blood, 111(8), pp. 4365-74. [CrossRef]
- Smith, S. J., Cases, S., Jensen, D. R., Chen, H. C., Sande, E., Tow, B., Sanan, D. A., Raber, J., Eckel, R. H. and Farese, R. V. (2000) ‘Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat’, Nat Genet, 25(1), pp. 87-90. [CrossRef]
- Liu, L., Trent, C. M., Fang, X., Son, N. H., Jiang, H., Blaner, W. S., Hu, Y., Yin, Y. X., Farese, R. V., Homma, S., Turnbull, A. V., Eriksson, J. W., Hu, S. L., Ginsberg, H. N., Huang, L. S. and Goldberg, I. J. (2014) ‘Cardiomyocyte-specific loss of diacylglycerol acyltransferase 1 (DGAT1) reproduces the abnormalities in lipids found in severe heart failure’, J Biol Chem, 289(43), pp. 29881-91. [CrossRef]
- Chavez, J. A., Siddique, M. M., Wang, S. T., Ching, J., Shayman, J. A. and Summers, S. A. (2014) ‘Ceramides and glucosylceramides are independent antagonists of insulin signaling’, J Biol Chem, 289(2), pp. 723-34. [CrossRef]
- Lenders, M. and Brand, E. (2021) ‘Fabry Disease: The Current Treatment Landscape’, Drugs, 81(6), pp. 635-645. [CrossRef]
- Bernardes, T. P., Foresto, R. D. and Kirsztajn, G. M. (2020) ‘Fabry disease: genetics, pathology, and treatment’, Rev Assoc Med Bras (1992), 66Suppl 1(Suppl 1), pp. s10-s16. [CrossRef]
- Germain, D. P. (2010) ‘Fabry disease’, Orphanet J Rare Dis, 5, pp. 30.
- Kovilakath, A. and Cowart, L. A. (2020) ‘Sphingolipid Mediators of Myocardial Pathology’, J Lipid Atheroscler, 9(1), pp. 23-49. [CrossRef]
- Machann, W., Breunig, F., Weidemann, F., Sandstede, J., Hahn, D., Köstler, H., Neubauer, S., Wanner, C. and Beer, M. (2011) ‘Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A’, Eur J Heart Fail, 13(3), pp. 278-83. [CrossRef]
- Liebau, M. C., Braun, F., Höpker, K., Weitbrecht, C., Bartels, V., Müller, R. U., Brodesser, S., Saleem, M. A., Benzing, T., Schermer, B., Cybulla, M. and Kurschat, C. E. (2013) ‘Dysregulated autophagy contributes to podocyte damage in Fabry’s disease’, PLoS One, 8(5), pp. e63506. [CrossRef]
- Birket, M. J., Raibaud, S., Lettieri, M., Adamson, A. D., Letang, V., Cervello, P., Redon, N., Ret, G., Viale, S., Wang, B., Biton, B., Guillemot, J. C., Mikol, V., Leonard, J. P., Hanley, N. A., Orsini, C. and Itier, J. M. (2019) ‘A Human Stem Cell Model of Fabry Disease Implicates LIMP-2 Accumulation in Cardiomyocyte Pathology’, Stem Cell Reports, 13(2), pp. 380-393. [CrossRef]
- De Francesco, P. N., Mucci, J. M., Ceci, R., Fossati, C. A. and Rozenfeld, P. A. (2011) ‘Higher apoptotic state in Fabry disease peripheral blood mononuclear cells.: effect of globotriaosylceramide’, Mol Genet Metab, 104(3), pp. 319-24. [CrossRef]
- De Francesco, P. N., Mucci, J. M., Ceci, R., Fossati, C. A. and Rozenfeld, P. A. (2013) ‘Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide’, Mol Genet Metab, 109(1), pp. 93-9. [CrossRef]
- Yogasundaram, H., Nikhanj, A., Putko, B. N., Boutin, M., Jain-Ghai, S., Khan, A., Auray-Blais, C., West, M. L. and Oudit, G. Y. (2018) ‘Elevated Inflammatory Plasma Biomarkers in Patients With Fabry Disease: A Critical Link to Heart Failure With Preserved Ejection Fraction’, J Am Heart Assoc, 7(21), pp. e009098. [CrossRef]
- Morales, L. E. (1996) ‘Gaucher’s disease: a review’, Ann Pharmacother, 30(4), pp. 381-8.
- Ivanova, M. (2020) ‘Altered Sphingolipids Metabolism Damaged Mitochondrial Functions: Lessons Learned From Gaucher and Fabry Diseases’, J Clin Med, 9(4). [CrossRef]
- Gözdaşoğlu, S. (2015) ‘Gaucher Disease and Gaucher Cells’, Turk J Haematol, 32(2), pp. 187-8. [CrossRef]
- Kurolap, A., Del Toro, M., Spiegel, R., Gutstein, A., Shafir, G., Cohen, I. J., Barrabés, J. A. and Feldman, H. B. (2019) ‘Gaucher disease type 3c: New patients with unique presentations and review of the literature’, Mol Genet Metab, 127(2), pp. 138-146. [CrossRef]
- Solanich, X., Claver, E., Carreras, F., Giraldo, P., Vidaller, A., Aguilar, R. and Cequier, A. (2012) ‘Myocardial infiltration in Gaucher’s disease detected by cardiac MRI’, Int J Cardiol, 155(1), pp. e5-6. [CrossRef]
- Vanier, M. T. (2013) ‘Niemann-Pick diseases’, Handb Clin Neurol, 113, pp. 1717-21. [CrossRef]
- Zervas, M., Somers, K. L., Thrall, M. A. and Walkley, S. U. (2001) ‘Critical role for glycosphingolipids in Niemann-Pick disease type C’, Curr Biol, 11(16), pp. 1283-7. [CrossRef]
- te Vruchte, D., Lloyd-Evans, E., Veldman, R. J., Neville, D. C., Dwek, R. A., Platt, F. M., van Blitterswijk, W. J. and Sillence, D. J. (2004) ‘Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport’, J Biol Chem, 279(25), pp. 26167-75. [CrossRef]
- Welch, C. L., Sun, Y., Arey, B. J., Lemaitre, V., Sharma, N., Ishibashi, M., Sayers, S., Li, R., Gorelik, A., Pleskac, N., Collins-Fletcher, K., Yasuda, Y., Bromme, D., D’Armiento, J. M., Ogletree, M. L. and Tall, A. R. (2007) ‘Spontaneous atherothrombosis and medial degradation in Apoe-/-, Npc1-/- mice’, Circulation, 116(21), pp. 2444-52. [CrossRef]
- Feng, B., Zhang, D., Kuriakose, G., Devlin, C. M., Kockx, M. and Tabas, I. (2003) ‘Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis’, Proc Natl Acad Sci U S A, 100(18), pp. 10423-8. [CrossRef]
- Afzali, M., Nakhaee, A., Tabatabaei, S. P., Tirgar-Fakheri, K. and Hashemi, M. (2013) ‘Aberrant promoter methylation profile of Niemann-pick type C1 gene in cardiovascular disease’, Iran Biomed J, 17(2), pp. 77-83. [CrossRef]
- Ma, W., Xu, J., Wang, Q., Xin, Y., Zhang, L., Zheng, X., Wang, H., Sun, K., Hui, R. and Huang, X. (2010) ‘Interaction of functional NPC1 gene polymorphism with smoking on coronary heart disease’, BMC Med Genet, 11, pp. 149. [CrossRef]
- Knapp, M., Lisowska, A., Knapp, P. and Baranowski, M. (2013) ‘Dose-dependent effect of aspirin on the level of sphingolipids in human blood’, Adv Med Sci, 58(2), pp. 274-81. [CrossRef]
- Spijkers, L. J., van den Akker, R. F., Janssen, B. J., Debets, J. J., De Mey, J. G., Stroes, E. S., van den Born, B. J., Wijesinghe, D. S., Chalfant, C. E., MacAleese, L., Eijkel, G. B., Heeren, R. M., Alewijnse, A. E. and Peters, S. L. (2011) ‘Hypertension is associated with marked alterations in sphingolipid biology: a potential role for ceramide’, PLoS One, 6(7), pp. e21817. [CrossRef]
- Daidone, M., Casuccio, A., Puleo, M. G., Del Cuore, A., Pacinella, G., Di Chiara, T., Di Raimondo, D., Immordino, P. and Tuttolomondo, A. (2024) ‘Mediterranean diet effects on vascular health and serum levels of adipokines and ceramides’, PLoS One, 19(5), pp. e0300844. [CrossRef]
- Mantovani, A. and Dugo, C. (2020) ‘Ceramides and risk of major adverse cardiovascular events: A meta-analysis of longitudinal studies’, J Clin Lipidol, 14(2), pp. 176-185. [CrossRef]
- Wang, D. D., Toledo, E., Hruby, A., Rosner, B. A., Willett, W. C., Sun, Q., Razquin, C., Zheng, Y., Ruiz-Canela, M., Guasch-Ferré, M., Corella, D., Gómez-Gracia, E., Fiol, M., Estruch, R., Ros, E., Lapetra, J., Fito, M., Aros, F., Serra-Majem, L., Lee, C. H., Clish, C. B., Liang, L., Salas-Salvadó, J., Martínez-González, M. A. and Hu, F. B. (2017) ‘Plasma Ceramides, Mediterranean Diet, and Incident Cardiovascular Disease in the PREDIMED Trial (Prevención con Dieta Mediterránea)’, Circulation, 135(21), pp. 2028-2040. [CrossRef]
- Yin, W., Li, F., Tan, X., Wang, H., Jiang, W., Wang, X., Li, S., Zhang, Y., Han, Q., Wang, Y. and Du, J. (2021) ‘Plasma Ceramides and Cardiovascular Events in Hypertensive Patients at High Cardiovascular Risk’, Am J Hypertens, 34(11), pp. 1209-1216. [CrossRef]
- Hilvo, M., Wallentin, L., Ghukasyan Lakic, T., Held, C., Kauhanen, D., Jylhä, A., Lindbäck, J., Siegbahn, A., Granger, C. B., Koenig, W., Stewart, R. A. H., White, H., Laaksonen, R. and Investigators, S. (2020) ‘Prediction of Residual Risk by Ceramide-Phospholipid Score in Patients With Stable Coronary Heart Disease on Optimal Medical Therapy’, J Am Heart Assoc, 9(10), pp. e015258. [CrossRef]
- Poss, A. M., Maschek, J. A., Cox, J. E., Hauner, B. J., Hopkins, P. N., Hunt, S. C., Holland, W. L., Summers, S. A. and Playdon, M. C. (2020) ‘Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease’, J Clin Invest, 130(3), pp. 1363-1376. [CrossRef]
- Saleem, M., Bandaru, V. V., Herrmann, N., Swardfager, W., Mielke, M. M., Oh, P. I., Shammi, P., Kiss, A., Haughey, N. J., Rovinski, R. and Lanctôt, K. L. (2013) ‘Ceramides predict verbal memory performance in coronary artery disease patients undertaking exercise: a prospective cohort pilot study’, BMC Geriatr, 13, pp. 135. [CrossRef]
- Pan, W., Yu, J., Shi, R., Yan, L., Yang, T., Li, Y., Zhang, Z., Yu, G., Bai, Y., Schuchman, E. H., He, X. and Zhang, G. (2014) ‘Elevation of ceramide and activation of secretory acid sphingomyelinase in patients with acute coronary syndromes’, Coron Artery Dis, 25(3), pp. 230-5. [CrossRef]
- Wittenbecher, C., Eichelmann, F., Toledo, E., Guasch-Ferré, M., Ruiz-Canela, M., Li, J., Arós, F., Lee, C. H., Liang, L., Salas-Salvadó, J., Clish, C. B., Schulze, M. B., Martínez-González, M. and Hu, F. B. (2021) ‘Lipid Profiles and Heart Failure Risk: Results From Two Prospective Studies’, Circ Res, 128(3), pp. 309-320. [CrossRef]
- Titievsky, L., Schuster, T., Wang, R., Younus, M., Palladino, A., Quazi, K., Wajnrajch, M. P., Hernandez, B., Becker, P. S., Weinreb, N. J., Chambers, C., Mansfield, R., Taylor, L., Tseng, L. J. and Kaplan, P. (2022) ‘Safety and effectiveness of taliglucerase alfa in patients with Gaucher disease: an interim analysis of real-world data from a multinational drug registry (TALIAS)’, Orphanet J Rare Dis, 17(1), pp. 145. [CrossRef]
- Weinreb, N. J., Camelo, J. S., Charrow, J., McClain, M. R., Mistry, P., Belmatoug, N. and investigators, I. C. G. G. I. G. R. N. (2021) ‘Gaucher disease type 1 patients from the ICGG Gaucher Registry sustain initial clinical improvements during twenty years of imiglucerase treatment’, Mol Genet Metab, 132(2), pp. 100-111. [CrossRef]
- Zimran, A., Durán, G., Giraldo, P., Rosenbaum, H., Giona, F., Petakov, M., Terreros Muñoz, E., Solorio-Meza, S. E., Cooper, P. A., Varughese, S., Alon, S. and Chertkoff, R. (2019) ‘Long-term efficacy and safety results of taliglucerase alfa through 5years in adult treatment-naïve patients with Gaucher disease’, Blood Cells Mol Dis, 78, pp. 14-21. [CrossRef]
- Chauhan, K., Olivares-Medina, C. N., Villagrana-Escareño, M. V., Juárez-Moreno, K., Cadena-Nava, R. D., Rodríguez-Hernández, A. G. and Vazquez-Duhalt, R. (2022) ‘Targeted Enzymatic VLP-Nanoreactors with β-Glucocerebrosidase Activity as Potential Enzyme Replacement Therapy for Gaucher’s Disease’, ChemMedChem, 17(19), pp. e202200384. [CrossRef]
- Martín-Banderas, L., Holgado, M. A., Durán-Lobato, M., Infante, J. J., Álvarez-Fuentes, J. and Fernández-Arévalo, M. (2016) ‘Role of Nanotechnology for Enzyme Replacement Therapy in Lysosomal Diseases. A Focus on Gaucher’s Disease’, Curr Med Chem, 23(9), pp. 929-52. [CrossRef]
- Banikazemi, M., Bultas, J., Waldek, S., Wilcox, W. R., Whitley, C. B., McDonald, M., Finkel, R., Packman, S., Bichet, D. G., Warnock, D. G., Desnick, R. J. and Group, F. D. C. T. S. (2007) ‘Agalsidase-beta therapy for advanced Fabry disease: a randomized trial’, Ann Intern Med, 146(2), pp. 77-86. [CrossRef]
- West, M., Nicholls, K., Mehta, A., Clarke, J. T., Steiner, R., Beck, M., Barshop, B. A., Rhead, W., Mensah, R., Ries, M. and Schiffmann, R. (2009) ‘Agalsidase alfa and kidney dysfunction in Fabry disease’, J Am Soc Nephrol, 20(5), pp. 1132-9. [CrossRef]
- Linhart, A., Dostálová, G., Nicholls, K., West, M. L., Tøndel, C., Jovanovic, A., Giraldo, P., Vujkovac, B., Geberhiwot, T., Brill-Almon, E., Alon, S., Chertkoff, R., Rocco, R. and Hughes, D. (2023) ‘Safety and efficacy of pegunigalsidase alfa in patients with Fabry disease who were previously treated with agalsidase alfa: results from BRIDGE, a phase 3 open-label study’, Orphanet J Rare Dis, 18(1), pp. 332. [CrossRef]
- Kuter, D. J., Mehta, A., Hollak, C. E., Giraldo, P., Hughes, D., Belmatoug, N., Brand, M., Muller, A., Schaaf, B., Giorgino, R. and Zimran, A. (2013) ‘Miglustat therapy in type 1 Gaucher disease: clinical and safety outcomes in a multicenter retrospective cohort study’, Blood Cells Mol Dis, 51(2), pp. 116-24. [CrossRef]
- Bennett, L. L. and Turcotte, K. (2015) ‘Eliglustat tartrate for the treatment of adults with type 1 Gaucher disease’, Drug Des Devel Ther, 9, pp. 4639-47. [CrossRef]
- van der Veen, S. J., Hollak, C. E. M., van Kuilenburg, A. B. P. and Langeveld, M. (2020) ‘Developments in the treatment of Fabry disease’, J Inherit Metab Dis, 43(5), pp. 908-921. [CrossRef]
- Baccam, G. C., Xie, J., Jin, X., Park, H., Wang, B., Husson, H., Ibraghimov-Beskrovnaya, O. and Huang, C. L. (2022) ‘Glucosylceramide synthase inhibition protects against cardiac hypertrophy in chronic kidney disease’, Sci Rep, 12(1), pp. 9340. [CrossRef]
- Jennemann, R., Volz, M., Bestvater, F., Schmidt, C., Richter, K., Kaden, S., Müthing, J., Gröne, H. J. and Sandhoff, R. (2021) ‘Blockade of Glycosphingolipid Synthesis Inhibits Cell Cycle and Spheroid Growth of Colon Cancer Cells In Vitro and Experimental Colon Cancer Incidence In Vivo’, Int J Mol Sci, 22(19). [CrossRef]
- Ivanova, M. M., Dao, J., Kasaci, N., Adewale, B., Nazari, S., Noll, L., Fikry, J., Sanati, A. H. and Goker-Alpan, O. (2021) ‘Cellular and biochemical response to chaperone versus substrate reduction therapies in neuropathic Gaucher disease’, PLoS One, 16(10), pp. e0247211. [CrossRef]
- Yam, G. H., Bosshard, N., Zuber, C., Steinmann, B. and Roth, J. (2006) ‘Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants’, Am J Physiol Cell Physiol, 290(4), pp. C1076-82. [CrossRef]
Figure 1.
Biosynthesis and catabolism of glycosphingolipids.
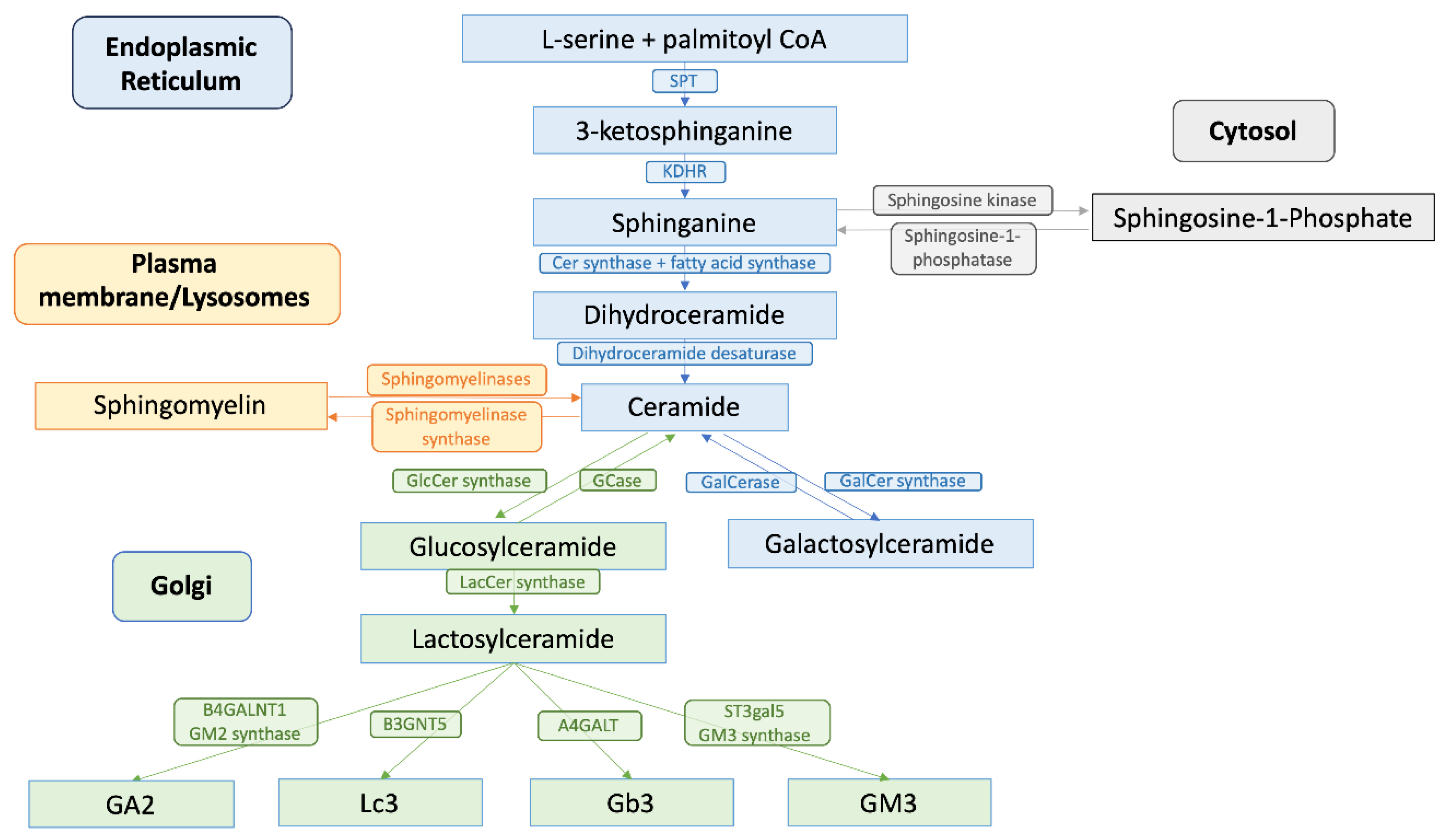
Figure 2.
Dysregulation of GSLs at MAMs leads to abnormal transfer of Ca2+ between the ER and the mitochondria. β-gal = beta-galactosidase, IP3R-1 = inositol 1,4,5-trisphosphate receptor type 1, grp75 = glucose-regulated protein 75, VDAC-1 = voltage-dependent anion-selective channel 1.
Figure 2.
Dysregulation of GSLs at MAMs leads to abnormal transfer of Ca2+ between the ER and the mitochondria. β-gal = beta-galactosidase, IP3R-1 = inositol 1,4,5-trisphosphate receptor type 1, grp75 = glucose-regulated protein 75, VDAC-1 = voltage-dependent anion-selective channel 1.
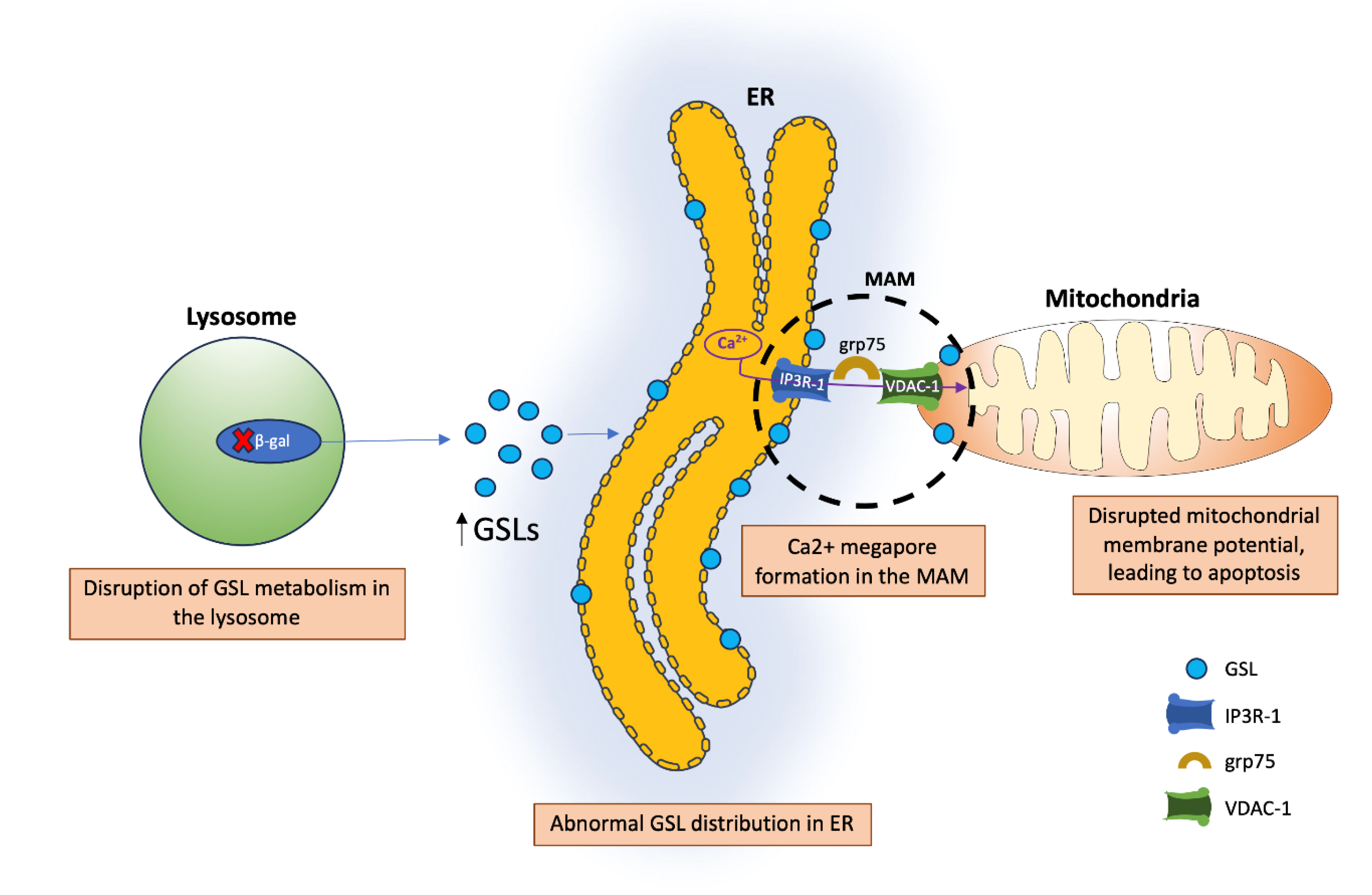
Figure 3.
Effects of GSLs on mitochondrial processes. ROS = reactive oxygen species, OXPHOS = ox-idative phosphorylation, mTOR = mechanistic target of rapamycin, Apaf-1 = apoptotic protease activating factor 1.
Figure 3.
Effects of GSLs on mitochondrial processes. ROS = reactive oxygen species, OXPHOS = ox-idative phosphorylation, mTOR = mechanistic target of rapamycin, Apaf-1 = apoptotic protease activating factor 1.
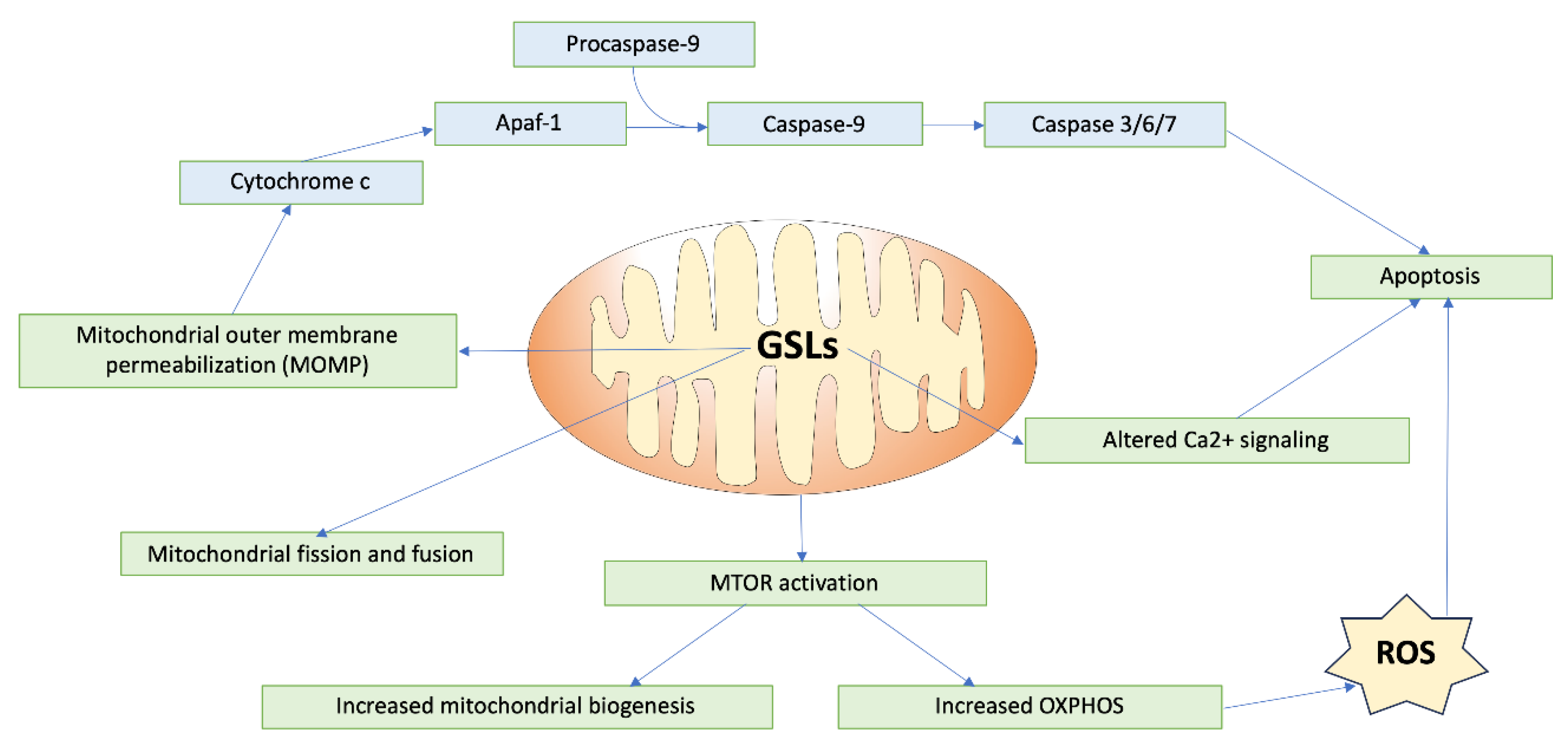
Figure 4.
GSL involved in atherosclerosis, leading to heart failure. HFpEF = heart failure with preserved ejection fraction, HFrEF = heart failure with reserved ejection fraction, LDL = low-density lipoprotein.
Figure 4.
GSL involved in atherosclerosis, leading to heart failure. HFpEF = heart failure with preserved ejection fraction, HFrEF = heart failure with reserved ejection fraction, LDL = low-density lipoprotein.
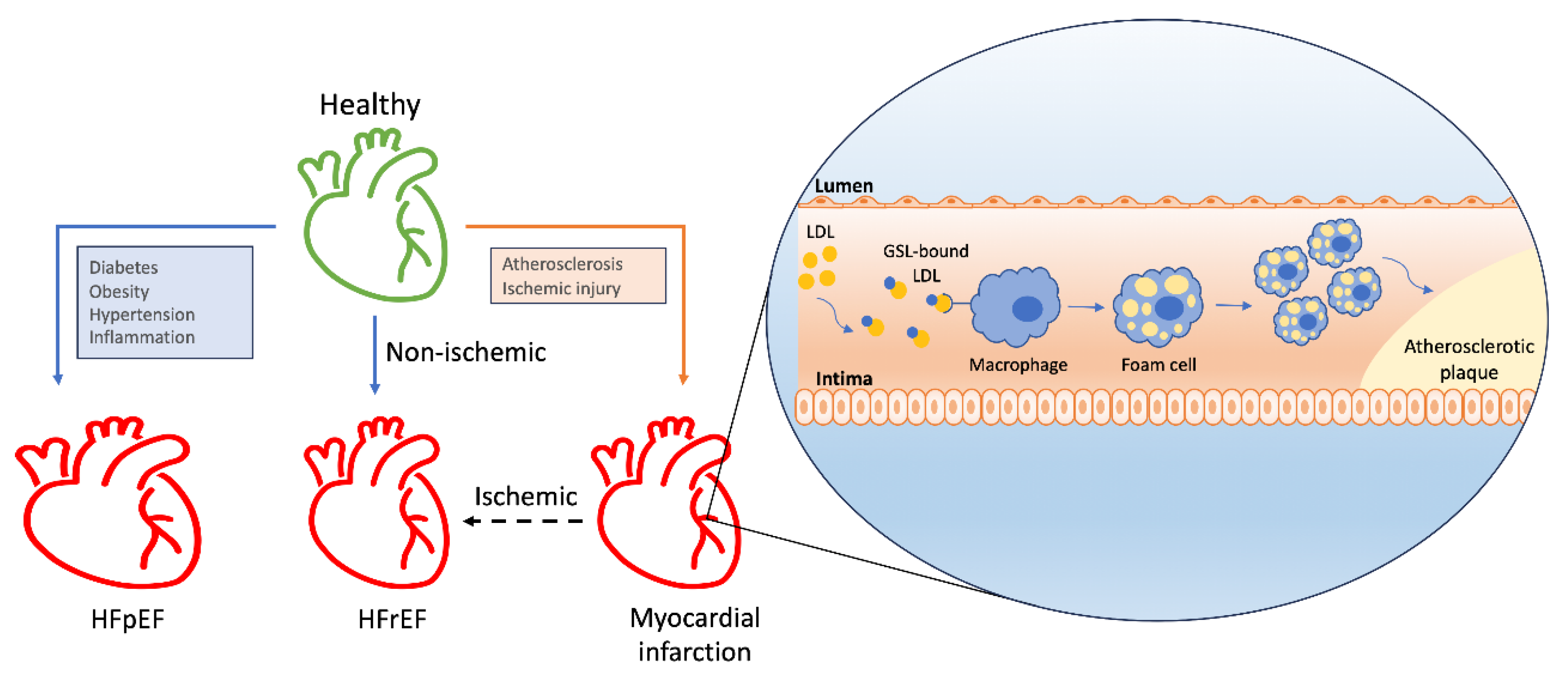
Figure 5.
LacCer activates NADPH oxidase to generate reactive oxygen species, leading to protein kinase-C activation, p44-mitogen activated protein kinase phosphorylation, and nuclear factor c-fos and c-jun expression, leading to cardiac hypertrophy. The gangliosides GD3 and GM1 interact with mitochondrial elements to induce caspase activation and set of a cascade of events leading to apoptosis. LacCer = lactosylceramide, NADPH = nicotinamide adenine dinucleotide phosphate, ROS = reactive oxygen species, PKC = protein kinase C, MEK = mitogen-activated protein kinase kinase (aka MAP2K), p42/44 MAPK = mitogen-activated protein kinases (aka Erk2 and Erk1), TFs = transcription factors, Apaf-1 = apoptotic protease activating factor 1, HFpEF = heart failure with preserved ejection fraction, HFrEF = heart failure with reduced ejection fraction.
Figure 5.
LacCer activates NADPH oxidase to generate reactive oxygen species, leading to protein kinase-C activation, p44-mitogen activated protein kinase phosphorylation, and nuclear factor c-fos and c-jun expression, leading to cardiac hypertrophy. The gangliosides GD3 and GM1 interact with mitochondrial elements to induce caspase activation and set of a cascade of events leading to apoptosis. LacCer = lactosylceramide, NADPH = nicotinamide adenine dinucleotide phosphate, ROS = reactive oxygen species, PKC = protein kinase C, MEK = mitogen-activated protein kinase kinase (aka MAP2K), p42/44 MAPK = mitogen-activated protein kinases (aka Erk2 and Erk1), TFs = transcription factors, Apaf-1 = apoptotic protease activating factor 1, HFpEF = heart failure with preserved ejection fraction, HFrEF = heart failure with reduced ejection fraction.
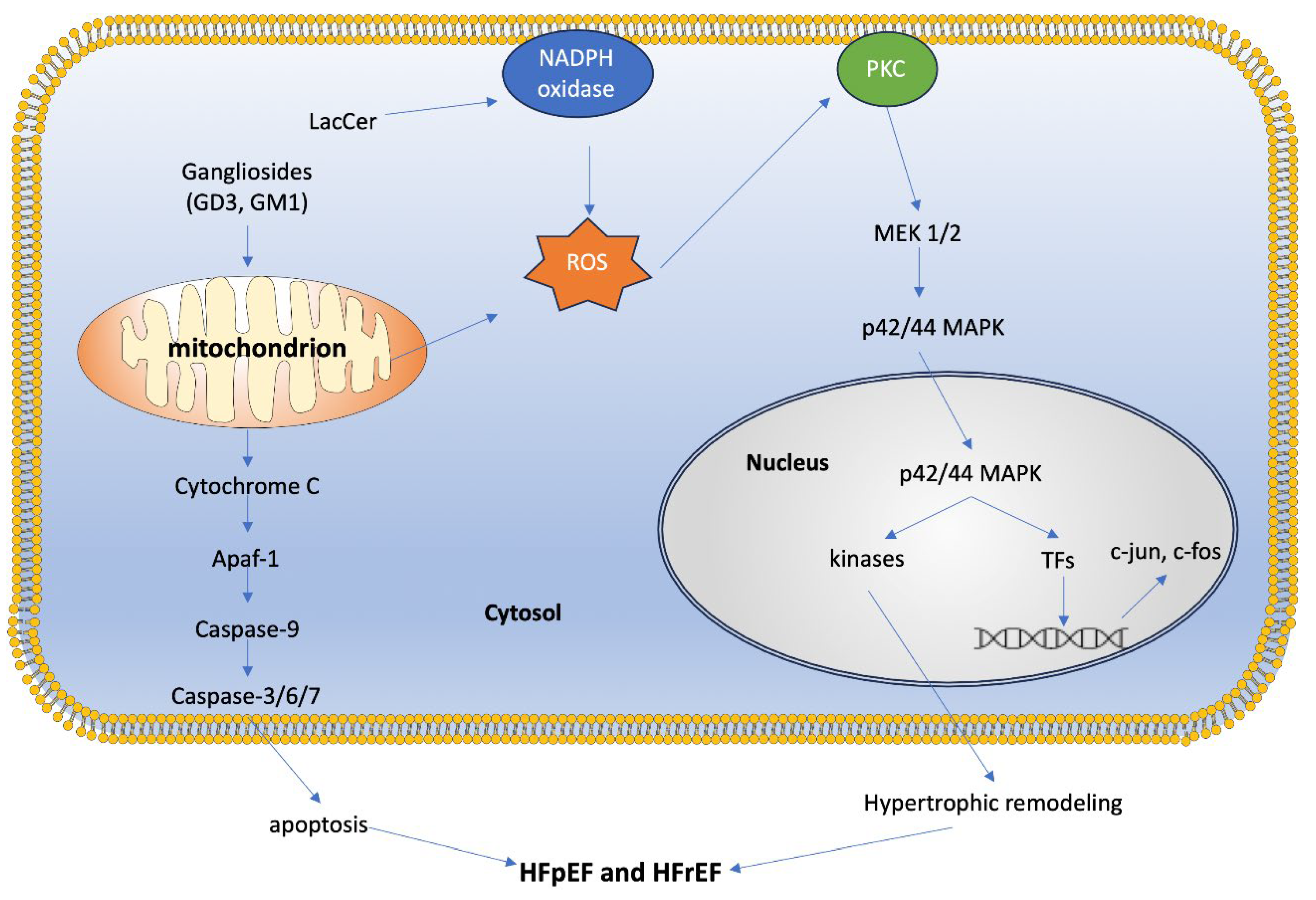

Table 1.
GSL classifications and core structures.
| Series | Abbreviation | Core Structure |
|---|---|---|
| Ganglio- | Gg | Galb3GalNAcb4Galb4Glc- |
| Globo- | Gb | GalNAcb3Gala4Galb4Glc- |
| Isoglobo- | iGB | GalNAcb3Gala3Galb4Glc- |
| Lacto- | Lc | Galb3GlcNAcb3Galb4Glc- |
| Neolacto- | nLc | Galb4GlcNAcb3Galb4Glc- |
| Mollu- | Mu | GlcNAcb2Mana3Manb4Glc- |
| Arthro- | At | GalNAcb4GlcNAcb3Manb4Glc- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



