Submitted:
06 September 2024
Posted:
09 September 2024
You are already at the latest version
Abstract
Malignant lymphomas, encompassing Hodgkin lymphomas (HL) and non-Hodgkin lymphomas (NHL), represent a significant category of hematological malignancies with diverse presentations and prognoses. This review synthesizes current understanding of malignant lymphomas, focusing on their classification, incidence, epidemiology, staging, histopathology, and evolving treatment strategies. Malignant lymphomas are broadly classified into HL and NHL, with NHL further subdivided into B-cell, T-cell, and NK-cell neoplasms. The emergence of grey zone lymphomas, which exhibit overlapping features and heightened aggressiveness, underscores the complexity of diagnosis and treatment. Epidemiologically, HL predominantly affects younger individuals, while NHL incidence increases with age, reflecting a global rise in both incidence and mortality rates. Advances in diagnostic techniques and treatments have led to improved outcomes, yet challenges persist, particularly in distinguishing between overlapping lymphoma types and managing increasing incidence rates. The management of HL and NHL involves a variety of treatment options tailored to the disease stage and individual patient characteristics. After diagnosis and staging, treatment regimens may include radiotherapy, chemotherapy (either alone or in combination), and immunotherapy. Monitoring patient response is crucial, with adjustments made in cases of therapeutic failure. Achieving remission—either partial or complete—depends on the normalization of biomarkers and imaging results, reduction of lymphoid organ size, and resolution of symptoms. Despite therapeutic advances, treatment complications, including short-term effects like myelosuppression and long-term issues such as cardiomyopathy and secondary cancers, remain significant. The future of HL and NHL treatment lies in refining these approaches, minimizing adverse effects, and enhancing efficacy through innovative therapies and personalized medicine strategies.
Keywords:
Malignant Lymphomas
; Hodgkin Lymphomas (HL)
; Non-Hodgkin Lymphomas (NHL)
; B-cell Neoplasms
; T-cell Neoplasms
; NK-cell Neoplasms
; Grey Zone Lymphomas
; Epidemiology
; Classification
; Staging
; Histopathology
; Radiotherapy
; Chemotherapy
; Immunotherapy
; Treatment Strategies
; Remission
; Treatment Complications
; Personalized Medicine
; Innovative Therapies
Introduction
Definition of Pathology and General Information
In the view of the authors of the work entitled “Clinical Hematology” (Berceanu et al., 1977), the concept of “pathology of the cellular immune system” does not represent a terminological or semiotic preference (etymological) of the researchers, but rather a synthesized expression encompassing the various diseases and conditions within this broad field that can affect human organisms. Moreover, this notion reflects the cumulative advancements, discoveries, and expanded knowledge regarding this complex cellular system. By focusing on the pathology known as “malignant lymphomas,” it can be asserted that this category includes the totality of primary nodal tumors, which are also referred to in the older specialized literature as lymphoproliferative or immunoproliferative diseases (Berceanu et al., 1977; Moraru, 1984; Oltean et al., 2009).
At the level of lymph nodes—secondary lymphoid organs situated either solitarily or in groups along lymphatic vessels—pathological changes may arise due to accumulations of substances (pigments, proteins, lipids) or regressive modifications such as atrophy. These changes are collectively referred to as metabolic lesions, and all structures may contribute to specific lymph node inflammations or lymphadenitis (e.g., sarcoidosis, toxoplasmosis, banal purulent lymphadenitis, dermatopathic lymphadenitis, the lymphadenopathy characteristic of AIDS, tuberculosis, pseudotuberculous lymphadenitis, reticular-abscessing lymphadenitis, angioimmunoblastic lymphadenopathy, syphilitic lymphadenitis, or rare forms such as “sinus histiocytosis with lymphadenopathy” and pseudo-sarcoidosis lesions) or non-specific lymphadenitis, which can lead to lesions such as lymphoid hyperplasia, reticulohistiocytic reactions, or nodal inflammations involving monocytes and granulocytes. Additionally, over the course of life, both benign and malignant tumors may develop (Moraru, 1984; Tasca, 1994).
An analysis of the nomenclature employed by specialists over time highlights the establishment of the term “malignant lymphomas,” which arose from the clear necessity of distinguishing neoplastic proliferations from the broader category of lymphoproliferative diseases, which also included benign proliferations of inflammatory origin, reactive to various antigenic stimuli. Malignant tumors (malignant lymphomas and, very rarely, angiosarcomas), characterized by rapid growth—evidenced by the increased frequency of mitoses, loss of growth control, and cellular disruption—are differentiated by their rate of proliferation. The majority of these neoplasms originate in the nodal parenchyma, and occasionally from histiocytes. Malignant lymphoproliferations exhibit considerable variability in cellular cycle dynamics, resulting in low mitotic indices (Moraru, 1984; Tasca, 1994, pp. 70-76, 240-247; Păun et al., 1997).
Classification
Malignant lymphomas, a type of malignant neoplasms, are divided into two main categories: Hodgkin lymphomas (HL) and non-Hodgkin lymphomas (NHL). According to the latest classifications issued by the World Health Organization (WHO) in 2017, there are over 80 types of mature lymphoid neoplasms. HL accounts for approximately 30-40% of all malignant lymphomas, while NHL occur in 60-70% of cases (Mihăescu et al., 2006; Oltean et al., 2009; De Leval & Jaffe, 2020).
These classifications are based on factors such as immunophenotype, morphology, genetic lesion types, molecular characteristics, cell types, and clinical manifestations. WHO members favor a division of malignant lymphoid neoplasms into three categories: HL, B-cell neoplasms, and T-cell or NK-cell lymphomas.
- Classification of NHL
In the classification process of NHL, several advanced techniques are employed, such as polymerase chain reaction (PCR), gene expression profiling using microarray analysis, human genome decoding, and procedures utilizing monoclonal antibodies with cellular phenotyping. These methods enable the identification of the proliferating lymphocyte population and the stage of differentiation and maturation. Additional tools include immunohistochemical tests and molecular genetics approaches. A primary classification of NHL can be made based on fundamental cellular structural elements: T-cell lymphomas (accounting for 10% of cases in Europe and the United States, and 20% in Asia), either helper or suppressor (rare), B-cell lymphomas (80-85%) , and NK-cell (natural killer) lymphomas, or lymphomas with large granular lymphocytes (De Leval & Jaffe, 2020; Oltean et al., 2009).
According to WHO, NHL can involve proliferations originating from two types of cells: precursor cells or mature cells, with the latter being classified as primarily extranodal, predominantly nodal, or predominantly disseminated. Based on the cellular proliferation patterns, NHL is categorized into peripheral lymphomas (1), central lymphomas (2), and lymphomas composed of “transformed” cells (3). Young lymphocytes (2) proliferate in central lymphoid organs, such as the thymus or bone marrow, while small antigen-stimulated lymphocytes undergo morpho-functional transformations leading to the appearance of effector cells involved in immune responses (3). Mature lymphocytes proliferate in peripheral lymphoid organs (1), including lymph nodes, Peyer’s patches, and the spleen (Jaffe et al., 2001; Oltean et al., 2009).
WHO classifies B-cell NHL as follows (WHO, 2017; De Leval & Jaffe, 2020; Swerdlow et al., 2017):
- ➢
- Predominantly disseminated
- ➢
- Primary extranodal
- ➢
- Primary nodal
Within the classification of the second type of T-cell or NK-cell neoplasms, the following types are included (WHO, 2017; De Leval & Jaffe, 2020):
- ➢
- Predominantly disseminated
- ➢
- Primary extranodal
- ➢
- Primary cutaneous
- ➢
- Predominantly nodal
- ●
- Classification of HL
WHO recognizes two major categories of HL: classic HL (CHL), which accounts for 95% of cases, and nodular lymphocyte-predominant HL (NLPHL), representing 5%.
CHL is characterized by the immunophenotypes CD15+, CD30+, CD20-/+, PAX5+, BCL6-, MUM1+, BOB1-, OCT2-, EBV+ (40%) and is subdivided into four histological types:
- ➢
- Mixed cellularity CHL (15-30%)
- ➢
- Lymphocyte depletion CHL (<1%)
- ➢
- Nodular sclerosis CHL (60-80%)
- ➢
- Lymphocyte-rich CHL (5%)
According to Bosch-Schips J., Granai M., Quintanilla-Martinez L., and Fend F., this classification is based on the analysis of tissue architecture, tumor cell density, frequency of Epstein-Barr virus (EBV) association, clinical progression, and epidemiology.
In contrast, the malignant cells in NLPHL have the following immunophenotypes: CD15-, CD30-, PAX5+, BCL6+, MUM1+, CD20+, BOB1+, EBV-, OCT2+, IgD+ (25%). NLPHL can be further classified into:
- ➢
- Nodular classical B-cell-rich lymphomas
- ➢
- Interconnected lymphomas
- ➢
- Lymphomas with prominent extranodular cells
(Jaffe et al., 2001; Oltean et al., 2009; Swerdlow et al., 2017; De Leval & Jaffe, 2020; Bosch-Schips et al., 2022)
- Classification of Grey Zone Lymphomas (GZL)
Malignant neoplasms situated at the intersection between the characteristics of CHL and diffuse large B-cell NHL (DLBCL) are extremely rare and fall under the spectrum termed “grey zone lymphomas” (GZL). These lymphomas exhibit heightened aggressiveness, leading to a reduced survival rate for diagnosed patients. Most cases strongly express CD20, CD30, PAX5, and MUM1, though in rare instances, they may lose CD20 expression (El-Sawalhy et al., 2021; Bosch-Schips et al., 2022).
The origin of HL in B cells explains the overlap between the characteristics of cHL and NHL/DLBCL or NLPHL, leading to the emergence of these intermediate lymphomas. The presence of different cell populations in the background, immunophenotypic aberrations, an increased number of Reed-Sternberg (RS) and Hodgkin cells, and cellular monotony contribute to the difficulty of establishing a clear diagnosis. According to Bosch-Schips et al., in the article “The Grey Zones of Classic Hodgkin Lymphoma”, the term “grey zone lymphomas” is primarily recommended for mediastinal neoplasms, while in other body regions, efforts are made to classify them into one of the major lymphoma categories, with additional characteristics specified (De Leval & Jaffe, 2020; Bosch-Schips et al., 2022).
Within this intersection, the following classes of grey zone lymphomas are recognized:
- ➢
- Unclassifiable B-cell lymphomas with intermediate features between cHL and DLBCL, both mediastinal and non-mediastinal.
- ➢
- Nodular lymphocyte-predominant lymphomas with unusual phenotypes.
- ➢
- Lymphomas resembling cHL, with T-cell and/or cytotoxic markers expression.
- ➢
- Epstein-Barr virus (EBV)-associated B-cell lymphoproliferative disorders with Hodgkin-like features, such as mucocutaneous ulcers, EBV-positive diffuse large B-cell lymphomas.
- ➢
- Mimics of unrelated entities, including chronic lymphocytic leukemia (CLL) and other indolent non-Hodgkin B-cell lymphomas with RS-like cells, anaplastic large cell lymphomas, post-transplant or iatrogenic lymphoproliferative disorders with cHL-like morphology, and T-cell lymphomas, including angioimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma (PTCL), NOS, with RS-like cells.
(Bosch-Schips et al., 2022)
Incidence and Epidemiology
The incidence of malignant lymphomas has shown an annual increase of 3-4% over the past four decades. In recent years, these values have stabilized globally, with the lowest incidence rates recorded in East Asia. The variability in frequency percentages and the lower accuracy over a prolonged period are attributed to factors such as the emergence of AIDS epidemics, aging populations, increased life expectancy, and prolonged exposure to carcinogens (Huh, 2012).
The incidence of NHL increases with advancing age, reaching its peak among individuals over the age of 45-50 years. In contrast, HL are more frequently diagnosed in younger individuals, with peak incidence rates occurring between the ages of 18 and 25 years, and again in adults aged 40-50 or 60 years. These findings are supported by research published in the course titled “Theoretical Hematological Foundations in Clinical Laboratory”. However, more recent studies, as noted in “Internal Medicine Course”, indicate that both HL and NHL predominantly affect males and boys, with 85% of HL patients being male (Oltean, 1996; Mihăescu et al., 2006; Oltean et al., 2009; De Leval & Jaffe, 2020).
HL have shown relatively stable population incidence rates over the past 20 years, with an incidence of 2-5 (~2.8) cases per 100,000 individuals in Europe and North America. In recent years, the global incidence of HL has declined by approximately 6.1% (Fitzmaurice & Naghavi, 2017).
Advancements in technology, numerous studies, and scientific progress in therapeutic approaches have contributed to a decrease in mortality from values exceeding 2 cases per 100,000 individuals to approximately 0.4-0.5 cases. Mortality and incidence rates exhibit an inverse proportional relationship (Oltean et al., 2009; Salati et al., 2014; Mihăilă, 2016).
HL exhibits a bimodal age distribution, with incidence peaks occurring during adolescence or up to the age of 30, and again in individuals over 50 years old. This bimodal distribution reflects distinct histological subtypes. Recent advances in molecular, cytological, genetic, cytogenetic, and immunohistological techniques have led to the conclusion that nodular sclerosis lymphomas are now diagnosed twice as frequently as mixed cellularity lymphomas among younger patients. However, the second peak, observed in older individuals, still shows a predominance of mixed cellularity subtype, while the nodular sclerosis subtype maintains a plateau in percentage or numerical terms. In young children aged 1 to 4 years, the nodular sclerosis subtype is the most commonly observed histological type. There is also a notable correlation between histological subtype and the level of economic and developmental status of a country. In less developed countries, lymphomas with lymphocytic depletion or mixed cellularity are more prevalent, whereas in developed nations, the nodular sclerosis subtype is more common. Although HL affects both genders, it predominantly occurs in males, with incidence rates approximately 1.5 to 2 times higher in men. A significant disparity is observed in boys during childhood, who constitute 85% of all male HL cases (Oltean et al., 2009; Dănaila & Dăscalescu, 2011).
According to Mihăilă R., 2016, HL is more common among individuals of higher intellectual status, only children, or those from higher socioeconomic backgrounds.
NHL are known for their high heterogeneity, with variations in prognosis, immunophenotyping, histology, and clinical classification. The probability of NHL increases with age, showing a higher incidence in elderly individuals. In contrast, HL displays an inverse relationship between age and incidence, with higher rates occurring in children and young adults, and a decrease in older age, particularly in developing countries (Mihăescu et al., 2006; Oltean et al., 2009; Dănaila & Dăscalescu, 2011).
NHL are more common in men, with a frequency exceeding 50% (Baeker Bispo, Pinheiro, & Kobetz, 2020). They also have a higher prevalence in the Caucasian population, which shows a greater vulnerability to these malignant lymphoproliferations compared to other racial groups. HL is predominantly observed in individuals of European descent, while cases among Asians are relatively rare (Oltean et al., 2009).
In contrast to HL, NHL have experienced an increase in both incidence and mortality rates globally, with an approximate annual rise of 4% since 1950 (Mihăilă, 2016). The recent upward trend, with 19 cases per 100,000 individuals reported in 2015 compared to 11.2 cases per 100,000 in earlier years, can be attributed to several factors. These include a greater number of environmental carcinogens, prolonged exposure to these carcinogens, and a higher incidence of immunodeficiency or the use of immunosuppressive treatments among individuals (Mihăescu et al., 2006; Ioniță et al., 2015).
Description and Histological Changes
HL, distinguished by the presence of RS cells in the spleen, lymph nodes, and other organs affected by malignant lymphoproliferations, occur in regions with heightened inflammatory responses. These neoplasms were first identified around 1832 by Thomas Hodgkin, who described a cohort of seven patients exhibiting splenomegaly and lymphadenopathy. Modern biomedical advancements have improved the curability of HL to approximately 80%. Histopathological and immunohistological analysis of affected anatomical structures, such as lymph nodes, reveals the presence of RS cells, Hodgkin cells, eosinophils, T and B lymphocytes, neutrophils, plasma cells, and macrophages.
HL can be defined as neoplasms of the lymphatic tissues originating from the germinal centers of B lymphocyte lines—regions where clonal proliferation occurs in affected individuals. These neoplasms are closely associated with a granulomatous inflammatory reaction (Thomas et al., 2004; Lupu et al., 2007; Oltean et al., 2009; Mihăilă, 2016).
RS cells are marked by various surface antigens including CD30 (a member of the tumor necrosis factor receptor family), CD25 (the receptor for IL-2, IL-5, IL-6, IL-9, IL-13), CD71 (also known as the transferrin receptor), and class II HLA antigens. The typical phenotype of these cells includes CD30, CD15, CD70, CD40, CD25, and KI-27. Markers such as CD15+, CD40+, and CD30/KI-1+ are specific to T cell lineage, while CD19+, CD22, and CD20+ are associated with B cell lineage. The origin of RS cells remains uncertain; although they are generally considered to arise from B lymphocytes, earlier theories proposed their development from granulocytes, reticulocytes, histiocytes, or immature lymphocytes (Poppema & Van Den Berg, 2000; Lupu et al., 2007; Mihăilă, 2016).
Recent research indicates that RS cells originate from pre-apoptotic germinal centers of B lymphocytes. These cells lose their B lymphocyte identity and exhibit numerous signaling pathway activation abnormalities (Thomas et al., 2004; Küppers & Hansmann, 2005).
Building on the broad classification of Hodgkin’s diseases into three categories—sarcoma, granuloma, and paragranuloma (Parker and Jackson, 1947)—more specific histopathological subvariants have been identified in recent decades (Table 1) (Butler & Lukes, 1966; REAL classification, 1994):
- ➢
- Malignant lymphomas with predominant or rich lymphocytic presence (PL)
- ➢
- Malignant lymphomas with lymphocytic depletion (DL)
- ➢
- Malignant lymphomas with mixed cellularity (MC)
- ➢
- Malignant lymphomas with nodular sclerosis (NS)
HL with lymphocytic predominance can be classified into both classic neoplasms (alongside DL, MC, and NS), characterized by the immunophenotype CD15+/CD20-/CD30+, and the nodular HL category, differentiated from the former by the presence of the immunophenotype CD15-/CD20+/CD30. The absence of CD15 and CD30 markers in the RS cells of the lymphocytic predominance histological subtype (PL) was demonstrated in a 1996 study, noting the only difference being the classification of these as either classic lymphomas (with these markers) or nodular lymphomas (negative for these markers) (Kanzler, Hansmann, & Klaus, 1996; Oltean et al., 2009).
Histopathological subtype analysis reveals that the DL variant is rare (<1% of all cases), often involving the bone marrow and retroperitoneal nodes, and is typically discovered at advanced stages (IIIB and IVB) due to the late appearance of symptoms. The PL subtype, found in a small percentage (5-10% of HL cases), is associated with early stages and responds very well to treatment but has the worst prognosis. The NS subtype (60-80%), common in younger individuals and characterized by nodules separated by collagen fibers organized in bands, is usually associated with early-stage discovery (stages I and II), with or without mediastinal involvement but affecting the lateral cervical lymph nodes. The MC subtype (15-30%) is primarily associated with advanced stages (III and IV) and has a poorer prognosis. Statistics show a higher prevalence of the NS and MC histological subtypes among individuals diagnosed with malignant HL (Lupu et al., 2007; Oltean et al., 2009).
For stage B HL, specific symptoms include pruritus (leading to hyperkeratosis and secondary infections), weight loss, fever, and heavy sweating. Clinical manifestations of the neoplasm involve progressive hypertrophy of lymphoid organs such as the spleen or lymph nodes. A specific type of fever known as Pel-Ebstein fever alternates between afebrile and febrile periods lasting one to three weeks, with other forms including irregular fevers, subfebrile states, and continuous fever (Oltean et al., 2009).
Histopathological analysis of “classic” HL lesions in affected lymphoid or extralymphatic organs and lymph nodes shows a shift from normal tissue to one characterized by granulomatous tissue (neutrophils, eosinophils, lymphocytes, plasma cells, fibroblasts, epithelioid cells, histiocytes) involved in inflammation, alongside proliferative clones implicated in malignancy. RS and Hodgkin cells predominate in these areas, undergoing various transformative processes. Specific mechanisms and viral involvement contribute to increased resistance to apoptosis (Thomas et al., 2004). Hodgkin cells proliferate rapidly, featuring small, regularly shaped nuclei with acidophilic nucleoli. In contrast, RS cells (CD15, CD30) have large, often multilobed or regular-shaped nuclei, very large basophilic nucleoli, loose chromatin, and abundant cytoplasm, with diameters ranging from 30 to 50 microns (Oltean et al., 2009).
In the histopathological examination of “nodular lymphocyte-predominant” HL (CD19+/CD20+, CD30-/CD15-), a complete or partial absence of RS cells is typically observed. These forms of malignant proliferations, which account for only 5% of all malignant lymphomas, are identifiable by the presence of “popcorn cells” (lymphohistiocytes). The lesions initially develop in the spleen, subsequently extending to the liver, and exhibit a degree of instability, possessing the capacity to transform and give rise to lymphomas of the lymphocyte-depleted (DL) or mixed-cellularity subtypes (MC) (Oltean et al., 2009).
NHL, defined as tumors of cellular systems (which include T, B, or NK (natural killer) lymphocytes and histiocytes), as well as their precursors within the immune system, occur in lymphoid organs and can be classified into low-, intermediate-, or high-grade malignancies. While low-grade lymphomas offer a favorable prognosis, intermediate-grade lymphomas are associated with a moderate prognosis, and highly malignant lymphoproliferations typically have a reserved to unfavorable outcome. Low-grade NHL include small lymphocytic lymphomas, mixed follicular lymphomas characterized by alternating large and small cleaved cells, and follicular lymphomas with a predominance of small cleaved cells. The spectrum of intermediate-grade NHL comprises three types of diffuse lymphomas—large cell, mixed cell (both small and large), and small cleaved cell (differentiated)—as well as follicular lymphomas with a predominance of large cells. High-grade NHL include large immunoblastic lymphomas with correspondingly large cells, Burkitt lymphoma, malignant lymphoblastic lymphomas, and non-Burkitt lymphomas with unclassified (undifferentiated) cells. (Oltean, 1996; Lupu, 2005; Mihăescu et al., 2006; Lupu et al., 2007)
The cellular components responsible for the formation of NHL, which are known for their high histopathological, clinical, and immunophenotypic heterogeneity, are found in the lymph nodes, the Waldeyer ring, or the spleen. (Oltean, 1996)
Similar to HL, NHL typically have an insidious onset, with patients generally feeling well. The early symptoms—often overlooked or deemed non-threatening—include weight loss, fever (not associated with an inflammatory process), and night sweats. Consequently, the majority of patients (overwhelmingly in the advanced stages III and IV) are diagnosed late, as the lymphocytes lead to a multicentric onset and continuous proliferation. The clinical onset manifests through the appearance of polyadenopathy or adenopathy with deep or superficial localization, often involving the spleen. Deep polyadenopathy is typically located in the mediastinal or subdiaphragmatic regions, affecting retroperitoneal or mesenteric lymph nodes. In about one-third of patients, the lymphoma originates in extraganglionic regions, with initial sites found in the stomach, skin, intestines, bones (often starting in the bone marrow and diagnosed in stage IV through lymphocytic infiltration), pancreas, lungs, thyroid, or the central nervous system. (Mihăescu et al., 2006; Lupu et al., 2007)
The Ann Arbor Staging System
The Ann Arbor staging system is a comprehensive classification method that evaluates the extent of lymphoid malignancies, such as lymphomas and leukemias. These malignancies vary in behavior, from indolent forms (like MALT lymphoma, associated with mucosa-associated lymphoid tissues) to aggressive, rapidly progressing types (such as Burkitt lymphoma). Leukemias are primarily recognized by their occurrence in the bone marrow and bloodstream, while lymphomas refer to malignant formations affecting lymph nodes and lymphoid organs. (Armitage, 2005)
The Ann Arbor staging system includes both histological and clinical classifications, based on observations of anatomical sites and the presence or absence of lymphoma-specific symptoms. Patients are categorized into four stages depending on the location of the affected sites relative to the diaphragm and the potential spread to extranodal sites (denoted by the addition of the letter “E” to the corresponding Roman numeral stage) (Figure 1).
In stage I, the patient presents with involvement of a single lymphatic region (I), or an affected organ or extranodal site (IE). According to the COTSWOLDS anatomical-clinical classification, this stage allows for the involvement of only one lymphoid structure, such as the Waldeyer ring, thymus, or spleen (Dănaila & Dăscalescu, 2011). Additionally, the Ann Arbor staging system in stage I includes an exception to stage IV, which represents the involvement of two adjacent territories (two contiguous lymph nodes) on the same side of the diaphragm.
Stage II involves the dissemination of cancer cells to at least two lymph node regions (II) on the same side of the diaphragm. An exception in this stage includes the involvement of hilar lymph nodes with bilateral distribution. This stage also encompasses cases where an anatomical region of lymph nodes is affected alongside an organ or extranodal site (IIE), with or without the involvement of additional lymph nodes on the same side of the diaphragm. The number of affected lymph node regions is quantified and indicated numerically (e.g., II4 indicates involvement of four lymph node regions).
In stage III, the lymph nodes and corresponding regions are affected on both sides of the diaphragm (III). This stage may include involvement of the spleen (IIIS), other extranodal areas connected to the affected lymph nodes (IIIE), or a combination of these features (IIIE,S). According to COTSWOLDS (1989), stage III1 indicates involvement of portal, celiac, hilar, or splenic lymph nodes, while stage III2 is characterized by involvement of mesenteric, iliac, and para-aortic lymph nodes (Oltean, 1996, pp. 144-145, Table 14.1).
Stage IV is marked by the dissemination of cancer to one or more extranodal tissues and organs, with or without associated lymph node involvement, or by the involvement of an extranodal organ directly related to the site of origin of the initial cancer cell, in the absence of involvement of any adjacent lymph nodes. When liver, pulmonary lymph nodes (multiple pulmonary metastases), or spinal marrow involvement is observed, the lymphoma is classified as stage IV upon biopsy confirmation. In cases where a primary extranodal lesion is involved or there is parenchymal extension between adjacent lymph nodes, the detection of tumorigenic processes or dissemination of cancer clones to the pericardium, pleura, liver, or marrow confirms a diagnosis of stage IV malignant lymphoma, with or without the presence of lymphadenopathy. However, this stage excludes involvement of a nearby adjacent lymph node.
Alongside these stages included in the clinical classification, the presence of form or substages A or B is noted. Substage A is characterized by the absence of symptoms, whereas substage B indicates the presence of symptoms such as weight loss (greater than 10% of body weight over approximately 6 months), significant night sweats, fever exceeding 38°C, and pruritus. Staging, including the determination of disease progression, is achieved through imaging studies, biological, biochemical, or laboratory tests, clinical examinations, anamnesis, biopsies, and, if necessary, other surgical interventions such as splenectomy or minimally invasive endoscopic techniques, including bronchoscopy. (Berceanu et al., 1977; Oltean, 1996; Armitage, 2005; Lupu, 2005; Lupu et al., 2007; Oltean et al., 2009; Dănaila & Dăscalescu, 2011)
A histological staging (HS) further specifies the absence (“-”) or presence (“+”) of lesions in various organs such as the skin (D+/-), liver (H+/-), bone marrow (M+/-), lungs (L+/-), bones (O+/-), lymph nodes (N+/-), or pleura (P+/-). (Berceanu et al., 1977)
In the Cotswolds staging system, an “X” is added when the tumor mass is notably large, exceeding 10 cm in diameter, or if it causes significant mediastinal widening, with the final measurement representing one-third or more of the recorded thoracic diameter. (Dănaila and Dăscalescu, 2011)
Although initially developed for staging HL, the Ann Arbor classification has served as the foundation for the development of the International Prognostic Index (IPI) (Figure 2, Figure 3). This index stratifies patients into risk categories based on their scores: low risk with 0 or 1 point, low-to-intermediate risk with 2 points, intermediate-to-high risk with 3 points, and high risk with a score between 4 and 5 points. The five key prognostic factors, each contributing one point to the total score, include: diagnosis of lymphoma at stage IV or III, age over 60 years, performance status greater than 2, involvement of more than two extranodal sites, and elevated lactate dehydrogenase (LDH) levels. The use of the IPI is recommended for predicting the likelihood of achieving remission, overall survival chances, and the maintenance of remission. (Armitage, 2005; El-Galaly, Gormsen, & Hutchings, 2018)
The performance index is assessed as follows (Oken, Creech, & Davis, 1982; Armitage, 2005):
- ■
- 0 : The individual is fully active and asymptomatic, capable of performing all activities without any limitations.
- ■
- 1: The individual is active but limited to light work, with restrictions from engaging in more physically demanding tasks.
- ■
- 2: The patient, while ambulatory, remains active for more than 50% of waking hours and is capable of performing any desired activities.
- ■
- 3: The patient’s activities are confined to self-care, spending more than 50% of waking hours either in bed or seated.
- ■
- 4: The patient is severely disabled, unable to care for themselves or move independently, requiring constant assistance and being either seated or bedridden.
- ■
- 5: This index denotes the patient’s death.
The comparative analysis of data published by James O. Armitage (“Staging Non-Hodgkin Lymphoma” - Armitage, 2005) reveals a higher likelihood of survival and remission for younger patients diagnosed with malignant lymphomas. For individuals diagnosed with stage IV lymphomas according to the Ann Arbor or COTSWOLD staging systems, data from an international study tracking the progression of 2,000 patients with aggressive NHL shows that while the response rates are similar across age groups (44% for those over 60 and 46% for younger individuals), the remission maintenance rate differs significantly by 18 percentage points (40% for older patients compared to 58% for younger patients). Furthermore, the survival rate supports the initial hypothesis of the study, indicating a greater resilience among younger patients, with a 6% difference between the two groups (26% for patients over 60 years old and 32% for those 60 years old or younger). However, the resilience of younger patients with malignant NHL is influenced by factors such as the origin of the primary site, gene or protein expression, and the presence of a bulky tumor, which may complicate treatment and necessitate the inclusion of radiotherapy alongside other therapeutic measures. Currently, there is no specific classification system for these variables. The determination of the presence or absence of the five risk factors, the disease stage (I, II, III, or IV), and the prognostic index (0, 1, 2, 3, 4, or 5) will play a crucial role in guiding medication choices and clinical recommendations.
Past studies have primarily used 67Ga (gallium-67) scintigraphy for restaging after a minimum of one treatment course. Currently, PET/CT scans utilizing FDG (18F) are the standard for staging procedures due to their increased accuracy. PET/CT or PET scans are preferred over CT scans because they offer higher sensitivity in detecting sites of proliferation, regions of metastasis, or in guiding biopsies in patients with indolent forms who are clinically suspected of having areas with aggressive, rapidly progressing disease (El-Galaly, Gormsen, and Hutchings, 2018).
The use of fluorodeoxyglucose (FDG) in positron emission tomography (PET) is based on the ability of proliferating lymphoma cells or cellular infiltrates associated with inflammatory processes to absorb this radiopharmaceutical, making them FDG-avid. However, there are exceptions; for instance, small lymphocytic lymphoma cells or MALT lymphomas are not considered FDG-avid according to some studies (El-Galaly, Gormsen, and Hutchings, 2018).
Treatment Options, Recommendations, and Complications
In the case of a HL diagnosis, after staging, specific tests, and establishing a prognosis, the patient will be recommended one of the following treatment options, considering their toxicity and the impact on the patient’s body: radiotherapy, chemotherapy, combination chemotherapy, combined chemotherapy and radiotherapy, maintenance treatments or treatments for complications, and immunotherapy. Patient monitoring is conducted to assess the effectiveness of the chosen treatment regimen. The patient may respond positively and achieve partial or complete remission. Additionally, if beneficial effects are not observed (“therapeutic failure”), this will lead to an automatic adjustment of the medication (Berceanu et al., 1977; Ioniță et al., 2015).
Achieving partial remission is determined based on the following conclusions: the patient exhibits abnormal values in certain biological and biochemical tests (fibrinogen, LDH, sedimentation rate) but shows the disappearance of all tumor markers and no new metastases. However, the volume of the affected lymphoid organs is reduced by 50% compared to their initial size. The desired stage at the end of treatment is complete remission, characterized by the normalization of mediastinal lymph nodes, lymphographic images, and biological test results (within normal limits), and, importantly, the reduction of hypertrophied lymphoid organs to their original size. This stage is accompanied by a negative bone biopsy, the inability to palpate the spleen, and lymph nodes measuring less than 1 cm or 1.5 cm (on CT, PET/CT), with the resolution of symptoms, radiological, and clinical signs (Berceanu et al., 1977; Imöhl and Schmidmaier, 2014).
Depending on the prognosis for the corresponding stage, as highlighted in Figure 4, the physician will recommend the following treatment regimens:
- For patients in the first category (IA and IIA): Two to four cycles of ABVD chemotherapy combined with irradiation of the affected areas. Alternatively, patients may receive only six cycles of ABVD or extensive radiotherapy, such as EF (extended field) or wide-field irradiation.
- For individuals in the second category (IIB and IB): A higher number of ABVD cycles is recommended, ranging from six to eight cycles, or four cycles if combined with radiotherapy of the affected area or extended field irradiation.
- In the exceptional case of a mediastinal tumor with a diameter greater than 10 cm or larger than one-third of the chest, the recommended therapeutic approaches include mantle radiotherapy (for tumors located or disseminated above the diaphragm, protecting the heart and lung parenchyma) and six cycles of ABVD.
Radiotherapy, the oldest therapeutic principle applied to HL, is used as a curative approach in most cases for stages IA, IB, IIA, IIB, and sometimes IIIA. Patients in advanced stages (III and IV) will undergo between six and eight cycles of ABVD, which may be increased by two additional cycles if imaging results are unfavorable, with the possibility of introducing the BEACOPP regimen. Due to high risks, these regimens may be combined with radiotherapy or the brief but effective Stanford V regimen may be considered. Stanford V is a first-line treatment for HL, involving intravenous administration of six drugs and one oral medication every other day, followed by radiotherapy if residual sites larger than 5 cm or splenic involvement (as evidenced by CT) are present (Berceanu et al., 1977; Abuzetun et al., 2009; Ioniță et al., 2015).
Megavoltage or Kilovoltage Radiotherapy is applied using wide fields (“extended field irradiation”) that encompass both affected and hypertrophied lymph nodes, as well as adjacent nodes. “Mantle Field” Irradiation is used for cancers located above the diaphragm and targets the axillae, mediastinum, and lateral cervical, para-aortic, and pelvic regions. “Inverted Y” Irradiation is used for cancers affecting subdiaphragmatic areas and focuses on inguinal, iliac, or lombo-aortic nodes. In the case of the first type of irradiation, organs within the irradiated extended areas—such as the kidneys, liver, spinal cord, lungs, and heart—are protected using special lead shields. Mantle field irradiation aims to protect the heart and lungs, while inverted Y irradiation protects the liver and kidneys. The standard doses of radiation vary between 3.5 and 4.4 Gy for affected lymph node regions and between 2.5 and 3 Gy for marginally involved, unaffected territories. These standard doses are adjusted in cases of aggressive lymphoproliferations with significant metastatic processes (Berceanu et al., 1977; Mihăescu et al., 2006; Ioniță et al., 2015).
Alternative medication options include autologous stem cell transplantation combined with polychemotherapy or hematopoietic stem cell transplantation combined with high-dose chemotherapy and growth factors. (Oltean, 1996; Mihăescu et al., 2006)
Chemotherapy is used in stages IIIB, IVB, and IVA. Monochemotherapy involves the use of a single cytotoxic agent in multiple doses or treatment cycles, such as inblastine, natulan, cyclophosphamide, or nitrogen mustard. Remission periods typically range from three to six months, occurring in 71% of patients treated with monochemotherapy. However, complete remission is achieved in only 26% of cases, while 45% of patients experience incomplete remission. (Berceanu et al., 1977)
Currently, combination therapies, which involve the use of at least two cytotoxic agents under the umbrella term polychemotherapy, are preferred for their ability to extend remission periods and enhance the efficacy of HL treatment (Table 2). Polychemotherapy operates by simultaneously targeting multiple biochemical and metabolic pathways, thereby intercepting cellular proliferation. These regimens typically include combinations of nucleic acid synthesis inhibitors such as BCNU, natulan, and CCNU; alkylating agents like nitrogen mustard and cyclophosphamide; corticosteroid hormones; and mitotic spindle poisons, including vinblastine, procarbazine, bleomycin, etoposide, and vincristine. The toxic agents have distinct actions on mitotic spindles: bleomycin causes single-strand DNA breaks, etoposide inhibits DNA synthesis and mitosis, procarbazine depolymerizes DNA, and vincristine inhibits mitosis. Furthermore, lymphocyte suppression is facilitated by bleomycin, etoposide, and vincristine (Imöhl and Schmidmaier, 2014).
Polychemotherapy and monochemotherapy are associated with a range of complications:
-
Late Complications:
- ○
- Infertility
- ○
- Secondary Acute Leukemias
- ○
- Cardiomyopathy induced by the use of Doxorubicin
- ○
- Pulmonary Fibrosis resulting from Bleomycin administration
- ○
- Neuropathies due to the heightened toxicity of Vincristine
-
Short-term Complications:
- ○
- Infections
- ○
- Alopecia
- ○
- Nausea, Vomiting, and General Malaise
- ○
- Myelosuppression
-
Radiotherapy also induces a spectrum of secondary effects:
- ○
- Nausea, Vomiting, Anorexia, Oral Dryness, and Cough
- ○
- Pharyngitis and Dermatitis
- ○
- Hematopoietic Suppression, including Thrombocytopenia, Myelosuppression, and Leukopenia
- ○
- Hepatitis
- ○
- Fever
- ○
- Hypothyroidism
- ○
- Exercise-Induced Dyspnea and Pneumonia
- ○
- Cardiopathy, including Acute Pericarditis and Coronary Artery Disease
- ○
- Nephritis
- ○
- Chronic Enteritis, potentially leading to stenosis
- ○
- Suppression of Genital Organ Functions
- ○
- Cardiac Tamponade
- ○
- Paresthesia of the Extremities caused by uncontrolled electric discharge (Lhermitte’s Syndrome)
- ○
- Secondary Neoplasms affecting the stomach, thyroid, breasts, or lungs
Surgical interventions may involve the resection of lymph node chains in cases where a tumor or refractory adenopathy is present, or the performance of a splenectomy. (Oltean, 1996)
For the treatment of malignant NHL (Table 3), early localized stages are typically managed with surgical interventions and radiotherapy, while stages IIIA, IIIB, IVA, and IVB are addressed with polychemotherapy. Low-risk NHL forms are treated with regimens such as R-COP and CVP, whereas aggressive forms require regimens like BACOP, R-CHOP, m-BACOD, and COMLA. High-grade malignancies are generally managed with the CHOP regimen. Advances in research have led to alternative treatment options, including the use of monoclonal antibodies, stem cell transplantation (both autologous and from circulating pluripotent stem cells), CAR-T cell therapy (particularly effective for large B-cell lymphomas enriched with T cells/histiocytes), and combinations of monoclonal antibodies with radioactive isotopes or cytokines. (Oltean, 1996; Mihăescu et al., 2006)
Prednisone is recognized as a glucocorticoid with anti-inflammatory and immunosuppressive properties, cyclophosphamide is classified as an alkylating agent, and rituximab functions as a monoclonal antibody targeting CD20. (Imöhl and Schmidmaier, 2014)
Conclusions
The study of malignant lymphomas reveals a complex landscape of classification and epidemiology. The division into HL and NHL, along with further subclassifications, reflects ongoing efforts to understand the heterogeneity of these malignancies. HL predominantly affect younger individuals, while NHL are more common in older adults and exhibit significant variability in clinical presentation and prognosis. The increasing incidence of both HL and NHL, particularly in the context of environmental and genetic factors, underscores the importance of ongoing research and advancements in treatment. The emergence of grey zone lymphomas highlights the need for precise diagnostic criteria to guide effective treatment. As scientific and technological advancements continue, the refinement of classification systems and therapeutic approaches will be crucial in improving patient outcomes and addressing the challenges posed by these complex diseases.
HL remains a complex disease with varied histopathological manifestations. The identification of specific RS cell markers and subtype-specific histological features plays a critical role in diagnosing and managing the disease. The nodular sclerosis subtype, with its favorable prognosis, contrasts sharply with the lymphocyte depletion subtype, which is associated with poor outcomes and advanced disease stages. Advances in imaging techniques, such as PET/CT, have further refined staging and treatment planning, highlighting the importance of subtype classification and accurate staging in improving patient outcomes. Future research should continue to explore the molecular mechanisms underlying these subtypes and their implications for targeted therapies.
Treatment of malignant lymphoma encompasses a multifaceted approach, combining traditional therapies with emerging innovations to address varying stages and patient responses. While therapies such as radiotherapy and chemotherapy remain cornerstone strategies, their efficacy is often accompanied by a spectrum of complications, from acute side effects to long-term health concerns. The balance between achieving remission and managing treatment-related toxicity is crucial. Recent advances, including stem cell transplantation and targeted immunotherapies, offer promising alternatives, though their integration into standard practice requires ongoing research and validation. The future of HL and NHL treatment is likely to involve a blend of conventional and novel therapies, aiming for increased specificity and reduced toxicity. Enhanced understanding and technological progress are expected to improve patient outcomes and survival rates, underscoring the importance of continued innovation and personalized treatment strategies.
References
- Abuzetun, J. Y.; et al. (2009) „The Stanford V regimen is effective in patients with good risk Hodgkin lymphoma but radiotherapy is a necessary component”, British Journal of Haematology, 144(4), pp. 531-537. [CrossRef]
- Armitage, J. O. (2005) „Staging in non-Hodgkin’s lymphomas”, CA: A Cancer Journal for Clinicians, 55(6), pp. 368-376. [CrossRef]
- Baeker Bispo, J. A. , Pinheiro, P. S. și Kobetz, E. K. (2020) „Epidemiology and etiology of leukemia and lymphoma”, Cold Spring Harbor Perspectives in Medicine, 10(6). [CrossRef]
- Berceanu, Ștefan et al. (1977) Hematologie clinica. EDITURA MEDICALĂ - BUCUREȘTI.
- Bosch-Schips, J.; et al. (2022) „The Grey Zones of Classic Hodgkin Lymphoma”, Cancers, 14(3). [CrossRef]
- Covic, M. (1976) Lucrări practice de biologie medicală. Iași.
- Covic, M. (1981) Curs de genetică medicală. Institutul de Medicină și Farmacie, Disciplina de Biologie si genetică medicală, Iași.
- Covic, M.; et al. (2017) Genetică Medicală. Editia a I. Iași: Polirom.
- Covic, M. , Stefănescu, D. și Sandovici, I. (2004) Genetică medicală. Editura Polirom.
- El-Galaly, T. C. , Gormsen, L. C. și Hutchings, M. (2018) „PET/CT for Staging; Past, Present, and Future”, Seminars in Nuclear Medicine, 48(1), pp. 4-16. [CrossRef]
- EL-Sawalhy, E.; et al. (2021) „Grey Zone Lymphoma, Diagnostic and Therapeutic Challenges: A Rare Case Report”, American Journal of Medical Case Reports, 9(5), pp. 298-300. [CrossRef]
- Fitzmaurice, C. și Naghavi, M. (2017) „Exclusion of Kaposi sarcoma from analysis of cancer burden - Reply”, JAMA Oncology, 3(10), pp. 1429-1430. [CrossRef]
- Imöhl, M. și Schmidmaier, R. (2014) Compendiu de hematologie. Editura FarmaMedia, Targu-Mures.
- Ioniță, H.; et al. (2015) Hematologie Clinică. Timișoara: Editura Victor Babeș.
- Jaffe, E. S.; et al. (2001) „World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Haematopopoietic and Lymphoid Tissues”, IARC Press:Lyon 2001.
- Kanzler, B. H.; Hansmann, M.; și Klaus, P. (1996) „Hodgkin and Reed-Sternberg Cells in Hodgkin’s Disease Represent the Outgrowth of a Dominant Tumor Clone Derived from (Crippled) Germinal Center B Cells”, J. Exp. Med. - The Rockefeller University Press, 184(October), pp. 1996-1505.
- Küppers, R.; și Hansmann, M. L. (2005) „The Hodgkin and Reed/Sternberg cell”, International Journal of Biochemistry and Cell Biology, 37(3), pp. 511-517. [CrossRef]
- De Leval, L.; și Jaffe, E. S. (2020) „Lymphoma classification”, Cancer Journal (United States), 26(3), pp. 176-185. [CrossRef]
- Lupu, A.-R. (2005) Hematologia in Practica Medicală. București: Editura Universitara „Carol Davila”.
- Lupu, A.-R.; et al. (2007) Patologia hematologica. București: Editura Universitara „Carol Davila”.
- Mihăescu, G. , Chifiriuc, C. și Dițu, L.-M. (2009) Imunobiologie. Editura Universității din București.
- Mihăescu, R.; et al. (2006) Bazele teoretice hematologice in laboratorul clinic - curs. Timișoara: LITO U.M.F. Timișoara.
- Mihăilă, R.-G. (2016) Noțiuni de hematologie clinică. Editura Universității „Lucian Blaga” din Sibiu.
- Ministerul Sanatatii (2021) Ghidul de practica medicala - aprobat prin ORDINUL nr. 219 din 23 februarie 2021, publicat în Monitorul Oficial, Partea I, nr. 189 din 25 februarie 2021. Valabil la: https://legislatie.just.ro/Public/DetaliiDocument/238473.
- Moraru, I. (1984) Imunologie (bazele moleculare ale starii de sanatate si boala). EDITURA MEDICALĂ - BUCUREȘTI.
- Oken, M. M. , Creech, R. H. și Davis, T. E. (1982) „Toxicology and response criteria of the Eastern Cooperative Oncology Group”, American Journal of Clinical Oncology: Cancer Clinical Trials, pp. 649-655. [CrossRef]
- Oltean, G. (1996) Aspecte diagnostice si terapeutice in bolile hematologice. Editura Tipomur.
- Oltean, G.; et al. (2009) Curs de medicina interna - Bolile hematologice. Litografia U.M.F Targu-Mures.
- Păun, R.; et al. (1997) Tratat de MEDICINA INTERNA - hematologie - partea I. EDITURA MEDICALĂ - BUCUREȘTI.
- Poppema, S. și Van Den Berg, A. (2000) „Interaction between host T cells and Reed-Sternberg cells in Hodgkin lymphomas”, Seminars in Cancer Biology, 10(5), pp. 345-350. [CrossRef]
- Salati, M.; et al. (2014) „Epidemiological overview of Hodgkin lymphoma across the Mediterranean basin”, Mediterranean Journal of Hematology and Infectious Diseases, 6(1), pp. 1-10. [CrossRef]
- Swerdlow, S. H.; et al. (2017) „World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues”, IARC Press: Lyon, France, 4th ed.
- Tasca, C. (1994) Curs de morfopatologie. seria Medi. București: Editura All.
- Thomas, R. K.; et al. (2004) „Part I: Hodgkin’s lymphoma - Molecular biology of Hodgkin and Reed-Sternberg cells”, Lancet Oncology, 5(1), pp. 11-18. [CrossRef]
Figure 1.
The Ann Arbor Staging System.
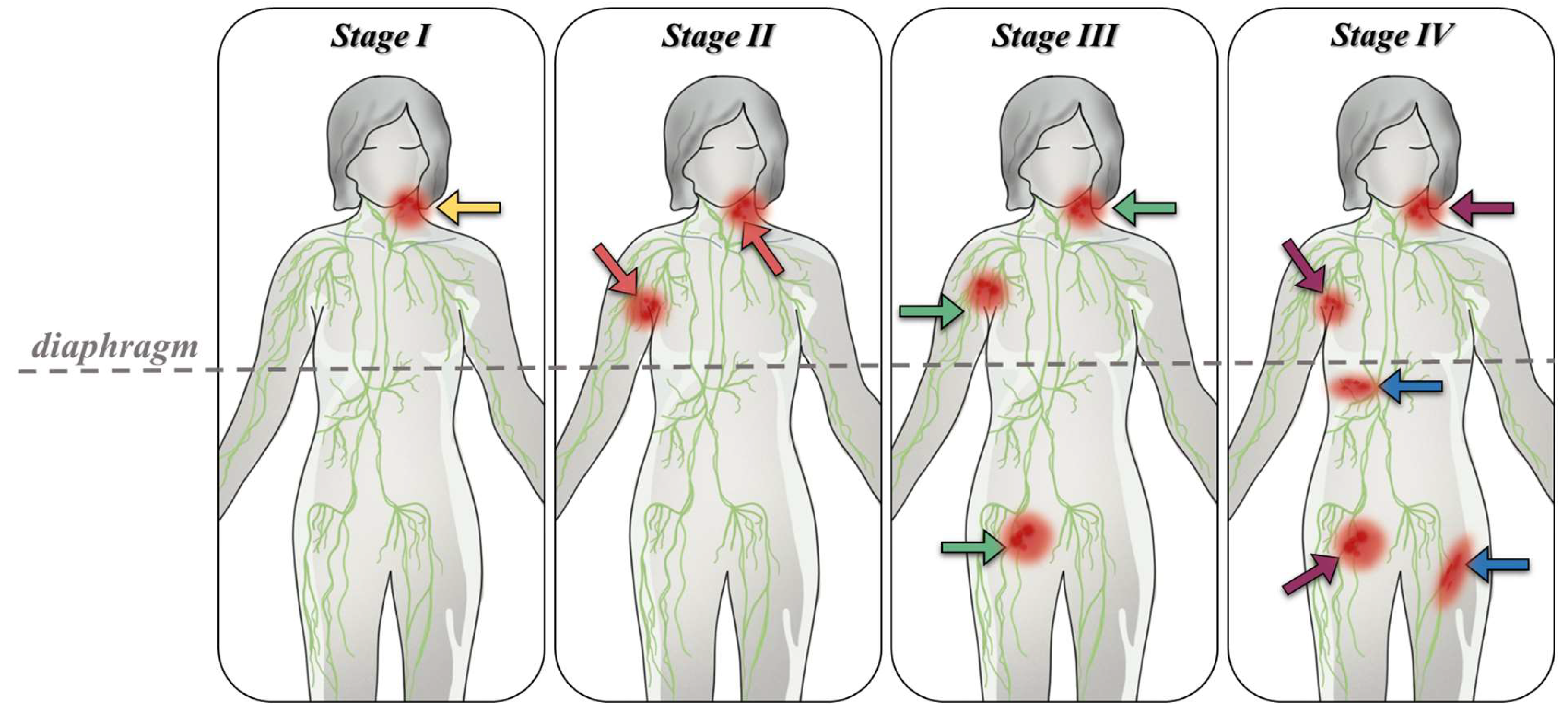
Figure 2.
International Prognostic Index for Individuals Over 60 Years of Age - James O. Armitage, MD, 2005 - According to Table 5, page 373.
Figure 2.
International Prognostic Index for Individuals Over 60 Years of Age - James O. Armitage, MD, 2005 - According to Table 5, page 373.
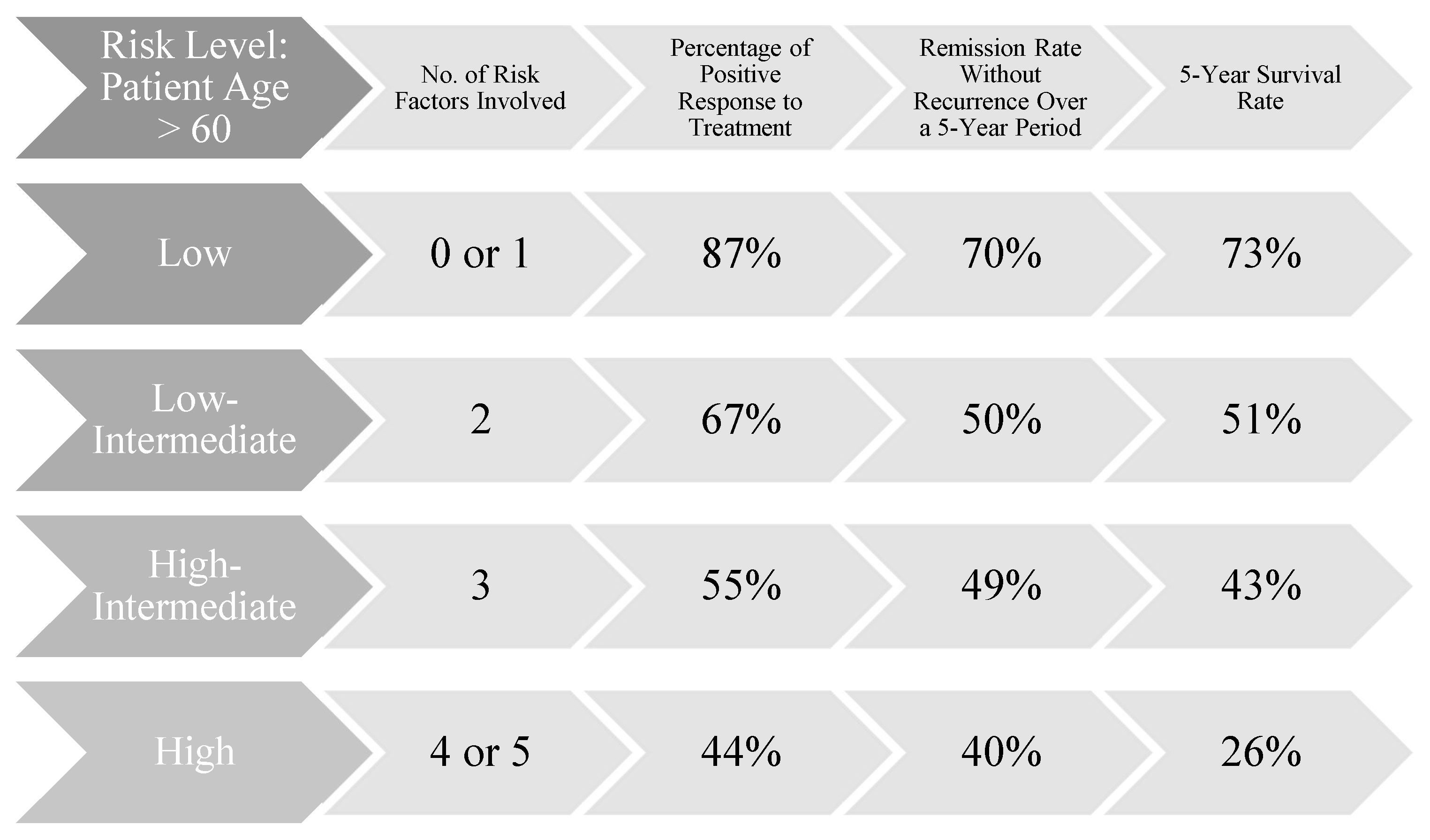
Figure 3.
International Prognostic Index for Individuals Under 60 Years of Age - James O. Armitage, MD, 2005 - According to Table 5, page 373.
Figure 3.
International Prognostic Index for Individuals Under 60 Years of Age - James O. Armitage, MD, 2005 - According to Table 5, page 373.
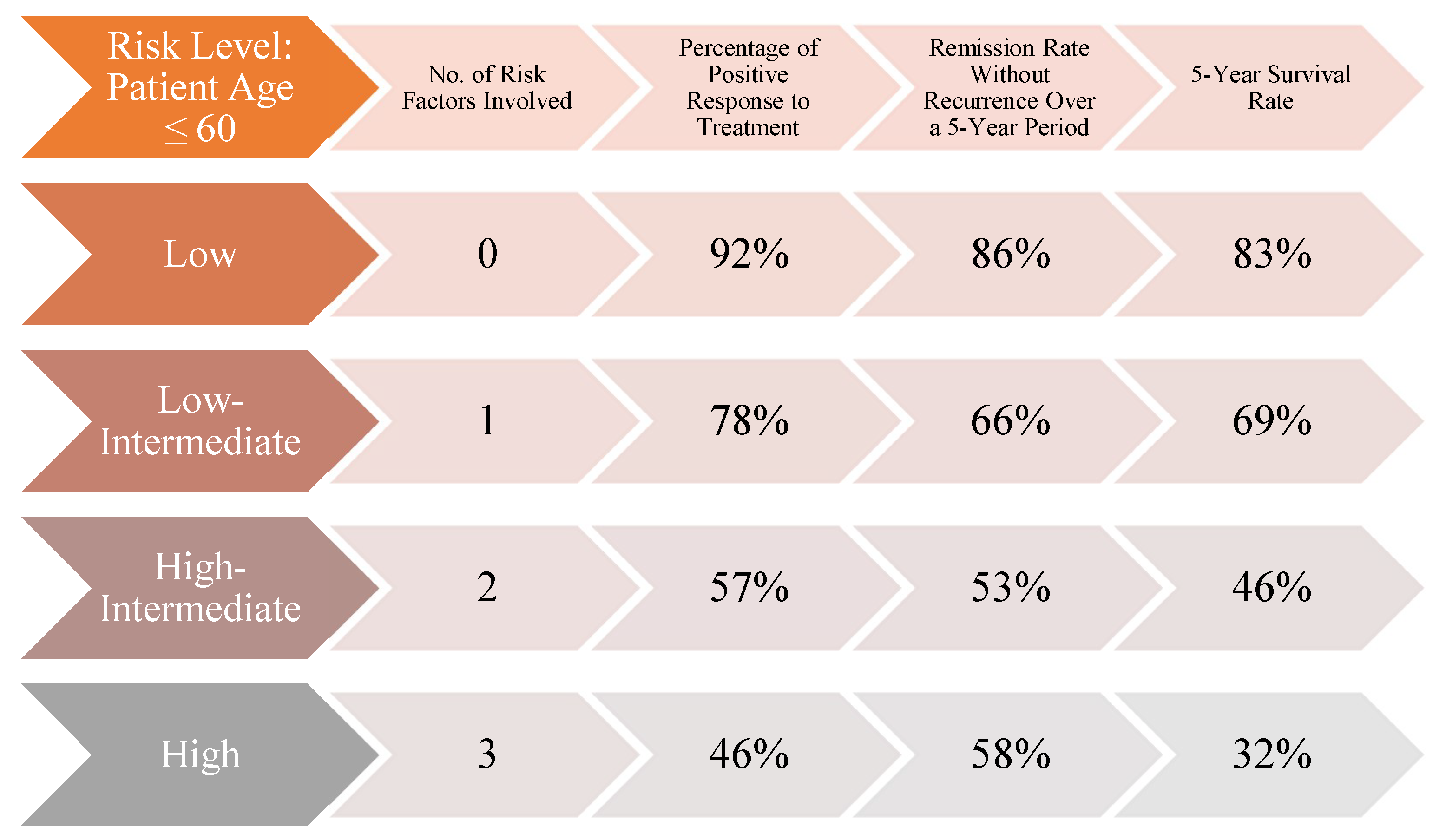
Figure 4.
Rata de curabilitate - „Hematologie clinică”- Ioniță H. et al., 2015, pag. 265-266.
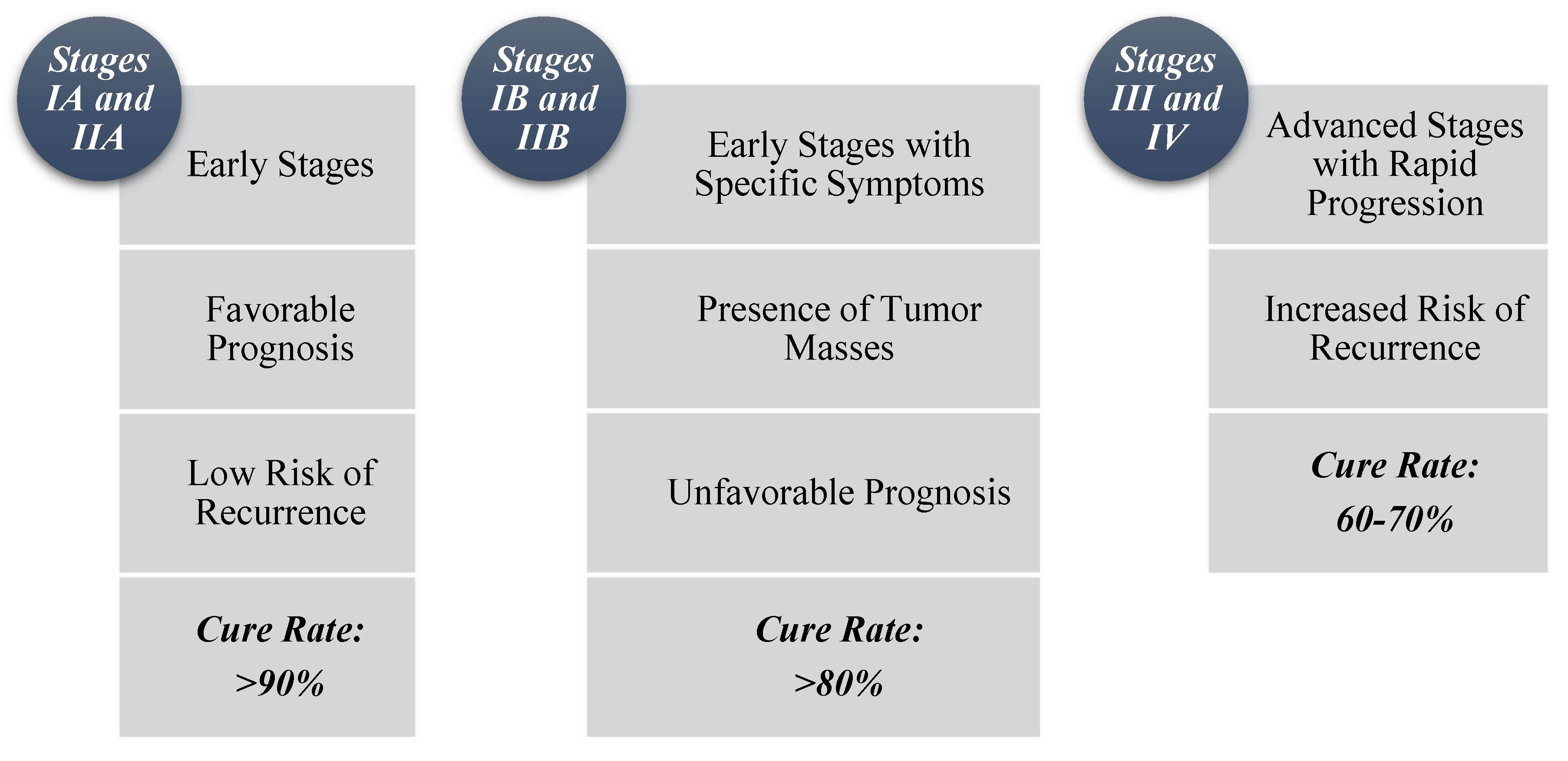

Table 1.
The characteristics of the histopathological subtypes of “CLASSIC” Hodgkin Lymphoma (CHL) - according to information taken from Oltean et al., 2009, p. 100 and Lupu et al.,2007, page 84.
Table 1.
The characteristics of the histopathological subtypes of “CLASSIC” Hodgkin Lymphoma (CHL) - according to information taken from Oltean et al., 2009, p. 100 and Lupu et al.,2007, page 84.
| Histologic Subtype | Histopathology | Statistics | Regions Involved |
|---|---|---|---|
|
Mixed Cellularity (MC) |
|
|
|
|
Nodular Sclerosis (NS) |
|
|
|
|
Lymphocyte Depletion (DL) |
|
|
|
|
Lymphocyte Predominant (PL) |
|
|
|
Table 2.
Types of Treatment Regimens Used for Treating Hodgkin Lymphoma.
| Polychemotherapy Regimens | Associated Substances - HL |
|---|---|
| MOPP |
|
| MVPP |
|
| COPP |
|
| VEBEP |
|
| CEVD |
|
| ABVD |
|
| BEACOPP |
|
| Stanford V |
|
Table 3.
Schemes of Chemotherapy Used in the Treatment of Non-Hodgkin Lymphomas - Oltean, 1996, Table 15.5, page 160.
Table 3.
Schemes of Chemotherapy Used in the Treatment of Non-Hodgkin Lymphomas - Oltean, 1996, Table 15.5, page 160.
| Polychemotherapy Regimens | Associated Substances - NHL |
|---|---|
| CVP |
|
| R-COP |
|
| CHOP +/- Bleo |
|
| BACOP |
|
| m-BACOD |
|
| MACOP-B |
|
| Pro-MACE |
|
| ESHAP |
|
| DNR-CAR |
|
| R-CHOP |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



