
Submitted:
04 September 2024
Posted:
10 September 2024
You are already at the latest version
Abstract
The organic material present at the same depth in the reservoir has the potential for conversion, as indicated by analyses conducted before and after combustion in the development of heavy oil. This paper examines the oxidation and pyrolysis reaction pathways of hydrocarbons, specifically benzaldehyde (C7H6O) and naphthalene (C10H8), before and after combustion using molecular dynamics simulations. The primary products formed under various temperature conditions include H2O, HO2, CO, and CO2. The discussion includes the number of molecules, such as HO and H, as well as their temperature variations. The activating group functions as an electron donor, while the inactivating group serves as an electron acceptor. The oxidation and pyrolysis reactions of naphthalene and the synthesis pathway of benzaldehyde are also explored. The C-C dissociation pathway in the early stages of combustion and the process of C-C bond synthesis in the later stages of the reactions are investigated through dynamic simulations at different temperatures: 3000 K, 3500 K, and 4000 K, with a particular focus on the reaction network at 4000 K. The application of the molecular reaction dynamics method to heavy oil combustion research is the primary objective of this work. This study aims to provide a novel approach to investigating hydrocarbon conversion at high temperatures and offers recommendations for enhanced oil recovery.
Keywords:
heavy oil: reaction path
; combustion reaction kinetics
; reaction rate
1. Introduction
Karamay heavy oil is not only abundant but also renowned for its unique rheological characteristics. In addition to typical features such as high density, viscosity, and a low freezing point—common in heavy oils from various oil fields—Karamay heavy oil is distinguished by its exceptionally low levels of asphaltene and sulfur. This specific type of heavy oil is highly sought after as a feedstock for producing premium lubricant base oils, resulting in a relatively higher market price compared to other crude oils. Karamay heavy oil reservoirs are typically located at depths of less than 1,000 meters and at lower temperatures, which contribute to their high viscosity under reservoir conditions. Consequently, thermal techniques such as cyclic steam stimulation, steam drive, and in situ combustion are employed to extract this heavy oil. Notably, the viscosity of Karamay heavy oil exhibits less variation with temperature at elevated levels, setting it apart from heavy oils found in other oil fields. While the distinct rheological properties of Karamay heavy oil are believed to be influenced by its chemical composition, further research is necessary to fully understand this phenomenon.
Olalekan [1] and colleagues researched new technologies for heavy oil extraction. Commercial production of heavy oil has heavily relied upon the steam injection method due to its effectiveness in reducing viscosity and improving mobility. However, due to uncertainty of oil price, high energy cost, water requirement, heat loss, and environmental issues, attention is being given to new thermal EOR technologies such as in situ steam generation using thermochemical fluid (TCF) injection. This technology essentially involves downhole generation of heat and pressure from exothermic chemical reactions. respectively. In comparison, from the injection of steam generated at 250°C, the pressure at the inlet of the core was 212 psi and the recovery factor was 71% OOIP. These results therefore confirm the increase in interest in the application of the TCF injection technology in heavy oil production.
Zhao [2] and other studies indicate that as the heating rate increases, the pyrolysis of oil shale occurs at higher temperature ranges. This trend is particularly pronounced with higher oil content. The stability of oil shale during pyrolysis is influenced by both oil content and the pyrolysis atmosphere. Specifically, higher oil content correlates with greater stability in the pyrolysis process. In a nitrogen atmosphere, the pyrolysis interval of oil shale is more concentrated, whereas exposure to air extends the pyrolysis interval and reduces the pyrolysis stability index. Furthermore, an increase in heating rate enhances the release characteristics of the products, which remain largely unaffected by oil content.
These ferroan carbonates are preferentially concentrated at distances ranging from 0.5 to 1.0 meters from sandstone-mudstone contacts. Kinetic modeling effectively reproduces the evolutionary pathways of various diagenetic minerals in the Es4s sandstones, which constitute the upper part of the fourth member of the Eocene Shahejie Formation. More importantly, the modeling results indicate that the dissolution of non-ferroan calcite cement does not improve overall reservoir quality, as it results in the precipitation of more stable ferroan calcite and ankerite cement [3].
Zhang [4] revealed that increased pressure enhances the generation of light components and accelerates the conversion of alkenes to alkanes and aromatics. The release temperature of gases increases under pressure, which promotes the production of alkane gas while simultaneously reducing hydrogen yield. As the pyrolysis temperature rises, the combustion performance of semi-coke deteriorates under low pressure; however, this trend is reversed under high pressure. Finally, a pyrolysis mechanism for the coupling of temperature and pressure in oil shale was proposed.
High temperatures induced various chemical and physical changes that led to the formation of the coking zone. Additionally, the small-aperture channels within the coking zone became concentrated with residual oil as a result of this formation process, which decreased fluidity and made production more challenging [5].
Lab-scale experiments and the analysis of core samples are the primary methods used to investigate compositional changes associated with in-situ combustion [6,7,8,9,10,11].
Furthermore, Figure 1 illustrates that benzaldehyde (C7H6O) was detected in the vicinity of the combustion interface (547.8 m). Simultaneously, naphthalene (C10H8) was identified in samples collected from the unswept area (<547.8 m). Zhao and colleagues [12] employed gas chromatography-mass spectrometry (GC-MS) analysis to confirm the presence of naphthalene in the unswept layers and benzaldehyde at the combustion interface. This finding supports the hypothesis that the pyrolysis and oxidation reactions involve C-O and O-H bonds. Collectively, these results demonstrate the potential for the conversion of benzaldehyde and naphthalene. However, there are limited studies in the existing literature that explore the reaction pathways and networks governing the molecular transition between these two compounds (benzaldehyde and naphthalene).
Enhancing oil recovery can be accomplished by gaining a deeper understanding of the mechanisms involved in the transformation of chemical molecules during the primary reactions associated with heavy oil thermal recovery. Research plays a crucial role in this process. Analyzing complex chemical networks in polymerization, combustion, and chemical engineering has always been essential for elucidating these mechanisms.
Most heavy oil combustion reaction mechanisms operate on a large scale and are primarily macroscopic in nature. The conversion between component molecules mainly involves the number of carbon atoms; however, the reaction pathways and networks between specific molecules have not been thoroughly investigated. By employing molecular dynamics methods to study the reaction processes involved in the transformation of heavy oil combustion, we can gain insights into various reactions occurring during combustion from the perspective of microscopic molecules.
Molecular dynamics methods can simulate networks consisting of thousands of reactions and hundreds of species. However, accurate modeling poses significant experimental and computational challenges. Reactive molecular dynamics (MD) has emerged as an essential tool for understanding these complex networks. In this study, we present a methodology for deriving both a network of reactions and its kinetic parameters from reactive MD simulations.
ReaxFF-MD, a method capable of addressing reactivity in computational calculations, has been proposed as a result of ongoing research developments [13,14]. In carbon/hydrogen/oxygen (C/H/O) fuel systems, such as polystyrene [15], cellulosic materials [16], styrene-butadiene [17], JP-10 [18], polycyclic aromatic hydrocarbons (PAHs) [19], hydrogen [20,21], and various other systems [22,23,24,25,26,27,28], ReaxFF-MD is recognized as a significant supplementary approach to experimental methods.
2. METHODOLOGY
2.1. ReaxFF-MD Reactive Force Field
ReaxFF-MD simulates the formation and breaking of chemical bonds by incorporating the concept of bond order, distinguishing it from non-reactive force fields. Each term, including bond order, is dependent on atomic connectivity. Non-bonded interactions, such as van der Waals and Coulomb forces, are considered between every pair of atoms due to their independent connections. Equation (1) [29] outlines the simulated system utilized by ReaxFF-MD to assess atomic charges through the Electronegativity Equalization Method [30].
Esystem=Ebond+Eover+Eangle+Etors+EvdWaals+Ecoulomb
2.2. Simulation Details.
To investigate the naphthalene oxidation process, we filled a cubic box with 20 naphthalene (C10H8) molecules and 240 oxygen (O2) molecules using Materials Studio 8.0. The volume of the box was calculated based on the initial number of molecules and the target density of 1.00 g/cm³.
We utilized Material Studio 8.0 to populate a cubic box with 20 naphthalene (C10H8) molecules and 240 oxygen (O2) molecules to investigate the naphthalene oxidation process. Based on the initial molecular count and the target density of 1.00 g/cm³, we calculated the box volume. The dimensions of the simulation boxes were measured in angstroms (Å). In the subsequent step, we assigned atomic velocities that conformed to a Gaussian distribution at the target temperature. The NVE ensemble, utilizing the Berendsen thermostat, was employed to maintain the system at the desired temperatures for one second. For molecular recognition, we established a C-C bond order cutoff of 0.55 Å, a C-O bond cutoff of 0.65 Å, and a time step of 0.1 fs. To ensure adequate collisions, the simulation temperatures were artificially elevated to 3000 K, 3500 K, and 4000 K. The high temperatures facilitated sufficient collisions between molecules and intermediates, thereby enhancing the simulation process. Within the temperature range of 3000 K to 4000 K, we conducted ten annealing simulations.
Compared to naphthalene, atomic charge analysis indicates that the activating group functions as an electron donor, while the inactivating group acts as an electron acceptor. The effects of substituents on naphthalene are primarily assessed through geometric and vibrational analyses. Furthermore, Natural Bond Orbital (NBO) studies have demonstrated that 2-naphthol esters possess the highest stability energy, which can be attributed to the increased electron density on the oxygen atom. A higher stability energy correlates with increased reactivity of the compound and a smaller band gap [33].
The species was analyzed, and the mechanisms and pathways of the oxidation process were investigated using the LAMMPS package (Large-scale Atomic/Molecular Massively Parallel Simulator) [34,35]. By assessing the number of reaction events that modify the chemical composition and current concentration of the respective reactants, these simulations can be utilized to compute rate constants with software such as the Chemical Trajectory Analyzer (CTY), which was employed in this study to analyze changes in chemical composition within a molecular dynamics (MD) trajectory [36,37].
3. Results and Discussion
A series of NVE-MD simulations of naphthalene oxidation at elevated temperatures (3000 K, 3500 K, and 4000 K) were conducted as described in the section on computational details. Additionally, we employed the CHO-2008 force field to simulate the same system in order to investigate the initiation mechanism and combustion dynamics. During the high-temperature simulations, Figure 2 illustrates the distribution of reactants, products, and intermediate species.
Several reactions, including pyrolysis, occur concurrently with the naphthalene oxidation reaction. This investigation examined both pyrolysis and oxidation reactions. It focused on the effect of temperature on the variation in the quantities of CO2, CO, H2O, and other products over time. As the temperature rises to 4000 K, the reaction rate of C10H8 in O2 oxidation reaches its peak, and at 60000 fs, the concentration of CO in the reaction exceeds that of CO2. Additionally, the number of small molecules and the diversity of free radical colors in the oxidation reaction significantly increase at 4000 K.
The three stages of the oxidation process can be identified based on the reaction rate of naphthalene oxidation. In the first stage, naphthalene reacts at a relatively slow rate, ranging from 10,000 fs to 40,000 fs, at temperatures of 3000 K, 3500 K, and 4000 K. Although the production of products is limited at this point, the temperature still influences the slope of the reaction curve. In the second stage, the number of products increases significantly, and the reaction rate of the reactants accelerates. Our simulations revealed the presence of various species, including highly unsaturated compounds, unstable radicals, alkanes, and alkenes. While the type of products remains unchanged in the third stage, the reaction rate does vary. The examination of Figure 2 highlights intriguing topics for further research, such as naphthalene's post-oxidation products and intermediates, their effects on the oxidation reaction, and the transformation of naphthalene into benzaldehyde. At 4000 K, the curve is notably steep due to the abundance of products. We intend to explore the kinetic details of naphthalene's oxidation reaction, particularly at 4000 K, given the active products and reactions involved.
3.1. Detailed Analysis of the Kinetic Mechanisms of the Oxidation Reaction of Naphthalene
The understanding of the complex pyrolysis process relies on the initiation of the MCH oxidation reaction, which determines the subsequent distributions of intermediates and products. Therefore, a comprehensive description of the initial reaction pathway is essential for elucidating the mechanisms of the MCH oxidation reaction.
Figure 2c illustrates the naphthalene oxidation process, which consists of three distinct stages. As previously mentioned, the time axis divisions for these stages remain consistent across various reaction temperature conditions. The rapid oxidation observed in Stage 1 can be attributed to the presence of highly reactive free radicals generated during the reactions [38]. This study identified several active free radicals, including H2O2, HO2, CH2O, among others. However, we were unable to identify the isomers present in the reaction products, which did not impact the overall objectives of the investigation. By analyzing the data collected at each stage, we were able to determine the reaction rates for the different phases of the process.
Naphthalene undergoes an oxidation reaction that produces carbon dioxide (CO2), water (H2O), carbon monoxide (CO), and hydrogen peroxide (HO2) as the principal products. These four products were analyzed in terms of their reaction types and rates. The highest reaction rate, along with a comparatively high number of reactions, is observed in the reactions listed below.
Naphthalene primarily dissociates through two types of reactions, as illustrated in Table 1: unimolecular decomposition and hydrogen abstraction reactions, both of which involve the dissociation of C-H and C-C bonds. The reactions listed in the table represent those with the highest frequency observed during the simulation process, and they predominantly contribute to the generation of the corresponding products. Naphthalene undergoes an oxidation reaction in which H-O and C-O bonds are formed. During the hydrogen abstraction reaction, the formation of C-O and O-H bonds occurs as part of the main product formation process.
3.2. Intermediate Reactions
Although naphthalene oxidation has been the subject of extensive research, a clear understanding of the intermediate processes has yet to emerge. Comprehending the intermediate mechanisms necessitates a thorough examination of the reactions related to the primary products. The reaction pathways for the main products were studied, and the proportions of reaction times for these products were calculated and documented. The primary forward and reverse reactions in each pathway were discussed, and the key reactions within the overall process were identified.
It is explained how free radicals influence the rate of reaction and synthesis activity. In Figure 2c, carbon dioxide (CO2) is identified as the most prevalent product resulting from oxidation. The changes in carbon dioxide concentrations throughout the reactions are highlighted. The time-dependent distributions of carbon dioxide are illustrated in Figure 2c, showing a dramatic increase in production between 0 and 40,000 femtoseconds (fs) during the reaction process. During this period, the production of carbon dioxide significantly exceeds its consumption. Figure 3a indicates that reversible reactions are present. Among the various positive reactions, the most frequent is the oxidation reaction of free radicals (CHO2 + HO → H2O + CO2), along with several pyrolysis reactions (C2O4 → CO2 + CO2). These reactions involve the dissociation of carbon-carbon (C-C) bonds and hydrogen abstraction. After 80,000 fs, the reaction rate of CO2 approaches equilibrium, at which point the consumption of carbon dioxide surpasses its production. Reversible reactions serve as the primary pathways for the consumption of CO2.
In the reactions illustrated in the figure, there are 10 positive reactions and 12 negative reactions. Among the positive reactions, the reaction with the highest frequency accounts for 38% (CHO2 + HO → H2O + CO2). In the negative reactions, the reaction with the highest frequency accounts for 34% (H2O + CO2 → CHO2).
In the oxidation reaction of naphthalene, the behavior of carbon monoxide (CO) undergoes significant changes. The production of CO has been continuously increasing during the time interval of 0 to 42,000 femtoseconds (fs), as illustrated in Figure 2c. The reaction with the highest frequency during the CO generation process is the pyrolysis reaction (CO3 → O2 + CO), depicted in Figure 3b. Conversely, the production of CO decreases during the period from 42,000 to 89,000 fs, indicating an increase in CO consumption during this stage. The reactions occurring during this interval are predominantly oxidation reactions. A comparison of the curves for CO and carbon dioxide (CO2) reveals an intersection point, where the production of CO2 is lower than that of CO within the range of 0 to 62,000 fs. Reason: The revisions enhance clarity, improve technical accuracy, and correct grammatical and punctuation errors while maintaining the original meaning of the text.
There are 16 positive reactions and 5 reverse reactions. Among the positive reactions, the one with the highest frequency accounts for 47% (CO3 → O2 + CO). In the case of reverse reactions, the reaction with the highest frequency constitutes 69% (O2 + CO → CO3).
In Figure 2c, there is a significant increase in H2O production during the initial stage of 0-30,000 fs. In the subsequent stages, H2O production remains relatively stable. In the positive reaction channel depicted in Figure 3c, the proportion of reaction counts reached 33% (H2O + HO + HO → H2O + H2O + O). Conversely, the reverse reaction accounts for 42% of the negative reaction channels. Overall, the ratio of positive reactions producing H2O is higher than that of the negative reaction channels. This disparity is one of the reasons for the relative stability in H2O production. Additionally, HO radicals participate in numerous reactions.
The generation curve data for the HO2 reaction is illustrated in the locally enlarged image in Figure 2c. After 35,000 fs, there is a significant increase in HO2 production. An analysis of the reaction channels presented in Figure 3d reveals that HO2 is involved in numerous oxidation reactions, as well as in the thermal decomposition of hydrocarbons and the dissociation of certain free radicals. These findings indicate that HO2 exhibits strong reactivity and can facilitate various reactions. Within the reaction channel for HO2, there are 11 forward reactions and 10 reverse reactions. The most prevalent reaction is the oxidation of hydrogen peroxide by oxygen (O2 + H2O2 → HO2 + HO2), with the reverse reaction occurring at a frequency that accounts for 27% of the total.
3.3. Detailed Reaction Map for Naphthalene Oxidation
Figure 4 illustrates reaction networks that have been suggested for the oxidation of naphthalene(C10H8) based on the outcomes of our simulation.
The impact of isomers was overlooked during the research on reaction networks. The reaction rate is high, and the reaction time is short, based on the simulation conditions, which include elevated temperatures. Within the reaction network, isomers are believed to have minimal influence. These networks provide a comprehensive explanation of the process that converts a C10H8 molecule into C7H6O. Four potential pathways are proposed for the formation of C7H6O.
The P1 pathway is the most straightforward of the four reaction pathways, as illustrated in Figure 4. The compound C7H4O is essential to the P1 reaction pathway and is involved in the P1-2, P1-4, and P1-5 reactions, among others. Additionally, C7H5O2 is critical to the P1-3, P1-6, and P2-5 reaction pathways, which presents a similar challenge. C7H5O2 is produced at the intersection of the P1 and P2 reaction pathways.
The P2 reaction pathway incorporates the products from both the P3 and P1 reaction pathways. C10H7O3 serves as the initial reactant in the P2 reaction pathway and plays a crucial role in the subsequent reactions. The P3 reaction pathway is relatively complex, encompassing numerous products, including eight smaller reaction pathways. The product that lies at the intersection of the P3 and P2 reaction pathways is C10H7O3.
The P4 reaction pathway is relatively short and is the shortest among all reaction pathways. It exhibits the highest conversion efficiency and a greater reaction rate. The P1, P2, and P3 reaction pathways encompass various types of reactions, including oxidation reactions, pyrolysis reactions, and hydrogen transfer reactions. In contrast, the P4 reaction pathway consists solely of pyrolysis reactions. The specific reactions and their corresponding rates involved in these four reaction pathways will be detailed in Table 2, Table 3 and Table 4.
Table 2 through Table 4 present the intermediate reactions for each reaction pathway, along with their corresponding reaction rate constants. The reaction map indicates that a total of 20 intermediate reactions are involved. There are three categories of statistical reactions: hydrogen abstraction, pyrolysis, and oxidation. In some of these reactions, hydroxyl (OH) and hydrogen (H) radicals play a significant role.
Based on the reaction network map and Table 2, we will discuss the characteristics of the P1 reaction pathway. This pathway consists of six reactions. The initial oxidation reaction, represented by the equation O2 + C10H8 → C10H8O2, exhibits the lowest reaction rate. The intermediate product, C7H4O, is formed through two reactions, P1-2 and P1-5, both of which involve the pyrolysis of C-C bond dissociation. Additionally, two reactions contribute to the formation of the product C7H5O2, specifically P1-3 and P2-5. The hydrogen elimination reaction occurs in the P1-4 pathway, resulting in the production of C9H5O2, as observed.
As a node in the reaction network, C7H5O2 has an alternative generation pathway, as illustrated in Figure 4. According to Table 3, three of the reactions are classified as pyrolysis reactions. Additionally, there is one oxidation reaction and one activation polymerization reaction. The oxidation reaction exhibits the lowest rate among these processes. The end products of the pyrolysis reaction (C8H7O2 → C7H5 + CH2O2), which results from the dissociation of C-C bonds, are C7H5 and CH2O2. This particular reaction has the highest reaction rate. A relatively high reaction rate is also observed when the free radical OH is involved.
Compared to the previously mentioned reaction pathway, the P3 reaction pathway is more complex, involving a total of nine reactions. Among these, the proportion of pyrolysis reactions is relatively high. The oxidation reaction exhibits the slowest rate among all the reactions. When examining the hydrogenation (C7H5O3 + H → C7H6O3) and dehydrogenation (C10H8O3 → H + C10H7O3) reactions, it is evident that the rate of the hydrogenation reaction is significantly higher than that of the dehydrogenation reaction. Conversely, the rate of the deoxidation reaction (C7H6O3 → C7H6O + O2) is greater than that of the oxidation reaction (O2 + C10H8O → C10H8O3).
As illustrated in Figure 4, the transition from C10H8 to C7H6O involves two critical products: C7H5O2 and C7H5O3. A comparison of the two reactions involving C7H5O2 reveals that the oxidation reaction (C7H5 + O2 → C7H5O2) has a relatively low rate, as shown in Table 4. In contrast, the formation of product C7H5O3 occurs at a higher rate during the pyrolysis reaction, which involves the participation of free radical OH. Among the two products mentioned, C7H5O2 reacts with free radical OH to produce C7H6O3, while C7H5O3 is formed through a hydrogenation reaction. Ultimately, C7H6O3 yields the target product, C7H6O, via a pyrolysis reaction (C7H6O3 → C7H6O + O2).
4. Conclusions
The ReaxFF molecular dynamics (MD) method was employed to investigate the distribution of products during the oxidative pyrolysis of naphthalene. Additionally, the reaction pathway and characteristics of oxidative pyrolysis were analyzed using the CTY method. The reaction products and rates exhibit distinct characteristics at varying temperatures. The reaction pathway network for the conversion of C10H8 to C7H6O primarily comprises three pathways, which include ring opening, carbon-carbon (C-C) bond cleavage, hydrogen transfer, and other related processes.
- Naphthalene undergoes oxidation at elevated temperatures using NVE-MD (microcanonical ensemble molecular dynamics) with a reactive force field. To obtain a comprehensive understanding of the oxidation process, simulations are conducted on a model system consisting of 20 naphthalene molecules. The primary species identified in the current simulations include CO2, CO, H2O, HO2, and others.
- The reaction rates of products in oxidation and pyrolysis reactions vary at different time stages. After the intersection of CO2 and CO, the yield of CO2 surpasses that of CO. This phenomenon can be attributed to insufficient collision contact in the early stages and more favorable reaction conditions in the later stages. In the reaction network from C10H8 to C7H6O, multiple types of reactions were identified across four reaction pathways, including dehydrogenation, hydrogenation, and small molecule activation reactions.
- The reaction network from C10H8 to C7H6O was examined. The findings of this investigation will enhance our understanding of the process. Among the reactions in the network, the hydrogenation reaction exhibits the highest reaction rate. Identifying the reaction network through conventional experiments is challenging due to the complexity of the reactions involved. In the study of oxidative pyrolysis reactions, computational simulations offer a novel research approach.
Author Contributions
Conceptualization, T.Y.; methodology, T.Y.; molecular dynamic simulation calculations, T.Y., H.W. and Z.S.; validation, T.Y., Z.S.; investigation, T.Y., L.C., and Z.S.; writing—original draft preparation, T.Y., H.L.; writing—review and editing, T.Y., L.L.; review and editing, T.Y., R.Z.; supervision, T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Karamay City - Innovative Talents Special Project (Nos. XQZX20220045).
Conflicts of Interest
The authors declare that none of their known financial conflicts or interpersonal connections could have influenced the work that was published in this paper
References
- Alade, O.S.; Hamdy, M.; Mahmoud, M.; Al Shehri, D.A.; Mokheimer, E.; Patil, S. Ayman Al-Nakhli. A preliminary assessment of thermochemical fluid for heavy oil recovery. J. Pet. Sci. Eng. 2020, 186, 106702. [Google Scholar] [CrossRef]
- Zhao, S.; Sun, Y.; Lü, X.; Li, Q. Energy consumption and product release characteristics evaluation of oil shale non-isothermal pyrolysis based on TG-DSC. J. Pet. Sci. Eng. 2020, 187, 106812. [Google Scholar] [CrossRef]
- Jia, Y.; Cao, Y.; Wang, H.; Ma, B. Influence of multiphase carbonate cementations on the Eocene delta sandstones of the Bohai Bay Basin, China. J. Pet. Sci. Eng. 2021, 205, 108866. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, W.; Pan, J.; Zhu, C.; Deng, S. In-situ pyrolysis of oil shale in pressured semi-closed system: Insights into products characteristics and pyrolysis mechanism. Energy 2024, 168, 129608. [Google Scholar] [CrossRef]
- Tao, L.; Hu, Z.; Xu, Z.; Zhang, X.; Ding, Y.; Wang, C.; Chen, D.; Li, S. Experimental investigation of in situ combustion (ISC) in heavy oil thermal recovery. Geoenergy Sci. Eng. 2024, 233, 212488. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Y.; Lu, N.; Chai, M.; Wang, S.; Feng, Q.; Chen, Z. Integration of ramped temperature oxidation and combustion tube tests for kinetic modeling of heavy oil in-situ combustion. Energy 2023, 274, 127435. [Google Scholar] [CrossRef]
- Jayaraman, K.; Kök, M.V.; Gökalp, I. Combustion mechanism and model-free kinetics of different origin coal samples: Thermal analysis approach. Energy 2020, 204, 117905. [Google Scholar] [CrossRef]
- Mahvelati, E.; Forcinito, M.; Fitschy, L.; Maesen, A. Three-dimensional CFD Model Development and Validation for Once Through Steam Generator (OTSG): Coupling Combustion Heat Transfer and Steam Generation. ChemEngineering 2022, 6, 23. [Google Scholar] [CrossRef]
- Askarova, A.; Popov, E.; Ursenbach, M.; Moore, G., Sudarshan Mehta and Alexey Cheremisin. Experimental Investigations of Forward and Reverse Combustion for Increasing Oil Recovery of a Real Oil Field. Energies 2020, 13, 4581.
- Huang, S.; Sheng, J.J.; Jiang, Q.; Liu, J. Screening of Spontaneous Ignition Feasibility During Air Injection EOR Process Based on Thermal Experiments. Screening of Spontaneous Ignition Feasibility During Air Injection EOR Process Based on Thermal Experiments. Energies 2019, 12, 3687. [Google Scholar] [CrossRef]
- Zhao RB.; Xia XT.; Luo WW.; et al. Alteration of heavy oil properties under in-situ combustion: a field study. Energy Fuels 2015, 29, 6839. [CrossRef]
- RB, Z.; CH, Z.; FX, Y.; MH, H.; PT, S.; Wang, Y.J. Influence of temperature field on rock and heavy components variation during in-situ combustion process. Fuel 2018, 230, 244. [Google Scholar]
- Sun, Z., Jincheng Ji and Weihua Zhu. Effects of Nanoparticle Size on the Thermal Decomposition Mechanisms of 3, 5-Diamino-6-hydroxy-2-oxide-4-nitropyrimidone through ReaxFF Large-Scale Molecular Dynamics Simulations. Molecules 2024, 29, 56.
- Sang.; Unocic, R.R.; Iacovella, C.R.; Gogotsi, Y., Adri, C. T. Van Duin and Peter, T. Cummings. An Atomistic Carbide-Derived Carbon Model Generated Using ReaxFF-Based Quenched Molecular Dynamics. J. Carbon Res. 2017, 3, 32.
- Thompson, M.W.; Dyatkin, B.; Wang, H.-W.; Turner, C.H.; Xiahan., *!!! REPLACE !!!*; Li, C.; Yang, Z.; Wu, X.; Shao, S. Xiangying Meng and Gaowu Qin. Reactive Molecular Dynamics Simulations of Polystyrene Pyrolysis. Int. J. Mol. Sci. 2023, 24, 16403. [Google Scholar]
- Fan, Y.; Li, Y.; Zhang, Y.; Shi, K. Mechanism Analysis of Ethanol Production from Cellulosic Insulating Paper Based on Reaction Molecular Dynamics. Polymers 2022, 14, 4918. [Google Scholar] [CrossRef]
- Deng, S.; Zhuo, H.; Wang, Y.; Leng, S.; Zhuang, G.; Zhong, X.; Wei, Z.; Yao, Z.; Wang, J. Multiscale Simulation on Product Distribution from Pyrolysis of Styrene-Butadiene Rubber. Polymers 2019, 11, 1967. [Google Scholar] [CrossRef]
- Liu, H.; Liang, J.; He, R.; Li, X.; Zheng, M.; Ren, C.; An, G.; Xu, X.; Zheng, Z. Overall mechanism of JP-10 pyrolysis unraveled by large-scale reactive molecular dynamics simulation. Combust. Flame 2022, 237, 111865. [Google Scholar] [CrossRef]
- Wang, Y.; Mao, Q.; Wang, Z.; Luo, K.H.; Zhou, L.; Wei, H. A ReaxFF molecular dynamics study of polycyclic aromatic hydrocarbon oxidation assisted by nitrogen oxides. Combust. Flame 2023, 248, 112571. [Google Scholar] [CrossRef]
- Cheng, T.; Jaramillo-Botero, A.; Goddard, W.A.; Sun, S. Adaptive accelerated ReaxFF reactive dynamics with validation from simulating hydrogen combustion. J. Am. Chem. Soc. 2014, 136, 9434. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Li, X.; Li, Z.; Xu, H.; Qing, M.-X.; Jiang, X.; Liu, L. The molecular evolution mechanism of direct pyrolysis and hydropyrolysis of Changqing petroleum coke was compared based on ReaxFF method. J. Mol. Struct. 2023, 1290, 135882. [Google Scholar] [CrossRef]
- T P, S.; van Duin, A.C.T.; Janik, M.J. Methane Activation at the Pd/CeO2 Interface. ACS Catal. 2017, 7, 327. [Google Scholar]
- Ostadhossein, A.; Rahnamoun, A.; Wang, Y.; Zhao, P.; Zhang, S.; V H, C.; van Duin, A.C.T. ReaxFF Reactive Force-Field Study of Molybdenum Disulfide (MoS2). J. Phys. Chem. Lett. 2017, 8, 631. [Google Scholar] [CrossRef]
- Yeon, J.; van Duin, A.C.T.; Kim, S. H. Effects of Water on Tribochemical Wear of Silicon Oxide Interface: Molecular Dynamics (MD) Study with Reactive Force Field (ReaxFF). Langmuir 2016, 32, 1018. [Google Scholar] [CrossRef]
- Yeon, J.; van Duin, A.C.T. ReaxFF Molecular Dynamics Simulations of Hydroxylation Kinetics for Amorphous and Nano-Silica Structure, and Its Relations with Atomic Strain Energy. J. Phys. Chem. C 2016, 120, 305. [Google Scholar] [CrossRef]
- Y K, S.; Gai, L.; Raman, S.; van Duin, A.C.T. Development of a ReaxFF Reactive Force Field for the Pt-Ni Alloy Catalyst. J. Phys. Chem. A. 2016, 120, 8044. [Google Scholar]
- Zhong, Q.; Mao, Q.; Xiao, J.; van Duin, A.C.T.; Jonathan, P. Mathews. ReaxFF simulations of petroleum coke sulfur removal mechanisms during pyrolysis and combustion. Combust. Flame 2018, 198, 146. [Google Scholar] [CrossRef]
- Liu, L.; Xu, H.; Zhu, Q.; Ren, H.; Li, X. Soot formation of n-decane pyrolysis: A mechanistic view from ReaxFF molecular dynamics simulation. Chem. Phys. Lett. 2020, 760, 137983. [Google Scholar] [CrossRef]
- Ashraf, C.; Shabnam, S.; Jain, A.; Xuan, Y.; van Duin, A.C.T. Pyrolysis of binary fuel mixtures at supercritical conditions: A ReaxFF molecular dynamics study. Fuel 2019, 235, 194. [Google Scholar] [CrossRef]
- Senftle; T.P., et al. The ReaxFF reactive force-field: development, applications and future directions. Npj Comput. Mater 2016, 2, 1.
- Huo, E.; Zhang, S.; Xin, L.; Wang, S.; Cai, S.; Zhang, L.; Bai, M. Pyrolysis mechanism study of n-heptane as an endothermic hydrocarbon fuel: A reactive molecular dynamic simulation and density functional theory calculation study. Comput. Theor. Chem. 2022, 1211, 113696. [Google Scholar] [CrossRef]
- Hirotoshi Hirai. Molecular dynamics simulations for initial formation process of polycyclic aromatic hydrocarbons in n-hexane and cyclohexane combustion. Chem. Phys. 2021, 548, 111225. [Google Scholar] [CrossRef]
- Density functional theory study of the influence of activating and deactivating groups on Naphthalene. Results Chem. 2022, 4, 10669.
- Alamfard, T.; Lorenz, T.; Breitkopf, C. Tommy Lorenz and Cornelia Breitkopf. Thermal Conductivities of Uniform and Random Sulfur Crosslinking in Polybutadiene by Molecular Dynamic Simulation. Polymers 2023, 15, 2058. [Google Scholar] [CrossRef]
- Orekhov, N.; Ostroumova, G.; Stegailov, V. High temperature pure carbon nanoparticle formation: Validation of AIREBO and ReaxFF reactive molecular dynamics. Carbon 2020, 270, 606. [Google Scholar] [CrossRef]
- Döntgen, M.; M D, P.-F.; Kröger L, C.; W A, K.; A E, I.; Leonhard, K. Automated discovery of reaction pathways, rate constants, and transition states using reactive molecular dynamics simulations. J. Chem. Theory Comput 2015, 11, 2517. [Google Scholar] [CrossRef] [PubMed]
- Döntgen, M.; Schmalz, F.; Kopp, W. A.; Kröger, L.C.; Leonhard, K. Automated Chemical Kinetic Modeling via Hybrid Reactive Molecular Dynamics and Quantum Chemistry Simulations. J. Chem. Inf. Model. 2018, 58, 1343. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Yuan, W.; Gaïl, S.; Li, W.; Zhao, L.; Yang, J.; Qi, F.; Li, Y.; Dagaut, P. Exploration on the combustion chemistry of p-xylene: A comprehensive study over wide conditions and comparison among C8H10 isomers. Combust. Flame 2024, 262, 113377. [Google Scholar] [CrossRef]
Figure 1.
Well J2107a core samples' morphology shows that they are oriented up-dip [12].
Figure 1.
Well J2107a core samples' morphology shows that they are oriented up-dip [12].

Figure 2.
The time evolution of O2 and C10H8 molecules during NVT-MD simulations of the oxidation process at temperatures of 3000 K, 3500 K, and 4000 K is illustrated (a. 3000 K, b. 3500 K, c. 4000 K).
Figure 2.
The time evolution of O2 and C10H8 molecules during NVT-MD simulations of the oxidation process at temperatures of 3000 K, 3500 K, and 4000 K is illustrated (a. 3000 K, b. 3500 K, c. 4000 K).
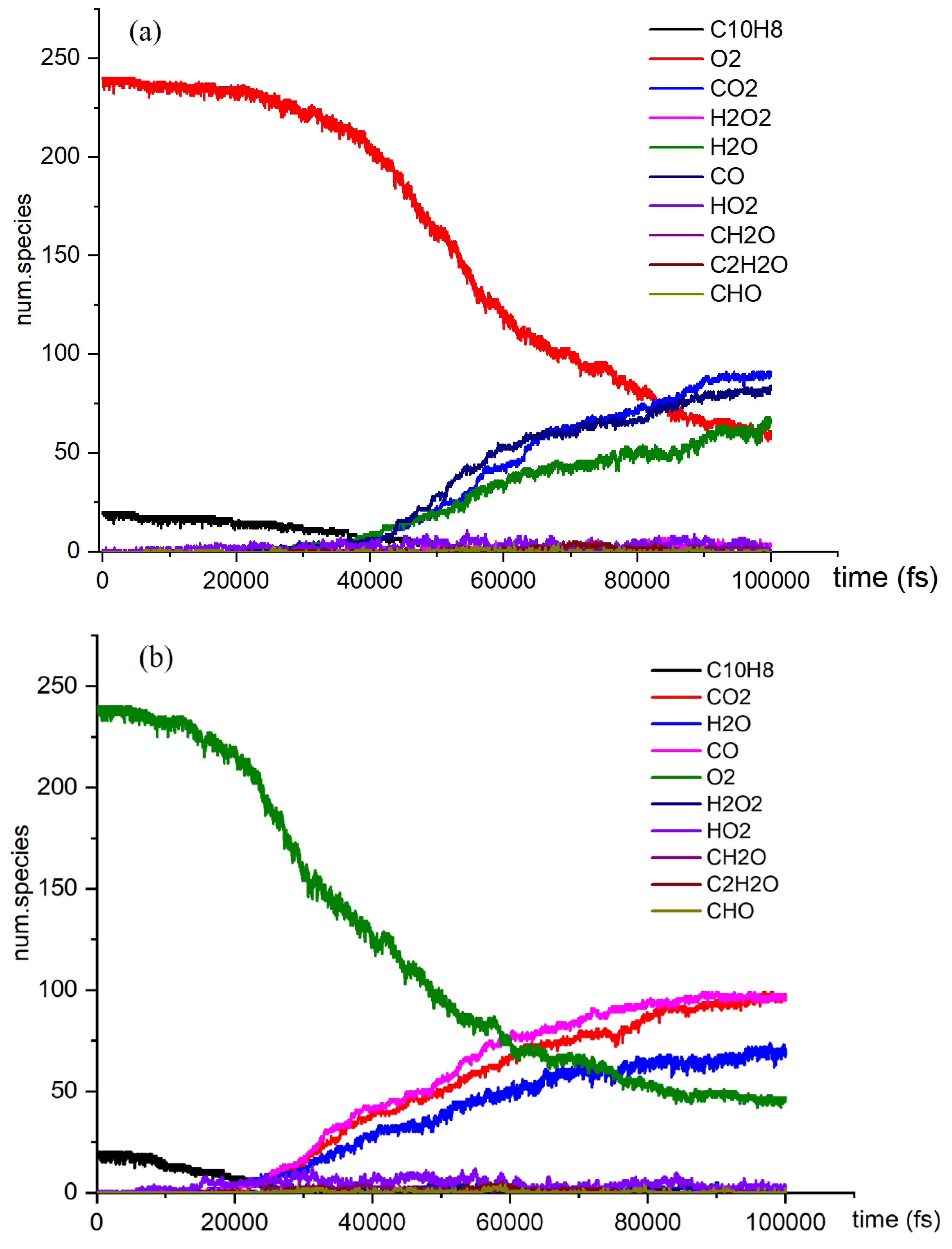
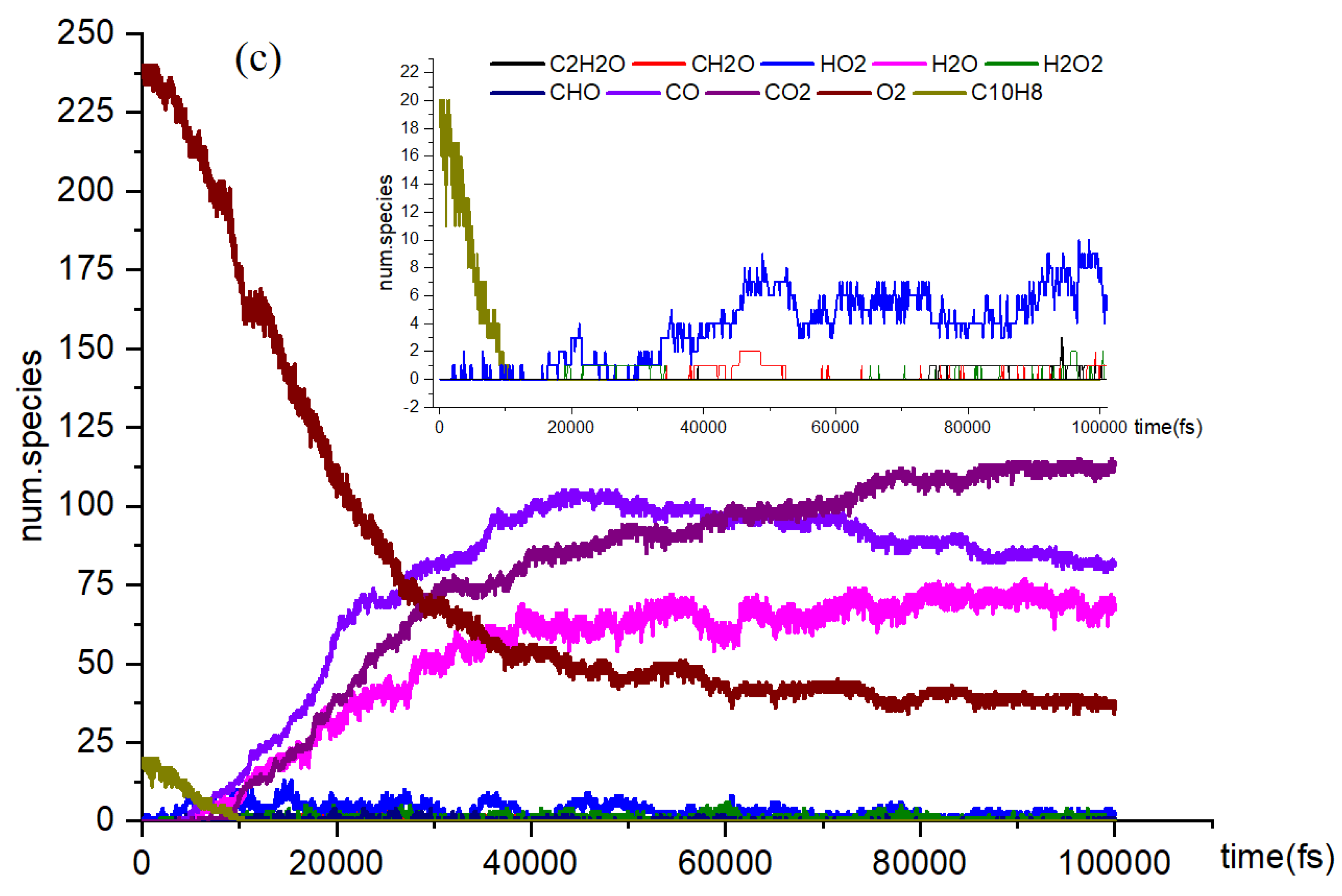
Figure 3.
(a) The primary CO2 reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively. (b) The primary CO reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively. (c) The primary H2O reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively. (d) The primary HO2 reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively.
Figure 3.
(a) The primary CO2 reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively. (b) The primary CO reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively. (c) The primary H2O reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively. (d) The primary HO2 reaction pathways. The production and consumption channels are represented by solid and medium-sized diamonds, respectively.
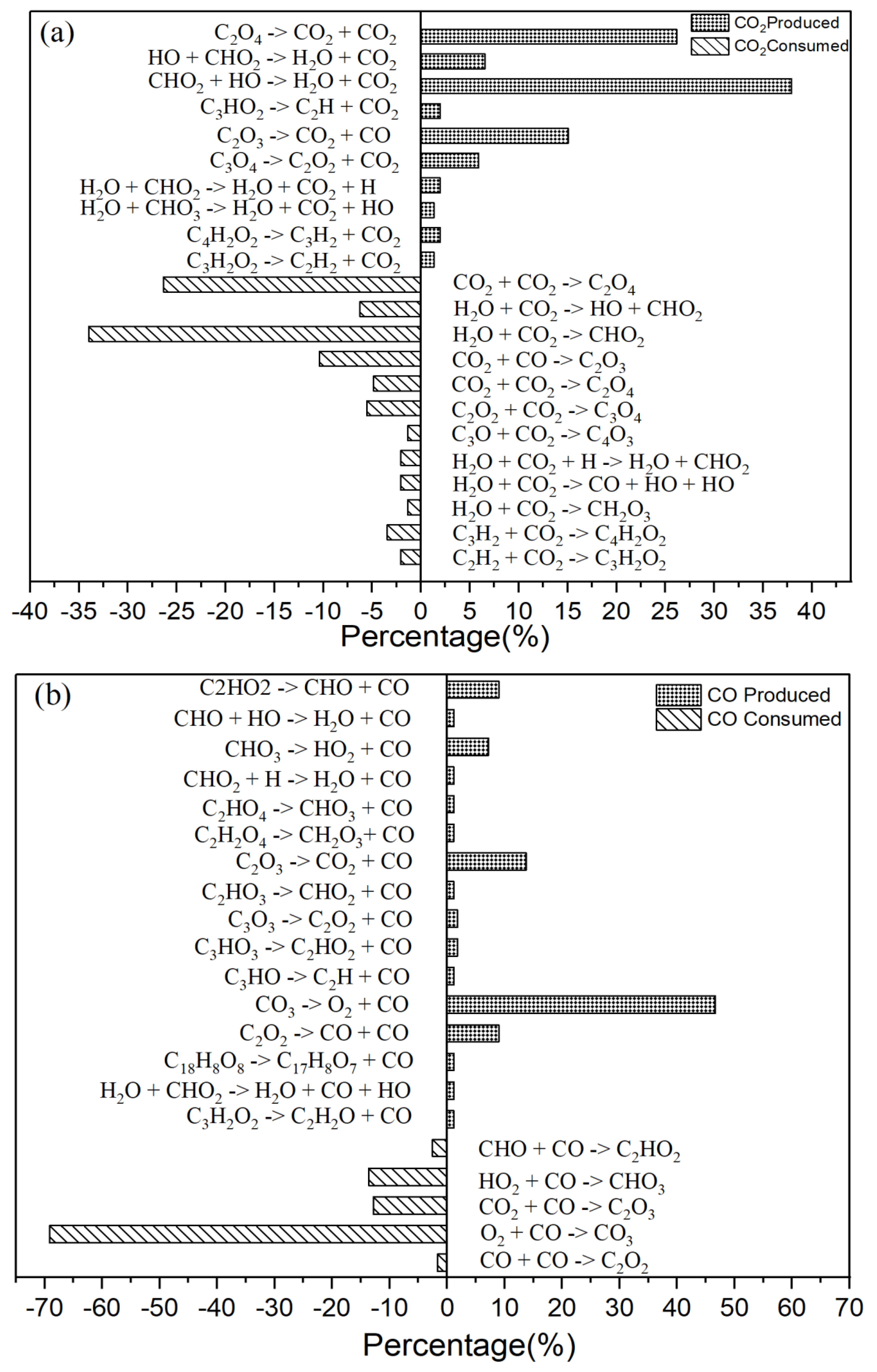
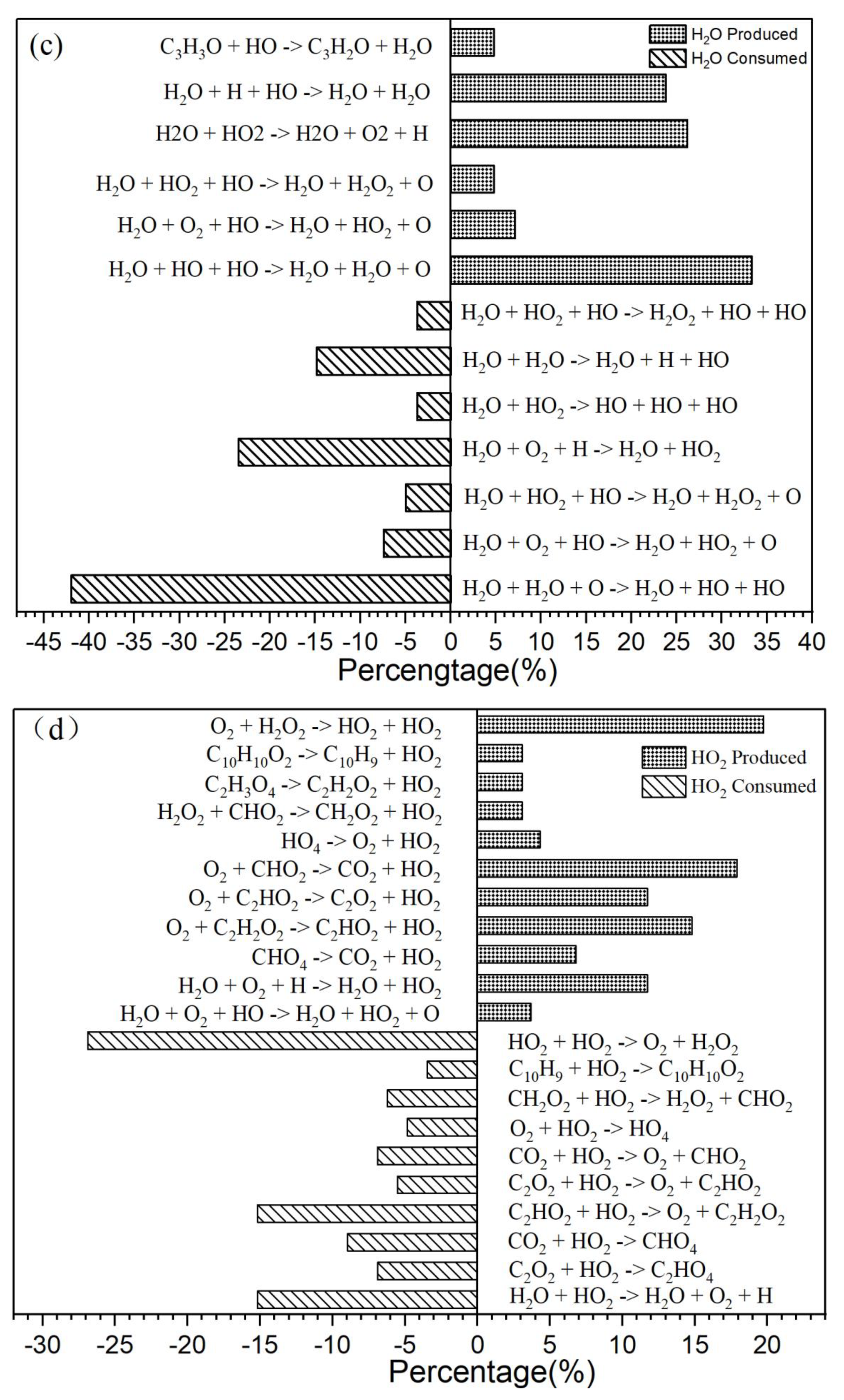
Figure 4.
Reaction map for the conversion of naphthalene (C10H8) to C7H6O.
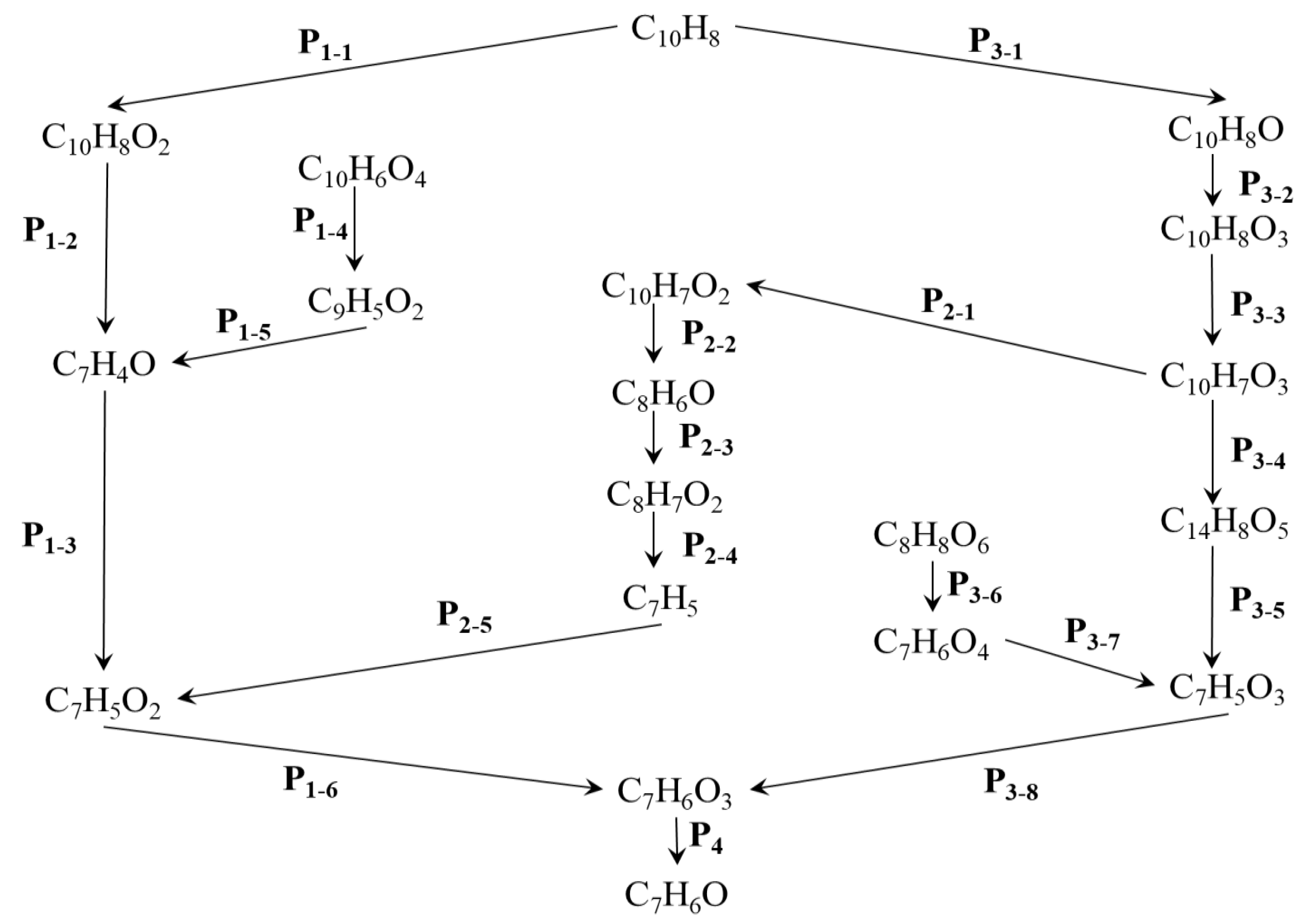

Table 1.
Main reactions in the reaction of naphthalene.
| Reactions | Rate Constants(s-1) | |
|---|---|---|
| 1 | CHO2 + HO → H2O + CO2 | 7.20×1010 |
| 2 | CO3 → O2 + CO | 4.04×1013 |
| 3 | H2O + HO2 → H2O + O2 + H | 7.39×108 |
| 4 | O2 + H2O2 → HO2 + HO2 | 5.41×109 |
Table 2.
Main reactions of the P1 reaction pathway.
| Path | Reactions | Rate Constants K (s-1) |
|---|---|---|
| P1-1 | O2 + C10H8 → C10H8O2 | 1.34×109 |
| P1-2 | C10H8O2 → C7H4O + C3H4O | 3.85×1011 |
| P1-3 | C7H4O + C2H2O2 → C2HO + C7H5O2 | 3.01×1013 |
| P1-4 | C10H6O4 → CO2 + C9H5O2 + H | 5.00×1013 |
| P1-5 | C9H5O2 → C2HO + C7H4O | 2.50×1013 |
| P1-6 | C7H5O2 + HO → C7H6O3 | 4.40×1012 |
Table 3.
Main reactions of the P2 reaction pathway.
| Path | Reactions | Rate Constants K (s-1) |
|---|---|---|
| P2-1 | C10H7O3 → C10H7O2 + O | 8.13×1011 |
| P2-2 | C10H7O2 → C8H6O + C2HO | 4.54×1012 |
| P2-3 | HO + C8H6O → C8H7O2 | 1.00×1013 |
| P2-4 | C8H7O2 → C7H5 + CH2O2 | 5.00×1013 |
| P2-5 | C7H5 + O2 → C7H5O2 | 5.51×1010 |
Table 4.
Main reactions of the P3 and P4 reaction pathway.
| Path | Reactions | Rate Constants K (s-1) |
|---|---|---|
| P3-1 | C10H7O + C10H8 → C10H8O + C10H7 | 1.44×1010 |
| P3-2 | O2 + C10H8O → C10H8O3 | 2.01×1011 |
| P3-3 | C10H8O3 → H + C10H7O3 | 7.69×1012 |
| P3-4 | C10H7O3 + C4HO2 → C14H8O5 | 1.51×1013 |
| P3-5 | C14H8O5 → C7H3O2 + C7H5O3 | 1.67×1013 |
| P3-6 | C8H8O6→ C7H6O4 + CHO + HO | 5.00×1012 |
| P3-7 | C7H6O4 → C7H5O3 + HO | 3.33×1013 |
| P3-8 | C7H5O3 + H → C7H6O3 | 6.02×1013 |
| P4 | C7H6O3 → C7H6O + O2 | 5.00×1013 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



