
Submitted:
09 September 2024
Posted:
10 September 2024
You are already at the latest version
Abstract
This study investigates the impact of single prolonged stress (SPS), a model of post-traumatic stress disorder (PTSD), on cardiovascular responses, hypothalamic paraventricular nucleus (PVN) activity, and vascular function to elucidate the mechanisms linking traumatic stress to hypertension. Although SPS did not directly cause chronic hypertension in male Sprague Dawley (SD) rats, it induced acute but transient increases in blood pressure and heart rate and significantly altered the expression of hypertension-associated genes, such as vasopressin, angiotensin II type 1 receptor (AT1R), and FOSL1 in the PVN. Notably, mitochondrial reactive oxygen species (mtROS) were predominantly elevated in the pre-autonomic regions of the PVN, colocalizing with AT1R- and FOSL1-expressing cells, suggesting that oxidative stress amplifies sympathetic activation and stress responses. SPS also triggered vascular inflammation, evidenced by increased mRNA levels of pro-inflammatory cytokines (TNFα and IL1β) and inducible nitric oxide synthase (iNOS) in the aorta, and impaired vascular reactivity to vasoconstrictor and vasodilator stimuli, reflecting compromised vascular function. These findings suggest that SPS sensitizes neuroendocrine, autonomic, and vascular pathways, creating a state of cardiovascular vulnerability that could pre-dispose individuals to hypertension when exposed to additional stressors. Understanding these mechanisms provides critical insights into the pathophysiology of stress-related cardiovascular disorders and underscores the need for targeted therapeutic interventions that address oxidative stress and modulate altered PVN pathways to mitigate the cardiovascular impact of PTSD and related conditions.
Keywords:
posttraumatic stress disorder
; single prolonged stress
; paraventricular nucleus
; oxidative stress
; vascular dysfunction
; vascular inflammation
; hypertension
1. Introduction
Post-traumatic stress disorder (PTSD) is a complex psychiatric condition associated with an increased risk of hypertension, a major risk factor for cardiovascular disease—the leading cause of death worldwide [1,2,3]. Studies, particularly among veterans, have demonstrated that individuals with PTSD are significantly more likely to develop hypertension compared to those without the disorder [4,5,6]. Despite this established association, the mechanisms linking PTSD to elevated blood pressure (BP) remain poorly understood. Understanding these mechanisms is critical, as it could inform the development of targeted interventions to prevent or mitigate the progression of hypertension and subsequent cardiovascular disease in individuals with PTSD.
The central nervous system plays a pivotal role in regulating BP and cardiovascular function by modulating sympathetic outflow and hormone levels [7,8] . Hyperactivity of sympathetic outflow and dysregulation of hormonal pathways are key factors in the development of hypertension and cardiovascular disease [9,10]. Among the brain regions involved, the hypothalamic paraventricular nucleus (PVN) is particularly important. The PVN integrates various neural and hormonal signals to modulate physiological processes, including cardiovascular regulation [11]. Two critical components within the PVN, vasopressin (AVP) and the angiotensin II type 1 receptor (AT1R), are essential in controlling BP and cardiovascular function [12,13]. AVP-producing neurons project to the posterior pituitary gland, releasing AVP into circulation to stimulate water reabsorption and vasoconstriction, thereby influencing BP. AT1R, a key component of the renin-angiotensin system (RAS), interacts with angiotensin II to regulate BP and the hypothalamic-pituitary-adrenal axis [13,14]. Dysregulation of these pathways in the PVN is hypothesized to link PTSD to cardiovascular dysfunction [15,16,17].
Oxidative stress, characterized by an imbalance between reactive oxygen species (ROS) production and antioxidant defenses, is another major contributor to hypertension [18]. Oxidative stress within the PVN has been linked to increased sympathetic nervous activity and is observed in various hypertension models [19,20,21]. Given the PVN’s involvement in stress responses, PTSD-induced anxiety may alter the expression of genes involved in BP regulation, such as AVP and AT1R, and elevate oxidative stress within the PVN, potentially contributing to hypertension.
Beyond central nervous system dysregulation, vascular dysfunction—including endothelial dysfunction and inflammation—plays a significant role in hypertension [22,23]. A key aspect of endothelial dysfunction is the reduction in nitric oxide (NO) bioavailability, primarily produced by endothelial nitric oxide synthase (eNOS). Insufficient NO impairs vasodilation, contributing to elevated BP [23]. Vascular inflammation further exacerbates this dysfunction by impairing NO production and increasing vascular resistance [22,24]. The interplay between neuroendocrine dysregulation, oxidative stress, and vascular dysfunction could form a mechanistic pathway linking PTSD to hypertension.
Given this background, we hypothesize that PTSD may contribute to the development of hypertension through the dysregulation of PVN activation and induction of vascular dysfunction. This study utilizes the single prolonged stress (SPS) paradigm, a widely accepted model of PTSD, to induce PTSD-related symptoms in normal Sprague Dawley (SD) rats [25]. Our primary objectives are to investigate whether SPS increases BP and alters the expression of key cardiovascular-related genes in the PVN, including AVP, AT1R, and Fos-like antigen 1 (FOSL1), a marker of chronic neuronal activation, and to examine the role of mitochondrial ROS (mtROS) in these processes. Additionally, we assess the impact of SPS on vascular health by evaluating vascular inflammation, eNOS abundance, and overall vascular function.
2. Materials and Methods
2.1. Animals
Adult male SD rats were obtained from Charles River Laboratories (Wilmington, MA, USA) and utilized in our breeding colony to generate offspring. The rats were housed under controlled conditions, with an ambient temperature ranging from 20 to 24°C and a 12-hour light-dark cycle. The rats were provided ad libitum access to food and water throughout the experimental period. All experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Michigan Technological University.
2.2. SPS Experimental Paradigm
SD rats were randomly assigned to either the control or SPS group. The SPS procedure was administered individually to each rat as described below. First, the rats were immobilized for 2 hours in plastic restraint bags (AIMS™ Rodent Restraint Bag). Immediately afterward, they were subjected to a 20-minute forced swim in a tank filled with water at approximately 25°C. The tank's depth was carefully calibrated so that the rats could not keep their noses above the water surface when their tails touched the bottom. Following the swim, a 10-minute recovery period was provided. Subsequently, the rats were exposed to diethyl ether in a sealed glass container until they lost consciousness. Finally, the rats were individually housed and left undisturbed for 7 days.
2.3. BP Measurement
8-week-old male SD rats were anesthetized, and telemetry transducers (HD-S10, Data Science, St. Paul, MN) were implanted into their femoral artery for BP and heart rate (HR) measurement [26]. Briefly, rats were anesthetized with isoflurane (2–3%), a catheter connected to a telemetry transmitter was inserted into the femoral artery, and the transmitter was placed subcutaneously. Following a recovery period of 1 week from the surgery, baseline recordings of mean arterial BP (MAP) and HR were obtained from conscious rats before initiating any treatment. Subsequent recordings were conducted on the initial day and 7-day intervals post-treatment.
2.4. mRNA Level Measurement in the PVN and Aorta
7 days post SPS, animals were euthanized with an overdose of isoflurane. Subsequently, their brains and aortas were removed and stored at −80°C until further use. The hypothalamic PVN was punched out to measure the mRNA levels of AVP, AT1R and FOSL1. The aorta was ground into powders with liquid nitrogen. RNA isolation was performed using the RNeasy Mini kit (Qiagen) following the manufacturer's instructions. Reverse transcription of RNA (200-400 ng from each sample) was carried out to synthesize cDNA, serving as a template in Real-Time PCR assays. TaqMan primers and probes specific for AVP (Rn00690189_g1), AT1R (Rn01435427_m1), FOSL1 (Rn00564119_m1), tumor necrosis factor alpha (TNFα, Rn99999017_m1), interleukin-1 beta (IL-1β, Rn00580432_m1), interleukin 6 (IL-6, Rn01410330_m1), inducible NOS (iNOS, Rn00561646_m1), nuclear factor NF-kB subunit NF-kB1 (Rn01399583_m1), and eNOS (Rn07312037_g1) were used to measure the mRNA levels of the genes of interest, with GAPDH (Rn01775763_g1) serving as the normalization reference.
2.5. Immunofluorescence of AVP, AT1R and FOSL1 in the PVN
Following SPS on day 7, animals underwent anesthesia with overdose of isoflurane, and then transcardial perfusion was carried out with cold PBS followed by paraformaldehyde (PFA) in 1× PBS. Post-perfusion, their brains were extracted, fixed in 4% PFA overnight, and preserved in 30% sucrose in 1×PBS until reaching the container's bottom. Afterwards, the brains were embedded in O.C.T. compound (Sakura Finetek), cryo-sectioned into 20 µm thick coronal sections containing PVN regions. Brain sections were then rinsed three times in PBS for 10 min each, permeabilized in cold methanol for 10 min and washed with PBS. Subsequent steps involved blocking with 5% horse serum for one hour, followed by incubation with primary antibodies including rabbit anti-FOSL1 (My BioSource, MBS3200399), rabbit anti-AVP (Cell Signaling, 50068) or guinea pig anti-AVP (Synaptic System, 403004), or rabbit anti-AT1R (Alomone Labs, AAR-011) in PBS containing 0.5% Triton X-100 and 5% horse serum for 24 hours at 4°C. The next day, the sections underwent three additional 10-min PBS washes before incubation with secondary antibodies (Alexa Fluor 488 donkey anti-rabbit IgG or Alexa Fluor 594 goat anti-guinea pig IgG) for one hour at room temperature. After additional PBS washes, the sections were mounted in Vectorshield, and fluorescent images were captured.
2.6. Intracerebroventricular (ICV) Injections and mtROS Measurement
Mitochondrial-targeting fluorescent probes (MitoProbe) were employed to determine mtROS levels of brain tissues [27]. Following SPS on day 7, rats received MitoProbe injection into the right lateral ventricle, following established protocols [26]. The ICV injections were guided by stereotaxic coordinates: 0.8-0.9 mm posterior to bregma, 1.4-1.8 mm lateral to the midline, and 3.2-3.8 mm below the dural surface. Injections were administered at a flow rate of 1 µL/min using an UltraMicroPump3 (World Precision Instruments). 3 hours after 10 µL MitoProbe injection (0.2 µM), an overdose of isoflurane was administered for euthanasia, followed by transcardial perfusion as described above. Immunofluorescence of AVP, AT1R and FOSL1 were performed in brain sections containing PVN, and fluorescent images were captured using confocal microscope (Olympus FV1000, Waltham, MA, USA).
2.7. BP and HR Response to Phenylephrine (PE), Sodium Nitroprusside (SNP) and Repeated Restraint
Intraperitoneal (i.p.) injections of 100 µg PE and 200 µg SNP were administered on day 7 following SPS, with a three-hour interval between each i.p. injection. Subsequently, 40-min restraint was performed on rats individually in their home cages. Their MAP and HR were recorded one hour before the initial experiment as a baseline and one hour after each subsequent experiment.
2.8. Statistical Analysis
Statistical comparisons were conducted using Prism 9 (GraphPad), with all data presented as mean ± SEM. The student’s t-test and two-way ANOVA test were applied to assess statistical significance. Differences for MAP and HR of each animal were calculated by subtracting baseline parameters. For experiments investigating the effect of PE, SNP and restraint, post-hoc analyses with Bonferroni multiple comparison tests were performed after establishing significant ANOVA results. The total fluorescent area was quantified using ImageJ software. A p-value of less than 0.05 was considered indicative of statistical significance.
3. Results
3.1. Acute but Transient Cardiovascular Responses to SPS
To assess the acute and longer-term effects of SPS on cardiovascular function, MAP and HR were measured in SPS and control rats 5 to 6 hours after stress exposure on day 1, and again on day 7 following a sensitization period (Figure 1 A and 1B). On day 1, SPS rats exhibited a significant acute increase (p < 0.01) in both MAP (ΔMAP: 8.3 mmHg) and HR (ΔHR: 32.8 beats/min) compared to control rats (ΔMAP: 2.3 mmHg, ΔHR: -1.5 beats/min). However, by day 7, the cardiovascular responses were no longer significantly different between the SPS and control groups, indicating that the initial cardiovascular effects of SPS were temporary. This finding highlights that SPS induces marked acute cardiovascular responses characterized by elevated MAP and HR immediately after exposure, but these changes do not persist beyond the acute phase. The results suggest that while SPS triggers significant immediate cardiovascular stress responses, these do not result in sustained hypertension or altered HR in the absence of additional stressors, emphasizing the need to consider both short- and long-term impacts of stress on cardiovascular health.
3.2. SPS Upregulates mRNA Expressions and Protein Activities of AVP, AT1R, and FOSL1 in the PVN
To assess the impact of SPS on the expression of genes involved in cardiovascular regulation within the PVN, mRNA levels of AVP, AT1R, and FOSL1 were evaluated in SPS-exposed and control rats on day 7 post-treatment. The results showed significant upregulation of AVP (3.9-fold), AT1R (1.6-fold), and FOSL1 (1.6-fold) mRNA levels in SPS rats compared to controls (p < 0.05; Figure 2A-C). Immunofluorescence analysis further confirmed increased protein levels of AVP, AT1R, and FOSL1 in the PVN of SPS-exposed rats, indicating enhanced expression at both the mRNA and protein levels (Figure 2D-I).
Detailed examination of PVN subregions, including the parvocellular (Pa), the intermediocellular (Me), and posterior magnocellular lateral (Ma) areas, revealed distinct patterns of expression [28,29]. The Pa and Me regions primarily contain pre-autonomic neurons involved in regulating sympathetic output, while the Ma region is rich in neuroendocrine neurons that secrete circulating hormones [28,30]. AVP immunoreactivity showed a significant 2.1-fold increase specifically in the magnocellular region (Figure 2D and 2G), suggesting a heightened neuroendocrine response to SPS, whereas no significant changes were observed in the pre-autonomic nuclei (Pa and Me).
Conversely, AT1R and FOSL1 exhibited significant increases across all PVN subregions, with particularly notable upregulation in the Pa and Me areas, which are critical for autonomic regulation (Figure 2E, 2F, 2H, and 2I). This regional specificity underscores the differential impact of SPS on the PVN, suggesting that SPS activates distinct subregions and enhances molecular pathways contributing to altered cardiovascular regulation.
These findings demonstrate that SPS significantly upregulates AVP, AT1R, and FOSL1 in the PVN, with enhanced protein activity in areas associated with both autonomic and neuroendocrine functions. The selective increase of AVP in the magnocellular region points to a specific neuroendocrine response, while the widespread upregulation of AT1R and FOSL1 suggests a broader activation of the PVN’s autonomic pathways. Together, these results highlight the role of SPS in modulating key molecular and regional responses within the PVN, potentially contributing to the cardiovascular dysregulation observed in conditions associated with traumatic stress.
3.3. SPS Elevates mtROS in AT1R- and FOSL1-Expressing Cells in the PVN
SPS significantly increases mtROS levels in the hypothalamic PVN, as evidenced by a substantial rise in fluorescence intensity compared to controls (control: 21,781 vs. SPS: 55,247, area: 0.04 mm², p < 0.05; Figure 3A-B). This increase reflects a notable elevation in oxidative stress within the PVN of SPS-exposed rats. Analysis of subregional distribution revealed that the elevated mtROS production was predominantly localized in the Pa and Me subregions, with minimal impact observed in the Ma region (Figure 3C-E). Notably, the most pronounced mtROS enrichment occurred in cells expressing AT1R and FOSL1 within the Pa and Me subregions, which are critical for autonomic regulation and neuroendocrine function. This colocalization suggests that SPS-induced oxidative stress specifically targets autonomic regulatory regions of the PVN, potentially contributing to dysregulated cardiovascular responses. In contrast, the colocalization of mtROS with AVP-expressing cells was less prominent, indicating a differential impact of SPS on various molecular pathways within the PVN. These findings underscore the critical role of oxidative stress in sensitizing the PVN’s autonomic and neuroendocrine functions, contributing to altered cardiovascular regulation in conditions such as PTSD. The elevation of mtROS in AT1R- and FOSL1-expressing cells highlights the importance of oxidative stress in the molecular and cellular alterations within the PVN following traumatic stress, providing a potential mechanistic link to the cardiovascular changes observed in PTSD and related disorders.
3.4. SPS Elevates mRNA Expression of Pro-Inflammatory Cytokines and Nitric Oxide Synthases in the Aorta of SD Rats
Figure 4 illustrates the impact of SPS on the mRNA expression of pro-inflammatory cytokines and NOS in the aorta of male SD rats. The analysis showed that SPS significantly increased mRNA levels of the pro-inflammatory cytokines TNFα (2.6-fold, p < 0.05) and IL-1β (4.4-fold, p < 0.05) compared to controls (Figure 4A-B). A dramatic 37-fold elevation in iNOS mRNA expression was also observed in the SPS group compared to controls (p < 0.001; Figure 4D), indicating a robust inflammatory response. However, mRNA levels of IL-6 and NF-κB were not significantly altered between SPS and control groups (Figure 4C and 4E). Additionally, SPS led to a significant 2-fold increase in eNOS mRNA expression compared to controls (p < 0.05; Figure 4F), suggesting potential changes in endothelial function.
These results indicate that SPS strongly upregulates key mediators of vascular inflammation and dysfunction in the aorta, including TNFα, IL-1β, and particularly iNOS, which is linked to severe inflammatory responses. The pronounced elevation of these cytokines, alongside iNOS, suggests that SPS triggers significant vascular inflammation, potentially contributing to endothelial damage and dysfunction. The absence of significant changes in IL-6 and NF-κB points to a selective activation of inflammatory pathways rather than a generalized inflammatory response.
These findings suggest that SPS-induced inflammatory and oxidative stress responses in the aorta contribute to vascular dysfunction, highlighting a potential link between traumatic stress and the development of cardiovascular diseases. The distinct upregulation of pro-inflammatory cytokines and NOS underscores the role of SPS in driving vascular pathology, providing insights into the mechanisms by which chronic stress may predispose individuals to cardiovascular complications.
3.5. SPS Attenuates Cardiovascular Responses to Vasoconstrictive, Vasodilatory, and Repeated Restraint Stress in Rats
SPS rats showed a significantly attenuated MAP response to PE compared to controls. After PE injection, SPS rats exhibited a gradual increase in ΔMAP, but the response was significantly lower (Δ23.01 ± 1.4 mmHg) compared to control rats (Δ33.55 ± 2.0 mmHg, p < 0.05; Figure 5A). There was no significant difference in HR response between the two groups following PE administration (Figure 5B). Similarly, SPS rats displayed a reduced MAP response to SNP. Within the first 3 minutes of SNP injection, ΔMAP decreased in both groups; however, SPS rats had a significantly attenuated response (Δ-12.87 ± 1.0 mmHg) compared to controls (Δ-22.75 ± 2.7 mmHg, p < 0.05; Figure 5C). The HR response to SNP was comparable between the two groups, showing no significant differences (Figure 5D).
The effects of repeated restraint stress on cardiovascular response were also assessed. During the 40-minute restraint, SPS rats had a significantly lower ΔMAP (23 ± 0.9 mmHg) compared to controls (30.59 ± 0.5 mmHg, p < 0.05; Figure 5E), even though both groups showed an increase in ΔMAP from their baseline levels. After restraint, the MAP of SPS rats remained elevated and returned to baseline levels more slowly than controls (SPS: 15.32 ± 1.6 mmHg vs. Control: 9.2 ± 1.5 mmHg, p < 0.05). The HR response remained similar across both groups throughout the restraint period (Figure 5F).
These results indicate that SPS impairs cardiovascular reactivity to both vasoconstrictive and vasodilatory stimuli as well as to repeated restraint stress. The attenuated MAP responses to PE and SNP suggest that SPS compromises vascular responsiveness, potentially due to altered autonomic regulation or impaired vascular smooth muscle function. The diminished MAP response during and after restraint stress further supports the idea that SPS disrupts adaptive cardiovascular responses to acute stressors. These findings highlight the potential role of SPS in contributing to vascular dysfunction and reduced cardiovascular adaptability in the face of acute and repeated stress, which could have implications for understanding the cardiovascular impacts of chronic stress exposure in clinical settings.
4. Discussion
The present study aimed to investigate the effects of SPS on cardiovascular responses, PVN activity, and vascular function, with a focus on elucidating the mechanisms through which stress contributes to hypertension. Our major findings indicate that SPS elicits acute but not sustained changes in BP and HR, alters hypertension-associated gene expressions in the PVN, increases mtROS levels in the PVN, and induces vascular inflammation as well as impaired vascular reactivity. These results suggest that traumatic stress may contribute to the progression of hypertension through persistent dysregulation of the PVN and vascular system.
Our data show that SPS led to heightened BP and HR responses on the day of stress exposure, reflecting an immediate cardiovascular response that aligns with previous studies demonstrating transient cardiovascular effects of acute stress [31,32,33,34]. However, these changes were not sustained beyond the sensitization period, indicating that SPS alone does not induce chronic cardiovascular alterations in healthy rats. This finding, combined with previous research showing that rodent models of PTSD exhibit increased susceptibility to Ang II-induced hypertension [35,36], has led us to hypothesize that while SPS does not directly cause long-term hypertension, it may prime the cardiovascular system for heightened reactivity to subsequent stressors, thereby predisposing individuals to hypertension development. To explore this, we compared the expression of hypertension-related genes in the PVN between SPS and control rats.
Our findings demonstrate that SPS significantly alters the expression of key genes involved in BP regulation within the PVN, including AVP, AT1R, and FOSL1, at both mRNA and protein levels (Figure 2). Notably, increased AVP expression in the magnocellular neurons suggests an enhanced neuroendocrine response that could lead to elevated AVP secretion, promoting water retention and vasoconstriction, both of which are associated with increasing BP [12]. We also observed an upregulation of AT1R and FOSL1 throughout the PVN region. PVN AT1R’s involvement in neurosecretion and autonomic control [13,14], coupled with the sustained activation of PVN neurons indicated by increased FOSL1, suggest that they may contribute to the heightened sympathetic nerve activity observed in PTSD and other stress-related conditions [37].
A key aspect of our findings is the significant increase in mtROS levels within the PVN following SPS, particularly in the Pa and Me regions involved in autonomic regulation (Figure 3). The colocalization of mtROS with AT1R- and FOSL1-expressing cells suggests that oxidative stress plays a critical role in amplifying sympathetic outflow and stress responses. This supports the notion that AT1R signaling can drive oxidative stress, which in turn enhances neuronal excitation and sympathetic nerve activity [38,39,40]. The preferential localization of mtROS effects in autonomic subregions rather than endocrine areas points to the selective vulnerability of sympathetic pathways to oxidative stress, contributing to sustained autonomic dysregulation.
SPS also induced significant changes in vascular function, characterized by increased expression of pro-inflammatory cytokines, including TNFα and IL-1β, in the aorta (Figure 4). These inflammatory mediators are known to impair endothelial function, fostering vascular inflammation and contributing to vascular dysfunction [22,41]. Notably, our results demonstrated significant increases in mRNA levels of both eNOS (2-fold) and iNOS (37-fold) in the aorta of SPS rats compared to controls. Both enzymes produce NO, a crucial signaling molecule that regulates blood vessel tone and BP. Under normal conditions, NO is primarily produced by endothelial cells via eNOS, facilitating vasodilation by relaxing smooth muscle cells in blood vessel walls, which lowers BP. In contrast, iNOS is typically induced by inflammatory stimuli and produces larger amounts of NO [42]. Although NO generally promotes vasodilation, its reactive nature means that excessive NO can lead to the formation of peroxynitrite, a reactive nitrogen species that causes oxidative stress [43]. This oxidative stress damages endothelial cells, impairs their ability to relax, and fosters vascular inflammation. As a result, blood vessel integrity is compromised, leading to increased vascular resistance, potentially contributing to hypertension and cardiovascular disease. Consistently, impaired vascular responses were observed in SPS rats during exposure to the vasoconstrictor PE and the vasodilator SNP (Figure 5). The reduced MAP responses suggest that SPS disrupts normal vascular function, impairing the ability of blood vessels to constrict or dilate effectively. This impaired reactivity, combined with increased oxidative stress and inflammation, highlights a potential mechanism by which chronic stress exposure may predispose individuals to cardiovascular diseases, including hypertension.
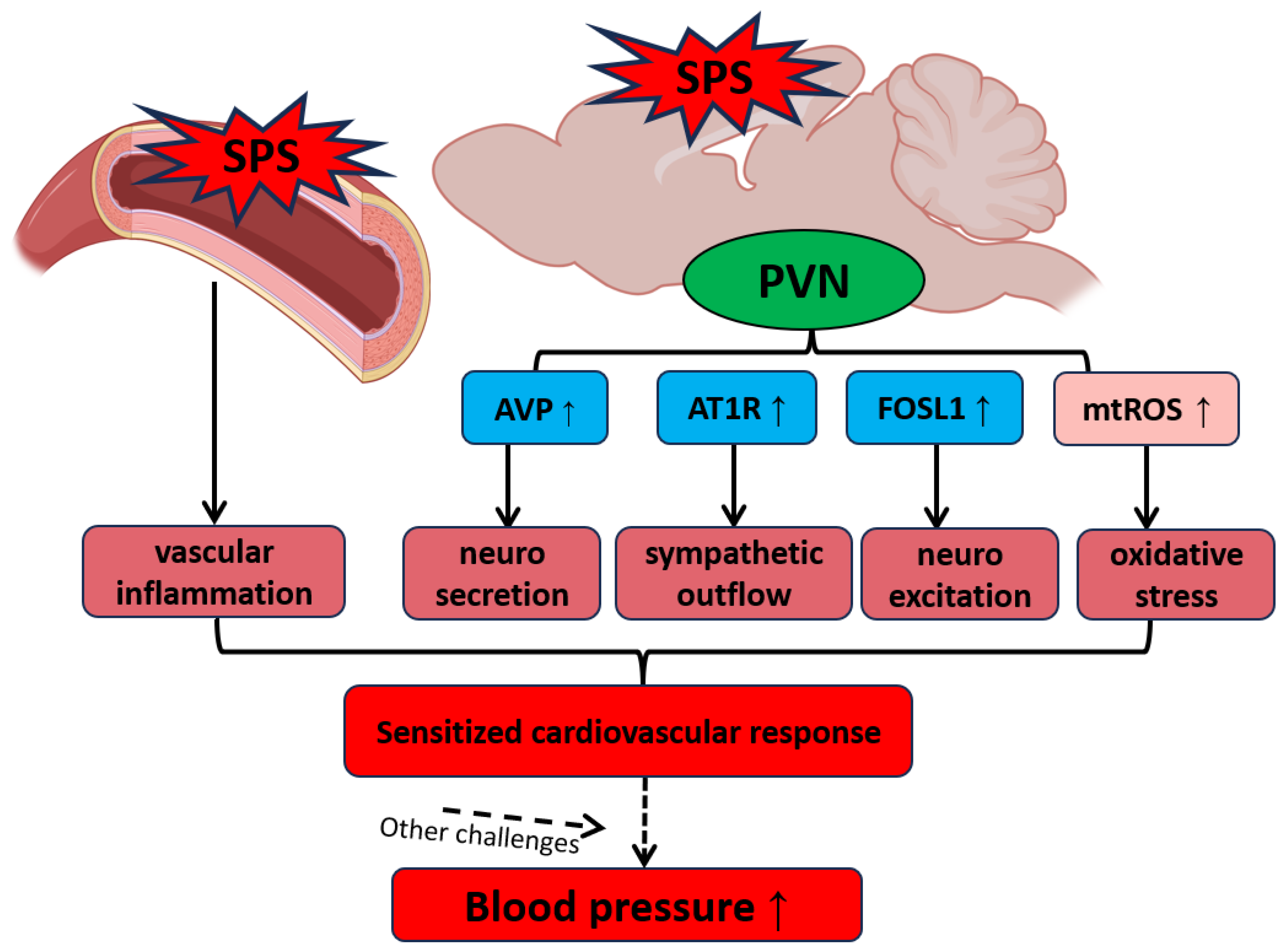
Our findings suggest a complex interplay between neuroendocrine and autonomic dysregulation within the PVN, amplified by oxidative stress and inflammation in both neural and vascular systems, as a key mechanism underlying PTSD-related hypertension (Figure 6). SPS-induced increases in mtROS as well as alterations in gene expression within the PVN and aorta enhance sympathetic outflow and disrupt vascular homeostasis, creating a state of cardiovascular vulnerability that may lead to hypertension under additional stress conditions. This hypothesized mechanism underscores the potential for traumatic stress to serve as a critical trigger for hypertension, particularly when combined with other cardiovascular stressors. Future research should focus on exploring therapeutic strategies targeting these stress-induced pathways to mitigate the long-term cardiovascular impact of PTSD and related disorders.
Conclusions
This study demonstrates that SPS induces acute but transient increases in BP and HR, disrupts the expression of hypertension-related genes in the PVN, elevates mtROS levels, and triggers vascular inflammation and impaired vascular reactivity. Although SPS does not directly cause chronic high BP, it induces persistent changes in the PVN and vascular system that may heighten susceptibility to hypertension and cardiovascular dysfunction when exposed to additional stressors. The findings emphasize the crucial role of oxidative stress, particularly mtROS, in driving sympathetic activation and stress responses, linking neuroendocrine, autonomic, and vascular dysregulation as key mechanisms underlying the cardiovascular effects of traumatic stress.
Clinically, these results suggest that traumatic stress may predispose individuals to cardiovascular diseases, especially under further stress exposure. Understanding these pathways provides critical insights into the pathophysiology of stress-related cardiovascular disorders such as PTSD, highlighting the need for targeted therapeutic strategies that address oxidative stress and modulate altered PVN pathways. Interventions that reduce oxidative stress, regulate neuroendocrine and autonomic dysfunction, or restore vascular health could lower the risk of cardiovascular complications in individuals exposed to chronic or traumatic stress, ultimately improving clinical outcomes in this vulnerable population. Future research should focus on developing and testing such targeted approaches to protect cardiovascular health in those affected by stress-related disorders.
Limitations
This study provides valuable insights into the cardiovascular effects of SPS, particularly focusing on PVN dysregulation, oxidative stress, and vascular inflammation. However, several limitations should be noted. First, the research was conducted on healthy male SD rats, which limits the generalizability of the findings to other populations, such as females and individuals with pre-existing health conditions. Future studies should include both sexes and models with underlying conditions to enhance applicability. Additionally, the study only examined effects within 7 days post-stress, leaving the long-term cardiovascular impact of SPS uncertain. Lon-term studies are necessary to determine whether the observed molecular changes lead to lasting cardiovascular dysfunction. Furthermore, in the experiment testing blood vessel function in response to vasoconstrictive and vasodilatory stimuli, PE and SNP should ideally be administered to conscious rats via a pre-implanted catheter directly into the bloodstream. However, i.p. injections were used in this study, which may have delayed drug absorption and caused additional stress, potentially affecting the BP response. Despite this, since all rats received the same treatment, the observations remain valuable for evaluating vascular function.
Author Contributions
Conceptualization, X.C., L.B. and Z.S.; methodology, X.C., X.Y., Q.-H.C., C.Yu., L.B. and Z.S.; data curation, X.C., and Z.S.; software, X.C.; validation, X.C. and Z.S.; investigation, X.C. and Z.S.; writing—original draft preparation, X.C., L.B. and Z.S.; writing—review and editing, X.C., X.Y., Q.-H.C., C.Yu., L.B. and Z.S.; supervision, Q.-H.C., C.Yu., L.B. and Z.S.; funding acquisition, Z.S., C.Yu. and L.B. All authors have read and agreed to the published version of the manuscript.
Funding
The project is supported by the NIH grant R01HL163159 (Z.S.), R15HL150703 (Z.S.) and R15NS133859 (C.Yu.), NIH (R15 EB035866, L.B), and an American Heart Association (AHA) grant 1807047 (L.B.).
Institutional Review Board Statement
The animal study was approved by the Institutional Animal Care and Use Committee (IACUC) at Michigan Technological University (Animal protocol number L0250).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to privacy.
Acknowledgments
The authors wish to acknowledge the critical support from the aforementioned funding bodies: the NIH and AHA. Moreover, the authors sincerely thank Dr. Rick Koubek for creating and maintaining an encouraging research environment that has significantly benefited our ongoing projects.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kibler, J.L.; Joshi, K.; Ma, M. Hypertension in relation to posttraumatic stress disorder and depression in the US National Comorbidity Survey. Behav Med 2009, 34, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, S.E. Hypertension and cardiovascular risk: General aspects. Pharmacol Res 2018, 129, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, F.D.; Whelton, P.K. High Blood Pressure and Cardiovascular Disease. Hypertension 2020, 75, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.M.; Brandt, C.; Buta, E.; Schwartz, J.; Bathulapalli, H.; Dziura, J.; Edmondson, D.E.; Haskell, S. Risk for Incident Hypertension Associated With Posttraumatic Stress Disorder in Military Veterans and the Effect of Posttraumatic Stress Disorder Treatment. Psychosom Med 2017, 79, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.E.; Marmar, C.; Ren, L.; Bertenthal, D.; Seal, K.H. Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. Jama 2009, 302, 489–492. [Google Scholar] [CrossRef]
- Howard, J.T.; Sosnov, J.A.; Janak, J.C.; Gundlapalli, A.V.; Pettey, W.B.; Walker, L.E.; Stewart, I.J. Associations of Initial Injury Severity and Posttraumatic Stress Disorder Diagnoses With Long-Term Hypertension Risk After Combat Injury. Hypertension 2018, 71, 824–832. [Google Scholar] [CrossRef]
- Chopra, S.; Baby, C.; Jacob, J.J. Neuro-endocrine regulation of blood pressure. Indian J Endocrinol Metab 2011, 15 Suppl 4, S281–288. [Google Scholar] [CrossRef]
- Stocker, S.D.; Ferreira, C.B.; Souza, G.; Abbott, S.B.G. Brain Pathways in Blood Pressure Regulation. Hypertension 2024, 81, 383–386. [Google Scholar] [CrossRef]
- Malpas, S.C. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 2010, 90, 513–557. [Google Scholar] [CrossRef]
- Szczepanska-Sadowska, E.; Czarzasta, K.; Cudnoch-Jedrzejewska, A. Dysregulation of the Renin-Angiotensin System and the Vasopressinergic System Interactions in Cardiovascular Disorders. Curr Hypertens Rep 2018, 20, 19. [Google Scholar] [CrossRef]
- Benarroch, E.E. Paraventricular nucleus, stress response, and cardiovascular disease. Clin Auton Res 2005, 15, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Japundžić-Žigon, N.; Lozić, M.; Šarenac, O.; Murphy, D. Vasopressin & Oxytocin in Control of the Cardiovascular System: An Updated Review. Curr Neuropharmacol 2020, 18, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Arnold, A.C. The renin-angiotensin system in cardiovascular autonomic control: recent developments and clinical implications. Clin Auton Res 2019, 29, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Elsaafien, K.; Kirchner, M.K.; Mohammed, M.; Eikenberry, S.A.; West, C.; Scott, K.A.; de Kloet, A.D.; Stern, J.E.; Krause, E.G. Identification of Novel Cross-Talk between the Neuroendocrine and Autonomic Stress Axes Controlling Blood Pressure. J Neurosci 2021, 41, 4641–4657. [Google Scholar] [CrossRef]
- Sipos, E.; Török, B.; Barna, I.; Engelmann, M.; Zelena, D. Vasopressin and post-traumatic stress disorder. Stress 2020, 23, 732–745. [Google Scholar] [CrossRef]
- Saavedra, J.M.; Ando, H.; Armando, I.; Baiardi, G.; Bregonzio, C.; Jezova, M.; Zhou, J. Brain angiotensin II, an important stress hormone: regulatory sites and therapeutic opportunities. Ann N Y Acad Sci 2004, 1018, 76–84. [Google Scholar] [CrossRef]
- Saavedra, J.M.; Benicky, J. Brain and peripheral angiotensin II play a major role in stress. Stress 2007, 10, 185–193. [Google Scholar] [CrossRef]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ Res 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Fujita, M.; Ando, K.; Nagae, A.; Fujita, T. Sympathoexcitation by oxidative stress in the brain mediates arterial pressure elevation in salt-sensitive hypertension. Hypertension 2007, 50, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Kishi, T.; Hirooka, Y. Oxidative stress in the brain causes hypertension via sympathoexcitation. Front Physiol 2012, 3, 335. [Google Scholar] [CrossRef]
- Hirooka, Y. Oxidative stress in the cardiovascular center has a pivotal role in the sympathetic activation in hypertension. Hypertens Res 2011, 34, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Savoia, C.; Sada, L.; Zezza, L.; Pucci, L.; Lauri, F.M.; Befani, A.; Alonzo, A.; Volpe, M. Vascular inflammation and endothelial dysfunction in experimental hypertension. Int J Hypertens 2011, 2011, 281240. [Google Scholar] [CrossRef]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv Exp Med Biol 2017, 956, 511–540. [Google Scholar] [CrossRef]
- Dinh, Q.N.; Drummond, G.R.; Sobey, C.G.; Chrissobolis, S. Roles of inflammation, oxidative stress, and vascular dysfunction in hypertension. Biomed Res Int 2014, 2014, 406960. [Google Scholar] [CrossRef] [PubMed]
- Lisieski, M.J.; Eagle, A.L.; Conti, A.C.; Liberzon, I.; Perrine, S.A. Single-Prolonged Stress: A Review of Two Decades of Progress in a Rodent Model of Post-traumatic Stress Disorder. Front Psychiatry 2018, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.J.; Fan, Y.; Jiang, E.; Zhu, F.; Larson, R.A.; Yan, J.; Li, N.; Chen, Q.H.; Shan, Z. Increased activity of the orexin system in the paraventricular nucleus contributes to salt-sensitive hypertension. Am J Physiol Heart Circ Physiol 2017, 313, H1075–h1086. [Google Scholar] [CrossRef]
- Yapici, N.B.; Mandalapu, S.; Gibson, K.M.; Bi, L. Targeted fluorescent probes for detection of oxidative stress in the mitochondria. Bioorg Med Chem Lett 2015, 25, 3476–3480. [Google Scholar] [CrossRef]
- Feetham, C.H.; O'Brien, F.; Barrett-Jolley, R. Ion Channels in the Paraventricular Hypothalamic Nucleus (PVN); Emerging Diversity and Functional Roles. Front Physiol 2018, 9, 760. [Google Scholar] [CrossRef]
- Kiss, J.Z.; Martos, J.; Palkovits, M. Hypothalamic paraventricular nucleus: a quantitative analysis of cytoarchitectonic subdivisions in the rat. J Comp Neurol 1991, 313, 563–573. [Google Scholar] [CrossRef]
- Chen, J.; Gomez-Sanchez, C.E.; Penman, A.; May, P.J.; Gomez-Sanchez, E. Expression of mineralocorticoid and glucocorticoid receptors in preautonomic neurons of the rat paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol 2014, 306, R328–340. [Google Scholar] [CrossRef]
- Farah, V.M.; Joaquim, L.F.; Bernatova, I.; Morris, M. Acute and chronic stress influence blood pressure variability in mice. Physiol Behav 2004, 83, 135–142. [Google Scholar] [CrossRef] [PubMed]
- do Vale, G.T.; Leoni, D.; Sousa, A.H.; Gonzaga, N.A.; Uliana, D.L.; La Gata, D.C.; Resstel, L.B.; Padovan, C.M.; Tirapelli, C.R. Acute restraint stress increases blood pressure and oxidative stress in the cardiorenal system of rats: a role for AT(1) receptors. Stress 2020, 23, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Irvine, R.J.; White, J.; Chan, R. The influence of restraint on blood pressure in the rat. J Pharmacol Toxicol Methods 1997, 38, 157–162. [Google Scholar] [CrossRef] [PubMed]
- McDougall, S.J.; Paull, J.R.; Widdop, R.E.; Lawrence, A.J. Restraint stress : differential cardiovascular responses in Wistar-Kyoto and spontaneously hypertensive rats. Hypertension 2000, 35, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Xue, J.; Yu, Y.; Wei, S.G.; Beltz, T.G.; Felder, R.B.; Johnson, A.K. Predator Scent-Induced Sensitization of Hypertension and Anxiety-like Behaviors. Cell Mol Neurobiol 2022, 42, 1141–1152. [Google Scholar] [CrossRef]
- Xue, B.; Yu, Y.; Wei, S.G.; Beltz, T.G.; Guo, F.; Felder, R.B.; Johnson, A.K. Stress-Induced Sensitization of Angiotensin II Hypertension Is Reversed by Blockade of Angiotensin-Converting Enzyme or Tumor Necrosis Factor-α. Am J Hypertens 2019, 32, 909–917. [Google Scholar] [CrossRef]
- Fu, Q. Autonomic dysfunction and cardiovascular risk in post-traumatic stress disorder. Auton Neurosci 2022, 237, 102923. [Google Scholar] [CrossRef]
- Su, Q.; Huo, C.J.; Li, H.B.; Liu, K.L.; Li, X.; Yang, Q.; Song, X.A.; Chen, W.S.; Cui, W.; Zhu, G.Q.; et al. Renin-angiotensin system acting on reactive oxygen species in paraventricular nucleus induces sympathetic activation via AT1R/PKCγ/Rac1 pathway in salt-induced hypertension. Sci Rep 2017, 7, 43107. [Google Scholar] [CrossRef]
- Braga, V.A.; Medeiros, I.A.; Ribeiro, T.P.; Franca-Silva, M.S.; Botelho-Ono, M.S.; Guimaraes, D.D. Angiotensin-II-induced reactive oxygen species along the SFO-PVN-RVLM pathway: implications in neurogenic hypertension. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas / Sociedade Brasileira de Biofisica... [et al.] 2011, 44, 871–876. [Google Scholar] [CrossRef]
- Burmeister, M.A.; Young, C.N.; Braga, V.A.; Butler, S.D.; Sharma, R.V.; Davisson, R.L. In vivo bioluminescence imaging reveals redox-regulated activator protein-1 activation in paraventricular nucleus of mice with renovascular hypertension. Hypertension 2011, 57, 289–297. [Google Scholar] [CrossRef]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.; Hayes, A.; Caprnda, M.; Petrovic, D.; Rodrigo, L.; Kruzliak, P.; Zulli, A. Inducible nitric oxide synthase: Good or bad? Biomed Pharmacother 2017, 93, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: regulation and function. Eur Heart J 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Effects of Single Prolonged Stress (SPS) on Physiological Responses in Male Sprague Dawley (SD) Rats on Day 1 and Day 7 Post-Treatment. Changes in mean arterial pressure (ΔMAP, A) and heart rate (ΔHR, B) were assessed in male SD rats with and without SPS on the initial day (Day 1) and seven days post-treatment (Day 7). Each data point represents individual values from one rat (n=4-8 per group). Statistical analysis was performed using a two-way unpaired Student’s t-test. Data are presented as mean ± SEM. * indicates p < 0.05, ** indicates p < 0.01.
Figure 1.
Effects of Single Prolonged Stress (SPS) on Physiological Responses in Male Sprague Dawley (SD) Rats on Day 1 and Day 7 Post-Treatment. Changes in mean arterial pressure (ΔMAP, A) and heart rate (ΔHR, B) were assessed in male SD rats with and without SPS on the initial day (Day 1) and seven days post-treatment (Day 7). Each data point represents individual values from one rat (n=4-8 per group). Statistical analysis was performed using a two-way unpaired Student’s t-test. Data are presented as mean ± SEM. * indicates p < 0.05, ** indicates p < 0.01.
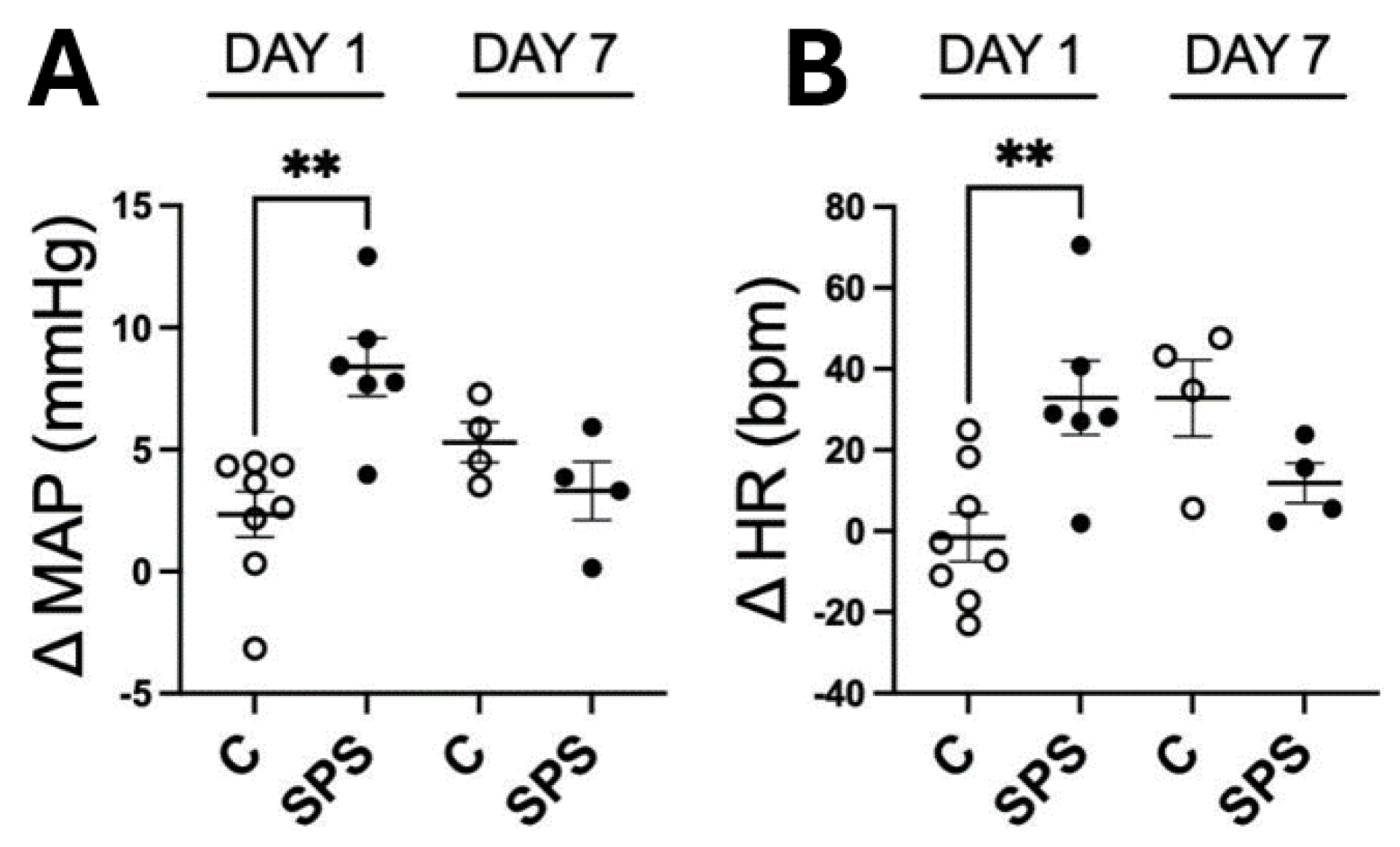
Figure 2.
SPS Increases mRNA Expression and Immunofluorescence of AVP, AT1R, and FOSL1 in the Hypothalamic Paraventricular Nucleus (PVN) of Male SD Rats. The mRNA levels of AVP (A), AT1R (B), and FOSL1 (C) were measured in the PVN of male SD rats with and without SPS exposure. mRNA expression was semi-quantified using real-time PCR and normalized to GAPDH, comparing control and SPS groups. Each data point represents individual values from one rat (n=7-8 per group). Statistical analysis was performed using a two-way unpaired Student’s t-test. (D-F) Representative fluorescent images show AVP (D, red), AT1R (E, green), and FOSL1 (F, green) within the PVN. Scale bar = 200 µm. The total fluorescent areas of AVP (G), AT1R (H), and FOSL1 (I) were quantified in the parvocellular (Pa), intermediocellular (Me), and posterior magnocellular lateral (Ma) subregions of the PVN and normalized to control values. Each data point represents a microscopic view of a PVN region. Statistical analysis was conducted using a one-way unpaired Student’s t-test. Data are presented as mean ± SEM. * indicates p < 0.05, ** p < 0.01.
Figure 2.
SPS Increases mRNA Expression and Immunofluorescence of AVP, AT1R, and FOSL1 in the Hypothalamic Paraventricular Nucleus (PVN) of Male SD Rats. The mRNA levels of AVP (A), AT1R (B), and FOSL1 (C) were measured in the PVN of male SD rats with and without SPS exposure. mRNA expression was semi-quantified using real-time PCR and normalized to GAPDH, comparing control and SPS groups. Each data point represents individual values from one rat (n=7-8 per group). Statistical analysis was performed using a two-way unpaired Student’s t-test. (D-F) Representative fluorescent images show AVP (D, red), AT1R (E, green), and FOSL1 (F, green) within the PVN. Scale bar = 200 µm. The total fluorescent areas of AVP (G), AT1R (H), and FOSL1 (I) were quantified in the parvocellular (Pa), intermediocellular (Me), and posterior magnocellular lateral (Ma) subregions of the PVN and normalized to control values. Each data point represents a microscopic view of a PVN region. Statistical analysis was conducted using a one-way unpaired Student’s t-test. Data are presented as mean ± SEM. * indicates p < 0.05, ** p < 0.01.
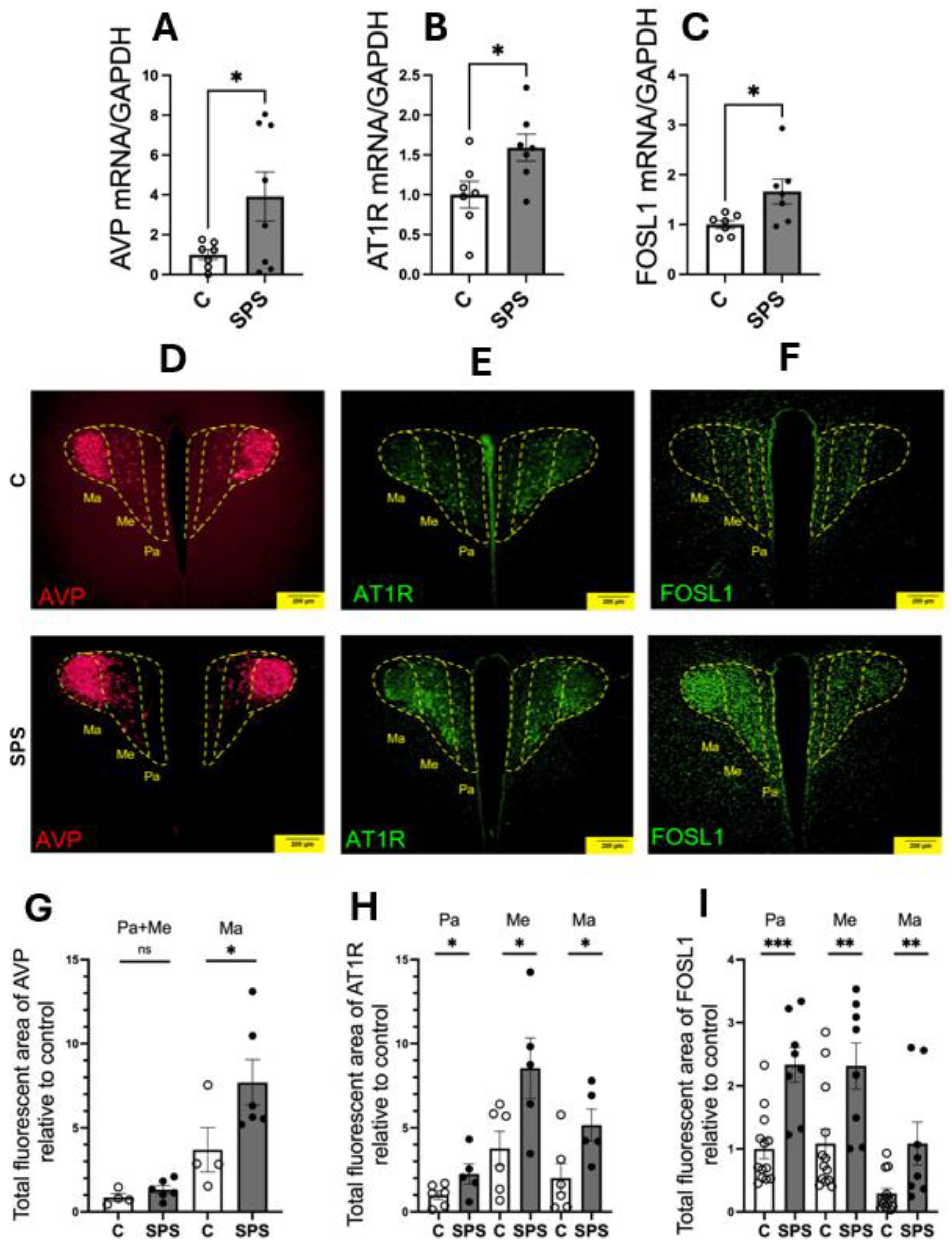
Figure 3.
SPS Increases Mitochondrial Reactive Oxygen Species (mtROS) and Its Colocalization with AT1R- and FOSL1-Expressing Cells in the PVN of SD Rats. (A) Representative fluorescent image showing mtROS (in red) within the PVN. Scale bar = 50 µm. (B) Quantification of the total fluorescent area of mtROS, comparing SPS and control rats. Data points represent average fluorescent intensities from multiple microscopic fields within the PVN region (n=4-5 per group). Each image area is 0.04 mm². Statistical analysis was conducted using a two-way unpaired Student’s t-test. (C-E) Representative fluorescent images depicting the distribution of mtROS (in red) across PVN subdivisions, including the Pa, Me, and Ma regions. Colocalization of mtROS with AVP (C), AT1R (D), and FOSL1 (E) was evaluated. Scale bar = 50 µm. Data are presented as mean ± SEM. * indicates p < 0.05.
Figure 3.
SPS Increases Mitochondrial Reactive Oxygen Species (mtROS) and Its Colocalization with AT1R- and FOSL1-Expressing Cells in the PVN of SD Rats. (A) Representative fluorescent image showing mtROS (in red) within the PVN. Scale bar = 50 µm. (B) Quantification of the total fluorescent area of mtROS, comparing SPS and control rats. Data points represent average fluorescent intensities from multiple microscopic fields within the PVN region (n=4-5 per group). Each image area is 0.04 mm². Statistical analysis was conducted using a two-way unpaired Student’s t-test. (C-E) Representative fluorescent images depicting the distribution of mtROS (in red) across PVN subdivisions, including the Pa, Me, and Ma regions. Colocalization of mtROS with AVP (C), AT1R (D), and FOSL1 (E) was evaluated. Scale bar = 50 µm. Data are presented as mean ± SEM. * indicates p < 0.05.
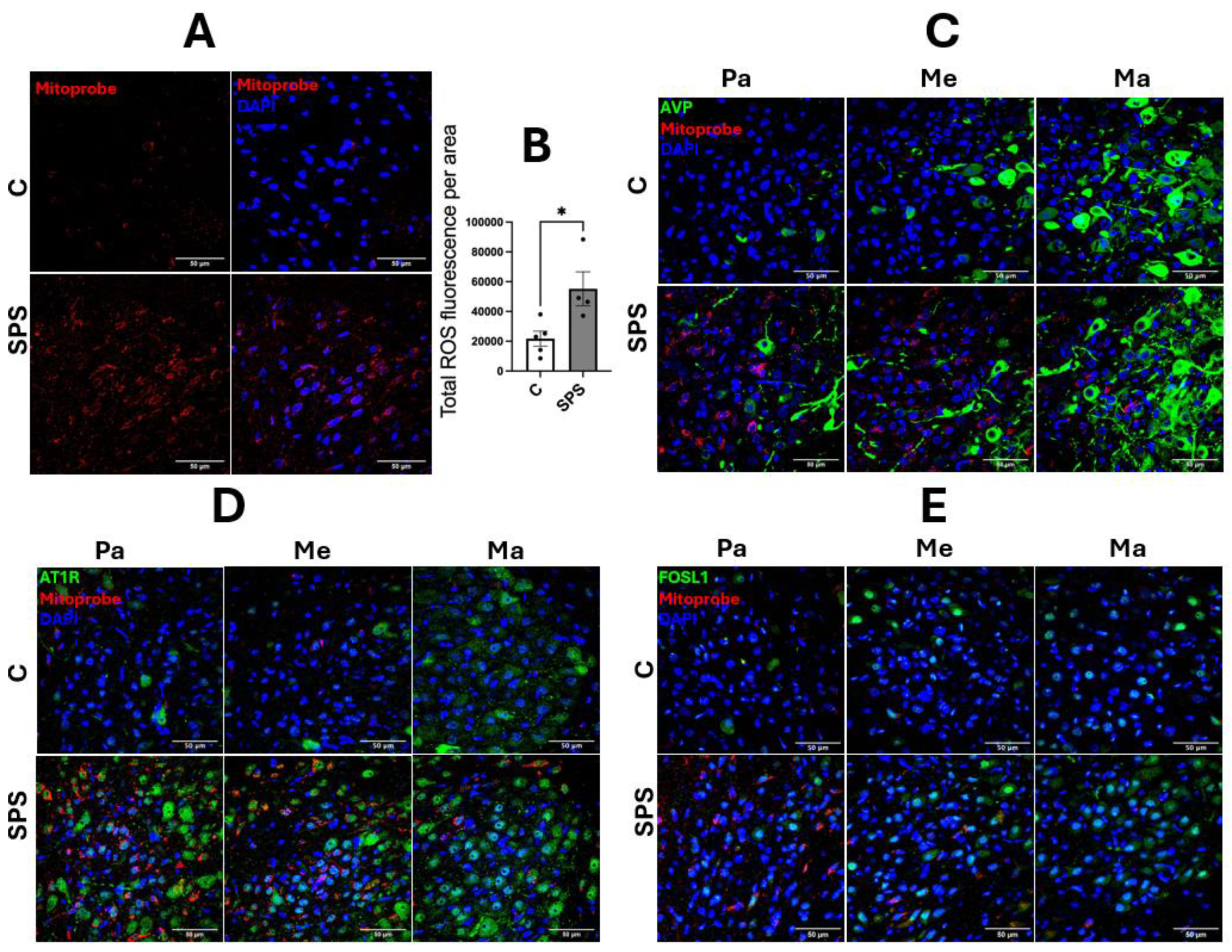
Figure 4.
SPS Increases mRNA Expression of Pro-Inflammatory Cytokines and Nitric Oxide Synthases in the Aorta of Male SD Rats. The mRNA levels of TNFα (A), IL-1β (B), IL-6 (C), iNOS (D), NF-κB (E), and eNOS (F) were measured in the aorta of male SD rats with and without SPS exposure. Gene expression was quantified using real-time PCR, with mRNA levels normalized to GAPDH. Statistical analysis was performed using a two-way unpaired Student’s t-test. Each data point represents an individual rat (n=3 per group). Data are presented as mean ± SEM. Statistical significance is indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 4.
SPS Increases mRNA Expression of Pro-Inflammatory Cytokines and Nitric Oxide Synthases in the Aorta of Male SD Rats. The mRNA levels of TNFα (A), IL-1β (B), IL-6 (C), iNOS (D), NF-κB (E), and eNOS (F) were measured in the aorta of male SD rats with and without SPS exposure. Gene expression was quantified using real-time PCR, with mRNA levels normalized to GAPDH. Statistical analysis was performed using a two-way unpaired Student’s t-test. Each data point represents an individual rat (n=3 per group). Data are presented as mean ± SEM. Statistical significance is indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001.
Figure 5.
Effects of Phenylephrine (PE), Sodium Nitroprusside (SNP), and 40-Minute Restraint on Male SD Rats with and without SPS. Panels (A-D) depict the cardiovascular responses of SPS and control rats (n=3 per group) 7 days post-SPS. Rats were administered 100 μg of PE, a vasoconstrictor, and 200 μg of SNP, a vasodilator, via intraperitoneal (i.p.) injection. MAP and HR were measured before, during, and after each injection, with a minimum interval of 3 hours between treatments. Panels (E-F) show the responses to a 40-minute restraint stress conducted 7 days post-SPS. Baseline BP and HR were recorded prior to the start of the restraint. A two-way ANOVA test was used to assess differences between SPS and control groups. Data are presented as mean ± SEM. * indicates p < 0.05.
Figure 5.
Effects of Phenylephrine (PE), Sodium Nitroprusside (SNP), and 40-Minute Restraint on Male SD Rats with and without SPS. Panels (A-D) depict the cardiovascular responses of SPS and control rats (n=3 per group) 7 days post-SPS. Rats were administered 100 μg of PE, a vasoconstrictor, and 200 μg of SNP, a vasodilator, via intraperitoneal (i.p.) injection. MAP and HR were measured before, during, and after each injection, with a minimum interval of 3 hours between treatments. Panels (E-F) show the responses to a 40-minute restraint stress conducted 7 days post-SPS. Baseline BP and HR were recorded prior to the start of the restraint. A two-way ANOVA test was used to assess differences between SPS and control groups. Data are presented as mean ± SEM. * indicates p < 0.05.
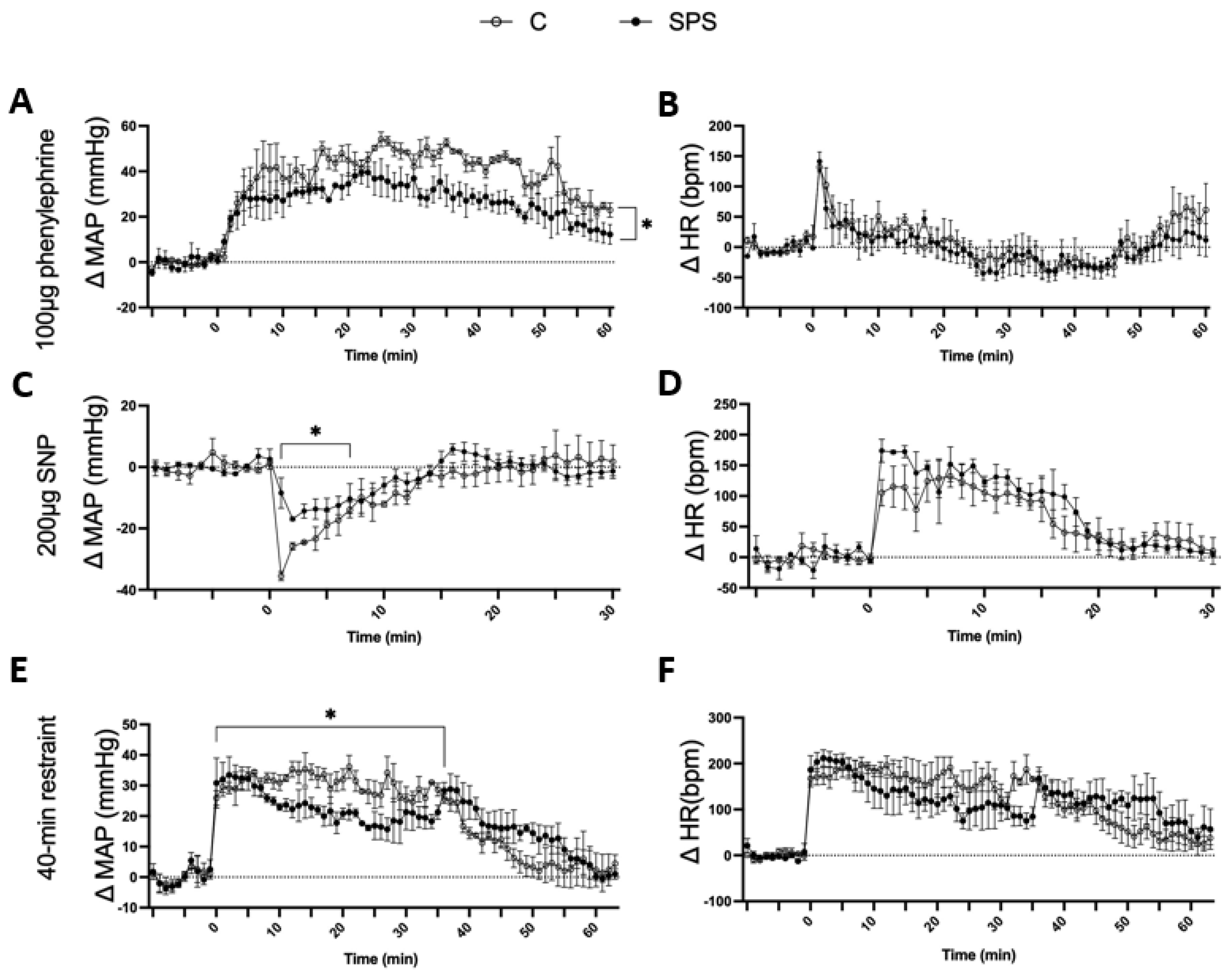
Figure 6.
Hypothesized Mechanisms of PTSD-Related Hypertension Involving PVN Activation and Vascular Dysfunction. Traumatic stress increases the levels of AVP, AT1R, FOSL1, and mtROS in the PVN, reflecting enhanced neurosecretory activity, heightened neuroexcitation, and increased oxidative stress within this brain region. These alterations, coupled with vascular dysfunction, sensitize the cardiovascular system, making it more reactive to additional stressors and contributing to the development and progression of hypertension.
Figure 6.
Hypothesized Mechanisms of PTSD-Related Hypertension Involving PVN Activation and Vascular Dysfunction. Traumatic stress increases the levels of AVP, AT1R, FOSL1, and mtROS in the PVN, reflecting enhanced neurosecretory activity, heightened neuroexcitation, and increased oxidative stress within this brain region. These alterations, coupled with vascular dysfunction, sensitize the cardiovascular system, making it more reactive to additional stressors and contributing to the development and progression of hypertension.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



