
Submitted:
10 September 2024
Posted:
10 September 2024
You are already at the latest version
Abstract
The past few decades have brought tremendous insight into the molecular and pathophysiological mechanisms responsible for thrombus generation. For the clinician it is usually sufficient to explain the incident deep vein thrombosis (DVT) with provoking factors such as trauma with vascular injury, immobilization, hormonal factors, or inherited or acquired coagulation defects. About half of the DVTs are, however, lacking such triggers and are called unprovoked. Venous stasis and hypoxia at the valve sinus level may start a chain of reactions. In order to better understand the mechanisms behind DVT and reach beyond the above-mentioned simplifications animal models and clinical epidemiological studies have informed of the complex interplay between leukocytes, platelets, endothelium, cytokines, complements and coagulation factors and inhibitors. These pathways and the interplay will be reviewed here.
Keywords:
venous thromboembolism
; hypoxia
; stasis
; cytokines
; neutrophil extracellular traps
; complements
1. Introduction
The pathophysiology of venous thrombosis has historically been explained with Virchow’s triad, which consists of hypercoagulability, venous stasis and injury to the endothelium of the blood vessels. Although the physician-scientist Rudolf Virchow studied venous thrombosis with its consequences, and coined the terms venous thrombosis and pulmonary embolism, he did not explicitly state that the components of the triad were the cause of venous thrombosis, as subsequently explained [1]. It was only about a hundred years later that Virchow’s triad started being quoted [2].
Studies in animal models have improved our understanding of the pathophysiology of thrombosis. The events on a molecular level during venous stasis have been investigated. Furthermore, the effects of hypoxemia have evoked substantial interest. Whole-genome sequencing and other molecular-genetic analyses have led to new important discoveries regarding thrombotic mechanisms. Although Rudolf Virchow ridiculed a French colleague for proposing that inflammation was causative in thrombosis [1], we now know better. Thromboinflammation has become a major player in the field.
2. The Basic Principles of Pathophysiology of Venous Thrombosis
2.1. Venous Stasis
Situations when venous stasis is implicated in the generation of thrombosis include deep vein thrombosis (DVT) in the lower extremity during immobilization, left atrial appendage thrombus formation in atrial fibrillation, left ventricle thrombosis due to akinetic myocardium and infarction, or due to actual obstruction of the blood flow such as external compression from an expansive process (typically a tumor) or from restricted venous lumen in catheter-related arm vein thrombosis or possibly also in recurrent DVT where there is residual thrombus or fibrotic scar from a prior event. On a molecular level there are a few possible mechanisms: hypoxemia, activated coagulation factors, and inflammatory reaction. In a dog model as well as in patients it has been shown that the blood within the pocket of the venous valves, also called valve sinus, became rapidly hypoxic during stasis and thrombus formation on the valve cusp could be demonstrated after 2 h of non-pulsatile flow [3].
2.1.1. Hypoxemia
Yan et al at Nigel Mackman’s laboratory used a murine lung thrombosis model and concluded that since an antibody against tissue factor (TF) prevented fibrin deposition during hypoxia, the expression of TF is an important component of the process [4]. Likewise, depletion of monocytes reduced fibrin accumulation in the hypoxic lung. The transcription factor early growth response-1 (Egr-1) seems to be an important modulator of the TF expression, because in homozygous Egr-1 null-mice there was no increase in TF antigen and no fibrin deposition during hypoxia [5]. The same research group subsequently demonstrated that protein kinase C (PKC) beta-isoform is upstream to Egr-1, since PKC beta null mice showed only minimal Egr-1 increase during hypoxia [6]. Following this, Lo et al in Taiwan found, using a bovine aortic endothelium model, that PKC alpha also is a mediator, and they further demonstrated that the signaling pathway goes from hypoxia triggering a temporary translocation of PKC alpha from cytosol to a membrane fraction, where it associates with Ras or Raf-1 or ERK1/ERK2 and eventually induces Egr-1 [7].
Another pathway is via the hypoxia-inducible factor-1α (HIF-1 α), which normally is hydroxylated and degraded, but under hypoxic conditions it becomes stabilized and able to mediate transcriptional activation of various genes, including those involved in angiogenesis, apoptosis, and metastases, and this is mediated by TF [8].
On the other side of the hemostatic mechanisms is the fibrinolysis, of which plasminogen activator inhibitor-1 (PAI-1) is a major factor for suppressing fibrinolysis and thereby increase the risk of thrombosis. Liao et al at Ann Arbor, Michigan, demonstrated in a murine macrophage model that the increase of PAI-1 in response to hypoxia is an effect of both increased transcription and improved mRNA stability [9]. This PAI-1 upregulation is mediated by HIF-1 α (possibly the dominant pathway), Egr-1 and CCAAT/enhancer binding protein alpha.
There are also report of increased levels of factors VIII, VIIa, XII, fibrinogen, endothelial protein C receptor, Annexin IV, thrombin-activatable fibrinolysis inhibitor, antiplasmin and platelet activation, as reviewed by Bikov et al [10].
The main hypoxia-induced pathways leading to increased thrombus formation are summarized in Figure 1.
High altitude-associated pulmonary embolism has been reported in at least two different contexts. In a retrospective study of 31,581 patients undergoing 1- to 2-level posterior lumbar fusion at a hospital attitude of >5000 feet above sea level and as many matched patients having the same surgery at <100 feet altitude, the rate of pulmonary embolism was 0.49% versus 0.35% resulting in an odds ratio (OR) of 1.38 (95% confidence interval [CI] 1.08-1.76) with no significant difference in DVT [11].
In a systematic literature review of sleep apnea and thrombosis 15 studies were identified and in 14 of those sleep apnea was an independent risk factor for DVT or for pulmonary embolism [12]. The risk for VTE was, as shown in 2 prospective studies, 2- to 3-fold higher compared to persons without sleep apnea [12]. A recent health care database study in France the time spent with oxygen saturation below 90% was an independent predictor for unprovoked VTE with a hazard ratio of 1.06 (95% CI, 1.01-1.02), or in other words, the more severe nocturnal hypoxia, the higher the risk for VTE [13]. Although ascent to high altitudes has been associated in several case reports and case series with development of pulmonary embolism, a systematic review of 2 studies was inconclusive regarding connection between hypoxia and thromboembolic events, and the authors speculated that exercise load, training status, mental stress and fluid status could be confounding factors in this context [14]. They found in the studies that explored hemostatic variables that there was increased thrombin generation but also decreased platelet activation, and at altitudes above 5,400 m there was activation of coagulation and at the same time increased fibrinolysis.
2.1.2. Reduced Clearance in Valve Pockets
Brooks et al reported in 2009 that the endothelium from valve sinus in the greater saphenous vein has higher levels of the endothelial protein C receptor (EPCR) and thrombomodulin and lower expression of von Willebrand factor (VWF) than in endothelium in the rest of this vein [15]. This implies that there under normal conditions is a dominant anticoagulant environment in the valve sinus. However, in a subsequent study from the same group, there was not a significant difference in expression of EPCR or thrombomodulin between endothelium in the valvular sinus versus in parts of the vein, thus only VWF levels differed [16]. The increase in VWF levels in the non-valvular endothelium was between 3-fold and 6.7-fold in different individuals.
It can then be hypothesized that during venous stasis there is a change in the balance towards hypercogulability. In a mouse model with stenosis causing 80-90% reduction of the venous lumen Brill et al and von Brühl et al reported that there was activation of the endothelium with release of VWF and P-selectin [17,18]. The latter will bind to P-selectin glycoprotein ligand-1 on leukocyte and VWF will bind with the GP1bα receptor on platelets that then become activated. The leukocytes, in turn, become activated and express more tissue factor. When the venous circulation is impaired, it is quite possible that these coagulation factors, activated platelets and leukocytes fail to clear and instead promote thrombin generation locally.
2.1.3. A Genetic Component
In a study with whole-exome sequencing, followed by gene-based collapsing analysis in 400 patients with VTE were compared with a large, general population cohort, and rare variants associated with VTE were identified [19]. As could be expected, 3 of the variants encoded the well-known natural anticoagulants protein C, protein S and antithrombin. The 4th gene was STAB2 stood out by having more than 4 times as many rare coding variants among patients with VTE than in the controls. The corresponding protein is Stabilin-2, which is a scavenger receptor on endothelial surface. The investigators found that all the patients with VTE and Stabilin-2 variants had impaired intracellular transport, for example of VWF. Indeed, individuals with STAB2 variants in an independent cohort had elevated VWF levels, which can contribute to thrombus formation.
2.2. Inflammation
2.2.1. Interleukins
The above described nuclear transcription factor Egr-1 regulates the expression of proteins such as interleukin (IL) 1β and CXCL2 that are involved in inflammatory processes. IL1β was demonstrated to be elevated in inflamed colon tissue, which in this study was experimental colitis, and when IL1β was injected to control animals it dose-dependently enhanced thrombus formation [20]. Furthermore, IL1β levels were higher in patients with COVID-19 that developed thrombotic events than in those without such complication [21]. In fact, a number of different ILs have been described to play a role in thrombus generation. The CD4+ Th17 cells produce IL9 and IL17A, which increase platelet aggregation rate, expression of P-selectin and development of thrombosis or acute coronary syndrome [22,23]. IL6 was recently reported to be elevated in cerebral venous sinus thrombosis and it was deemed useful for differentiating the condition from anatomical variants [24]. IL6 was also shown to be independently associated with high FVIII, which is a well-described risk factor for thrombosis [25]. IL18 was elevated in patients with ankle fracture, who developed DVT compared to those who did not [26].
The mechanism of increased VTE risk from air pollution also seems to be mediated by IL-6. In a mouse model with intratracheal instillation of 10 g of particulate matter with diameter smaller than 10 m (PM10), there was activation of coagulation, as measured by shortened bleeding time, prothrombin time and activated partial thromboplastin time [27]. This was explained by increased levels of fibrinogen and coagulation factor II, VIII and X, whereas there was no platelet activation, as assessed with flow cytometry. The IL-6 level in bronchoalveolar fluid was also increased, but when macrophages were depleted the response to particulate matter and the thrombotic tendency were reduced. Furthermore, when IL-6 knockout mice were exposed to the particulate matter there was no shortening of the bleeding time or evidence of activation of coagulation.
Mast cells and release of histamine also seem to play a role in air pollution-associated hypercoagulability, as shown in a hamster model [28]. When 50 g of diesel exhaust particles were instilled into the trachea, there was airway inflammation and release of histamine in broncho-alveolar lavage and in plasma. A small venous thrombosis, induced before the exposure, increased in size. Pretreatment with dexamethasone prevented these alterations. A similar study was performed in 20 human volunteers, who were exposed to 350 mg/m3 of dilute diesel exhaust [29]. Soluble P-selectin and soluble CD40 ligand increased significantly after 6 h compared to after inhalation of filtered air, which served as control, but there was no significant increase in IL-6 or C-reactive protein. In this study there was evidence of platelet activation, based on increased platelet-neutrophil and platelet-monocyte aggregates in flow cytometry. Thrombus formation was assessed ex vivo in a Badimon chamber and it increased significantly by 27% at 2 h after inhalation of diesel exhaust compared to inhalation of filtered air.
These experimental studies are supported by an epidemiological study in Santiago, Chile [30].
Hospitalizations for DVT or pulmonary embolism during 2001-2005 were compared against local data on air pollution from O3, NO2, SO2, CO, PM10 and PM 2.5. There were significant associations between hospitalization for VTE – stronger for PE than for DVT – and increased levels of these air pollutants. A systematic literature review identified 11 studies that overall suggested an association between particulate matter and VTE, but due to great variability between the studies in methodology, measurement of effects and duration of follow-up, the authors were unable to perform a meta-analysis to provide quantitative data [31].
2.2.2. Neutrophil Extracellular Traps
Activated platelets have been described to release inorganic polyphosphate (polyP), which in turn activates factor XII and thereby the contact activation pathway, leading to fibrin formation but also the kallikrein-kinin system with release of bradykinin, resulting in inflammatory reactions [32]. Neutrophils that become activated by damaged endothelium or by activated platelets will then release tissue factor as well as neutrophil extracellular traps (NETs) [33]. The NETs consist of a network of DNA strings and associated histones and a number of neutrophil granule proteins, e.g. elastase, lactoferrin, myeloperoxidase and α1-antitrypsin. Charles Esmon’s group reported that histones have cytotoxic effects on endothelium in vitro and when administered in vivo the histones caused vacuolated endothelium as well as micro- and macrovascular thrombosis in mice [34]. Co-administration of histones with activated protein C prevented death and when the protein C activation was blocked death occurred with sublethal doses of histones.
Nucleosomes consist of DNA segments coiled around a core of histone and are derived from NETs. In 149 patients that were diagnosed with DVTs, nucleosomes and elastase-α1-antitrypsin complexes were elevated compared to 183 controls, consisting of patients with suspected DVT but negative ultrasound examination [35]. The median nucleosome level in patients with DVT was 17 U/mL (interquartile range [IQR] 9-35) and in controls 9 U/mL (IQR 5-17), p<0.001, and the level of elastase-α1-antitrypsin complexes was 53 ng/mL (IQR 43-71) in those with DVT and 45 ng/mL (IQR 33-55) in controls, p<0.001.
Myeloperoxidase-DNA complexes are generated during the formation of NETs and can serve as a biomarker for NETs. A recent study demonstrated elevated plasma levels of myeloperoxidase-DNA complexes in patients with arterial as well as with venous thrombosis, although increased level was not predictive of future cardiovascular events or death [36].
NETs can activate the intrinsic pathway directly via activation of factor XII or via release of polyP from trapped and thereby activated platelets. A possible sequence of events, starting from impaired venous flow and involving NET formation is shown in Figure 2. Furthermore, leukocyte elastase, released from activated neutrophils, is known to cleave tissue factor pathway inhibitor (TFPI) by proteolysis of the polypeptide linking Kunitz-1 with Kunitz-2 domain and thereby promote the extrinsic pathway of coagulation [37]. Activated platelets and endothelial cells release protein disulphide isomerase, which can alter the tissue factor conformation to allow decryption and activation and further enhance the extrinsic pathway of coagulation [38].
2.2.3. Genetic Susceptibility
In a genome-wide association scan of samples from 5,862 patients with history of venous thrombosis and 7,112 controls the HIVEP1 locus (chromosome 6p24.1) was identified as a susceptibility locus for the disease [40]. The corresponding protein binds to DNA sequences in the promoter and enhancer regions of inflammatory target genes, resulting in transcriptional regulation. Presence of the HIVEP1 rs169613C allele demonstrated an association with increased VTE risk (OR 1.20; 95% CI, 1.13-1.27).
2.2.4. Complement System
In the context of damaged endothelium, neutrophils are recruited and activated, resulting in release of various proteolytic enzymes and NETs and a complex cross-talk between the complement and contact coagulation pathways as recently reviewed [41]. This cross-talk between the complement system and coagulation represents another mechanism for VTE that has received attention recently. In a prospective Danish study with over 80,000 persons complement C3 levels were higher already at baseline among those that developed VTE during follow-up (n=1176) versus those that did not; hazard ratio for the 3rd tertile of C3 was 1.58 (95% CI, 1.33-1.87) [42]. Subsequently, Hansen’s group in Tromsø, Norway showed that increased levels of the terminal complement complex C5b-9 predicted unprovoked VTE with an OR in the highest quartile of C5b-9 of 1.74 (95% CI, 1.10-2.78) compared to age-and sex-matched controls [43]. The same group also reported an association between C5 and future VTE risk and this became stronger for unprovoked versus provoked VTE [44]. They subsequently found that an overactive complement system due to insufficient regulation in patients with C1 inhibitor deficiency, who suffer from hereditary angioedema, was also associated with increased risk of VTE and in humans as well as in a mouse model there was an increase of prothrombin fragment 1+2, thrombin-antithrombin complex and thrombin generation, and in the mice also more frequent VTE but not arterial thrombosis [45].
2.3. Cancer and Thrombosis
2.3.1. Risk Factors of Cancer-Associated Thrombosis
Patients with active cancer have a 20% risk of being diagnosed with VTE and an additional proportion of patients are found to have occult thrombosis or pulmonary embolism on autopsy [46,47]. Thrombosis is the second most common cause of death in patients with cancer [48], and if a patient with cancer develops VTE, the prognosis and survival is worse than in patients without cancer [49]. Cancer also increases the risk of recurrent VTE and at the same time the risk of bleeding complications [50].
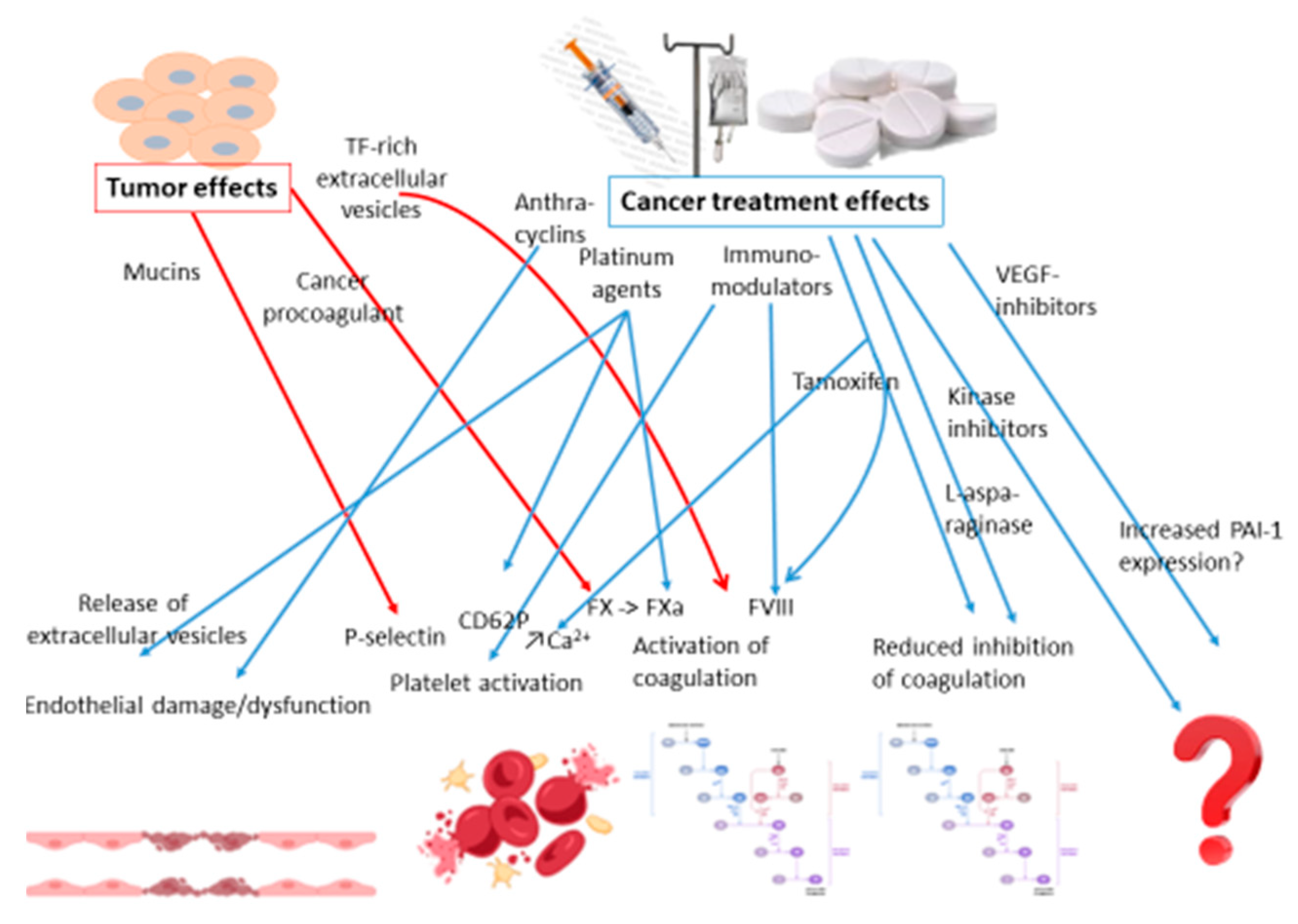
There are a multifold of risk factors associated with VTE in patients with cancer. At a high level they can be divided into patient related, treatment related and cancer related risk factors [51]. The patient related factors are typically the same as in patients without cancer, such as older age and immobility. As for the treatment related factors, surgery with immobilization and tissue damage and central venous catheters with partial obstruction of the vein are not much different either from patients without cancer. The tumor- and treatment-related thrombogenic mechanisms are summarized in Figure 3.
2.3.2. Pharmaceutical Treatments for Cancer and Risk of Thrombosis
Chemotherapeutic agents have been used alone or in combinations for cancer therapy for about 80 years, with nitrogen mustard as the first agent. This was followed by the development of antifolates, such as methotrexate, which does not appear to have any toxic effect on endothelium [52]. In the 1950s development of antimetabolites started with 6-mercaptopurine and 5-fluorouracil (5-FU). The latter has been associated with cardiotoxicity and coronary thrombosis [53] or left ventricle thrombosis [54]. Cwikiel et al showed that when 5-FU was incubated with endothelial cells the DNA synthesis decreased significantly [52]. The same group demonstrated that 5-FU could disrupt the endothelial cell barrier, thereby exposing subendothelial matrix, with ensuing platelet aggregation [52,55]. It has, however, not been convincingly shown that treatment with 5-FU is associated with an increased risk of VTE.
The 1960s saw the development of anthracyclines, starting with daunorubicin, isolated from a Streptomyces strain. Subsequently, doxorubicin from a mutated variant, and analogues were developed. The anti-cancer mechanism of anthracyclines is by intercalation between adjacent DNA base pairs and inhibits topoisomerase II and thereby also the DNA-repair capacity. The anthracyclines are thrombogenic by several effects, that includes activation of coagulation, as evidenced by an increase in D-dimers and thrombin-antithrombin complexes, and this increase could be prevented with a prophylactic dose of LMWH [56]. A research group at McMaster University, Canada, led by Patricia Liaw showed that in endothelial cells that were exposed to doxorubicin there was down-regulation of the mRNA of endothelial protein C-receptor (EPCR) and EPCR shedding, resulting in decreased EPCR levels on the endothelial surface and reduced activation of protein C [57]. They thereafter demonstrated that anthracyclines increased the exposure of phosphatidylserine and expression of TF on endothelial cells as well as on monocytes [58], which was also shown by Nigel Mackman’s group [59]. Subsequently, they found that anthracyclines, as well as 5-FU, given to healthy mice increased the release of cell-free DNA, which correlated with an increase in thrombin-antithrombin complexes and in thrombin generation [60]. Others have reported that patients treated with anthracyclines have decreased vasomotor reactivity, implying that the endothelial function was impaired compared to controls [61]. All these prothrombotic alterations have clinical importance, since in a study of patients with multiple myeloma receiving two different chemotherapy regimens, one of which included doxorubicin, the latter was associated with a 6.5-fold increase in VTE [62]. L-asparaginase was shown in 1963 to have antileukemic effect and was approved for medical use in the U.S. in 1978. It is used as first line treatment for acute lymphoblastic leukemia. Side-effects involving reduced level of antithrombin and development of VTE was reported already in 1979 [63]. A meta-analysis of 17 studies including 1752 patients treated with L-asparaginase reported a VTE incidence of 5.2% (95% CI: 4.2-6.4) [64]. Jacqueline Conard and others studied the mechanism for this and demonstrated that there is a decrease of antithrombin, protein C, protein S, FIX, FX, prothrombin and fibrinogen in plasma, with the natural anticoagulant reduction dominating initially, generating this prothrombotic state [65,66,67].
The anticancer properties of cisplatin were accidentally discovered and the agent was introduced clinically in the 1970s. The mechanism of action of cisplatin and its analogs carboplatin, oxaliplatin and nedaplatin is through crosslinking of DNA, resulting in apoptosis of the tumor cells. Evidence of a procoagulant activity of cisplatin was demonstrated in 1990, based on TF-like activity from exposed monocytoid cells [68]. Later, Lechner et al found that cisplatin caused release from endothelium of highly procoagulant microparticles, but the ensuing thrombin generation was driven by phospholipids rather than TF [69]. Other reported procoagulant effects of cisplatin are upregulation of FXa and increased formation of thrombin on platelets [70]. A meta-analysis of 38 trials with 8,216 patients showed that chemotherapy that included cisplatin increased the risk of VTE more than two-fold to 1.92% compared to 0.79% in controls, and the risk seems to be dose-dependent [71].
Hormonal agents for cancer therapy were developed based on early observations of regression of breast cancer and prostate cancer after removal of the ovaries or testicles, respectively. Thus, in 1941 Huggins and Hodges initiated treatment of patients with prostate cancer with orchiectomy or estrogen [72]. The goal is to achieve anti-androgen effect, with luteinizing hormone-releasing hormone (LHRH) synthetic agonists, gonadotropin-releasing hormone antagonsists (GnRH) and, in order to block the adrenal androgen production, the use of steroidal or non-steroidal antiandrogens, typically in combination [73]. Hormonal therapies are also very effective for receptor-positive breast cancer, starting with tamoxifen, first produced in 1962, and with the mechanism of action through its anti-estrogenic effect in breasts, although it has estrogenic effects in the uterus and liver. It is thus a selective estrogen receptor modulator (SERM). The effects of tamoxifen on hemostasis are, similar to those of estrogen, prothrombotic with reduction of the natural anticoagulant levels (antithrombin, protein C, total protein S) and increase of coagulation factor activity (factors VIII, IX, VWF) [74], and increased activated protein C resistance [75]. Tamoxifen also contributes to platelet activation via activation of phospholipase Cγ and phosphoinositide-3-kinase, which results in release of intracellular free Ca2+ from the endoplasmatic reticulum [76]. Aromatase inhibitors prevent the enzyme from converting androgen into estrogen in adipose tissue and are also used for the management of breast cancer. Anastrozole and letrozole belong to the family of selective aromatase inhibitors. A meta-analysis of 25 studies found that compared to the VTE prevalence of 0.5% in the general population, aromatase inhibitors had a VTE prevalence of 2.95%, although that was lower than for tamoxifen, OR 0.61 (95% CI, 0.37-1.00), driven by lower risk for DVT (OR 0.68) but not for pulmonary embolism (OR 1.01) [77]. Although there is no evidence of aromatase inhibitor-associated changes in levels of coagulation factors and inhibitors [78], an in vitro study showed that anastrozole decreased P-selectin expression and induced platelet aggregation and fibrin network formation [79].
Immunomodulators include thalidomide, lenalidomide and pomalidomide. Although none of these agents seems to have any effect on coagulation on their own, when given together with dexamethasone for treatment of multiple myeloma, the CD62P increased on platelets and the closure time in the Platelet Function Analyzer-100 shortened, as well as increased levels of F VIII and soluble thrombomodulin [80]. In a meta-analysis of 3,322 patients with multiple myeloma, treatment with thalidomide, dexamethasone and the combination increased the risk of VTE 2.6, 2.8 and 8-fold, respectively, whereas concomitant prophylactic dose anticoagulation eliminated that increase in risk [81]. The VTE risk is similar with the second generation immunomodulators.
Growth factors that are targeted with monoclonal antibodies for cancer therapy include epidermal growth factor and its receptor (EGFR) and vascular endothelial growth factor (VEGF, anti-angiogenic agents). A meta-analysis of 17 studies on the anti-EGFR agents cetuximab and panitumumab showed a 50% increase in risk of VTE compared to those receiving other regimens [82], but the mechanism for hypercoagulability has not been elucidated. Although the VEGF-directed antibody bevacizumab was shown to increase expression of PAI-1 and the thrombus development in femoral vein injury or inferior vena cava obstruction in a mouse model [83], the use of bevacizumab in 10 randomized clinical trials or of aflibercept in a meta-analysis did not reveal increased risk of VTE [84,85].
Kinase inhibitors include receptor tyrosine kinase inhibitors (RTKI) and cyclin-dependent kinase inhibitors (CDK). Whereas the first generation TKI, imatinib, was not associated with increased risk of VTE, the second generation agents, the breakpoint cluster region protein-tyrosine kinase protein ABL1 derivatives, such as ponatinib increased expression of VWF and platelet adhesion with microvascular angiopathy [86]. In a meta-analysis of randomized controlled trials the second generation TKIs (ponatinib, nilotinib, dasatinib) were mainly associated with arterial thromboembolism, but there was also a 3-fold increase in venous occlusive events compared to imatinib [87]. Of the CDK inhibitors it seems like mainly abemaciclib is associated with an increased risk of VTE but the pathophysiology behind this effect has not been revealed [88].
2.3.3. Cancer Related Factors
Adenocarcinoma that secrete mucin seem to be more thrombogenic than for example squamous cell carcinoma [89]. These abnormally glycosylated mucins carry binding sites for selectins and interact with L-selectins (on leukocytes) and P-selectins (on platelets), resulting in platelet aggregation and thrombin generation [90]. Important selectin ligands on tumor cells and secreted mucin are sialyl-Lewis (SLe) SLex and SLea [91].
Cancer procoagulant (CP) is a cysteine proteinase from malignant tissues and it can directly activate F X to Xa [92], and it also promotes cancer metastasis. CP is expressed in a wide range of tumor cells.
Tissue factor is overexpressed in many types of cancer and plays a major role in the progression of cancer. The mechanism for TF in cancer-associated thrombosis is probably via the shedding of TF-rich extracellular vesicles from the tumor cells [93].
Finally, the tumor can by compression of a vein or by intravascular invasion cause stasis and thrombus formation.
3. Future Research
The presentation of DVT differs between patients. Why do only about half of the patients get embolization to the lungs? Why do some develop hormone-associated thrombosis in the leg and others in the cerebral venous sinus? What factors decide that when the thrombus stops growing? Why do some develop post-thrombotic syndrome or pulmonary hypertension? These are important clinical questions, but they can only be answered by further studies to better understand the pathophysiologic mechanisms.
4. Conclusions
The principles of pathophysiology of VTE have become much more complex than the classic Virchow’s triad. Venous stasis with hypoxia or accumulation of activated coagulation factors, platelets, leukocytes and activated endothelium may initiate inflammatory processes that play a major role in thrombus generation. The latter may involve release of interleukins, NETs, and activated complement factors. In thromboinflammation there is evidence of activation of both the extrinsic and intrinsic pathways, as well as reduced inhibition due to cleavage of TFPI. During the past decade several genetic variants have been reported to have association with risk for VTE, and some of those findings have increased our understanding of additional mechanisms for thrombosis. Rather than assigning a single mechanism to the generation of thrombosis, there is even in the most straight-forward cases of DVT a complex chain of pathophysiologic events involving cell cooperation and multiple biochemical pathways. Fortunately, prevention or treatment of VTE usually only requires inhibition or blockage at one point such as inhibition of a specific coagulation factor. In more severe forms of thromboinflammation, such as severe sepsis, antiphospholipid syndrome, preeclampsia or Behҁet’s disease there might be a need for combination therapies to control the unrestrained thrombus generation.
Author Contributions
Conceptualization, Sam Schulman; methodology, Sam Schulman; software Jamilya Khizroeva, Victoria Bitsadze; validation, Alexander Makatsariya and Daredzhan Kapanadze; writing—original draft preparation, Sam Schulman. All authors have read and agreed to the published version of the manuscript.
Funding
Sam Schulman has received a research grant from Octapharma and honoraria from Alexion, Bayer, Boehringer Ingelheim, Daiichi-Sankyo, Octapharma, Sanofi, Servier, Bristol-Myers Squibb, Pfizer, and Hemostasis Reference Laboratory. FY has no conflicts of interest to declare.
Acknowledgments
None.
Conflicts of Interest
“The authors declare no conflicts of interest.”
References
- Bagot, C.N.; Arya, R. Virchow and his triad: a question of attribution. Br. J. Haematol. 2008, 143, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Wessler, S. Thrombosis in the presence of vascular stasis. Am. J. Med. 1962, 33, 648–666. [Google Scholar] [CrossRef] [PubMed]
- Hamer, J.D.; Malone, P.C.; A Silver, I. The PO2 in venous valve pockets: Its possible bearing on thrombogenesis. Br. J. Surg. 1981, 68, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.-F.; Mackman, N.; Kisiel, W.; Stern, D.M.; Pinsky, D.J. Hypoxia/Hypoxemia-Induced Activation of the Procoagulant Pathways and the Pathogenesis of Ischemia-Associated Thrombosis. Arter. Thromb. Vasc. Biol. 1999, 19, 2029–2035. [Google Scholar] [CrossRef]
- Yan, S.-F.; Zou, Y.S.; Gao, Y.; Zhai, C.; Mackman, N.; Lee, S.L.; Milbrandt, J.; Pinsky, D.; Kisiel, W.; Stern, D. Tissue factor transcription driven by Egr-1 is a critical mechanism of murine pulmonary fibrin deposition in hypoxia. Proc. Natl. Acad. Sci. USA 1998, 95, 8298–8303. [Google Scholar] [CrossRef]
- Yan, S.F.; Lu, J.; Zou, Y.S.; Kisiel, W.; Mackman, N.; Leitges, M.; Steinberg, S.; Pinsky, D.; Stern, D. Protein kinase C-beta and oxygen deprivation. A novel Egr-1-dependent pathway for fibrin deposition in hypoxemic vasculature. J Biol Chem 2000, 275, 11921–11928. [Google Scholar]
- Lo, L.W.; Cheng, J.J.; Chiu, J.J.; Wung, B.S.; Liu, Y.C.; Wang, D.L. Endothelial exposure to hypoxia induces Egr-1 expression involving PKCalpha-mediated Ras/Raf-1/ERK1/2 pathway. J Cell Physiol 2001, 188, 304–312. [Google Scholar] [CrossRef]
- Hsieh, K.Y.; Wei, C.K.; Wu, C.C. YC-1 prevents tumor-associated tssue factor expression and procoagulant activity in hypoxic conditions by inhibiting p38/NF-kB signaling pathway. Int J Mol Sci 2019, 20, 244. [Google Scholar] [CrossRef]
- Liao, H.; Hyman, M.C.; Lawrence, D.A.; Pinsky, D.J. Molecular regulation of the PAI-1 gene by hypoxia: contributions of Egr-1, HIF-1alpha, and C/EBPalpha. FASEB J 2007, 21, 935–949. [Google Scholar] [CrossRef]
- Bikov, A.; Meszaros, M.; Schwarz, E.I. Coagulation and Fibrinolysis in Obstructive Sleep Apnoea. Int. J. Mol. Sci. 2021, 22, 2834. [Google Scholar] [CrossRef]
- Donnally, C.J.; Vakharia, A.M.; Sheu, J.I.; Vakharia, R.M.; Damodar, D.; Shenoy, K.; Gjolaj, J.P. High altitude is an independent risk factor for developing a pulmonary embolism, but not a deep vein thrombosis following a 1- to 2-level lumbar fusion Global Spine Journal 2019, 9, 729–734.
- Mattiuzzi, C.; Franchini, M.; Lippi, G. Sleep apnea and venous thromboembolism. A systematic review。 Thromb. Haemost. 2015, 114, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Trzepizur, W.; Gervès-Pinquié, C.; Heudes, B.; Blanchard, M.; Meslier, N.; Jouvenot, M.; Kerbrat, S.; Le Mao, R.; Magois, E.; Racineux, J.-L.; et al. Sleep Apnea and Incident Unprovoked Venous Thromboembolism: Data from the Pays de la Loire Sleep Cohort. Thromb. Haemost. 2022, 123, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Treml, B.; Wallner, B.; Blank, C.; Fries, D.; Schobersberger, W. The influence of environmental hypoxia on hemostasis-a systematic review Front Cardiovasc Med 2022, 9, 813550.
- Brooks, E.G.; Trotman, W.; Wadsworth, M.P.; Taatjes, D.J.; Evans, M.F.; Ittleman, F.P.; Callas, P.W.; Esmon, C.T.; Bovill, E.G. Valves of the deep venous system: an overlooked risk factor. Blood 2009, 114, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Trotman, W.E.; Taatjes, D.J.; Callas, P.W.; Bovill, E.G. The endothelial microenvironment in the venous valvular sinus: thromboresistance trends and inter-individual variation. Histochem. 2011, 135, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Köllnberger, M.; Wakefield, T.W.; Lämmle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor–mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef]
- Von Brühl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Desch, K.C.; Ozel, A.B.; Halvorsen, M.; Jacobi, P.M.; Golden, K.L.; Underwood, M.; Germain, M.; Tregouet, D.-A.; Reitsma, P.H.; Kearon, C.; et al. Whole-exome sequencing identifies rare variants in STAB2 associated with venous thromboembolic disease. Blood 2020, 136, 533–541. [Google Scholar] [CrossRef]
- Yoshida, H.; Russell, J.; Senchenkova, E.Y.; Almeida Paula, L.D.; Granger, D.N. Interleukin-1beta mediates the extra-intestinal thrombosis associated with experimental colitis. Am J Pathol 2010, 177, 2774–2781. [Google Scholar] [CrossRef]
- El-Ghani, S.E.-S.A.; Hamed, R.M.R.; Eid, R.A.; Ibrahim, A.Y.M.; Abdel-Hamid, H.M.; Abdelrahman, W.; Ibrahim, R.E.; Abdel-Aziz, M.M.; Mohamed, M.S. Serum interleukin 1β and sP-selectin as biomarkers of inflammation and thrombosis, could they be predictors of disease severity in COVID 19 Egyptian patients? (a cross-sectional study). Thromb. J. 2022, 20, 71. [Google Scholar] [CrossRef]
- Feng, Y.; Yu, M.; Zhu, F.; Zhang, S.; Ding, P.; Wang, M. IL-9 Promotes the Development of Deep Venous Thrombosis by Facilitating Platelet Function. Thromb. Haemost. 2018, 118, 1885–1894. [Google Scholar] [CrossRef]
- Ding, P.; Zhang, S.; Yu, M.; Feng, Y.; Long, Q.; Yang, H.; Li, J.; Wang, M. IL-17A promotes the formation of deep vein thrombosis in a mouse model. Int. Immunopharmacol. 2018, 57, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Pan, L.; Lan, D.; Chen, Z.; Wang, Z.; Zou, M.; Meng, R. Inflammatory Markers Differentiate Cerebral Venous Sinus Thrombosis from Mimics. Thromb. Haemost. 2022, 123, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Bittar, L.F.; Mazetto, B.d.M.; Orsi, F.L.A.; Collela, M.P.; De Paula, E.V.; Annichino-Bizzacchi, J.M. Long-term increased factor VIII levels are associated to interleukin-6 levels but not to post-thrombotic syndrome in patients with deep venous thrombosis. Thromb. Res. 2014, 135, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.Q.; Tang, J.; Zhang, Z.X.; Bian, J. Correlation of interleukin-18 and high-sensitivity c-reactive protein with perioperative deep vein thrombosis in patients with ankle fracture Ann Vasc Surg 2019, 54, 282–289.
- Mutlu, G.M.; Green, D.; Bellmeyer, A.; Baker, C.M.; Burgess, Z.; Rajamannan, N.; Christman, J.W.; Foiles, N.; Kamp, D.W.; Ghio, A.J.; et al. Ambient particulate matter accelerates coagulation via an IL-6–dependent pathway. J. Clin. Investig. 2007, 117, 2952–2961. [Google Scholar] [CrossRef]
- Nemmar, A.; Hoet, P.H.; Vermylen, J.; Nemery, B.; Hoylaerts, M.F. Pharmacological Stabilization of Mast Cells Abrogates Late Thrombotic Events Induced by Diesel Exhaust Particles in Hamsters. Circulation 2004, 110, 1670–1677. [Google Scholar] [CrossRef]
- Lucking, A.J.; Lundback, M.; Mills, N.L.; Faratian, D.; Barath, S.L.; Pourazar, J.; Cassee, F.R.; Donaldson, K.; Boon, N.A.; Badimon, J.J.; et al. Diesel exhaust inhalation increases thrombus formation in man. Eur. Hear. J. 2008, 29, 3043–3051. [Google Scholar] [CrossRef]
- Dales, R.E.; Cakmak, S.; Vidal, C.B. Air pollution and hospitalization for venous thromboembolic disease in Chile. J. Thromb. Haemost. 2010, 8, 669–674. [Google Scholar] [CrossRef]
- Franchini, M.; Mengoli, C.; Cruciani, M.; Bonfanti, C.; Mannucci, P.M. Association between particulate air pollution and venous thromboembolism: A systematic literature review. Eur. J. Intern. Med. 2015, 27, 10–13. [Google Scholar] [CrossRef]
- Müller, F.; Mutch, N.J.; Schenk, W.A.; Smith, S.A.; Esterl, L.; Spronk, H.M.; Schmidbauer, S.; Gahl, W.A.; Morrissey, J.H.; Renné, T. Platelet Polyphosphates Are Proinflammatory and Procoagulant Mediators In Vivo. Cell 2009, 139, 1143–1156. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Wagner, D.D. Neutrophil Extracellular Trap (NET) Impact on Deep Vein Thrombosis. Arter. Thromb. Vasc. Biol. 2012, 32, 1777–1783. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- van Montfoort, M.L.; Stephan, F.; Lauw, M.N.; Hutten, B.A.; Van Mierlo, G.J.; Solati, S.; Middeldorp, S.; Meijers, J.C.; Zeerleder, S. Circulating Nucleosomes and Neutrophil Activation as Risk Factors for Deep Vein Thrombosis. Arter. Thromb. Vasc. Biol. 2013, 33, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Bressan, A.; Faggin, E.; Donato, M.; Tonon, L.; Buso, R.; Nardin, C.; Tiepolo, M.; Cinetto, F.; Scarpa, R.; Agostini, C.; et al. NETosis in Acute Thrombotic Disorders. Semin. Thromb. Hemost. 2023, 49, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, D.A.; Wun, T.C.; Likert, K.M.; Broze, G.J., Jr. The effect of leukocyte elastase on tissue factor pathway inhibitor. Blood 1992, 79, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.A.; Pendurthi, U.R.; Rao, L.V.M. Role of Cell Surface Lipids and Thiol-Disulphide Exchange Pathways in Regulating the Encryption and Decryption of Tissue Factor. Thromb. Haemost. 2019, 119, 860–870. [Google Scholar] [CrossRef]
- Levy, J.H.; Iba, T.; Olson, L.B.; Corey, K.M.; Ghadimi, K.; Connors, J.M. COVID-19: Thrombosis, thromboinflammation, and anticoagulation considerations. International Journal of Laboratory Hematology 2021, 43, (S1), 29–35. [Google Scholar] [CrossRef]
- Morange, P.-E.; Bezemer, I.; Saut, N.; Bare, L.; Burgos, G.; Brocheton, J.; Durand, H.; Biron-Andreani, C.; Schved, J.-F.; Pernod, G.; et al. A Follow-Up Study of a Genome-wide Association Scan Identifies a Susceptibility Locus for Venous Thrombosis on Chromosome 6p24.1. Am. J. Hum. Genet. 2010, 86, 592–595. [Google Scholar] [CrossRef]
- Pryzdial, E.L.G.; Leatherdale, A.; Conway, E.M. Coagulation and complement: Key innate defense participants in a seamless web. Front. Immunol. 2022, 13, 918775. [Google Scholar] [CrossRef]
- Nørgaard, I.; Nielsen, S.F.; Nordestgaard, B.G. Complement C3 and High Risk of Venous Thromboembolism: 80517 Individuals from the Copenhagen General Population Study. Clin. Chem. 2016, 62, 525–534. [Google Scholar] [CrossRef]
- Høiland, II; Liang, R.A.; Braekkan, S.K.; Pettersen, K.; Ludviksen, J.K.; Latysheva, N.; Snir, O.; Ueland, T.; Hindberg, K.; Mollnes, T.E., et al. Complement activation assessed by the plasma terminal complement complex and future risk of venous thromboembolism. J Thromb Haemost 2019, 17, 934–943.
- Skjeflo, E.W.; Brækkan, S.K.; Ludviksen, J.K.; Snir, O.; Hindberg, K.; Mollnes, T.E.; Hansen, J.-B. Elevated plasma concentration of complement factor C5 is associated with risk of future venous thromboembolism. Blood 2021, 138, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Kawano, T.; Wan, J.; Tanratana, P.; Polai, Z.; Shim, Y.J.; Snir, O.; Brækkan, S.K.; Dhrolia, S.; Kasthuri, R.R.; et al. C1 inhibitor deficiency enhances contact pathway mediated activation of coagulation and venous thrombosis. Blood 2023, 141, 2390–2401. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.K.; Wun, T.; Harvey, D.; Zhou, H.; White, R.H. Incidence of Venous Thromboembolism and Its Effect on Survival Among Patients With Common Cancers. Arch. Intern. Med. 2006, 166, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Carrier, M.; Ay, C.; Di Nisio, M.; Hicks, L.K.; Khorana, A.A.; Leavitt, A.D.; Lee, A.Y.Y.; Macbeth, F.; Morgan, R.L.; et al. American Society of Hematology 2021 guidelines for management of venous thromboembolism: prevention and treatment in patients with cancer. Blood Adv. 2021, 5, 927–974. [Google Scholar] [CrossRef]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Kuderer, N.M.; Lyman, G.H. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J. Thromb. Haemost. 2007, 5, 632–634. [Google Scholar] [CrossRef]
- Levitan, N.; Dowlati, A.; Remick, S.C.; Tahsildar, H.I.; Sivinski, L.D.; Beyth, R.; Rimm, A.A. Rates of Initial and Recurrent Thromboembolic Disease Among Patients with Malignancy Versus Those without Malignancy: Risk Analysis Using Medicare Claims Data. Medicine 1999, 78, 285–91. [Google Scholar] [CrossRef]
- Prandoni, P.; Lensing, A.W.A.; Piccioli, A.; Bernardi, E.; Simioni, P.; Girolami, B.; Marchiori, A.; Sabbion, P.; Prins, M.H.; Noventa, F.; et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood 2002, 100, 3484–3488. [Google Scholar] [CrossRef]
- Ikushima, S.; Ono, R.; Fukuda, K.; Sakayori, M.; Awano, N.; Kondo, K. Trousseau's syndrome: cancer-associated thrombosis. Jpn J Clin Oncol 2016, 46, 204–208. [Google Scholar] [CrossRef]
- Cwikiel, M.; Eskilsson, J.; Albertsson, M.; Stavenow, L. The influence of 5-fluorouracil and methotrexate on vascular endothelium. An experimental study using endothelial cells in the culture. Ann. Oncol. 1996, 7, 731–737. [Google Scholar] [CrossRef]
- Sorrentino, M.F.; Kim, J.; Foderaro, A.E.; Truesdell, A.G. 5-fluorouracil induced cardiotoxicity: Review of the literature. Cardiol. J. 2012, 19, 453–457. [Google Scholar] [CrossRef]
- Kumar, D.; Warsha, F.; Mehta, A.; Deepak, V.; Jawad, W. 5-Fluorouracil Induced Takotsubo Cardiomyopathy Complicated by Left Ventricular Thrombosis. Cureus 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Kinhult, S.; Albertsson, M.; Eskilsson, J.; Cwikiel, M. Antithrombotic treatment in protection against thrombogenic effects of 5-fluorouracil on vascular endothelium: A scanning microscopy evaluation. Scanning 2001, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Weitz, I.C.; Israel, V.K.; Waisman, J.R.; Presant, C.A.; Rochanda, L.; Liebman, H.A. Chemotherapy-Induced Activation of Hemostasis: Effect of a Low Molecular Weight Heparin (Dalteparin Sodium) on Plasma Markers of Hemostatic Activation. Thromb. Haemost. 2002, 88, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Woodley-Cook, J.; Shin, L.Y.; Swystun, L.; Caruso, S.; Beaudin, S.; Liaw, P.C. Effects of the chemotherapeutic agent doxorubicin on the protein C anticoagulant pathway. Mol. Cancer Ther. 2006, 5, 3303–3311. [Google Scholar] [CrossRef]
- Swystun, L.L.; Shin, L.Y.Y.; Beaudin, S.; Liaw, P.C. Chemotherapeutic agents doxorubicin and epirubicin induce a procoagulant phenotype on endothelial cells and blood monocytes. J. Thromb. Haemost. 2009, 7, 619–626. [Google Scholar] [CrossRef]
- Boles, J.C.; Williams, J.C.; Hollingsworth, R.M.; Wang, J.G.; Glover, S.L.; Owens, A.P., 3rd; Barcel, D.A.; Kasthuri, R.S.; Key, N.S.; Mackman, N. Anthracycline treatment of the human monocytic leukemia cell line THP-1 increases phosphatidylserine exposure and tissue factor activity. Thromb Res 2012, 129, 197–203. [Google Scholar] [CrossRef]
- Swystun, L.L.; Mukherjee, S.; Liaw, P.C. Breast cancer chemotherapy induces the release of cell-free DNA, a novel procoagulant stimulus. J Thromb Haemost 2011, 9, 2313–2321. [Google Scholar] [CrossRef]
- Chow, A.Y.; Chin, C.; Dahl, G.; Rosenthal, D.N. Anthracyclines Cause Endothelial Injury in Pediatric Cancer Patients: A Pilot Study. J. Clin. Oncol. 2006, 24, 925–928. [Google Scholar] [CrossRef]
- Zangari, M.; Barlogie, B.; Thertulien, R.; Jacobson, J.; Eddleman, P.; Fink, L.; Fassas, A.; Van Rhee, F.; Talamo, G.; Lee, C.-K.; et al. Thalidomide and Deep Vein Thrombosis in Multiple Myeloma: Risk Factors and Effect on Survival. Clin. Lymphoma 2003, 4, 32–35. [Google Scholar] [CrossRef]
- Anderson, N.; Lokich, J.J.; Tullis, J.L. L-asparaginase effect on antithrombin-III levels. Med Pediatr Oncol 1979, 7, 335–340. [Google Scholar] [CrossRef]
- Caruso, V.; Iacoviello, L.; Di Castelnuovo, A.; Storti, S.; Mariani, G.; de Gaetano, G.; Donati, M.B. Thrombotic complications in childhood acute lymphoblastic leukemia: a meta-analysis of 17 prospective studies comprising 1752 pediatric patients. Blood 2006, 108, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Conard, J.; Cazenave, B.; Maury, J.; Horellou, M.H.; Samama, M. L-asparaginase, antithrombin III, and thrombosis. Lancet 1980, 1, 1091. [Google Scholar] [CrossRef] [PubMed]
- Conard, J.; Horellou, M.H.; Van Dreden, P.; Potevin, F.; Zittoun, R.; Samama, M. Decrease in protein C in L-asparaginase-treated patients. Br. J. Haematol. 1985, 59, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Hunault-Berger, M.; Chevallier, P.; Delain, M.; Bulabois, C.-E.; Bologna, S.; Bernard, M.; Lafon, I.; Cornillon, J.; Maakaroun, A.; Tizon, A.; et al. Changes in antithrombin and fibrinogen levels during induction chemotherapy with L-asparaginase in adult patients with acute lymphoblastic leukemia or lymphoblastic lymphoma. Use of supportive coagulation therapy and clinical outcome: the CAPELAL study. Haematologica 2008, 93, 1488–1494. [Google Scholar] [CrossRef]
- Wheeler, H.R.; Geczy, C.L. Induction of macrophage procoagulant expression by cisplatin, daunorubicin and doxorubicin. Int. J. Cancer 1990, 46, 626–632. [Google Scholar] [CrossRef]
- Lechner, D.; Kollars, M.; Gleiss, A.; Kyrle, P.A.; Weltermann, A. Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J. Thromb. Haemost. 2007, 5, 2445–2452. [Google Scholar] [CrossRef]
- Ma, R.; Bi, Y.; Kou, J.; Zhou, J.; Shi, J. Enhanced procoagulant activity of platelets after chemotherapy in non-small cell lung cancer. Cancer Biol. Ther. 2017, 18, 627–634. [Google Scholar] [CrossRef]
- Seng, S.; Liu, Z.; Chiu, S.K.; Proverbs-Singh, T.; Sonpavde, G.; Choueiri, T.K.; Tsao, C.-K.; Yu, M.; Hahn, N.M.; Oh, W.K.; et al. Risk of Venous Thromboembolism in Patients With Cancer Treated With Cisplatin: A Systematic Review and Meta-Analysis. J. Clin. Oncol. 2012, 30, 4416–4426. [Google Scholar] [CrossRef]
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer: I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. J. Urol. 2002, 168, 9–12. [Google Scholar] [CrossRef]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2018, 22, 24–38. [Google Scholar] [CrossRef]
- Cosman, F.; Baz-Hecht, M.; Cushman, M.; Vardy, M.D.; Cruz, J.; Nieves, J.; Zion, M.; Lindsay, R. Short-term effects of estrogen, tamoxifen and raloxifene on hemostasis: a randomized-controlled study and review of the literature. Thromb. Res. 2005, 116, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rühl, H.; Schröder, L.; Müller, J.; Fimmers, R.; Sukhitashvili, S.; Welz, J.; Kuhn, W.C.; Oldenburg, J.; Rudlowski, C.; Pötzsch, B. Tamoxifen induces resistance to activated protein C. Thromb. Res. 2014, 133, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.P.; Chegini, H.A.; Vishneski, S.R.; Weatherman, R.V.; Blackmore, P.F.; Dobrydneva, Y. Tamoxifen promotes superoxide production in platelets by activation of PI3-Kinase and NADPH oxidase pathways. Thromb. Res. 2012, 129, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-J.; Jung, E.-A.; Kim, Z.; Kim, B.-Y. Risk of Cardiovascular Events and Lipid Profile Change in Patients with Breast Cancer Taking Aromatase Inhibitor: A Systematic Review and Meta-Analysis. Curr. Oncol. 2023, 30, 1831–1843. [Google Scholar] [CrossRef]
- Blondon, M.; Bodmer, A.; Thouvenin, L.; Lecompte, T.; Righini, M.; Fontana, P.; Casini, A. Differential impact of tamoxifen and aromatase inhibitors on thrombin generation: the prospective HEMOBREAST cohort. Blood Adv. 2022, 6, 2884–2892. [Google Scholar] [CrossRef]
- Pather, K.; Dix-Peek, T.; Duarte, R.; Chetty, N.; Augustine, T. Breast cancer cell-induced platelet activation is compounded by tamoxifen and anastrozole in vitro. Thromb. Res. 2019, 177, 51–58. [Google Scholar] [CrossRef]
- Robak, M.; Treliński, J.; Chojnowski, K. Hemostatic changes after 1 month of thalidomide and dexamethasone therapy in patients with multiple myeloma. Med Oncol. 2012, 29, 3574–3580. [Google Scholar] [CrossRef]
- El Accaoui, R.; Shamseddeen, W.; Taher, A. Thalidomide and thrombosis. A meta-analysis。 Thromb. Haemost. 2007, 97, 1031–1036. [Google Scholar] [CrossRef]
- Miroddi, M.; Sterrantino, C.; Simmonds, M.; Caridi, L.; Calapai, G.; Phillips, R.S.; Stewart, L.A. Systematic review and meta-analysis of the risk of severe and life-threatening thromboembolism in cancer patients receiving anti-EGFR monoclonal antibodies (cetuximab or panitumumab). Int. J. Cancer 2016, 139, 2370–2380. [Google Scholar] [CrossRef]
- Chen, N.; Ren, M.; Li, R.; Deng, X.; Li, Y.; Yan, K.; Xiao, L.; Yang, Y.; Wang, L.; Luo, M.; et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol. Cancer 2015, 14, 1–7. [Google Scholar] [CrossRef]
- Hurwitz, H.I.; Saltz, L.B.; Van Cutsem, E.; Cassidy, J.; Wiedemann, J.; Sirzén, F.; Lyman, G.H.; Rohr, U.-P. Venous Thromboembolic Events With Chemotherapy Plus Bevacizumab: A Pooled Analysis of Patients in Randomized Phase II and III Studies. J. Clin. Oncol. 2011, 29, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Kanukula, R.; Ganta, S.; Sirumalla, Y.; Salam, A.; Baddam, R.; Pasupuleti, B.C. Risk of Venous Thromboembolic Events in Patients With Cancer Treated With Aflibercept: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Am. J. Ther. 2019, 26, e549–e552. [Google Scholar] [CrossRef] [PubMed]
- Latifi, Y.; Moccetti, F.; Wu, M.; Xie, A.; Packwood, W.; Qi, Y.; Ozawa, K.; Shentu, W.; Brown, E.; Shirai, T.; et al. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood 2019, 133, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Haguet, H.; Douxfils, J.; Mullier, F.; Chatelain, C.; Graux, C.; Dogné, J.-M. Risk of arterial and venous occlusive events in chronic myeloid leukemia patients treated with new generation BCR-ABL tyrosine kinase inhibitors: a systematic review and meta-analysis. Expert Opin. Drug Saf. 2016, 16, 5–12. [Google Scholar] [CrossRef]
- Thein, K.Z.; Htut, T.W.; Ball, S.; Swarup, S.; Sultan, A.; Oo, T.H. Venous thromboembolism risk in patients with hormone receptor-positive HER2-negative metastatic breast cancer treated with combined CDK 4/6 inhibitors plus endocrine therapy versus endocrine therapy alone: a systematic review and meta-analysis of randomized controlled trials. Breast Cancer Res Treat 2020, 183, 479–487. [Google Scholar]
- Blom, J.W.; Osanto, S.; Rosendaal, F.R. The risk of a venous thrombotic event in lung cancer patients: higher risk for adenocarcinoma than squamous cell carcinoma. J. Thromb. Haemost. 2004, 2, 1760–1765. [Google Scholar] [CrossRef]
- Wahrenbrock, M.; Borsig, L.; Le, D.; Varki, N.; Varki, A. Selectin-mucin interactions as a probable molecular explanation for the association of Trousseau syndrome with mucinous adenocarcinomas. J. Clin. Investig. 2003, 112, 853–862. [Google Scholar] [CrossRef]
- Varki, A.; Kannagi, R.; Toole, B.; Stanley, P. Glycosylation changes in cancer. 2017/01/01 ed.; Cold Sprin Hardor Laboratory Press: Cold Spring Harbor (NY), 2017; Vol. Chapter 47, p 597-609.
- Gordon, S.G.; Franks, J.J.; Lewis, B. Cancer procoagulant A: A factor X activating procoagulant from malignant tissue. Thromb. Res. 1975, 6, 127–137. [Google Scholar] [CrossRef]
- Rondon, A.M.R.; Kroone, C.; Kapteijn, M.Y.; Versteeg, H.H.; Buijs, J.T. Role of tissue factor in tumor progression and cancer-associated thrombosis. Semin Thromb Hemost 2019, 45, 396–412. [Google Scholar] [CrossRef]
Figure 1.
The main hypoxia-induced pathways of thrombus formation.
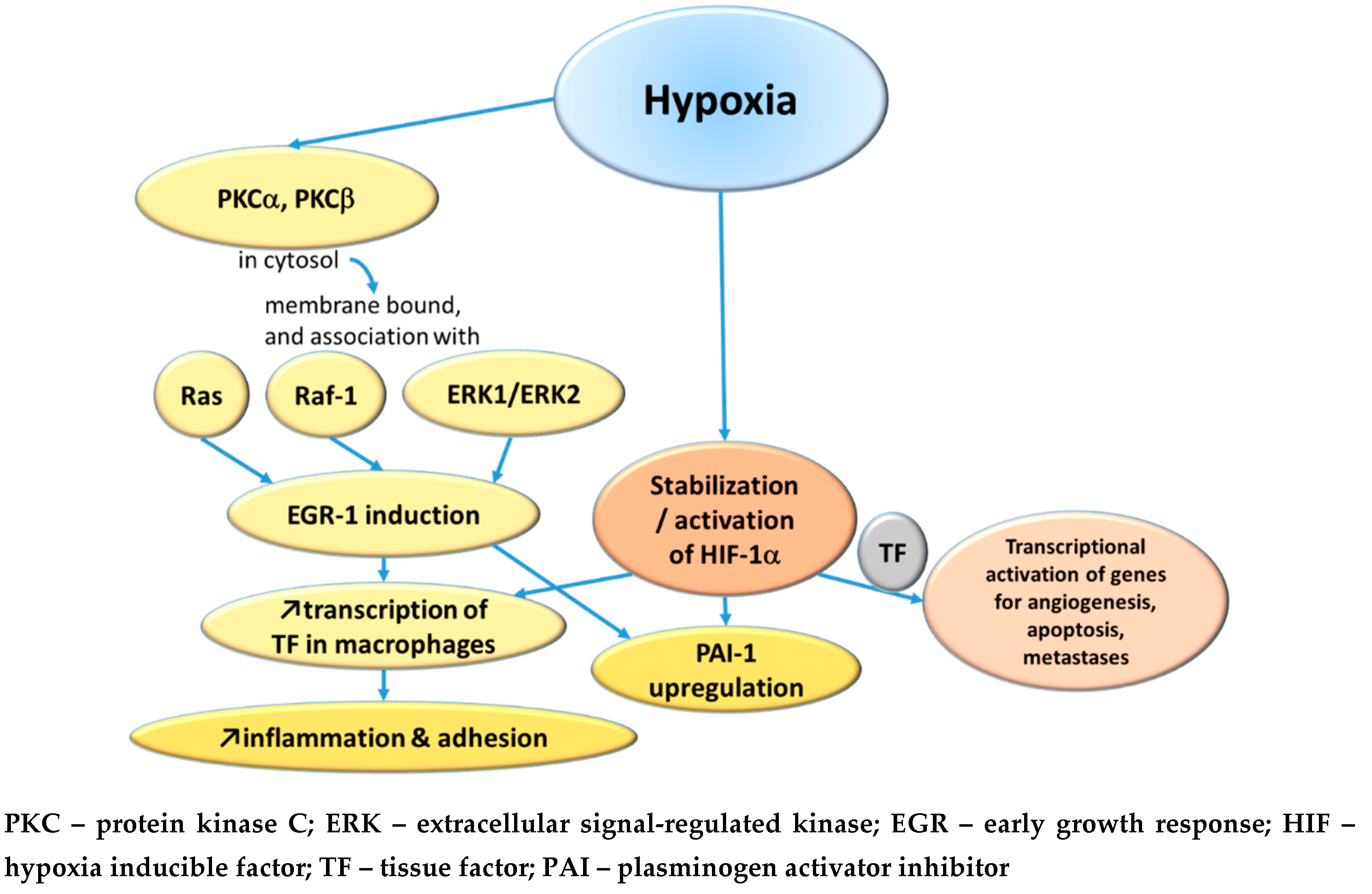
Figure 2.
Coagulation pathways activation through NETs release and platelets activation.
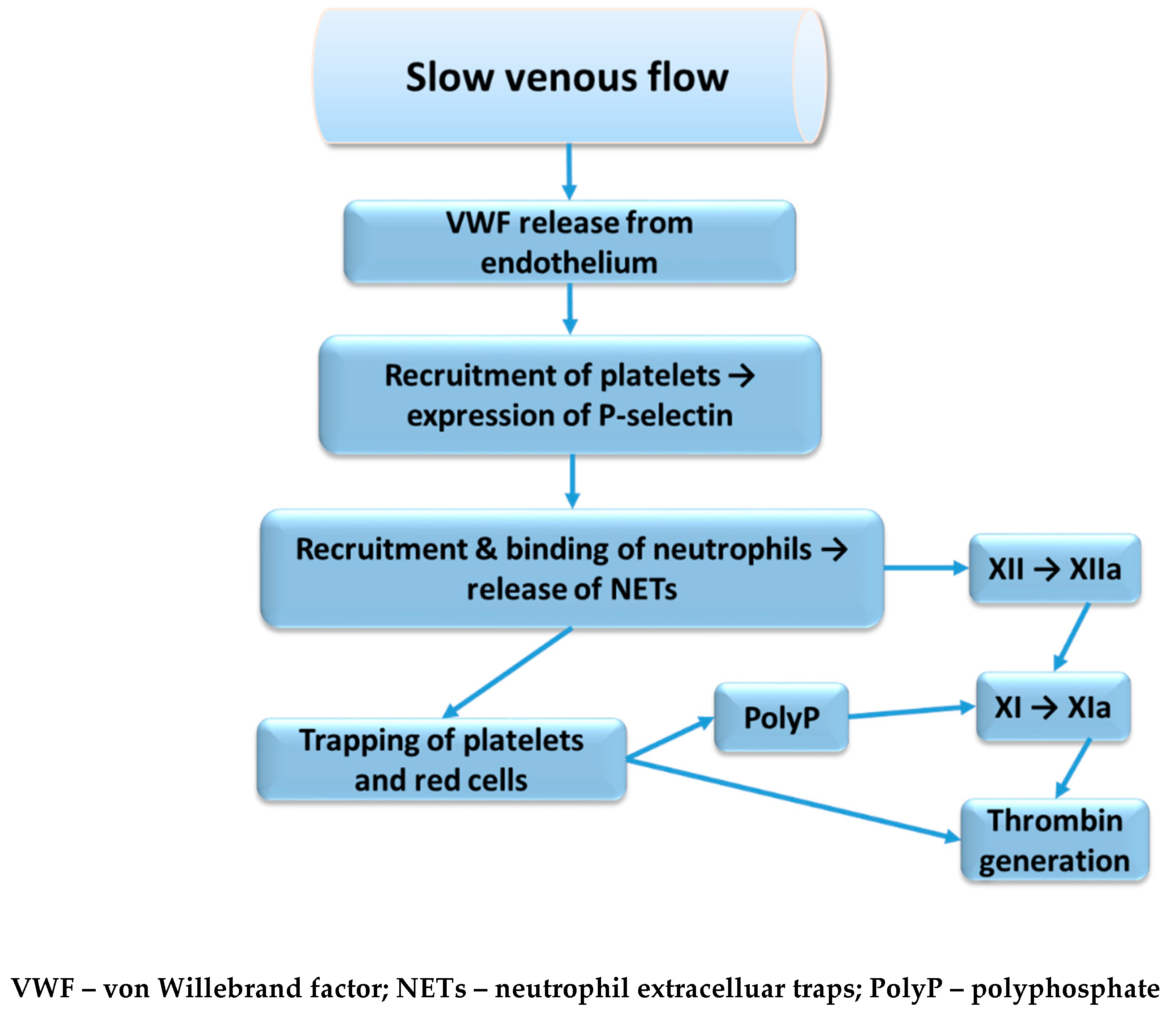
Figure 3.
The tumor- and treatment-related thrombogenic mechanisms.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



