
Submitted:
10 September 2024
Posted:
11 September 2024
You are already at the latest version
Abstract
Transcriptomes from stem-inoculated Scots pine saplings were analyzed to identify unique and enriched H. annosum transcripts in the early stages of infection. Comparing different time points since inoculation identified 131 differentially expressed H. annosum gene with p values ≤ 0.01. Our research supports the results of previous studies on the Norway spruce – Heterobasidion annosum s.l. pathosystem, indicating the role of carbohydrate- and lignin degradation genes in pathogenesis at different timepoints post inoculation and the role of lipid metabolism genes (including but not limited to the delta-12 fatty acid desaturase gene previously reported to be an important factor). The results of this study indicate that malic enzyme could be a potential gene of interest in the context of H. annosum virulence.
Keywords:
plant pathology
; root rot
; pathogen gene expression
; early stage infection
1. Introduction
Scots pine (Pinus sylvestris L.) is the most widely distributed pine species and is found throughout Eurasia [1]. It is a species of major economic importance, widely used in timber, pulp, and paper production [2]. It is also a keystone species providing stability for large ecosystems [3,4] and has been a dominant species for millennia [5].
One of the main causes of mortality of pine, excluding bark beetles – genera Ips De Geer, 1775 and Tomicus Latreille, 1802 [6,7], are Heterobasidion complex fungi, in particular, the basidiomycete Heterobasidion annosum (Fr.) Bref., which causes root rot [8]. In the European forest sector, the Heterobasidion complex causes combined economic losses in the hundreds of millions of euros annually [9]. While the exact proportion of the losses specifically related to Scots pine is not known, given the widespread distribution of Scots pine in European forests and the high economic value of Scots pine, the economic impact of this pathosystem on Scots pine is significant.
Currently, mitigation of Heterobasidion root rot includes forest management activities (monitoring, creation of sustainable forests, sanitary felling, etc.) [10,11,12,13]. One of the strategies for the protection of renewed pine stands is the use of biological control agents based on the fungus Phlebiopsis gigantea (Fr.) Jülich [14,15,16]. Breeding for resistance or tolerance is promising, as genetic components for H. annosum resistance have been determined [17], Scots pine clones with varying resistance against H. annosum have been described [18] and the heritability and genetic gain values for breeding for resistance against root rot have been calculated [19].
To increase the efficiency of breeding programs, genetic mechanisms and candidate genes for resistance need to be identified, thus, studies of gene expression changes in the host in response to inoculation and related genetic testing have been performed for Scots pine reacting to H. annosum, and also for related pathosystems [20,21,22,23,24]. Genetic virulence factors of H. annosum have also been studied [25,26]. A study combining genomic and transcriptome approaches identified genes linked with secreted proteins as potential virulence factors (besides genes involved in oxidation- reduction processes and genes encoding domains relevant to transcription factors) [27]. In the same study secretome annotation and analysis in the pathogen-host interactions database [28] showed that the most virulent H. parviporum isolate was found to contain many carbohydrate-active enzyme genes for cell wall degradation and an increased amount of secreted proteins for lignin degradation. Proteins of the reactive oxygen species-scavenging system and proteases were identified as an important part of the secretome. This study also identified cytochrome P450 proteins as significant for pathogenesis, which could be explained by the involvement of cytochrome P450 gene family members in detoxification of substances from the environment and synthesis of fungal toxins [29]. However, the study of Zeng et al. (2018) did not identify a specific gene as the sole determinant for variations in virulence between different H. parviporum isolates.
Recently, dual transcriptome studies have provided information on host and pathogen transcriptomes simultaneously [30,31,32,33]. However, not all of these studies focus on the host-pathogen interaction [31]. The dual transcriptome studies are also possible only if high number of reads can be obtained from samples to provide sufficient amount of pathogen transcriptome reads, as the proportion of transcripts from the pathogen can be below 1% [32]. No dual transcriptome studies on the H. annosum – P. sylvestris pathosystem have been published and, to best of our knowledge, there are no investigations providing data on the H. annosum (sensu stricto) transcriptome during infection of Scots pine. However, in a study by Lunden et al., 2015 about inoculation of Norway spruce with H. annosum s.s., delta-12 fatty acid desaturase and clavaminate synthase were indicated as potential virulence factors [33]. Polysaccharide-degrading enzymes and lignin-degrading enzymes have long been regarded as important for pathogenesis [34]. Expression of these genes was also detected in the dual transcriptome experiment by Kovalchuk et al., 2019 [30]. The same study also mentions that seven Heterobasidion genes classified as lipases are expressed, but no further details were provided in that study. Expression of lipid metabolism genes (encoding lipases, malic enzyme) was detected in Fusarium circinatum Nirenberg & O'Donnell while infecting Pinus species [32]. In addition to expression of polysaccharide- and lignin-degrading enzyme genes, accumulation of toxins and oxalate have been reported in host tissues colonized by Heterobasidion [34].
Previous research has shown that analysis of fungal transcriptomes enables understanding of adaptations by pathogens to different host species [32]. Therefore, accumulation of in planta gene expression data for H. annosum can enable analysis of the variation of defense strategies within-the host species by inoculation experiments with genetically characterized fungal isolates. This could be used as a basis for the identification of molecular markers for resistance-oriented forest tree breeding. In addition, comparison of fungal gene expression between isolates could provide insights into differences in aggressiveness or virulence, providing a better understanding of the role of genetic variation of pathogens in the development of plant diseases. This investigation provides an initial assessment of time post inoculation-dependent differentially expressed pathogen genes from one to four weeks post inoculation. This assessment is based on mapping a large amount of reads to the H. annosum transcriptome (> 11 million). This provides information about genes and pathways important in early pathogenesis, and demonstrates the feasibility of the utilized sequencing technology.
2. Materials and Methods
One year old Scots pine saplings were inoculated with a Latvian isolate (ID HA2) of H. annosum s.s.. The saplings were obtained from the Latvian State Forests seedling nursery in Kalsnava, Latvia, and were grown from mixed origin improved seed material. For inoculation, a wooden plug grown-through with the H. annosum isolate was placed on an area of the stem with the bark surface removed. Samples for RNA extraction were collected at one, two, three, and four weeks post inoculation and stored at -80 °C until RNA extraction. Four biological replicate samples were collected for each time point. RNA extraction was performed following a CTAB-based protocol [35]. The sampling site included the inoculation site. These time points were chosen, firstly, to allow time for the pathogen to grow into the host tissue and form biomass to increase the amount of obtainable reads and, secondly, to allow partial comparison with previous studies (Lundén et al., 2015 [33] and Zamora-Ballesteros et al., 2021 [32]) which used 5 and 4 days post inoculation as their only time points, respectively. In context of infection progression, in the related Picea abies (L.) H. Karst. – H. parviporum Niemelä & Korhonen pathosystem, germ tubes form in roots within 24 h after spore adhesion, colonization of cortical tissue happens 24 – 48 post inoculation, and the endodermis is reached at 72 h [36]. By 12 to 15 days post inoculation stelar cells are deteriorated but plant responses involving papillae formation and lignification in cortical and endodermis tissue in roots can be observed [36]. Thus, the selected time points represent early infection and later stages.
RNA extraction was followed by quality control of the RNA samples using the Agilent 2100 Bioanalyzer and RNA 6000 nano kit (Agilent, Cat. No. 5067-1511) for RNA integrity determination and quantitation using the Qubit spectrofluorometer and Qubit RNA BR Assay Kit (Thermo Fisher Scientific, Cat. No. Q10210). The obtained material was quite degraded. RIN values ranged from 1.9 to 4.7 (average 2.6) and three samples didn’t produce a RIN value but were included as good results were obtained using qPCR to detect the presence of H. annosum RNA in the total RNA samples. Considering that short read (100bp) sequencing was used, thus reducing the influence of degraded RNA, sequencing libraries were prepared with all samples. RNA quality control data are provided in Supplementary Table S1.
To confirm presence of H. annosum RNA in the extracted RNA samples, one-step RT-qPCR with H. annosum-specific primers targeting the laccase gene was performed. The GoTaq® 1-Step RT-qPCR System (Promega, Cat. No. A6021) was used with the forward primer: 5'-CCAGAAAGTAGACAATTATTGGATTCG-3', and reverse primer: 5’-GAGTTGCGGCCATTATCGA-3’ [37]. Reaction mixture per sample was: 10 µl GoTaq qPCR Master Mix (2X); 0.4 µl GoScript RT Mix for 1-Step RT-qPCR (50X); 0.33 µl CXR Reference Dye; final concentration of each primer 200 nM; nuclease-free water to a volume of 20 µl. Thermal cycling profile: 37 °C 15 min, 95 °C 10 min, followed by 40 cycles of 95 °C 10 s, 60 °C 30 s, 72 °C 30 s, then followed by a melt curve stage during which the temperature is increased from 60 °C to 95 °C in 0.3 °C intervals. One-step RT-qPCR was performed in an Applied Biosystems StepOnePlus qPCR machine. To avoid the influence of primer dimers on the results, melting curve peak height for the specific qPCR product was used instead of Ct value. This approach was possible because none of the reactions reached plateau phase. Peak height of the specific product was divided by RNA concentration to obtain a ratio characterizing the proportion of H. annosum RNA in the RNA sample.
Transcriptome sequencing libraries were created using the MGIEasy RNA Directional Library Prep Set (MGI Tech Co., Cat. No. 1000006385) following the manufacturer’s protocol. 500 ng of total RNA was used for each library. Ribosomal RNA was depleted by use of the Qiagen QIAseq FastSelect –rRNA Plant Kit (Cat. No. 334315). Incubation was performed as described in Table 4 of the QIAseq® FastSelect™ Handbook (according to treatment of RIN < 3 samples). The reaction mixture for this step was as following: 10 µl of RNA sample containing 500 ng of RNA, 4 µl fragmentation buffer (MGI), 4 µl directional RT Buffer 1 (MGI), 1 µl diluted directional RT buffer 2 (MGI), 1 µl QIAseq FastSelect −rRNA Plant reagent. RT enzyme mix (MGI) was only added after this incubation, before the reverse transcription step. The rest of the library preparation protocol was not modified from the manufacturers’ protocol. DNBSEQ-G400RS High-throughput Sequencing Set (MGI Tech Co., Cat. No. 1000016950) was used for sequencing. Sequencing was performed on a DNBSEQ G400 sequencer (MGI Tech Co.) by the Latvian Biomedical Research and Study Centre (Riga, Latvia) on May 20, 2023. The manufacturer’s standard protocol was used for sequencing.
The CLC Genomics Workbench v.23 was used for data import and CLC Genomics Workbench v.21.0.5 (including the Blast2GO commercial plugin v.1.21.14 (BioBam Bioinformatics S.L.)) was used for data analysis and annotation. Data analysis workflow: demultiplexing (automatically performed by the sequencer), trimming (settings: quality limit: 0.05; max. number of ambiguous nucleotides: 2; no adapter trimming; trim homopolymers; no terminal nucleotide removal; max. Length: 150), RNA-seq analysis (settings: no spike-in controls; mismatch cost: 2; insertion cost: 3; deletion cost: 3; length fraction: 0.8; similarity fraction: 0.8; no global alignment; strand specific: both; library type: bulk; max. number of hits for a read: 10; count paired reads as two: no; expression value: total counts; create reads track: yes), differential gene expression analysis (settings: technology – whole transcriptome RNA-Seq; filter on average expression for FDR correction: No; metadata table: yes; test differential expression due to: WPI (weeks post inoculation); while controlling for: not set; comparisons: all group pairs), selection of the differentially expressed genes to perform annotation using the Blast2GO plugin (settings: blast program: blastx-fast; Blast DB = nr; mapping using Goa version 2022_08). Mapping of reads to a reference transcriptome of H. annosum was performed [38,39] (settings: masking mode – no masking; update contigs: no; match score: 1; mismatch cost: 2; cost of insertions and deletions: linear gap cost; insertion cost: 3; deletion cost: 3; length fraction 0.5; similarity fraction: 0.8; no global alignment; auto-detect paired distances; non-specific match handling: map randomly) to be able to calculate the percentage of H. annosum reads. The reads were also mapped against the transcriptome of P. sylvestris from Wachowiak et al., 2015 [40] using the same settings.
A gene (transcript) was considered differentially expressed if the comparison between time points produced a statistically significantly different count of reads mapped to the transcript in question with the p value below 0.01. Results are expressed as fold change comparing different time points (with four biological replicates for each time point). Calculations of the differential gene expression related parameters were performed using the differential gene expression analysis tool in the CLC genomic workbench software, see paragraphs 31.6 and 31.6.4 in the manual [41] for more details.
For visualization of the differential expression, the online tool for up to 6-group Venn diagram creation InteractiVenn [42]. Visualization of other data was performed using the tools in CLC software and Blast2GO plugin.
3. Results
3.1. Sequencing Statistics
3.36 × 109 reads were obtained from the 16 sequenced libraries, of which ~ 11.25 million reads mapped to the H. annosum transcriptome. A summary of the quantity of read mapping to H. annosum and P. sylvestris transcriptomes from each sequenced library is provided in Table 1.
3.84 million H. annosum reads were obtained from material taken one week post inoculation. Material taken at two to four weeks post inoculation produced, respectively, 2.54, 3.05 and 1.82 million H. annosum reads. This shows that sufficient amount of reads for statistical analysis is obtained regardless the low RIN values of the samples. Mapping against a transcriptome of P. sylvestris produced hits for, on average, 42.51 % of the reads, depending on the library.
The high proportion of reads not mapping to P. sylvestris can be explained with the limited nature of the reference transcriptome. A Scots pine reference genome is currently not available and this influences the annotation and quality control of any Scots pine transcriptome generated before the availability of a high quality reference genome.
3.2. Most Transcribed Genes in Each Time Point
Based on the number of reads mapping to H. annosum reference transcripts, we determined the 100 most transcribed genes at each time point (Supplementary File 1).
Frequency of level 7 biological process gene ontology (GO) terms were similar between all timepoints. The unique GO terms for each timepoint are summarized in Table 2.
50 of the most highly expressed genes were shared between all timepoints, and each timepoint had 50 unique genes.
3.3. Differential Gene Expression
All timepoints were compared against all other timepoints, resulting in six statistical comparisons for differential gene expression (DGE). The main focus of this study was the analysis of early stage infection transcripts, therefore, for visualizations, the transcriptome one week after inoculation was compared with the other time points (Figure 1). A Venn diagram of the comparison between all timepoints is provide in Supplementary Figure S1.
The most up- and downregulated transcripts comparing one and two weeks, one and three weeks, and one and four weeks post inoculation are presented in Table 3, Table 4 and Table 5, respectively. In the Venn diagram above the gene upregulated in all the comparisons is annotated as malic enzyme.

Table 3.
Twelve (10 annotated) most upregulated and six downregulated transcripts (1 WPI vs 2 WPI).
| Mapping Reference ID | Annotation | Fold change | P-value | |
|---|---|---|---|---|
| Upregulated | ||||
| CCPA1999.b1 | Hypothetical protein HETIRDRAFT_426980 | 181.85 | 4.72E-05 | |
| CCPB2345.b1 | Carotenoid ester lipase precursor | 145.16 | 2.55E-04 | |
| CCOZ2064.b1 | ATP-utilizing phosphoenolpyruvate carboxykinase | 64.23 | 9.74E-05 | |
| CCPA2867.g1 | Aldo/keto reductase | 45.65 | 1.76E-04 | |
| CCPB993.g1 | Terpenoid cyclases/protein prenyltransferase alpha-alpha toroid | 43.52 | 5.23E-03 | |
| CCPC5739.g1 | NAD-P-binding protein | 34.14 | 1.76E-03 | |
| CCPC2435.b1 | ---Na---* | 33.93 | 2.87E-04 | |
| CCPA4098.g1 | GPI mannosyltransferase 3 | 29.59 | 2.71E-03 | |
| CCPB1601.b1 | ---Na--- | 29.53 | 8.77E-04 | |
| CCOZ1600.b1 | Methionine adenosyltransferase | 28.24 | 4.23E-03 | |
| CCPC3360.b1 | Isocitrate lyase | 27.32 | 9.33E-05 | |
| CCPA4929.b1 | Alpha/beta hydrolase | 25.73 | 5.18E-04 | |
| Downregulated | ||||
| CCPC8078.b1 | Transcription regulator | –23.45 | 7.99E-03 | |
| CCOZ5192.g1 | Dnaj-domain-containing protein | –20.35 | 3.12E-03 | |
| CCPB3914.b1 | Hypothetical protein HETIRDRAFT_426907 | –18.18 | 4.34E-03 | |
| CCOZ3764.b1 | Negative regulator of differentiation 1 | –12.93 | 9.40E-03 | |
| CCPC2832.b1 | Ornithine decarboxylase antizyme-domain-containing protein | –12.24 | 8.22E-03 | |
| CCPA3492.b1 | Hypothetical protein HETIRDRAFT_477666 | –8.36 | 3.06E-03 | |
* ---Na--- – no annotation.
Table 4.
Ten most upregulated and eleven (10 annotated) most downregulated transcripts (1 WPI vs 3 WPI).
Table 4.
Ten most upregulated and eleven (10 annotated) most downregulated transcripts (1 WPI vs 3 WPI).
| Mapping Reference ID | Annotation | Fold change | P-value |
|---|---|---|---|
| Upregulated | |||
| CCPC2187.b1 | Malic enzyme | 61.69 | 2.06E-04 |
| CCOZ3444.b1 | Heat shock protein 70 | 55.59 | 3.80E-03 |
| CCPB993.g1 | Terpenoid cyclases/protein prenyltransferase alpha-alpha toroid | 44.46 | 1.40E-03 |
| CCPC2829.b1 | Pali-domain-containing protein | 32.82 | 2.45E-04 |
| CCPA4010.b1 | Fatty acid desaturase-domain-containing protein | 27.64 | 4.38E-03 |
| CCPC4213.b1 | Hypothetical protein HETIRDRAFT_325943 | 25.72 | 5.16E-03 |
| 11E44-04-08 | Predicted protein | 24.54 | 8.11E-04 |
| CCPC4213.g1 | Hypothetical protein HETIRDRAFT_325943 | 24.37 | 6.08E-03 |
| CCPA4569.b1 | Hypothetical protein HETIRDRAFT_441917 | 23.75 | 3.45E-04 |
| CCPC6772.g1 | Putative BAG domain containing protein | 23.30 | 7.21E-03 |
| Downregulated | |||
| CCPC3479.g1 | Groes-like protein | –41.47 | 1.64E-03 |
| CCPA5017.g1 | Protein arginine N-methyltransferase | –39.95 | 3.01E-04 |
| CCOZ3601.b1 | Secy protein | –27.45 | 6.26E-03 |
| 16D10 | HSP20-like chaperone | –25.79 | 2.78E-03 |
| CCOZ4082.b1 | Glucoamylase | –23.26 | 2.19E-03 |
| CCPA3011.b1 | Cell division control/GTP binding protein | –20.94 | 8.14E-03 |
| CCPA3961.g1 | Glutamate decarboxylase | –18.18 | 7.47E-03 |
| CCPC7984.b1 | ---Na--- | –17.48 | 5.70E-03 |
| D69E9 | 40S ribosomal protein S26 | –16.73 | 3.76E-03 |
| CCPB4097.b1 | Hypothetical protein HETIRDRAFT_439855 | –14.76 | 4.80E-03 |
| CCPC6286.b1 | Leucine aminopeptidase | –13.02 | 7.79E-03 |
Table 5.
Ten most upregulated and seven downregulated transcripts (1 WPI vs 4 WPI).
| Mapping Reference ID | Annotation | Fold change | P-value |
|---|---|---|---|
| Upregulated | |||
| CCPA575.b1 | Delta-12 fatty acid desaturase | 48.12 | 7.94E-05 |
| CCPA1686.b1 | Polysaccharide lyase family 1 protein | 47.55 | 4.24E-03 |
| CCPA1999.b1 | Hypothetical protein HETIRDRAFT_426980 | 46.41 | 1.60E-04 |
| 10F24-03-16 | Elongase of fatty acids ELO | 45.14 | 9.47E-03 |
| CCPC993.b1 | Erylysin B | 38.75 | 5.07E-03 |
| CCPC1268.b1 | Delta-12 fatty acid desaturase protein | 38.60 | 1.89E-03 |
| CCPA3999.b1 | Fatty acid desaturase-domain-containing protein | 35.32 | 4.51E-03 |
| CCPC5436.b1 | Hypothetical protein EW146_g3762 | 33.84 | 4.25E-03 |
| CCPA5235.g1 | Hypothetical protein HETIRDRAFT_468348 | 33.44 | 3.52E-03 |
| CCPC8046.b1 | Delta-12 fatty acid desaturase | 33.02 | 1.59E-04 |
| Downregulated | |||
| D128H9 | RS27A protein | –128.69 | 2.57E-04 |
| CCPA2234.b1 | Hypothetical protein HETIRDRAFT_409605 | –50.89 | 7.84E-04 |
| CCOZ3606.b1 | Glycoside hydrolase superfamily | –21.73 | 5.62E-03 |
| CCPB3914.b1 | Hypothetical protein HETIRDRAFT_426907 | –20.09 | 3.36E-03 |
| CCPA5017.g1 | Protein arginine N-methyltransferase | –18.89 | 5.61E-03 |
| CCPA2400.b1 | General substrate transporter | –13.93 | 8.68E-03 |
| CCPB4930.b1 | Family 43 glycosylhydrolase | –11.52 | 3.40E-03 |
Tables showing most up- and downregulated transcripts for rest of DGE comparisons are provided as Supplementary File 2. Complete expression table is provided in Supplementary File 3.
4. Discussion
Differences among the libraries in the proportion of H. annosum reads might indicate biologically relevant information such as the proportion of living pathogen in the tissue from which RNA was extracted but other explanations like sampling effects, host genotype effects, unequal rRNA depletion or induced host rRNA production at some time points could influence the results. This could also be an indication of a varying positive effect from the inoculum plug still providing resources to the pathogen but this hypothesis was not explicitly analyzed in this study. If one outlier (library with largest difference from average percentage of H. annosum reads per group) is removed per time point, a strong correlation (R2 = 0.8116) between longer time since inoculation and lower proportion of H. annosum reads is observed. Yet, given the many factors influencing the proportion of H. annosum reads, we refrain from conclusions.
Most of the differentially expressed genes lack detailed annotations, however, some of the genes showing the highest upregulation during infection might have a role in disturbing host cell integrity (e.g. carotenoid ester lipase precursor, which is secreted and can cross the cell wall, [43], and erylysin B [44]), or can affect lipid metabolism (e.g.carotenoid ester lipase precursor, fatty acid desaturase-domain-containing protein, delta-12 fatty acid desaturase [33], elongase of fatty acids ELO, malic enzyme [45]), terpene metabolism (terpenoid cyclases/protein prenyltransferase alpha-alpha toroid), are involved in oxidation-reduction processes (aldo/keto reductase [46]), cellular growth (GPI mannosyltransferase 3 [47]), cell development and metabolism control (methionine adenosyltransferase (synonym for S-adenosylmethionine synthetase) [48]) and stress tolerance (heat shock protein 70). Increased expression of delta-12 fatty acid desaturases and related protein genes is especially pronounced comparing 1 WPI to 4 WPI. This could be related to lignin degradation [49], especially in the context of elevated transcription of polysaccharide lyase [50]. The initial stage of infection requires the fungal hyphae to penetrate host cell walls to colonize the tissue. As the structural integrity of conifer tissue is mainly ensured by cellulose, hemicellulose and lignin, polysaccharide-degrading enzymes are needed to penetrate the tissue and to access nutrition sources away from potential competitors on tissue surface. Several microorganisms in soil and tissue surfaces can negatively affect the growth of H. annosum, therefore the ability to quickly penetrate cell walls and to colonize tissues is important for survival of the fungus and successful infection of the host [34].
At one week post inoculation, a terpenoid cyclase gene is more highly expressed than at two or three weeks post inoculation. However, as terpenoids represent the most diverse and abundant class of natural products and have high functional diversity[51,52], any suggestions about the possible role in the infection process would be highly speculative.
Genes downregulated during early stage infection have roles in transcription and translation (transcription regulator, 40S ribosomal protein S26, RS27A protein), polyamine regulation (ornithine decarboxylase antizyme-domain-containing protein [53]), protein folding as chaperones (groes-like protein, HSP20-like chaperone), cell development / signaling (Cell division control/GTP binding protein). Downregulation of the negative regulator of differentiation 1 one week after inoculation could result in positive regulation of differentiation.
Clearly, improved characterization of the proteins encoded by the differentially expressed genes is needed to obtain a better understanding of the molecular processes affecting H. annosum pathogenicity. However, the results obtained about annotated genes suggest that delta-12 fatty acid desaturases could be a virulence factor. The delta-12 fatty acid desaturase gene expression in H. annosum – Norway spruce pathosystem were detected by Lundén et al. (2015) [33]. These authors suggest a central role of this enzyme in sustaining of fungal growth. Considering the extremely small amount of H. annosum transcripts in the work of Lundén et al. and the small proportion of H. annosum reads in our work, identification of delta-12 fatty acid desaturase gene as important for the pathogen indicates that this gene plays an important role during infection. An increase in the expression of other transcripts influencing lipid metabolism was also observed (malic enzyme, elongase of fatty acids and an unspecified type of fatty acid desaturase-domain-containing protein). The gene for malic enzyme was the only differentially up-regulated gene comparing the one week post inoculation time point against all other time points. This concurs with findings showing the role of fungal lipids and lipid biosynthesis proteins in plant-pathogen interactions as potential virulence factors [54,55]. Malic enzyme is potentially highly interesting in this context. As reviewed by Voreapreeda et al., (2013) [56], there are different types of malic enzymes (EC 1.1.1.38 – EC 1.1.1.40), but the malic enzyme gene identified in our study (sequence ID: CCPC2187.b1 [39]) was not categorized to a specific group. However, activity of malic enzyme can drastically change the lipid content of oleaginous fungi [57]. Furthermore, a study investigating the H. annosum transcriptome during saprotrophic growth on Scots pine bark, sapwood and heartwood detected down-regulation of malic enzyme during growth on bark and heartwood chips [58]. This may suggest that malic enzyme activity is needed specifically when invading a living host, however further research is needed, as in the previous study, the time post treatment was 3 months, in contrast to one week in our study.
The observed downregulation of glycoside- and glycosyl hydrolase genes at 1 WPI compared to 4 WPI might seem contra-intuitive, as enzymes of these groups and carbohydrate-active enzymes in general have been recognized as one of the main components facilitating fungal invasion [59,60]. Glycoside- and glycosyl hydrolases have a role in degradation of plant cell wall components, thus it could be expected to see increased activity of these genes at earlier stages of infection. However, this class of proteins is vast and complex [61], and therefore these could be family members with other functions. Members of these groups could be involved in fungal cell wall remodeling, putting it in context with increased fungal growth at later stages of infection.
Up-regulation of specific transcription factors sensitive to fungal or plant hormones was not observed, except for one transcription regulator at 1 WPI with limited information about this protein. In general, the biological functions of most highly expressed genes specific to 1 WPI indicates attempts to mitigate oxidative stress and other elements involved in plant defenses. Response to oxidative stress could be linked with protection from reactive oxygen species involved in host defense responses [62]. Alternatively, it could also be associated in the pathogen’s own development-related cell signaling [63] or both. Other biological processes of 1WPI-specific transcripts are related to metabolism, gene expression and response to heat, osmotic and oxidative stress. In addition, the glyoxylate cycle can also facilitate reduction of oxidative stress [64]. In a broad sense, these transcriptome changes suggest that the pathogen is employing protective measures against plant defenses.
Considering the high proportion of differentially expressed genes lacking detailed annotations, there are still many unknown factors and genes that are involved in the pathogenicity mechanism. More research on the molecular biology of H. annosum is needed to gain better understanding of virulence factors. Further research using genetically identical host plants to obtain a high-quality dual transcriptome of H. annosum – P. sylvestris interaction at different time points post inoculation can provide additional information about the interactions between the pathogen and host during the infection process.
5. Conclusions
This study indicates that carbohydrate- and lignin degradation gene transcription is essential for the pathogenicity of H. annosum s.s. against P. sylvestris, similarly to the results reported by Kovalchuk et al. [30] in a study on the interaction between Norway spruce and H. annosum s.l.. In addition, results from this study suggest that lipid metabolism has a significant role starting from week two post inoculation. Our data suggests that a number of lipid metabolism pathway genes, in addition to the delta-12 fatty acid desaturase gene, are involved in pathogenicity. Many of the differentially expressed genes were not annotated, and improved annotation of a reference transcriptome of H. annosum would provide additional insights into processes occurring during the early stage infection in the H. annosum-P. sylvestris pathosystem.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Table S1: RNA quality control data; Figure S1: Venn diagram of all compared timepoints.
Author Contributions
Conceptualization, M.R., D.R. and V.Š.; methodology, M.R., D.R. and V.Š.; software, M.R., and V.Š.; validation, M.R., and V.Š.; formal analysis, M.R., and V.Š.; investigation, M.R., and V.Š.; resources, M.R., and V.Š.; data curation, M.R., and V.Š.; writing—original draft preparation, M.R., and V.Š.; writing—review and editing, M.R., D.R. and V.Š.; visualization, M.R., and V.Š.; supervision, D.R. and V.Š; project administration, M.R., and V.Š.; funding acquisition, M.R., and V.Š. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by European Regional Development Fund, grant number 1.1.1.2/VIAA/4/20/686. The APC was funded by Latvian State Forest Research Institute “Silava”.
Data Availability Statement
The transcriptome sequencing reads have been deposited to NCBI SRA archive with the BioProject ID PRJNA985902 and accession numbers SRR25032047 – SRR25032062 for the individual sequencing libraries.
Acknowledgments
we would like to thank D. Fridmanis lab at the Latvian Biomedical Research and Study Centre for sequencing of our libraries and Ina Baļķe for consultations during library preparation. We would also like to thank Nadezda Cistjakova and Andis Slaitas from MGI Latvia for technical support. We would also like to thank phytopathology department of Latvian State Forest Research Institute “Silava” for the H. annosum isolate used in this study. We are thankful to prof. Fred O. Asiegbu from University of Helsinki for consultations regarding experimental design and required sequencing throughput.
Conflicts of Interest
The authors declare no conflict of interest.
References
- San-Miguel-Ayanz, J.; de Rigo, D.; Caudullo, G.; Houston Durrant, T.; Mauri, A. European Atlas of Forest Tree Species. Publication Office of the European Union, 2016, 202. ISSN: 9279528335.
- Farjon, A. A Handbook of the World’s Conifers. Brill, 2010, 1–1111. [CrossRef]
- Gonzalez Diaz, P. Development and Maintenance of Genetic Diversity in Scots Pine, Pinus sylvestris (L.); Ph.D. Thesis, University of Stirling, Stirling, U.K., 2018.
- Tyrmi, J.S.; Vuosku, J.; Acosta, J.J.; Li, Z.; Sterck, L.; Cervera, M.T.; Savolainen, O.; Pyhäjärvi, T. Genomics of Clinal Local Adaptation in Pinus Sylvestris under Continuous Environmental and Spatial Genetic Setting. G3 Genes, Genomes, Genet. 2020, 10, 2683–2696. [Google Scholar] [CrossRef]
- Hsieh, S.; Uchman, A. Spatially Associated or Composite Life Traces from Holocene Paleosols and Dune Sands Provide Evidence for Past Biotic Interactions. Sci. Nat. 2023, 110. [Google Scholar] [CrossRef]
- Siitonen, J. Ips Acuminatus Kills Pines in Southern Finland. Silva Fenn. 2014, 48. [Google Scholar] [CrossRef]
- Hlávková, D.; Doležal, P. Cambioxylophagous Pests of Scots Pine: Ecological Physiology of European Populations—A Review. Front. For. Glob. Chang. 2022, 5, 864651. [Google Scholar] [CrossRef]
- Garbelotto, M.; Gonthier, P. Biology, Epidemiology, and Control of Heterobasidion Species Worldwide. Annu. Rev. Phytopathol. 2013, 51, 39–59. [Google Scholar] [CrossRef]
- Asiegbu, F.O.; Adomas, A.; Stenlid, J. Conifer Root and Butt Rot Caused by Heterobasidion Annosum (Fr.) Bref. s.L. Mol. Plant Pathol. 2005, 6, 395–409. [Google Scholar] [CrossRef]
- Samils, N. Monitoring the Control Methods of Heterobasidion Annosum s.l. Root Rot. Ph.D. Thesis, Swedish University of Agricultural Sciences, Uppsala, Sweden, 2008, ISBN: 9789185913800.
- Piri, T. Silvicultural Control of Heterobasidion Root Rot in Norway Spruce Forests in Southern Finland : Regeneration and Vitality Fertilization of Infected Stands; Ph.D. Thesis, University of Helsinki, Finnish Forest Research Institute, Vantaa Research Centre, Finland, 2003.
- Piri, T. Response of Compensatory-Fertilized Pinus Sylvestris to Infection by Heterobasidion Annosum. Scand. J. For. Res. 2010, 15, 218–224. [Google Scholar] [CrossRef]
- Rönnberg, J.; Berglund, M.; Johansson, U.; Cleary, M. Incidence of Heterobasidion Spp. Following Different Thinning Regimes in Norway Spruce in Southern Sweden. For. Ecol. Manage. 2013, 289, 409–415. [Google Scholar] [CrossRef]
- Blomquist, M.; Cleary, M.; Sherwood, P.; Pinto, W.; Herrera, S.L.; Marčiulynienė, D.; Elsafy, M.; Bakal, I.; Nilsson, A.; Rönnberg, J. The Potential of Biological Control against Heterobasidion Root Rot Is Not Realized in Practical Forestry. For. Ecol. Manage. 2023, 531, 120778. [Google Scholar] [CrossRef]
- Kenigsvalde, K.; Brauners, I.; Zaļuma, A.; Jansons, J.; Gaitnieks, T. Biological Protection of Conifers against Heterobasidion Infection – Interaction between Root-Rot Fungus and Phlebiopsis Gigantea. Research for Rural Development 2017. [Google Scholar] [CrossRef]
- Piri, T.; Saarinen, M.; Hamberg, L.; Hantula, J.; Gaitnieks, T. Efficacy of Biological and Chemical Control Agents against Heterobasidion Spore Infections of Norway Spruce and Scots Pine Stumps on Drained Peatland. J. Fungi 2023, 9, 346. [Google Scholar] [CrossRef]
- Korshikov, I.I.; Demkovich, A.E. Genetic Features of Root Fungus-Resistant Scotch Pine Trees in Artificial Stands in the Steppe Zone of Ukraine. Cytol. Genet. 2008, 42, 323–328. [Google Scholar] [CrossRef]
- Vasiliauskas, A. Šakninės Pinties (Heterobasidion Annosum (Fr.) Bref.) Paplitimas Pušynuose, Įveistuose Žemės Ūkio Naudmenuose, Kovos Su Ja Priemonės Ir Kovos Rezultatai [Root Rot Caused by Heterobasidion Annosum in Pinus Sylvestris Plantations on Former Agricultural Land]. Miskininkyste 2001, 2, 42–59. [Google Scholar]
- Rieksts-Riekstiņš, R.; Zeltiņš, P.; Baliuckas, V.; Bruna, L.; Zaluma, A.; Kapostiņš, R. Pinus Sylvestris Breeding for Resistance against Natural Infection of the Fungus Heterobasidion Annosum. Forests 2020, 11, 23. [Google Scholar] [CrossRef]
- Adomas, A.; Heller, G.; Li, G.; Olson, Å.; Chu, T.M.; Osborne, J.; Craig, D.; Van Zyl, L.; Wolfinger, R.; Sederoff, R.; et al. Transcript Profiling of a Conifer Pathosystem: Response of Pinus Sylvestris Root Tissues to Pathogen (Heterobasidion Annosum) Invasion. Tree Physiol. 2007, 27, 1441–1458. [Google Scholar] [CrossRef]
- Mukrimin, M.; Kovalchuk, A.; Ghimire, R.P.; Kivimäenpää, M.; Sun, H.; Holopainen, J.K.; Asiegbu, F.O. Evaluation of Potential Genetic and Chemical Markers for Scots Pine Tolerance against Heterobasidion Annosum Infection. Planta 2019, 250, 1881–1895. [Google Scholar] [CrossRef]
- Hernandez-Escribano, L.; Visser, E.A.; Iturritxa, E.; Raposo, R.; Naidoo, S. The Transcriptome of Pinus Pinaster under Fusarium Circinatum Challenge. BMC Genomics 2020, 21, 1–18. [Google Scholar] [CrossRef]
- Šķipars, V.; Ruņģis, D. Transcript Dynamics in Wounded and Inoculated Scots Pine. Int. J. Mol. Sci. 2021, 22, 1–20. [Google Scholar] [CrossRef]
- Liu, M.; Wang, K.; Haapanen, M.; Ghimire, R.P.; Kivimäenpää, M.; Asiegbu, F.O. Analysis of Transcriptome and Terpene Constituents of Scots Pine Genotypes Inherently Resistant or Susceptible to Heterobasidion Annosum. Front. Plant Sci. 2022, 13, 947734. [Google Scholar] [CrossRef]
- Dalman, K.; Himmelstrand, K.; Olson, Å.; Lind, M.; Brandström-Durling, M.; Stenlid, J. A Genome-Wide Association Study Identifies Genomic Regions for Virulence in the Non-Model Organism Heterobasidion Annosum s.s. PLoS One 2013, 8, e53525. [Google Scholar] [CrossRef]
- Adomas, A.; Eklund, M.; Johansson, M.; Asiegbu, F.O. Identification and Analysis of Differentially Expressed CDNAs during Nonself-Competitive Interaction between Phlebiopsis Gigantea and Heterobasidion Parviporum. FEMS Microbiol. Ecol. 2006, 57, 26–39. [Google Scholar] [CrossRef]
- Zeng, Z.; Sun, H.; Vainio, E.J.; Raffaello, T.; Kovalchuk, A.; Morin, E.; Duplessis, S.; Asiegbu, F.O. Intraspecific Comparative Genomics of Isolates of the Norway Spruce Pathogen (Heterobasidion Parviporum) and Identification of Its Potential Virulence Factors. BMC Genomics 2018, 19, 1–21. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-Base: The Pathogen–Host Interactions Database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.E.; Lee, Y.W.; Son, H. Fungal Cytochrome P450s and the P450 Complement (CYPome) of Fusarium Graminearum. Toxins 2018, 10. [Google Scholar] [CrossRef]
- Kovalchuk, A.; Zeng, Z.; Ghimire, R.P.; Kivimäenpää, M.; Raffaello, T.; Liu, M.; Mukrimin, M.; Kasanen, R.; Sun, H.; Julkunen-Tiitto, R.; et al. Dual RNA-Seq Analysis Provides New Insights into Interactions between Norway Spruce and Necrotrophic Pathogen Heterobasidion Annosum s.l. BMC Plant Biol. 2019, 19, 1–17. [Google Scholar] [CrossRef]
- Wen, Z.; Zeng, Z.; Ren, F.; Asiegbu, F.O. The Conifer Root and Stem Rot Pathogen (Heterobasidion Parviporum): Effectome Analysis and Roles in Interspecific Fungal Interactions. Microorg. 2019, 7, 658. [Google Scholar] [CrossRef]
- Zamora-Ballesteros, C.; Pinto, G.; Amaral, J.; Valledor, L.; Alves, A.; Diez, J.J.; Martín-García, J. Dual Rna-Sequencing Analysis of Resistant (Pinus Pinea) and Susceptible (Pinus Radiata) Hosts during Fusarium Circinatum Challenge. Int. J. Mol. Sci. 2021, 22, 5231. [Google Scholar] [CrossRef]
- Lundén, K.; Danielsson, M.; Durling, M.B.; Ihrmark, K.; Nemesio Gorriz, M.; Stenlid, J.; Asiegbu, F.O.; Elfstrand, M. Transcriptional Responses Associated with Virulence and Defence in the Interaction between Heterobasidion Annosum s.s. and Norway Spruce. PLoS One 2015, 10, e0131182. [Google Scholar] [CrossRef]
- Asiegbu, F.O.; Johansson, M.; Woodward, S.; Hüttermann, A. Biochemistry of the Host - Parasite Interaction; In Heterobasidion Annosum : Biology, Ecology, Impact and Control; Woodward, S.; Stenlid, J.; Karjalainen, R.; Hüttermann, A., Eds.; CABI Publishing: Wallingford, U.K., 1998; pp 176-180.
- Doyle, J.J.; Doyle, J.L. Isolation of Plant DNA from Fresh Tissue. Focus 1987, 12, 13–15. [Google Scholar]
- Asiegbu, F.; Daniel, G.; Johansson, M. Studies on the Infection of Norway Spruce Roots by Heterobasidion Annosum. Canad. J. Bot. 1993, 71, 1552–1561. [Google Scholar] [CrossRef]
- Karlsson, M.; Hietala, A.M.; Kvaalen, H.; Solheim, H.; Olson, Å.; Stenlid, J.; Fossdal, C.G. Quantification of Host and Pathogen DNA and RNA Transcripts in the Interaction of Norway Spruce with Heterobasidion Parviporum. Physiol. Mol. Plant Pathol. 2007, 70, 99–109. [Google Scholar] [CrossRef]
- Olson, Å.; Aerts, A.; Asiegbu, F.; Belbahri, L.; Bouzid, O.; Broberg, A.; Canbäck, B.; Coutinho, P.M.; Cullen, D.; Dalman, K.; et al. Insight into Trade-off between Wood Decay and Parasitism from the Genome of a Fungal Forest Pathogen. New Phytol. 2012, 194, 1001–1013. [Google Scholar] [CrossRef]
- https://genome.jgi.doe.gov/portal/Hetan2/Hetan2.download.html (accessed on 28 June 2023).
- Wachowiak, W.; Trivedi, U.; Perry, A.; Cavers, S. Comparative Transcriptomics of a Complex of Four European Pine Species. BMC Genomics 2015, 16, 234. [Google Scholar] [CrossRef]
- CLC Genomics Workbench User Manual for CLC Genomics Workbench 23.0.5 2023, 1165.
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinformatics 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Brutyn, M.; D’Herde, K.; Dhaenens, M.; Rooij, P. Van; Verbrugghe, E.; Hyatt, A.D.; Croubels, S.; Deforce, D.; Ducatelle, R.; Haesebrouck, F.; et al. Batrachochytrium Dendrobatidis Zoospore Secretions Rapidly Disturb Intercellular Junctions in Frog Skin. Fungal Genet. Biol. 2012, 49, 830–837. [Google Scholar] [CrossRef]
- Panevska, A.; Hodnik, V.; Skočaj, M.; Novak, M.; Modic, Š.; Pavlic, I.; Podržaj, S.; Zarić, M.; Resnik, N.; Maček, P.; et al. Pore-Forming Protein Complexes from Pleurotus Mushrooms Kill Western Corn Rootworm and Colorado Potato Beetle through Targeting Membrane Ceramide Phosphoethanolamine. Sci. Reports 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Wynn, J.P.; Hamid, A.B.A.; Ratledge, C. The Role of Malic Enzyme in the Regulation of Lipid Accumulation in Filamentous Fungi. Microbiology 1999, 145, 1911–1917. [Google Scholar] [CrossRef]
- Chang, Q.; Griest, T.A.; Harter, T.M.; Mark Petrash, J. Functional Studies of Aldo-Keto Reductases in Saccharomyces Cerevisiae. Biochim. Biophys. Acta 2007, 1773, 321–329. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, H.; Zhao, G.; Yang, J.; Luo, Y.; Sun, S.; Wang, Z.; Li, S.; Jin, C. Genetical and O-Glycoproteomic Analyses Reveal the Roles of Three Protein O-Mannosyltransferases in Phytopathogen Fusarium Oxysporum f.sp. Cucumerinum. Fungal Genet. Biol. 2020, 134. [Google Scholar] [CrossRef]
- Gerke, J.; Bayram, Ö.; Braus, G.H. Fungal S-Adenosylmethionine Synthetase and the Control of Development and Secondary Metabolism in Aspergillus Nidulans. Fungal Genet. Biol. 2012, 49, 443–454. [Google Scholar] [CrossRef]
- Watanabe, T.; Tsuda, S.; Nishimura, H.; Honda, Y.; Watanabe, T. Characterization of a Delta12-Fatty Acid Desaturase Gene from Ceriporiopsis Subvermispora, a Selective Lignin-Degrading Fungus. Appl. Microbiol. Biotechnol. 2010, 87, 215–224. [Google Scholar] [CrossRef]
- Van Den Brink, J.; De Vries, R.P. Fungal Enzyme Sets for Plant Polysaccharide Degradation. Appl. Microbiol. Biotechnol. 2011, 91, 1477. [Google Scholar] [CrossRef]
- Huang, Z.Y.; Ye, R.Y.; Yu, H.L.; Li, A.T.; Xu, J.H. Mining Methods and Typical Structural Mechanisms of Terpene Cyclases. Bioresour. Bioprocess. 2021, 8, 1–27. [Google Scholar] [CrossRef]
- González-Hernández, R.A.; Valdez-Cruz, N.A.; Macías-Rubalcava, M.L.; Trujillo-Roldán, M.A. Overview of Fungal Terpene Synthases and Their Regulation. World J. Microbiol. Biotechnol. 2023, 39, 194. [Google Scholar] [CrossRef]
- Palanimurugan, R.; Scheel, H.; Hofmann, K.; Dohmen, R.J. Polyamines Regulate Their Synthesis by Inducing Expression and Blocking Degradation of ODC Antizyme. EMBO J. 2004, 23, 4857–4867. [Google Scholar] [CrossRef]
- Beccaccioli, M.; Reverberi, M.; Scala, V. Fungal Lipids: Biosynthesis and Signalling during Plant-Pathogen Interaction. Front. Biosci. - Landmark 2019, 24, 172–185. [Google Scholar] [CrossRef]
- Cardoza, R.E.; McCormick, S.P.; Lindo, L.; Mayo-Prieto, S.; González-Cazón, D.; Martínez-Reyes, N.; Carro-Huerga, G.; Rodríguez-González, Á.; Proctor, R.H.; Casquero, P.A.; et al. Effect of Farnesol in Trichoderma Physiology and in Fungal-Plant Interaction. J. fungi 2022, 8. [Google Scholar] [CrossRef]
- Vorapreeda, T.; Thammarongtham, C.; Cheevadhanarak, S.; Laoteng, K. Repertoire of Malic Enzymes in Yeast and Fungi: Insight into Their Evolutionary Functional and Structural Significance. Microbiology 2013, 159. [Google Scholar] [CrossRef]
- Hao, G.; Chen, H.; Wang, L.; Gu, Z.; Song, Y.; Zhang, H.; Chen, W.; Chen, Y.Q. Role of Malic Enzyme during Fatty Acid Synthesis in the Oleaginous Fungus Mortierella Alpina. Appl. Environ. Microbiol. 2014, 80, 2672. [Google Scholar] [CrossRef]
- Raffaello, T.; Chen, H.; Kohler, A.; Asiegbu, F.O. Transcriptomic Profiles of Heterobasidion Annosum under Abiotic Stresses and during Saprotrophic Growth in Bark, Sapwood and Heartwood. Environ. Microbiol. 2014, 16, 1654–1667. [Google Scholar] [CrossRef]
- Looi, H.K.; Toh, Y.F.; Yew, S.M.; Na, S.L.; Tan, Y.C.; Chong, P.S.; Khoo, J.S.; Yee, W.Y.; Ng, K.P.; Kuan, C.S. Genomic Insight into Pathogenicity of Dematiaceous Fungus Corynespora Cassiicola. PeerJ 2017, 5. [Google Scholar] [CrossRef]
- Gong, L.; Liu, Y.; Xiong, Y.; Li, T.; Yin, C.; Zhao, J.; Yu, J.; Yin, Q.; Gupta, V.K.; Jiang, Y.; et al. New Insights into the Evolution of Host Specificity of Three Penicillium Species and the Pathogenicity of P. Italicum Involving the Infection of Valencia Orange (Citrus Sinensis). Virulence 2020, 11, 748–768. [Google Scholar] [CrossRef]
- Mewis, K.; Lenfant, N.; Lombard, V.; Henrissat, B. Dividing the Large Glycoside Hydrolase Family 43 into Subfamilies: A Motivation for Detailed Enzyme Characterization. Appl. Environ. Microbiol. 2016, 82, 1686. [Google Scholar] [CrossRef]
- Sahu, P.K.; Jayalakshmi, K.; Tilgam, J.; Gupta, A.; Nagaraju, Y.; Kumar, A.; Hamid, S.; Singh, H.V.; Minkina, T.; Rajput, V.D.; et al. ROS Generated from Biotic Stress: Effects on Plants and Alleviation by Endophytic Microbes. Front. Plant Sci. 2022, 13. [Google Scholar] [CrossRef]
- Heller, J.; Tudzynski, P. Reactive Oxygen Species in Phytopathogenic Fungi: Signaling, Development, and Disease. Annu. Rev. Phytopathol. 2011, 49, 369–390. [Google Scholar] [CrossRef]
- Ahn, S.; Jung, J.; Jang, I.A.; Madsen, E.L.; Park, W. Role of Glyoxylate Shunt in Oxidative Stress Response. J. Biol. Chem. 2016, 291, 11928. [Google Scholar] [CrossRef]
Figure 1.
Venn diagram showing differential gene expression (DGE) between 1 WPI and other time points.
Figure 1.
Venn diagram showing differential gene expression (DGE) between 1 WPI and other time points.
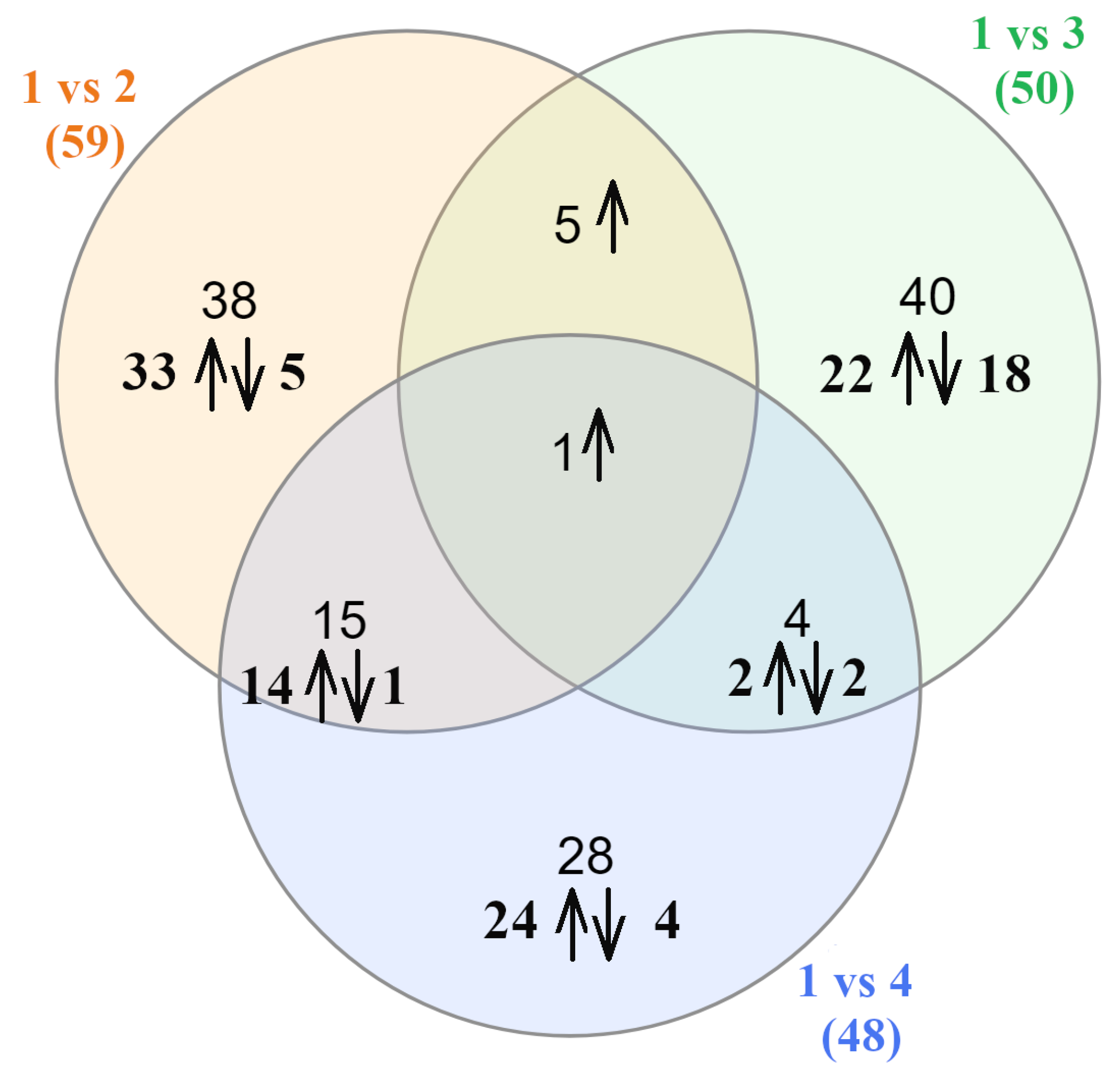
Table 1.
Sequencing statistics – read mapping against H. annosum and P. sylvestris transcriptomes.
| Sample name1 | Total reads | Percentage of reads mapping to H. annosum transcriptome | Percentage of reads mapping to P. sylvestris transcriptome |
|---|---|---|---|
| 1_5 | 330514752 | 0.32 | 26.69 |
| 1_9 | 148181900 | 0.47 | 69.69 |
| 1_10 | 127532184 | 0.73 | 44.92 |
| 1_14 | 265534720 | 0.44 | 45.81 |
| 2_4 | 302911430 | 0.17 | 26.15 |
| 2_10 | 200927014 | 0.36 | 54.40 |
| 2_14 | 138048364 | 0.12 | 23.87 |
| 2_15 | 297177502 | 0.38 | 42.04 |
| 3_3 | 183571350 | 0.65 | 55.76 |
| 3_5 | 175591306 | 0.38 | 57.62 |
| 3_7 | 214847606 | 0.28 | 41.78 |
| 3_12 | 176541044 | 0.32 | 29.99 |
| 4_1 | 163295084 | 0.27 | 56.09 |
| 4_3 | 123870986 | 0.52 | 49.52 |
| 4_10 | 400583236 | 0.11 | 23.70 |
| 4_13 | 115595890 | 0.24 | 32.19 |
1 First symbol in the name represents weeks after inoculation.
Table 2.
Unique GO terms (level 7) for the top 100 expressed transcripts at each time point.
| Biological process | Unique for timepoint |
|---|---|
| Histidine biosynthetic process | 1 WPI |
| Arginine biosynthetic process | 1 WPI |
| Coenzyme A metabolic process | 1 WPI |
| Response to heat | 1 WPI |
| Response to oxygen-containing compound | 1 WPI |
| Isoprenoid biosynthetic process | 1 WPI |
| Response to osmotic stress | 1 WPI |
| Response to oxidative stress | 1 WPI |
| Glyoxylate cycle | 1 WPI |
| Carboxylic acid metabolic process | 1 WPI |
| Protein-containing complex assembly | 1 WPI |
| S-adenosylmethionine biosynthetic process | 1 WPI |
| Proton transmembrane transport | 2 WPI |
| Negative regulation of protein modification process | 2 WPI |
| Negative regulation of phosphate metabolic process | 2 WPI |
| DNA-templated transcription | 2 WPI |
| Macroautophagy | 2 WPI |
| Regulation of translation | 2 WPI |
| Cellular response to amino acid starvation | 2 WPI |
| Regulation of protein dephosphorylation | 2 WPI |
| Positive regulation of transcription by RNA polymerase II | 2 WPI |
| Acetyl-coa biosynthetic process | 3 WPI |
| Citrate metabolic process | 3 WPI |
| Cellular biosynthetic process | 4 WPI |
| Protein import into mitochondrial matrix | 4 WPI |
| Signal transduction | 4 WPI |
| Ergosterol biosynthetic process | 4 WPI |
* WPI – weeks post inoculation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



