
Submitted:
11 September 2024
Posted:
11 September 2024
You are already at the latest version
Abstract
Microglial cells are highly specialized central nervous system (CNS) cells that can promote the inflammation and neurodegeneration seen in Multiple Sclerosis (MS). MS is a neurodegenerative autoimmune disease characterized by inflammation, demyelination, and axonal degeneration. In MS patients, demyelination is associated with activated microglia. Endothelin-1 (ET-1) is a potent vasoconstrictor that induces severe and prolonged cerebral vasoconstriction and inflammation. However, the mechanism of how ET-1 activates a proinflammatory response in the CNS is unknown. To investigate ET-1’s role in microglia activation, HMC3 cells were treated with ET-1 in the presence or absence of endothelin receptor B antagonist, BQ788. TNFα and IL-6 levels were measured using ELISA. Nitric Oxide (NO) production was measured using Griess Reagent. Reactive oxygen species (ROS) production was measured using the MUSE Oxidative Stress kit. ET-1 increases TNFα levels by 56% (p= 0.0003) and IL-6 levels by 86% (p= 0.0111) in HMC3 cells, and it was decreased to basal levels in the presence of BQ788. NO and ROS production is induced by ET-1 (p<0.05), and treatment with BQ788 was able to decrease them. ET-1 increases STAT-1 activation by 3.5 folds compared to control (p <0.0001) in microglia cells. Moreover, to study ET-1 levels in MS, we used C57BL/6 mice brains with or without induced experimental autoimmune encephalomyelitis (EAE). The brain ET-1 levels were measured by qPCR and ELISA. We found that the ET-1 gene and protein were upregulated by 1.5 folds (p= 0.0199) in EAE mice compared to the control. These data suggest that in vitro administration of ET-1-activated microglia significantly increased inflammatory cytokine levels and NO and ROS formation. This novel mechanism of microglial cell activation will provide key information to understand MS pathogenesis.
Keywords:
endothelin
; inflammation
; microglia
; multiple sclerosis
; reactive oxygen species
1. Introduction
Microglia are the resident cells of the innate immune system in the central nervous system (CNS) [1] and provide the defense against invading pathogens and during inflammation. Their primary role is to maintain cellular, synaptic, and myelin homeostasis during the normal function of the CNS. [2] Microglia readily become activated through stimuli of injury or infection and release cytokines, induce phagocytosis, and direct cytotoxicity. Microglia are the major source of reactive oxygen species (ROS) and Nitric Oxide (NO) in the CNS. [3] Microglia are very diverse, and during the past decade, they have emerged as a critical target in neurodegenerative and neuroinflammatory diseases like Multiple Sclerosis (MS). [4]
MS is an autoimmune neurodegenerative disorder of the CNS characterized by inflammation, demyelination, and axonal degeneration. [5] Currently, there is no cure for this condition, and around 2 million people worldwide suffer from it. [4] MS patient immunopathogenesis has been described as heterogeneous due to various types of immune cells participating in neuroinflammation, including T cells, B cells, microglia, and macrophages. [6,7,8,9] Microglial activation has been linked to active demyelination and neurodegeneration, leading to the accumulation of macrophages in the affected tissue and contributing to MS pathophysiology. Microglia are involved in the production of proinflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor (TNF), and chemokines in MS patients. Moreover, activated microglia account for the features of MS pathological findings presenting major histocompatibility complex (MHC) class I and II molecules to T cells and activating the immune response. [10]
Endothelin-1 (ET-1) is a regulator of MHC promoter activity. [11] ET-1 is a potent vasoconstrictor that induces severe and prolonged cerebral vasoconstriction in MS via its two receptors ETRA and ETRB. [12] ET-1 has been associated with cerebral hypoperfusion and reduced extraocular blood flow in MS patients. [12,13,14] Astrocytic ET-1 overexpression directly stimulates nearby microglial cells and macrophages, resulting in the production and secretion of proinflammatory cytokines; this same astrocytic ET-1 causes severe breakdown of the blood-brain barrier (BBB). [13]
Although ET-1’s role in inflammation has been described, there is no evidence that indicates the mechanism by which ET-1 activates microglial cells. We hypothesize that ET-1 is responsible for the differentiation of microglial cells into their pro-inflammatory form, accounting for MS pathophysiology and disease progression. ET-1 will stimulate proinflammatory cytokines, NO, and ROS. In this study, we used HMC3 cell lines to investigate the ET-1 role in microglia activation.
2. Materials and Methods
2.1. Cell Culture
Human microglial clone 3 cell line, HMC3 (ATCC, CRL-3304, RRID:CVCL_II76), were maintained in MEM with 10% FBS (Sigma Aldrich) and 100 U/ml penicillin/streptomycin (Life Technologies) at 37°C in a 5% CO2-humidified atmosphere. Briefly, twelve hours before treatment, cells were serum starved in EMEM with 0.2% FBS. At the time of treatment, cells were washed with PBS and incubated with vehicle, ET-1, or BQ788 in EMEM with 0.2% FBS. The cells were harvested for analysis after 24-hour incubation.
2.2. RNA Extraction and Quantitative Real-Time PCR
Total RNA was prepared with 1 mL of TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to make 20 µL of cDNA from 2 µg of RNA. Gene expression was analyzed using real-time PCR using TaqMan gene expression assay for EDN, EDNRA, EDNRB, NOS2, and GAPDH (Applied Biosystems) in a StepOne Plus from ABI. The ΔΔ cycle threshold method was used to determine mRNA levels. Gene expression was normalized to GAPDH levels.
2.3. IL-6 ELISA
To determine the concentration of IL-6 in cell culture media of control or treated HMC3 cells, an ELISA kit from R&D systems was used following the manufacturer’s instructions.
2.4. Determination of ROS Generation
ROS generation of HMC3 cells was measured by the Muse Oxidative stress kit using the Muse cell analyzer (Millipore, Billerica, MA, USA) fluorescent-based analysis. The manufacturer specific protocol was followed for the assay. In brief, HMC3 cells were treated with 100 nM ET-1 with or without BQ788 treatment and incubated for 24 h. 1 X 107 cells/mL samples were prepared in 1 X assay buffer and treated with Oxidative stress reagent, based on dihydroethidium (DHE) used to detect ROS that is oxidized with superoxide anion to procedure the DNA-binding fluorophore ethidium bromide which intercalates with DNA resulting in red fluorescence.
2.5. Measurement of Nitric Oxide Levels
Nitric oxide levels in the culture supernatant were evaluated by measuring NO2, a major stable product of nitric oxide. Briefly, 50µL of each supernatant was mixed with an equal amount of the Griess solution (0.1% N-(1-naphthyl) ethyl-enediamine and 1% p-aminobenzenesulfonamide in 5% ortho-phosphoric acid) at room temperature for 30 min. Absorbance was read at 540nm using a microplate spectrophotometer. Sodium nitrite was used for the standard curve.
2.6. Detection of STAT-1 Activation
The signal transducer and activator of transcription 1 (STAT-1) activation in HMC3 cells were measured by the Muse® STAT1 Activation Dual Detection Kit using the Muse cell analyzer (Millipore, Billerica, MA, USA). The manufacturer specific protocol was followed for the assay. In brief, HMC3 cells were treated with 100 nM ET-1 with or without BQ788 treatment and incubated for 24 h. Cells were fixed for 5 min and then permeabilized for 5 min. The antibody cocktail was added to the samples and incubated at room temperature and dark for 30 min. Samples were analyzed using the Muse cell analyzer.
2.7. Mice EAE Induction
Brain tissue from experimental autoimmune encephalomyelitis (EAE) mice was purchased from Hooke Laboratories (MA, USA). As described previously, EAE was induced in female C57BL/6 mice (RRID:MGI:2159769). [15] Briefly, 12 female C57BL/6 mice 8 to 12 weeks of age were acclimated to the facility for at least seven days and divided into two groups (EAE and control). EAE was induced by injecting an emulsion containing 1 mg MOG35-55/mL and 5 mg killed Mycobacterium tuberculosis H37Ra/mL subcutaneously at the upper and lower back. This emulsion was supplemented with 100 ng pertussis toxin administered on days 0 and 1. Control mice were injected using the Hooke Control Kit. Mice are scored daily for EAE severity on a scale of 0-5 in 0.5-unit increments. Mice were euthanized at the peak of the disease. Mice were immediately perfused with PBS; then, the brain was isolated and snap-frozen in liquid nitrogen.
2.8. ET-1 ELISA
Tissue samples were homogenized in the cold using a Bullet Blender Storm 24 (Next Advance) in RIPA containing protease inhibitors (Roche) at a 10:1 ratio of buffer volume to tissue wet weight. The homogenate was centrifuged at 17,000 g for 20 min at 4°C, and the supernatant was removed and saved. An ELISA kit from Invitrogen was used to determine the concentration of ET-1 in mice tissue, following the manufacturer’s instructions.
2.9. Statistical Analysis
Data were analyzed by one-way or two-way ANOVA with Bonferroni post-test or Student’s t-test when appropriate. The p-value was set to be <0.05. Data are expressed as mean ± SD of (n) independent experiments unless otherwise stated.
3. Results
3.1. ET-1 induces NO production by HMC3 cells
The human microglia cell line, HMC3, was used to study the effect of ET-1 in microglia activation. HMC3 cells showed an increase in NO concentration when treated with ET-1. When treated with ET-1 and BQ788 (ETRB antagonist), NO concentration showed no significant changes compared to controls (Figure 1A). HMC3 cells showed an increase in inducible nitric oxide synthase (iNOS) expression when treated with ET-1. When treated with ET-1 and BQ788, iNOS expression showed no significant changes compared to controls (Figure 1B).
3.2. ET-1 Induces ROS Production by HMC3 Cells
HMC3 cells treated with ET-1 showed a significant increase in ROS production when compared to controls. HMC3 cells treated with ET-1 and BQ788 showed no significant change in ROS production compared to controls. When compared with controls, HMC3 cells treated with BQ788 show no significant increase in ROS production (Figure 2).
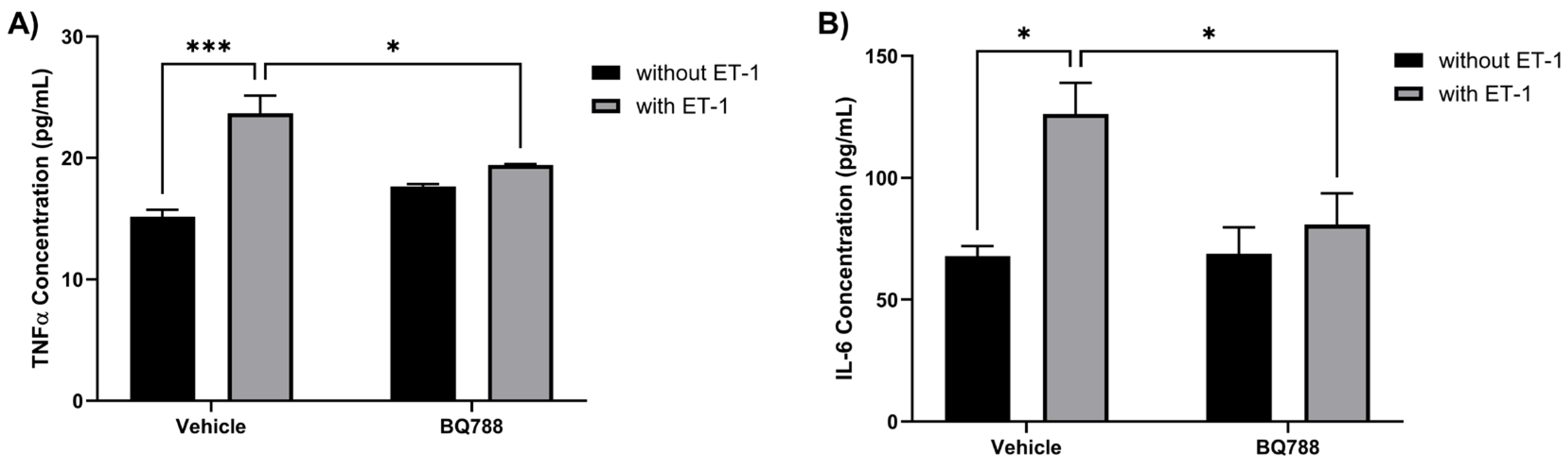
3.3. ET-1 Increases the Secretion of Proinflammatory Cytokines by HMC3 Cells
HMC3 cells treated with ET-1 showed an increase in TNFα concentrations when compared to controls (Figure 3A). HMC3 treated with both ET-1 and BQ788 showed no significant change in concentration of TNFα when compared with control. HMC3 cells treated with ET-1 showed an increase in IL-6 concentrations when compared to controls (Figure 3B). HMC3 treated with ET-1 and BQ788 showed no significant change in the concentration of IL-6 compared with the control.
Figure 2.
ET-1 induces ROS production by microglia cells. HMC3 cells were incubated for 24 hours in the absence or presence of 100 nM ET-1 with or without BQ788 (1uM) and analyzed by flow cytometric assay. Results represent the mean ± SD of n=4. *, p < 0.05 100nM ET-1 versus vehicle and **, p < 0.01 100nM ET-1 versus 100nM ET-1 with 1uM BQ-788.
Figure 2.
ET-1 induces ROS production by microglia cells. HMC3 cells were incubated for 24 hours in the absence or presence of 100 nM ET-1 with or without BQ788 (1uM) and analyzed by flow cytometric assay. Results represent the mean ± SD of n=4. *, p < 0.05 100nM ET-1 versus vehicle and **, p < 0.01 100nM ET-1 versus 100nM ET-1 with 1uM BQ-788.
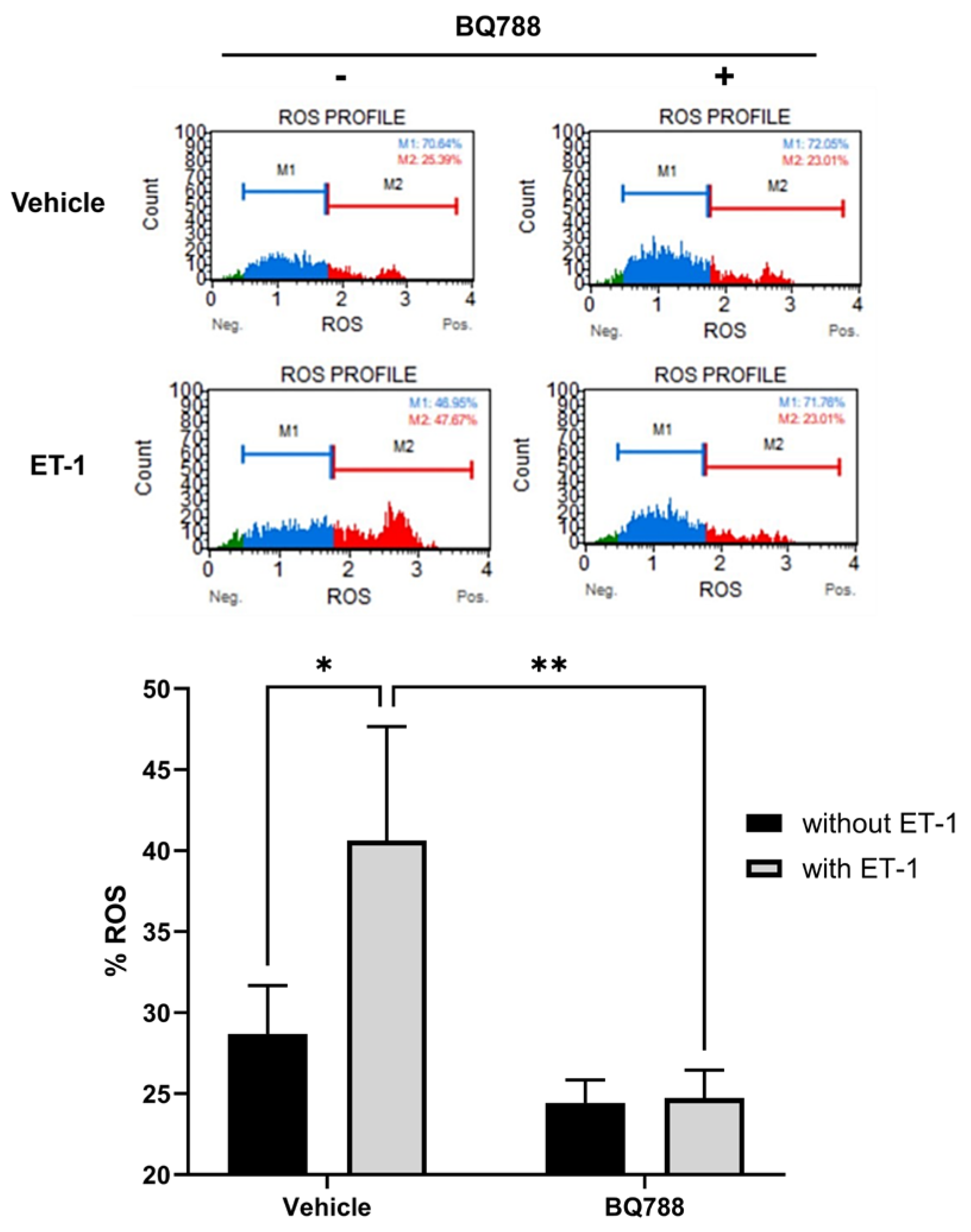
Figure 3.
ET-1 increases the secretion of proinflammatory cytokines by HMC3 cells. HMC3 cells were incubated for 24 hours in the absence or presence of 100 nM ET-1 with or without BQ788. Supernatants were collected to determine secretion levels of TNFα (A) and IL-6 (B) and measured by ELISA assay. Results represent the mean ± SD of n=4. *p < 0.05, **p < 0.01, ***p<0.001.
Figure 3.
ET-1 increases the secretion of proinflammatory cytokines by HMC3 cells. HMC3 cells were incubated for 24 hours in the absence or presence of 100 nM ET-1 with or without BQ788. Supernatants were collected to determine secretion levels of TNFα (A) and IL-6 (B) and measured by ELISA assay. Results represent the mean ± SD of n=4. *p < 0.05, **p < 0.01, ***p<0.001.
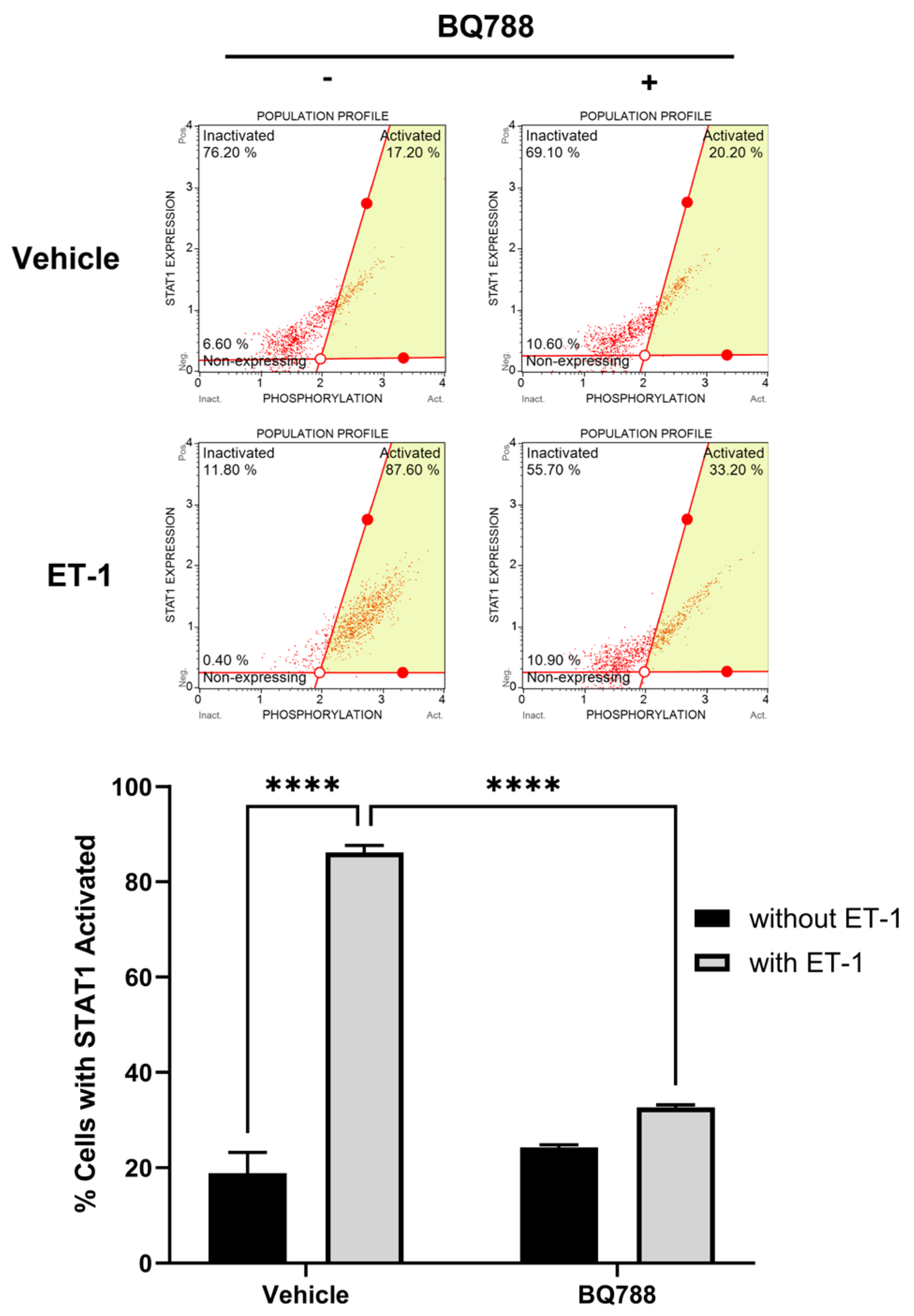
3.4. ET-1 Activates STAT-1 Pathway
Human microglia cells treated with ET-1 for 24h showed an increase in STAT-1 activation (phosphorylation) when compared to controls (Figure 4). This activation was decreased when cells were treated with BQ788, which showed an activation by ET-1 receptor B.
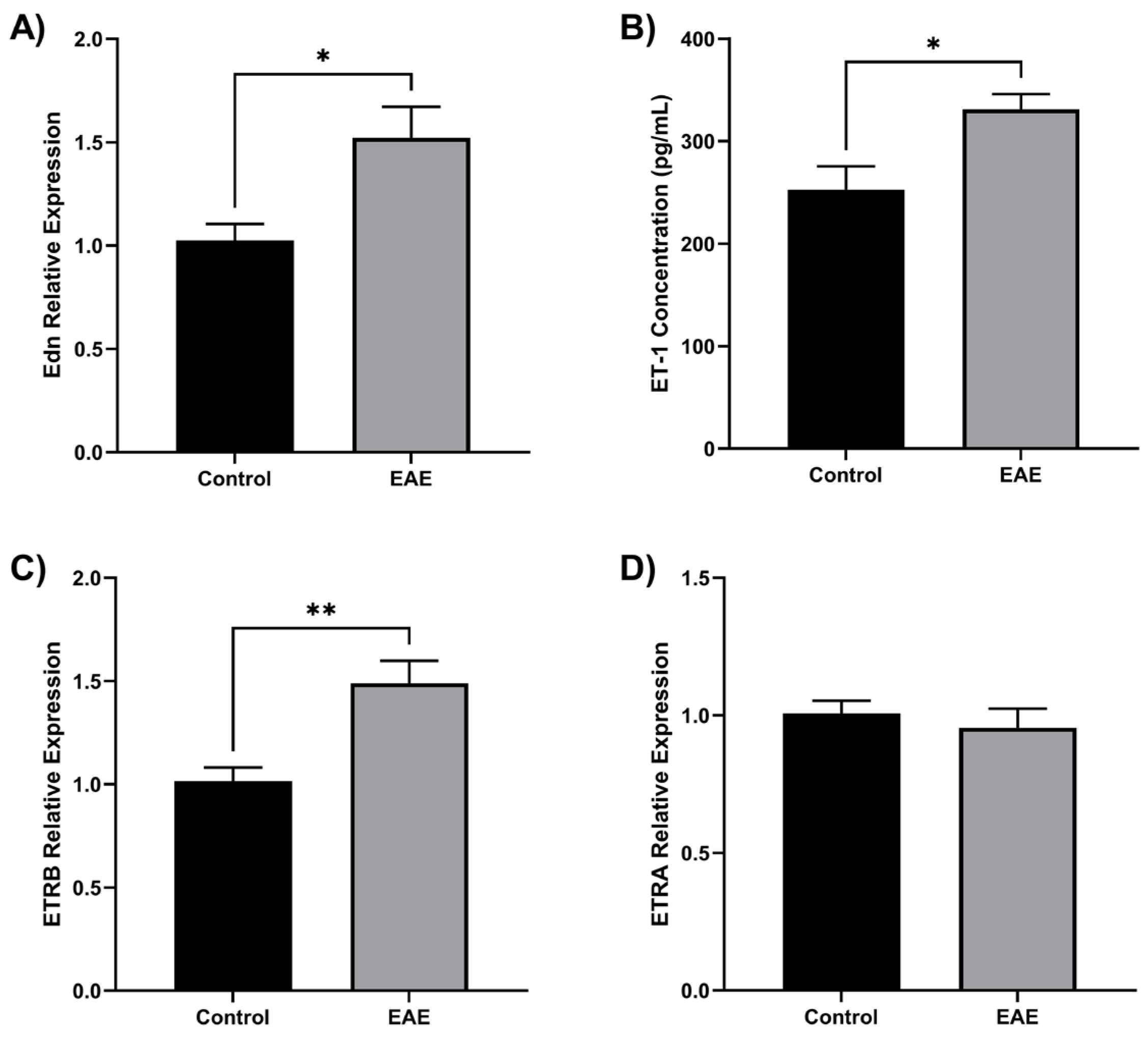
3.5. ET-1 Is Overexpressed in EAE Mice
The results showed that ET-1 is overproduced in EAE mice and contributes to the development of the disease. Specifically, EAE-immunized mice showed increased levels of Edn1 gene expression (Figure 5A) and ET-1 concentration (Figure 5B) compared to control mice, and the ET-1 receptor ETRB was overexpressed in EAE mice (Figure 5C), while ETRA was found in similar levels in both groups (Figure 5D). 3.1. Subsection
4. Discussion
Our study provides evidence that ET-1 activates microglia cells and produces inflammation. Treatment of ET-1 in HMC3 cells resulted in an overproduction of proinflammatory factors as cytokines. We found a decrease in the pro-inflammatory response when treating HMC3 cells with BQ788, an ETRB receptor blocker. By seeing activation in STAT1, we suggest that the release of these inflammatory mediators occurs due to the binding of ET-1 on ETRB, which activates the STAT1 pathway. An elevation of ET-1 and ETRB in transgenic mice brains induced with EAE demonstrates the key importance of ET-1 and ETRB in MS. Blockage of ETRB could represent an innovative approach in the development of new pharmacological therapies that can treat the inflammatory state and flares in MS patients.
Figure 5.
ET-1 is overproduced in EAE mice. Twelve female C57BL/6 mice were divided into EAE (n=6) and control (n=6). EAE was induced by injecting MOG35-55/mL and killed Mycobacterium tuberculosis H37Ra/mL. Mice were euthanized at the peak of the disease, and the brain was isolated. (A) Endothelin 1 gene (Edn) was overexpressed in the EAE brain compared to control (*p < 0.05). (B) Using ELISA, it was found that EAE brains have an elevated ET-1 concentration (*p < 0.05). (C) The ETRB gene was overexpressed in the EAE brain compared to the control (**p < 0.01). (D) The ETRA gene has no change in the EAE brain. Gene expression was quantified by qRT-PCR using Taqman Edn, Ednra, and Ednrb probes.
Figure 5.
ET-1 is overproduced in EAE mice. Twelve female C57BL/6 mice were divided into EAE (n=6) and control (n=6). EAE was induced by injecting MOG35-55/mL and killed Mycobacterium tuberculosis H37Ra/mL. Mice were euthanized at the peak of the disease, and the brain was isolated. (A) Endothelin 1 gene (Edn) was overexpressed in the EAE brain compared to control (*p < 0.05). (B) Using ELISA, it was found that EAE brains have an elevated ET-1 concentration (*p < 0.05). (C) The ETRB gene was overexpressed in the EAE brain compared to the control (**p < 0.01). (D) The ETRA gene has no change in the EAE brain. Gene expression was quantified by qRT-PCR using Taqman Edn, Ednra, and Ednrb probes.
ET-1 levels have been shown to be elevated in various cerebral diseases. [16,17] Because of its vasoconstrictive characteristic, it has been related to cerebrovascular accidents. Elevated ET-1 levels can disrupt cerebral circulation, causing hypoperfusion and leaving the brain more susceptible to ischemia. [18] Past evidence suggests ET-1 is involved in MS pathogenesis. A previous study reported a significant increase of ET-1 in the cerebrospinal fluid of patients suffering from MS.[11] Additionally, in MS patients, ET-1 levels were higher in blood drawn from the internal jugular vein and a peripheral vein, suggesting ET-1 was released from the brain into the circulation mainly by astrocytes. [12] Furthermore, our study demonstrates an overproduction of ET-1 in EAE mice, therefore confirming the role of ET-1 in MS disease pathophysiology.
ET-1 is normally produced by cells, primarily by endothelial cells, macrophages, neurons, and astrocytes in the brain. It acts through two types of receptors: ETRA and ETRB. In this study, we found an overexpression of ETRB in EAE mice. This receptor is expressed by many tissues, including endothelium, brainstem neurons, and glia. [19] Dysregulation of the endothelin system, including abnormalities in ETRB signaling, has been implicated in the pathogenesis of several diseases, such as pulmonary hypertension and neurodegenerative diseases, such as Alzheimer’s disease. [20,21] ETRB has been shown to play a role in neuroinflammation through astrocyte activation. [22] In this study, we demonstrate the role of ETRB in the activation of microglia cells by ET-1.
Microglial cell activation can damage the myelin sheath. [23] The production of ROS and NO radicals from microglia leads to the development of brain oxidative stress. Studies have suggested that oligodendrocytes, the myelin makers of the CNS, are vulnerable to oxidative burst in activated microglia, causing loss and apoptosis of oligodendrocytes and promoting demyelination in MS. [23] ROS can also induce damage to neurons and other cells in the CNS, contributing to the neurodegeneration observed in MS. [24] We show that ET-1 induced the production of ROS from microglia cells through ETRB, potentially contributing to the MS pathophysiology. In MS, NO production is primarily attributed to the activity of iNOS, which is upregulated in microglia during inflammation. ET-1 induced the gene expression of iNOS in microglia cells, increasing the NO production. Increased NO production by microglia can contribute to neuroinflammation and neurotoxicity. [25]
Microglia produce proinflammatory cytokines that contribute to the inflammatory response and tissue damage seen in MS. The release of proinflammatory cytokines enhances the inflammatory response by stimulating the chemotaxis of more inflammatory cells, including neutrophils and other phagocytes. [19] MS is associated with increased IL-6 and TNFα production in the brain. [26,27] IL-6 and TNFα may contribute to the inflammation and immune response seen in MS and are associated with disease activity, severity, and progression. [28] We found that ET-1 increases the production of IL-6 and TNFα from microglia cells, consequently contributing to neuroinflammation.
STAT-1 is a protein that plays a crucial role in cell signaling and immune responses. Over-activation of STAT-1 signaling contributes to several physiological changes leading to MS. [29] Previously, it was found that there is an increased expression of STAT-1 proteins and mRNAs in the CNS during the acute phase of disease in EAE mice. [30] Another study found a strong up-regulation of pSTAT-1 in MS patients in relapse compared with patients in remission and controls. [31] In our study, it was found that ET-1 increases pSTAT-1 through ETRB in microglia cells. The up-regulation of pSTAT-1 is associated with the production of proinflammatory cytokines.
5. Conclusions
In conclusion, we found that ET-1 activates microglial cells through ETRB and STAT-1 phosphorylation. ET-1 increased the production of ROS, NO, and proinflammatory cytokines such as TNFα and IL-6 in microglia cells. In addition, we found elevated levels of ET-1 and ETRB in EAE mice. These findings account for MS pathophysiology and disease progression. These findings demonstrate that ETRB antagonists could be used as a potential therapy to treat MS. ETRB blockage could limit the stated effects of ET-1 on microglia cells to reduce inflammation and demyelination in MS. Further studies on ETRB antagonists need to be done to develop new therapies that can improve the wellbeing and reduce the disease progression and relapses of patients with MS.
Author Contributions
Conceptualization, YIN; methodology, YIN, JA, DCR, JM, and CA; formal analysis, YIN, JA, and SB; investigation, YIN; writing—original draft preparation, YIN, SB, and JG; writing—review and editing, YIN, SB and JG; supervision, YIN; funding acquisition, YIN. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the San Juan Bautista School of Medicine Pilot Projects Program to YIN.
Data Availability Statement
The datasets generated and analyzed during the current study are available in Mendeley Data, http://dx.doi.org/10.17632/2gtcvmf4t2.1. .
Acknowledgments
We thank the San Juan Bautista School of Medicine for its support and our laboratory staff for their time, effort, and input during this project. We also want to thank Dr. Estela S. Estape for her feedback during the write-up of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest. .
References
- Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441-468. [CrossRef]
- Correale J. The role of microglial activation in disease progression. Mult Scler. 2014;20(10):1288-1295. [CrossRef]
- Merson TD, Binder MD, Kilpatrick TJ. Role of cytokines as mediators and regulators of microglial activity in inflammatory demyelination of the CNS. Neuromolecular Med. 2010;12(2):99-132. [CrossRef]
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017;16(11):877-897. [CrossRef]
- Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622-1636. [CrossRef]
- Disanto G, Morahan JM, Barnett MH, Giovannoni G, Ramagopalan SV. The evidence for a role of B cells in multiple sclerosis. Neurology. 2012;78(11):823-832. [CrossRef]
- Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10(12):1252-1259. [CrossRef]
- Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis [published correction appears in Immunology. 2008 Nov;125(3):438]. Immunology. 2008;125(2):161-169. [CrossRef]
- Kebir H, Ifergan I, Alvarez JI, et al. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol. 2009;66(3):390-402. [CrossRef]
- Almolda B, González B, Castellano B. Activated microglial cells acquire an immature dendritic cell phenotype and may terminate the immune response in an acute model of EAE. J Neuroimmunol. 2010;223(1-2):39-54. [CrossRef]
- Inostroza-Nieves Y, Rivera A, Romero JR. Blockade of endothelin-1 receptor B regulates molecules of the major histocompatibility complex in sickle cell disease. Front Immunol. 2023;14:1124269. Published 2023 Feb 28. [CrossRef]
- Monti L, Morbidelli L, Bazzani L, Rossi A. Influence of Circulating Endothelin-1 and Asymmetric Dimethylarginine on Whole Brain Circulation Time in Multiple Sclerosis. Biomark Insights. 2017;12:1177271917712514. Published 2017 Jun 6. [CrossRef]
- D’haeseleer M, Beelen R, Fierens Y, et al. Cerebral hypoperfusion in multiple sclerosis is reversible and mediated by endothelin-1. Proc Natl Acad Sci U S A. 2013;110(14):5654-5658. [CrossRef]
- Guo Y, Chung SK, Siu CW, et al. Endothelin-1 overexpression exacerbate experimental allergic encephalomyelitis. J Neuroimmunol. 2014;276(1-2):64-70. [CrossRef]
- Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. 1995;25(7):1951-1959. [CrossRef]
- Nie XJ, Olsson Y. Endothelin peptides in brain diseases. Rev Neurosci. 1996;7(3):177-186. [CrossRef]
- Schinelli S. The brain endothelin system as potential target for brain-related pathologies. Curr Drug Targets CNS Neurol Disord. 2002;1(6):543-553. [CrossRef]
- Faraco G, Moraga A, Moore J, Anrather J, Pickel VM, Iadecola C. Circulating endothelin-1 alters critical mechanisms regulating cerebral microcirculation. Hypertension. 2013;62(4):759-766. [CrossRef]
- Kowalczyk A, Kleniewska P, Kolodziejczyk M, Skibska B, Goraca A. The role of endothelin-1 and endothelin receptor antagonists in inflammatory response and sepsis. Arch Immunol Ther Exp (Warsz). 2015;63(1):41-52. [CrossRef]
- Titus A, Marappa-Ganeshan R. Physiology, Endothelin. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK551627/.
- Koyama Y. Endothelin ETB Receptor-Mediated Astrocytic Activation: Pathological Roles in Brain Disorders. Int J Mol Sci. 2021;22(9):4333. Published 2021 Apr 21. [CrossRef]
- Morga E, Faber C, Heuschling P. Stimulation of endothelin B receptor modulates the inflammatory activation of rat astrocytes. J Neurochem. 2000;74(2):603-612. [CrossRef]
- Luo C, Jian C, Liao Y, et al. The role of microglia in multiple sclerosis. Neuropsychiatr Dis Treat. 2017;13:1661-1667. Published 2017 Jun 26. [CrossRef]
- Kempuraj D, Thangavel R, Natteru PA, et al. Neuroinflammation Induces Neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1):1003.
- Brown GC, Vilalta A. How microglia kill neurons. Brain Res. 2015;1628(Pt B):288-297. [CrossRef]
- Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta. 2016;1863(6 Pt A):1218-1227. [CrossRef]
- Grzegorski T, Iwanowski P, Kozubski W, Losy J. The alterations of cerebrospinal fluid TNF-alpha and TGF-beta2 levels in early relapsing-remitting multiple sclerosis. Immunol Res. 2022;70(5):708-713. [CrossRef]
- Imitola J, Chitnis T, Khoury SJ. Cytokines in multiple sclerosis: from bench to bedside. Pharmacol Ther. 2005;106(2):163-177. [CrossRef]
- Kumar N, Sharma N, Mehan S. Connection between JAK/STAT and PPARγ Signaling During the Progression of Multiple Sclerosis: Insights into the Modulation of T-Cells and Immune Responses in the Brain. Curr Mol Pharmacol. 2021;14(5):823-837. [CrossRef]
- Maier J, Kincaid C, Pagenstecher A, Campbell IL. Regulation of signal transducer and activator of transcription and suppressor of cytokine-signaling gene expression in the brain of mice with astrocyte-targeted production of interleukin-12 or experimental autoimmune encephalomyelitis. Am J Pathol. 2002;160(1):271-288. [CrossRef]
- Frisullo G, Angelucci F, Caggiula M, et al. pSTAT1, pSTAT3, and T-bet expression in peripheral blood mononuclear cells from relapsing-remitting multiple sclerosis patients correlates with disease activity. J Neurosci Res. 2006;84(5):1027-1036. [CrossRef]
Figure 1.
ET-1 induces NO production by MHC3 cells. (A) Microglia cells were treated with ET-1 in the absence and presence of BQ788, and NO levels were measured using Griess Reagent. ET-1 treatment increases levels of NO compared to vehicle (p < 0.01, n=4). Co-treatment with ET-1 and BQ788 results in significantly lower NO production compared to ET-1 alone (p < 0.001, n=4). (B) RNA was extracted from microglia cells treated with ET-1 in the absence and presence of BQ788. Gene expression was quantified by qRT-PCR using a Taqman iNOS probe. Data represent mean ± SD (n = 4). *p < 0.05, **p < 0.01, ***p<0.001, ****p<0.0001.
Figure 1.
ET-1 induces NO production by MHC3 cells. (A) Microglia cells were treated with ET-1 in the absence and presence of BQ788, and NO levels were measured using Griess Reagent. ET-1 treatment increases levels of NO compared to vehicle (p < 0.01, n=4). Co-treatment with ET-1 and BQ788 results in significantly lower NO production compared to ET-1 alone (p < 0.001, n=4). (B) RNA was extracted from microglia cells treated with ET-1 in the absence and presence of BQ788. Gene expression was quantified by qRT-PCR using a Taqman iNOS probe. Data represent mean ± SD (n = 4). *p < 0.05, **p < 0.01, ***p<0.001, ****p<0.0001.
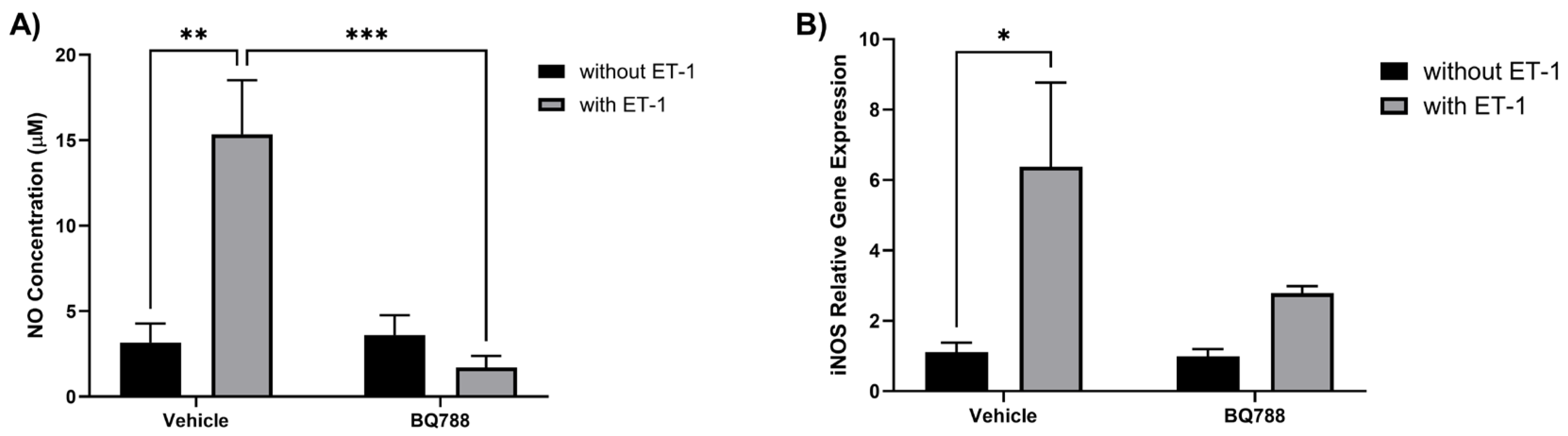
Figure 4.
ET-1 activates the STAT-1 pathway. HMC3 cells were treated with 100 nM ET-1 with or without BQ788 treatment and incubated for 24 h. Samples were analyzed using a flow cytometry assay for p-STAT1. Results represent the mean ± SD of n=4. *p < 0.05, **p < 0.01, ***p<0.001, ****p<0.0001.
Figure 4.
ET-1 activates the STAT-1 pathway. HMC3 cells were treated with 100 nM ET-1 with or without BQ788 treatment and incubated for 24 h. Samples were analyzed using a flow cytometry assay for p-STAT1. Results represent the mean ± SD of n=4. *p < 0.05, **p < 0.01, ***p<0.001, ****p<0.0001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



