
Submitted:
10 September 2024
Posted:
12 September 2024
You are already at the latest version
Abstract
The bioremediation induced by bacteria offers a promising alternative for the contamination of aromatic hydrocarbons, due to their metabolic processes suitable for the removal of these pollutants, as many of them are carcinogenic molecules and dangerous to human health. Our research focused on isolating a bacterium from the rhizosphere of the tara tree with the ability to degrade polycyclic aromatic hydrocarbons, using draft genomic sequencing and computational analysis. Enterobacter sp. strain UNJFSC 003 possesses 4,460 protein‐coding genes, 2 rRNA genes, 77 tRNA genes, and a GC content of 54.38%. A taxonomic analysis of our strain revealed that it has an average nucleotide identity of 87.8% <95%, indicating that it is a new endemic enterobacterium. Additionally, pangenomic analysis with 15 strains demonstrated that our strain has a phylogenetic relationship with strain FDAARGOS 1428 (E. cancerogenus), with a total of 381 core genes and 4,778 accessory genes. Orthologous methods predicted that strain UNJFSC 003 possesses genes with potential for use in hydrocarbon bioremediation. Genes were predicted in the subpathways for the degradation of homoprotocatechuate and phenylacetate, primarily located in the cytoplasm. Studies conducted through molecular modeling and docking revealed the affinity of the predicted proteins in the degradation of benzo[a]pyrene in the homoprotocatechuate subpathway, specifically hpcB, which has enzymatic activity as a dioxygenase, and hpcC, which functions as an aldehyde dehydrogenase. This study provides information on native strains from the Lomas de Lachay with capabilities for the bioremediation of aromatic hydrocarbons and other compounds.
Keywords:
NGS
; genomics
; microbiology
; benzo[a]pyrene
; hydrocarbon and bioremediation
; Molecular Docking
1. Introduction
Hydrocarbon pollution in the environment occurs through human activities or accidentally, resulting in environmental contamination [1]. Exposure to hydrocarbons can lead to a wide range of health conditions, increasing morbidity and affecting human health [2]. Hydrocarbons are classified into two groups: aliphatic and aromatic. The term "aliphatic hydrocarbon" refers to saturated compounds, such as alkanes or paraffins, which do not have double or triple bonds. "Unsaturated aliphatic hydrocarbons" have one or more double bonds (alkenes) or one or more triple bonds (alkynes). "Aromatic hydrocarbons" exhibit properties associated with the benzene ring and are known as polycyclic aromatic hydrocarbons (PAHs) [3]. Due to rapid industrialization and large-scale anthropogenic activities, the level of hydrocarbon pollution continues to increase, which is concerning for the environment. There are many techniques for hydrocarbon remediation, but the use of microorganisms has several advantages, such as cost-effectiveness, minimal or no by-products, and more [4].
Microbes are primarily used for their rapid growth and ease of manipulation [5]. The phyla Proteobacteria and Bacteroidetes, which are generally gram-negative bacteria, have potential for hydrocarbon degradation [6]. Proteobacteria constitute one of the largest and most versatile phyla in the bacterial domain. Single bacteria or consortia of Proteobacteria have been utilized for hydrocarbon degradation, demonstrating their effectiveness in remediating these compounds [7,8]. In the last decade, several microorganisms capable of degrading PAHs have been identified and characterized. Additionally, metabolic enzymes have been isolated from microorganisms for degrading various PAH compounds, identifying diverse novel metabolic pathways, including studies on PAH catabolic operons [9].
In this study, de novo genome sequencing was performed on an enterobacterium isolated from soil samples of the rhizosphere of tara plant (Caesalpinia espinosa) from the Lomas de Lachay National Reserve. Genes involved in the degradation of aromatic hydrocarbons were identified. In addition, a proteome prediction was conducted followed by the construction of three-dimensional molecular structures of putative proteins involved in aromatic hydrocarbon bioremediation. This involved deep learning modeling for molecular docking with the benzopyrene molecule to assess their potential interaction against this compound. To the best of our knowledge, this work provides the first report on bacteria isolated from the Lomas de Lachay reserve and on the genomics and molecular biology of Enterobacter sp. UNJFSC003 in response to polycyclic hydrocarbons.
2. Materials and Methods
2.1. Isolation of Strain, Extraction of DNAg and Genome Sequencing
The sample was collected in Lomas de Lachay (11°22'39.2"S, 77°21'34.6"W), Province of Huaura, Lima, Peru. One kilogram of soil was collected in sterile bags at a depth of 30 cm and 4 cm from the root of the tara plant (Caesalpinia espinosa). To isolate the bacteria, 1 g of soil was mixed with 9 mL of sterile 0.5% sodium chloride (NaCl), serial dilutions were prepared, and these dilutions were plated on Tryptic Soy Agar (TSA) medium. The plates were incubated at 30°C for 48 hours. To purify the bacteria, subculturing was performed on TSA medium. Colony morphology on the plate and Gram staining were identified. Additionally, biochemical tests were conducted to establish its identity.
The pure sample was selected for DNA extraction using the commercial EZ-10 Spin Column Bacterial Genomic DNA Miniprep Kit (Bio Basic, Canada). Genome sequencing was performed using the Illumina NovaSeq system at a foreign company (Macrogen, Republic of Korea). The read quality was assessed using FastQC v0.12.1 [10]. Additionally, the reads were trimmed and filtered (Q-25) using trimmomatic v0.36 [11] and fastp v0.20.1 [12] with default parameters.
2.2. Genome Assembly, Annotation and Analysis Functional
To obtain the genome of Enterobacter sp. strain UNJFSC 003, a de novo assembly was performed using the Unicycler v0.5.0 assembly algorithm [13]. This assembly algorithm was selected due to its distinct methodological approaches, which allow for the optimization of the SPAdes assembly and thus obtain longer contigs used for this genome. After this, we used QUAST v5.2.0 [14] to evaluate the assembly statistics. Subsequently, the assembly was subjected to the BUSCO v5.2.2 strategy [15] to assess the completeness of our genome assembly compared to three other strains deposited in the NCBI Datasets Genome (Figure 1a), using previous scripts (https://github.com/GianmarcoCastillo/Genome-UNJFSC003/tree/main). Additionally, MiGA v1.3.9.0 was used to evaluate the quality, contamination, and identification of the genome [16].
Genome annotation was performed using Prokka v1.14.5 [17] and RAST [18], and a genomic map was created using the Proksee tool [19]. Moreover, a pie chart of the genome subsystem was created from the annotation results using the matplotlib v3.8.4 [20] and pandas v1.5.3 packages in Python v3.10.12. The predicted genes were functionally characterized using the COG (Cluster of Orthologous Genes) tool and orthology assignment through the eggNOG-Mapper tool [21], and pathway reconstruction was performed using the KEGG (Kyoto Encyclopedia of Genes and Genomes) [22] and BlastKOALA [23].
2.3. Comparative Genome Analysis, Confirmation Molecular and Pan Genome
In the genome comparison and molecular confirmation of Enterobacter sp. UNJFSC003, the whole-genome sequence (WGS) of the isolate was used. An average nucleotide identity (ANI) analysis was performed between Enterobacter sp. UNJFSC003 and 15 other Enterobacter strains obtained from the NCBI genome database. This analysis was calculated using the fastANI v1.34 tool [24]. Subsequently, a heatmap of clusters of the 16 genomes was generated using the ANIclustermap v1.3.0 tool [25] to assess the diversity of taxonomic and phylogenetic lineages among the genomes. A pangenome analysis of 16 different Enterobacter strains was conducted using Roary v3.13.0 [26], utilizing GFF3 files obtained from Prokka. For this analysis, the following parameters were used: a BLASTP identity threshold of 90%, clustering in groups of 100,000 sequences, and separation of paralogs.
2.4. Prediction of Hydrocarbon-Degrading Genes and Enzymes
In the prediction of genes and enzymes involved in hydrocarbon degradation, the A Curate Hydrocarbon Aerobic Degradation Enzymes and Genes Database (HADEG) [27] was used. This database contains experimentally validated protein and gene sequences involved in the aerobic degradation of hydrocarbons. The analysis used the .faa files obtained from Prokka and the tool proteinortho v6.0.33 [28] to detect orthologous proteins capable of degrading hydrocarbons within the genome of Enterobacter sp. UNJFSC003.
2.5. In Silico Protein-Protein Interaction and Subcellular Localization of Hydrocarbon-Degrading Proteins
The protein interaction network was evaluated by subjecting the proteins involved in aromatic hydrocarbon degradation to the STRING database with a high confidence score threshold (> 0.7) [29]. The database includes both direct (physical) and indirect (functional) associations through computational predictions. The interaction results of all proteins were visualized using Cytoscape v3.10.2 [30]. For the protein localization within the cell of the Enterobacter sp. UNJFSC003 genome, the bioinformatics tool PSORT v3.0.3 [31] was employed. The results were visualized in a heatmap using the Pheatmap package [32] in R v4.4.0 [33].
2.6. Molecular Modeling, Model Validation, and Molecular Docking
The proteins presumed to be involved in aromatic hydrocarbon degradation, specifically the hpc gene cluster predicted by HADEG, were selected. Functional analysis of these hpc proteins was conducted by classifying them into families and predicting domains and active sites in each protein using the bioinformatics tool InterProScan [34]. For the 3D molecular structure modeling, proteins were modeled using the trRosetta server [35]. The models were validated using PDBsum [36], ERRAT [37], and ProSA Web [38]. Additionally, signal peptide analysis was performed using SignalP v.6 [39] for each predicted protein, and physicochemical analyses were conducted using the Expasy ProtParam tool [40]. These analyses are described in Table 4. The molecular docking analysis involved blind docking using the AutoDock Vina v1.1.2 software [41], facilitated by the graphical interface AutodockTools v1.5.6 [42]. This approach was used to study protein interactions and conduct preliminary analysis with the ligand benzopyrene (https://pubchem.ncbi.nlm.nih.gov/compound/2336) sourced from PubChem. Beforehand, hydrogens were added to the protein, charges were assigned using the Kollman method, and non-polar hydrogens were subsequently removed. The grid coordinates used for all proteins were as follows: x=16.305, y=20.737, z=1.165, with dimensions of 126 in each direction (x, y, z) and a spacing of 0.631 Å. Additionally, for the ligand molecules, energy minimization was performed beforehand using Avogadro v1.2.0 [43]. Hydrogens were added, Gasteiger charges were assigned, and non-polar hydrogens were subsequently removed. The results of the molecular docking were visualized using PyMOL v3.0.2 [44].
3. Results
3.1. Isolation of the Strain and Genome Characteristics of Enterobacter sp. UNJFSC 003
The pure strain was successfully isolated on TSA culture medium. Additionally, the colony exhibited a transparent color and a star-shaped form. The Gram stain revealed that it was a Gram-negative bacterium. The biochemical profile included a growth assay [45] on MacConkey and eosin methylene blue (EMB) media, fermentation of glucose (+), gas production (+), oxidase negative (-), catalase positive (+), and motility test (+).
The total number of raw reads was 10,914,547 sequences, with an average length of 151 base pairs (bp), a GC content of 54%, and a total sequencing output of 1.6 gigabytes (GB). The results of the cut made can be seen in Table 1.
The de novo assembly was conducted with the unicycler program because it can assemble Illumina read assemblies by working as a SPAdes optimizer, obtaining better results from N50 and longer contings. We tested different k-mers (from 27 to 127). In the assembly quality assessment (Quast), we obtained statistical results of 51 contigs (>= 0 bp) with a GC content of 54.38%. The longest contig obtained was 4,663,955 base pairs (>= 50,000 bp). Additionally, the N50 result was 415,254 and the L50 was 3. In the taxonomic classification analysis of the Enterobacter sp. UNJFSC003 genome assembly conducted by MiGA, the results indicate that our strain belongs to the family Enterobacteriaceae with a p-value of 0.0025. It is likely to belong to the genus Enterobacter (p-value: 0.02) and possibly to the species E. asburiae NZ CP065693 with an average nucleotide identity (ANI) of 87.85%. Additionally, MiGA provided other results such as the consistency and quality of essential genes, with a genome quality value of 93.6%, completeness of 98.1%, and contamination of 0.9% (Table 2). Finally, in the genome annotation, the following results were obtained: a total genome size of 4,798,267 base pairs (bp), 4,460 protein-coding sequences, 2 rRNAs, 77 tRNAs, and 1 tmRNA. Additionally, functional analysis of genes and orthologous proteins yielded a total of 2,671 orthologous proteins and 1,487 KEGG-annotated metabolic proteins obtained through prior analysis using KOALA-KEGG. Other results include 4,349 orthologous gene clusters, 4,186 Pfam annotations, 2,540 gene ontology assignments, 77 carbohydrate-active enzymes, and reconstruction of metabolic networks (BiGG) with a total of 1,064 nodes. These results were summarized using the EggNOG-Mapper online tool and are presented in Table 2.
The genome sequencing data is available in GenBank at NCBI under the accession number JBAKHK000000000.
3.2. Comparative Analysis of the Complete Genome of Enterobacter sp. UNJFSC 003
The comparative analysis of the complete genome based on Average Nucleotide Identity (ANI) of 16 different Enterobacter strains showed that Enterobacter sp. UNJFSC003 was closely related to nine Enterobacter strains with ANI values ranging from 87.8 to 87.3 as the upper limit, and the other 5 strains had ANI values ranging from below 86.5 to 80.9 (Figure 2 and Table 3). Our strain UNJFSC003 and the Crenshaw strain of E. asburiae with GenBank accession GCA_016027695 had a maximum ANI value of 87.8%. This ANI value indicates a relationship between our strain and this bacterium with an ANI value below 95%, considering that the threshold for classifying strains is (≥) 95% ANI [46]. Based on this, we can suggest that our strain is a new native Enterobacter isolated from soil samples from Lomas de Lachay [47,48].
3.3. Analysis of the Pangenome of Enterobacter sp. UNJFSC 003
The pangenome model developed with the participation of 16 Enterobacter strains, including 12 Enterobacter strains, 3 strains of other Enterobacteriaceae species, and our strain, indicated a close genetic relationship between UNJFSC003 and FDAARGOS-1 (E. cancerogenus). Both strains grouped into a single node, indicating their similarity in identical gene clusters. Ten other Enterobacter strains with less variation in genes were observed in the matrix and separated into another node. The three Enterobacteriaceae strains, including Leclercia adecarboxylata strain ATCC2316, Klebsiella spallanzanii strain SB6411, and Citrobacter freundii ATCC8090, exhibited greater variation, along with Enterobacter soli strain ATCC-BAA-2. The pangenome of the 16 Enterobacter strains consists of 381 core genes, 75 soft core genes, 4778 shell genes, and 38044 cloud genes, totaling 43278 genes. Interestingly, Enterobacter sp. UNJFSC003 harbors unique gene clusters presumably present with E. cancerogenus strain code FDAARGOS-1 (Figure 3).
3.4. The Strain UNJFSC 003 Harbors Genes Encoding Enzymes for the Bioremediation of Hydrocarbons
The HADEG database identified our strain as a potential organism for developing hydrocarbon bioremediation methods, finding two types of hydrocarbons: alkanes (a total of 5) and aromatics (a total of 16). Additionally, two 2 enzymes for biosurfactant production, all determined through orthology analysis, were identified (Figure 4). The orthology analysis performed by proteinortho with the HADEG database identified proteins involved in the metabolic sub-pathways of the genome. This detailed and specific analysis identified two different types of hydrocarbons (alkanes and aromatics) and biosurfactants, as well as the specific number of enzymes for the bioremediation of aromatic hydrocarbons and alkanes (Figure 4).
3.5. In Silico Interaction Protein-Protein and Heat-Map of Proteins Involved in Bioremediation
The analysis of protein-protein interactions indicated possible interactions among the various proteins involved in hydrocarbon bioremediation. Indeed, the interaction among enzymes involved in the bioremediation of aromatic hydrocarbons ensures their mutual connection for each different HC, such as hpc genes totaling 6 in the homoprotocatechuate pathways, paa genes totaling 6 in the phenylacetate pathway, and hpa genes in aerobic alkane degradation (Figure 5a). In the localization of the proteins involved in hydrocarbon bioremediation, it can be observed that 17 proteins predicted by PSORT are located in the cytoplasm and 6 are located in different sites of the bacterial cell (Figure 5b).
3.6. Molecular Modelling and Docking Analysis
The three-dimensional molecular structures of 6 proteins were constructed using deep modeling and Rosetta. These proteins are related to the bioremediation of aromatic hydrocarbons, specifically the proteins predicted for the bioremediation of aromatic hydrocarbons involved in the subpathways of homoprotocatechuate. According to the validation, all the models can be considered reliable, as the Z-score, Ramachandran analysis, and ERRAT provided satisfactory or good scores.In this context, the best model corresponds to hpcB, based on the results obtained: 95.4% in the Ramachandran plot, a Z-score of -11.36, and an ERRAT score of 97.87. On the other hand, hpcD and hpcE correspond to the worst models, with Ramachandran plot results of 94.6%, a Z-score of 4.79, and an ERRAT score of 83.46.
On the other hand, none of the proteins have shown a signal peptide, indicating that they are all intracellular, with each protein containing between 126 and 488 amino acids. Additionally, all the obtained models displayed a monomeric folded structure. In the six predicted proteins, all possess active sites for the degradation of polycyclic aromatic hydrocarbons (Table 4).

Table 4.
Evaluation of hpc Protein Models from Enterobacter sp. UNJFSC003: Physicochemical Analysis and Signal Peptide.
Table 4.
Evaluation of hpc Protein Models from Enterobacter sp. UNJFSC003: Physicochemical Analysis and Signal Peptide.
| Modelling Server | Protein Modelling | Errat | Error/Warning/Plass | R. Plot% | Z-score | SignalP | Num. aa | pI | Mol Weight | GRAVY |
|---|---|---|---|---|---|---|---|---|---|---|
| trRosseta | hpcB | 97.87 | 2/4/3 | 95.4% | -11.36 | No | 283 | 5.72 | 31721.04 | -0.228 |
| trRosseta | hpcC | 93.38 | 2/4/3 | 94.0% | -11.41 | No | 488 | 6.25 | 53130.86 | -0.112 |
| trRosseta | hpcD | 83.49 | 1/3/4 | 94.6% | -4.79 | No | 126 | 5.82 | 14336.35 | -0.202 |
| trRosseta | hpcE | 92.44 | 3/2/4 | 90.6% | -9.65 | No | 425 | 5.03 | 46227.37 | -0.146 |
| trRosseta | hpcG | 93.44 | 0/4/4 | 91.8% | -6.87 | No | 267 | 5.69 | 29481.68 | -0.081 |
| trRosseta | hpcH | 96.85 | 0/4/5 | 94.7% | -8.81 | No | 265 | 5.73 | 28175.33 | 0.107 |
The hpcB model has a topology of 16 alpha helices, 23 beta sheets, and 33 coils. In the respective molecular docking analysis, this predicted protein obtained an energy value of -10.1 kcal/mol, interacting with the amino acids TRP58, ASN67, LEU121, LEU123, and GLU124; On the other hand, the hpcC model has a topology of 14 alpha helices, 19 beta sheets, and 34 coils. In the respective molecular docking analysis, this predicted protein obtained a value of -10.7 kcal/mol, interacting with the amino acids GLN95, LEU267, PHE268, SER272, and GLN316 And does not form any hydrogen bonds; on the other hand, hpcD has a topology of 3 alpha helices, 6 beta sheets, and 10 coils. In the respective molecular docking analysis, this predicted protein obtained an energy value of -9.7 kcal/mol, interacting with the amino acids GLY81, GLU82, PHE85, and PHE105; in the other analyses, hpcE has a topology of 18 alpha helices, 12 beta sheets, and 32 coils. In its molecular docking analysis, this protein obtained a value of -12.0 kcal/mol, interacting with the amino acids TYR28, ASN29, THR30, TYR103, ARG315, and TYR314; additionally, hpcG has a topology of 11 alpha helices, 10 beta sheets, and 19 coils. In the respective molecular docking analysis, this predicted protein obtained an energy value of -9.1 kcal/mol, interacting with the amino acids TYR128, ASP181, LEU185, HIS210, THR161, and despite having a histidine amino acid, it does not form any hydrogen bonds; Finally, in the group of proteins that degrade homoprotocatechuate, hpcH has a topology of 12 alpha helices, 8 beta sheets, and 21 coils. In the respective molecular docking analysis, this predicted protein obtained an energy value of -9.0 kcal/mol, interacting with the amino acids TRP19, GLY21, LEU212, and VAL234. Similar to hpcG, it also does not form any hydrogen bonds despite the presence of histidine.
4. Discussion
The microorganisms with efficient biodegradation of aromatic hydrocarbons, belonging to Enterobacter sp. and E. xianfagensis [49,50], have been reported for their ability to degrade both aromatic hydrocarbons and petroleum. This includes real-time experimental methods and computational molecular docking. Additionally, studies have explored their production of biosurfactants like rhamnolipids, as seen in E. asburiae [51]. In another study, E. ludwigii strains found as endophytes in plants were discovered capable of degrading hydrocarbons from the rhizosphere [52]. In this study, the strain of Enterobacter belonging to the species E. ludwigii possesses specific genes for the degradation of both alkanes and aromatic hydrocarbons, as well as genes involved in biosurfactant production. Thus, Enterobacter sp. UNJFSC003 exhibited a variety of enzymatic activities, including dioxygenase activity attributed to the LigB domain in hpcB. This family of enzymes performs the cleavage of aromatic rings as a key function in the degradation of these compounds. They also participate in the metabolism of halogenated aromatic compounds [53], in addition, aldehyde dehydrogenase in hpcC functions enzymatically by oxidizing a broad spectrum of aliphatic and aromatic aldehydes, utilizing NAD+ and NADP+ as cofactors. These enzymes play pivotal roles in different stages of hydrocarbon degradation [54], These enzymes also play roles in the degradation of compounds such as pesticides and other toxic substances [55]. The enzymatic function of CHMI (5-carboxymethyl-2-hydroxymuconate isomerase) in the decomposition of aromatic compounds is studied within the context of hpcD. The structure of this protein was predicted using artificial intelligence through AlphaFold 2, developed by DeepMind [56], user is studying bioremediation of aromatic compounds in Pseudomonas aeruginosa PAO1. Additionally, they conducted an experimental study cloning the gene in Escherichia coli to produce large quantities of this protein and determine its properties [57]. Finally, the hydrolase activity in hpcE involves two present domains of hydrolase, whereas hpcG has a single present domain of hydrolase. The latter catalyzes the hydration of a carbon-carbon double bond without the assistance of any cofactor. The crystal structure of hpcG features the FAH fold, which is quite common among enzymes involved in the degradation of aromatic compounds [58], Lastly, hpcH also obtained a functional domain in the degradation of aromatic compounds according to the analyses conducted by InterProScan [59]. These studies were conducted using the proteome obtained from annotation developed by Prokka, orthology methods employing the Proteinortho tool, the HADEG database, and active sites identified by InterProScan.
In the present study, our bacteria isolated from soil samples from Lomas de Lachay was observed to form star-shaped colonies and was classified as Gram-negative by Gram staining. Its genome was sequenced using the Illumina NovaSeq system (Macrogen, Rep. of Korea). Subsequent taxonomic identity analysis characterized this bacterium as belonging to the genus E. asburiae with an ANI value of 87.85%. However, taxonomic classification of a microbial strain in the genomic era generally requires sharing an average nucleotide identity (ANI) value of >95% across multiple aligned genes [60]. This suggests that our isolated strain does not belong to the genus E. asburiae but may instead represent a new Enterobacter species. It maintains a close nucleotide content relationship with E. asburiae and possesses unique gene clusters similar to those found in E. cancerogenus, as observed in the pangenome analysis results with 16 Enterobacter strains. In another study, the genome of strain WCHECI-C4 has an ANI of only 93.34%, leading to its classification as a new species within the genus Enterobacter, named E. chengduensis sp. nov. [61]. The draft genome of strain UNJFSC003 has a size of 4,798,267 base pairs, 4,460 protein-coding genes, and a GC content of 54.38%. This GC content aligns with findings from the work of [62]. Enterobacter sp. UNJFSC003 showed a genome completeness of 98.1%, contamination of 0.9%, and quality of 93.6%. In another study, Enterobacter sp. strain RIT-637 with antibacterial activities showed a genome completeness of 99.96% and contamination of 2.08% [63]. A total of 2671 genes associated with KEGG pathways were obtained. This represents a significantly larger number of genes compared to the study by Indugo [64], who found a total of 1567 genes in KEGG pathways. The classification of 2540 orthologous genes, 77 active enzymes in carbohydrates, 1064 metabolic networks in total, and 4329 clusters of orthologous genes. Interestingly, studies by Szczerba [65] found a new strain of the genus Enterobacter isolated from the cow rumen, E. aerogenes LU2, with protein-coding genes involved in carbohydrate metabolism pathways, amino acid transport, carbon source transport, and nitrogen source transport. Furthermore, the genome of Enterobacter sp. UNJFSC003 showed capabilities for hydrocarbon bioremediation and production of low molecular weight (LMW) biosurfactants similar to emulsan. Microbial biosurfactants are produced as secondary metabolites or through enzymatic processes, well-known for their ability to reduce surface tension [66]. Due to the analyses conducted, we found biotechnological potential in this genus of bacteria. In the study by Peng [67], they isolated Enterobacter sp. T2 from contaminated sludge capable of rapidly degrading tetrabromobisphenol (TBBPA) via a key protein, haloacid dehalogenase, responsible for TBBPA degradation. In another study by El-belgati [68], they observed highly significant positive correlations between genetic expressions and increased concentrations of heavy metals, indicating that gene expression was induced by higher concentrations of heavy metals. These facts indicate the great potential that Enterobacter strains have for degrading hydrocarbons, heavy metals, among other compounds, thanks to studies using next-generation sequencing technologies.
Through the analysis of hydrocarbon biodegradation, the results suggested the efficiency of our strain UNJFSC003, identifying enzymes related to hydrocarbon bioremediation, including alkanes and aromatics. Such result is associated with the hydrocarbon degradation effect employed by 3 strains of Pseudomonadota bacteria isolated from a century-old abandoned oil exploration well, analyzing their proteome and using the same gene and enzyme database for aerobic hydrocarbon degradation, finding similar types of hydrocarbons present in our study, among other types of hydrocarbons [69]. Here, the in silico analysis of the putative protein interaction networks involved in hydrocarbon bioremediation, using the STRING database, revealed potential links between the homoprotocatechuate degradation pathway, specifically hpcB, and the hydroxyphenylacetate degradation system, hpaB and hpaC. An additional connection was found between the catechol degradation pathway, specifically pcaF, and all predicted proteins in the phenylacetate degradation pathway. Additionally, the two biosurfactant-producing proteins interacted with each other without revealing links to other proteins, maintaining their phylogenetic coexistence. Interactions of proteins involved in bioremediation have already been reported in other bacteria isolated from wastewater, such as Bacillus cereus AA-18 [70]. Additionally, another study on cadmium bioremediation by Khan [71] shows a similar interaction network to the hpc cluster in our study, involving binding proteins such as YfiY, yfmF, and yfmE. In the subcellular prediction made by the online tool PSORTb v3.0, 17 proteins were located in the cytoplasm, while the rest were found in various cellular regions. There are many methods for predicting the subcellular localization of microbial proteins, ranging from genome-based predictions using PSORTB to metagenome-based predictions with PSORTm. These methods also include the identification of new markers in aquatic and terrestrial microorganisms [72,73].
The molecular models of key enzymes (Table 4 and Figure 6) involved in the bioremediation of aromatic hydrocarbons showed highly conserved amino acids in the active sites. These results, along with the Ramachandran plot percentage, support the reliability of the modeled structures. Furthermore, all models did not form hydrogen bonds but showed high affinity energy with the benzopyrene molecule at the active sites of all modeled proteins through deep modeling, with affinity energy values ranging from -9.0 kcal/mol to -12.0 kcal/mol. This provides information about the structure and function of the hpc enzyme cluster. Therefore, the 3D models of hpc cluster enzymes strongly support their function in the bioremediation of aromatic hydrocarbons, as annotated from the genes of Enterobacter sp. UNJFSC003.
Combining genomic analysis and molecular modeling, this research has contributed to a better understanding of the great potential of strain UNJFSC 003 isolated from soil samples from Lomas de Lachay. Genomic analysis provides information about the genes involved in the bioremediation of aromatic hydrocarbons, alkanes, and biosurfactant production. Lou [74] revealed information about the genome of various genetic functionalities, specifically the metabolism of phenanthrene and pyrene degradation in 2 new bacterial strains, Klebsiella michiganensis EF4 and K. oxytoca ETN19, isolated from a soil sample contaminated with PAHs. In the molecular docking analysis, benzopyrene was used as the ligand. Benzopyrene is listed among the priority pollutant polycyclic aromatic hydrocarbons by the United States Environmental Protection Agency [75]. Benzopyrene has been detected in industrial waste, diesel exhaust gases, charcoal-based foods, and elevated levels of cigarette smoke, and it is also known as a recalcitrant molecule [76]. Gram-negative bacteria with the ability to degrade polycyclic aromatic compounds have already been reported, such as in the study by Wang & Tam [77], where the bacterium Cycloclasticus was confirmed to be a ubiquitous degrader of marine PAHs, even in subterranean marine environments. In fact, a limited number of previous studies have reported PAH degradation capabilities in the phylum Proteobacteria, including pathogenic bacteria.
5. Conclusions
We evaluated the bioremediation capability of aromatic hydrocarbons by Enterobacter sp. UNJFSC003 through computational methods. This strain was isolated from a soil sample from the rhizosphere of the tara plant (Caesalpinia espinosa) in the Lomas de Lachay. The findings revealed genes with the ability to degrade aromatic hydrocarbons, alkanes, and produce biosurfactants. The analysis of the draft genome revealed these genes, as well as a new species of Enterobacter due to the results showing an average nucleotide identity (ANI) of less than 95%. In the pangenome analysis, it was revealed that our strain UNJFSC003 harbors unique gene clusters similar to those found in strain FDAARGOS-1, with a total of 381 core genes and 4778 shell genes. The 3D structural analysis of modeled proteins from the hpc gene cluster related to the bioremediation of aromatic hydrocarbons has shown highly conserved regions, particularly in their active sites, strongly supporting their predicted functions in the degradation of polycyclic aromatic compounds. The computational prediction of all modeled monomers by deep learning using trRosetta is also noteworthy, specifically hpcB and hpcC. This study provides information to obtain, in the future, a better experimental understanding of native strains from the Lomas de Lachay and their capability in the bioremediation of aromatic hydrocarbons and other compounds.
Author Contributions
Conceptualization, G.C., S.C-L., C.A. and P.R-G.; Data curation, G.C., S.C-L., C.A. and P.R-G; Formal analysis, G.C., C.A. and P.R-G.; Funding acquisition, S.C-L. and C.A.; Investigation, G.C., S.C-L., C.A. and P.R-G.; Methodology, G.C., S.C-L., C.A. and P.R-G.; Project administration, S.C-L., C.A. and P.R-G.; Resources, S.C-L., C.A. and P.R-G.; Software, G.C. and P.R-G.; Supervision, S.C-L, C.A. and P.R-G; Validation, G.C. and P.R-G.; Visualization, G.C., S.C-L., C.A. and P.R-G.; Writing – original draft, G.C. and P.R-G.; Writing – review & editing, S.C-L., C.A. and P.R-G. All authors have read and agreed to the published version of the manuscript.
Funding
This research work has been financed by the Universidad Nacional Toribio Rodriguez de Mendoza de Amazonas.
Data Availability Statement
The sequence reads generated in this study have been submitted to the Sequence Read Archive (SRA) under the accession number SRR28705977. The assembled genome has been deposited to NCBI GenBank under the accession number JBAKHK000000000, the BioProject accession number PRJNA1076189 and the Biosample accession number SAMN39934077. The PROKKA annotation files have been published on Zenodo (DOI: https://doi.org/10.5281/zenodo.11069170).
Acknowledgments
We thank the Universidad Nacional Toribio Rodriguez de Mendoza de Amazonas for funding all the research.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- Singh, K. & Chandra, S. (2014). Treatment of petroleum hydrocarbon polluted environment through bioremediation: a review. Pak J Biol Sci. 1:17. 1-8. [CrossRef]
- Tormoehlen, L.M., Tekulve, K.J. & Ñañagas, K.A. (2014). Hydrocarbon toxicityÑ A review. Clinical Toxiclogy. 52, 479-489.
- Volkman, J.K. (1998). Hydrocarbons . In: Geochemistry. Encyclopedia of Earth Science. Springer, Dordrecht. [CrossRef]
- Das, T. & Dash. H.R. (2014). Microbial Bioremediation: A Potential Tool for Restoraton of Contaminated Areas. Microbial Biodegradation and Biorremediation. Pages 1-21. [CrossRef]
- Ayilara, M.S. & Babalola, O.O. (2023). Bioremediation of environmental wastes: the role of microorganisms. Front. Agron., Plant-Soil Interactions. Vol5. [CrossRef]
- Castillo-Rogel, R.T., More-Calero, F.J., La-Torre, M.C., Fernandez-Ponce, J.N. & Mialhe-Matonnier, E.L. (2020). Aislamiento de bacterias con potencial biorremediador y analisis de comunidades bacterianas de zona impactada por derrame de petroleo encondorcanqui- Amazonas - Peru. Rev. invest. Altoand. Vol.22 no.3. [CrossRef]
- Emenike, C.U., Agamuthu, P., Fauziah, S.H. et al. Enhanced Bioremediation of Metal-Contaminated Soil by Consortia of Proteobacteria. Water Air Soil Pollut 234, 731 (2023). [CrossRef]
- Nyoyoko, V.F. (2022). Chapter 13 - Proteobacteria response to heavy metal pollution stress and their bioremediation potential. Cost Effective Technologies for Solid Waste and Wastewater Treatment. Pages 147-159. [CrossRef]
- Thacharodi, A., Hassan, S., Singh, T., Mandal, R., Chinnadurai, J., Khan, H.A., Hussain, M.A., Brindhadevi, K. & Pugazhendhi, A. (2023). Biormediation of polycyclic aromatic hydrocarbons: Anupdated microbiological review. Chemosphere. Vol328. 138498. [CrossRef]
- Andrews S. 2010. FastQC: una herramienta de control de calidad para datos de secuencias de alto rendimiento . Disponible en: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: un recortador flexible para datos de secuencias de Illumina. Bioinformática 30:2114–2120.
- Chen. 2023. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2: e107. [CrossRef]
- Wick, R.R., Judd, L.M., Gorrie, C.L., & Holt, K.E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Computational Biology, 13(6), e1005595. [CrossRef]
- Gurevich, A., Saveliev, V., Vyahhi, N., & Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29(8), 1072–1075. [CrossRef]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO online supplementary information: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212.
- Rodriguez-R, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., & Konstantinidis, K. T. (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Research, 46(W1), W282–W288. [CrossRef]
- Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics, 30(14), 2068–2069. [CrossRef]
- Aziz., R.K., Bartels, D., Best, A.A., DeJongh, M., Disz, T., Edwards, R.A., Formsma, K., Gerdes, S., Glass, E.M., Kubal, M., Meyer, F., Olsen, G.J., Olson, R., Osterman, A.L., Overbeek, R.A., McNeil, L.K., Paarmann, D., Paczian, T., Parrello, B., Pusch, G.D., Reich, C., Stevens, R., Vassieva, O., Vonstein, V., Wilke, A. & Zagnitko, O. (2008). The RAST Server: Rapid Annotations using Subsystems Technology. , 9(1), 75–0. [CrossRef]
- Grant, J.R., Enns, E., Marinier, E., Manda, A., Herman, E.K., Chen, C. Graham, M.,Domselaar, G.V., Stothard, P. & Notes, A. (2023). Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Research. Vol 51. W484-W492. [CrossRef]
- Hunter, J.D. "Matplotlib: A 2D Graphics Environment", Computing in Science & Engineering, vol. 9, no. 3, pp. 90-95, 2007.
- Cantalapiedra, C.P., Hernandez-Plaza, A., Letunic, I., Bork, P., Jaime Huerta-Cepas. 2021. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular Biology and Evolution, msab293. [CrossRef]
- Kanehisa, M. & Goto, S. (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes, Nucleic Acids Research, Volume 28, Issue 1, 1 January 2000, Pages 27–30. [CrossRef]
- Kanehisa, M., Sato, Y. & Morishima, K. (2016) .BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. Journal of Molecular Biology, Vol-428, 726-731. [CrossRef]
- Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114. [CrossRef]
- Shimoyama Y. ANIclustermap: a tool for drawing ANI clustermap between all-vs-all microbial genomes. 2022. https://github.com/moshi4/ANIclustermap.
- Page, A.J., Cummins, C.A., Hunt, M., Wong, V.K., Reuter, S., Holden-Matth T.G., Fookes, M., Falush, D., Keane, J.A. & Parkhill, J. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics, 31(22), 3691–3693. [CrossRef]
- Rojas-Vargas, J., Castelán-Sánchez, H.G., Pardo-López, L. (2023) HADEG: A curated hydrocarbon aerobic degradation enzymes and genes database. Computational Biology and Chemistry. [CrossRef]
- Lechner, M., Findeisz, S., Steiner, L., Marz, M., Stadler, P. F., & Prohaska, S. J. (2011). Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC bioinformatics, 12(1), 124.
- Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, Hachilif, R., Gable, A.L., Fang, T., Doncheva, N.T., Pyysalo, S., Bork, P., Jensen, L.J. & Mering, C.v. (2023). The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Research, 51(W), W638-W646. [CrossRef]
- Otasek D, Morris JH, Bouças J, Pico AR, Demchak B. Cytoscape Automation: empowering workflow-based network analysis. Genome Biol. 2019 Sep 2;20(1):185. PMID: 31477170; PMCID: PMC6717989. [CrossRef]
- Yu, N.Y., Wagner, J.R., Laird, M.R., Melli, G., Rey, S., Lo, R. & Brinkman, F.S.L. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics, 26(13), 1608–1615. [CrossRef]
- Kolder R. (2018). pheatmap: Pretty Heatmaps. R package version 4.4.0.
- R Core Team (2024). _R: A Language and Environment for Statistical Computing_. R Foundation for Statistical Computing, Vienna, Austria. <https://www.R-project.org/>.
- Blum, M., Chang, H.Y., Chuguransky, S., Grego, T., Kandasaamy, S., Mitchell, A. Nuka, G., Paysan-Lafosse, T., Qureshi, M., Raj, S., Richardson, L., Salazar, G.A., Williams, L., Bork, P., Bridge, A., Gough, J., Haft, D.H., Letunic, I., Marchler-Bauer, A., Mi, H., Natale, D.A., Necci, M., Orengo, C.A., Pandurangan, A.P., Rivoire, C., Sigrist, C.J.A., Sillitoe, I., Thanki, N., Thomas, P.D., Tosatto, S.C.E., Wu, C.H., Bateman, A. & Finn, R.D. (2020). The InterPro protein families and domains database: 20 years on. Nucleic Acids Research, (), gkaa977–. [CrossRef]
- Du Z, Su H, Wang W, Ye L, Wei H, Peng Z, Anishchenko I, Baker D, Yang J. The trRosetta server for fast and accurate protein structure prediction. Nat Protoc. 2021 Dec;16(12):5634-5651. [CrossRef]
- de Beer TA, Berka K, Thornton JM, Laskowski RA. PDBsum additions. Nucleic Acids Res. 2014 Jan;42(Database issue):D292-6. [CrossRef]
- Colovos, C. & Todd O.Y. (1993). Verification of protein structures: Patterns of nonbonded atomic interactions. , 2(9), 1511–1519. [CrossRef]
- Wiederstein, M., Sippl, M. J. (2007). ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research, 35(Web Server), W407–W410. [CrossRef]
- Teufel, F., Armenteros, A.J.J., Johansen, A.R. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol 40, 1023–1025 (2022). [CrossRef]
- Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A.; Protein Identification and Analysis Tools on the Expasy Server; (In) John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press (2005). pp. 571-607.
- Trott, O., A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading, Journal of Computational Chemistry 31 (2010) 455-461.
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S. and Olson, A. J. (2009) Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J. Computational Chemistry 2009, 16: 2785-91.
- Hanwell, M.D., Curtis, D.E., Lonie, D.C. et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 4, 17 (2012). [CrossRef]
- Schrödinger, L. & DeLano, W. (2024). PyMOL. Retrieved from http://www.pymol.org/pymol.
- Farmer JJ, Davis BR, Hickman-Brenner FW, McWhorter A, Huntley-Carter GP, Asbury MA, Riddle C, Wathen-Grady HG, Elias C, Fanning GR. 1985. Biochemical identification of new species and biogroups of Enterobacteriaceae isolated from clinical specimens. J Clin Microbiol 21:. [CrossRef]
- Rosselló-Móra R, Amann R. 2015. Past and future species definitions for bacteria and archaea. Syst Appl Microbiol 38:209–216.
- Anzuay, M.S., Prenollio, A., Ludueña, L.M. et al. Enterobacter sp. J49: A Native Plant Growth-Promoting Bacteria as Alternative to the Application of Chemical Fertilizers on Peanut and Maize Crops. Curr Microbiol 80, 85 (2023). [CrossRef]
- Tovar C, Sánchez Infantas E, Teixeira Roth V (2018) Plant community dynamics of lomas fog oasis of Central Peru after the extreme precipitation caused by the 1997-98 El Niño event. PLoS ONE 13(1): e0190572. [CrossRef]
- Muneeswari, R., Iyappan, S., Swathi, KV., Sudheesh, KP., Rajesh, T., Sekaran G. & Ramani, K. (2021). Genomic characterization of Enterobacter xiangfangensis STP-3: Application to real time petroleum oil sludge bioremediation. Microbiological Research, ScienceDirect. Vol-253. [CrossRef]
- Ya-Ming C., Li X. & Ling-Yun J. (2011). Analysis of PCBs degradation abilities of biphenyl dioxygenase derived from Enterobacter sp. LY402 by molecular simulation. , 29(1), 90–98. [CrossRef]
- Hošková, M., Ježdík, R., Schreiberová, O., Chudoba, J., Šír, M., Čejková, A., Masák, J., Jirků, V. & Řezanka, T. (2015). Structural and physiochemical characterization of rhamnolipids produced by Acinetobacter calcoaceticus, Enterobacter asburiae and Pseudomonas aeruginosa in single strain and mixed cultures. Journal of Biotechnology, 193(), 45–51. [CrossRef]
- Yousaf, S., Afzal, M., Reichenauer, T. G., Brady, C. L., & Sessitsch, A. (2011). Hydrocarbon degradation, plant colonization and gene expression of alkane degradation genes by endophytic Enterobacter ludwigii strains. Environmental Pollution, 159(10), 2675–2683. [CrossRef]
- Broderick, J.B. (1999). Catechol dioxygenases. Essays In Biochemestry, Vol. 34. [CrossRef]
- Baburam, C., Feto, N.A. Mining of two novel aldehyde dehydrogenases (DHY-SC-VUT5 and DHY-G-VUT7) from metagenome of hydrocarbon contaminated soils. BMC Biotechnol 21, 18 (2021). [CrossRef]
- Gangola, S., Bhandari, G., Joshi, S., Sharma, A., Simsek, H. & Bhatt, P. (2023). Esterase and ALDH dehydrogenase-based pesticide degradation by Bacillus brevis 1B from a contaminated environment. Environmental Research, ScienceDirect. Vol-231. [CrossRef]
- Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS (2016). Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. (2016). [CrossRef]
- Roper, D.I., Cooper, R.A. (1990). Purification, some properties and nucleotide sequence of 5-carboxymethyl-2-hydroxymuconate isomerase of Escherichia coli C. Febs Letters. Vol 266, Pages: 63-66. [CrossRef]
- Izumi, A., Rea, D., Adachi, T., Unzai, S., Sam-Yong, P., Roper, D.I. and Tame, J.R.H. (2007). Structure and Mechanism of HpcG, a Hydratase in the Homoprotocatechuate Degradation Pathway of Escherichia coli. JMB. Vol 370. Pages: 899-911. [CrossRef]
- Blum, M., Chang, H.Y., Chuguransky, S., Grego, T., Kandasaamy, S., Mitchel, A., Nuka, G., Lafosse, T.P., Qureshi, M., Raj, S., Richardson, L., Salazar, G.A., Williams, L., Bork, P., Bridge, A., Gough, J., Haft, D.H., Letunic, I., Marchler-Bauer, A., Mi, H., Natale, D.A., Necci, M., Orengo, C.A., Pandurangan, A., Rivoire, C., Sigrist, C.J.A., Silitoe, I., Thanki, N., Thomas, P.D., Tosatto, S.C.E., Wu, C.H., Bateman, A. & Finn, R.D. (2021). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. Vol, 49. Pages D344-D354. [CrossRef]
- Thompson, C.C., Chimetto, L., Edwards, R.A., Swings, J., Stackebrandt, E. & Thompson, F.L. (2013). Microbial genomic taxonomy. BMC Genomics, 14(1), 913–. [CrossRef]
- Wu, W., Feng, Y. & Zong, Z. (2018). Characterization of a strain representing a new Enterobacter species, Enterobacter chengduensis sp. nov.. Antonie van Leeuwenhoek, (), –. [CrossRef]
- Ho, N.R., Kondiah, K., De Maayer, P. & Putonti, C. (2018). Complete Genome Sequence of Enterobacter xiangfangensis Pb204, a South African Strain Capable of Synthesizing Gold Nanoparticles. Microbiology Resource Announcements, 7(22), –. [CrossRef]
- Schroeter, M,N., Gazali, S.J., Parthasarathy, A., Wadsworth, C.B. & Rezende, R. (2021). Isolation, Whole-Genome Sequencing, and Annotation of Three Unclassified Antibiotic-Producing Bacteria, Enterobacter sp. Strain RIT 637, Pseudomonas sp. Strain RIT 778, and Deinococcus sp. Strain RIT 780. ASM Journals, Microbiology Resource Announcements. Vol.10.
- Indugu, N., Sharma, L., Jackson, C.R. & Singh P. (2020). Whole-Genome Sequence Analysis of Multidrug-Resistant Enterobacter hormaechei Isolated from Imported Retail Shrimp. Microbiol Resour Announc. Vol.9. [CrossRef]
- Szczerba, H., Komon-Janczara, E., Krawczyk, M., Dudziak, K., Nowak, A., Kuzdraliński, A., Waśko, A. & Targónski, Z. (2020). Genome analysis of a wild rumen bacterium Enterobacter aerogenes LU2 - a novel bio-based succinic acid producer. Sci Rep 10, 1986 (2020). [CrossRef]
- Jahan, R., Bodratti, A.M., Tsianou, M. & Alexandridis, P. (2020). Biosurfactants, natural alternatives to synthetic surfactants: Physicochemical properties and applications. Advances in Colloid and Interface Science. Vol-275. 102061. [CrossRef]
- Peng, X., Zheng, Q., Liu, L., He, Y., Li, T. & Jia, X. Efficient biodegradation of tetrabromobisphenol A by the novel strain Enterobacter sp. T2 with good environmental adaptation: Kinetics, pathways and genomic characteristics. Journal of Hazardous Materials, ScienceDirect. Vol-429. [CrossRef]
- El-Beltagi, H.S., Halema, A., Almutairi, Z.M., Almutairi, H.H., Elarabi, N., Abdelhadi, A.A., Henawy, A.R. & Abdelhaleem, H.A.R. 2024. Draft genome analysis for Enterobacter kobei, a promising lead bioremediation bacterium. Front. Bioeng. Biotechnology. Vol-11. [CrossRef]
- Camacho, J., Mesen-Porras, E., Rojas-Gatjens, D., Perez-Pantoja, D., Puente-Sanchez, F. & Chavarria, M. (2024). Draft genome sequence of three hydrocarbon-degrading Pseudomonadota strains isolated from an abandoned century-old oil exploration well. ASM Journals, Environmental Microbiology, Vol. 13. [CrossRef]
- Amin, A., Naveed, M., Sarwar, A., Rasheed, S., Saleem, H.G.M., Latif, Z. & Bechthold, A. (2022). In vitro and in silico Studies Reveal Bacillus cereus AA-18 as a Potential Candidate for Bioremediation of Mercury-Contaminated Wastewater. Front. Microbiol. Vol-13. [CrossRef]
- Khan, A., Gupta, A., Singh, P., Mishra, A. K., Ranjan, R. K., & Srivastava, A. (2019). Siderophore-assisted cadmium hyperaccumulation in Bacillus subtilis. International Microbiology. [CrossRef]
- Gardy, J.L., Laird, M.R., Chen, F., Rey, S., Walsh, C.J., Ester, M. & Brinkman, F.S.L. (2005). PSORTb v.2.0: expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Comparative Study. Vol, 21. Pages 617-623. [CrossRef]
- Peabody, M.A., Lau, W.Y.V., Hoad, G.R., Jia, B., Maguire, F., Gray, K.L., Beiko, R.G., Brinkman, F.S.L. (2020). PSORTm: a bacteria and archaeal protein subcellular localization prediction tool tor metagenomics data. Bioinformatics. Vol-36. Pages: 3043-3048. [CrossRef]
- Lou, Feiyue., Okoye, C.O.. Gao, L., Jiang, H., Wu, Y., Wang, Y., Li, X. & Jiang, J. (2023). Whole-genome sequence analysis reveals phenanthrene and pyrene degradation pathways in newly isolated bacteria Klebsiella michiganensis EF4 and Klebsiella oxytoca ETN19. Microbiological Research. Vol-273. 127410. [CrossRef]
- Louvado, A., Gomes, N.G.M., Simões, N.M.Q., Almeida, A., Cleary, D.F.R. & Cunha, A. (2015). Polycyclic aromatic hydrocarbons in deep sea sediments: Microbe-pollutant interactions in a remote enviroment. Science of the Total Environment. Vol 526, Pages: 312-328. [CrossRef]
- Bukowska, B., Mokra, K. & Michalowicz, J. (2022). Benzo[a]pyrene—Environmental Occurrence, Human Exposure, and Mechanisms of Toxicity. Int J Mol Sci, 23, 6348. [CrossRef]
- Wang, Y.F. & Tam, N.F.Y. (2011). Microbial community dynamics and biodegradation of polycyclic aromatic hydrocarbons in polluted marine sediments in Hong Kong. Marine Pollution Bulletin. Vol-63. Pages: 424-430. [CrossRef]
Figure 1.
Genomic Results of Enterobacter sp. a) Comparison of BUSCO analysis between UNJFSC003 and the strains CCA6, Crenshaw, and FDAARGOS1428 b) Genomic map of Enterobacter sp. UNJFSC003. c) Subsystem distribution in UNJFSC003.
Figure 1.
Genomic Results of Enterobacter sp. a) Comparison of BUSCO analysis between UNJFSC003 and the strains CCA6, Crenshaw, and FDAARGOS1428 b) Genomic map of Enterobacter sp. UNJFSC003. c) Subsystem distribution in UNJFSC003.
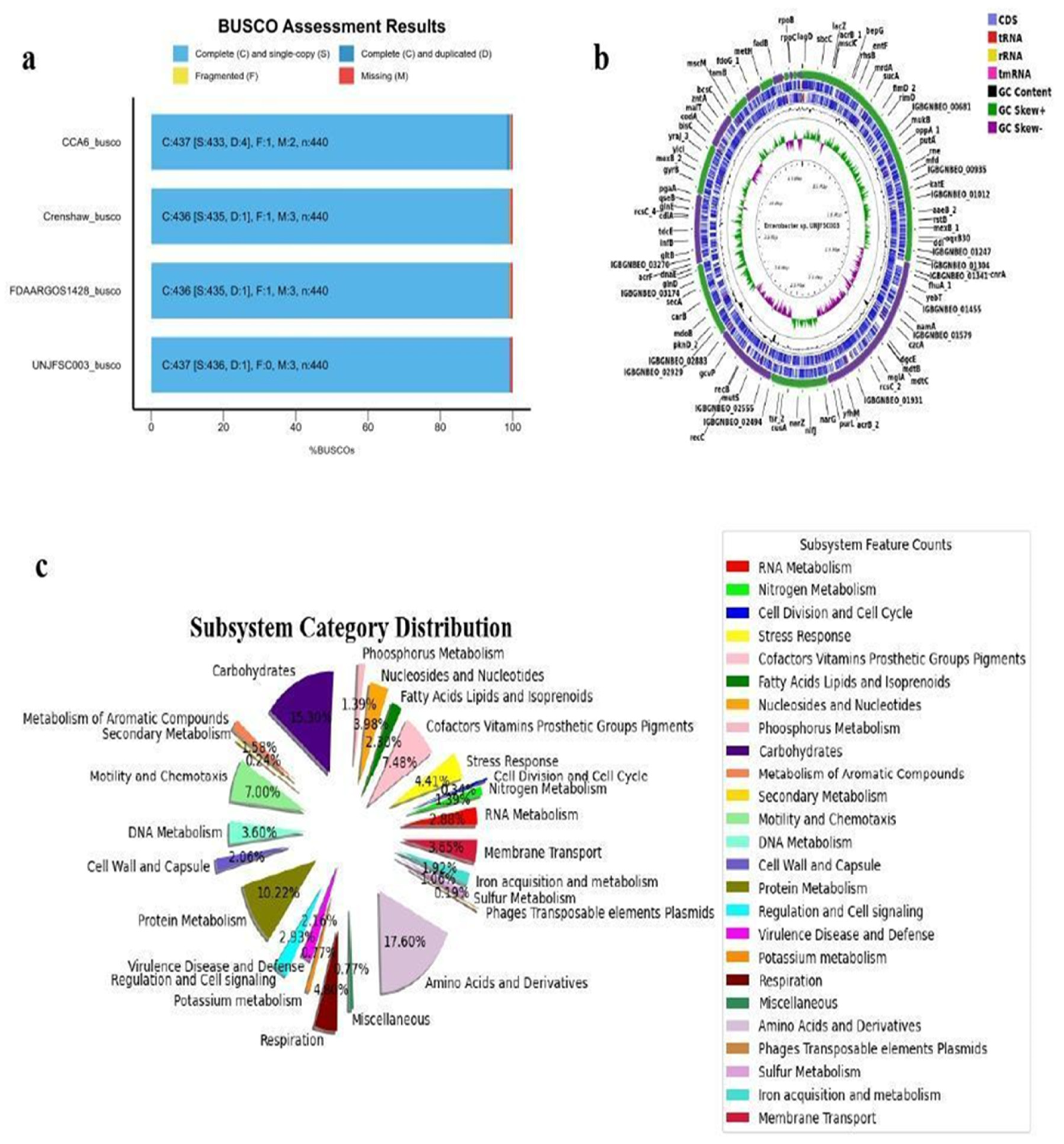
Figure 2.
a) The heatmap of Average Nucleotide Identity (ANI) values the isolated genome of Enterobacter sp. UNJFSC003 against 15 reference genomes. The numbers represent ANI (%) values between genome sequences. The NCBI GenBank reference strains used in this study are listed in Table 3.
Figure 2.
a) The heatmap of Average Nucleotide Identity (ANI) values the isolated genome of Enterobacter sp. UNJFSC003 against 15 reference genomes. The numbers represent ANI (%) values between genome sequences. The NCBI GenBank reference strains used in this study are listed in Table 3.
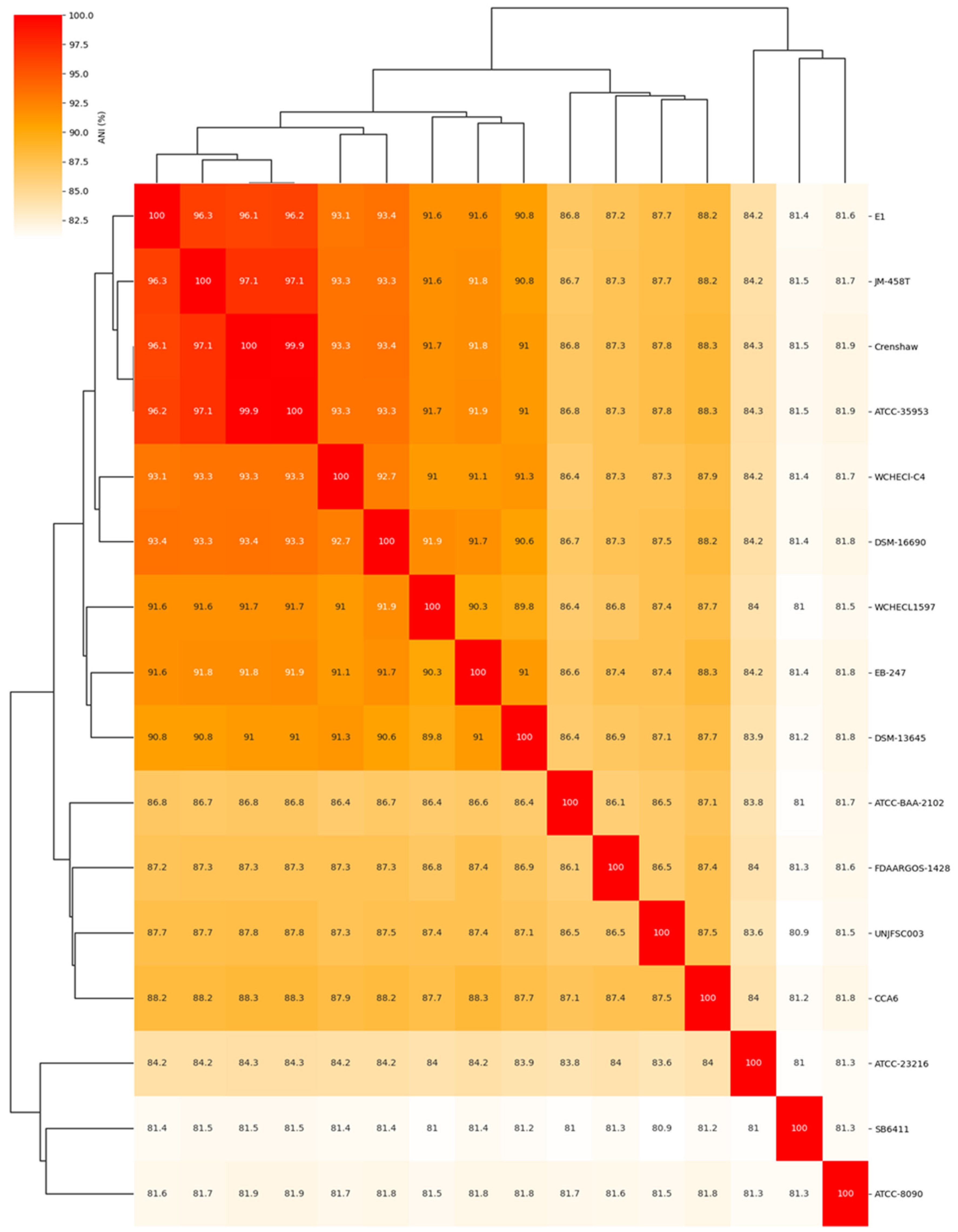
Figure 3.
The pangenome of 16 Enterobacter strains was determined using the Roary matrix. A total of 33,385 orthologous protein-coding genes were found. a) Heatmap showing the presence (dark blue) or absence (light blue) of genes in each of the 16 strains. On the left is a phylogeny constructed based on core genes, and on the right are the genome strain names. b) Histogram showing the distribution of genomes per gene.
Figure 3.
The pangenome of 16 Enterobacter strains was determined using the Roary matrix. A total of 33,385 orthologous protein-coding genes were found. a) Heatmap showing the presence (dark blue) or absence (light blue) of genes in each of the 16 strains. On the left is a phylogeny constructed based on core genes, and on the right are the genome strain names. b) Histogram showing the distribution of genomes per gene.
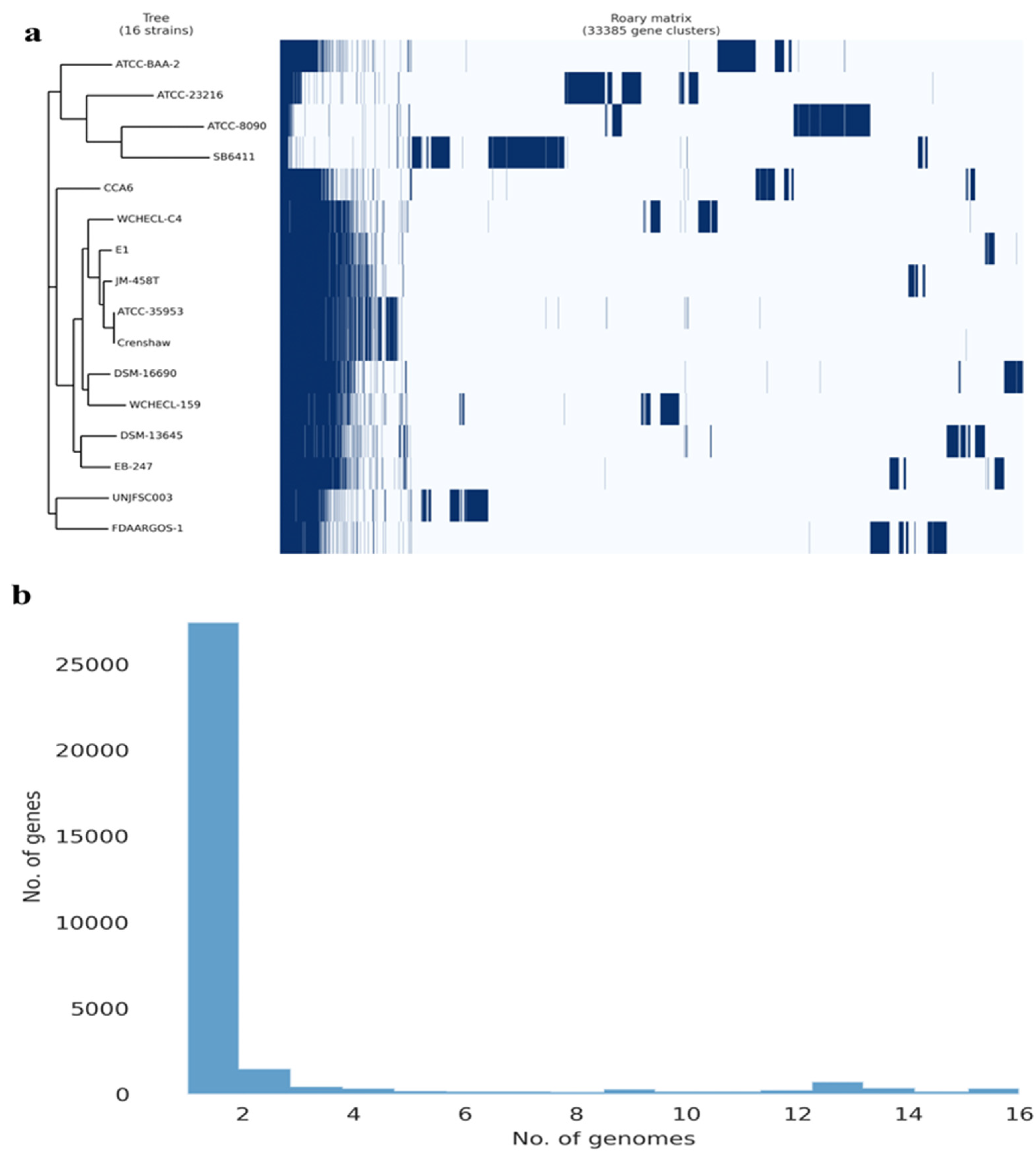
Figure 4.
Prediction of enzymes in hydrocarbon degradation by HADEG. a) Heatmap of all 21 enzymes of UNJFSC003 involved in the degradation of hydrocarbons, including alkanes, aromatics, and biosurfactants. b) Bubble chart showing the distribution of sub-pathways of the HADEG-validated enzymes. c) Histogram displaying the number of enzymes for each type of hydrocarbon and biosurfactant.
Figure 4.
Prediction of enzymes in hydrocarbon degradation by HADEG. a) Heatmap of all 21 enzymes of UNJFSC003 involved in the degradation of hydrocarbons, including alkanes, aromatics, and biosurfactants. b) Bubble chart showing the distribution of sub-pathways of the HADEG-validated enzymes. c) Histogram displaying the number of enzymes for each type of hydrocarbon and biosurfactant.
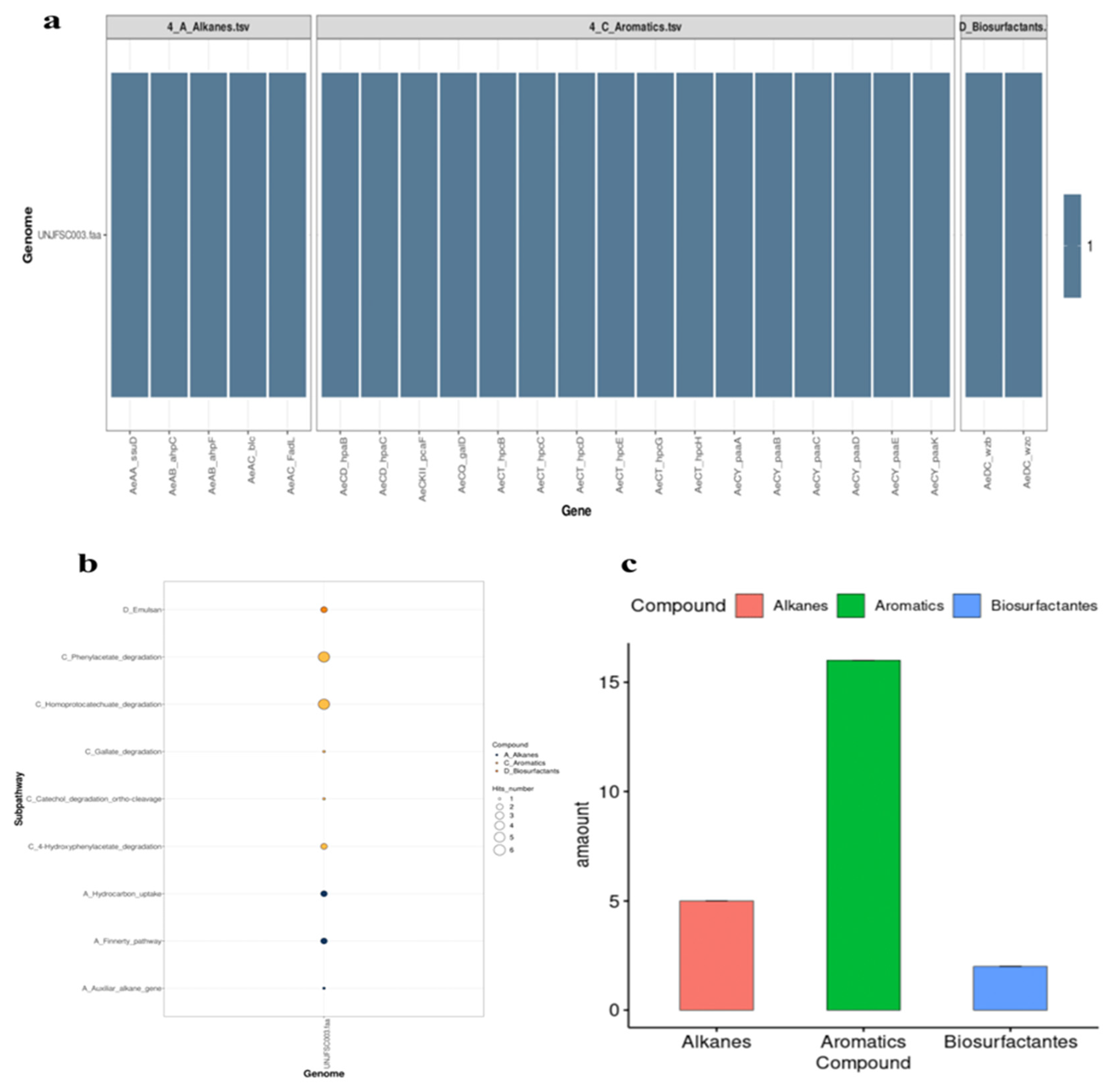
Figure 5.
(a) Heat map showing the subcellular localization of the 21 enzymes. (b) Predicted network interactions of proteins involved in hydrocarbon degradation, with protein clusters marked in green (hpc and hpa), yellow (paa and pca), and red representing the two biosurfactant proteins. (c) Model of hpcB. (d) Model of hpcC. (e) Model of hpcD. (f) Model of hpcE. (g) Model of hpcG. (h) Model of hpcH.
Figure 5.
(a) Heat map showing the subcellular localization of the 21 enzymes. (b) Predicted network interactions of proteins involved in hydrocarbon degradation, with protein clusters marked in green (hpc and hpa), yellow (paa and pca), and red representing the two biosurfactant proteins. (c) Model of hpcB. (d) Model of hpcC. (e) Model of hpcD. (f) Model of hpcE. (g) Model of hpcG. (h) Model of hpcH.
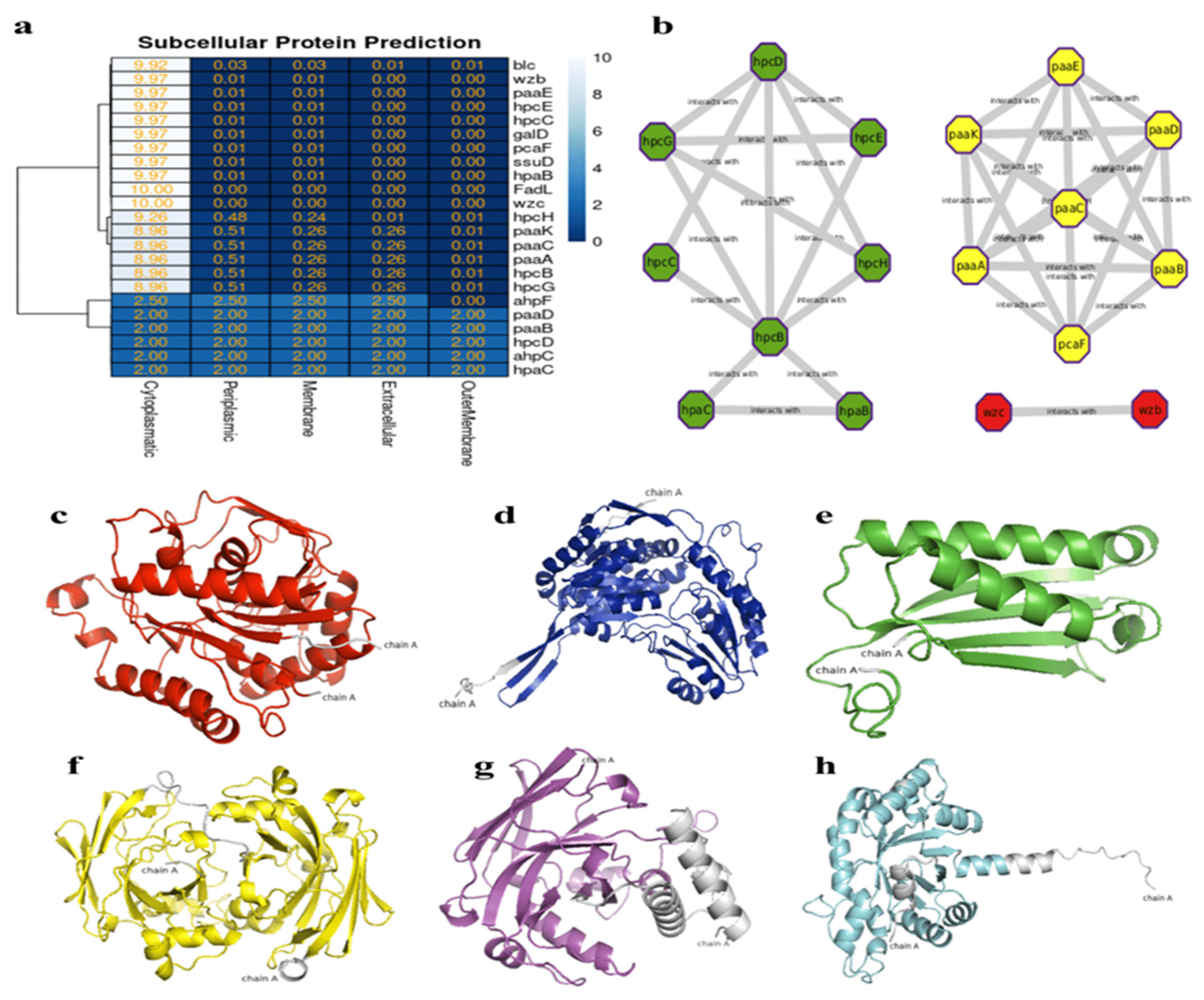
Figure 6.
Results of molecular docking with the HAP (benzo[a]pyrene) molecule: a) Active site amino acids of hpcB. b) Active site amino acids of hpcC. c) Active site amino acids of hpcD. d) Active site amino acids of hpcE. e) Active site amino acids of hpcG. f) Active site amino acids of hpcH.
Figure 6.
Results of molecular docking with the HAP (benzo[a]pyrene) molecule: a) Active site amino acids of hpcB. b) Active site amino acids of hpcC. c) Active site amino acids of hpcD. d) Active site amino acids of hpcE. e) Active site amino acids of hpcG. f) Active site amino acids of hpcH.
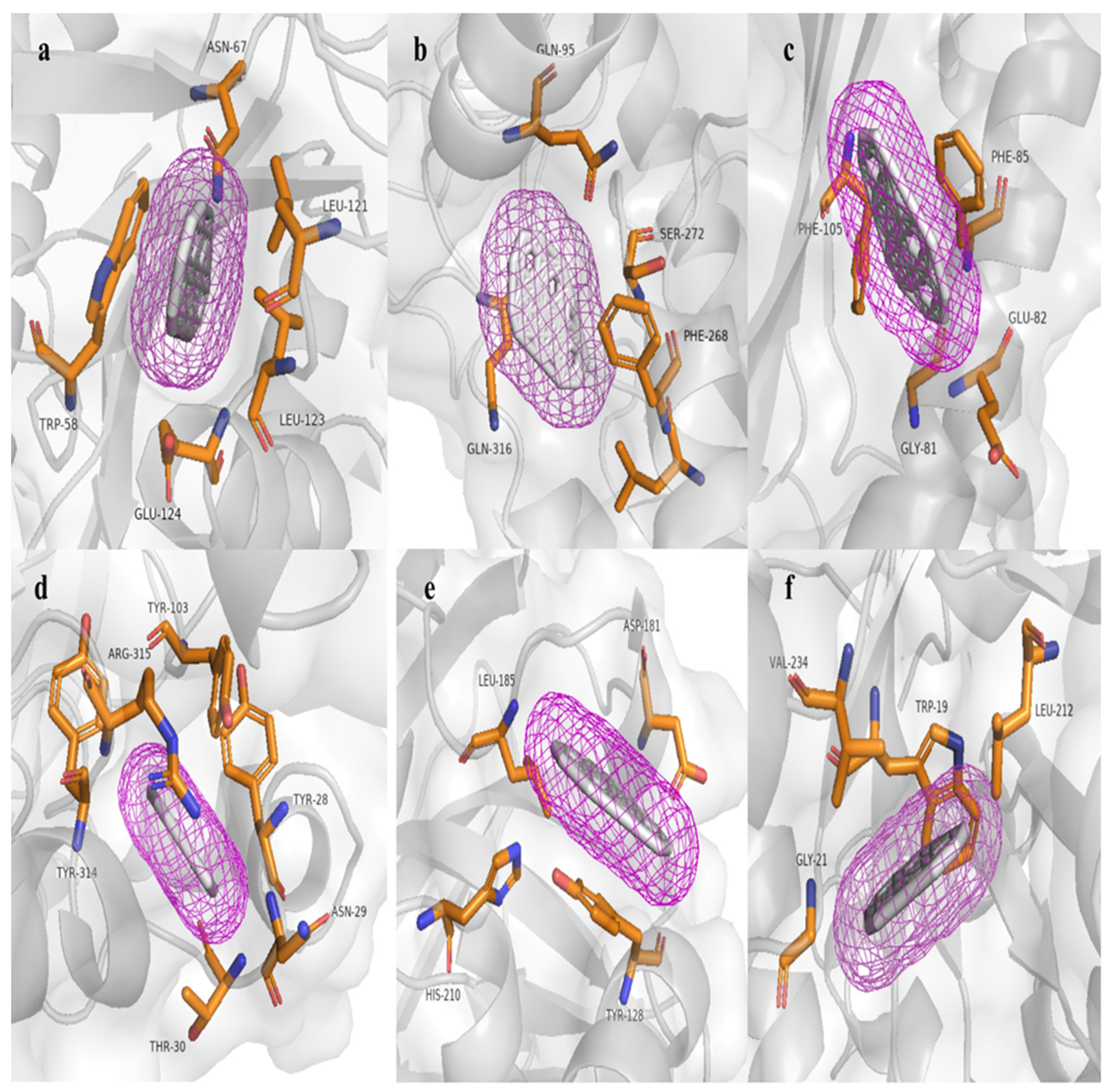
Table 1.
Trimming illumina reads.
| Quality control FastQ | Results |
|---|---|
| High-quality readings | 10,613,744 (97,24%) |
| Readings due to low quality | 19.324 (0,18%) |
| Contained too much pollution | 336 reads (0,001539%) |
| Readings too short | 5.8520(0,026808%) |
| Cut-out adapters | Yes |
Table 2.
Genome properties and characteristics of the draft genome of Enterobacter sp. UNJFSC 003.
| Properties and characteristics | Total |
|---|---|
| Sequence size Genome | 4,798,267 |
| No of Scaffolds | |
| Contigs >= 0 bp/1000bp/50000bp | 51/28/12 |
| N50/L50 | 415,254/3 |
| No of CDS | 4460 |
| No of rRNA/tRNA/tmRNA | 2/77/1 |
| GC% | 54.38 |
| KEEG Mapper Reconstruction | |
| KEEG Orthology (KO) | 2671 |
| Metabolism protein | 1487 |
| Genetic information processing | 690 |
| Signaling and cellular processes | 829 |
| Carbohydrate Metabolism | 318 |
| Amino acid Metabolism | 157 |
| Nucleotide Metabolism | 104 |
| Metabolism of cofactors and vitamins | 134 |
| Energy Metabolism | 101 |
| EggNOG-Mapper | |
| COG | 4349 |
| Pfam | 4186 |
| GO | 2540 |
| CAZy | 77 |
| BIGG | 1064 |
Table 3.
Reference genomes of enterobacteria from GenBank, strain code, scientific name, and FastANI results.
Table 3.
Reference genomes of enterobacteria from GenBank, strain code, scientific name, and FastANI results.
| Genomes NCBI | FAST ANI | ||||
|---|---|---|---|---|---|
| Strain/ | GENBANK | SCIENTIFIC NAME | ANI score | Fragment lenght | Total Fragment |
| Crenshaw | GCA_016027695.1 | Enterobacter asburiae | 87.8055 | 1250 | 1586 |
| ATCC 35953 | GCA_001521715.1 | Enterobacter asburiae | 87.8536 | 1241 | 1586 |
| JM-458T.1 | GCA_900180435.1 | Enterobacter asburiae | 87.8124 | 1282 | 1586 |
| E1 | GCA_008364625.1 | Enterobacter dykesii | 87.8021 | 1262 | 1586 |
| DSM 16690 | GCA_001729805.1 | Enterobacter roggenkampii | 87.6385 | 1258 | 1586 |
| CCA6 | GCA_009176645.1 | Enterobacter oligotrophicus | 87.547 | 1218 | 1586 |
| WCHECL1597 | GCA_002939185.1 | Enterobacter sichuanensis | 87.326 | 1248 | 1586 |
| WCHECl-C4 | GCA_001984825.2 | Enterobacter chengduensis | 87.3321 | 1295 | 1586 |
| DSM-13645 | GCA_001729765.1 | Enterobacter kobei | 87.1635 | 1222 | 1586 |
| FDAARGOS 1428 | GCA_019047785.1 | Enterobacter cancerogenus | 86.5672 | 1214 | 1586 |
| EB-247 | GCA_900324475.1 | Enterobacter bugandensis | 87.4329 | 1290 | 1586 |
| ATCC BAA-2102 | GCA_001654845.1 | Enterobacter soli | 86.5295 | 1222 | 1586 |
| ATCC 23216 | GCA_000735515.1 | Leclercia adecarboxylata | 83.6121 | 1043 | 1586 |
| SB6411 | GCA_902158555.1 | Klebsiella spallanzanii | 80.9556 | 840 | 1586 |
| ATCC 8090 | GCA_011064845.1 | Citrobacter freundii | 81.5917 | 820 | 1586 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



