
Submitted:
12 September 2024
Posted:
13 September 2024
You are already at the latest version
Abstract
When we grow old, we experience loss of muscle strength and power, a condition commonly referred to as sarcopenia (ICD-10-CM code (M62.84)). Sarcopenia leads to disability in 5-10% of the population. In this review we emphasise that it is not a sudden event, but a deterioration that occurs over time but is only recognised as a disease when it becomes clinically manifest in the 6th-7th decade of life. Evidence from animal studies, elite athletes and longitudinal population studies all confers that once sarcopenia is manifested, the underlying process has been ongoing for decades. We present various hypotheses about the underlying cause(s) of this process and their supporting evidence, i.e. does this disease have a neurogenic, a myogenic or a scaffold-related origin? We briefly review various proposals to alleviate sarcopenia, including stem cell rejuvenation, combating scaffold degeneration, reducing senescent cell burden, skeletal muscle secretomes and muscle innervation. We conclude that although there are potential target candidates and ongoing preclinical and clinical trials with drug treatments, the only evidence-based intervention today is exercise. We present different execrcise programmes and discuss to what extent the inter-individual susceptibility to developing sarcopenia is due to our genetic predisposition or lifestyle factors.
Keywords:
dynapenia
; muscle fiber atrophy
; motor unit
; ageing
; senescence
1. Popular Summary
In this review, we present the current evidence on the background to age-related muscle weakness (sarcopenia), which affects around 5-10% of older people in society, and how we can help to intervene in the disease process and improve the quality of life of those affected. We will discuss what is currently known about the cause of this disease. Is it due to age-related changes in the muscle fibres, the muscle organ or the innervation of the muscle? We explain the efforts to alleviate sarcopenia by restoring a more youthful skeletal musculature and the drug treatments currently being tested in preclinical and clinical trials. However, we conclude that there is no biochemical treatment available today that can halt the condition, and that the only evidence-based intervention that can slow the progression of sarcopenia is exercise. Finally, we discuss whether the large differences between people in the severity of sarcopenia depend on their background or lifestyle.
2. Growing Old
In the last two centuries, life expectancy at birth has almost doubled, while the years in health have not improved likewise (discussed in [1]). It is widely believed that underlying the deterioration in health at older ages is the cumulative detrimental effects of aging at the cellular, systems and organismal levels. The challenge we now face is therefore to extend the healthy lifespan through evidence-based lifestyle recommendations and rational biomedical interventions.
Aging causes the characteristic phenotypic changes that we are all familiar with. These include the loss of integrity of the connective tissue, which leads among other things to a decrease in lung compliance and a stiffening of the blood vessels, which affects blood pressure and the strain on the heart. In the musculoskeletal system, there is a loss of bone (osteopenia and osteoporosis), joint cartilage and the innervation, function and mass of skeletal muscles (sarcopenia). These changes hinder movement and balance and drastically increase the risk of falls and bone fractures. At the same time, there is an accumulation of waste products. These are not sudden processes, but start slowly and accelerate with age, but are not closely linked to chronological age. The genetic predisposition of the individual in combination with random and fixed environmental factors is reflected in a considerable variability in the biological age of people of the same chronological age [2,3] (discussed in[4]).
The decline in function and structural integrity of the aged phenotype is driven by two main processes: Failure to replace worn-out cells due to senescence and/or depletion of stem (progenitor) cells(reviewed in [5]), and inadequate machinery to maintain cellular and extracellular homeostasis(reviewed in [6] [4,7,8]).
3. Loosing Muscle Strength and Mass
As we enter middle age and beyond, most of us find that the speed of muscle contraction, strength and mass decline with age. The loss of strength and mass is often collectively referred to as sarcopenia (definition guidelines for diagnostic criteria have been published and revised by European, Asian and North American working groups (the EWGSOP2[9,10], the Asian Working Group for Sarcopenia (AWGS)[11], and the FNIH[12]), a deterioration that becomes clinically significant in the sixth decade of life with an annual increase of 0.5-2 %. In advanced age, sarcopenia can be the main or sole cause of disability in daily life and has its own disease code (WHO [[13]]; ICD-10-CM code (M62.84)[14]). The prevalence of sarcopenia among elderly varies between populations but is usually within the range of 5-10%. As discussed elsewhere, the prevalence of sarcopenia may increase in parallel with the concurrent digitalization where people tend to be stationary in front of their digital devices and physical labour is robotized. Here we will use the term sarcopenia in its broader definition and not refer to dynapenia as a separate entity. Muscle strength and contraction speed (velocity) precedes the loss of muscle mass (for references see[4]), changes that are already detectable beyond the age of 30 in elite athletes[15,16,17,18,19] and this is also evident in population wide cohorts ([20](Figure 1). Initially, the pace of decay is small (<1% per year) but later the loss accelerates (~1-2% per year) and by the time untrained people notice it, it has already been going on for decades (idem). We will refer to the early phase as the preclinical phase while the overt condition is referred to as the clinical phase of sarcopenia (Figure 1; [21]).
There is still a lack of both evidence and scientific consensus on the cause(s) of sarcopenia (for references see [1,22,23,24,25,26]. Below is a brief description of aging in the neuromuscular system (with references to more detailed accounts).
3.1. The Motor Unit
Motor neurons (MNs) in the spinal cord and brainstem of the central nervous system trigger the contraction of skeletal muscle fibers by sending signals through the neuromuscular junctions (NMJ). A motor neuron innervates a number of myofibers (between ~8 and >1000 depending on muscle type and type of MU), which together form a motor unit (MU). All myofibers of a MU are of the same type: slow (type I; S-type MU) in contraction speed (twitch time) or fast contracting (type II), and the latter type of MU (F-type MU) contains often many more myofibers. The MUs of a muscle are the building blocks of graded muscle contraction. The main mechanism by which we regulate muscle strength is therefore the recruitment and de-recruitment of MUs. The timing and precision of these processes are crucial for all body movements and for balancing the pull of gravity on body mass. A second mechanism by which we can modulate muscle tension is by increasing the MN firing frequency (up to the fusion rate) until a maximum is reached (Tetani). This mechanism is dependent on the firing and impulse conduction properties of the MNs. Recordings from humans and animal models show that the number of MUs per muscle continuously decreases during aging and in parallel the surviving MUs increase in size. The interpretation of these observations is that the myofibers are denervated due to the degeneration of motor axons and in parallel some of the denervated myofibers are reinnervated by nearby intact motor axons (increase in MU size) (Figure 2) [22,24,25,27,28], the loss will accelerate at later ages due to limitation of the re-innervation capacity (idem).
Observations in the peripheral and central nervous system have shown that axonal degeneration with atrophy and dystrophy - in both cases the axon terminals at synaptic contacts are lost - is widespread and more conspicuous than the loss of neurons during aging (reviewed in [29]). This process is called “dying-back" of neurons and projecting neurons, such as MNs, appear to be more susceptible than short circuit neurons (idem). The systems affected include the descending bulbospinal aminergic systems, which act as amplifiers for MN excitability ([30,31,32,33,34,35]. Thus, MN innervation of myofibers is vulnerable and impaired during aging, and the available evidence also supports the notion that age-related degeneration of some of the gain-setting systems of MN excitability inherent in the nervous system may impair the firing probability and firing frequency of aged MNs[36,37].
It is not yet clear whether the loss of MUs during aging is solely due to the inability to maintain innervation by the motoneurons, or whether this process is also driven by changes in the myofibers and/or the local environment (muscle scaffold), or a combination of both. Another unresolved question is why the loss of MU preferentially affects the fast-twitch MUs, while the slow-twitch MUs increase in size. These two questions need to be clarified [38]. Importantly, aging-induced remodelling of the MU population of a muscle affects also individuals that exercise at a very high level throughout life[39,40,41,42,43]. The remodelling of the MU population leads to a general slowing of motor behavior and a decrease in muscle strength and power output. In the initial phase, the total muscle mass is not affected to the same extent, while in the advanced age the mass also decreases (Figure 1).
Observations made over the past decades show that as we age, we lose some myofibers while many other myofibers atrophy, a process that, for unknown reasons, affects fast-contracting myofibers (type II) more than slow-contracting fibres (type I). Microscopic observations in aged sarcopenic muscle reveal an increased variability in myofiber size with clusters of severely atrophied fibres and an increase in interstitial tissue. In advanced age, the adaptive (hypertrophy) response of myofibers to exercise is blunted, a change attributed to impaired anabolic response and impaired recruitment of myocytes from the local stem cell pool (satellite cell; SC, niche) (Figure 3; discussed in[4,5,44]). Aged myofibers exhibit increased lipid content and dysfunctional mitochondria; signs of impaired autophagy-lysosomal and proteasomal degradation of worn-out cellular components with an accumulation of polyubiquitinated proteins and lipofuscin (Figure 3; [7,8]).
The replacement and maintenance mechanisms described above are crucial for the adaptive and regenerative capacity of skeletal muscles. While MNs rely solely on maintaining their functionality as we age, skeletal muscle is also able to adapt and regenerate by increasing fibre size through anabolism and the recruitment of locally available stem cells (satellite cells, SC). However, as we age, the pool of progenitor cells (satellite cells, SC) is depleted and many of the remaining SC do not respond to stimuli to proliferate and supply the myofiber with additional myocytes to grow[45]. Furthermore, the intrinsic machineries for anabolism and catabolism in myofibers appear to be dysregulated. Thus, despite the myofiber atrophy seen in aged muscle, the level of the key scaffold that drives myofiber hypertrophy (mTORC1) is increased and not decreased [46,47], likewise the main machinery to remove targeted protein (through proteasomal degradation) is also increased but dysregulated[8,48]. These disturbances of the resources and machineries critical for myofiber adaptations may explain why aged myofiber show poor or blunted adaptive responses to stimuli such as exercise (see also below).
Myofibers also secrete molecules (secretome; e.g. chemokines, myokines and trophic factors) by which it communicate not only with adjoining myofibers, SCs and axons of peripheral motor nerves, but also with the immune system, bone tissue, the muscle stroma cells including resident macrophages[49,50,51,52,53,54,55]. The secretome has attracted considerable interest in the search for useful targets to impede the sarcopenic process (see below), especially those that are secreted following exercise (exerkines; idem).
3.2. The Muscle Scaffold
The connective tissue forms the structure and the molecular environment (scaffold, stroma) for the skeletal muscle cells and the pathways for the vessels and the innervation of the muscle organ (for a detailed account see [56,57,58,59,60,61,62,63]). In sarcopenia research, much less attention has been paid to the scaffold than to the myofibers and their innervation. The scaffold is colonised by stromal cells derived from fibro-adipogenic mesenchymal precursor cells (idem). In skeletal muscle, it is mesodermal fibroblasts that are adapted to the skeletal muscle organ and produce the ECM, which consists of proteins. The proteins form fibrils by cross-linking and fibrils combine to form fibres with a complex 3D structure that can grow into sheaths (fascial structures) that divide a skeletal muscle into tensile compartments in which the sarcolemma of a single myofiber is surrounded by a bilayered basal lamina (endomysium), groups of muscle fibres are separated by septal sheaths (perimysium, Figure 1 A-B) and the entire muscle is enveloped by the muscle fascia (epimysium). The ECM of these units has a distinctly different composition, reflecting their respective function (idem). At one or both ends of a muscle, the ECM connects the muscle to a tendon. The tendon anchors the muscle to the skeleton. The composition of the ECM of a tendon differs from the ECM of the muscle and is formed by tenoblasts of mesenchymal origin [64,65]. Together, the ECM of the muscle and the tendon transmit the tension generated by the contraction of the myofibers to the bone. It is the structure of the muscle ECM that allows the myofibers in multi-pennate muscles to pull in concert on the tendon.
Molecules secreted by fibroblasts (secretome; chemokines, myokines and trophic factors) communicate not only with myofibers, SCs and axons of peripheral motor nerves, but also with the immune system and resident macrophages ([56,66]), and bone tissue (bone-muscle crosstalk, [55,67]). The remodelling of the ECM occurs slowly and under normal conditions is controlled by the resident fibroblasts. Fibroblasts produce both the ECM and the proteases that digest it (mainly matrix metalloproteases MMPs ([56,63]). Since ECM proteins are long-lived, they are targets for environmentally induced secondary modifications such as glycosylation. Such adducts can impair the proper function of the ECM [61]. With increasing age, changes (fibrosis) occur in the ECM, which lead to increased stiffness and fragility of the tendon and muscle compartments ([57,61,62,68]).
It is hypothesised that this contributes to the age-related impairment of myocyte renewal from SCs. Changes in the ECM may also play a role in the failure to reinnervate denervated myofibers by collateral sprouting in advanced age. The fibrosis seen in sarcopenic muscles indicates an imbalance in the production, (re)assembly and degradation of ECM components ([57,69,70]). In conclusion, the muscle scaffold is overly affected by the sarcopenic process and may play an important role on the progression of this condition.
4. Cellular Senescence
Cellular senescence is a term introduced half a century ago to describe the loss of the ability of cells to replicate that occurs in cell cultures [71]. Such cells no longer respond to environmental stimuli and therefore no longer contribute to tissue function in vivo. More recently, it has been found that the number of senescent cells increases with age and that they become a burden on the tissue due to their space requirements and - perhaps more importantly - their secretome- referred to as SAPS (senescence associated protein secretory (cell) phenotype) [72,73]. It has been hypothesized that the age-related accumulation of senescent cells is due to an increase in the rate at which cells become senescent and/or a decrease in the ability of the immune system's natural killers (NK) cells to remove these cells [74]. Thus, one attempt to improve the regenerative and adaptive capacity of the tissue in old age is to find an intervention that promotes the removal of senescent cells [74].
4.1. Interventions to Impede Sarcopenia
Exercise
If we do not use the muscles in everyday life, we will lose strength and muscle mass [75]. As we get older, however, we lose muscle strength despite normal daily activity. Not even top athletes who train at a high level throughout their lives are spared the age-related decline in muscle function. However, people who exercise regularly are more successful at maintaining their mobility and independence as they age than those who lead sedentary lives [16] [76](for a more detailed discussion on this topic, see[4,77,78,79]).
The only evidence-based intervention to combat the decline in muscle function and mass is the prescription of exercise, and this also applies to frail individuals suffering from multiple age-related comorbidities. The uptake of exercise in older age is not without its challenges and needs to be individualized to optimize the cost-benefit ratio. For untrained, frail individuals, balance and flexibility of the body should be trained first and then regular endurance and strength exercises should be introduced. Even at low intensity and low volume (e.g. once a week), most people make progress within a few weeks. Initially through improved postural and neuromuscular control and later, depending on the type and dose of training, also in endurance, strength and muscle mass [80,81,82]. In advanced age, it is perhaps most important to avoid periods of detraining to minimize deconditioning of neuromuscular control and muscle mass [83]. With increasing age, it becomes more and more difficult to recover from phases of immobilization (deconditioning). Such episodes should be kept as short as possible and supplemented by a re-training program [4]. In recent decades there has been a lively debate about the relative benefits of different forms of exercise (endurance training vs. strength training), the intensity and frequency of training and the amount of exercise. Endurance training (aerobic exercise) (ET) improves cardiovascular function and oxygen uptake (VO2), cellular respiration (mitochondrial function and mtDNA copy number), but has a lesser effect on overall muscle strength and mass [78,84,85,86]. The multisystemic effect of ET counteracts several of the observed consequences of aging, such as mitochondrial dysfunction, low-level inflammation, decreased aerobic capacity (cardiovascular and pulmonary systems), and changes in connective tissue (e.g., stiffening of arteries and cardiac and pulmonary compliance, and fibrosis of the muscle scaffold). Therefore, lifelong ET is a measure to counteract the consequences of aging at a systemic level [87,88,89]. Regular endurance training is less efficient in preventing the loss of muscle strength and mass in old age, and ET used as a mobilization measure in older people does not lead to a significant increase in muscle mass or strength [90].
Resistance training (RT) is an effective way of improving strength and muscle mass [76,80,81,82,91]. The response to RT in terms of improving strength and myofiber hypertrophy depends on the duration, frequency and intensity of training, with the current prevailing view being the more the better. However, high intensity and very large amounts of RT can be counterproductive in older and frail individuals [81,91]. Although strength increases with RT dose, the response of myofibers to hypertrophy is lower and sometimes blunted in the elderly, indicating impaired myofiber adaptive potential (see above and [4,92,93]). Therefore, in older people, improving strength rather than muscle mass is a better measure of the success of RT. From the above, it seems logical that training programs to combat the occurrence and progression of sarcopenia should be based on both ET and RT, as they have complementary effects on the aging process and a large number of studies point to the dual benefits of simultaneous ET and RT [94,95] [86,96,97,98,99,100,101].
In conclusion, simultaneous training of ET and RT is currently the best measure to prevent the progression of sarcopenia and physical disability in older people of both sexes. Although lifelong training is preferable, it is recommended to start training also in frail elderly to slow down the progressive deconditioning of skeletal muscles. Especially in the frail elderly, these efforts should begin with balance, flexibility and coordination training, followed by strength and endurance training, with all components adapted to the subject's situation. Even with a low frequency, e.g. one session per week, most people will see improvements. There exist guidelines on recommended physical activity, like “The 2018 physical activity guidelines for Americans” and studies have shown that those that comply with the guide-line recommendations have a significant survival benefit[102]. The frequency and intensity of RT and ET sessions can be continuously adjusted as progress is made. Three sessions per week (totally ~150 min at moderate level) is a generally recommended value for repetitive exercise. Overtraining should be avoided, while targeted retraining after episodes of immobilisation is strongly recommended. Balance, coordination, endurance, muscle strength and power are useful measures, while muscle mass/myofiber size is a less appropriate measure in advanced age.
4.2. Biochemical Approaches to Halt Sarcopenia
Although exercise is an effective measure to slow down sarcopenia, it does not eliminate it and there are situations where exercise is not enough. With prolonged or repeated deconditioning, recovery may be inadequate, leading to physical disability and rapid progression of sarcopenia. Therefore, strategies to rejuvenate the adaptive potential of myofibres and mimetics that could replace or enhance the effect of ET/RT sessions to bridge episodes of immobilisation are being intensively searched for, but have not yet resulted in breakthroughs for clinical practise. These strategies include stem cell transplantation, adjuvant endocrine therapy, facilitating the removal of senescent cells and the use of molecules secreted by exercised myofibres (secretome)[45,49,103,104,105,106,107,108].
Rejuvenation of the adaptive regeneration capacity by reconstitution of SC cells has not been successful so far (discussed in [5]). Rejuvenation of SC in vitro for subsequent autologous re-transplantation could be an alternative but has not yet been tested in humans [106]. Another approach is to reduce the burden of senescent cells and their SAPS. Here, trials with tyrosine kinase inhibitors such as quersetin and dastinib (drugs already used to treat certain cancers) and/or the flavonoid polyphenol fesetin show promising results in animal models, but have yet to be tested in humans ([72,73] ,reviewed in[109] [110]). Another strategy in this context is neutralising antibodies against circulating levels of interleukin IL-6 [70] (thought to be responsible for the low tissue inflammation that is a known component of the ageing phenotype) or myostatin [111] (a prominent trigger of myofibre atrophy via the TGFβ-activin-Smad pathway), both of which are components of the myofibre secretome [51]. However, this strategy is unlikely to be applicable to large numbers of people or over a prolonged period of time. Furthermore, data from experimental studies suggest that IL-6 secreted by myofibres plays an important role in enhancing the myotrophic response to exercise [112,113].
The secretome of myofibres is very diverse and more than 600 proteins have been identified to date. The content of the secretome is context-dependent [50,51] and at least 30 proteins are induced by exercise, such as irisin, S100 (A and/or B) and apelin. Apelin a secreted myokine that acts on the g-protein-coupled APJ receptor, has attracted considerable attention as a potential mediator of myofibre adaptation to exercise [49,52,104,114,115,116] and clinical trials may follow. In this context, it is interesting to note that apelin protects the brain from cytotoxic damage by inducing brain-derived neurotrophic factor (BDNF) [117]. BDNF belongs to the family of neurotrophins (NTs), another group of proteins that increase in myofibres in response to exercise [108,118]. NTs are of interest not only because they play an important role during development and possibly also in the regeneration of myofibres due to their effect on SCs, but also because they support motoneurons and show characteristic expression changes during ageing [27,103,119]. For example, satellite cells of extraocular eye muscles, muscles that escape age-related loss of function and mass, express higher levels of NT and their receptors compared to limb muscles [120]. The means by which NT signalling can be promoted in the muscles, apart from training, remains however to be clarified.
Endocrine therapy with growth hormones, gonadal steroids or androgens has been widely discussed and tested in humans, but the risks associated with these molecules may outweigh the benefits of their use.
Efforts to spare muscles through shorter unloading phases with partial mimetics of calorie restriction (CR; the gold standard for slowing ageing) such as metformin (a drug used to treat type II diabetes) and rapamycin-like molecules (inhibitor of the mTORC1 complex) appear to be promising [46,121].
In summary, research efforts to date have not been successful in establishing a biochemical treatment to preserve neuromuscular function in situations of inactivity and ageing.
5. Concluding Remarks
The adult lifespan trajectory of physical performance, with a peak at around 30 years of age, followed by a decline that is initially slow but aggravates with age, appears to be universal for humans and suggests that the trajectory is constitutive and not acquired. Furthermore, no data are available to help us understand the extent to which the inter-individual variation in the progression (rate of decline) and extent of sarcopenia has a background in genetic variation. In general, it is estimated that heredity accounts for only 25% of the variability in human lifespan (see above). Data on muscle mass and strength show a greater influence of heredity, estimated at ≥50% [122,123,124]. However, cross-generational studies on the relationship between genetic variations and the development and progression of sarcopenia are lacking but would be extremely valuable to rationally design future interventions at the individual level.
To make further progress in our knowledge of the causes of sarcopenia, we need longitudinal observations that identify the inflexion point at which MU number and the speed and force of MU contraction begin to decline, as well as the events in the nervous system, myofibers, and muscle skeleton (ECM) that precede this moment. Therefore, we should not focus on those who already have manifest or incipient sarcopenia, but on adults who are about to pass the peak of their physical performance. In the later stages of sarcopenia progression, it is difficult to distinguish cause from consequence. As has already been emphasised, myofibers and their innervation depend on a well-maintained ECM to preserve their adaptive and regenerative capacity. The ECM has not received the same attention as myofibers, myofiber innervation and resident myocyte precursors, i.e. SCs. Simultaneous consideration of the ECM in future studies on sarcopenia is urgently needed. Similar mechanisms may also be active in the motor neurons and myofibers as well as in the cells of the muscle scaffold. The timing of events and rate of progression between the different cell and tissue types may be due to differences in their susceptibility rather than differences in mechanisms. A common mechanism is suggested by the fact that the universal gold standard for delaying the onset of ageing in all physiological systems of an organism is caloric (dietary) restriction (CR; [108]). As discussed elsewhere [109], CR is not a realistic intervention in humans, but it may help us find molecules that act as CR mimetics.
Author Contributions
Brun Ulfhake made the illustrations and wrote the first draft. Both authors read and contributed to the final manuscript.
Funding
The study was funded by Karolinska Institutet, Sweden, and the National research council (VR) grant No 2020-02009-3. This review was made independently of grant providing bodies which did not partake in decision to publish, or preparation of the manuscript..
Competing Interests
The author declares no conflict of interest.
References
- Gustafsson T, Ulfhake B. Sarcopenia: What Is the Origin of This Aging-Induced Disorder? Front Genet. 2021;12:688526. Epub 20210702. PubMed PMID: 34276788; PubMed Central PMCID: PMCPMC8285098. [CrossRef]
- Wijsman CA, Rozing MP, Streefland TC, le Cessie S, Mooijaart SP, Slagboom PE, et al. Familial longevity is marked by enhanced insulin sensitivity. Aging Cell. 2011;10(1):114-21. Epub 2010/11/13. PubMed PMID: 21070591. [CrossRef]
- Kaplanis J, Gordon A, Shor T, Weissbrod O, Geiger D, Wahl M, et al. Quantitative analysis of population-scale family trees with millions of relatives. Science. 2018;360(6385):171-5. Epub 2018/03/03. PubMed PMID: 29496957; PubMed Central PMCID: PMCPMC6593158. [CrossRef]
- Gustafsson T, Ulfhake B. Sarcopenia: What Is the Origin of This Aging-Induced Disorder? Frontiers in Genetics. 2021;12(914). [CrossRef]
- Etienne J, Liu C, Skinner CM, Conboy MJ, Conboy IM. Skeletal muscle as an experimental model of choice to study tissue aging and rejuvenation. Skelet Muscle. 2020;10(1):4. Epub 20200207. PubMed PMID: 32033591; PubMed Central PMCID: PMCPMC7007696. [CrossRef]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194-217. Epub 2013/06/12. PubMed PMID: 23746838; PubMed Central PMCID: PMCPMC3836174. [CrossRef]
- Altun M, Grönholdt-Klein, M., Wang, L. and Ulfhake, B. Cellular Degradation Machineries in Age-Related Loss of Muscle Mass (Sarcopenia). 2012. In: Senescence [Internet]. InTech; [269-86]. Available from: http://www.intechopen.com/articles/show/title/cellular-degradation-machineries-in-age-related-loss-of-muscle-mass-sarcopenia.
- Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, et al. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem. 2010;285(51):39597-608. Epub 2010/10/14. [CrossRef]
- M110.129718 [pii]. PubMed PMID: 20940294; PubMed Central PMCID: PMC3000941.
- Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing. 2010;39(4):412-23. Epub 2010/04/16. [CrossRef]
- 10.1093/ageing/afq034. PubMed PMID: 20392703; PubMed Central PMCID: PMC2886201.
- Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, et al. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing. 2019;48(1):16-31. Epub 2018/10/13. PubMed PMID: 30312372; PubMed Central PMCID: PMCPMC6322506. [CrossRef]
- Chen LK, Woo J, Assantachai P, Auyeung TW, Chou MY, Iijima K, et al. Asian Working Group for Sarcopenia: 2019 Consensus Update on Sarcopenia Diagnosis and Treatment. J Am Med Dir Assoc. 2020;21(3):300-7.e2. Epub 20200204. PubMed PMID: 32033882. [CrossRef]
- Studenski SA, Peters KW, Alley DE, Cawthon PM, McLean RR, Harris TB, et al. The FNIH Sarcopenia Project: Rationale, Study Description, Conference Recommendations, and Final Estimates. The Journals of Gerontology: Series A. 2014;69(5):547-58. [CrossRef]
- A LIFECOURSE APPROACH TO HEALTH. WHO/NMH/HPS/00.2.
- Cao L, Morley JE. Sarcopenia Is Recognized as an Independent Condition by an International Classification of Disease, Tenth Revision, Clinical Modification (ICD-10-CM) Code. J Am Med Dir Assoc. 2016;17(8):675-7. PubMed PMID: 27470918. [CrossRef]
- Trappe S. Master athletes. Int J Sport Nutr Exerc Metab. 2001;11 Suppl:S196-207. PubMed PMID: 11915921.
- Ganse B, Kleerekoper A, Knobe M, Hildebrand F, Degens H. Longitudinal trends in master track and field performance throughout the aging process: 83,209 results from Sweden in 16 athletics disciplines. Geroscience. 2020;42(6):1609-20. Epub 2020/10/14. PubMed PMID: 33048301. [CrossRef]
- Baker AB, Tang YQ. Aging performance for masters records in athletics, swimming, rowing, cycling, triathlon, and weightlifting. Exp Aging Res. 2010;36(4):453-77. PubMed PMID: 20845122. [CrossRef]
- Lazarus NR, Harridge SDR. Declining performance of master athletes: silhouettes of the trajectory of healthy human ageing? J Physiol. 2017;595(9):2941-8. Epub 20170118. PubMed PMID: 27808406; PubMed Central PMCID: PMCPMC5407960. [CrossRef]
- Gava P, Kern H, Carraro U. Age-associated power decline from running, jumping, and throwing male masters world records. Exp Aging Res. 2015;41(2):115-35. PubMed PMID: 25724012. [CrossRef]
- Westerståhl M, Jansson E, Barnekow-Bergkvist M, Aasa U. Longitudinal changes in physical capacity from adolescence to middle age in men and women. Sci Rep. 2018;8(1):14767. Epub 20181003. PubMed PMID: 30283061; PubMed Central PMCID: PMCPMC6170499. [CrossRef]
- GrönholdtKlein M, Gorzi A, Wang L, Edström E, Rullman E, Altun M, et al. Emergence and Progression of Behavioral Motor Deficits and Skeletal Muscle Atrophy across the Adult Lifespan of the Rat. Biology (Basel). 2023;12(9). Epub 20230828. PubMed PMID: 37759577; PubMed Central PMCID: PMCPMC10526071. [CrossRef]
- Larsson L, Degens H, Li M, Salviati L, Lee YI, Thompson W, et al. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol Rev. 2019;99(1):427-511. Epub 2018/11/15. PubMed PMID: 30427277; PubMed Central PMCID: PMCPMC6442923 drugs are held by Mayo Clinic. This work has been revised by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic conflict of interest policies. No other conflicts of interest, financial or otherwise, are declared by the authors. [CrossRef]
- Hepple RT, Rice CL. Innervation and neuromuscular control in ageing skeletal muscle. J Physiol. 2016;594(8):1965-78. PubMed PMID: 26437581; PubMed Central PMCID: PMC4933121. [CrossRef]
- Tintignac LA, Brenner HR, Ruegg MA. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol Rev. 2015;95(3):809-52. PubMed PMID: 26109340. [CrossRef]
- Willadt S, Nash M, Slater C. Age-related changes in the structure and function of mammalian neuromuscular junctions. Ann N Y Acad Sci. 2018;1412(1):41-53. Epub 2018/01/02. PubMed PMID: 29291259. [CrossRef]
- Gutmann E. Age changes in the neuromuscular system [by] E. Gutmann and V. Hanzlikova. Hanzlikova V, editor. Bristol: Scientechnica; 1972.
- Edström E, Altun M, Bergman E, Johnson H, Kullberg S, Ramírez-León V, et al. Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol Behav. 2007;92(1-2):129-35. Epub 2007/06/26. PubMed PMID: 17585972. [CrossRef]
- Rowan SL, Rygiel K, Purves-Smith FM, Solbak NM, Turnbull DM, Hepple RT. Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PLoS One. 2012;7(1):e29082. Epub 20120103. PubMed PMID: 22235261; PubMed Central PMCID: PMCPMC3250397. [CrossRef]
- Cowen T, Ulfhake B, King RHM. Aging in the Peripheral Neurvous System. Peripheral Neuropathy. 1. Philadelphia: Elsevier Saunders; 2005. p. 483-507.
- Johnson H, Ulfhake B, Dagerlind A, Bennett GW, Fone KC, Hökfelt T. The serotoninergic bulbospinal system and brainstem-spinal cord content of serotonin-, TRH-, and substance P-like immunoreactivity in the aged rat with special reference to the spinal cord motor nucleus. Synapse. 1993;15(1):63-89. Epub 1993/09/01. PubMed PMID: 7508641. [CrossRef]
- Kudina LP, Andreeva RE. Repetitive doublet firing in human motoneurons: evidence for interaction between common synaptic drive and plateau potential in natural motor control. J Neurophysiol. 2019;122(1):424-34. Epub 20190605. PubMed PMID: 31166815. [CrossRef]
- Fuglevand AJ, Dutoit AP, Johns RK, Keen DA. Evaluation of plateau-potential-mediated 'warm up' in human motor units. J Physiol. 2006;571(Pt 3):683-93. Epub 20060119. PubMed PMID: 16423860; PubMed Central PMCID: PMCPMC1805803. [CrossRef]
- Alaburda A, Perrier JF, Hounsgaard J. Mechanisms causing plateau potentials in spinal motoneurones. Adv Exp Med Biol. 2002;508:219-26. PubMed PMID: 12171115. [CrossRef]
- Ramírez-León V, Kullberg S, Hjelle OP, Ottersen OP, Ulfhake B. Increased glutathione levels in neurochemically identified fibre systems in the aged rat lumbar motor nuclei. Eur J Neurosci. 1999;11(8):2935-48. PubMed PMID: 10457189. [CrossRef]
- Kullberg S, Ramírez-León V, Johnson H, Ulfhake B. Decreased axosomatic input to motoneurons and astrogliosis in the spinal cord of aged rats. J Gerontol A Biol Sci Med Sci. 1998;53(5):B369-79. PubMed PMID: 9754135. [CrossRef]
- Orssatto LBR, Borg DN, Pendrith L, Blazevich AJ, Shield AJ, Trajano GS. Do motoneuron discharge rates slow with aging? A systematic review and meta-analysis. Mech Ageing Dev. 2022;203:111647. Epub 20220223. PubMed PMID: 35218849. [CrossRef]
- Orssatto LBR, Rodrigues P, Mackay K, Blazevich AJ, Borg DN, Souza TRd, et al. Intrinsic motor neuron excitability is increased after resistance training in older adults. Journal of Neurophysiology. 2023;129(3):635-50. PubMed PMID: 36752407. [CrossRef]
- Gutman B, Hanzlikova V. Age changes in the neuromuscular system. Scientechnica Ltd, Bristol. 1972:1-20.
- Power GA, Allen MD, Gilmore KJ, Stashuk DW, Doherty TJ, Hepple RT, et al. Motor unit number and transmission stability in octogenarian world class athletes: Can age-related deficits be outrun? J Appl Physiol (1985). 2016;121(4):1013-20. Epub 2016/11/01. PubMed PMID: 27013605; PubMed Central PMCID: PMCPMC5142311. [CrossRef]
- Korhonen MT, Cristea A, Alén M, Häkkinen K, Sipilä S, Mero A, et al. Aging, muscle fiber type, and contractile function in sprint-trained athletes. J Appl Physiol (1985). 2006;101(3):906-17. Epub 2006/05/13. PubMed PMID: 16690791. [CrossRef]
- Drey M, Sieber CC, Degens H, McPhee J, Korhonen MT, Müller K, et al. Relation between muscle mass, motor units and type of training in master athletes. Clin Physiol Funct Imaging. 2016;36(1):70-6. Epub 2014/10/28. PubMed PMID: 25345553. [CrossRef]
- Piasecki M, Ireland A, Coulson J, Stashuk DW, Hamilton-Wright A, Swiecicka A, et al. Motor unit number estimates and neuromuscular transmission in the tibialis anterior of master athletes: evidence that athletic older people are not spared from age-related motor unit remodeling. Physiol Rep. 2016;4(19). Epub 2016/10/04. PubMed PMID: 27694526; PubMed Central PMCID: PMCPMC5064139. [CrossRef]
- Chakravarty EF, Hubert HB, Lingala VB, Fries JF. Reduced Disability and Mortality Among Aging Runners: A 21-Year Longitudinal Study. Archives of Internal Medicine. 2008;168(15):1638-46. [CrossRef]
- Franco I, Fernandez-Gonzalo R, Vrtacnik P, Lundberg TR, Eriksson M, Gustafsson T. Healthy skeletal muscle aging: The role of satellite cells, somatic mutations and exercise. Int Rev Cell Mol Biol. 2019;346:157-200. Epub 2019/05/28. PubMed PMID: 31122394. [CrossRef]
- Gopinath SD, Rando TA. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 2008;7(4):590-8. Epub 20080628. PubMed PMID: 18462272. [CrossRef]
- Joseph GA, Wang SX, Jacobs CE, Zhou W, Kimble GC, Tse HW, et al. Partial Inhibition of mTORC1 in Aged Rats Counteracts the Decline in Muscle Mass and Reverses Molecular Signaling Associated with Sarcopenia. Mol Cell Biol. 2019;39(19). Epub 20190911. PubMed PMID: 31308131; PubMed Central PMCID: PMCPMC6751631. [CrossRef]
- Fuqua JD, Lawrence MM, Hettinger ZR, Borowik AK, Brecheen PL, Szczygiel MM, et al. Impaired proteostatic mechanisms other than decreased protein synthesis limit old skeletal muscle recovery after disuse atrophy. J Cachexia Sarcopenia Muscle. 2023;14(5):2076-89. Epub 20230714. PubMed PMID: 37448295; PubMed Central PMCID: PMCPMC10570113. [CrossRef]
- Edstrom E, Altun M, Hagglund M, Ulfhake B. Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61(7):663-74. PubMed PMID: 16870627.
- Magliulo L, Bondi D, Pini N, Marramiero L, Di Filippo ES. The wonder exerkines-novel insights: a critical state-of-the-art review. Mol Cell Biochem. 2022;477(1):105-13. Epub 20210923. PubMed PMID: 34554363; PubMed Central PMCID: PMCPMC8755664. [CrossRef]
- Cornish SM, Bugera EM, Duhamel TA, Peeler JD, Anderson JE. A focused review of myokines as a potential contributor to muscle hypertrophy from resistance-based exercise. European Journal of Applied Physiology. 2020;120(5):941-59. [CrossRef]
- Mancinelli R, Checcaglini F, Coscia F, Gigliotti P, Fulle S, Fanò-Illic G. Biological Aspects of Selected Myokines in Skeletal Muscle: Focus on Aging. Int J Mol Sci. 2021;22(16). Epub 20210807. PubMed PMID: 34445222; PubMed Central PMCID: PMCPMC8395159. [CrossRef]
- Kwon JH, Moon KM, Min KW. Exercise-Induced Myokines can Explain the Importance of Physical Activity in the Elderly: An Overview. Healthcare (Basel). 2020;8(4). Epub 20201001. PubMed PMID: 33019579; PubMed Central PMCID: PMCPMC7712334. [CrossRef]
- Son JS, Chae SA, Testroet ED, Du M, Jun HP. Exercise-induced myokines: a brief review of controversial issues of this decade. Expert Rev Endocrinol Metab. 2018;13(1):51-8. Epub 20171219. PubMed PMID: 30063442. [CrossRef]
- Barbalho SM, Prado Neto EV, De Alvares Goulart R, Bechara MD, Baisi Chagas EF, Audi M, et al. Myokines: a descriptive review. J Sports Med Phys Fitness. 2020;60(12):1583-90. Epub 20200623. PubMed PMID: 32586076. [CrossRef]
- Kirk B, Feehan J, Lombardi G, Duque G. Muscle, Bone, and Fat Crosstalk: the Biological Role of Myokines, Osteokines, and Adipokines. Curr Osteoporos Rep. 2020;18(4):388-400. PubMed PMID: 32529456. [CrossRef]
- Plikus MV, Wang X, Sinha S, Forte E, Thompson SM, Herzog EL, et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. 2021;184(15):3852-72. PubMed PMID: 34297930; PubMed Central PMCID: PMCPMC8566693. [CrossRef]
- Chapman MA, Meza R, Lieber RL. Skeletal muscle fibroblasts in health and disease. Differentiation. 2016;92(3):108-15. Epub 20160606. PubMed PMID: 27282924; PubMed Central PMCID: PMCPMC5079803. [CrossRef]
- Contreras O, Rossi FMV, Theret M. Origins, potency, and heterogeneity of skeletal muscle fibro-adipogenic progenitors-time for new definitions. Skelet Muscle. 2021;11(1):16. Epub 20210701. PubMed PMID: 34210364; PubMed Central PMCID: PMCPMC8247239. [CrossRef]
- Esteves de Lima J, Relaix F. Master regulators of skeletal muscle lineage development and pluripotent stem cells differentiation. Cell Regen. 2021;10(1):31. Epub 20211001. PubMed PMID: 34595600; PubMed Central PMCID: PMCPMC8484369. [CrossRef]
- McKee TJ, Perlman G, Morris M, Komarova SV. Extracellular matrix composition of connective tissues: a systematic review and meta-analysis. Sci Rep. 2019;9(1):10542. Epub 20190722. PubMed PMID: 31332239; PubMed Central PMCID: PMCPMC6646303. [CrossRef]
- Birch HL. Extracellular Matrix and Ageing. Subcell Biochem. 2018;90:169-90. PubMed PMID: 30779010. [CrossRef]
- Csapo R, Gumpenberger M, Wessner B. Skeletal Muscle Extracellular Matrix - What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front Physiol. 2020;11:253. Epub 20200319. PubMed PMID: 32265741; PubMed Central PMCID: PMCPMC7096581. [CrossRef]
- Karamanos NK, Theocharis AD, Piperigkou Z, Manou D, Passi A, Skandalis SS, et al. A guide to the composition and functions of the extracellular matrix. Febs j. 2021;288(24):6850-912. Epub 20210323. PubMed PMID: 33605520. [CrossRef]
- Kirkendall DT, Garrett WE. Function and biomechanics of tendons. Scand J Med Sci Sports. 1997;7(2):62-6. PubMed PMID: 9211605. [CrossRef]
- Kannus P. Structure of the tendon connective tissue. Scand J Med Sci Sports. 2000;10(6):312-20. PubMed PMID: 11085557. [CrossRef]
- Wang X, Sathe AA, Smith GR, Ruf-Zamojski F, Nair V, Lavine KJ, et al. Heterogeneous origins and functions of mouse skeletal muscle-resident macrophages. Proc Natl Acad Sci U S A. 2020;117(34):20729-40. Epub 20200813. PubMed PMID: 32796104; PubMed Central PMCID: PMCPMC7456122. [CrossRef]
- Buvinic S, Balanta-Melo J, Kupczik K, Vásquez W, Beato C, Toro-Ibacache V. Muscle-Bone Crosstalk in the Masticatory System: From Biomechanical to Molecular Interactions. Front Endocrinol (Lausanne). 2020;11:606947. Epub 20210301. PubMed PMID: 33732211; PubMed Central PMCID: PMCPMC7959242. [CrossRef]
- Chapman MA, Mukund K, Subramaniam S, Brenner D, Lieber RL. Three distinct cell populations express extracellular matrix proteins and increase in number during skeletal muscle fibrosis. Am J Physiol Cell Physiol. 2017;312(2):C131-c43. Epub 20161123. PubMed PMID: 27881411; PubMed Central PMCID: PMCPMC5336596. [CrossRef]
- Gumpenberger M, Wessner B, Graf A, Narici MV, Fink C, Braun S, et al. Remodeling the Skeletal Muscle Extracellular Matrix in Older Age-Effects of Acute Exercise Stimuli on Gene Expression. Int J Mol Sci. 2020;21(19). Epub 20200925. PubMed PMID: 32992998; PubMed Central PMCID: PMCPMC7583913. [CrossRef]
- Wan M, Gray-Gaillard EF, Elisseeff JH. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 2021;9(1):41. Epub 20210910. PubMed PMID: 34508069; PubMed Central PMCID: PMCPMC8433460. [CrossRef]
- Hayflick L, Moorhead PS. Exp Cell Res. 1961;25. [CrossRef]
- Moiseeva V, Cisneros A, Sica V, Deryagin O, Lai Y, Jung S, et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature. 2023;613(7942):169-78. Epub 20221221. PubMed PMID: 36544018; PubMed Central PMCID: PMCPMC9812788. [CrossRef]
- Zhu Y. Aging Cell. 2015;14. [CrossRef]
- Arora S. Med. 2021;2. [CrossRef]
- Twomey LT, Taylor JR. Old age and physical capacity: use it or lose it. Aust J Physiother. 1984;30(4):115-20. PubMed PMID: 25026448. [CrossRef]
- Churchward-Venne TA, Tieland M, Verdijk LB, Leenders M, Dirks ML, de Groot LC, et al. There Are No Nonresponders to Resistance-Type Exercise Training in Older Men and Women. J Am Med Dir Assoc. 2015;16(5):400-11. Epub 20150221. PubMed PMID: 25717010. [CrossRef]
- Phu S, Boersma D, Duque G. Exercise and Sarcopenia. J Clin Densitom. 2015;18(4):488-92. Epub 20150610. PubMed PMID: 26071171. [CrossRef]
- McKendry J, Breen L, Shad BJ, Greig CA. Muscle morphology and performance in master athletes: A systematic review and meta-analyses. Ageing Res Rev. 2018;45:62-82. Epub 20180430. PubMed PMID: 29715523. [CrossRef]
- Kirkendall DT, Garrett WE, Jr. The effects of aging and training on skeletal muscle. Am J Sports Med. 1998;26(4):598-602. PubMed PMID: 9689386. [CrossRef]
- Deschenes MR, Kraemer WJ. Performance and physiologic adaptations to resistance training. Am J Phys Med Rehabil. 2002;81(11 Suppl):S3-16. PubMed PMID: 12409807. [CrossRef]
- Polito MD, Papst RR, Farinatti P. Moderators of strength gains and hypertrophy in resistance training: A systematic review and meta-analysis. J Sports Sci. 2021;39(19):2189-98. Epub 20210512. PubMed PMID: 33977848. [CrossRef]
- Borde R, Hortobágyi T, Granacher U. Dose-Response Relationships of Resistance Training in Healthy Old Adults: A Systematic Review and Meta-Analysis. Sports Med. 2015;45(12):1693-720. PubMed PMID: 26420238; PubMed Central PMCID: PMCPMC4656698. [CrossRef]
- Wall BT, Dirks ML, van Loon LJ. Skeletal muscle atrophy during short-term disuse: implications for age-related sarcopenia. Ageing Res Rev. 2013;12(4):898-906. Epub 2013/08/21. PubMed PMID: 23948422. [CrossRef]
- McKendry J, Shad BJ, Smeuninx B, Oikawa SY, Wallis G, Greig C, et al. Comparable Rates of Integrated Myofibrillar Protein Synthesis Between Endurance-Trained Master Athletes and Untrained Older Individuals. Front Physiol. 2019;10:1084. Epub 20190830. PubMed PMID: 31543824; PubMed Central PMCID: PMCPMC6728413. [CrossRef]
- Grosicki GJ, Gries KJ, Minchev K, Raue U, Chambers TL, Begue G, et al. Single muscle fibre contractile characteristics with lifelong endurance exercise. J Physiol. 2021;599(14):3549-65. Epub 20210615. PubMed PMID: 34036579. [CrossRef]
- Tseng BS, Marsh DR, Hamilton MT, Booth FW. Strength and aerobic training attenuate muscle wasting and improve resistance to the development of disability with aging. J Gerontol A Biol Sci Med Sci. 1995;50 Spec No:113-9. PubMed PMID: 7493203.
- Gu Q, Wang B, Zhang XF, Ma YP, Liu JD, Wang XZ. Chronic aerobic exercise training attenuates aortic stiffening and endothelial dysfunction through preserving aortic mitochondrial function in aged rats. Exp Gerontol. 2014;56:37-44. Epub 20140305. PubMed PMID: 24607516. [CrossRef]
- Mikkelsen UR, Couppé C, Karlsen A, Grosset JF, Schjerling P, Mackey AL, et al. Life-long endurance exercise in humans: circulating levels of inflammatory markers and leg muscle size. Mech Ageing Dev. 2013;134(11-12):531-40. Epub 20131125. PubMed PMID: 24287006. [CrossRef]
- Fleenor BS. Large elastic artery stiffness with aging: novel translational mechanisms and interventions. Aging Dis. 2013;4(2):76-83. Epub 20121211. PubMed PMID: 23696949; PubMed Central PMCID: PMCPMC3659253.
- Marcell TJ, Hawkins SA, Wiswell RA. Leg strength declines with advancing age despite habitual endurance exercise in active older adults. J Strength Cond Res. 2014;28(2):504-13. PubMed PMID: 24263662. [CrossRef]
- Straight CR, Fedewa MV, Toth MJ, Miller MS. Improvements in skeletal muscle fiber size with resistance training are age-dependent in older adults: a systematic review and meta-analysis. J Appl Physiol (1985). 2020;129(2):392-403. Epub 20200723. PubMed PMID: 32702280; PubMed Central PMCID: PMCPMC7473942. [CrossRef]
- Alian S, Baker RG, Wood S. Rural casualty crashes on the Kings Highway: A new approach for road safety studies. Accid Anal Prev. 2016;95(Pt A):8-19. PubMed PMID: 27372441. [CrossRef]
- Lundberg TR, Gustafsson T. Fibre hypertrophy, satellite cell and myonuclear adaptations to resistance training: Have very old individuals reached the ceiling for muscle fibre plasticity? Acta Physiol (Oxf). 2019;227(1):e13287. Epub 20190513. PubMed PMID: 31009166. [CrossRef]
- Ferketich AK, Kirby TE, Alway SE. Cardiovascular and muscular adaptations to combined endurance and strength training in elderly women. Acta Physiol Scand. 1998;164(3):259-67. PubMed PMID: 9853013. [CrossRef]
- Irving BA, Lanza IR, Henderson GC, Rao RR, Spiegelman BM, Nair KS. Combined training enhances skeletal muscle mitochondrial oxidative capacity independent of age. J Clin Endocrinol Metab. 2015;100(4):1654-63. Epub 20150119. PubMed PMID: 25599385; PubMed Central PMCID: PMCPMC4399307. [CrossRef]
- Lundberg TR, Fernandez-Gonzalo R, Gustafsson T, Tesch PA. Aerobic exercise does not compromise muscle hypertrophy response to short-term resistance training. J Appl Physiol (1985). 2013;114(1):81-9. Epub 2012/10/30. PubMed PMID: 23104700. [CrossRef]
- Cadore EL, Izquierdo M. How to simultaneously optimize muscle strength, power, functional capacity, and cardiovascular gains in the elderly: an update. Age (Dordr). 2013;35(6):2329-44. Epub 20130104. PubMed PMID: 23288690; PubMed Central PMCID: PMCPMC3825007. [CrossRef]
- Cadore EL, Menger E, Teodoro JL, da Silva LXN, Boeno FP, Umpierre D, et al. Functional and physiological adaptations following concurrent training using sets with and without concentric failure in elderly men: A randomized clinical trial. Exp Gerontol. 2018;110:182-90. Epub 20180613. PubMed PMID: 29908345. [CrossRef]
- Leuchtmann AB, Mueller SM, Aguayo D, Petersen JA, Ligon-Auer M, Flück M, et al. Resistance training preserves high-intensity interval training induced improvements in skeletal muscle capillarization of healthy old men: a randomized controlled trial. Sci Rep. 2020;10(1):6578. Epub 20200420. PubMed PMID: 32313031; PubMed Central PMCID: PMCPMC7171189. [CrossRef]
- Wilhelm EN, Rech A, Minozzo F, Botton CE, Radaelli R, Teixeira BC, et al. Concurrent strength and endurance training exercise sequence does not affect neuromuscular adaptations in older men. Exp Gerontol. 2014;60:207-14. Epub 20141113. PubMed PMID: 25449853. [CrossRef]
- Moghadam BH, Bagheri R, Ashtary-Larky D, Tinsley GM, Eskandari M, Wong A, et al. The Effects of Concurrent Training Order on Satellite Cell-Related Markers, Body Composition, Muscular and Cardiorespiratory Fitness in Older Men with Sarcopenia. J Nutr Health Aging. 2020;24(7):796-804. PubMed PMID: 32744578. [CrossRef]
- Zhao M, Veeranki SP, Magnussen CG, Xi B. Recommended physical activity and all cause and cause specific mortality in US adults: prospective cohort study. Bmj. 2020;370:m2031. Epub 20200701. PubMed PMID: 32611588; PubMed Central PMCID: PMCPMC7328465. [CrossRef]
- Stanga S, Boido M, Kienlen-Campard P. How to Build and to Protect the Neuromuscular Junction: The Role of the Glial Cell Line-Derived Neurotrophic Factor. Int J Mol Sci. 2020;22(1). Epub 2020/12/31. PubMed PMID: 33374485; PubMed Central PMCID: PMCPMC7794999. [CrossRef]
- Besse-Patin A, Montastier E, Vinel C, Castan-Laurell I, Louche K, Dray C, et al. Effect of endurance training on skeletal muscle myokine expression in obese men: identification of apelin as a novel myokine. Int J Obes (Lond). 2014;38(5):707-13. Epub 20130827. PubMed PMID: 23979219. [CrossRef]
- Zofkie W, Southard SM, Braun T, Lepper C. Fibroblast growth factor 6 regulates sizing of the muscle stem cell pool. Stem Cell Reports. 2021;16(12):2913-27. Epub 20211104. PubMed PMID: 34739848; PubMed Central PMCID: PMCPMC8693628. [CrossRef]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med. 2014;20(3):255-64. Epub 20140216. PubMed PMID: 24531378; PubMed Central PMCID: PMCPMC3949152. [CrossRef]
- Dorsey SG, Lovering RM, Renn CL, Leitch CC, Liu X, Tallon LJ, et al. Genetic deletion of trkB.T1 increases neuromuscular function. Am J Physiol Cell Physiol. 2012;302(1):C141-53. Epub 20111005. PubMed PMID: 21865582; PubMed Central PMCID: PMCPMC3328911. [CrossRef]
- Gyorkos AM, Spitsbergen JM. GDNF content and NMJ morphology are altered in recruited muscles following high-speed and resistance wheel training. Physiol Rep. 2014;2(2):e00235. Epub 20140225. PubMed PMID: 24744904; PubMed Central PMCID: PMCPMC3966253. [CrossRef]
- Wan M, Gray-Gaillard EF, Elisseeff JH. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Research. 2021;9(1):41. [CrossRef]
- Wong, Carissa (2024-05-15). "How to kill the 'zombie' cells that make you age". Nature. 629 (8012): 518–520.
- Becker C, Lord SR, Studenski SA, Warden SJ, Fielding RA, Recknor CP, et al. Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015;3(12):948-57. Epub 20151027. PubMed PMID: 26516121. [CrossRef]
- Leuchtmann AB, Furrer R, Steurer SA, Schneider-Heieck K, Karrer-Cardel B, Sagot Y, et al. Interleukin-6 potentiates endurance training adaptation and improves functional capacity in old mice. J Cachexia Sarcopenia Muscle. 2022. Epub 20220222. PubMed PMID: 35191221. [CrossRef]
- Chowdhury S, Schulz L, Palmisano B, Singh P, Berger JM, Yadav VK, et al. Muscle-derived interleukin 6 increases exercise capacity by signaling in osteoblasts. J Clin Invest. 2020;130(6):2888-902. PubMed PMID: 32078586; PubMed Central PMCID: PMCPMC7260002. [CrossRef]
- Vinel C, Lukjanenko L, Batut A, Deleruyelle S, Pradere JP, Le Gonidec S, et al. The exerkine apelin reverses age-associated sarcopenia. Nat Med. 2018;24(9):1360-71. Epub 2018/08/01. PubMed PMID: 30061698. [CrossRef]
- Chen YY, Chiu YL, Kao TW, Peng TC, Yang HF, Chen WL. Cross-sectional associations among P3NP, HtrA, Hsp70, Apelin and sarcopenia in Taiwanese population. BMC Geriatr. 2021;21(1):192. Epub 20210320. PubMed PMID: 33743591; PubMed Central PMCID: PMCPMC7980650. [CrossRef]
- Castan-Laurell I, Dray C, Valet P. The therapeutic potentials of apelin in obesity-associated diseases. Mol Cell Endocrinol. 2021;529:111278. Epub 20210407. PubMed PMID: 33838166. [CrossRef]
- Saral S, Topçu A, Alkanat M, Mercantepe T, Akyıldız K, Yıldız L, et al. Apelin-13 activates the hippocampal BDNF/TrkB signaling pathway and suppresses neuroinflammation in male rats with cisplatin-induced cognitive dysfunction. Behav Brain Res. 2021;408:113290. Epub 20210415. PubMed PMID: 33845103. [CrossRef]
- Roh HT, Cho SY, So WY. A Cross-Sectional Study Evaluating the Effects of Resistance Exercise on Inflammation and Neurotrophic Factors in Elderly Women with Obesity. J Clin Med. 2020;9(3). Epub 20200320. PubMed PMID: 32244926; PubMed Central PMCID: PMCPMC7141497. [CrossRef]
- Mrówczyński W. Health Benefits of Endurance Training: Implications of the Brain-Derived Neurotrophic Factor-A Systematic Review. Neural Plast. 2019;2019:5413067. Epub 2019/07/26. PubMed PMID: 31341469; PubMed Central PMCID: PMCPMC6613032. [CrossRef]
- Carrero-Rojas G, Benítez-Temiño B, Pastor AM, Davis López de Carrizosa MA. Muscle Progenitors Derived from Extraocular Muscles Express Higher Levels of Neurotrophins and their Receptors than other Cranial and Limb Muscles. Cells. 2020;9(3):747. PubMed PMID:. [CrossRef]
- Petrocelli JJ, McKenzie AI, de Hart N, Reidy PT, Mahmassani ZS, Keeble AR, et al. Disuse-induced muscle fibrosis, cellular senescence, and senescence-associated secretory phenotype in older adults are alleviated during re-ambulation with metformin pre-treatment. Aging Cell. 2023;22(11):e13936. Epub 20230724. PubMed PMID: 37486024; PubMed Central PMCID: PMCPMC10652302. [CrossRef]
- Stewart CE, Rittweger J. Adaptive processes in skeletal muscle: molecular regulators and genetic influences. J Musculoskelet Neuronal Interact. 2006;6(1):73-86. PubMed PMID: 16675891.
- Seeman E, Hopper JL, Young NR, Formica C, Goss P, Tsalamandris C. Do genetic factors explain associations between muscle strength, lean mass, and bone density? A twin study. Am J Physiol. 1996;270(2 Pt 1):E320-7. PubMed PMID: 8779955. [CrossRef]
- Huygens W, Thomis MA, Peeters MW, Vlietinck RF, Beunen GP. Determinants and upper-limit heritabilities of skeletal muscle mass and strength. Can J Appl Physiol. 2004;29(2):186-200. PubMed PMID: 15064427. [CrossRef]
Figure 1.
Shows a schematic representation of the changes in normalized physical performance over the lifespan. The age at which peak performance is reached depends on the type of activity but is usually in the range of 25-35 years (dashed vertical line). Three curves are shown, representing very physically active individuals (green line), a person with 2-4 hours of leisure-time physical activity per week (blue line) and a person with a sedentary lifestyle (red line). What all three phenotypes have in common is that peak performance is reached at around 25-30 years of age and that the rate of loss is initially low, but accelerates later on. The preclinical phase continues when daily activities are not affected, while the need to moderate physical activities to cope with the loss of muscle mass and strength marks the entry into the clinical phase (the upper horizontal dashed line). As the process progresses, it eventually leads to disability (lower horizontal dashed line). Note that the range of phenotypes in the population is the product of genotype and lifestyle. The changes in skeletal muscle during these phases were visualized in HTx-stained sections of an animal model of human sarcopenia with normal histology during development and growth, essentially maintained myofiber size during the preclinical phase of sarcopenia, and muscle fiber atrophy and other abnormalities in the clinical phase of this disease (see text for more information).
Figure 1.
Shows a schematic representation of the changes in normalized physical performance over the lifespan. The age at which peak performance is reached depends on the type of activity but is usually in the range of 25-35 years (dashed vertical line). Three curves are shown, representing very physically active individuals (green line), a person with 2-4 hours of leisure-time physical activity per week (blue line) and a person with a sedentary lifestyle (red line). What all three phenotypes have in common is that peak performance is reached at around 25-30 years of age and that the rate of loss is initially low, but accelerates later on. The preclinical phase continues when daily activities are not affected, while the need to moderate physical activities to cope with the loss of muscle mass and strength marks the entry into the clinical phase (the upper horizontal dashed line). As the process progresses, it eventually leads to disability (lower horizontal dashed line). Note that the range of phenotypes in the population is the product of genotype and lifestyle. The changes in skeletal muscle during these phases were visualized in HTx-stained sections of an animal model of human sarcopenia with normal histology during development and growth, essentially maintained myofiber size during the preclinical phase of sarcopenia, and muscle fiber atrophy and other abnormalities in the clinical phase of this disease (see text for more information).
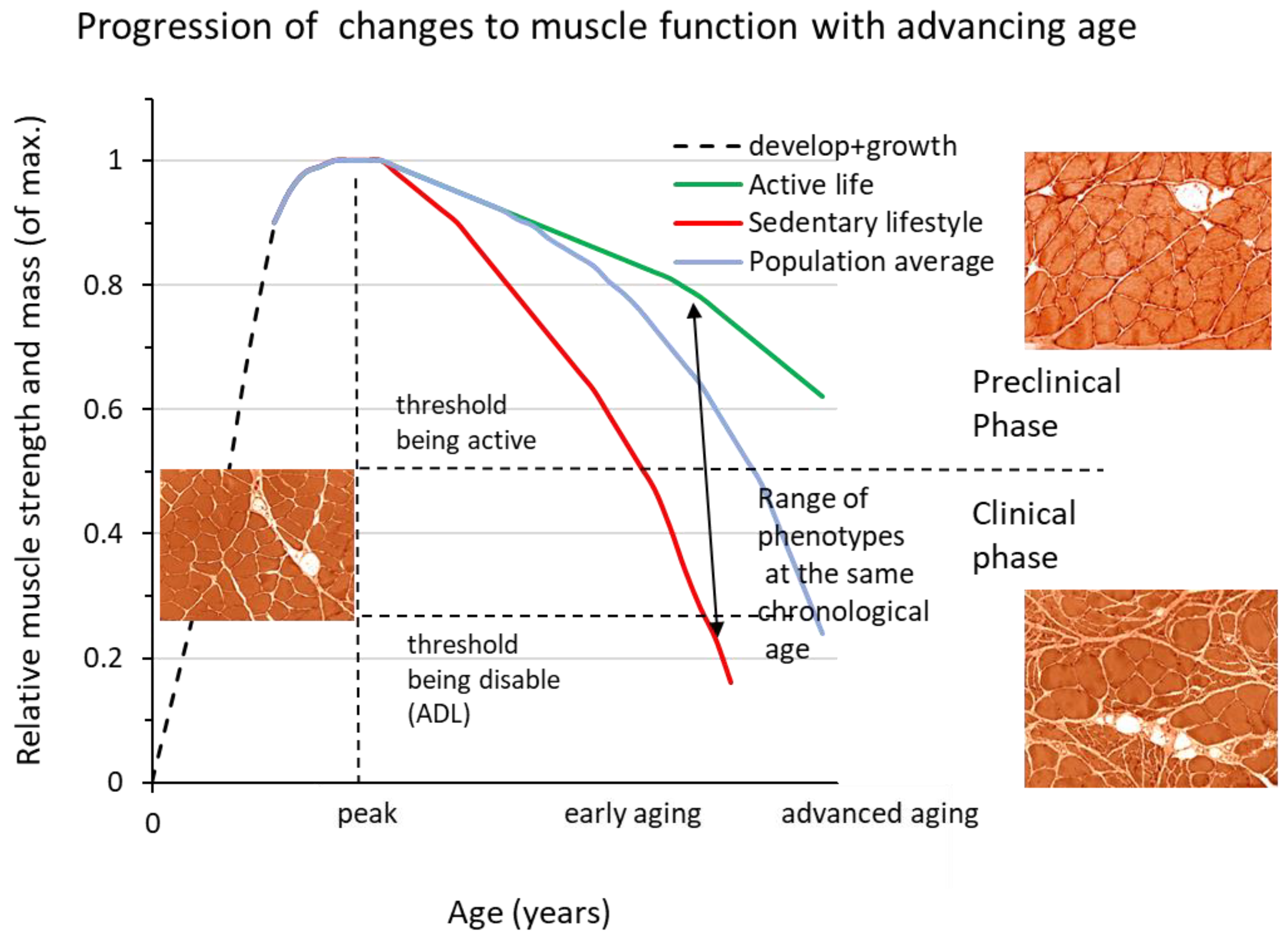
Figure 2.
In this schematic example, a fast myofiber expressing type II myosin is denervated due to axon atrophy. In the denervated state, fast (and slow) myofibers often express both type I and type II myosins, plus embryonic myosin as a sign of denervation. When the fast myofiber is reinnervated by a slow MN, it switches to type I myosin as the predominant form, while residual amounts of type II myosin can often be detected. However, the expression of embryonic myosin is suppressed. We refer to this phenotype as hybrid myofiber. During aging, the frequency of hybrid myofibers increases from <1% to several percent of the myofiber population.
Figure 2.
In this schematic example, a fast myofiber expressing type II myosin is denervated due to axon atrophy. In the denervated state, fast (and slow) myofibers often express both type I and type II myosins, plus embryonic myosin as a sign of denervation. When the fast myofiber is reinnervated by a slow MN, it switches to type I myosin as the predominant form, while residual amounts of type II myosin can often be detected. However, the expression of embryonic myosin is suppressed. We refer to this phenotype as hybrid myofiber. During aging, the frequency of hybrid myofibers increases from <1% to several percent of the myofiber population.
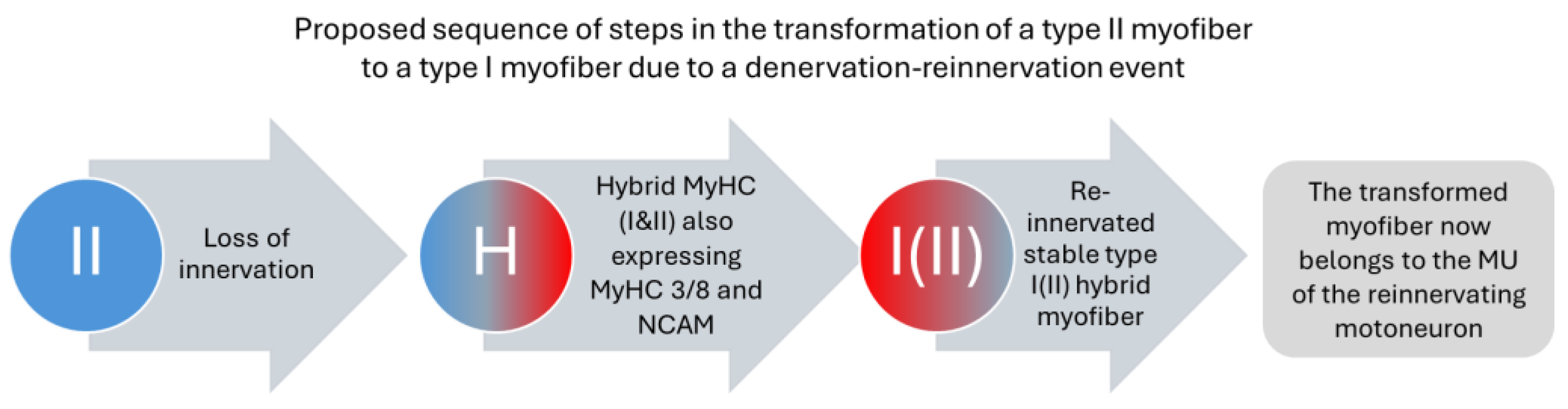
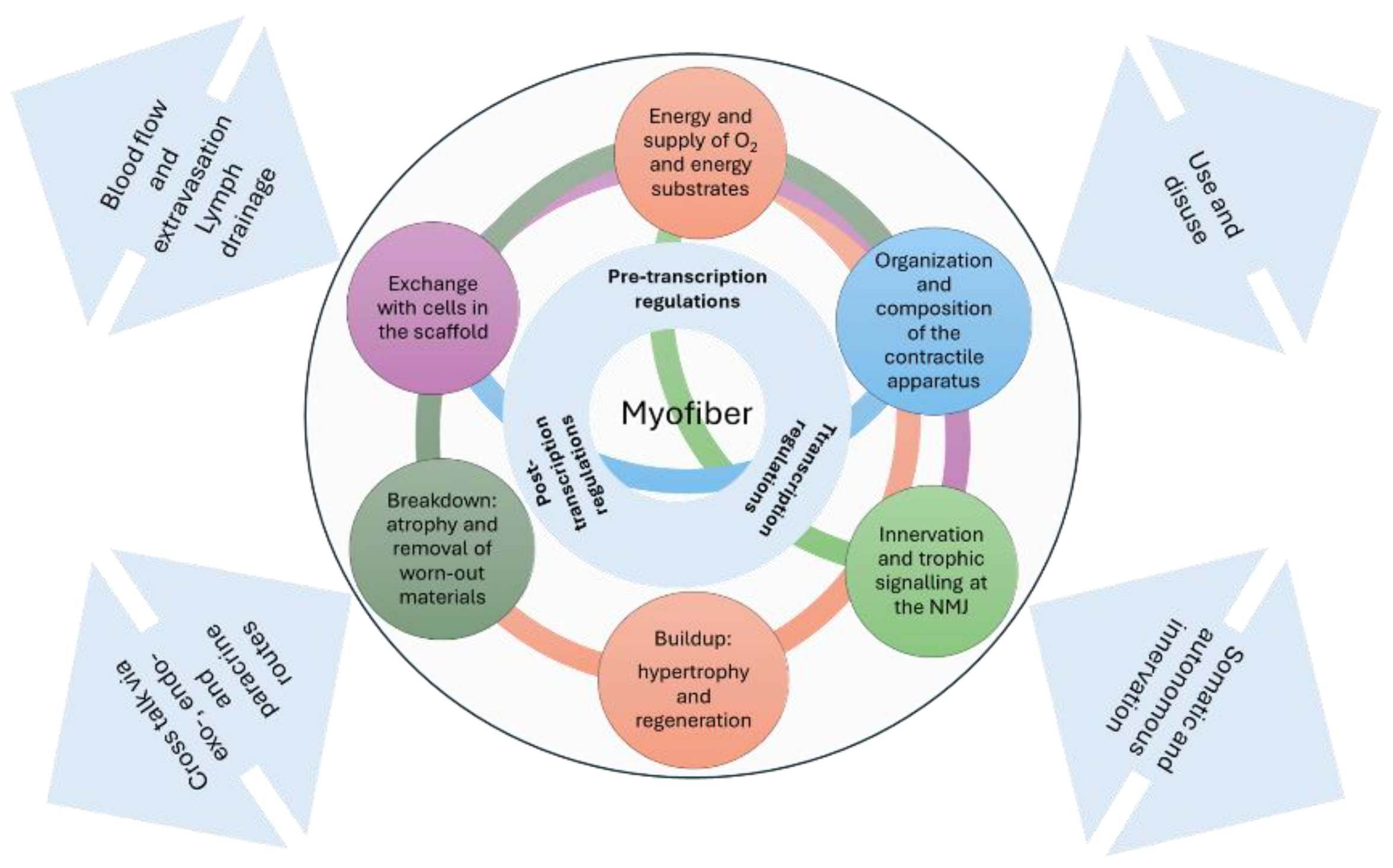
Figure 3.
Schematic representation of the myofiber's inherent tools that enable adaptive and regenerative responses. The interface with the tissues involved in balance and movement processes and the system level are indicated by bidirectional arrows.
Figure 3.
Schematic representation of the myofiber's inherent tools that enable adaptive and regenerative responses. The interface with the tissues involved in balance and movement processes and the system level are indicated by bidirectional arrows.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



