
Submitted:
12 September 2024
Posted:
13 September 2024
You are already at the latest version
Abstract
Arabidopsis and other model plants can help improve crops by providing a reference framework for assembling omics data from plants. Arabidopsis is extensively studied and has a well-curated and annotated genome, making it an excellent reference organism for comparative analysis. When there is no reference genome of the organism being investigated, using the genome of Arabidopsis may be an option thanks to the extensive knowledge we have about it. Crop improvement can help to increase crop yields, improve crop quality, and enhance resistance to pests, diseases, and environmental stresses. It can also help to reduce the use of pesticides and fertilizers, which can have negative impacts on the environment and human health. Therefore, crop improvement is essential to ensure food security and sustainability in the face of global challenges such as climate change, land degradation, and water scarcity. The assembly process of genomic, transcriptomic, and metabolomic data from plants using carefully selected information on Arabidopsis can lead to a deeper understanding of organisms and their potential for genetic improvement for crops. However, the successful integration of data requires expertise and experience to design more effective and precise crop improvement strategies.
Keywords:
Arabidopsis (Arabidopsis thaliana)
; Bioinformatics
; Genetic improvement
; Omics databases
; Crops
1. Introduction
Some of the issues facing humanity in the twenty-first century include the rising global food demand, climate change, and the rapid expansion of the world's population. Following the green revolution and its controversial effects on the use and implementation of chemical fertilizers and pesticides, which in some cases had detrimental effects on human and global health [1]. A significant portion of the environmental preservation challenges mostly relate to food production. One of the most promising approaches for improving plant health involves the use of biotechnological tools like biolistic or CRISPR-Cas9 and the in-depth study of genes and metabolic pathways [2]. These two biotechnological strategies together have produced significant plant breeding tools that could aid in increasing the production of food and protecting the environment.
This exchange of genes between species is a major challenge for plant breeding, but this section poses a scientific, moral, and ethical dilemma. The term "transgenic plant" has historically been used to refer to any plant that has undergone artificial genetic modification; however, this term is now beginning to be differentiated, depending mainly on the source of the external genetic material [3]. According to this point of view, we currently refer to transgenesis when the genetic material to be inserted into the recipient plant comes from an evolutionary distant organism, or even from a different kingdom. Like Bt corn, which was modified through the introduction of a plasmid containing genes from a bacterium called Bacillus thuringiensis [4].
Contrarily, if the genetic material derives from a sexually compatible or closely related species, it is referred to as cisgenesis, and when traits are transferred between varieties of plants that are members of the same species, it is referred to as intragenesis [5,6]. Based on these viewpoints, it is possible to improve plant species safely and, given the potential for gene transfer in the plants employed, to carry out these transfers naturally. This phase involves the utilization of computational biology, bioinformatics, and in silico technologies that enable in-depth research at the genomic, epigenetic, gene expression, and metabolic pathway levels [2]. Finding metabolic pathways or specific genes with potential for plant improvement has become possible because of this information. This makes it possible to develop genetic improvement plans with greater precision and the potential to develop plant varieties quickly.
Due to the reduction in the cost of massive sequencing, or NGS, and the expanding development of bioinformatics, databases have been constructed with information on important plant species with an interest in agriculture, forestry, as well as model organisms [7]. Many parts of the world are currently experiencing droughts, so analyzing this data and identifying potential metabolic pathways that help plants in resisting high temperatures or dry soils may provide solutions. On the other hand, analyzing individual genes can support finding potential genes linked to diseases or pest susceptibility. The ability to safely silence them based on knowledge of their roles would result in disease- and pest-resistant plants, in addition to the traditional methods of transferring important genes between plant varieties and more contemporary methods like gene editing [8]. The use of omics data from Arabidopsis species as a reference in de novo omics and multi-omics research of plant species with an interest in agriculture and/or forestry will be discussed in this review.
2. Initial Processing of Omics Data and Databases of Model and Reference Plants
As we have indicated, crop genetic improvement programs around the world are currently promoting the integration of omics science, bioinformatics, biotechnology, and agronomic disciplines in the development of improved crops. One of the main difficulties in this approach is to relate the omics results obtained with each type of data and to integrate this information. To properly develop this process, several levels of data processing must be performed. From basic quality control of sequencing products to the exploration of metabolic and regulatory systems. In this section, we discuss the many applications of omics databases from Arabidopsis and other model plants for the study of crops or plants of economic importance.
2.1. De Novo Assembly of NGS Products and Other Types of Omics Information
The use of NGS techniques to speed up the breeding process is commonly suggested in current plant breeding programs today. These strategies are finally included in the objectives of plant breeding projects as useful tools for prospecting the goals of these projects [9]. Most plant species of commercial interest have already had their genomes partially or completely sequenced. In the best situation, it might be possible to have a reference transcriptome, genome, and even metabolome of the plant being studied [10]. In the worst case, the only available information is individual sequences of the studied species. This will make it more difficult to process the NGS data and, as a result, assemble genomes, transcriptomes, and other omics data correctly. It is possible to assemble the NGS reads using only mathematical parameters with a variety of bioinformatics tools. With its single processor version, ABySS is helpful for assembling helps assemble genomes up to 100 bases in length. furthermore, having a Trans-ABySS version for transcriptome assembly. Other such examples are EDENA, aTRAM, and EULER, which can de novo assembly short NGS reads. Long reads from PacBio or Oxford Nanopore NGS, which can have higher error rates than short reads from Illumina, can be assembled using software like CANU, FALCON, and HGAP, among others.
Although becoming accurate and flexible, mathematical models have limitations that are inherent to biology itself. For example, mathematical models often ignore biological compartments like the nucleus or mitochondria and instead, assume that all components in a reaction are equally accessible [11]. The set of NGS readings shows an analogous pattern, since the models occasionally underrepresented some repetitive sequences which could be related to crucial regulatory roles. In this case, the assembly is statistically correct from a mathematical perspective but is incomplete and diverged from the expected behavior from a biological perspective [12]. For this reason, it is suggested that the de novo assembly use the Arabidopsis thaliana genome as a reference (Figure 1). The biological approach provided by the Arabidopsis genome adjusts the mathematical model and brings the results closer to biological reality, even in some cases where there is a significant evolutionary distance between the researched plant and Arabidopsis [13].
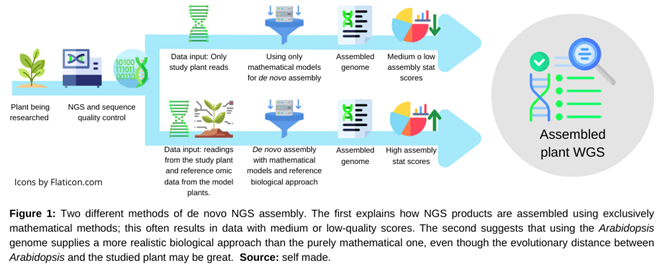
Figure 1.
Recent research has shown that using the Arabidopsis genome as a reference improves the efficacy of de novo assembly compared to experiments that did not use a reference genome [14]. This approach may be employed with a more focused strategy, such as an evolutionary adaptation, to improve plants with commercial value. Hypothetically, genome sequencing of cacao plants endemic to the Moskitia region (Cocoa Vavilov Center) in Honduras, could allow for the discovery of genes that provide information to improve cocoa crops. Vavilov centers are composed of individuals who have the highest diversity and ancestry within that plant species, resulting in the ideal candidates for discovering ancestral genes for plant improvement. These individuals' genomes can be explored to discover information useful to agronomy, biotechnology, and botany [15]. Inbreeding domestication techniques and conventional breeding have been observed to lead to the silencing or deletion of various genes that enable plants to resist attack by pests and diseases [16].
The genomes of plants with higher levels of ancestry carry genes and metabolic pathways that may be related to their evolutionary origins because native plants are more interrelated to the plants that gave rise to commercial crops [17]. Ironically, while the genomes of some crops are available, curated, and annotated, it would be necessary to use the omics information of domesticated plants as a reference for assembling the genomes of these crops' ancestral plants. The main goal of plant breeding programs soon will be to produce plants that are resilient to a variety of environmental conditions and supply sustenance. The development of novel plant varieties will accelerate if high-quality genetic information from ancestor plants is available, this is because selection and transfer of genes and metabolic pathways will be more efficacious [18,19].
Although they are comparable, the main difference between this method and reference genome assembly is the genome used as the reference. The reference genome assembly method makes use of a previously assembled genome as a scaffold [14]. Ideally, this genome should come from the species under study or from one that is closely related to it. In this way, the model is adjusted using experimental curated data of the species that serves as a reference. In the second method, the reference genome comes from a different or evolutionarily distant species. In contrast to the earlier case, the goal of the Arabidopsis genome is to provide a biological reference framework that aids in adjusting the mathematical model and not an assembly scaffold, thus, the method still is essentially a de novo assembly [20].
When working with a species of agricultural or forestry interest at the genomic level for the first time, or when exploring plant alternatives with the potential to become commercial species, this method can be applied [13]. For example, to identify a new tuber species with nutritional potential but low yield. It is possible that the species being researched lacks the species being researched may lack an evolutionary-related organism with a genome that can serve as a reference. A de novo assembly is typically employed in this situation [21]. This assembly will avoid losing some features of the genomic organization shared by many plant species by using the Arabidopsis genome as a reference, which, if merely a mathematical model were used, would be lost.
The data will be analyzed from a strictly mathematical perspective, ignoring some biological peculiarities, to determine its probability of being aligned and oriented by the theoretical reality. In some instances, this implies that the assembled genome may have underrepresented genes because of the similarity of some conserved regions in distinct genes [22]. A biological model that includes the possibility of these isoforms or genetic variations is employed to solve the problem. When there is no reference genome of the organism being investigated, using the genome of Arabidopsis may be an option because of the extensive knowledge we have about it and its importance as a model organism.
The information included in transcriptomes and metabolomes shows a similar pattern. Transcriptomes, in this example, show the degree of gene expression over a period and space. To make accurate comparisons, one would need a reference that was generated at a similar and comparable period and location as the transcriptome under study [23]. Therefore, to determine the amount of Resistance Genes (R-Gene) expression, it is important to evaluate the gene sets of both the host and the pathogen at different time points and in different tissues to predict the gene variations in expression [24,25,26,27]. From a practical standpoint, it is not possible to have reference transcriptomes for every scenario. Nonetheless, there are alternatives that some alternatives can be used to improve the algorithms that allow us to identify and measure sequences of interest. These options, however, can function more as an adaptation to the mathematical model than as a reference framework. There are already about 20,000 Arabidopsis RNA-seq libraries deposited in open databases (http://ipf.sustech.edu.cn/pub/athrna/), this data provides an excellent platform for comparison in a variety of situations, including those affecting transcriptional regulation, tissue specificity, stress responses, and dynamics of gene development [28].
Regardless of the type of information one is working with, supplying a reference is necessary when analyzing omics data. In some cases, such as human exomes, enough information is available to produce precise and biologically coherent assemblies, but in other cases, using a de novo assembly is the only option. In these cases, it will always be preferable to adjust the algorithms and mathematical models using data from a reference organism or an extensively studied as a reference. The results of this de novo assembly approach are more accurate and biologically plausible than using mathematical models alone through the scoring system. Assembling genomic, transcriptomic, metabolomic, etc., data from plants using the available and carefully selected information on Arabidopsis can help to perform this procedure with greater precision and a closer approximation to biological reality. This can lead to a deeper understanding of the organisms and an evaluation of their potential for genetic improvement or crop use.
2.2. Annotations of Crops Omics Information without Reference Genome
Once a genome has been assembled, it is usually essential to interpret the generated sequences. Sequence annotation is a procedure that often involves analyzing sequences throughout many databases in hopes of finding as much information as possible about these sequences [29]. The selection of the database(s) that will be used for the annotation, as well as the software used to conduct the procedure, are common steps in the annotation process (Figure 2). Using specific databases that reduce the errors that can be produced when comparing a genome against all the possible known sequences is a widely used method to increase the pressure on these annotations [30]. In some cases, the results of annotation against the universe of sequences' data can lead to comparisons with distant species' sequences, which, regardless of their high statistical quality value, ignore the approach to biological reality yet again. The accuracy of the annotations and, thus, the quality of the process, can be improved by using data from model organisms. Additionally, this method can use more plant databases as a reference for the accuracy of the information it delivers.
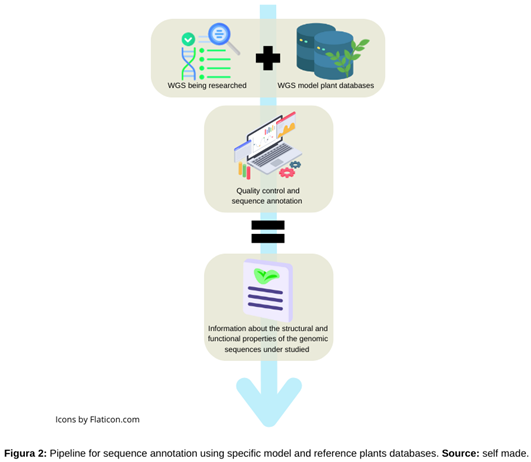
Figure 2.
For example, it is typical to search specific molecular patterns conserved in a class of genes when researching for R-Genes in a plant species' genome. Some of these patterns, however, are present in other taxonomic groups and can be related to proteins that, in some cases, have distinct functions [31]. For instance, NBS-LRR-like proteins have a significant role in immunity in both plants and widely diverse organisms like mammals. Consequently, it might be more useful to annotate the sequences using databases of plant models and reference organisms to identify the R-Genes more precisely in plants' WGS [32]. By restricting the data for comparison, search models like Hidden Markov Model (HMM) or Artificial Neural Networks (ANN) also adjust to the biological reality of plants.
The Arabidopsis thaliana genome assembly (TAIR 10.1) is available on the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/genome/4), Ensemble Plant (http://plants.ensembl.org/Arabidopsis_thaliana/Info/Index), and TAIR (https://www.arabidopsis.org/index.jsp) website where tools for bioinformatics analysis can also be found, among other places. In addition to these databases, several commercially important plants with accessible reference genomes include rice (http://rice.uga.edu/), wheat (https://www.wheatgenome.org/), maize (https://www.maizegdb.org/), potato and tomato (https://solgenomics.net), arabica coffee (https://coffeegenome.ucdavis.edu/), sugar cane (https://sugarcane-genome.cirad.fr/), banana (https://banana-genome-hub.southgreen.fr/), and citrus (https://www.citrusgenomedb.org/), among others. On the other hand, we may require more than one type of software, depending on the type of annotation we want.
Another example is the study of metabolic pathways that help break the dormancy of some seeds of economically useful plant species, such as Coyol (Acrocomia aculeata). In this example, the main goal is to determine which metabolites in Coyol seeds help to break the dormancy. For this purpose, transcriptomic and metabolomic data and their integration can be used, and which genes are expressed at the time of latency break can be determined by transcription [33]. This would allow researchers to find the proteins and enzymes needed to produce these metabolites. Also, it will be possible to find the concentration of these metabolites and their effects at distinct stages of seed germination by metabolomics [34]. Like other palms, Coyol has a very low germination rate compared to the amount of fruit it produces. Nevertheless, they should be considered important plants because of their cultural use and potential for biofuel production [35]. Identification of metabolites that break seed dormancy could help the breeding of endangered species and domestication of wild species for cultural purposes, with increased long-term survival.
Applications in structural and functional biology can help both from annotations of genes and proteins. It can help explain proteins and genes and their interaction and control by defining the molecular function, cellular location, or biological process in which they are involved. They even allow the calculation of evolutionary distances between proteins and genes in comparison to other members of the same plant species or in relation to a specific protein family. In general, annotations depend on the goal of the project and the type of data that can be accessed. Predictions based on information now known about a particular protein type, gene, domain, etc., will occasionally be the results, not just annotations. The system must perform as predicted because there is no in vivo evidence to contradict what the simulations show. The predictions can be considered curated annotations after this information has been verified by experiments. And yet, the simulations contain a significant statistical component that underpins the results.
2.3. Using Data from Arabidopsis to Mapping Metabolic Pathways in Plants
A considerable amount of knowledge about metabolic pathway genes has been accumulated through biochemical and genetic approaches [36]. This increasing information of biological data facilitates the discovery of new metabolic pathways by using mathematical modeling approaches to select candidate genes involved in general and specific functions [37]. Numerous biological metabolic networks in various organisms can be built and analyzed thanks to the development of bioinformatics tools and the accessibility of relevant information in databases [38] (Figure 3).
The databases contain a wide range of species that can be investigated for data, including information at the exon, transcriptional, and gene levels. This data is input into further investigations, enabling the use of different bioinformatics tools for functional analysis such as modeling of signaling pathways [39]. Tools for visualizing and analyzing metabolic pathways include databases, software, and software- packages. These instruments can be used to determine the enzymes and metabolites engaged in a certain pathway, to forecast the impacts of genetic or environmental changes on pathway activity, and to produce hypotheses regarding the roles of as-yet-uncharacterized enzymes or metabolites [40].
While functional analysis tools use a wide range of methodologies, they can be categorized into three main groups: over-representation analysis, functional class scoring, and pathway topology [39]. The R package DOSE (https://bioconductor.org/packages/release/bioc/html/DOSE.html), which is designed for DO-based semantic similarity measurement and enrichment analysis, is one example of a tool that fits within these categories. Pathview is a set of tools for pathway-based data integration and visualization (https://bioconductor.org/packages/release/bioc/html/pathview.html). Likewise, the cluster Profiler package (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) provides methods for analyzing and displaying the functional profiles of genes and gene clusters. Some examples of web-based pathway tools include KEGG (https://www.genome.jp/kegg/), MetaCyc (https://metacyc.org/), and Reactome (https://reactome.org/). These tools are widely used in the field of systems biology to study the complex interactions between genes, proteins, and metabolites that underlie cellular metabolism [40]. A pipeline for investigating metabolic pathways can be built using these strategic techniques in conjunction, and it might include:
- Collect information on relevant metabolites, enzymes, and pathways from a variety of sources, including literature, experimental data, and pathway databases [38].
- Using metabolic mapping tools for building a metabolic pathway map that includes all the metabolites and enzymes involved in the pathway [38]. It involves obtaining and compiling data on biochemical reactions from current sources, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG), to discover the functional annotation of genes [38,41].
- Making predictions regarding the roles of uncharacterized enzymes or metabolites while using pathway tools to examine the pathway map, identify important enzymes and metabolites, and predict the effects of genetic or environmental changes on pathway activity [41].
- Using the pathway information to develop new strategies against affections that are associated with dysregulated metabolic pathways [42].
The advancement of computational methods and the availability of multi-omics data have made it possible to predict the metabolic pathways of important plant chemicals. To better understand the genes involved in the generation and modification of plant metabolites, which is important in increasing plant productivity and quality, it complements conventional genetic and/or biochemical approaches.
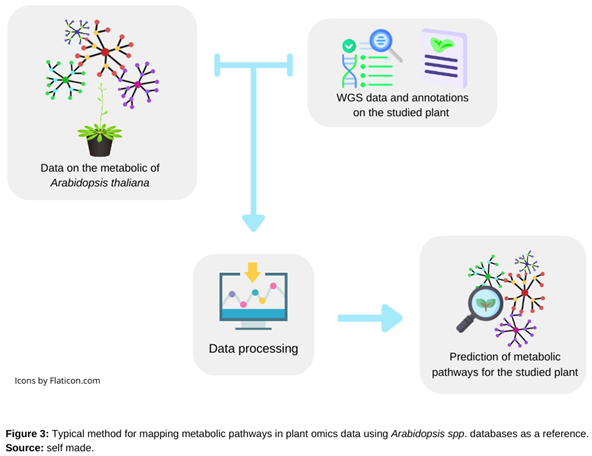
Figure 3.
3. Approaches to Data Integration for Plant Genetic Improvement
The complexity of this last step depends significantly on the skills of data analysts, plant geneticists, and experts in other areas of crop biology, making it probably the most difficult part of the entire procedure. This stage involves integrating the data obtained via different omics techniques and proving how it interacts to produce a specific phenotype. This stage similarly depends extensively on all the earlier analyses, which emphasizes the significance of including a biological element that adjusts the mathematical models and improves the precision of the information obtained.
A regulatory element that activates or silences a gene of interest can be found by integrating genomes and measuring levels of genetic expression from transcriptomes of plants. These regulatory elements can then be used to modify biological processes because they affect the metabolic pathways in which the gene and its products are involved. Finding genes and regulatory areas that can be used as plant breeding elements requires understanding how metabolic pathways are affected by genes and how they function as regulators. Studies that employ epigenetic data can more fully understand how specific genes are silenced in crops and, so, how to prevent this from happening. Incorporating proteomic and metabolomic data, on the other hand, can support experimental validation of bioinformatic simulations and predictions. Through the identification and measurement of proteins and metabolites in plant samples, it would be possible to demonstrate how genetic modifications in plants affect them, helping to fulfill the goals of the plant breeding planning.
Finally, the data integration process will be found by the plant breeding project's final objectives as well as any prospective biotechnological tools accessible for crop modification. This aspect must be considered since some plants cannot be modified genetically using certain techniques. For example, not all plants can be infected by Agrobacterium tumefaciens, a vector commonly used in plant genetic improvement. On the other hand, it is also important to consider the previous information that is currently accessible with the goal of using techniques and experiments that allow collecting supplementary information to carry out the data integration. In this context, it is also essential to consider the accessible computer equipment, as the analyses often call for servers or specialized computers that have the right hardware for processing the calculations conducted throughout the analyses. The interaction between these components and the expertise and experience of the specialists determines whether the integration is successful.
4. Conclusions
The development of better crop varieties could be accelerated using multi-omics and systems biology approaches. It is possible to increase the precision of predictions generated by bioinformatics tools mostly due to the integration of different omics data. Final analysis results will serve as a goal for genetic and biotechnological researchers, enabling them to improve plants in a way that is more precise and safer for both humans and the environment. These predictions will become more accurate as the amount of information they incorporate increases. Therefore, using Arabidopsis reference databases can help to improve the quality of analysis results when there is a lack of information from a closely related species to the cultivar that needs improvement, assisting finally, in the identification of data-integration patterns that allow for the selection of genes with potential for genetic improvement. In conclusion, to design more effective and precise crop breeding strategies, new professionals with experience in omics sciences, bioinformatics, and computational biology must be integrated into the process of crop genetic improvement.
Author Contributions
“Conceptualization, A.O.; writing—original draft preparation, A.O. and M.M.; writing—review and editing, A.O. and M.M.
Funding
This research received no external funding
Acknowledgments
We gratefully acknowledge the generous support of Dr. Lourdes Enriquez and Dr. Gustavo Fontecha in the final review of this review. We also thank the School of Microbiology and the Microbiology Research Institute of the National Autonomous University of Honduras for allowing us to participate
Conflicts of Interest
The authors declare they have no conflict of interest in this review writing.
References
- FAO The State of Food and Agriculture 2020. Overcoming water challenges in Agriculture. 2020.
- Pazhamala, L.T.; Kudapa, H.; Weckwerth, W.; Millar, A.H.; Varshney, R.K. Systems biology for crop improvement. Plant Genome 2021, 14. [Google Scholar] [CrossRef]
- Jhansi Rani, S.; Usha, R. Transgenic plants: Types, benefits, public concerns and future. J Pharm Res 2013, 6, 879–83. [Google Scholar] [CrossRef]
- Koch, M.S.; Ward, J.M.; Levine, S.L.; Baum, J.A.; Vicini, J.L.; Hammond, B.G. The food and environmental safety of Bt crops. Front Plant Sci 2015, 06. [Google Scholar] [CrossRef]
- Rousselière, D.; Rousselière, S. Is biotechnology (more) acceptable when it enables a reduction in phytosanitary treatments? A European comparison of the acceptability of transgenesis and cisgenesis. PLoS One 2017, 12, e0183213. [Google Scholar] [CrossRef]
- Mullins, E.; Bresson, J.; Dalmay, T.; Dewhurst, I.C.; Epstein, M.M.; Firbank, L.G.; et al. Updated scientific opinion on plants developed through cisgenesis and intragenesis. EFSA Journal 2022, 20. [Google Scholar] [CrossRef]
- Lai, K.; Lorenc, M.T.; Edwards, D. Genomic Databases for Crop Improvement. Agronomy 2012, 2, 62–73. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, F.; Zhang, J.; Liu, H.; Rahman, S.; Islam, S.; et al. Application of CRISPR/Cas9 in Crop Quality Improvement. Int J Mol Sci 2021, 22, 4206. [Google Scholar] [CrossRef]
- Zhu, X.-G.; Lynch, J.P.; LeBauer, D.S.; Millar, A.J.; Stitt, M.; Long, S.P. Plants in silico : why, why now and what?-an integrative platform for plant systems biology research. Plant Cell Environ 2016, 39, 1049–57. [Google Scholar] [CrossRef]
- Weckwerth, W.; Ghatak, A.; Bellaire, A.; Chaturvedi, P.; Varshney, R.K. PANOMICS meets germplasm. Plant Biotechnol J 2020, 18, 1507–25. [Google Scholar] [CrossRef]
- Torres, N.V.; Santos, G. The (Mathematical) Modeling Process in Biosciences. Front Genet 2015, 6. [Google Scholar] [CrossRef]
- Vittadello, S.T.; Stumpf, M.P.H. Open problems in mathematical biology. Math Biosci 2022, 354, 108926. [Google Scholar] [CrossRef]
- Lischer, H.E.L.; Shimizu, K.K. Reference-guided de novo assembly approach improves genome reconstruction for related species. BMC Bioinformatics 2017, 18, 474. [Google Scholar] [CrossRef]
- Bao, E.; Jiang, T.; Girke, T. AlignGraph: algorithm for secondary de novo genome assembly guided by closely related references. Bioinformatics 2014, 30, i319–28. [Google Scholar] [CrossRef]
- Tang, H.; Sezen, U.; Paterson, A.H. Domestication and plant genomes. Curr Opin Plant Biol 2010, 13, 160–6. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A. Genetics and Consequences of Crop Domestication. J Agric Food Chem 2013, 61, 8267–76. [Google Scholar] [CrossRef]
- Scossa, F.; Brotman, Y.; de Abreu e Lima, F.; Willmitzer, L.; Nikoloski, Z.; Tohge, T.; et al. Genomics-based strategies for the use of natural variation in the improvement of crop metabolism. Plant Science 2016, 242, 47–64. [Google Scholar] [CrossRef]
- Wang, X.; Wang, S.; Lin, Q.; Lu, J.; Lv, S.; Zhang, Y.; et al. The wild allotetraploid sesame genome provides novel insights into evolution and lignan biosynthesis. J Adv Res 2022. [CrossRef]
- Li, Z.; Wang, J.; Fu, Y.; Jing, Y.; Huang, B.; Chen, Y.; et al. The Musa troglodytarum L. genome provides insights into the mechanism of non-climacteric behaviour and enrichment of carotenoids. BMC Biol 2022, 20, 186. [Google Scholar] [CrossRef]
- Gnerre, S.; Lander, E.S.; Lindblad-Toh, K.; Jaffe, D.B. Assisted assembly: how to improve a de novo genome assembly by using related species. Genome Biol 2009, 10, R88. [Google Scholar] [CrossRef]
- Sohn, J.; Nam, J.-W. The present and future of de novo whole-genome assembly. Brief Bioinform 2016, bbw096. [Google Scholar] [CrossRef]
- Schatz, M.C.; Witkowski, J.; McCombie, W.R. Current challenges in de novo plant genome sequencing and assembly. Genome Biol 2012, 13, 243. [Google Scholar] [CrossRef]
- Cavill, R.; Jennen, D.; Kleinjans, J.; Briedé, J.J. Transcriptomic and metabolomic data integration. Brief Bioinform 2016, 17, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Deng, Z.-N.; Wu, K.-C.; Malviya, M.K.; Solanki, M.K.; Verma, K.K.; et al. Transcriptome analysis reveals a gene expression pattern that contributes to sugarcane bud propagation induced by indole-3-butyric acid. Front Plant Sci 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Han, S.; Yuan, M.; Ma, X.; Hagan, A.; He, G. Transcriptomic analyses reveal the expression and regulation of genes associated with resistance to early leaf spot in peanut. BMC Res Notes 2020, 13, 381. [Google Scholar] [CrossRef]
- Ji, X.; Liu, T.; Xu, S.; Wang, Z.; Han, H.; Zhou, S.; et al. Comparative transcriptome analysis reveals the gene expression and regulatory characteristics of broad-spectrum immunity to leaf rust in a wheat–Agropyron cristatum 2P addition line. Int J Mol Sci 2022, 23, 7370. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, C.; Yang, H.; Kuang, R.; Liu, K.; Huang, B.; et al. Comparative transcriptomics and genomic analyses reveal differential gene expression related to Colletotrichum brevisporum resistance in papaya (Carica papaya L.). Front Plant Sci 2022, 13. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, F.; Yu, Y.; Feng, L.; Jia, J.; Liu, B.; et al. A Comprehensive Online Database for Exploring ∼20,000 Public Arabidopsis RNA-Seq Libraries. Mol Plant 2020, 13, 1231–3. [Google Scholar] [CrossRef]
- Ejigu, G.F.; Jung, J. Review on the computational genome annotation of sequences obtained by Next-Generation Sequencing. Biology (Basel) 2020, 9, 295. [Google Scholar] [CrossRef]
- Salzberg, S.L. Next-generation genome annotation: we still struggle to get it right. Genome Biol 2019, 20, 92. [Google Scholar] [CrossRef]
- Petrey, D.; Fischer, M.; Honig, B. Structural relationships among proteins with different global topologies and their implications for function annotation strategies. Proceedings of the National Academy of Sciences 2009, 106, 17377–82. [Google Scholar] [CrossRef]
- Anthoney, N.; Foldi, I.; Hidalgo, A. Toll and Toll-like receptor signalling in development. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-X.; Chen, D.; Zhao, Y.; Zhang, X.-Y.; Zhao, M.; Peng, R.; et al. RNA-seq analysis reveals key genes associated with seed germination of Fritillaria taipaiensis P.Y.Li by cold stratification. Front Plant Sci 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Lyv, Y.; Zheng, W.; Yang, C.; Li, Y.; Wang, X.; et al. Comparative metabolomics reveals two metabolic modules affecting seed germination in rice (Oryza sativa). Metabolites 2021, 11, 880. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.V.; Sanjinez-Argandoña, E.J.; Linzmeier, A.M.; Cardoso, C.A.L.; Macedo, M.L.R. Food value of mealworm grown on Acrocomia aculeata pulp flour. PLoS One 2016, 11, e0151275. [Google Scholar] [CrossRef]
- Jensen, L.M.; Halkier, B.A.; Burow, M. How to discover a metabolic pathway? An update on gene identification in aliphatic glucosinolate biosynthesis, regulation and transport. Biol Chem 2014, 395, 529–43. [Google Scholar] [CrossRef]
- Toubiana, D.; Puzis, R.; Wen, L.; Sikron, N.; Kurmanbayeva, A.; Soltabayeva, A.; et al. Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data. Commun Biol 2019, 2, 214. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.S.; Shah, F.F.M.; Mohamad, M.S.; Hamran, N.L.; Salleh, A.H.M.; Deris, S.; et al. Database and tools for metabolic network analysis. Biotechnology and Bioprocess Engineering 2014, 19, 568–85. [Google Scholar] [CrossRef]
- Meeta Mistry, Mary Piper, Jihe Liu, Radhika Khetani. Differential gene expression workshop lessons from HCBC (first release). Zenodo 2021.
- Wang, L.; Dash, S.; Ng, C.Y.; Maranas, C.D. A review of computational tools for design and reconstruction of metabolic pathways. Synth Syst Biotechnol 2017, 2, 243–52. [Google Scholar] [CrossRef]
- Wang, P.; Schumacher, A.M.; Shiu, S.-H. Computational prediction of plant metabolic pathways. Curr Opin Plant Biol 2022, 66, 102171. [Google Scholar] [CrossRef]
- Mulvihill, M.M.; Nomura, D.K. Metabolomic strategies to map functions of metabolic pathways. American Journal of Physiology-Endocrinology and Metabolism 2014, 307, E237–44. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



