Submitted:
11 September 2024
Posted:
14 September 2024
You are already at the latest version
Abstract
A new tetracyclic morpholine-fused [5,1-c]-triazole, (4aS,5S,6aR,10aR,10bR)-5-methoxy-9,9-dimethyl-4a,5,6a,7,10a,10b-hexahydro-12H-[1,3]dioxino[4',5':5,6]pyrano[4,3-b][1,2,3]triazolo[1,5-d][1,4]oxazine, was synthesized via a five-step sequence starting from methyl α-D-glucopyranoside by using as a key step an intramolecular copper(I) cat-alyzed alkyne-azide cycloaddition (CuAAC). The synthesized compound was fully characterized by 1H and 13C NMR, FT-IR, and HRMS.
Keywords:
1
; 2
; 3-triazoles
; click chemistry
; carbohydrates
; morpholine-fused [5
; 1-c]-triazole
1. Introduction
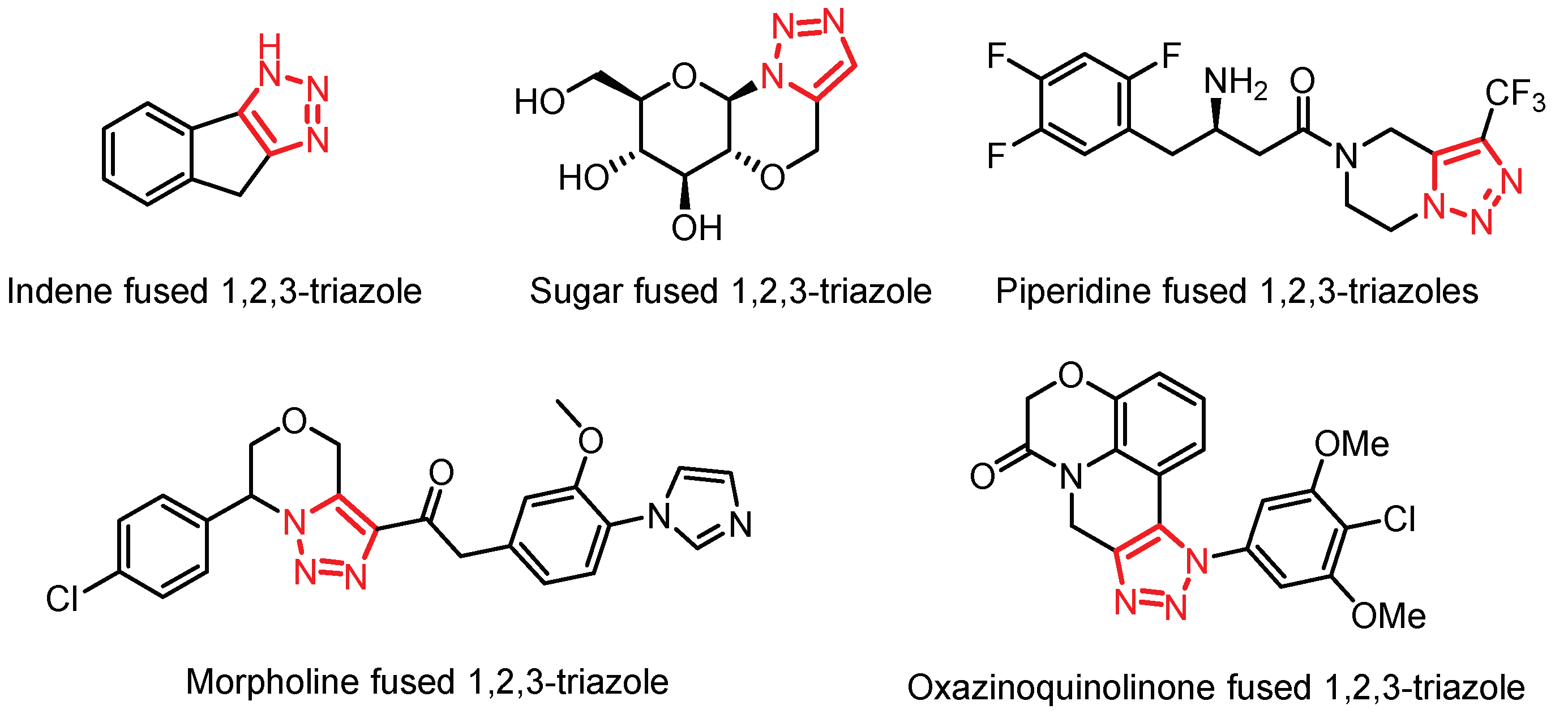
The development of fused heterocyclic systems has gained great attention in the field of medicinal chemistry [1] and materials science [2]. Fused heterocycles are found in many natural products, pharmaceutical and synthetic compounds. Particularly, compounds containing fused 1,2,3-triazoles have shown some interesting biological applications. For example, as an antidiabetic [3], in the treatment of Alzheimer's disease [4], as anticancer [5] or antitumor agents [6], etc. (Figure 1). Therefore, developing efficient strategies for the access to fused 1,2,3-triazoles continues to be of great interest [7].
In the field of click chemistry, the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction is a powerful, highly reliable, selective, and atom-efficient reaction that allows easy access to 1,4-disusbtituted 1,2,3-triazoles [8]. Often, this reaction involves the use of mild reaction conditions and readily available starting materials and reagents, so it is not surprising that triazoles have been extensively used in many fields of interest such as medicinal chemistry [9], materials science [10], and catalysis [11]. On the other hand, the synthesis of 1,5-disubstituted 1,2,3-triazoles and their applications have been much less studied in comparison to their regioisomeric counterpart. The Ru-catalyzed azide-alkyne cycloaddition (RuAAC) reaction is one of the most widely used strategies for preparing 1,5-disubstituted triazoles [12]. However, the cost of the catalyst and the potential toxicity of ruthenium salts have forced the search for new synthetic strategies to accessing these triazoles. Due to the widespread applicability of 1,2,3-triazole-containing heterocycles, and by taking advantage of the inherent strain in the 1,2,3-triazole fused cyclic system, organic chemists have been able to develop methodologies for the synthesis of 1,5-disubstituted-1,2,3-triazole fused heterocycles via intramolecular azide-alkyne cycloaddition reactions [13] Nucleotides and carbohydrates are substrates that stand out for their versatility in synthesizing fused triazoles [14]. Several oxa- [15], aza- [16] and carba-ring [17] fused triazoles from carbohydrates have been reported utilizing thermal- or Cu(I)-catalyzed intramolecular azide-alkyne cycloaddition reactions, where the key intermediate for its synthesis is the formation of an azido alcohol.
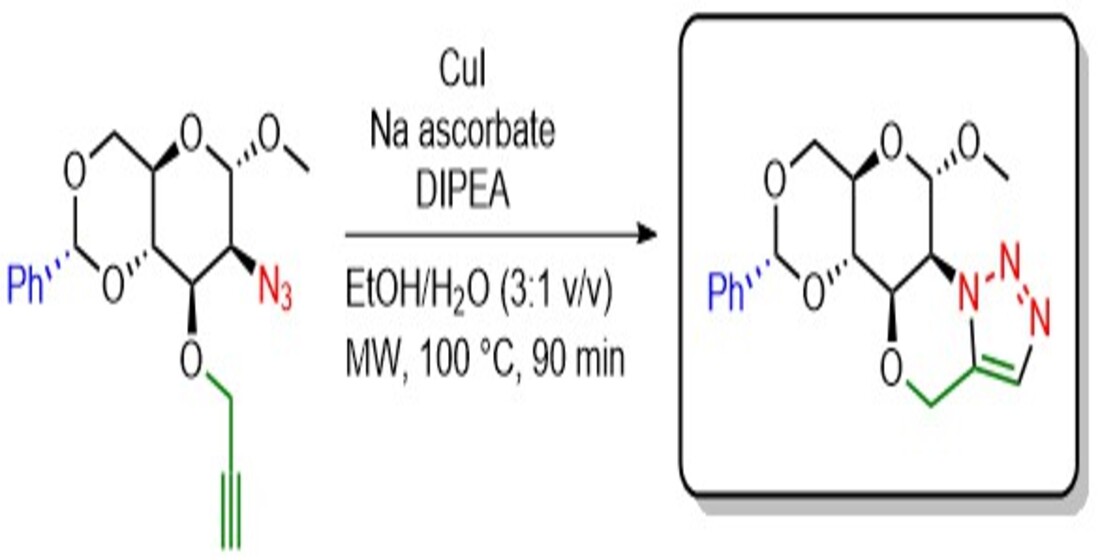
Organic azides have been extensively used in organic synthesis as precursors for numerous nitrogen-containing molecules [18]. Particularly, 1,2-azido alcohols have been widely employed in organic synthesis for the preparation of 1,2-amino alcohols as well as 1,2,3-triazole fused heterocycles. For example, glycosyl 1,2-azido alcohols have been used as starting materials for preparing fused bicyclic molecules containing a morpholine-fused triazole [19]. Triazolo-morpholine 0moiety is a highly attractive bicyclic system due to its wide range of biological applications, especially when conjugated to carbohydrates, as their presence has been shown to enhance enzyme inhibitory activity by increasing interactions with the biomolecular target [20]. Herein, we report the synthesis of a new morpholino-fused [5,1-c]-triazole 6, by a five-step sequence starting from methyl α-D-glucopyranoside 1. This is the first tetracyclic morpholino-fused triazole system containing a hexose.
2. Results and Discussion
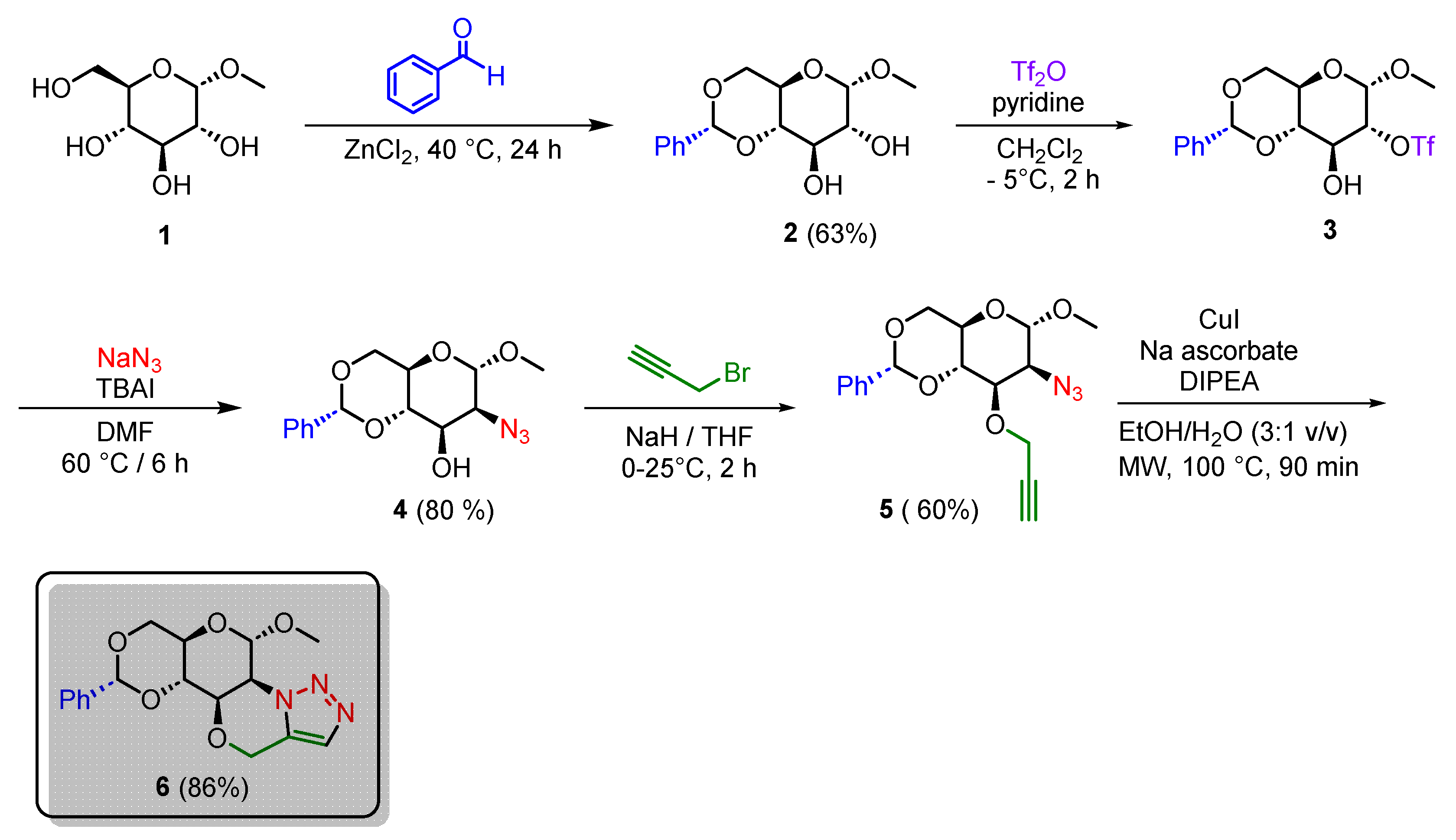
Tiwari et.al., reported the synthesis of bicyclic compounds based on morpholine-fused triazole via a thermal intramolecular azide–alkyne cycloaddition reaction from glycosyl 1,2-azido alcohols and propargyl bromide [21]. Based on their synthetic pathway, the replacement of monosaccharides of five carbon atoms (pentoses) with methyl-4,6-O-benzylidene-α-D-glucopyranoside 2 (a hexose) to obtain a tetracyclic morpholine-fused triazole was envisioned (Scheme 1).
The synthetic strategy begins with the commercially available methyl-α-D-glucofuranose 1. First, compound 1 was subjected to a benzylidene acetal protection of the 1,3-diol by using powdered zinc chloride and benzaldehyde for 40 hours at room temperature to afford the methyl-4,6-O-benzylidene-α-D-glucopyranoside 2 in a 63% yield. [24] Compound 2 was next reacted with trifluoromethanesulfonic anhydride (Tf2O) in the presence of pyridine at – 5°C for 2 hours to produce the trifluoromethanesulfonic ester 3. [25] The subsequent azidation of the triflated sugar 3 with sodium azide and tetrabutylammonium iodide (TBAI) in dimethyl formamide (DMF) at 80 °C for 3 hours resulted in the methyl 2-azido-2-deoxy-4,6-O-benzylidene-α-D-mannopyranoside 4 in a moderate 48 % yield after purification using column chromatography. [26] Once the 1,2-azido alcohol 4 was achieved, the next step was the introduction of the terminal alkyne necessary for synthesizing the morpholine-fused triazole. The O-propargylation of the secondary hydroxyl group of the 1,2-azido alcohol 4 was performed using propargyl bromide and sodium hydride in dry tetrahydrofuran (THF) to afford the propargylated sugar 5 in a 60 % yield. Finally, for the synthesis of target compound 6 we used a copper(I) catalyzed intramolecular alkyne-azide cycloaddition by using the azido-alkyne 4, copper iodide, sodium ascorbate, and diisopropylethylamine (DIPEA) at 100°C with microwave heating for 90 min in ethanol–water (3:1 v/v) as solvent. To our delight, under these conditions, the tetracyclic morpholine-fused [5,1-c]triazole 6 was satisfactorily obtained in 86% yield as a white solid.
The chemical structure of compound 6 was confirmed by NMR, IR, and HRMS analyses. The 1H NMR spectrum of compound 6 exhibited a singlet of one proton observed at δ 7.60 ppm assigned to the triazole-proton. A broad singlet at δ 5.57 ppm confirmed the presence of the anomeric proton, while appearance of a quartet at δ 5.10 ppm is attributed to the OCH2 protons of the triazolo morpholine ring. In addition, two triplet signals at δ 3.81 and δ 3.70 and the three multiplets from δ 4.74 to δ 3.95 ppm displayed the remaining carbohydrate protons, while the singlet at δ 3.57 ppm assigned to the methoxy group and two signals of aromatic protons at δ 7.50 and δ 7.40 ppm confirmed the occurrence of the intramolecular cycloaddition. 13C NMR of compound 6 showed two resonances at δ 130.6 and δ 128.9 ppm corresponding to the triazole-carbons. Finally, the molecular ion peak at m/z 346.14321 [M + H]+ confirms the synthesis of the morpholine-fused [5,1-c]triazole 6.
3. Materials and Methods
All reagents were purchased from commercial suppliers and used without further purification. Solvents used in the syntheses were of technical grade and freshly distilled prior to use. Melting points were obtained on a Fisher-Johns apparatus and are uncorrected. NMR spectra were recorded on Bruker Ascend-400 (400 MHz) and Bruker Avance DMX-400 (400 MHz) spectrometers in CDCl3, and chemical shifts are given in ppm, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br s = broad singlet), coupling constant (J) with TMS as the reference. Mass spectra were recorded on mass spectrometer model micrOTOF II (Bruker Daltonics Inc.) using the Compass platform (otofControl and DataAnalysis from Bruker Daltonics Inc.). Spectra were acquired in positive mode with a capillary voltage of 4500 V, nebulizer gas: 0.5 Bar, drying gas 4.0 L/min and a drying temperature of 150 C. Microwave irradiation experiments were performed using a Discover System (CEM Corporation, Matthews, NC, USA) single-mode microwave with standard sealed microwave glass vials. The precursor 2 [22] and 4 [23,24] were prepared according to the previously reported procedures.
Synthesis de Methyl 2-azido-4,6-O-benzyliden-2-deoxy-3-O-propargyl- a-D-glycopyranoside 5
To a solution of azide 4 (0.200 g, 0.650 mmol) in dry THF (5 mL), and under nitrogen atmosphere was added NaH (0.018 g, 0.780 mmol, 60% dispersion in oil) at 0 °C followed by the dropwise addition of propargyl bromide (0.058 mL, 0.650 mmol). The resulting mixture was allowed to warm up at room temperature and was kept under stirring for 2 hours. Finally, the reaction was quenched carefully with H2O (5 mL), the solvent was evaporated, and the residue was diluted with EtOAc (30 mL), washed with brine (1x60 mL), dried with anhydrous Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography on silica gel using eluents (7:1 Hex-EtOAc) to give propargylated product 5 (0.130 g, 60% yield) as a yellow oil. [α]D20 -30 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3) δ: 7.53 – 7.47 (m, 2H), 7.45 – 7.35 (m, 3H), 5.62 (s, 1H), 4.71 (d, J = 1.7 Hz, 1H), 4.50 (dd, J = 16.0, 2.4 Hz, 1H), 4.40 (dd, J = 15.9, 2.4 Hz, 1H), 4.31 – 4.20 (m, 2H), 4.14 (dd, J = 3.9, 1.5 Hz, 1H), 4.11 – 4.02 (m, 1H), 3.88 – 3.81 (m, 2H), 3.41 (s, 3H), 2.51 (d, J = 2.4 Hz, 1H). 13C RMN (101 MHz, CDCl3) δ 137.25, 128.93, 128.31(2C), 126.15(2C), 101.63, 101.11, 79.51, 78.06, 77.59, 75.65, 69.65, 68.87, 63.79, 58.13, 56.51. HRMS (ESI-TOF) Calculated for C17H21N3O5 [M + 2H]+ 347.14812, found 347.14321.
Synthesis Morpholine-fused [5,1-c]triazole 6
the propargyl carbohydrate 5 (0.100 g, 0.289 mmol was placed in a microwave tube equipped with magnetic stirrer), and then was dissolved in 5 mL of a solution of EtOH-H2O (2:1). After that, sodium ascorbate (0.011 g, 0.057 mmol), copper iodide (0.016 g, 0.086 mmol), and DIPEA (0.019 mL, 0.111 mmol) were added. The resulting mixture was heated under microwave irradiation (40 W, 100 °C) for 90 min. Finally, the reaction mixture was diluted with ethyl acetate and filtered through a short plug of silica, the resulting organic phase was dried over Na2SO4 and concentrated at reduced pressure. The resulting residue was purified by flash chromatography on silica gel using a mixture of Hex-EtOAc (1:1) to give morpholine-fused [5,1-c]-triazole 6 (0.086 g, 86% yield) as a white solid, m. p. 198-200°C. [α]D20
-70 (c 1, CHCl3). 1H RMN (400 MHz, CDCl3) δ: 7.60 (s, 1 H), 7.50 (d, J =5.1, 1.6, Hz, 2 H), 7.40 ( d, J = 5.0, 1.4, Hz, 3 H), 6.00 (s, 1H), 5.57 (br s, 1H), 5.10 (q, J= 1.5 Hz, 2 H), 4.74 – 4.66 (m, 2 H), 4.32 (dd, J = 10.2, 4.9, 1.5 Hz, 1 H), 4.08 – 3.95 (m, 1 H ), 3.81 (t, J = 9.3 Hz, 1Hj), 3.70 ( t, J = 10.4, 1.6 Hz, 1H), 3.57 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 136.87, 130.57, 129.51, 128.94, 128.52(2C), 126.37(2C), 102.31, 98.16, 72.76, 69.34, 68.79, 62.13, 58.38, 57.02, 55.71. HRMS (ESI-TOF) Calculated for C17H20N3O5 [M + H]+ 346.1397, found 346.1398.
4. Conclusions
The synthesis of a new tetracyclic morpholine-fused [5,1-c]-triazole was performed successfully through a five-step sequence starting from methyl α-D-glucopyranoside by using as a key step an intramolecular copper(I) catalyzed alkyne-azide cycloaddition with a 28% overall yield. The title product could be used as a starting point for the synthesis of morpholine-fused [5,1-c]-triazolyl glycoconjugates, since hydrolysis of the benzylidene acetal will allow regeneration of the 1,3-diol, which can be functionalized in several ways. In addition, the reported morpholine-fused [5,1-c]-triazole could be consider for further in vitro studies because it contains a pharmacophoric moiety.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Copies of 1H-NMR, 13C-NMR spectra for the new product 6.
Author Contributions
Synthesis and characterization, R.C.S., E.R.D., A.S.E.; investigation and methodology, G.E.N.S., L.L.R. and A.S.E.; conceptualization and writing—original draft preparation, R.C.S and A.S.E.; funding acquisition and writing—review and editing, R.C.S., A.S.E., and G.E.N.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
All authors gratefully acknowledge to Silvano Cruz Gregorio and Mónica A. Rincón-Guevara for their NMR and HRMS acquisitions, respectively.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Shiro, T.; Fukaya, T.; Tobe, M. The chemistry and biological activity of heterocycle-fused quinolinone derivatives: A review, Eur. J. Med. Chem. 2015, 97, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Zhang, Q.; Shreeve, J.M. Fused heterocycle-based energetic materials (2012–2019). J. Mater. Chem. A, 2020; 8, 4193–4216. [Google Scholar]
- Shan, Z.; Peng, M.; Fan, H.; Lu, Q.; Lu, P.; Zhao, C.; Chen, Y. Discovery of potent dipeptidyl peptidase IV inhibitors derived from β-aminoamides bearing substituted [1,2,3]-triazolopiperidines for the treatment of type 2 diabetes, Bioorg. Med. Chem. Lett. 2011, 21, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, B.; Steele, C.; Hardick, D.; Dale, M.; Pomel, V.; Quattropani, A.; Beher, D. Fused Triazole Derivatives as Gamma Secretase Modulators. Eur. Pat. Appl 2014, EP 2687528 A1. [Google Scholar]
- Narsimha, S.; Battula, K.S.; Nukala, S.K.; Gondru, R.; Reddy, Y.N. Nagavelli, V.R. One-pot synthesis of fused benzoxazino[1,2,3]triazolyl[4,5-c]quinolinone derivatives and their anticancer activity. RSC Adv., 2016, 6, 74332–74339. [Google Scholar] [CrossRef]
- Antonini, I.; Santoni, G.; Lucciarini, R.; Amantini, C.; Sparapani, S.; Magnano, A. Synthesis and Biological Evaluation of New Asymmetrical Bisintercalators as Potential Antitumor Drugs. J. Med. Chem. 2006, 49, 7198–7207. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Ray, K. Synthesis of 1,2,3-Triazole-Fused Heterocycles via Intramolecular Azide–Alkyne Cycloaddition Reactions. Synthesis 2011, 23, 3767–3783. [Google Scholar] [CrossRef]
- Vala, D.P.; Vala, R.M.; Patel, H.M. Versatile Synthetic Platform for 1,2,3-Triazole Chemistry. ACS Omega 2022, 7, 36945–36987. [Google Scholar] [CrossRef]
- Matin, M.M. , Matin P.; Rahman, Md.R., Ben, H.T.; Almalki, F.A.; Mahmud, S., Ghoneim, M.M.; Alruwaily, M.; Alshehri, S. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Front. Mol. Biosci. 2022, 9, 1–8. [Google Scholar] [CrossRef]
- Huo, J.; Hu, H.; Zhang, M.; Hu, X.; Chen, M.; Chen, D.; Liu, J.; Xiao, G.; Wanga, Y.; Wen, Z. A mini review of the synthesis of poly-1,2,3-triazole-based functional materials. RSC Adv., 2017, 7, 2281–2287. [Google Scholar] [CrossRef]
- Joseph, M.C.; Swarts, A.J.; Mapolie, S.F. Transition metal complexes of click-derived 1,2,3-triazoles as catalysts in various transformations: An overview and recent developments. Coord. Chem. Rev. 2023, 493, 215317. [Google Scholar] [CrossRef]
- Johansson, J.R.; Beke-Somfai, T.; Stålsmeden, A.S.; Kann, N. Ruthenium-Catalyzed Azide Alkyne Cycloaddition Reaction: Scope, Mechanism, and Applications. Chem. Rev. 2016, 116, 14726–14768. [Google Scholar] [CrossRef] [PubMed]
- P, R.; Thomas, J.; Dehaen, W.; John, J. Advances in the Synthesis of Fused 1,2,3-Triazoles via a MCR-Intramolecular Azide-Alkyne Cycloaddition Approach. Molecules 2023, 28, 308. [Google Scholar] [CrossRef] [PubMed]
- Pathak, T.; Bose, A. 1,5-disubstituted 1,2,3-triazolylated carbohydrates and nucleosides. Carbohydr. Res. 2024, 541, 109126. [Google Scholar] [CrossRef] [PubMed]
- Hotha, S.; Anegundi, R.I.; Natu, A.A. Expedient synthesis of 1,2,3-triazole-fused tetracyclic compounds by intramolecular Huisgen (click) reactions on carbohydrate-derived azido-alkynes, Tetrahedron Lett. 2005, 46, 4585–4588.
- Adhikary, N. D.; Chattopadhyay, P. Design and synthesis of 1,2,3-triazole-fused chiral medium-ring benzo-heterocycles, scaffolds mimicking benzolactams. J. Org. Chem. 2012, 77, 5399–5405. [Google Scholar] [CrossRef]
- Panday, N.; Meyyappan, M.; Vasella, A. A comparison of glucose- and glucosamine-related inhibitors: probing the interaction of the 2-hydroxy group with retaining β-glucosidases. Helv. Chim. Acta 2000, 83, 513–538. [Google Scholar] [CrossRef]
- Nayl, A.A.; Aly, A.A.; Arafa, W.A.A.; Ahmed, I.M.; Abd-Elhamid, A.I.; El-Fakharany, E.M.; Abdelgawad, M.A.; Tawfeek, H.N.; Bräse, S. Azides in the Synthesis of Various Heterocycles. Molecules 2022, 27, 3716. [Google Scholar] [CrossRef]
- Mishra, K. B.; Tiwari, V. K. Click Chemistry Inspired Synthesis of Morpholine-Fused Triazoles. J. Org. Chem. 2014, 79, 5752–5762. [Google Scholar] [CrossRef]
- Reddy, Y. S.; John Pal, A. P.; Gupta, P.; Ansari, A. A.; Vankar, Y. D. Ceric Ammonium Nitrate-Catalyzed Azidation of 1,2-Anhydro Sugars: Application in the Synthesis of Structurally Diverse Sugar-Derived Morpholine 1,2,3-Triazoles and 1,4-Oxazin-2-ones. J. Org. Chem. 2011, 76, 5972. [Google Scholar] [CrossRef]
- Mishra, K. B.; Shasi, S.; Tiwari, V. K. Metal free synthesis of morpholine fused [5,1-c] triazolyl glycoconjugates via glycosyl azido alcohols. RSC Adv., 2015, 5, 86840. [Google Scholar] [CrossRef]
- Espinoza-Vázquez, A.; Cervantes, R. M. A.; Negrón-Silva, G. E.; Rodríguez Gómez F., J. , Palomar, P. M.; Lomas, R. L., Ángeles Beltrán D., Pérez Martínez, D. Carbohydrates as Corrosion Inhibitors of API 5L X70 Steel Immersed in Acid Medium. Int. J. Electrochem. Sci 2019, 14, 9206–9220. [Google Scholar] [CrossRef]
- Knapp, S.; Kukkola, P. J.; Sharma, S.; Dhar, T. G. M.; Naughton, A. B. J. , Amino Alcohol and Amino Sugar Synthesis by Benzoylcarbamate Cyclization. J. Org. Chem. 1990, 55, 5700–5710. [Google Scholar] [CrossRef]
- Popelová, A.; Kefurt, K.; Hlaváckova, M.; Moravcová, J. A concise synthesis of 4-nitrophenyl-2-azido-2-deoxy and 2-acetamido-2-deoxy-d-mannopyranosides. Carbohydr. Res. 2005, 340, 161–166. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Examples of pharmacology active fused 1,2,3-triazoles.
Scheme 1.
Synthetic route for the synthesis of tetracyclic morpholine-fused[5,1-c] triazole 6.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



