
Submitted:
14 September 2024
Posted:
16 September 2024
You are already at the latest version
Abstract
Salmonella species are important foodborne pathogens worldwide. Salmonella pathogenicity is associated with multiple virulence factors and enhanced antimicrobial resistance. To determine the molecular characteristics and genetic correlations of Salmonella, 24 strains of Salmonella isolated from different sources (raw poultry, human stool, and food) in the Wenzhou area were investigated to determine the distribution of antimicrobial resistance and virulence determinants using whole-genome sequencing (WGS). Aminoglycoside-resistance genes were detected in all samples. Over half of the samples found antimicrobial resistance genes (ARGs) and point mutations for several clinically frequently used antibiotic, β-lactamases, tetracyclines, and fluoroquinolones. Of these strains, 62.5% were predicted to be multidrug-resistant (MDR). The quinolone-modifying enzyme gene aac(6')-Ib-cr, detected in five samples (S1–S4 and S10), were located on integrons. Analysis of Salmonella pathogenicity island (SPI) profiles suggests that serotypes with close genetic relationships share the same distribution of virulence factors, revealing a link between genotype and SPI profiles. CgMLST analysis indicated that five isolates S14–S18 were closely related to strains originating from the UK, suggesting that they may have been introduced from the UK. Data from this study may enrich the molecular traceability database for Salmonella and provide a basis for effective public health policies.
Keywords:
Salmonella
; antimicrobial resistance
; pathogenicity islands
; cgMLST
; Whole genome sequencing
1. Introduction
Salmonella is a facultative anaerobic Gram-negative bacteria belonging to the Enterobacteriaceae family. It is an important zoonotic foodborne pathogen found in animal products such as poultry, pork, and eggs [1,2]. More than 90 million infections and 150,000 deaths are recorded each year due to the consumption of Salmonella-contaminated meat worldwide [3]. Salmonella is the second most common bacterial cause of foodborne illnesses globally each year, after Campylobacter. Furthermore, non-typhoidal Salmonella enterica predominates in deaths caused by foodborne diarrhoeal disease agents worldwide [4]. These results indicate that Salmonella infections contribute significantly to the global burden of foodborne illnesses. The genus Salmonella can be divided into the following two species: Salmonella bongori and Salmonella enterica. The former is rarely associated with human infections. The latter include the following six subspecies: S. enterica subsp. enterica (I), S. enterica subsp. salamae (II), S. enterica subsp. arizonae (IIIa), S. enterica subsp. diarizonae (IIIb), S. enterica subsp. houtenae (IV), and S. enterica subsp. indica (VI) [5]. Within these subspecies, strains can be further classified into serotypes, and more than 2600 serotypes have been described based on combinations of different somatic (O) and flagellar (H) antigens using the Kauffman–White scheme [6]. More than half of these serotypes belong to S. enterica subsp. enterica, which is the main cause of gastroenteritis in warm-blooded animals [7]. Other Salmonella subspecies (II–VI) and S. bongori are associated with diseases in cold-blooded organisms and occasionally cause systemic diseases in humans. Different Salmonella serotypes exhibit different genetic characteristics, host specificity, pathogenicity, and antimicrobial resistance, leading to the development of distinct epidemiological and clinical symptoms [8,9].
The pathogenicity of Salmonella is associated with multiple virulence factors and antimicrobial resistance [10]. Salmonella uses multiple virulence determinants such as flagella, capsules, plasmids, adhesion systems, and the type 3 secretion system (T3SS) encoded by Salmonella pathogenicity island (SPI) to colonize the host by attaching, invading, and bypassing the gastrointestinal defense mechanisms of the hosts [9]. The SPI is an unstable chromosomally located DNA segment containing virulence-related genes. To date, 17 SPIs (1-17) have been recognized. Of these, the virulence genes involved in the intestinal phase of infection were located in SPI-1 and SPI-2; the remaining SPIs are involved in physiological processes, such as Salmonella survival in host cells, magnesium and iron uptake, multi-antimicrobial resistance, and the development of systemic infections [11,12]. In addition to SPI, Salmonella virulence-related genes are distributed in virulence plasmids, bacteriophages, flagella, and enterotoxins, which play various physiological roles in the host [13]. Understanding Salmonella virulence islands and the composition of virulence genes is essential for predicting their pathogenic potential. Recently, Salmonella resistance has become an increasing problem due to the misuse of antibiotics in poultry farming and clinical medicine. Recent study hasreported that non-typhoidal Salmonella is resistant to almost all classes of antibiotics, which significantly limits the choice of effective drugs and poses a considerable challenge in the treatment of salmonellosis [14]. Mobile elements, such as plasmids and integrons, play an important role in Salmonella antimicrobial resistance because they mediate the horizontal transfer of antimicrobial resistance genes (ARGs) to exacerbate the spread of resistance [9,15,16]. Ongoing research on Salmonella resistance can guide drug selection and inform the development of new drugs.
Previous study has reported that Salmonella can be transmitted across geographic regions through contaminated food and can form independent evolutionary branches in different regions, thereby increasing the genetic diversity of Salmonella populations [17]. Therefore, studying the genetic relationships among Salmonella species is very important. Several molecular typing methods have been used in epidemiological studies of Salmonella to determine the genetic relationships between isolates, e.g., pulsed-field gel electrophoresis, enterobacterial repetitive intergenic consensus polymerase chain reaction, and multiple-locus variable number tandem repeat analysis (Ngoi et al., 2015). Currently, whole-genome sequencing (WGS) is an accurate tool for predicting the genetic characterization of bacterial strains as well as their potential pathogenicity and antimicrobial resistance [18,19]. WGS can also provide insights into the genomes of pathogens, including information on species, serotypes, resistance and virulence factors, pathogenicity mechanisms, and their evolution in pathogens [20,21].
Wenzhou is an important commercial city on the southeastern coast of China, located near Shanghai, with frequent import and export trade. An in-depth understanding of the molecular characteristics of Salmonella and an assessment of the risk of importation can be effective in preventing and controlling infections with this pathogen. In this study, 24 Salmonella strains isolated from different sources (human stool, poultry, and food) in Wenzhou were analyzed for antimicrobial resistance, potential pathogenicity, and genetic diversity using WGS. These results may serve as a reference for the prevention and control of bacterial diseases.
2. Methods
2.1. Salmonella Isolation and DNA Extraction
In August 2020, 24 Salmonella isolates were collected from raw poultry, human stools, and food by the Wenzhou Center for Disease Control and Prevention (CDC), Wenzhou, China. Sterile containers were used during sampling, and the samples were transported to the Wenzhou CDC following aseptic procedures. All samples were delivered to the CDC in ice packs within four hours. Amongst 18 strains isolated from raw poultry (S1–S3, S5–S13, and S19–S24), five strains were isolated from human stool (S14–S18) and one strain was isolated from gluten (for food) (S4) (Table 1). Strains were centrifuged at 6010g for 1 min (Allegra 64R Benchtop Centrifuge, Beckman Coulter, USA), pellets with genomic DNA was extracted from Salmonella using a bacterial genomic DNA extraction kit GK1072 (Shanghai Jereh Bioengineering Co., Ltd., Shanghai, China) and whole-genome sequencing was performed.
2.2. WGS and Assembly
Salmonella genomes were sequenced on the Illumina NovaSeq platform using 2 × 150 bp paired ends according to the manufacturer’s protocols (Illumina TruSeq Nano DNA LT) at a commercial company (Shanghai Personal Biotechnology Co., Ltd.). The Illumina TruSeq DNA Sample Preparation Guide was used to construct a genomic sequencing library. After the libraries were qualified, they were sequenced using Illumina NovaSeq according to the effective concentration. The results were stored in a paired-end FASTQ format, which contained sequence information of sequencing reads and corresponding sequencing quality information. To ensure the quality of the subsequent information analysis, AdapterRemoval v2.2.2 was used to remove adapter contamination from the downlinked data, and SOAPec v2.03 was used to quality correct all raw reads based on Kmer frequency. Illumina reads were assembled denovo using SPAdes v3.12.0 to obtain genome sequence contigs. This step was bp corrected using Pilon v1.18.
2.3. Bioinformatics Analyses
For further genome analysis, raw reads were submitted to Enterobase [22]. Preliminary analysis and identification of isolates were based on information from the S. enterica multilocus sequence typing (MLST) database (http://mlst.warwick.ac.uk/mlst/dbs/Senterica), using an in silico model of the S. enterica MLST method. Seven Salmonella housekeeping loci (aroC, dnaN, hemD, hisD, purE, sucA, and thrA) were used for this analysis and were also used to assign numbers to sequence types. In silico serotyping was performed using the SeqSero1.2 tool (https://cge.food.dtu.dk/services/SeqSero/) [23]. We also used the Enterobase and cgMLST schemes to construct a phylogenetic tree. In total, 3002 loci were used for cgMLST analysis. A Grape Tree was constructed using the neighbor-joining (NJ) algorithm and visualized using iTOL (https://itol.embl.de/).
Detection of acquired ARGs and chromosomal point mutations in the genome was performed using ResFinder 4.1 (https://cge.food.dtu.dk/services/ResFinder/) [24,25,26]. PlasmidFinder 2.0.1 was used to identify plasmids in the genome (https://cge.food.dtu.dk/services/PlasmidFinder/) with a minimum threshold of 95% identity and 60% coverage [27]. Integron Finder (version 2.0.1) was used, followed by BLASTp analysis to identify the I integron in the genome data [28]. Genomes were annotated for virulence factors using the Virulence Factor Database (VFDB) Online Annotation (http://www.mgc.ac.cn/VFs/)[29]. SPIFinder was performed using the local command line version of SPIFinder of the Center for Genomic Epidemiology for Salmonella available at (https://cge.food.dtu.dk/services/SPIFinder/) with a minimum threshold of 95% identity and 60% coverage [30].
2.4. Data Availability
Sequencing data for all 24 Salmonella strains were deposited in NCBI under Bioproject PRJNA831306. Accession numbers were JALPKT000000000–JALPLQ000000000 (Table 2).
The 314 S. Liverpool (Table. S3) and 13 S. Typhimurium (Table. S4) strains used for cgMLST analyses, respectively, were obtained from the EnteroBase database (https://enterobase.warwick.ac.uk/species/index/senterica).
3. Results
3.1. Genomic Features of the Salmonella Strains
After quality control filtering, the percentage of bases with identification accuracies above 99% (Q20) and 99.9% (Q30) exceeded 97% and 92%, respectively, indicating the high reliability of the sequencing data. A total of 1713 contigs were generated by assembling, and per genome contained contigs varying from 32 to 240. Among the 24 strains, genome sizes were roughly equal and ranged from 4.58 to 5.08 Mb with GC content of 51.9–52.3%. Functional annotation predicated that that these strains had 4289–5108 genes (Table. S1).
3.2. The Serotyping of Salmonella Isolates Using the MLST Typing
Among 24 Salmonella isolates, ten serotypes were identified. Salmonella Typhimurium was the most prevalent serotype (n = 7, 29.2%), followed by Liverpool (n = 5,20.8%), London (n = 4,16.7%), and Goldcoast (n = 2, 8.3%). Only one isolate was present for each of the following serotypes: Javiana, Kentucky, Corvallis, Meleagtidis, Infantis, and Anatum (Table 1).
Based on the available data in the S. enterica MLST database, 12 distinct sequence types were recognized; three different sequence types were identified as ST19, ST34, and ST1544 in S. Typhimurium, and the other serotypes corresponded to a unique sequence type. The sequence types included ST155, ST19, ST34, ST1544, ST358, ST1959, ST463, ST1541, ST198, ST64, ST32, and ST9074 (Table 1).
3.3. ARG
Among the 24 strains, aminoglycoside-resistance genes showed the highest frequency (100%,24/24), followed by β-lactamases (50%,12/24), tetracyclines (50%,12/24), chloramphenicol (45.83%,11/24), sulfonamides (41.67%,10/24), fluoroquinolones (37.5%,9/24); quaternary ammonium compounds (37.5%,9/24), rifampicin (29.17%,7/24), trimethoprim (29.17%,7/24), macrolides (16.67%,4/24), fosfomycin (8.33%,2/24), and lincomycin (4.17%,1/24) (Figure 1). Different serotypes exhibit differences in antimicrobial resistance. All 24 isolates possessed at least one ARG, of which 15 contained more than 3 ARGs and 10 contained more than 5 ARGs. Aminoglycoside-resistance genes show diversity, including aac(3)-Id, aac(3)-IId, aac(3)-IV, aac(6’)-Iaa, aadA1 , aadA16, aadA17, aadA2, aadA7, aph(3’)-Ia, aph(3’‘)-Ib, aph(4)-Ia, aph(6)-Id, rmtB, and aac(6’)-Ib-cr. Five isolates carried the quinolone-modifying enzyme gene aac(6’)-Ib-cr. In total, 12 isolates carried β-lactamase genes, all belonging to extended-spectrum-β-lactamase (ESBL) genotypes, including TEM, CTX-M, and OXAgenotypes. One S. Kentucky strain had TEM combined with the CTX-M dual genotype, and one S. Typhimurium strain had TEM combined with the OXA dual genotype. Six isolates carried both qnr and ESBL genes. S. Kentucky carried up to 12 classes of ARGs. Lnu(F) and fosA3 genes were found only in S. Kentucky. FosA7 was detected in one S. Meleagridis strain (Table 1).
In total, 24 point mutations were detected in gyrA and parC associated with quinolone resistance in 24 salmonella strains. No mutations were detected in gyrB or parE. The most common mutation in gyrA was S83Y, followed by S83F, D87N, and D87Y. The most common mutation in parC was T57S, followed by S80I. Among the 20 isolates with point mutations detected, seven strains carried plasmid-mediated qnr genes, and four strains of S. London carried both qnr and aac(6’)-Ib-cr genes (Table 1), suggesting that there is an extended spectrum of resistance to quinolones. A strain of S. Kentucky with four point mutations also carried the qnrS1 gene.
3.4. Plasmid and I Integron
In total, 29 bacterial plasmids were identified in 24 strains. S9 contains the sulfonamide resistance gene sul2 and streptomycin resistance genes aph(6)-Id and aph(3’‘)-Ib. Ten isolates did not carry any plasmids, 14 isolates had at least one plasmid, and nine isolates carried two or more plasmid types. IncFIB was the dominant incompatibility group. 58% of the genomes contained plasmids (Table 2).
Five complete class I integrons have been identified. All four strains of S. London contain one I integron carrying the same gene cassette arrays aadA16- dfrA27- ARR-3- aac(6’)-Ib-cr. A strain of S. Infantis was found to have one I integron carrying the gene cassette aadA1. A CALIN-type integron in the form of a gene cassette was found in one S. Typhimurium, carrying the gene cassette aac(6’)-Ib-cr - blaOXA-1- catB3 - ARR- 3 (Table 2).
3.5. Virulence Genes
The results of the online annotation of virulence factors for the 24 isolates using the virulence factor database VFBD are shown in Table. S2. Some genes (95/234) were conserved among all the isolates. The phage-associated gene sodCI, prophage-encoded gene gogB, macrophage-inducible gene mig-5, plasmid-mediated genes spvB, spvD, rck, and plasmid-encoded fimbrial gene sefABCD were detected only in S. Typhimurium. In particular, S. Typhimurium, numbered S9, lacked all virulence plasmid-associated genes. S. Liverpool lacked the multiple fimbrial operons stf, sti, stj, stk, lpf, peg, and tcf, which may affect the ability of bacteria to invade and colonize. S. Liverpool also lacked the bacterial effector avrA. Additionally, the cytolethal-distending toxin cdtB was detected in one S. Javanica and two S. Goldcoast. The yersiniabactin (ybt) gene was detected in a strain of S. Infantis (Table. S2).
3.6. The SPI Profiles
All 24 Salmonella isolates carried the same five SPIs, namely SPI-1, SPI-2, SPI-3, SPI-5, and SPI-9. SPI–4 was detected in all isolates except for two strains of S. Typhimurium of the ST34 genotype. SPI–8 was detected in only one strain of S. Corvallis and one strain of S. Meleagridis. SPI-11 was detected in only one strain of S. Javiana. Nine SPI profiles were identified, named P1 to P9, and most ST strains corresponded to the same SPI profiles. One strain of S. Corvallis and one strain of S. Meleagridis with similar phylogenetic affinities shared the same SPI profile. A strain with genotype ST1544 in S. Typhimurium has the same SPI profile as S. London. Additionally, the three sequence types of S. Typhimurium, ST19, ST34, and ST1544, corresponded to three SPI profiles, P6, P9, and P8, respectively (Figure 2).
3.7. Phylogenetic Relationship of Salmonella strains by cgMLST Analysis
The assignment of serotypes to the four main clusters was consistent with the global phylogenetic tree. All the remaining serotypes were unique. Four clusters contained the main serotypes detected, namely S. Typhimurium, Liverpool, London, and Goldcoast, respectively defined as C1, C2, C3 and C4. A sub-clustering was noted within the cluster C3 and two sub-clustering within the cluster C4 (Figure 2) providing evidence of their genetic diversity.
4. Discussion
In this study, a collection of 24 S. enterica strains was studied through WGS and subsequent bioinformatics analysis to determine the distribution of serotypes, sequence types, virulence genes, ARGs, plasmid sequences, integron sequences, and SPIs.
4.1. Serotyping and MLST
Relevant studies have confirmed the high accuracy of WGS-based serotyping [31]. It has been widely accepted as a promising tool for high-resolution typing of enteric pathogens [32]. It is gradually replacing the traditional methods of foodborne pathogen typing [33,34]. Our study identified the presence of three major serotypes based on the WGS data, namely S. Typhimurium (7/24;29.2%), S. Liverpool (5/24;20.8%), and S. London (4/24;16.7%). Previous studies have shown that S. Typhimurium and S. London are the most important bacterial types in Wenzhou [35,36,37], which is consistent with the results of the present study. However, these studies did not report on the emergence of S. Liverpool. Based on Enterobase cgMLST with Hierarchical Clustering (HierCC), S. Liverpool in our study appeared to be related to strains prevalent to the UK and USA. They were found simultaneously in both countries, and it is speculated that they may share a common ancestor. S. Liverpool in our study appeared to be more closely related to several strains in the UK, suggesting that it may have been introduced from the UK (see Figure 3 and Table. S3). As Salmonella can be transmitted through contaminated food in different regions, the risk of Salmonella imports has increased in recent years with the increase in global trade. The establishment of an international surveillance network would be beneficial in controlling the risk of international transmission of Salmonella.
The results of MLST of Salmonella strains based on seven housekeeping genes showed that none of the serotypes had multiple MLST types, except for S. Typhimurium. ST19 and ST34 are the two most common sequence types reported in multinational studies [38,39,40]. In contrast, ST1544 has been reported in only a few studies. In addition, based on the cgMLST with HierCC phylogenetic tree, the ST1544 strain in our study from raw poultry was associated with the prevalence of five strains isolated from wild animals in Yangzhou, China, two strains from human and food samples in Vietnam, and two strains from environmental samples in Cambodia (Figure 4 and Table. S4).
Our study reports for the first time the occurrence of ST1544 in the Wenzhou region, increasing the population diversity of S. Typhimurium in the region. Owing to extensive host contamination, its genetic characteristics for contaminating different hosts are unknown. In the future, it may develop into a novel contaminant in Southeast Asia and pose a threat to the coastal cities in eastern China.
4.2. Antimicrobial Resistance Determinants
ARGs analysis showed that these strains may be resistant to multiple antibiotics. High frequency of detection of aminoglycosides, tetracyclines, β-lactamases, chloramphenicol, and sulfonamides ARGs in the isolates from poultry samples, probably because amoxicillin, florfenicol, ciprofloxacin, sulfonamide, and streptomycin have been widely used to prevent or treat bacterial diseases in most poultry farms in China. The proportion of ESBL-producing strains reached 50%, which was significantly higher than that reported by Chen Sucai et al. in Wenzhou [41]. Notably, ESBLs can hydrolyze a variety of antibiotics such as penicillin, cephalosporins, and aztreonam. Secondly, its presence is more likely to lead to cross-resistance in bacteria [42,43], probably because the plasmids encoding ESBLs also carry resistance genes for quinolones, aminoglycosides, and other antibiotics [44]. However, all ESBL-producing strains in our study were multidrug-resistant (MDR), with aminoglycoside, quinolone, tetracycline, and chloramphenicol-resistance genes. A previous study showed that when CTX-M-55 was combined with TEM, it significantly reduced susceptibility toward piperacillin–tazobactam and cefotetan. When CTX-M-55, TEM, and SHV genes were present together, the strains were 100% resistant to both antibiotics [45]. This suggests that strains containing dual ESBL genotypes or above cansignificantly decrease antibiotic susceptibility and broaden the spectrum of antimicrobial resistance.
The high level of resistance to fluoroquinolones in strains can be attributed to the accumulation of point mutations in genes encoding cellular topoisomerases and the acquisition of several auxiliary mechanisms, such as efflux-pump-encoding qepA and oqxAB, Qnr proteins, and the aminoglycoside acetyltransferase aac(6′)-Ib-cr, that increase the level of expressed resistance [46]. Although these plasmid-mediated quinolone resistance determinants confer low levels of resistance toward quinolones and fluoroquinolones, they can assist in the emergence of other chromosomally encoded quinolone resistance mechanisms [47]. The aminoglycoside acetyltransferase aac(6’)-Ib-cr is a variant of aac(6’)-Ib that induces resistance toward aminoglycosides and fluoroquinolones. The remaining three appeared at high frequencies in our study, except for the efflux pump. However, the frequency of point mutations was as high as 83% (20/24), and S. London also contained aac(6’)-Ib-cr and qnrB6. S. Kentucky had most point mutations and contained qnrS1 This indicates that the fluoroquinolone resistance situation in our study is critical, especially for S. London and S. Kentucky. We recommend the rational use of fluoroquinolones in poultry farming and clinical practice to prevent the acceleration of this resistance mechanisms in the future. Furthermore, for the treatment of diseases caused by ciprofloxacin-resistant Salmonella, a third-generation cephalosporin, such as cefotaxime or ceftriaxone, is preferred [48]. Second, ciprofloxacin-resistant strains were predicted to carry ESBL at a frequency of 50% (11/22) in our study. Thus,it means that the scope of clinical drugs is further decreased, and new drugs may need to be developed to address this situation in the future.
In addition to ARGs, mobile genetic elements such as plasmids and integrons play a key role in the spread and persistence of antimicrobial resistance [49]. IncFIB (also known as the ColV plasmid) was the most common plasmid type in our study and may be related to virulence plasmids. A previous study found that when S. Kentucky acquired the IncFIB plasmid, it increased its ability to colonize the chicken cecum and caused significant extra-intestinal disease [50]. Here, IncFIB plasmids did not carry resistance genes, which may be due to the lack of such genes in virulence plasmids or the inability of the draft genome to assemble complete plasmid sequences [27]. Although most plasmids do not encode known ARGs, they can bind to other transposable elements to form MDR clusters, facilitating their spread [51].
Integrons can trap exogenous resistance genes, causing the spread of resistance genes among bacterial populations. Our results showed that 100% of S. London contains a complete class 1 integron regardless of the source. Each integron carried the same gene cassette arrays aadA16- dfrA27- ARR-3- aac(6’)-Ib-cr, which is associated with resistance toward aminoglycoside, trimethoprim, rifampicin, and fluoroquinolone. This indicates that the contribution of integrons to MDR in S. London is significant; therefore, the spread of resistance at the genetic level in S. London is concerning. Second, the quinolone-modifying enzyme genes aac(6’)-Ib-cr detected in 5 samples were all located on the integrons, suggesting that the integron is highly capable of capturing and propagating the aac(6’)-Ib-cr gene.
4.3. Virulence Determinants
The pathogenicity of Salmonella is closely associated with its virulence genes. A cluster of relatively concentrated virulence genes constitutes the SPI. At least 23 SPIs have been identified in the genus Salmonella, which plays a crucial role in the pathogenesis of the strains [52]. SPI-1 to SPI-5 were highly conserved. SPI-1 and SPI-2 were the two main virulence determinants of S. enterica and encoded the type III secretion system (T3SS). SPI-1 is required for host cell invasion and the induction of macrophage apoptosis; SPI-2 is required for Salmonella survival within macrophages and causes systemic infection; SPI-3 and SPI-4 are both associated with intracellular survival; and SPI-5 is associated with host cell invasion and inflammatory diseases. Other SPIs are less well studied than SPI-1 to SPI-5 but may also have specific functions, such as invasion and colonization [53,54]. In our study, the investigation of 24 strains showed that five SPIs, namely SPI-1, SPI-2, SPI-3, SPI-5, and SPI-9, were conserved in all strains. A previous study showed the prevalence of SPI-1 to SPI-5, SPI-13, and SPI-14 and the absence of SPI-7, SPI-8, and SPI-15 in all non-Salmonella typhi isolates. Nevertheless, SPI-8 was detected in one strain of S. Meleagridis and one strain of S. Corvallis in our study. To the best of our knowledge, this is the first study to report SPI-8 in these two serotypes.
Our research showed that the SPI profiles (P1–P9) varied according to the cgMLST clusters (Figure 2). For example, the dominant SPI profiles, P4, P5, and P8, were represented among clusters carrying S. Goldcoast (ST358), Liverpool (ST1959), and London (ST155), respectively. More importantly, the three STs in S. Typhimurium corresponded to three SPI profiles, suggesting that SPI profiles are likely to influence the classification of Salmonella genotypes. Furthermore, a strain of S. Corvallis and a strain of S. Meleagridis with similar phylogenetic relationships shared the same SPI profiles, indicating that serotypes with close genetic relationships may have the same distribution of virulence factors. The more closely related S. London and S. Typhimurium possessed broader SPI profiles, suggesting that they may have greater pathogenic potential.
Fimbriae are the most common adhesion systems that play a major role in the pathogenesis of Salmonella [55]. It has been shown that fimbriae represent a source of diversity among Salmonella serotypes, which is differentially expressed across serotypes and found in specific patterns [56]. In our study, the fimbrial operons bcf, fim, inv, csg, stb, and sth were present in all isolates. These genes may be part of a core gene that is key for Salmonella to invade the host and cause infection. Other operons lpf, peg, saf, ste, stf, and sti exist depending on serotypes (Table. S2). Virulence plasmids of Salmonella are important for systemic infection in animal models. Spv genes may accelerate Salmonella growth in host cells and affect the interactions between Salmonella and the host immune system [57]. In our study, the spv gene was found only in S. Typhimurium, which may also be responsible for making it more pathogenic. Moreover, the phage-associated gene sodCI, prophage-encoding gene gogB, macrophage-inducible gene mig-5, plasmid-mediated gene rck, and plasmid-encoded fimbrial gene sefABCD were only detected in S. Typhimurium. These results indicate that S. Typhimurium is the most virulent strains of Salmonella causing poultry infections and threatening food safety in Wenzhou, China.
4.4. Genetic Diversity of the Salmonella Isolates
In our study, genetic relationships between the strains were constructed using cgMLST on the Enterobase platform. Construction of phylogenetic clusters of isolates based on 3002 core-genome loci. This clustering by serotype was also presented in the study of Hassena et al. [18]. Due to the dispersion of serotypes, more detailed clustering could not be observed. However, the sub-clustering of S. Typhimurium and S. London indicated that these two serotypes have richer genetic diversity in the region. In particular, more distant nodal linkages were observed in S. Typhimurium, suggesting greater allelic variation between strains.
The cgMLST scheme uses a consistent set of conserved loci and allele assignments with the advantage of being easily and consistently applied across laboratories and jurisdictions. In addition, EnteroBase supports HierCC, a new approach that supports the analyses of population structures based on cgMLST at multiple-level resolutions. EnteroBase reported the most reliable Salmonella-specific subset of the HierCC clusters [22]. In our study, S. Liverpool, first identified in the Wenzhou area, appears to be closely related to several strains in the UK (HC20), suggesting that it may have been introduced from the UK (Figure 3). The discovery of this important transmission route would be beneficial for monitoring Salmonella in Wenzhou.
5. Conclusions
In this study, we explored the association between Salmonella serotypes and antimicrobial resistance, genotypes and pathogenic potential and confirmed the presence of an imported risk. Salmonella contains a variety of ARGs, SPIs, virulence plasmids, multidrug resistance plasmids, phages, and integrons that influence its classification of Salmonella and shed light on the causes of the severity of this bacterial disease. Hence, obtaining the genome sequence of Salmonella will not only help to improve the reproducibility and accessibility of genomic analyses, but also contribute to future surveillance and epidemiological investigations of salmonellosis. The data obtained in this study can provide reference for the prevention and control of bacterial diseases and dynamic monitoring.
Author Contributions
Yafang Jin: Conceptualization, Data curation, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. Yi Li: Formal analysis, Data curation, Resources, Writing – review & editing. Shaojie Huang: Formal analysis, Data curation, Resources, Writing – review & editing. Chengji Hong: Writing – review & editing. Xucong Feng: Writing – review & editing. Huidi Cai: Writing – review & editing. Yanmei Xia: Writing – review & editing. Shengkai Li: Writing – review & editing, Visualization. Leyi Zhang: Conceptualization, Supervision, Project administration. Yongliang Lou: Conceptualization, Supervision, Project administration. Wanchun Guan: Conceptualization, Data curation, Formal analysis, Supervision, Project administration.
Funding
This study was supported by the National Major Infectious Disease Prevention Projects (2018ZX10201001), the Wenzhou Key Laboratory of Sanitary Microbiology, the Key Discipline of Zhejiang Province in Medical Technology (First Class, Category A), the Key Discipline of Zhejiang Province in Biology (First Class, Category B).
Acknowledgments
We thank anonymous reviewers whose comments and suggestions helped improve this manuscript.
Conflicts of Interest
The authors had no conflicts of interest to declare.
References
- Aduah, M.; Adzitey, F.; Amoako, D.G.; Teye, G.A.; Shariff, A.H.M.; Huda, N. Low Prevalence of Antibiotic-Resistant Salmonella enterica in Ready-to-Eat (RTE) Meats in Ghana Is Associated with Good Vendors’ Knowledge of Meat Safety. Foods. 2021, 10, 1011. [Google Scholar] [CrossRef] [PubMed]
- Clavijo, V.; Baquero, D.; Hernandez, S.; Farfan, J.; Arias, J.; Arévalo, A.; Donado-Godoy, P.; Vives-Flores, M. Phage cocktail SalmoFREE® reduces Salmonella on a commercial broiler farm. Poultry science. 2019, 98, 5054–5063. [Google Scholar] [CrossRef]
- Majowicz, S.E.; Musto, J.; Scallan, E.; Angulo, F.J.; Kirk, M.; OBrien, S.J.; Jones, T.F.; Fazil, A.; Hoekstra, R.M. The global burden of nontyphoidal Salmonella gastroenteritis. Clinical Infectious Diseases. 2010, 50, 882–889. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO estimates of the global burden of foodborne diseases: foodborne disease burden epidemiology reference group 2007–2015. Available online: https://apps.who.int/iris/bitstream/handle/10665/199350/9789241565165_eng.pdf?sequence=1 (accessed on November 13).
- Grimont, P.A.D.; Weill, F.X. Antigenic formulae of the Salmonella serovars. WHO collaborating centre for reference and research on Salmonella. 2007, 9, 1–166. [Google Scholar]
- Issenhuth-Jeanjean, S.; Roggentin, P.; Mikoleit, M.; Guibourdenche, M.; De Pinna, E.; Nair, S.; Fields, P.I.; Weill, F.-X. Supplement 2008–2010 (no. 48) to the white–Kauffmann–Le minor scheme. Research in microbiology. 2014, 165, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Guibourdenche, M.; Roggentin, P.; Mikoleit, M.; Fields, P.I.; Bockemühl, J.; Grimont, P.A.D.; Weill, F.-X. Supplement 2003-2007 (No. 47) to the White-Kauffmann-Le Minor scheme. Research in Microbiology. 2010, 161, 26–29. [Google Scholar] [CrossRef]
- Fierer, J.; Guiney, D.G. Diverse virulence traits underlying different clinical outcomes of Salmonella infection. Journal of Clinical Investigation. 2001, 107, 775–780. [Google Scholar] [CrossRef]
- Jajere, S.M. A review of Salmonella enterica with particular focus on the pathogenicity and virulence factors, host specificity and antimicrobial resistance including multidrug resistance. Veterinary World. 2019, 12. [Google Scholar] [CrossRef]
- Ngoi, S.T.; Teh, C.S.J.; Chai, L.C.; Thong, K.L. Overview of Molecular Typing Tools for The Characterization of Salmonella enterica in Malaysia. Biomedical and Environmental Sciences. 2015, 28, 764. [Google Scholar] [CrossRef]
- Kombade, S.; Kaur, N. Pathogenicity Island in Salmonella. IntechOpen. 2021. [Google Scholar] [CrossRef]
- Siriken, B. Salmonella pathogenicity islands. Mikrobiyoloji Bulteni. 2013, 47, 181–188. [Google Scholar] [CrossRef] [PubMed]
- van Asten, A.J.; van Dijk, J.E. Distribution of “classic” virulence factors among Salmonella spp. FEMS Immunology and Medical Microbiology. 2005, 44, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Xiang, Y.; Qiu, S. Resistance in Enteric Shigella and nontyphoidal Salmonella: emerging concepts. Current Opinion In Infectious Diseases. 2023, 36, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gao, S.; Chang, Y.; Su, M.; Xie, Y.; Sun, S. Occurrence and Characterization of Salmonella Isolated from Large-Scale Breeder Farms in Shandong Province, China. BioMed Research International. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Inbaraj, S.; Agrawal, R.K.; Thomas, P.; Mohan, C.; R.K, S.A.; Verma, M.R.; Chaudhuri, P. Antimicrobial resistance in Indian isolates of non typhoidal Salmonella of livestock, poultry and environmental origin from 1990-2017. Comparative Immunology, Microbiology and Infectious Diseases. 2022, 80. [Google Scholar] [CrossRef]
- Li, B.; Yang, X.; Tan, H.; Ke, B.; He, D.; Wang, H.; Chen, Q.; Ke, C.; Zhang, Y. Whole genome sequencing analysis of Salmonella enterica serovar Weltevreden isolated from human stool and contaminated food samples collected from the Southern coastal area of China. International Journal of Food Microbiology. 2018, 266, 317–323. [Google Scholar] [CrossRef]
- Hassena, A.B.; Haendiges, J.; Zormati, S.; Guermazi, S.; Gdoura, R.; Gonzalez-Escalona, N.; Siala, M. Virulence and resistance genes profiles and clonal relationships of non-typhoidal food-borne Salmonella strains isolated in Tunisia by whole genome sequencing. International Journal of Food Microbiology. 2020, 337. [Google Scholar] [CrossRef]
- Pornsukarom, S.; Vliet, A.H.M.v.; Thakur, S. Whole genome sequencing analysis of multiple Salmonella serovars provides insights into phylogenetic relatedness, antimicrobial resistance, and virulence markers across humans, food animals and agriculture environmental sources. BMC Genomics. 2018, 19. [Google Scholar] [CrossRef]
- Punina, N.V.; Makridakis, N.M.; Remnev, M.A.; Topunov, A.F. Whole-genome sequencing targets drug-resistant bacterial infections. Hum Genomics. 2015, 9. [Google Scholar] [CrossRef]
- Ibrahim, G.M.; Morin, P.M. Salmonella Serotyping Using Whole Genome Sequencing. Front Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.-F.; Mohamed, K.; Fan, Y.; Achtman, M.; Brown, D.; Chattaway, M.; Dallman, T.; Delahay, R.; Kornschober, C. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome research. 2020, 30, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yin, Y.; Jones, M.B.; Zhang, Z.; Kaiser, B.L.D.; Dinsmore, B.A.; Fitzgerald, C.; Fields, P.I.; Deng, X. Salmonella Serotype Determination Utilizing High-Throughput Genome Sequencing Data. Journal of Clinical Microbiology. 2015, 53, 1685. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. Journal of Antimicrobial Chemotherapy. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. Journal of Antimicrobial Chemotherapy. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009, 10. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemothererapy. 2014, 58. [Google Scholar] [CrossRef] [PubMed]
- Cury, J.; Jove, T.; Touchon, M.; Neron, B.; Rocha, E.P. Identification and analysis of integrons and cassette arrays in bacterial genomes. Nucleic acids research. 2016, 44, 4539–4550. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Research. 2005, 33, D325–D328. [Google Scholar] [CrossRef]
- Roer, L.; Hendriksen, R.S.; Leekitcharoenphon, P.; Lukjancenko, O.; Kaas, R.S.; Hasman, H.; Aarestrup, F.M. Is the Evolution of Salmonella enterica subsp. enterica Linked to Restriction-Modification Systems? MSystems. 2016, 1, e00009-00016. [Google Scholar] [CrossRef]
- Zhang, L.; Shen, Q.; Zhang, C.; Zhao, Q.; Cui, M.; Li, T.; Cheng, M. Predictive analysis of whole genome sequencing for Salmonella serotype and antimicrobial resistance phenotypes. Acta Microbiologica Sinica. 2021, 61, 4038–4047. [Google Scholar] [CrossRef]
- Lindsay, C.; Flint, J.; Lilly, K.; Hope, K.; Wang, Q.; Howard, P.; Sintchenko, V.; Durrheim, D.N. Retrospective use of whole genome sequencing to better understand an outbreak of Salmonella enterica serovar Mbandaka in New South Wales, Australia. Western Pacific Surveillance and Response Journal. 2017, 9, 20–25. [Google Scholar] [CrossRef]
- Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Crombé, F.; Rauw, K.D.; Piérard, D.; Marchal, K.; Vanneste, K.; Roosens, N.H.C.; et al. The Benefits of Whole Genome Sequencing for Foodborne Outbreak Investigation from the Perspective of a National Reference Laboratory in a Smaller Country. Foods. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.J.; Neale, B.M.; Huang, H.; Werling, D.M.; Dong, S.; Abecasis, G.; Arguello, P.A.; Blangero, J.; Boehnke, M.; Daly, M.J.; et al. Whole genome sequencing in psychiatric disorders: the WGSPD consortium. Nature Neuroscience. 2018, 21. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Zhang, L.; Li, Y.; Hu, Y.; Wu, Y. Contamination and characteristics of foodborne Salmonella in Wenzhou. Chinese Journal of Food Hygiene. 2019, 31, 461–464. [Google Scholar] [CrossRef]
- GUO, S.; LIN, D.; WANG, L.; HU, H. Monitoring the Results of Foodborne Diseases in Sentinel Hospitals in Wenzhou City, China from 2014 to 2015. Iranian Journal of Public Health. 2018, 47, 674–681. [Google Scholar]
- Lin, D.; Wang, L.; Shan, R.; Cai, Y.; Gao, S.; Li, Y.; Zhang, L.; Sun, B.; Shang, G.; Wu, K. Epidemiological analysis of 317 cases with foodborne diseases in Wenzhou in 2014. Chinese Preventive Medicine. 2017, 18. [Google Scholar] [CrossRef]
- Sun, J.; Ke, B.; Huang, Y.; He, D.; Li, X.; Liang, Z.; Ke, C. The molecular epidemiological characteristics and genetic diversity of salmonella typhimurium in Guangdong, China, 2007-2011. PLoS One. 2014, 9, e113145. [Google Scholar] [CrossRef]
- Arai, N.; Sekizuka, T.; Tamamura-Andoh, Y.; Barco, L.; Hinenoya, A.; Yamasaki, S.; Iwata, T.; Watanabe-Yanai, A.; Kuroda, M.; Akiba, M.; et al. Identification of a Recently Dominant Sublineage in Salmonella 4,[5],12:i:- Sequence Type 34 Isolated From Food Animals in Japan. Front Microbiol. 2021, 12, 690947. [Google Scholar] [CrossRef]
- Carroll, L.M.; Pierneef, R.; Mathole, M.; Matle, I. Genomic Characterization of Endemic and Ecdemic Non-typhoidal Salmonella enterica Lineages Circulating Among Animals and Animal Products in South Africa. Frontiers in Microbiology. 2021, 4, 748611. [Google Scholar] [CrossRef]
- Chen, S.; Shen, L. Distribution of serotypes and drug resistance of ESBLs-producing Salmonella in Wenzhou. Disease Surveillance. 2019, 34, 62–65. [Google Scholar] [CrossRef]
- Farrag, H.A.; Abdallah, N.; Shehata, M.M.K.; Awad, E.M. Natural outer membrane permeabilizers boost antibiotic action against irradiated resistant bacteria. Journal of Biomedical Science. 2019, 26, 69. [Google Scholar] [CrossRef]
- Hassuna, N.A.; Khairalla, A.S.; Farahat, E.M.; Hammad, A.M.; Abdel-Fattah, M. Molecular characterization of Extended-spectrum β lactamase- producing E. coli recovered from community-acquired urinary tract infections in Upper Egypt. Scientific Reports. 2020, 10, 2772. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Sirous, M.; Javaherizadeh, H.; Motamedifar, M.; Saki, M.; Veisi, H.; Ebrahimi, S.; SeyedMohammadi, S.; Hashemzadeh, M. Antibiotic resistance pattern and molecular characterization of extended-spectrum β-lactamase producing enteroaggregative Escherichia coli isolates in children from southwest Iran. Infection and Drug Resistance. 2018, 8, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Gou, J.; Guo, X.; Cao, Z.; Li, Y.; Jiao, H.; He, X.; Ren, Y.; Tian, F. Genetic contexts related to the diffusion of plasmid-mediated CTX-M-55 extended-spectrum beta-lactamase isolated from Enterobacteriaceae in China. Annals of Clinical Microbiology and Antimicrobials. 2018, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Mechanisms of resistance and clinical relevance of resistance to β-lactams, glycopeptides, and fluoroquinolones. Mayo Clinic Proceedings. 2012, 87, 198–208. [Google Scholar] [CrossRef]
- Poirel, L.; Cattoir, V.; Nordmann, P. Plasmid-Mediated Quinolone Resistance; Interactions between Human, Animal, and Environmental Ecologies. Frontiers in Microbiology. 2012, 3, 24. [Google Scholar] [CrossRef]
- Dutta, P.; Mitra, U.; Dutta, S.; De, A.; Chatterjee, M.K.; Bhattacharya, S.K. Ceftriaxone therapy in ciprofloxacin treatment failure typhoid fever in children. Indian Journal of Medical Research. 2001, 113, 210–213. [Google Scholar] [PubMed]
- Li, S.; Zhang, S.; Baert, L.; Jagadeesan, B.; Ngom-Bru, C.; Griswold, T.; Katz, L.S.; Carleton, H.A.; Deng, X. Implications of mobile genetic elements for Salmonella enterica single-nucleotide polymorphism subtyping and source tracking investigations. Applied environmental microbiology. 2019, 85, e01985-01919. [Google Scholar] [CrossRef]
- Johnson, T.J.; Thorsness, J.L.; Anderson, C.P.; Lynne, A.M.; Foley, S.L.; Han, J.; Fricke, W.F.; McDermott, P.F.; White, D.G.; Khatri, M. Horizontal gene transfer of a ColV plasmid has resulted in a dominant avian clonal type of Salmonella enterica serovar Kentucky. PLoS One. 2010, 5, e15524. [Google Scholar] [CrossRef]
- Miriagou, V.; Carattoli, A.; Fanning, S. Antimicrobial resistance islands: resistance gene clusters in Salmonella chromosome and plasmids. Microbes and Infection. 2006, 8, 1923–1930. [Google Scholar] [CrossRef]
- Hayward, M.R.; Jansen, V.A.; Woodward, M.J. Comparative genomics of Salmonella enterica serovars Derby and Mbandaka, two prevalent serovars associated with different livestock species in the UK. BMC Genomics. 2013, 14, 365. [Google Scholar] [CrossRef] [PubMed]
- Solnick, J.V.; Young, G.M. Chapter 10 - Bacterial Pathogenicity Islands and Infectious Diseases. Horizontal Gene Transfer. 2002, 111–121. [Google Scholar] [CrossRef]
- Hensel, M. Evolution of pathogenicity islands of Salmonella enterica. International Journal of Medical Microbiology. 2004, 294, 95–102. [Google Scholar] [CrossRef]
- Dufresne, K.; Daigle, F. Salmonella fimbriae: What is the clue to their hairdo. Current Topics in Salmonella Salmonellosis. 2017, 59–79. [Google Scholar] [CrossRef]
- Dufresne, K.; Saulnier-Bellemare, J.; Daigle, F. Functional analysis of the chaperone-usher fimbrial gene clusters of Salmonella enterica serovar Typhi. Frontiers in cellular infection microbiology. 2018, 8, 26. [Google Scholar] [CrossRef]
- Gulig, P.A.; Danbara, H.; Guiney, D.G.; Lax, A.J.; Norel, F.; Rhen, M. Molecular analysis of spv virulence genes of the Salmonella virulence plasmids. Molecular microbiology. 1993, 7, 825–830. [Google Scholar] [CrossRef]
Figure 1.
The frequency of ARGs in different drug classes and the correlation between serotype and drug class were described. ARGs of 24 Salmonella isolated from Wenzhou are obtained using ResFinder. The shades of color in the legend represent the presence (1) absence (0) of that class of genes.
Figure 1.
The frequency of ARGs in different drug classes and the correlation between serotype and drug class were described. ARGs of 24 Salmonella isolated from Wenzhou are obtained using ResFinder. The shades of color in the legend represent the presence (1) absence (0) of that class of genes.
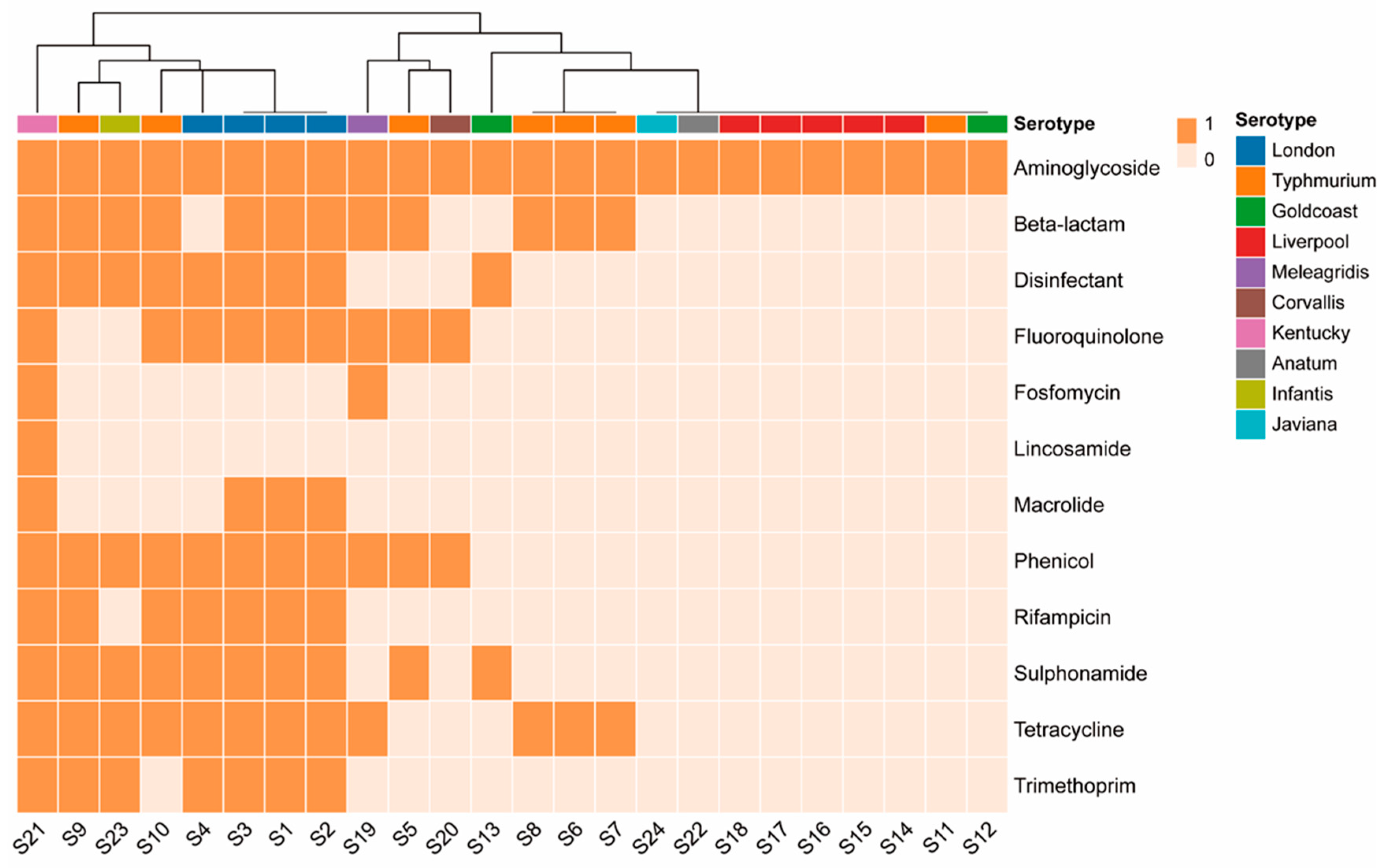
Figure 2.
cgMLST tree and SPI profiles of 24 Salmonella isolates in this study. cgMLST tree using the 3002 locus cgMLST scheme provided by EnteroBase. Assinment of serotypes into four main clusters contained the main serotypes detected; S. Typhimurium, Liverpool, London and Goldcoast, respectively defined as C1, C2, C3 and C4. SPI profiles were obtained using the SPIFinder. Varied according to cgMLST clusters, nine SPI profiles were identified named P1 to P9.
Figure 2.
cgMLST tree and SPI profiles of 24 Salmonella isolates in this study. cgMLST tree using the 3002 locus cgMLST scheme provided by EnteroBase. Assinment of serotypes into four main clusters contained the main serotypes detected; S. Typhimurium, Liverpool, London and Goldcoast, respectively defined as C1, C2, C3 and C4. SPI profiles were obtained using the SPIFinder. Varied according to cgMLST clusters, nine SPI profiles were identified named P1 to P9.
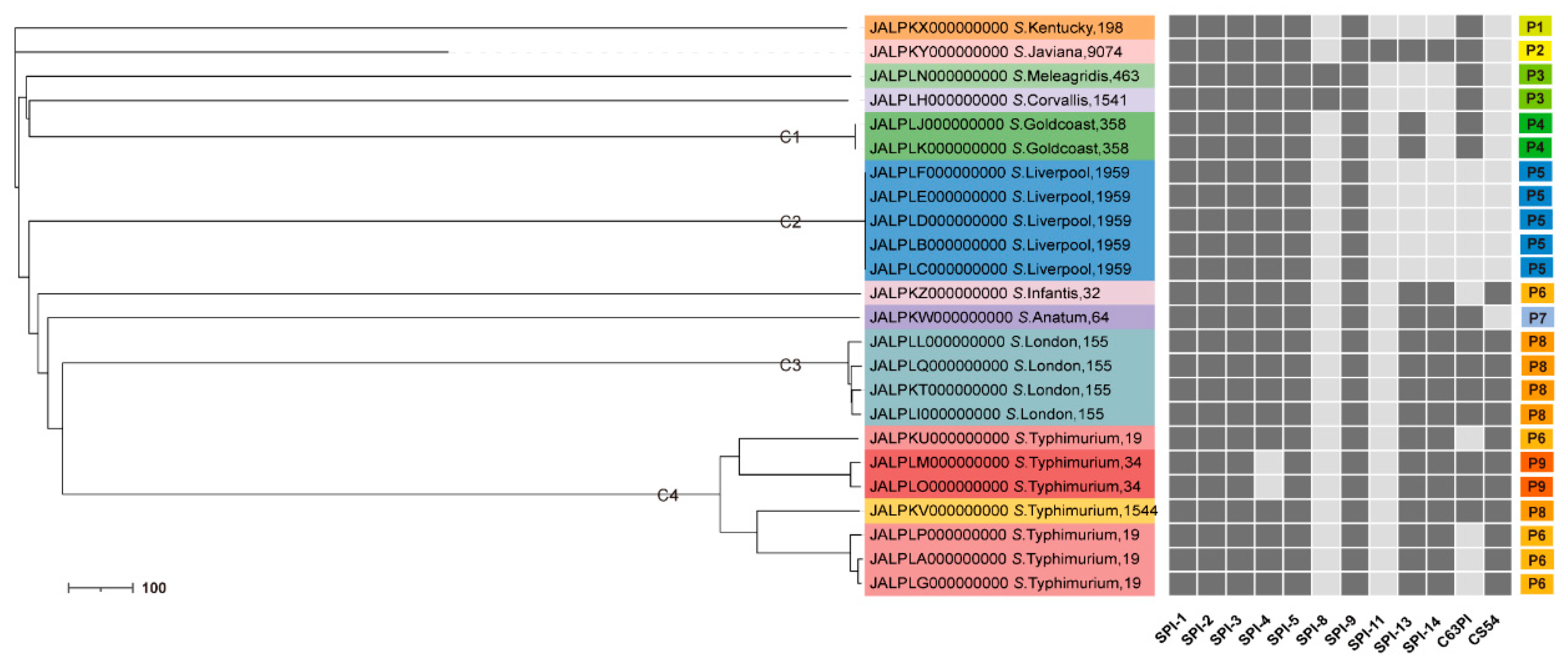
Figure 3.
Phylogenetic relationships among the S. Liverpool(ST1959)by cgMLST analysis based on country of origin (A) and HC20 (68) of HierCC (B). Minimum spanning tree of 314 S. Liverpool strains was generated from cgMLST+HierCC data using the 3002 locus cgMLST scheme provided by EnteroBase. The isolates within this analysis were from S14-S18 (position of the red circle in the picture) and its closely related strains. A: The colors distinguish the countries of origin of the strains, with the majority coming from the United States, followed by the United Kingdom. The red rectangular dashed boxes indicate the clustering of the five S. Liverpool strains (S14-S18) in our study with strains from the UK and the US. B: All strains were obtained by screening for the HC20 (68) cutoff value. Thus, all strains differed by no more than 20 alleles. C: A magnified view of the red rectangular dashed boxes in Figure A. In the magnified view, they appear to be closer to the UK strains.
Figure 3.
Phylogenetic relationships among the S. Liverpool(ST1959)by cgMLST analysis based on country of origin (A) and HC20 (68) of HierCC (B). Minimum spanning tree of 314 S. Liverpool strains was generated from cgMLST+HierCC data using the 3002 locus cgMLST scheme provided by EnteroBase. The isolates within this analysis were from S14-S18 (position of the red circle in the picture) and its closely related strains. A: The colors distinguish the countries of origin of the strains, with the majority coming from the United States, followed by the United Kingdom. The red rectangular dashed boxes indicate the clustering of the five S. Liverpool strains (S14-S18) in our study with strains from the UK and the US. B: All strains were obtained by screening for the HC20 (68) cutoff value. Thus, all strains differed by no more than 20 alleles. C: A magnified view of the red rectangular dashed boxes in Figure A. In the magnified view, they appear to be closer to the UK strains.
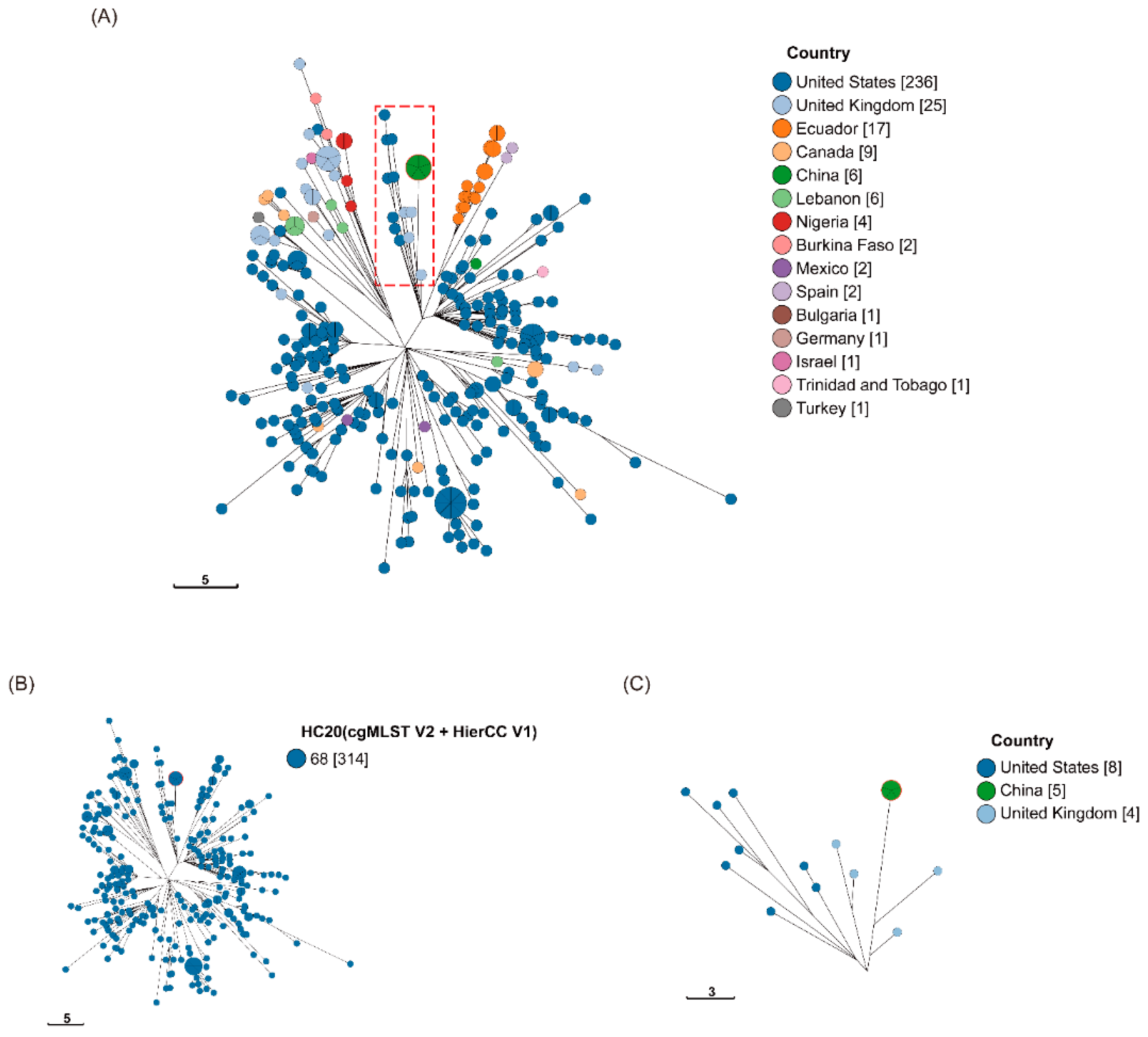
Figure 4.
Phylogenetic relationships among the S. Typhimurium(ST1544)by cgMLST analysis based on country of origin (A) and HC100 (23441) of HierCC (B). Minimum spanning tree of 13 S. Typhimurium strains was generated from cgMLST+HierCC data using the 3002 locus cgMLST scheme provided by EnteroBase. The isolates within this analysis were from S11 (position of the red circle in the picture) and its closely related strains. A: The colors distinguish the countries of origin of the strains: China, dark blue; Cambodia, light blue; Vietnam, orange. B: All strains were obtained by screening for the HC100 (23441) cutoff value. Thus, all strains differed by no more than 100 alleles, suggesting that ST1544 in our study was associated with the prevalence of these strains.
Figure 4.
Phylogenetic relationships among the S. Typhimurium(ST1544)by cgMLST analysis based on country of origin (A) and HC100 (23441) of HierCC (B). Minimum spanning tree of 13 S. Typhimurium strains was generated from cgMLST+HierCC data using the 3002 locus cgMLST scheme provided by EnteroBase. The isolates within this analysis were from S11 (position of the red circle in the picture) and its closely related strains. A: The colors distinguish the countries of origin of the strains: China, dark blue; Cambodia, light blue; Vietnam, orange. B: All strains were obtained by screening for the HC100 (23441) cutoff value. Thus, all strains differed by no more than 100 alleles, suggesting that ST1544 in our study was associated with the prevalence of these strains.
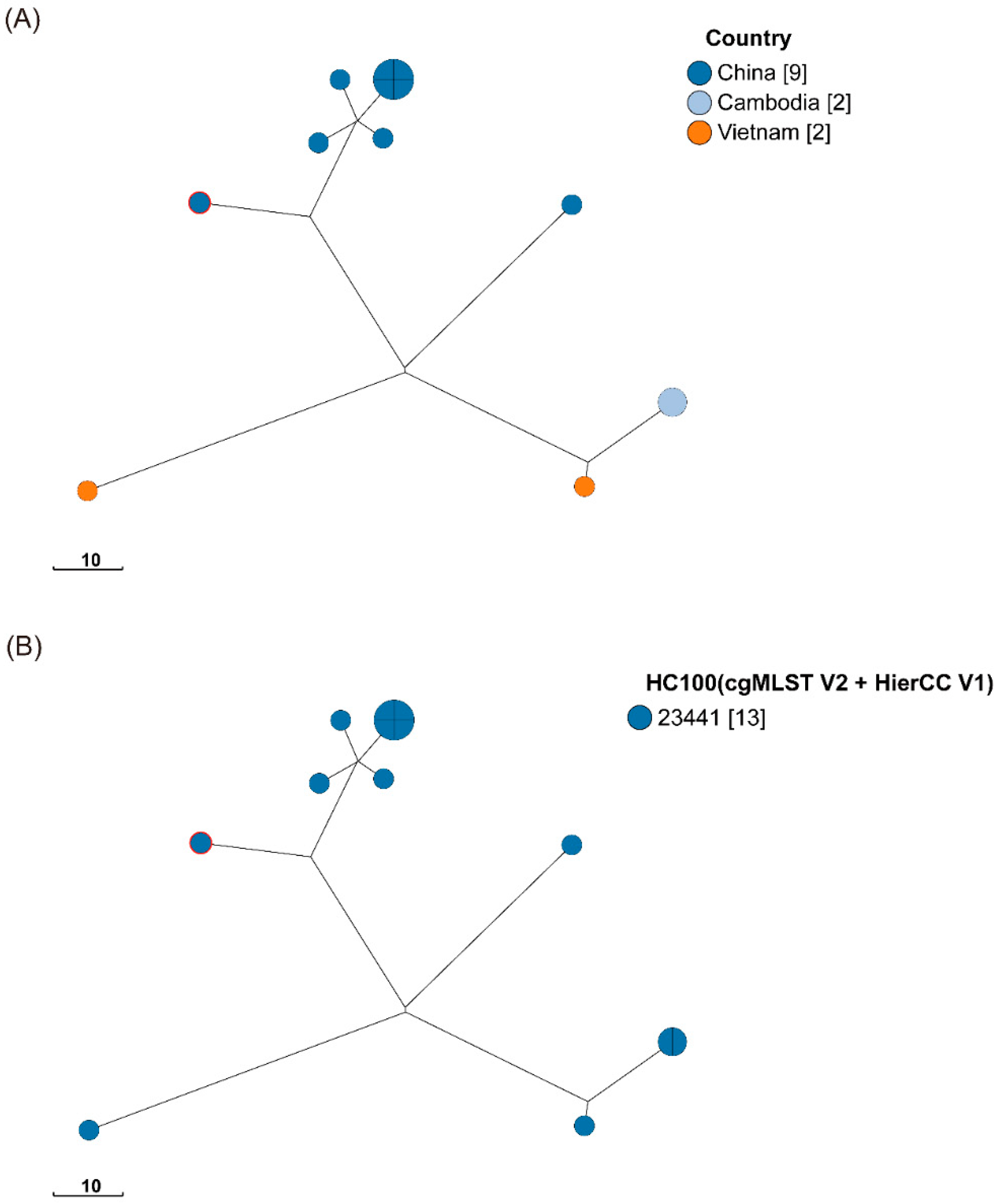

Table 1.
Sources, serotype and antimicrobial resistance genes (ARGs) of Salmonella (Location *Wenzhou, Year *2020, Species *S. enterica).
Table 1.
Sources, serotype and antimicrobial resistance genes (ARGs) of Salmonella (Location *Wenzhou, Year *2020, Species *S. enterica).
| Sample | Genome accession | Source | serotype | ST | ARG | QRDR | |
|---|---|---|---|---|---|---|---|
| GyrA | ParC | ||||||
| S1 | JALPKT000000000 | raw poultry | London | 155 | aac(3)-Iid, aac(6’)-Iaa, aac(6’)-Ib-cr, aadA16, aph(3’‘)-Ib, aph(6)-Id, blaTEM-1B, qnrB6, tet(A), sul1, sul2, catA2, floR, ARR-3, dfrA27, qacE, mph(A) | - | T57S |
| S2 | JALPLI000000000 | raw poultry | London | 155 | aac(3)-Iid, aac(6’)-Iaa, aac(6’)-Ib-cr,aadA16, aph(3’‘)-Ib, aph(6)-Id, blaTEM-1B, qnrB6, tet(A), sul1, sul2, floR, ARR-3, dfrA27, qacE, mph(A) | - | T57S |
| S3 | JALPLL000000000 | raw poultry | London | 155 | aac(3)-Iid, aac(6’)-Iaa, aac(6’)-Ib-cr,aadA16, aph(3’‘)-Ib, aph(6)-Id, blaTEM-1B, qnrB6, tet(A), sul1, sul2, floR, ARR-3, dfrA27, qacE, mph(A) | - | T57S |
| S4 | JALPLQ000000000 | food | London | 155 | aac(6’)-Iaa, aac(6’)-Ib-cr, aadA16, aph(3’‘)-Ib, aph(6)-Id, qnrB6, tet(A), sul1, sul2, floR, ARR-3, dfrA27, qacE | - | T57S |
| S5 | JALPKU000000000 | raw poultry | Typhimurium | 19 | aac(6’)-Iaa, blaCTX-M-65, OqxA, OqxB, qnrS2, sul1, floR | - | - |
| S6 | JALPLA000000000 | raw poultry | Typhimurium | 19 | aac(6’)-Iaa, aph(3’‘)-Ib, aph(6)-Id, blaTEM-1B, tet(A) | S83Y | - |
| S7 | JALPLG000000000 | raw poultry | Typhimurium | 19 | aac(6’)-Iaa, aph(3’‘)-Ib, aph(6)-Id, blaTEM-1B, tet(A) | S83Y | - |
| S8 | JALPLP000000000 | raw poultry | Typhimurium | 19 | aac(6’)-Iaa, aph(3’‘)-Ib, aph(6)-Id, blaTEM-1B, tet(A) | S83Y | - |
| S9 | JALPLM000000000 | raw poultry | Typhimurium | 34 | aac(3)-Iid, aac(6’)-Iaa, aph(3’‘)-Ib, aph(6)-Id, blaTEM-1B, tet(B), sul1, sul2, floR, ARR-3, dfrA27, qacE | - | - |
| S10 | JALPLO000000000 | raw poultry | Typhimurium | 34 | aac(3)-IV, aac(6’)-Iaa, aac(6’)-Ib-cr, aph(3’‘)-Ib, aph(4)-Ia, aph(6)-Id, blaOXA-1, blaTEM-1B, tet(B), sul1, sul2, catB3, ARR-3, qacE | - | - |
| S11 | JALPKV000000000 | raw poultry | Typhimurium | 1544 | aac(6’)-Iaa | - | - |
| S12 | JALPLJ000000000 | raw poultry | Goldcoast | 358 | aac(6’)-Iaa | - | T57S |
| S13 | JALPLK000000000 | raw poultry | Goldcoast | 358 | aac(6’)-Iaa, aac(3)-Iid, sul1 , qacE, fosA7 | - | T57S |
| S14 | JALPLB000000000 | human stool | Liverpool | 1959 | aac(6’)-Iaa | - | T57S |
| S15 | JALPLC000000000 | human stool | Liverpool | 1959 | aac(6’)-Iaa | - | T57S |
| S16 | JALPLD000000000 | human stool | Liverpool | 1959 | aac(6’)-Iaa | - | T57S |
| S17 | JALPLE000000000 | human stool | Liverpool | 1959 | aac(6’)-Iaa | - | T57S |
| S18 | JALPLF000000000 | human stool | Liverpool | 1959 | aac(6’)-Iaa | - | T57S |
| S19 | JALPLN000000000 | raw poultry | Meleagridis | 463 | aac(6’)-Iaa, aph(3’)-Ia, blaTEM-1A, OqxA, OqxB, qnrS1, tet(A), floR | - | T57S |
| S20 | JALPLH000000000 | raw poultry | Corvallis | 1541 | aac(6’)-Iaa, qnrS1, floR | - | T57S |
| S21 | JALPKX000000000 | raw poultry | Kentucky | 198 | aac(3)-Id, aac(3)-Iid, aac(6’)-Iaa, aadA17, aadA7, aph(3’)-Ia, rmtB, blaCTX-M-55, blaTEM, qnrS1, tet(A), sul1, floR, ARR-2, dfrA14, qacE, mph(A), fosA3, lnu(F) | S83F, D87N | T57S,S80I |
| S22 | JALPKW000000000 | raw poultry | Anatum | 64 | aac(6’)-Iaa | - | T57S |
| S23 | JALPKZ000000000 | raw poultry | Infantis | 32 | aac(3)-IV, aac(6’)-Iaa, aadA1, aph(4)-Ia, blaCTX-M-65, tet(A), sul1, floR, dfrA14, qacE | D87Y | T57S |
| S24 | JALPKY000000000 | raw poultry | Javiana | 9074 | aac(6’)-Iaa | - | T57S |
ST: sequence type, QRDR: Quinolone resistance determining region.
Table 2.
Plasmid and I Integron of 24 Salmonella isolates.
| Sample | serotype | plasmids | Gene Found on plasmids | presence of Class I Integron | Gene Cassette Found on Integron |
|---|---|---|---|---|---|
| S1 | London | IncFIB(K) | - | 1 | aadA16, dfrA27, ARR-3 , aac(6’)-Ib-cr |
| S2 | London | IncFIB(K) | - | 1 | aadA16, dfrA27, ARR-3 , aac(6’)-Ib-cr |
| S3 | London | IncFIB(K), IncI1-I(Alpha) | - | 1 | aadA16, dfrA27, ARR-3 , aac(6’)-Ib-cr |
| S4 | London | IncFIB(K) | - | 1 | aadA16, dfrA27, ARR-3 , aac(6’)-Ib-cr |
| S5 | Typhimurium | IncHI2A, IncHI2, IncFIB(S), IncFII(S) |
- | - | - |
| S6 | Typhimurium | IncFIB(S), IncFII(S) | - | - | - |
| S7 | Typhimurium | IncFIB(S), IncFII(S) | - | - | - |
| S8 | Typhimurium | IncFIB(S), IncFII(S) | - | - | - |
| S9 | TyphimuriumTyphimurium | IncQ1 | sul2,aph(6)-Id,aph(3’‘)-Ib | - | - |
| S10 | Typhimurium | IncHI2A, IncHI2, IncFIB(S), IncFII(S), IncQ1 |
- | CALIN | aac(6’)-Ib-cr |
| S11 | Typhimurium | IncFIB(S), IncFII(S) | - | - | - |
| S12 | Goldcoast | - | - | - | - |
| S13 | Goldcoast | - | - | - | - |
| S14 | Liverpool | - | - | - | - |
| S15 | Liverpool | - | - | - | - |
| S16 | Liverpool | - | - | - | - |
| S17 | Liverpool | - | - | - | - |
| S18 | Liverpool | - | - | - | - |
| S19 | Meleagridis | IncFIA(HI1), IncFIB(K) | - | - | - |
| S20 | Corvallis | - | - | - | - |
| S21 | Kentucky | - | - | - | - |
| S22 | Anatum | IncFII(p96A), Col(pHAD28), Col440II |
- | - | - |
| S23 | Infantis | IncFIB(pN55391) | - | 1 | aadA1 |
| S24 | Javiana | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



