
Submitted:
13 September 2024
Posted:
16 September 2024
You are already at the latest version
Abstract
Cellular therapies exhibit immense potential in treating complex diseases with sustained response. Manufacture of cell therapies involves purification and engineering of specific cells from a donor or patient to achieve a therapeutic response upon injection. Magnetic cell sorting targeting the presence or absence of surface markers is commonly used for upfront purification. However, emerging research is showing that optimal therapeutic phenotypes are characterized not only by the presence or absence of specific antigens but also on antigen density. Unfortunately, current cell purification tools like magnetic or fluorescence activated cell sorting lack the resolution to differentiate populations based on antigen density while maintaining scalability. Utilizing a technique known as Digital Magnetic Sorting (DMS) we demonstrate proof of concept for a scalable, magnetic based approach to fractionate cell populations based on antigen density level. Targeting CD4 on human leukocytes, DMS demonstrated fractionation into CD4High T cells and CD4Low monocytes/neutrophils as quantified by flow cytometry and single cell RNA sequencing. DMS also demonstrated high throughput processing at throughputs 5-10X faster than flow cytometry. We believe DMS can be leveraged and scaled to enable antigen density-based sorting in cell therapy manufacturing, leading to the production of more potent and sustainable cellular therapies.
Keywords:
Cell Therapy Manufacturing
; Digital Magnetic Sorting
; Antigen Density
1. Introduction
Adoptive cell therapies taking advantage of engineered Chimeric Antigen Receptors (CAR) or T Cell Receptors (TCR) have shown incredible potential as “living drugs” that achieve personalized immunotherapies for cancer patients [1]. As the field expands beyond liquid tumors and into other disease indications, the need to develop therapies based on more well defined and specialized cell populations is growing [2,3]. Typically, manufacturing of cellular therapies requires the enrichment of specific cell types from a donor or patient before genetic engineering and expansion. In many cases a specific phenotype drives the therapeutic response, for example: the CD34+ CD90+ stem cell subpopulation shows superior engraftment compared to single positive CD34 hematopoietic stem cells [4]. However, a cell population’s functional performance is not only characterized by the presence or absence of markers but also by subsets defined by surface marker density. For example, CD4 is highly expressed on certain T cell populations (~98K antigens/cell) but is also expressed at low level on some monocytes and neutrophils (~50-20K antigens/cell) [5,6,7,8]. Additionally, natural killer cell populations can be differentiated into CD56Hi and CD56Low which correlate to cytokine activity or higher cytotoxicity phenotypes [9]. For engineered CART cells, having sufficient CART antigen density is a critical metric of therapeutic performance where too little antigen density leads to insufficient anti-tumor activity and too much can lead to tonic signaling and off target effects [10,11]. The ability to fractionate cell populations by both antigen presence and density would be a significant capability for cell therapy production and expand the field’s capabilities. However current cell isolation methods cannot achieve the scale required for cell therapy manufacturing while also having sufficient resolution to differentiate variable antigen density. Magnetic activated cell sorting (MACS) is a standard technique in cell therapy manufacturing, but its binary nature prevents it from reliably differentiating high vs low antigen populations. Fluorescence activated cell sorting (FACS) has sufficient resolution to differentiate high vs low expressing populations but lacks the capability to scale to manufacturing levels because it is an intrinsically serial process where one cell is measured at a time. Some microfluidic technologies are in development for variable antigen isolation but also face scalability challenges, are susceptible to clogging, and are too complex or expensive to manufacture under cGMP [12].
Digital Magnetic Sorting (DMS), also known as ratcheting cytometry, utilizes single use cartridges containing arrays of ferromagnetic microstructures that can isolate different populations of cells based on the level of magnetic content bound to the surface of each cell [13,14,15,16,17] while operating in a parallel fashion like standard MACS. By generating a cycling magnetic field, provided by the DMS benchtop instrument, cells can be captured and fractionated into discrete populations based on magnetic intensity of each cell, similar to how FACS sorts based on light intensity.
In this work, we evaluated the ability for DMS and a superparamagnetic bead cocktail to fractionate CD4+ cells into CD4Hi and CD4Low populations corresponding to T cells and monocytes & neutrophils. It has been well documented that CD4 is highly expressed on CD4 T Helper Cells and expressed at lower levels on monocytes and some neutrophils [5,6,7,8]. Leveraging DMS’ multiplexed sorting capabilities, we enriched CD4Hi cells into a High Magnetic Fraction (HMF) which could be differentiated from CD4Low cells in the Low Magnetic Fraction (LMF) within the DMS cartridge. The capability for DMS to fractionate cells based on antigen expression at throughputs conducive to cell therapy process development and manufacturing (108 to 109 target cells/hr) and can potentially expand the capabilities of cell therapy manufacturing to generate therapeutic cells based on antigen level specific cell populations and improve therapeutic efficacy or even procure optimized subpopulations of engineered cells based on a threshold of a specific marker.
2. Materials and Methods
2.1. Biological Samples
Human buffy coat samples were procured from San Diego Blood Bank (CAT # WB-BC-1F). Ficoll gradient separation (Ficoll-Paque+, Cytivia) was performed using standard protocol to procure all leukocytes. Leukocyte samples were then cryopreserved in aliquots of 10M cells/vial and stored in a -80ºC freezer until used.
2.2. Magnetic Labeling CD4Hi CD4Low Populations
Leukocyte samples were thawed and resuspended in working buffer (PBS-Calcium containing 0.5% BSA + 0.1mM EDTA) to a volume of 500µL. 50µL of Ferrologix proprietary CD4 magnetic bead cocktail (SKU: DMS-0-CD4) was added to samples ranging from 10M to 40M cells. The samples were placed on a rotor at 18RPM in the fridge (4ºC) for 30 minutes.
2.3. DMS of High & Low Populations
After magnetic labeling, all magnetic cells were selected using a quadrupole magnet separator (Biomagnetic Solutions QP-5) to select all CD4+ cells (a process we call “preselection”). Cells were allowed to magnetically precipitate for 5 minutes and washed with 2mL of working buffer. The cells were then removed from the magnetic separator and resuspended in 1mL of working buffer. The sample was then loaded onto a Ferrologix DMS cartridge (SKU: DMS-0-CRT) primed with working buffer and separated under the “Dynamic Load” program at a magnetic separation frequency of 20Hz for a 30 min duration. After DMS processing, the cartridge extraction lock valve was actuated to separate the HMF from LMF fractions. The cells were then eluted from the DMS cartridge via extraction ports [18].
2.4. Flow Cytometry, Cell Counting, & Viability
Antibodies that were used were from Biolegend. FITC anti-human CD14 Antibody (CAT # 367116), PE anti-human CD4 Antibody (CAT # 317410), and APC anti-human CD45 Antibody (CAT #304012). SYTOX™ Advanced™ Dead Cell Stain Kit (CAT #S10274) was acquired from Thermo Fisher Scientific to determine the viability of cells post-DMS. Cells were counted using immunofluorescent microscopy using a Keyence microscope. Each field of view is assessed using two optical channels: the bright field channel, which displays an image of the captured cells, and the viability channel, which employs a fluorescently labeled cell viability stain (Calcein AM). Calcein AM green was used to determine how many cells were in each sample for each step of the protocol. Flow cytometry data was analyzed with FlowJo per the gating strategy in Appendix A Figure A4.
2.5. Single Cell RNA Seq of High & Low Populations
Cells were processed into HMF and LMF fractions and titrated to a concentration of 10K cells/µL. The cells were then added into a Chromium™ Next GEM Chip (10X Genomics) and processed through a Chromium Controller for generation of single cell RNA seq libraries. Following the manufacturers protocol, cDNA libraries were generated and sequenced on an Illumina Sequencer. FASTQ files were aligned and analyzed via 10X Cloud Analysis System. Loupe Browser files were analyzed using standard quality control methods with Q30 scores >90%. Cell categorization was performed using 10X recommended gene list for leukocytes. Differential CD4 expression between LMF and HMF was also performed for different cell categories.
3. Results
3.1. DMS Workflow
Digital magnetic sorting (aka ratcheting cytometry) is a novel technology which can quantitatively sort magnetically labeled cells based on their surface-level expression correlating to the number of beads present on a cell [13,14,15,16,17]. Magnetically tagged cells travel across a chip composed of ferromagnetic microstructures that are subjected to a directionally cycled magnetic field. The DMS benchtop system contains a rotating magnetic wheel composed of rare earth magnets which drive magnetic particles across the ferromagnetic structure to enrich and transport cells when an instantaneous field is applied. In this way, the microstructure array generates an oscillating microscale field which causes magnetized cells to continuously transport across the chip/cartridge. By introducing a gradient in the microstructure pitch (i.e., the distance between ferromagnetic elements) target cells will separate and trap at increasing distant spatial locations across the chip contingent upon increasing amounts of magnetic content on its surface (Figure A1). Depending on the number of particles bound to the cell, the cells will snap to the corresponding location or specific critical pitch at a given field frequency [16]. Leveraging DMS’ quantitative magnetic sorting capabilities, cells can be targeted with superparamagnetic particles such that cells will become magnetized in proportion to their antigen density, I.E. higher antigen density cells will become more magnetized than lower antigen density cells and can be differentiated using DMS [17].
The DMS technology is automated through a benchtop instrument with single use cartridge format as shown in Figure 1. The benchtop instrument contains an automated wheel of rare earth magnets (not shown) to drive magnetic separations as well as an on-board peristaltic pump to perform automated fluid handling operations and is controlled via a simple graphic user interface on a tablet computer (not shown). The DMS cartridge consists of a plastic clamshell assembly which enclosed a magnetic gradient microstructure chip to form a sterile cartridge (See Supplemental Figure A1).
Samples of magnetized cells of up to 5mL can be loaded into a Luer Lock reservoir and processed through the cartridge where target cells are first pulled onto the magnetic chip loading zone and separated vertically into extraction regions. Actuatable valves, known as extraction locks, can be dropped to sequester different cell fractions corresponding to on chip locations and extracted via Luer Lock ports (Figure 1B). Specifically, this DMS cartridge fractionates magnetized cells into two fractions: High Magnetic Fraction (HMF) and Low Magnetic Fraction (HMF). The LMF fraction corresponds to magnetic microelement pitch ranges between 10µm and 34µm while the HMF fraction corresponds to microelement pitch ranges between 36µm and 50µm. Under a 20Hz magnetic frequency the LMF corresponds to an iron oxide content per cell of 3.5 to 56 picograms per cell and the HMF corresponds to an iron oxide content per cell of 64 to 135 picograms under a 20Hz separation (Appendix A Figure A2 & A3). In this way, DMS can fractionate populations based on high vs low magnetic content which correlates with antigen density.
3.2. DMS Fractionation of CD4Hi & CD4Low Cell Types
Figure 2 shows the flow cytometry outputs before separation and after separation into High Magnetic Fraction (HMF) targeting CD4Hi and Low Magnetic Fraction (LMF) targeting CD4Low. Initially, the population consisted of CD45+ 6.62% CD4Hi T cells and 52.6% CD45+ CD4Low monocytes. Note that the CD4Hi and CD4Low populations can be readily observed in the sample corresponding to the T cell and monocyte populations (Figure 2A). In this way flow cytometry gates were established to delineate the Hi/Low fractions. The leukocyte sample was then magnetically tagged using a proprietary superparamagnetic bead cocktail targeting CD4. Following magnetic tagging, CD4+ cells (both High & Low) were concentrated using a standard quad pole tube magnet to enrich all labeled cells. After enrichment of all CD4+ cells the sample was processed through the DMS workflow as shown in Figure 1B.
After elution from the cartridge and cells were quantified via flow cytometry, the HMF fraction showed high enrichment of the CD45+ CD4Hi T cell fraction of 81.8% ±4.1 of total cells within the CD4Hi gate (Figure 2B). The LMF consisted of both CD4+ T Cells and CD4+ monocytes but had significantly lower CD4 expression where 78.8% ±4.3% of cells fell within the CD4Low gate (Figure 2C). Histogram plots of CD4 expression between the HMF and LMF show statistically significant difference in CD4 expression as quantified by ANOVA differential analysis (p value < 0.0001) and over an 0.6 log difference in peak-to-peak expression which correlates with the log difference in the unsorted sample. These data show that the DMS system is capable of rapidly fractionating populations based on antigen density in a single step and exhibits significant advantages over traditional, binary magnetic sorting methods. In total 3 donors were processed in triplicate with DMS high/low sorting to fractionate different antigen density populations. Figure 3 shows the summarized results of the % of cells within each CD4Hi/CD4Low flow cytometry gate within the HMF and LMF extractions.
3.3. Single Cell RNA Seq Confirms Antigen Density Separation
CD4 Hi/Low cells sorted by DMS were processed for single-cell RNA sequencing to corroborate flow cytometry findings for cell population fractionation and examine gene expression. DMS processing into HMF and LMF fractions was performed in the same manner as previously described and then added directly into a 10X Genomics chip. Three conditions were processed through the single cell RNA seq workflow: (1) unsorted control, (2) LMF, and (3) HMF. Figure 4 shows tSNE plots before and after DMS processing into LMF and HMF populations. Initially the sample consisted of standard cell populations with monocytes & neutrophils at a 54% frequency and total T cells at 32% (CD3+ T Cells: 20% and.
Other T Cells (Natural Killer - NK and Gamma Delta T Cell - GDT: 12%) which was consistent with flow cytometry controls. After DMS, the LMF contained mostly monocytes and neutrophils (~78%), some T cells and subpopulations (11%, 6%), and a few B cells (4%). The HMF consisted largely of T cells (52%) and subpopulations (4%, 10%) as well monocytes & neutrophils (31%), and very few B Cells (1.5%). This shows that the DMS system is fractionating populations which correlate with published literature with CD4Hi T Cells and CD4Low monocytes [5,6,7,8,9].
In depth gene expression analysis on T cell markers and monocyte & neutrophil markers show a significant difference between the HMF and LMF in terms of population fractionation. Figure 5a shows a comparative violin plot between LMF and HMF visualizing aggregated gene expression for multiple T cell markers. These markers correlate with T cells and their subpopulations specifically, T Cells (CD3), Naïve CD4+ T Cells (ANK3 & DOCK9), and natural killer & gamma delta T cells (GNLY & FGFBP2) [19]. As shown the mean gene expression of T cell markers in the HMF is significantly higher than the LMF (p < 0.0001) meaning the HMF is enriching for T cell populations. Conversely, Figure 5b shows that the LMF is enriching monocyte and neutrophil populations all with statistically significant differences (p value ranges from <0.05 to <0.0001). This data confirms that the DMS process is successfully fractionating cell types correlating with CD4Hi and CD4Low expression in established literature. This data in conjunction with the flow cytometry findings suggests that the DMS system is indeed fractionating for CD4Hi and CD4Low populations and agrees with previously published datasets. It is of note that measurement of CD4 surface marker expression with RNA sequencing data cannot be directly correlated with surface protein levels [20].
3.4. Cell Processing Throughput
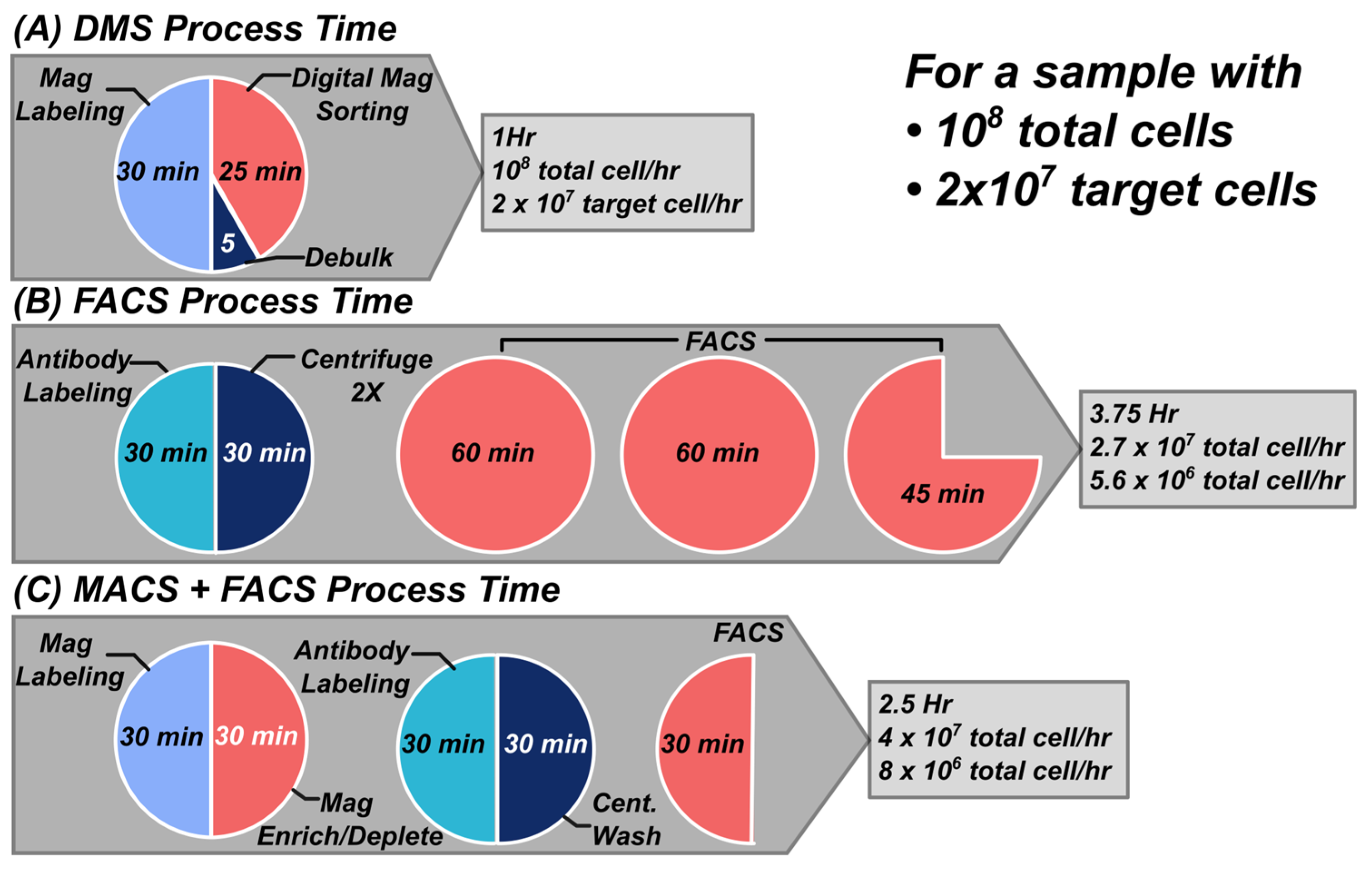
Because of its parallelized nature and streamlined sample processing steps (centrifugation free), DMS can achieve higher throughput compared to traditional methods for antigen density-based separations such as FACS. Quantification of DMS throughout was characterized by evaluating the total target cells separated over the entire process (magnetic labeling, magnetic preselection/de-bulking, and DMS). Figure 6 below shows a throughput breakdown from sample procurement to completion assuming a sample of 108 total cells and a target fraction of 2x107 target cells. Briefly, a single benchtop DMS cartridge can process 108 total cells containing 2x107 target cells and fractionate them into High/Low populations between 50 and 60 minutes (30 min magnetic bead labeling, 5 min magnetic preselection/de-bulking, and a 20-to-30-minute separation. Assuming a 60 minute total process time, this yields a 108 total cell per hour throughput and a 2x107 target cell per hour throughput. Compared to flow cytometry (Figure 6B) a sample containing 108 cells would take ~3.75 hrs (30 minutes for antibody staining, 2 x 15min centrifugation and washing steps, and 2.75 hrs for FACS sorting assuming 10k cells/sec throughput) [21] yielding a 2.7x107 total cell per hour throughput.
The main challenge with using FACS to sort large cell quantities is that both target and non-target cells are sorted, in this case ~80% of sorting time is spent on non-target cells. Often magnetic enrichment or depletion steps are used to debulk non-target cells before running on FACS [22,23,24]. In this case a 108 cell sample with 2x107 target cells would take approximately 2.5 hrs (30 min magnetic labeling and 30 min magnetic pulldown or column sort followed by standard FACS antibody staining and washing 60 minutes, and FACS sorting 30 minutes assuming 10k cells/sec throughput on 2x107 target cells). This combined magnetic enrichment/depletion and FACS yields a total cell throughput of 4x107 total cells per hour throughput or 8x108 target cells per hour assuming 2x107 target cells in the starting fraction. Although a FACS + MACS method enhances throughput, it necessitates multiple additional sample processing steps, which can significantly reduce target cell yield and increases the costs of consumables and reagents. This suggests that the benchtop DMS system can achieve 2X to 3X throughput compared to FACS based techniques in a single process workflow and can be readily scaled to larger footprints to accommodate larger cell quantities.
4. Discussion
In this study, we investigated the potential of Digital Magnetic Sorting (DMS) to fractionate cell populations based on CD4 antigen density. Our results provide strong evidence of the successful fractionation of CD4+ cells into CD4Hi T cells and CD4Low monocytes/neutrophils, confirming the robustness of DMS in accurately sorting cell subsets based on antigen density. Furthermore, the findings were confirmed by multiple assay approaches, specifically flow cytometry and single cell RNA sequencing, both which show fractionation of expected CD4Hi T cells and CD4Low monocytes/neutrophil populations and are in agreement with established literature [5,6,7,8]. The ability to fractionate cell populations based on antigen density holds at manufacturing scale throughput shows significant promise for improving cell therapy manufacturing. Even at benchtop scale, the DMS platform can process at throughputs of 108 total cells per hour, capable of processing 108 total cells from sample to sorted cells in less than 60 minutes, which is significantly faster than FACS systems. Assuming a FACS sort of 10K cells/second [21], 108 total cells would take ~3.75 hrs to process, showing that DMS alone is >3X faster. When you take into account the staining and multiple washing and centrifugation steps required for FACS (~ 2 hrs), a benchtop DMS system shows a speed improvement of ~5X compared to FACS. DMS’ throughput is comparable to widely utilized magnetic sorting systems such as the Miltenyi ® AutoMACS or StemCell Technologies® Robosep. However, the key differentiator for DMS is the capability to fractionate based on antigen density which is not currently possible with binary magnetic sorters.
Figure 7.
Comparison of DMS and other cell sorting footprints. MACS GMP scale systems for cell therapy production can accommodate large cell quantities but cannot fractionate subpopulations based on antigen density. FACS sorters have quantitative gating capabilities but are too low throughput to accommodate cell therapy production scale. DMS offers a scalable approach to sorting based on antigen density by cartridge parallelization. A single DMS benchtop sorter can achieve a 108 cell/hr throughput in a single run. The scaled DMS GMP system has 8 parallelized cartridges which operate in semicontinuous batch mode to achieve a total throughput of.
Figure 7.
Comparison of DMS and other cell sorting footprints. MACS GMP scale systems for cell therapy production can accommodate large cell quantities but cannot fractionate subpopulations based on antigen density. FACS sorters have quantitative gating capabilities but are too low throughput to accommodate cell therapy production scale. DMS offers a scalable approach to sorting based on antigen density by cartridge parallelization. A single DMS benchtop sorter can achieve a 108 cell/hr throughput in a single run. The scaled DMS GMP system has 8 parallelized cartridges which operate in semicontinuous batch mode to achieve a total throughput of.

While a benchtop DMS system is capable to operate at cell therapy process development scales, which generally consist of cell quantities of 107 to 108 total cells [23], cell therapy manufacturing operates at much larger cell quantities of 109 to 1011 total cells [25]. Fortunately, the scaling strategy for DMS to operate at cell therapy manufacturing levels is relatively straight forward where a parallelized cartridge approach can be adopted. Indeed, Ferrologix is actively developing a manufacturing scale GMP compliant DMS system which consists of multiple cartridges processing samples in parallel [26]. The DMS chip has a small footprint (2.5cm x 6.5cm) which can be readily scaled to larger footprints for parallel sorting of higher cell quantities. Therefore a 25 cm x 6.5cm chip can process 109 total cells in a single run or a whole leukopak (~2 x 109 total cells) in two batch runs. A throughput of ~109 cells/hr is comparable to current cell therapy manufacturing magnetic sorting systems but would add the capability to fractionate based on antigen density.
In the larger landscape of cell therapy manufacturing, cost of production is a major obstacle for wide scale deployment. Indeed, one of the major cost drivers for cell therapy production is the procurement of cGMP grade materials [27], of which genetic modification reagents are the costliest (Example, GMP Viral Vector, CrispR/CAS9 reagents). One potential solution to reducing costs of cell therapy production is to engineer lower quantities of highly specific cell populations to achieve the same or improved therapeutic response. Recent research in cell therapy production also supports this approach where lower numbers of higher quality cells are achieving superior response to traditional heterogenous population approaches and are much lower cost to produce [28,29,30]. One specific example in support of the lower quantity and higher specialization argument is in production of natural killer (NK) cell therapies. It has been well established that NK cells split into different phenotypes based on antigen density where CD56Hi NK cells exhibit higher cytokine activity and cytotoxicity [31] and have higher anti-tumor activity [32]. Isolation and engineering CD56Hi NK cell subpopulation, which represent approximately 10% of the total circulating NK cell fraction, could substantially reduce the total cell quantity in the end therapeutic and therefore the required quantity of reagents needed to produce the cell therapy. Using DMS to fractionate NK cells based on CD56 antigen density can enable the isolation of specific subpopulations with higher therapeutic potential while also reducing the reagent cost to manufacture. CD56Hi/Low fractionation represents one specific example where DMS can potentially improve therapeutic potency and cost reduction with many other potential applications in production of engineered T cells or stem cell therapeutics.
5. Conclusions
In this work, DMS demonstrated successful fractionation of cell populations based on CD4Hi & CD4Low antigen density using differential magnetic binding per cell as a rapid and scalable method for differentiating cell sup-populations. DMS separations characterized by both flow cytometry and single cell RNA seq show differential CD4 expression between HMF and LMF fractions and cell population compositions that are consistent with established literature on CD4Hi/Low leukocyte cell types. The benchtop DMS system also demonstrated higher throughput compared to FACS systems with a clear scaling strategy to accommodate cell quantities associated with cell therapy manufacturing This capability to fractionate cell types by antigen density in a scalable manner can enable novel precision medicine applications including specific examples in use today including natural killer cell subpopulation enrichment, stem cell subpopulation fractionation, or even enrichment of engineered cells after transduction. Our hope is to utilize this novel technology to reduce production cost of cell therapies by providing cell therapy developers a tool to isolate lower numbers of highly specialized cell types which can be engineered more efficiently and with lower reagent consumption.
6. Patents
Ferrologix has and continues to develop a patent portfolio on its innovations. Please see US20220241797A1.
Author Contributions
Conceptualization, C.M and T.T. methodology, S.B.C.; software, S.R..; validation, S.B.C, A.B..; formal analysis, S.B.C; investigation, S.B.C & A.B.; resources, C.M.; data curation, C.M..; writing—original draft preparation, C.M.; writing—review and editing, S.B.C; visualization, S.B.C; supervision, C.M. & T.T.; project administration, T.T.; funding acquisition, T.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the National Institute of General Medical Sciences (NIGMS), grant number RGM149063A. The APC was funded by Ferrologix, Inc.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Protocols are available on the Cancer Nanotechnology Laboratory (caNanoLab) database.
Acknowledgments
This publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). This work was also supported in part by Federal SBIR Award RGM149063A.
Conflicts of Interest
The authors, Coleman Murray and Timothy Tiemann disclose they are equity shareholders in Ferrologix, Inc. This work was funded through federal SBIR awards of which the funders (Dept of Health and Human Services) had no role in the design of the study; in the collection, analyses, or interpretation of data; or in the decision to publish the results.
Appendix A
Figure A1.
Primer on Digital Magnetic Sorting (DMS) also known as Ratcheting Cytometry. (A) The DMS benchtop instrument accepts a cartridge into a “saddle” which is then hooked to tubing & peristaltic pump to draw fluid through the cartridge. Internal and centered with the cartridge is a rotating magnetic wheel in radial Halbach array orientation. This rotating wheel drives the digital magnetic sorting behavior across the magnetic chip within the cartridge. (B) The cartridge itself consist of a buffer/sample input reservoir, a flexible extraction lock valve to sequester LMF and HMF regions in the chip, a three way leur lock valve to connect to the peristaltic pump, and leur lock extraction ports to remove cells. (C) The DMS cartridge is a clamshell assembly containing the DMS magnetic chip which is secured to the cartridge chamber via a backplate and die cut double sided adhesive strip. (D) The DMS chip itself is a 25 x 75mm glass substrate with regions of high-density Nickel/Iron micro-bars with a 5µm width, 25mm length, and a thickness between 3 and 5µm. The first region is the LMF and is 25 x 25mm with a microbar pitch of 10µm. This region is directly connected into the HMF region which consists of multiple 2mm long x 25mm wide sections of increasing pitch. The first region has a 12µm pitch and then increments by 2µm with each 2mm section (12µm, 14µm, 16µm… 50µm). (E) In response to a rotating magnetic field generated by the DMS benchtop instrument, the microbars magnetize in alignment to the bulk field, modifying the magnetic potential energy landscape and introducing potential wells in which superparamagnetic particles/cells migrate. As the wheel is cycled, particles/cells will follow the potential energy wells and jump from bar to bar against gravity based on size and bound iron oxide content. Using a chip consisting of microstructure arrays with gradient horizontal pitch, cells with different levels of magnetic binding will traverse the array until reaching their critical pitch, where they will collect and oscillate. Magnetized cells with increasing magnetic content will have correspondingly higher critical pitches and can therefore be separated. This results in temporally stable distributions of cells based on levels of magnetic content per cell or particles per cell. In this way cell populations can be differentiated based on their level of magnetic binding.
Figure A1.
Primer on Digital Magnetic Sorting (DMS) also known as Ratcheting Cytometry. (A) The DMS benchtop instrument accepts a cartridge into a “saddle” which is then hooked to tubing & peristaltic pump to draw fluid through the cartridge. Internal and centered with the cartridge is a rotating magnetic wheel in radial Halbach array orientation. This rotating wheel drives the digital magnetic sorting behavior across the magnetic chip within the cartridge. (B) The cartridge itself consist of a buffer/sample input reservoir, a flexible extraction lock valve to sequester LMF and HMF regions in the chip, a three way leur lock valve to connect to the peristaltic pump, and leur lock extraction ports to remove cells. (C) The DMS cartridge is a clamshell assembly containing the DMS magnetic chip which is secured to the cartridge chamber via a backplate and die cut double sided adhesive strip. (D) The DMS chip itself is a 25 x 75mm glass substrate with regions of high-density Nickel/Iron micro-bars with a 5µm width, 25mm length, and a thickness between 3 and 5µm. The first region is the LMF and is 25 x 25mm with a microbar pitch of 10µm. This region is directly connected into the HMF region which consists of multiple 2mm long x 25mm wide sections of increasing pitch. The first region has a 12µm pitch and then increments by 2µm with each 2mm section (12µm, 14µm, 16µm… 50µm). (E) In response to a rotating magnetic field generated by the DMS benchtop instrument, the microbars magnetize in alignment to the bulk field, modifying the magnetic potential energy landscape and introducing potential wells in which superparamagnetic particles/cells migrate. As the wheel is cycled, particles/cells will follow the potential energy wells and jump from bar to bar against gravity based on size and bound iron oxide content. Using a chip consisting of microstructure arrays with gradient horizontal pitch, cells with different levels of magnetic binding will traverse the array until reaching their critical pitch, where they will collect and oscillate. Magnetized cells with increasing magnetic content will have correspondingly higher critical pitches and can therefore be separated. This results in temporally stable distributions of cells based on levels of magnetic content per cell or particles per cell. In this way cell populations can be differentiated based on their level of magnetic binding.
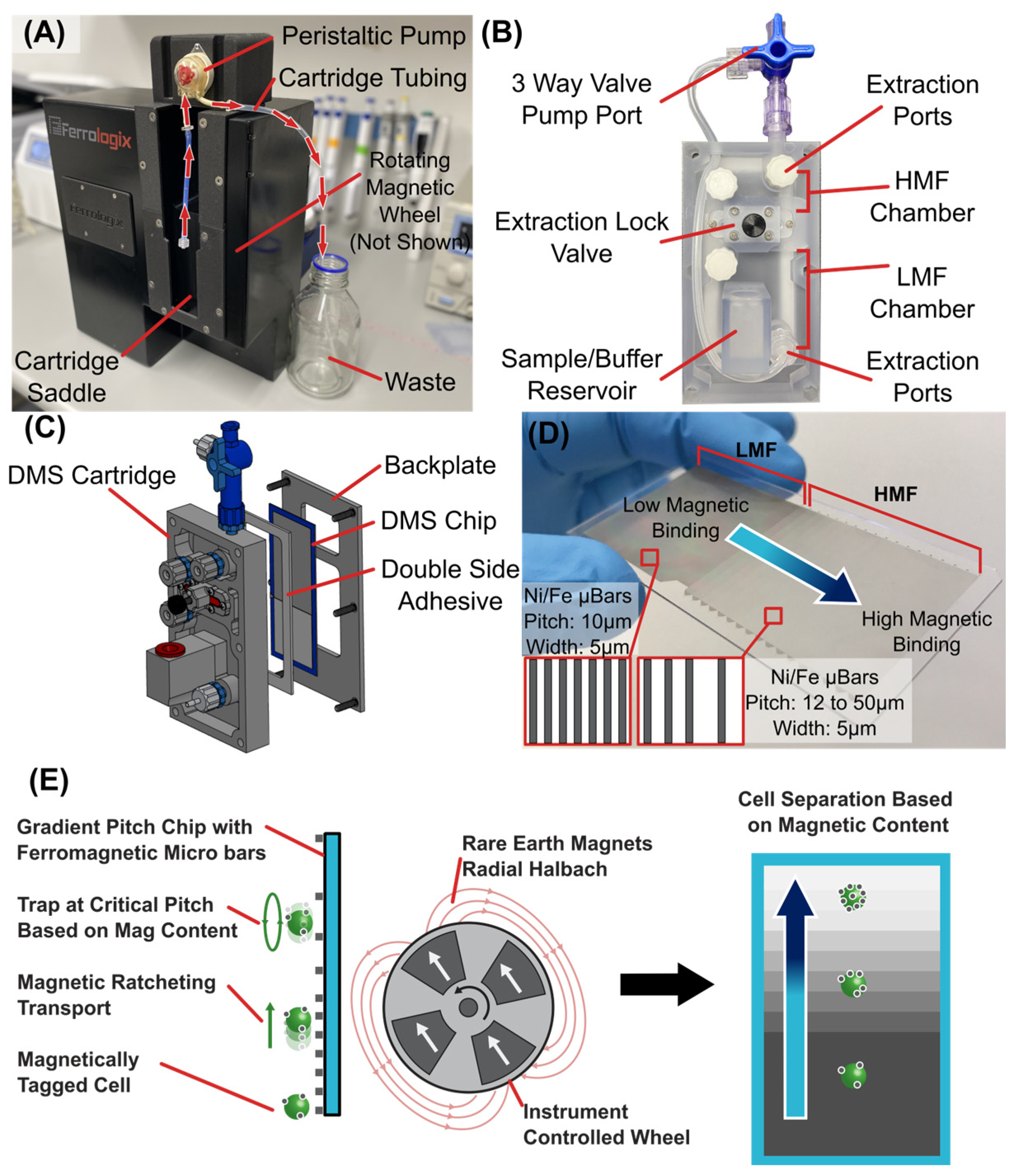
Figure A2.
Numerical Modeling of Magnetic Content per Cell. We have already demonstrated in previous work (C Murray et. al., Small, 2015) that cells transport and concentrate onto magnetic pitch ranges depending on the microelement pitch, cell bound magnetic content, and the magnetic field frequency. (a) Using the numerical model developed previously, we can determine what level of iron oxide content per cell corresponds to a specific magnetic microstructure pitch under a given magnetic frequency. (b) The predictive model matches well with multiple empirical datasets where individual cells were fluorescently imaged through DMS chips to quantify the number of bound magnetic beads per cell with known levels of magnetic content per bead.
Figure A2.
Numerical Modeling of Magnetic Content per Cell. We have already demonstrated in previous work (C Murray et. al., Small, 2015) that cells transport and concentrate onto magnetic pitch ranges depending on the microelement pitch, cell bound magnetic content, and the magnetic field frequency. (a) Using the numerical model developed previously, we can determine what level of iron oxide content per cell corresponds to a specific magnetic microstructure pitch under a given magnetic frequency. (b) The predictive model matches well with multiple empirical datasets where individual cells were fluorescently imaged through DMS chips to quantify the number of bound magnetic beads per cell with known levels of magnetic content per bead.
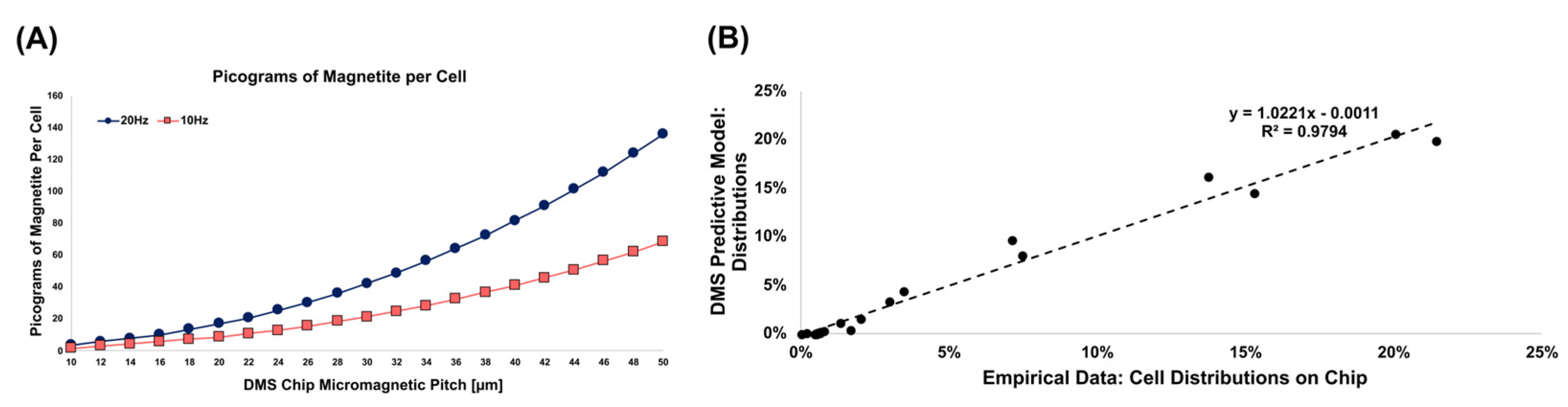
Figure A3.
T Cell and Monocyte Distributions on the DMS chip. Two key system and cartridge design parameters needed to be considered for fractionating the High and Low antigen density populations: Primarily the frequency of the magnetic field that the magnetic cartridge was subjected to and the micromagnetic pitch range that the cells magnetically equilibrate to. From numerical modeling we know the ranges that cells separate to with varying magnetic contents per cell. Empirical separations needed to be carried out to correlate CD4 surface expression on human cell populations with iron oxide content and ultimately, micropillar pitch ranges and frequencies on the DMS cartridge. To achieve this, we procured pre fractionated PBMC samples which were split into T cells and monocytes (custom order from AllCells, Inc). Using established assays from pervious work (C. Murray, et.al. SLAS Technology, 2017) we stained the T cells with calcein AM red and the monocytes with calcein AM green to create a two-population mixture. After magnetic labeling and separation on a DMS chip we fluorescently imaged the chip (a) to determine where the populations equilibrated under various magnetic labeling conditions and magnetic frequencies. (b) Image analysis enabled us to develop distributions of T cells vs monocytes and determine that a 20Hz separation frequency and a cutoff off pitch of 34µm enabled maximum separability between the CD4Hi T Cells and CD4Low monocytes/neutrophils. T cells (red) separated above the 34µm pitch at a 20Hz frequency while the monocytes were mainly retained in the bottom, below the 34µm pitch. This experiment set allowed us to optimize DMS separation parameters and cartridge designs to determine physical cartridge cutoff locations (extraction lock location and separation frequency).
Figure A3.
T Cell and Monocyte Distributions on the DMS chip. Two key system and cartridge design parameters needed to be considered for fractionating the High and Low antigen density populations: Primarily the frequency of the magnetic field that the magnetic cartridge was subjected to and the micromagnetic pitch range that the cells magnetically equilibrate to. From numerical modeling we know the ranges that cells separate to with varying magnetic contents per cell. Empirical separations needed to be carried out to correlate CD4 surface expression on human cell populations with iron oxide content and ultimately, micropillar pitch ranges and frequencies on the DMS cartridge. To achieve this, we procured pre fractionated PBMC samples which were split into T cells and monocytes (custom order from AllCells, Inc). Using established assays from pervious work (C. Murray, et.al. SLAS Technology, 2017) we stained the T cells with calcein AM red and the monocytes with calcein AM green to create a two-population mixture. After magnetic labeling and separation on a DMS chip we fluorescently imaged the chip (a) to determine where the populations equilibrated under various magnetic labeling conditions and magnetic frequencies. (b) Image analysis enabled us to develop distributions of T cells vs monocytes and determine that a 20Hz separation frequency and a cutoff off pitch of 34µm enabled maximum separability between the CD4Hi T Cells and CD4Low monocytes/neutrophils. T cells (red) separated above the 34µm pitch at a 20Hz frequency while the monocytes were mainly retained in the bottom, below the 34µm pitch. This experiment set allowed us to optimize DMS separation parameters and cartridge designs to determine physical cartridge cutoff locations (extraction lock location and separation frequency).
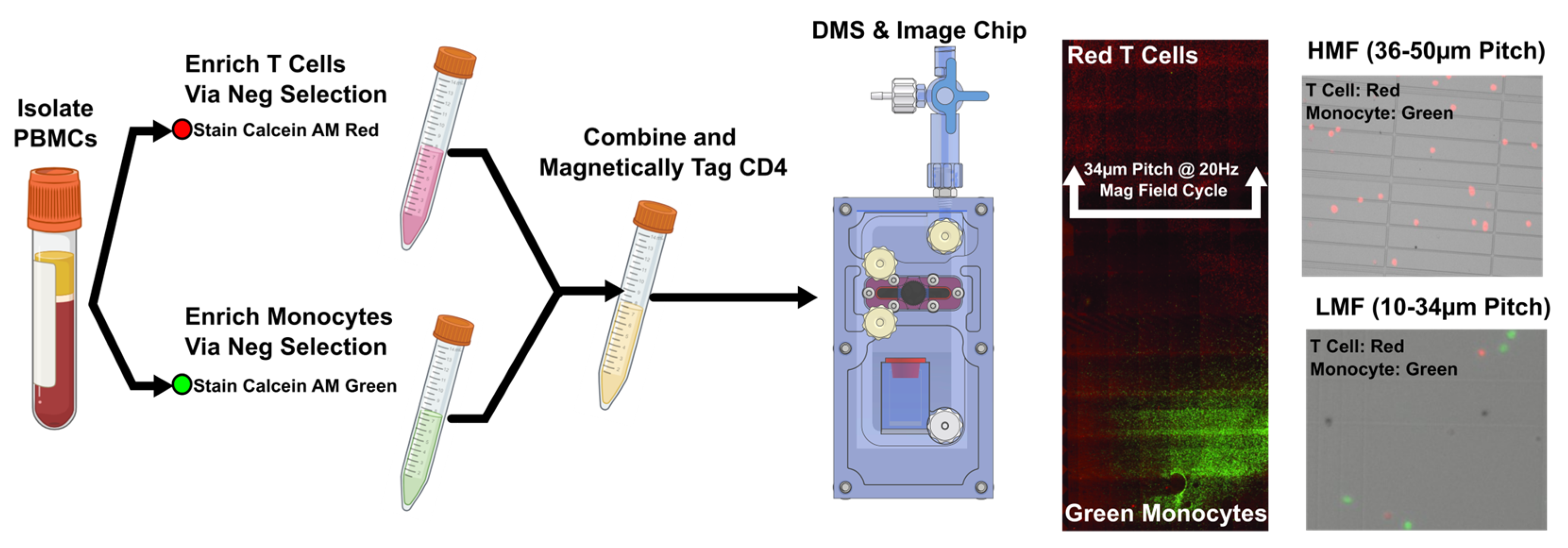
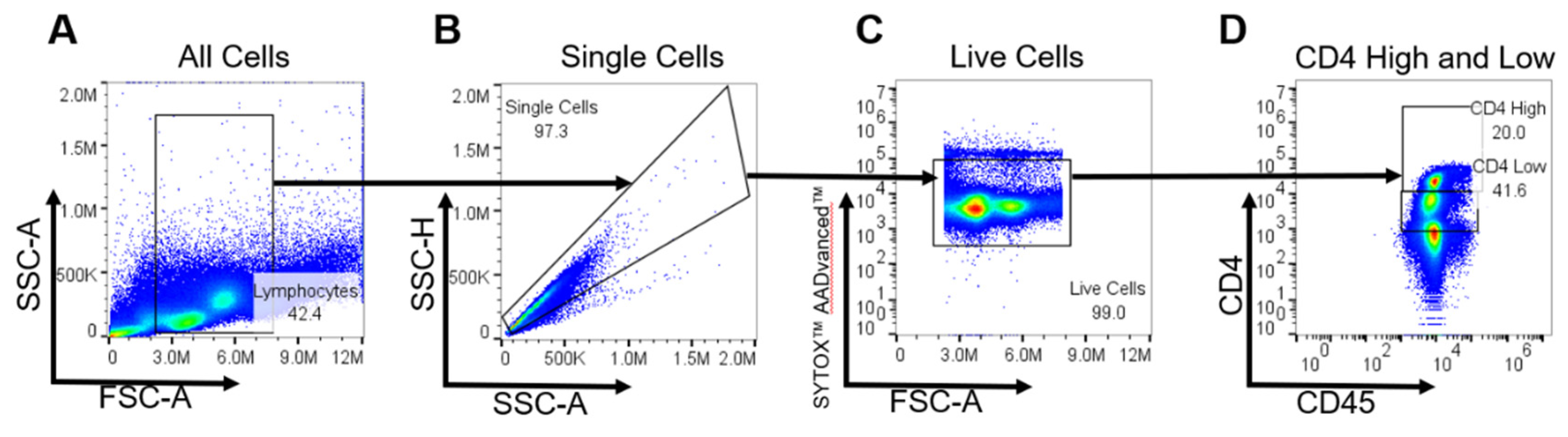
Figure A4.
(A) Initial gating on lymphocytes was performed on the native sample, utilizing forward scatter (FSC) and side scatter (SSC) parameters to segregate the lymphocyte population based on their intrinsic physical properties.(B) The 'Single Cells' gate was established to ensure analysis of individual cell events, thereby excluding multi-cell aggregates and enhancing the accuracy of the subsequent gating steps.(C) Viability assessment was conducted using Thermo Fisher Scientific’s SYTOX™ AADvanced™ Dead Cell Stain Kit (Catalog No. S10349). This step involved gating live cells to exclude non-viable cells, ensuring the analysis focused solely on live cellular constituents. (D) Discrimination of CD4Hi and CD4Low cell populations was then performed. CD4Hi cells, typically representative of T cell populations, were gated based on their characteristic expression patterns. In contrast, CD4Low cells, correlating with expected monocyte populations, were gated accordingly. This distinction was pivotal in reflecting the anticipated frequencies of these cell types within peripheral blood mononuclear cells (PBMCs).
Figure A4.
(A) Initial gating on lymphocytes was performed on the native sample, utilizing forward scatter (FSC) and side scatter (SSC) parameters to segregate the lymphocyte population based on their intrinsic physical properties.(B) The 'Single Cells' gate was established to ensure analysis of individual cell events, thereby excluding multi-cell aggregates and enhancing the accuracy of the subsequent gating steps.(C) Viability assessment was conducted using Thermo Fisher Scientific’s SYTOX™ AADvanced™ Dead Cell Stain Kit (Catalog No. S10349). This step involved gating live cells to exclude non-viable cells, ensuring the analysis focused solely on live cellular constituents. (D) Discrimination of CD4Hi and CD4Low cell populations was then performed. CD4Hi cells, typically representative of T cell populations, were gated based on their characteristic expression patterns. In contrast, CD4Low cells, correlating with expected monocyte populations, were gated accordingly. This distinction was pivotal in reflecting the anticipated frequencies of these cell types within peripheral blood mononuclear cells (PBMCs).
References
- Jackson HJ, et. al “Driving CAR T-cells forward.” Nat Rev Clin Oncol. March 2016. doi:10.1038/nrclinonc.2016.36. [CrossRef]
- Sommermeyer et. al. “Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo.” Leukemia. 2016 Feb;30(2):492-500.
- Koehl U, et. al “Ex vivo expansion of highly purified NK cells for immunotherapy after haploidentical stem cell transplantation in children.” Klin Padiatr. 2005 Nov-Dec;217(6):345-50.
- Radtke, S., Pande, D., Cui, M., Perez, A. M., Chan, Y. Y., Enstrom, M., Schmuck, S., Berger, A., Eunson, T., Adair, J. E., & Kiem, H. P. (2020). Purification of Human CD34+CD90+ HSCs Reduces Target Cell Population and Improves Lentiviral Transduction for Gene Therapy. Molecular therapy. Methods & clinical development, 18, 679–691. https://doi.org/10.1016/j.omtm.2020.07.010. [CrossRef]
- Filion LG, et. al “Detection of surface and cytoplasmic CD4 on blood monocytes from normal and HIV-1 infected individuals.” J Immunol Methods. 1990 Dec 31;135(1-2):59-69. doi: 10.1016/0022-1759(90)90256-u. PMID: 1703191. [CrossRef] [PubMed]
- Zhen, A., Krutzik, S. R., Levin, B. R., Kasparian, S., Zack, J. A., & Kitchen, S. G. (2014). CD4 ligation on human blood monocytes triggers macrophage differentiation and enhances HIV infection. Journal of virology, 88(17), 9934–9946. https://doi.org/10.1128/JVI.00616-14. [CrossRef]
- Kazazi, F., Mathijs, J. M., Foley, P., & Cunningham, A. L. (1989). Variations in CD4 expression by human monocytes and macrophages and their relationships to infection with the human immunodeficiency virus. The Journal of general virology, 70 ( Pt 10), 2661–2672. https://doi.org/10.1099/0022-1317-70-10-2661. [CrossRef]
- Biswas, P., Mantelli, B., Sica, A., Malnati, M., Panzeri, C., Saccani, A., Hasson, H., Vecchi, A., Saniabadi, A., Lusso, P., Lazzarin, A., & Beretta, A. (2003). Expression of CD4 on human peripheral blood neutrophils. Blood, 101(11), 4452-4456. https://doi.org/10.1182/blood-2002-10-3056. [CrossRef]
- Ziegler, S., et al. CD56 Is a Pathogen Recognition Receptor on Human Natural Killer Cells. Sci Rep 7, 6138 (2017). https://doi.org/10.1038/s41598-017-06238-4. [CrossRef]
- Majzner RG, et. al “Tuning the Antigen Density Requirement for CAR T-cell Activity.” Cancer Discov. 2020 May;10(5):702-723. doi: 10.1158/2159-8290.CD-19-0945. Epub 2020 Mar 19. PMID: 32193224; PMCID: PMC7939454. [CrossRef] [PubMed]
- Chen, X. et al Rational Tuning of CAR Tonic Signaling Yields Superior T-Cell Therapy for Cancer. bioRxiv 2020.10.01.322990.
- Wang, Z., et al. Efficient recovery of potent tumour-infiltrating lymphocytes through quantitative immunomagnetic cell sorting. Nat Biomed Eng 6, 108–117 (2022).
- Bshara-Corson, S., Vukovic, L., Verhagen, A., Tiemann, T., Murray, C. “ Rare Cell Purification Using Ferrologix’s Digital Magnetic Sorting” Annual Conference of the Society for Laboratory Automation and Screening. February 6, 2022. Boston, MA, USA.
- Adeyiga, O. B., Murray, C., Muñoz, H. E., Escobar, A., & Di Carlo, D. (2021). Magnetic microparticle concentration and collection using a mechatronic magnetic ratcheting system. PloS one, 16(2), e0246124. https://doi.org/10.1371/journal.pone.0246124. [CrossRef]
- Murray, C., Miwa, H., Dhar, M., Park, D. E., Pao, E., Martinez, J., Kaanumale, S., Loghin, E., Graf, J., Raddassi, K., Kwok, W. W., Hafler, D., Puleo, C., & Di Carlo, D. (2018). Unsupervised capture and profiling of rare immune cells using multi-directional magnetic ratcheting. Lab on a chip, 18(16), 2396–2409. https://doi.org/10.1039/c8lc00518d. [CrossRef]
- Murray, C., Pao, E., Tseng, P., Aftab, S., Kulkarni, R., Rettig, M., & Di Carlo, D. (2016). Quantitative Magnetic Separation of Particles and Cells Using Gradient Magnetic Ratcheting. Small (Weinheim an der Bergstrasse, Germany), 12(14), 1891–1899. https://doi.org/10.1002/smll.201502120. [CrossRef]
- Murray, C., Pao, E., Jann, A., Park, D. E., & Di Carlo, D. (2018). Continuous and Quantitative Purification of T-Cell Subsets for Cell Therapy Manufacturing Using Magnetic Ratcheting Cytometry. SLAS technology, 23(4), 326–337. https://doi.org/10.1177/2472630317748655. [CrossRef]
- Ferrologix, Inc. Digital Magnetic Sorting. synthesia.io. https://share.synthesia.io/8704ea27-e45b-46e4-ae29-5d36bf204dfe.
- Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., Sivertsson, Å., Kampf, C., Sjöstedt, E., Asplund, A., Olsson, I., Edlund, K., Lundberg, E., Navani, S., Szigyarto, C. A., Odeberg, J., Djureinovic, D., Takanen, J. O., Hober, S., Alm, T., … Pontén, F. (2015). Proteomics. Tissue-based map of the human proteome. Science (New York, N.Y.), 347(6220), 1260419. https://doi.org/10.1126/science.1260419. [CrossRef]
- Li, J., Zhang, Y., Yang, C., & Rong, R. (2020). Discrepant mRNA and Protein Expression in Immune Cells. Current genomics, 21(8), 560–563.
- Flow cytometry University of Iowa Helathcare. Cell Sorting | Flow Cytometry. (n.d.). https://medicine.uiowa.edu/flowcytometry/cell-sorting. Accessed 10/31/2023.
- Miltenyi. (n.d.). autoMACS ® Pro Separator user manual. https://www.miltenyibiotec.com/upload/assets/IM0001886.PDF . Accessed 10/31/2023.
- Miltenyi. (n.d.). CliniMACS Prodigy® User Manual. https://static.miltenyibiotec.com/asset/150655405641/document_h0fd9i4al17q32uhfuf4h7lu25?content-disposition=inline. Accessed 10/31/2023.
- Valli, H., Sukhwani, M., Dovey, S. L., Peters, K. A., Donohue, J., Castro, C. A., Chu, T., Marshall, G. R., & Orwig, K. E. (2014). Fluorescence- and magnetic-activated cell sorting strategies to isolate and enrich human spermatogonial stem cells. Fertility and sterility, 102(2), 566–580.e7. https://doi.org/10.1016/j.fertnstert.2014.04.036. [CrossRef]
- Pigeau, G. M., Csaszar, E., & Dulgar-Tulloch, A. (2018). Commercial Scale Manufacturing of Allogeneic Cell Therapy. Frontiers in medicine, 5, 233. https://doi.org/10.3389/fmed.2018.00233. [CrossRef]
- Ferrologix, Inc. (2021). High Throughput & Quantitative Cell Purification for Immunotherapy Manufacture (SBIR Grant No. 5R44CA228844-03). U.S. Department of Health and Human Services.
- Ten Ham, R. M. T., Hövels, A. M., Hoekman, J., Frederix, G. W. J., Leufkens, H. G. M., Klungel, O. H., Jedema, I., Veld, S. A. J., Nikolic, T., Van Pel, M., Zwaginga, J. J., Lin, F., de Goede, A. L., Schreibelt, G., Budde, S., de Vries, I. J. M., Wilkie, G. M., Dolstra, H., Ovelgönne, H., Meij, P., … Hoefnagel, M. H. N. (2020). What does cell therapy manufacturing cost? A framework and methodology to facilitate academic and other small-scale cell therapy manufacturing costings. Cytotherapy, 22(7), 388–397. https://doi.org/10.1016/j.jcyt.2020.03.432. [CrossRef]
- Song, H. W., Prochazkova, M., Shao, L., Traynor, R., Underwood, S., Black, M., Fellowes, V., Shi, R., Pouzolles, M., Chou, H. C., Cheuk, A. T., Taylor, N., Jin, P., Somerville, R. P., Stroncek, D. F., Khan, J., & Highfill, S. L. (2024). CAR-T cell expansion platforms yield distinct T cell differentiation states. Cytotherapy, 26(7), 757–768. https://doi.org/10.1016/j.jcyt.2024.03.003Bruce. [CrossRef]
- de Macedo Abdo, L., Barros, L. R. C., Saldanha Viegas, M., Vieira Codeço Marques, L., de Sousa Ferreira, P., Chicaybam, L., & Bonamino, M. H. (2020). Development of CAR-T cell therapy for B-ALL using a point-of-care approach. Oncoimmunology, 9(1), 1752592. https://doi.org/10.1080/2162402X.2020.1752592. [CrossRef]
- Ghassemi, S., Durgin, J. S., Nunez-Cruz, S., Patel, J., Leferovich, J., Pinzone, M., Shen, F., Cummins, K. D., Plesa, G., Cantu, V. A., Reddy, S., Bushman, F. D., Gill, S. I., O'Doherty, U., O'Connor, R. S., & Milone, M. C. (2022). Rapid manufacturing of non-activated potent CAR T cells. Nature biomedical engineering, 6(2), 118–128. https://doi.org/10.1038/s41551-021-00842-6. [CrossRef]
- Poli, A., Michel, T., Thérésine, M., Andrès, E., Hentges, F., & Zimmer, J. (2009). CD56bright natural killer (NK) cells: an important NK cell subset. Immunology, 126(4), 458–465. https://doi.org/10.1111/j.1365-2567.2008.03027.x. [CrossRef]
- Wagner, J. A., Rosario, M., Romee, R., Berrien-Elliott, M. M., Schneider, S. E., Leong, J. W., Sullivan, R. P., Jewell, B. A., Becker-Hapak, M., Schappe, T., Abdel-Latif, S., Ireland, A. R., Jaishankar, D., King, J. A., Vij, R., Clement, D., Goodridge, J., Malmberg, K. J., Wong, H. C., & Fehniger, T. A. (2017). CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. The Journal of clinical investigation, 127(11), 4042–4058. https://doi.org/10.1172/JCI90387. [CrossRef]
Figure 1.
(a) DMS benchtop system houses a closed loop controlled internal magnetic wheel of rare earth magnets arranged in a Halbach array orientation to generate a cycling magnetic field and a peristaltic pump to perform automated sample processing through the DMS cartridge. The DMS cartridge is loaded into the benchtop system and connected to the peristaltic pump. A tablet with simple graphic user interface walks the user through a standardized protocol. (b) The DMS cartridge workflow consists of: (1) buffer priming to fill the cartridge with working buffer. (2) Sample injection where the magnetically tagged cell sample is circulated through the base of the cartridge chamber where magnetized cells are captured and pulled onto the micromagnetic substrate. (3) DMS processing where cells are transported vertically across the micromagnetic substrate and separate into high magnetic and low magnetic fractions. (4) After separation, a flexible “extraction lock” valve is actuated to separate the High Magnetic Fraction from the Low Magnetic Fraction. Each fraction can be eluted from the cartridge via Leur Lock ports.
Figure 1.
(a) DMS benchtop system houses a closed loop controlled internal magnetic wheel of rare earth magnets arranged in a Halbach array orientation to generate a cycling magnetic field and a peristaltic pump to perform automated sample processing through the DMS cartridge. The DMS cartridge is loaded into the benchtop system and connected to the peristaltic pump. A tablet with simple graphic user interface walks the user through a standardized protocol. (b) The DMS cartridge workflow consists of: (1) buffer priming to fill the cartridge with working buffer. (2) Sample injection where the magnetically tagged cell sample is circulated through the base of the cartridge chamber where magnetized cells are captured and pulled onto the micromagnetic substrate. (3) DMS processing where cells are transported vertically across the micromagnetic substrate and separate into high magnetic and low magnetic fractions. (4) After separation, a flexible “extraction lock” valve is actuated to separate the High Magnetic Fraction from the Low Magnetic Fraction. Each fraction can be eluted from the cartridge via Leur Lock ports.
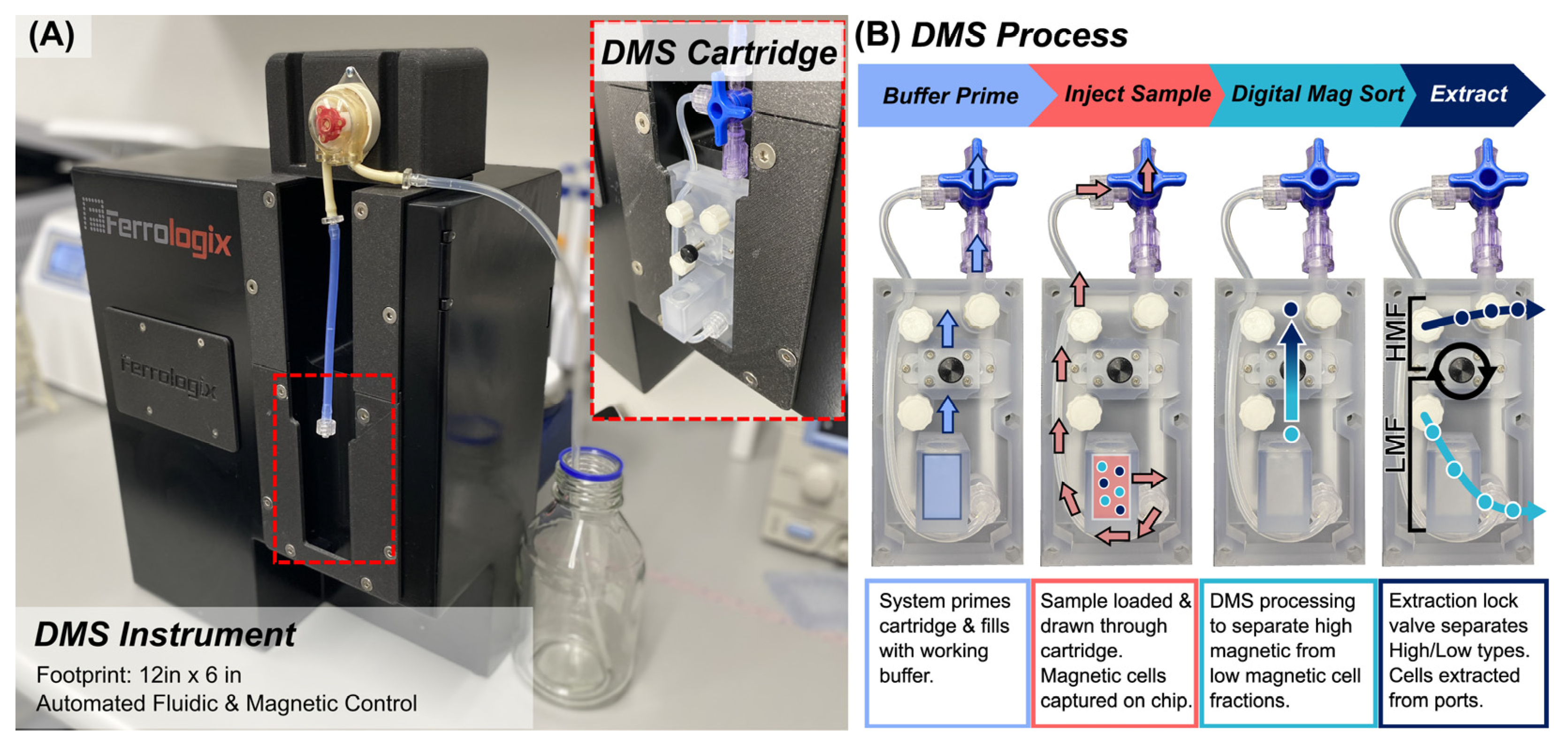
Figure 2.
Flow cytometry analysis of leukocytes processed through the DMS Hi/Low workflow. (a) Before separation, the sample was observed to have CD4Hi and CD4Low populations with >99% of the cells being CD45+. The peak-to-peak difference between CD4Hi and CD4Low populations was observed to be approximately 0.6 Log. (B) The HMF demonstrated enrichment of CD4Hi cells with 81.8% ±4.1 (N=3 trials) of cells occupying the CD4Hi gate while the (c) The LMF showed enrichment of CD4Low cells with 78.8% ±4.3% (N=3 trials). (d) Histogram plots of CD4 expression between HMF & LMF show significant peak expression with a peak-to-peak difference that correlates with the original sample. ANOVA analysis of the HMF and LMF fractions showed statistically significant differences of CD4 expression (p value < 0.0001).
Figure 2.
Flow cytometry analysis of leukocytes processed through the DMS Hi/Low workflow. (a) Before separation, the sample was observed to have CD4Hi and CD4Low populations with >99% of the cells being CD45+. The peak-to-peak difference between CD4Hi and CD4Low populations was observed to be approximately 0.6 Log. (B) The HMF demonstrated enrichment of CD4Hi cells with 81.8% ±4.1 (N=3 trials) of cells occupying the CD4Hi gate while the (c) The LMF showed enrichment of CD4Low cells with 78.8% ±4.3% (N=3 trials). (d) Histogram plots of CD4 expression between HMF & LMF show significant peak expression with a peak-to-peak difference that correlates with the original sample. ANOVA analysis of the HMF and LMF fractions showed statistically significant differences of CD4 expression (p value < 0.0001).

Figure 3.
Multi-donor fractionation performance of the DMS system of CD4Hi & CD4Low populations into the High Magnetic Fraction (HMF) and Low Magnetic Fraction (LMF) respectively. Donor samples had varying ratios of CD4(-), CD4Hi, & CD4Low phenotypes typically split between 40 to 50% CD4(-) 40 to 50% CD4Low, and 5 to 10% CD4Hi. Phenotypic gating based of CD4 expression was carried out using flow cytometry of samples before separation and in the LMF & HMF fractions. Separations across multiple donors show high enrichment of CD4Hi cells in the HMF (77.4 ± 4.9% donor average) and high enrichment of CD4Low cells in the LMF (79.9 ± 7.5% donor average). All DMS fractionations show a >90% purity of CD4(+) cells in both HMF and LMF fractions.
Figure 3.
Multi-donor fractionation performance of the DMS system of CD4Hi & CD4Low populations into the High Magnetic Fraction (HMF) and Low Magnetic Fraction (LMF) respectively. Donor samples had varying ratios of CD4(-), CD4Hi, & CD4Low phenotypes typically split between 40 to 50% CD4(-) 40 to 50% CD4Low, and 5 to 10% CD4Hi. Phenotypic gating based of CD4 expression was carried out using flow cytometry of samples before separation and in the LMF & HMF fractions. Separations across multiple donors show high enrichment of CD4Hi cells in the HMF (77.4 ± 4.9% donor average) and high enrichment of CD4Low cells in the LMF (79.9 ± 7.5% donor average). All DMS fractionations show a >90% purity of CD4(+) cells in both HMF and LMF fractions.
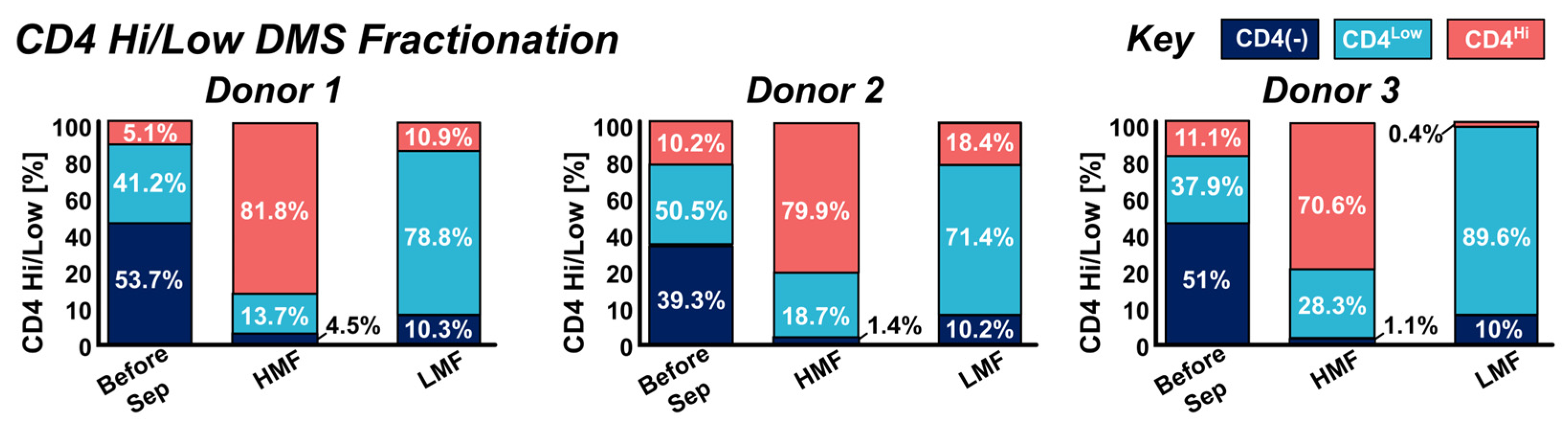
Figure 4.
Single cell RNA seq using a 10X GEM Chip was performed with leukocyte samples before separation and after fractionation into LMF and HMF. (a) Before separation, leukocytes exhibited expected populations of B Cells, Monocytes/Neutrophils, T Cells & subtypes, and other various sub-populations. (b) tSNE plot of LMF shows enrichment of monocytes (78%) and some T cells (11%). (c) The HMF shows enrichment of T cells (52%) and subpopulations (~14%) with some monocytes and neutrophils (31%). This data confirms that the DMS system is fractionating populations which have been shown to correlate with CD4 High/Low expression and aligns with published data and scientific publications.
Figure 4.
Single cell RNA seq using a 10X GEM Chip was performed with leukocyte samples before separation and after fractionation into LMF and HMF. (a) Before separation, leukocytes exhibited expected populations of B Cells, Monocytes/Neutrophils, T Cells & subtypes, and other various sub-populations. (b) tSNE plot of LMF shows enrichment of monocytes (78%) and some T cells (11%). (c) The HMF shows enrichment of T cells (52%) and subpopulations (~14%) with some monocytes and neutrophils (31%). This data confirms that the DMS system is fractionating populations which have been shown to correlate with CD4 High/Low expression and aligns with published data and scientific publications.
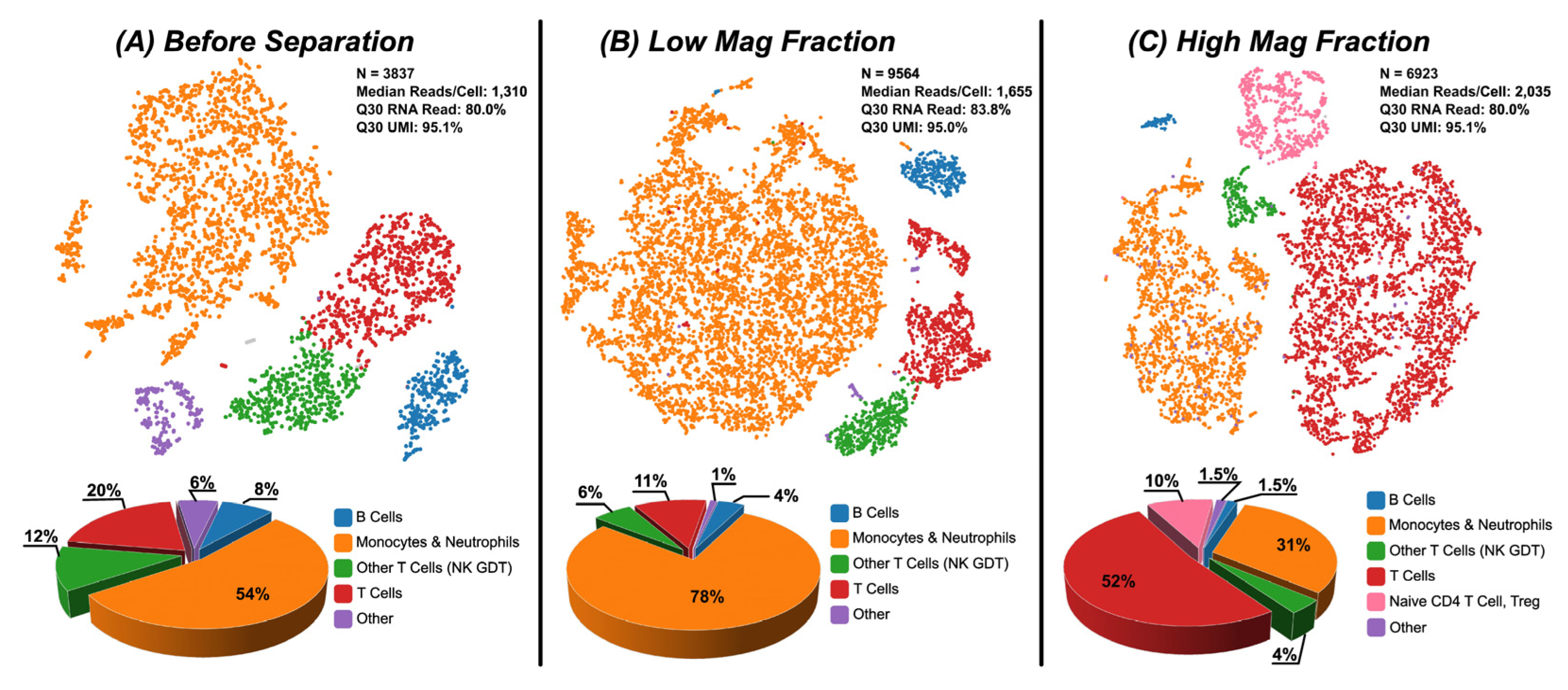
Figure 5.
Single cell RNA sequencing of LMF and DMS fractions. (a) cells in HMF show significantly higher expression of multiple T cell markers compared to the LMF (b) which showed significantly higher expression of monocyte and neutrophil markers. These data shows that the HMF preferentially enriching T cell populations while the LMF is preferentially enriching for monocyte and neutrophil populations.
Figure 5.
Single cell RNA sequencing of LMF and DMS fractions. (a) cells in HMF show significantly higher expression of multiple T cell markers compared to the LMF (b) which showed significantly higher expression of monocyte and neutrophil markers. These data shows that the HMF preferentially enriching T cell populations while the LMF is preferentially enriching for monocyte and neutrophil populations.
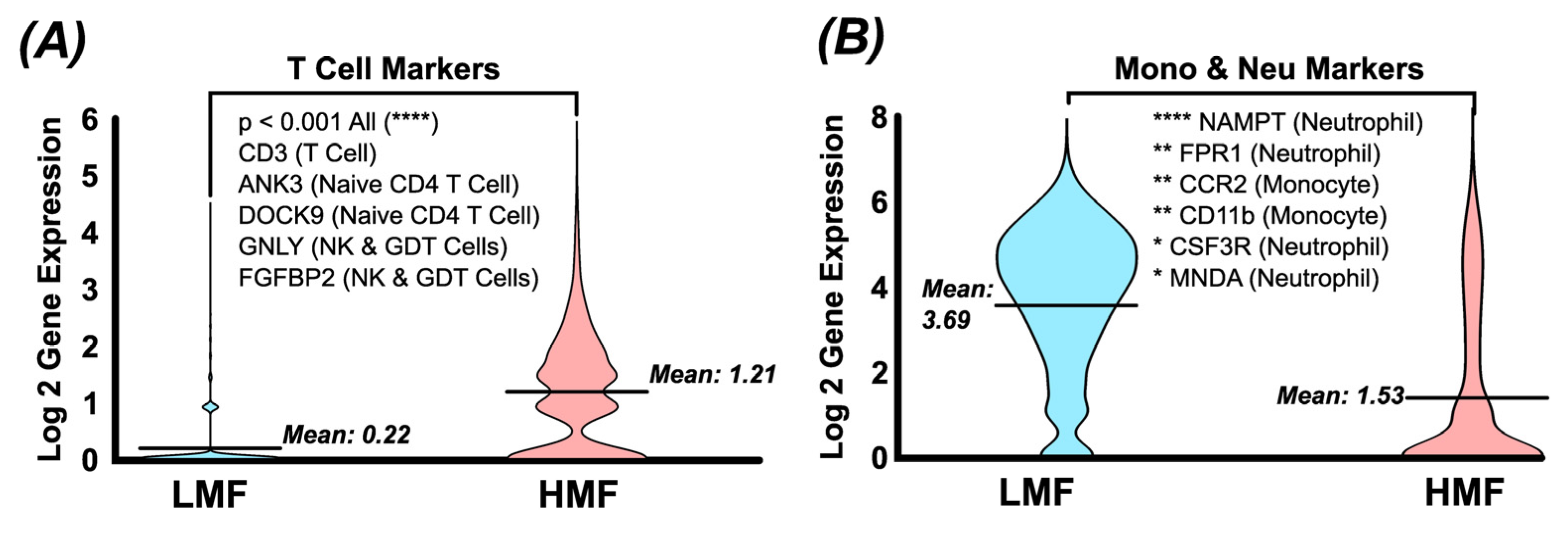
Figure 6.
Throughput comparison between DMS, FACS and Magnetic Enrichment/Depletion then FACS. (A) Due to its parallelized nature, DMS can achieve higher throughput processing of antigen density subpopulations. DMS is centrifugation free and requires a 30 min magnetic labeling, a rapid magnetic debulking or preselection followed by antigen density fractionation via DMS. (B) Contrastingly, FACS workflows require all cells in a sample to be analyzed and sorted which leads to long sort times which negatively impact viability and throughput. A FACS workflow requires antibody labeling and multiple centrifugation/wash steps before proceeding to FACS. Assuming a 10K cell per second throughput, a sample of 108 cells would take about 2.7 hrs to complete. (C) Often times magnetic enrichment or depletion is performed to debulk a sample before FACS. First magnetic enrichment/depletion is performed by magnetic labeling and pulldown via bulk magnet or column. Then the sample is processed for FACS isolation. While this FACS + MACS method increases throughput it requires the addition of multiple sample processing steps which reduces target cell yield and increases consumable & reagent cost significantly.
Figure 6.
Throughput comparison between DMS, FACS and Magnetic Enrichment/Depletion then FACS. (A) Due to its parallelized nature, DMS can achieve higher throughput processing of antigen density subpopulations. DMS is centrifugation free and requires a 30 min magnetic labeling, a rapid magnetic debulking or preselection followed by antigen density fractionation via DMS. (B) Contrastingly, FACS workflows require all cells in a sample to be analyzed and sorted which leads to long sort times which negatively impact viability and throughput. A FACS workflow requires antibody labeling and multiple centrifugation/wash steps before proceeding to FACS. Assuming a 10K cell per second throughput, a sample of 108 cells would take about 2.7 hrs to complete. (C) Often times magnetic enrichment or depletion is performed to debulk a sample before FACS. First magnetic enrichment/depletion is performed by magnetic labeling and pulldown via bulk magnet or column. Then the sample is processed for FACS isolation. While this FACS + MACS method increases throughput it requires the addition of multiple sample processing steps which reduces target cell yield and increases consumable & reagent cost significantly.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



