
Submitted:
13 September 2024
Posted:
16 September 2024
You are already at the latest version
Abstract
Vitamin D-dependent rickets type 1A (VDDR1A) is a rare autosomal recessive disorder, caused by pathogenic variants in the CYP27B1 gene, typically characterized by growth failure, rickets, leg bowing, fracture, seizures, hyperparathyroidism, hypocalcemia, high alkaline phosphatase, high or normal 25(OH)D3, and low 1,25(OH)2D3. We studied two siblings in a Mexican family with an atypical form of VDDR1A. In addition to the typical features of VDDR1A, the proband showed cafe au lait spots, small teeth and grayish sclera, with hypophosphatemia, normocalcemia and normal 25(OH)D3, the proband´s brother showed grayish sclera. Proband underwent Next Generation Sequencing. Sanger sequencing was performed in the proband, his brother, the parents and 100 healthy controls for validation of detected variant. Both brothers presented a recurrent variant NM_000785.3; c.1319_1325dupCCCACCC and a novel nonsense variant NM_000785.3; c.227G>A in the CYP27B1 gene. Calcitriol treatment had better response in proband´s younger brother. We describe the first Mexican family with an atypical form of VDDR1A associated with novel nonsense variant, the results contribute to the phenotypic spectrum and increases the pool of pathogenic variants in CYP27B1. Data suggesting that nonsense-truncating variants play a significant role in the severity of VDDR1A.
Keywords:
Vitamin D-deficient rickets type 1A
; CYP27B1 gene
; compound nonsense heterozygous
; hypophosphatemia
; atypical
; calcitriol
1. Introduction
Vitamin D-dependent rickets type 1A (VDDR1A; OMIM 264700) is a heterogeneous autosomal recessive disorder, caused by pathogenic variants in the Cytochrome P450 Family 27 Subfamily B Member 1 gene (CYP27B1; OMIM 609506) (NM_000785.3) on chromosome 12q13, which encodes a polypeptide of 508 amino-acids, the enzyme 25-hydroxyvitamin D3-1α-hydroxylase (NP_000776.1) (1,2). CYP27B1 gene defect result in bad conversion of 25-hydroxyvitamin D3 [25(OH)D3] to the active form 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] at renal level (3). 57% of cases described in the literature have a family history of rickets (3). VDDR1A manifests around 6 months to 2 years of age with similar symptoms to nutritional vitamin D-deficient rickets, such as short stature, deformities, bone fractures, hypotonia/weakness or seizures. Radiographic images show rickets (4,5). Biochemically present elevated parathyroid hormone (PTH) and alkaline phosphatase (ALP) with normo or hypocalcemia (6). The 25(OH)D3 levels can be normal or elevated, while 1,25(OH)2D3 can be normal or low. Treatment consists of calcitriol, calcium and/or phosphate supplementation (4). Eighty-one different pathogenic variants on CYP27B1 gene have been reported; around 28.6% are protein truncating (7,8). Hypophosphatemia has been observed constantly in VDDR1A cases (1,9,10,11,12), however, has not been considered relevant for the initial diagnostic, leading inexperienced doctors to confusion and delay in diagnosis (1). Hypophosphatemic rickets have been linked to seven gene mutations (13). Here, we described two brothers with a novel compound nonsense variant in the CYP27B1 gene associated to atypical features of VDDR1A. Truncating variants play a significant role in the severity of VDDR1A. This study emphasizes the importance of early diagnosis and treatment to limit complications in patients.
2. Case Reports
2.1. Patient 1
The 6-year-10-month-old Mexican proband is the first child of healthy, young-unrelated parents at delivery. No history of VDDR1A or malformations was mentioned in family members. The mother denied exposure to teratogens or diseases during pregnancy. He proband was born by vaginal delivery at 39 weeks of gestation with a weight of 3,260 g (25th percentile) and length of 51 cm (75–50th percentile). Occipitofrontal circumference (OFC) was not registered. Apgar’s scores were 8, 7 at 5 and 10 minutes. His language and motor development has been normal. At 3 months of age, he presented fracture in the tibiae with conservative management. At 15 months of age, he presented with a fracture in the right femur due to a fall from a height of 80 cm, was treated surgically.
Physical examination at 6 years-10 months of age, had a weight of 19,250 kg (10th centile), length of 103 cm (<3rd centile; -5.6 SD [standard deviation]), and OFC of 53.5 cm (75th centile). He had frontal bossing, midface hypoplasia, blue-grayish sclera (Figure 2B), small teeth, slight pectum carinatum, a widening right wrist and bilateral genu varum. Café aut lait greater than 1 cm were observed on the thorax and abdomen. External genitalia were Tanner I. X-rays showed a thickened short rib, a bilateral cystic femoral head, shortening of long bones with decreased density, irregular widening of the metaphysis, and curved diaphysis (Figure 1A-D).
Initial biochemical study at 13 months of age showed hypophosphatemia, normocalcemia and normal 25(OH)D3 levels, a diagnosis of hypophosphatemic rickets was suspected. More study at 14 months showed high levels of PTH and ALP, while, the 1,25(OH)2D3 was low. Thus, a diagnosis of vitamin D-dependent rickets was made. The proband began treatment with calcitriol at a dose of 0.5 μg/day and phosphate supplementation at a standard dose. After 2 months of treatment (16 months old), phosphorus and 1,25(OH)2D3 parameters returned to normal levels, calcium was low, while PTH and ALP remained high, calcium supplementation was added at dose of 500 mg/day. The genetic study at 32 months old confirmed the VDDR1A diagnostic, the biochemical parameters showed normal calcium, phosphorus and 1,25(OH)2D3; PTH and ALP remained high, calcitriol dose was changed to 1.5 μg/day. At 67 months old, PTH normalized, ALP was observed in mild high levels until the last evaluation (82 months of old). His stature is short with curved tibias. Renal tubular acidosis was ruled out in view of negative history of polyuria and normal pH in blood gas. The abdominal ultrasound and echocardiogram were normal. Biochemical parameters and radiographic changes of the proband before and after calcitriol treatment are shown in Table 1 and Figure 1A-D, respectively.
2.2. Patient 2
The proband’s brother, a 3-year-old male, is the second child of healthy parents, aged 28 and 34 years at delivery. Pregnancy was uneventful without prenatal exposure to teratogens or maternal illness. He was born by vaginal delivery at 37 weeks of gestation with a weight of 3,135 g (25–10th centile), length of 52 cm (75th centile), and OFC of 34 cm (25–10th centile). Apgar’s scores were 81, 95, without special neonatal management. On last physical examination at 3-year-old, he had a weight of 16 kg (75–50th percentile), length of 90 cm (25-10th percentile; -1.5 SD), and OFC of 51 cm (50th centile). His motor-intellectual development has been normal. Sanger sequencing at 2 months old was positive for the same mutation as his brother, he was treated with calcitriol dose of 1.0 μg/day from the third month old. After 16 months post-treatment ALP remained slightly above of upper limit, whereas, the calcium, phosphorus, 1,25(OH)2D3, and PTH levels were normal (Table 1). Renal tubular acidosis was ruled out. Radiographic images at one year of age were observed with minimal changes (Figure 1E). At 3-year-old his growth is normal, without fracture or body deformities The renal ultrasound, thyroid profile, expanded metabolic screening were normal.
3. DNA Samples and Next-Generation Sequencing
Written informed consent was obtained of all participants, for genetic analysis and publication of the results. This study required hospital ethics committee approval in accordance with the Declaration of Helsinki and local/national guidelines (approval number: DI/23/501/04/32).
Genomic DNA were extracted from leukocytes of patients, the parents, and 100 healthy controls using a blood DNA extraction kit according to the protocol provided by the manufacturer (Promega inc, Madison, WI, USA).
Next-generation sequencing (NGS) analysis was carried out in the proband. To capture the exonic regions of interest, the TruSight One Sequencing panel was used, which includes 4811 genes related to various genetic bone disorders or hypophosphatemic rickets. Paired end libraries were generated, and the Illumina HiSeq 2000 Sequencer (Illumina Inc., San Diego, CA, USA) was used for the massive sequencing of these libraries (2 x150bp). Next, bioinformatic analysis of the obtained data was performed using FastQC for the quality control of the reads, BWA for the mapping of the reads, GATK for the identification of the variants, and SnpEff for the verification of the impact of the variants. Subsequently, the variants in the exonic and splice site regions were prioritized using Clinvar database, HGMD, OMIM phenotypes for clinical correlation and Mendelian inheritance patterns. The allelic frequency of the variants was carried out with 1000 genomes and genomAD. The in-silico prediction of the effect of the variant at the protein level was analyzed with Polyphen and SHIFT.
4. Variant’s Validation
Validation of the variants was performed by Sanger sequencing from the proband (patient 1), his brother, their parents and 100 healthy controls using an automated sequencer (ABI3500; PE Biosystems, Foster City, CA). The primers for amplifying the CYP27B1 gene were forward 5′-CAGGTATCCAAGTGTCCGCT-3′ and reverse 5′-GATAGTTTCGGGACCCGCAG-3′ for exon 2 and forward 5′-CACTCTGTGTCACTATGCCAC-3′and reverse 5-GAAGATTCATTCTACCAGGTC-3′ for exon 8.
5. Statistical Analysis
Continuous variables are presented as means ± standard deviation. After verifying normal distribution, T of student (two-sided) and ANOVA of repeated measures was used to compare paired variables of changes in biochemical parameters at before and after calcitriol treatment. A p < 0.05 was considered statistically significant. SPSS version 24.0 statistical software (SPSS Inc., Chicago, IL 2016) was used for statistical analysis.
6. Genetic and Biochemical Results
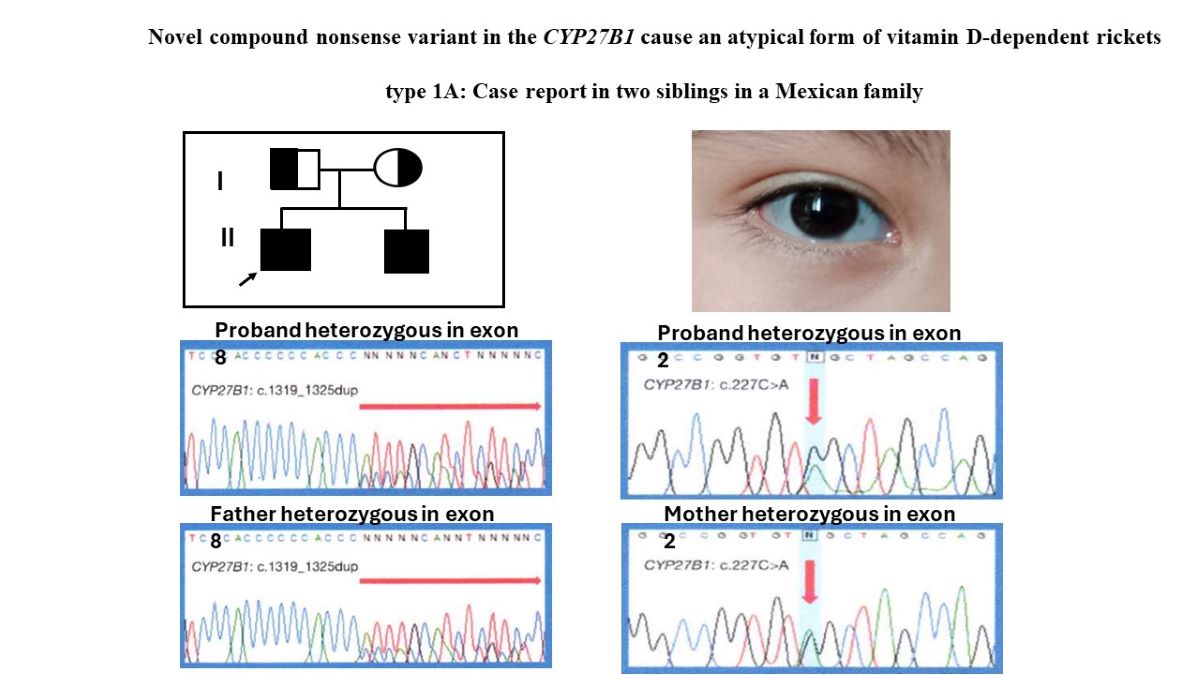
A heterozygous pathogenic variant previously reported c.1319_1325dupCCCACCC (p.Phe443Profs*24) (Figure 2C) was detected in exon 8 (9,11,12,14,15) and a novel heterozygous nonsense variant, c.227G>A (p.Trp76*) was located in exon 2 of both brothers (Figure 2D). The father was a heterozygous carrier for the variant in exon 8 (Figure 2E), whereas the mother was a heterozygous carrier for the exon 2 (Figure 2F). The c.227G>A (p.Trp76*) variant has not been described in the literature or in databases or population registries. SnpEff showed a high impact on protein switching, an allele frequency of 0.0 was obtained by 1000 genomes and genomAD. In silico predictors suggested that c.227G>A (p.Trp76*) is a deleterious change. This change was not found in 100 healthy normal controls. The truncation mutation p.Trp76* was situated in the N-terminal end of the CYP27B1 protein, losing the entire region of the binding pocket to the substrate downstream (16).
Biochemical parameters of the proband and his brother were compared before and after of calcitriol treatment during 53 and 16 months of follow-up, respectively. Significant statistical differences were observed for calcium, phosphorus, ALP, PTH, while for 25(OH)D3 and 1,25(OH)2D3 were not observed in the proband when were analyzed with repeated measures ANOVA, however, when T-student´s test for paired samples was applied, statistically significant differences were found to be present for 1,25(OH)2D3. In the proband´s brother, the calcium, PTH, 1,25(OH)2D3 were observed with statistically significant differences in the analysis with T student for paired samples (Table 1).
7. Discussion
This study describes two Mexican brother with VDDR1A and a novel c.227G>A (p.Trp76*) heterozygous nonsense variant, compound with the recurrent c.1319_1325dupCCCACCC (p.phe443Profs*24) in the CYP27B1 gene. Our proband presented atypically café au lait spots, small teeth, bossing frontal and grayish sclera. Initially hypophosphatemia, normocalcemia and normal 25(OH)D3 called our attention. The proband showed a partial response while his brother youngest had a good response to calcitriol treatment. A brief literature review of the VDDR1A cases with CYP27B1 pathogenic variants showed clinically: 59 % failure to thrive/ growth retardation, 38-41 % delayed motor milestones/ unable to walk, 13 % poor feeding, leg bowing 27 %, seizure 16.5 %, fracture 13.6 %; biochemically: hypocalcemia 75%, high ALP 92%, high 25(OH)D3 42%, low 1,25(OH)2D3 45%, high PTH 91%, and hypophosphatemia 68-83% (1,9,10,11,12). The cafe au lait spots seen in our proband were previously reported in an 11-month-old girl, the authors commented that it could be to low levels of 25(OH)D3 (17), however, in our proband it was normal. Small teeth observed in our proband, was documented in 81% of adults and 40% of children related to delay in dentition (11) or enamel hypoplasia (10). Bossing frontal was observed in a case (7). Finally, blue-grey sclera seen in our proband could be considered relevant because has not been reported in VDDR1A cases and can be confused with imperfect osteogenesis. It is noteworthy that hypophosphatemia, an initial biochemical result in our patients, has been observed constantly in retrospective studies of VDDR1A cases (11), however, has not been significantly considered as an initial indicator of VDDR1A often delaying diagnosis (1). Hypophosphatemia in VDDR1A result from elevated PTH and renal phosphate excretion (9). Others pathogenic variants in PHEX, FGF23, CLCN5, DMP1 ENPP1 SCL34A3, VDR, CYP2R1 genes can manifest hypophosphatemic rickets (13). VDDR1A have complete penetrance but clinical heterogeneity can be observed with the same mutation in CYP27B1 (8,11,18). Interesting, some cases may recover from the loss of CYP27B1 enzyme function, probably due to 1α-hydroxylase activity exerted by a non-CYP27B1 enzyme (15,16).
The best of our knowledge, around than 81 pathogenic variants (58 % missense, 23% frameshift, 9.8% nonsense have been identified in the CYP27B1 gene in patients with VDDR1A worldwide; 64.4% were homozygous, 38 % harbored compound heterozygous (7,8). The recurrent pathogenic variant c.1319_1325dup is for first time reported in the Mexican population, Previously, the case of a Mexican child with typical features was described but without genetic study (9), Cases previous in homozygous or compound state with others variants can be observed in Table 2 (3,7,10,12,15,16,21). To the best of our knowledge, this is the first study reporting the combination of the variant c.1319_1325dup compound with a nonsense variant, both result in a truncating protein, which is explained by a RNA degraded by the nonsense mechanism, causing severe inactivation of the 1α-hydroxylase enzyme (5). Our proband with the heterozygous c.1319_1325dup (p.phe443Profs*24) and the c.227G>A (p.Trp76*) variants presented with hypophosphatemia, normocalcemia without seizures, with fractures, severe growth retardation (height –5.6 SD at 86 months old), but café au lait spots, and small teeth are atypical features and the blue-gray sclera is observed for the first time, supporting the clinical heterogeneity for these variants.
Doses of calcitriol ranging from 0.5 to 2 μg/day or 0.008 to 0.40 μg/kg/day have been used to treatment of VDDR1A (11,16,19). Our proband was treated with calcitriol at doses of 0.5-1.5 μg/day, after 53 months of follow-up all biochemical parameters normalized, except for ALP that remained slightly high (Table 1), similar behavior was seen in the proband’s brother after 19-month follow-up, this indicative yet of pathological activity. However, in our proband radiographic images showed partial improvement (Figure 1A, B,C,D), clinically remained with short stature and a curved tibia. In the proband´s brother the imaging studies showed mild changes of cupping and fraying without widening (Figure 1E), the stature is normal (-1.5 SD), without deformities or nephrocalcinosis. Our proband had good compliance with his treatment, the partial response to treatment could be explained by the delay in management and the presence of truncated variants as previously observed, where adults or children older than 12 years with truncating variant and delayed treatment were seen to be more severe than those without this variant type, which led to permanent short stature and deformities (1,11). Another study of 12 children that presented truncating by the frameshift variant in at least one allele, were observed with delayed growth when were treated late and did not have correct compliance (21). In the treatment of a 13-month-old girl, a good biochemical response at 10 months post-treatment was observed, however she presented multiple fractures but growth or deformity data were not reported (7). In our case 2, the diagnosis at 3 moths and early treatment helped a good biochemical response, limitation of rickets without delayed growth, fractures or deformities. Therefore, according to this and previous studies the truncated variants suggest give a more severe phenotype, but a diagnosis and early treatment with a correct compliance and an adequate dose are important factors that must be taken care of for a better health prognosis in the patients with VDDR1A.
8. Conclusion
Here, we describe a VDDR1A Mexican family with atypical features associated with a novel compound nonsense variant, the results contribute to the phenotypic spectrum and increases the pool of pathogenic variants in CYP27B1. Truncating variants play a significant role in the severity of VDDR1A. Patient with rickets, initial hypophosphatemia, normocalcemia, and normal 25(OH)D3, CYP27B1 should be investigated as a possible diagnosis. These results can be useful in the genetic counselling of patients and we emphasize the importance of early diagnosis and treatment to limit complications in patients.
Author Contributions
Jaime Toral Lopez Physical examination, Clinical characterization, Methodology, Writing—review and editing. Cesar Candia Tenopala Physical examination, Clinical characterization, Biochemical data analysis and treatment control. Luz Maria Gonzalez Huerta Analyzed NGS data, designed primers and analyzed Sanger Sequencing. Methodology, Writing—review and editing. Alix Daniela Reyes Mosqueda Analyzed radiological images, Miguel Ángel Fonseca Sánchez obtaining genetic material. All authors read and approved the final manuscript.
Funding
The authors have no funding sources to declare.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by Ethics Committee OF Hospital general de México “Dr. Eduardo Liceaga with of registration DI/23/501/04/32, date: august 21th of 2023.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data generated or analyzed during this study is included in the final published article.
Acknowledgments
The authors would like to thank all the participants.
Conflicts of Interest
The authors declare that there is no conflict of interest.
References
- Fraser D, Kooh SW, Kind HP, Holick MF, Tanaka Y, DeLuca HF. Pathogenesis of hereditary vitamin-D-dependent rickets. An inborn error of vitamin D metabolism involving defective conversion of 25-hydroxyvitamin D to 1 alpha,25-dihydroxyvitamin D. N Engl J Med 1973;289:817-822. [CrossRef]
- Labuda, M.; Morgan, K.; Glorieux, F.H. Mapping autosomal recessive vitamin D dependency type I to chromosome 12q14 by linkage analysis. Am J Hum Genet 1990, 47, 28–36. [Google Scholar] [PubMed]
- Dodamani MH, Sehemby M, Memon SS, Sarathi V, Lila AR, Chapla A, Bhandare VV, Patil VA, Shah NS, Thomas N, Kunwar A, Bandgar TR. Genotype and phenotypic spectrum of vitamin D dependent rickets type 1A: our experience and systematic review. J Pediatr Endocrinol Metab 2021; 34:1505-1513. [CrossRef]
- Kitanaka S, Takeyama K, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa Y, Niimi H, Yanagisawa J, Tanaka T, Kato S. Inactivating mutations in the 25-hydroxyvitamin D3 1alpha-hydroxylase gene in patients with pseudovitamin D-deficiency rickets. N Engl J Med 1998; 338:653-661. [CrossRef]
- Wang JT, Lin CJ, Burridge SM, Fu GK, Labuda M, Portale AA, Miller WL. Genetics of vitamin D 1alpha-hydroxylase deficiency in 17 families. Am J Hum Genet 1998; 63:1694-1702. [CrossRef]
- Miller WL, Portale AA. Vitamin D 1 alpha-hydroxylase. Trends Endocrinol Metab 2000; 11:315-319. [CrossRef]
- Babiker AM, Al Gadi I, Al-Jurayyan NA, Al Nemri AM, Al Haboob AA, Al Boukai AA, Al Zahrani A, Habib HA. A novel pathogenic mutation of the CYP27B1 gene in a patient with vitamin D-dependent rickets type 1: a case report. BMC Res Notes 2014; 7:783. [CrossRef]
- Dhull RS, Jain R, Deepthi B, Cheong HI, Saha A, Mehndiratta M, Basu S. Vitamin D-dependent rickets (VDDR) type 1: case series of two siblings with a CYP27B1 mutation and review of the literature. J Bras Nefrol 2020; 42:494-497. [CrossRef]
- Velásquez-Jones, L.; Medeiros, M.; Valverde-Rosas, S.; Jiménez-Triana, C.; Del Moral-Espinosa, I.; Romo-Vázquez, J.C.; Franco-Alvarez, I. Seguimiento a largo plazo de un paciente con raquitismo dependiente de vitamina D tipo I [Long term follow up of a patient with type I vitamin D-dependent rickets]. Bol Med Hosp Infant Mex. 2015, 72, 190–194. [Google Scholar] [PubMed]
- Dursun F, Özgürhan G, Kırmızıbekmez H, Keskin E, Hacıhamdioğlu B. Genetic and Clinical Characteristics of Patients with Vitamin D Dependent Rickets Type 1A. J Clin Res Pediatr Endocrinol 2019;11:34-40. [CrossRef]
- Edouard T, Alos N, Chabot G, Roughley P, Glorieux FH, Rauch F. Short- and long-term outcome of patients with pseudo-vitamin D deficiency rickets treated with calcitriol. J Clin Endocrinol Metab 2011; 96:82-89. [CrossRef]
- Tahir S, Demirbilek H, Ozbek MN, Baran RT, Tanriverdi S, Hussain K. Genotype and Phenotype Characteristics in 22 Patients with Vitamin D-Dependent Rickets Type I. Horm Res Paediatr 2016;85:309-317. [CrossRef]
- Zou M, Guven A, BinEssa HA, Al-Rijjal RA, Meyer BF, Alzahrani AS, Shi Y. Molecular Analysis of CYP27B1 Mutations in Vitamin D-Dependent Rickets Type 1A: c.590G > A (p.G197D) Missense Mutation Causes a RNA Splicing Error. Front Genet 2020; 11:607517. [CrossRef]
- Yamazaki M, Michigami T. Osteocytes and the pathogenesis of hypophosphatemic rickets. Front Endocrinol (Lausanne) 2022; 13:1005189. [CrossRef]
- Li Y, Yuan X, Chen R, Lin X, Shangguan H, Yang X, Zhang Y. Clinical and genetic analysis of two Chinese families with vitamin D-dependent rickets type IA and follow-up. Orphanet J Rare Dis 2020; 15:273. [CrossRef]
- Durmaz E, Zou M, Al-Rijjal RA, Bircan I, Akçurin S, Meyer B, Shi Y. Clinical and genetic analysis of patients with vitamin D-dependent rickets type 1A. Clin Endocrinol (Oxf) 2012 Sep;77(3):363-369. [CrossRef]
- Yamamoto K, Masuno H, Sawada N, Sakaki T, Inouye K, Ishiguro M, Yamada S. Homology modeling of human 25-hydroxyvitamin D3 1alpha-hydroxylase (CYP27B1) based on the crystal structure of rabbit CYP2C5. J Steroid Biochem Mol Biol 2004;89-90:167-171. [CrossRef]
- Koek WN, Zillikens MC, van der Eerden BC, van Leeuwen JP. Novel Compound Heterozygous Mutations in the CYP27B1 Gene Lead to Pseudovitamin D-Deficient Rickets. Calcif Tissue Int 2016; 99:326-331. [CrossRef]
- Özcabı B, Tahmiscioğlu Bucak F, Jaferova S, Oruç Ç, Adrovic A, Ceylaner S, Ercan O, Evliyaoğlu O. A Case of Vitamin D-Dependent Rickets Type 1A with a Novel Mutation in the Uzbek Population. J Clin Res Pediatr Endocrinol 2016; 8:484-489. [CrossRef]
- Hu WW, Ke YH, He JW, Fu WZ, Wang C, Zhang H, Yue H, Gu JM, Zhang ZL. A novel compound mutation of CYP27B1 in a Chinese family with vitamin D-dependent rickets type 1A. J Pediatr Endocrinol Metab 2014; 27:335-341. [CrossRef]
- Lin Y, Guan Z, Mei H, Zhang W, Zhou Z, Su L, Cheng J, Zheng R, Liang C, Cai Y, Yin X, Wu D, Liu L, Zeng C. Clinical characteristics and long-term outcomes of 12 children with vitamin D-dependent rickets type 1A: A retrospective study. Front Pediatr 2022; 10:1007219. [CrossRef]
Figure 1.
Shows X-rays of the proband and his brother before and after calcitriol treatment. (A) The proband at 10-month-old with discrete cupping and widened metaphysis (white arrows). (B) Shows the proband at 2 year and 6-month-old (18 months post-treatment) with wide anterior rib ridge, irregularity of costal bodies associated with osteopenia (small white circles), humerus with diffuse osteopenia (big white circle), increased amplitude of the growth plate, at the level of the radius and ulna with frayed, cupped metaphysis and membranous ossification (white arrows) consistent with rickets. (C) Proband at the 3-year-old and (D) at 4 year-6-moths-old shows partial recoveries with diaphyseal slope (white arrowhead), cupping, widening and fraying at metaphysis of long bones (white arrows). Qualitative improvement in bone density is observed (black circle). (E) Shows X-ray of the proband’s brother at 9 months post- treatment with minimal changes in the bone density, cupping or fraying without widening in the metaphysis of humerus, femur, radius and ulna (white arrows) are observed.
Figure 1.
Shows X-rays of the proband and his brother before and after calcitriol treatment. (A) The proband at 10-month-old with discrete cupping and widened metaphysis (white arrows). (B) Shows the proband at 2 year and 6-month-old (18 months post-treatment) with wide anterior rib ridge, irregularity of costal bodies associated with osteopenia (small white circles), humerus with diffuse osteopenia (big white circle), increased amplitude of the growth plate, at the level of the radius and ulna with frayed, cupped metaphysis and membranous ossification (white arrows) consistent with rickets. (C) Proband at the 3-year-old and (D) at 4 year-6-moths-old shows partial recoveries with diaphyseal slope (white arrowhead), cupping, widening and fraying at metaphysis of long bones (white arrows). Qualitative improvement in bone density is observed (black circle). (E) Shows X-ray of the proband’s brother at 9 months post- treatment with minimal changes in the bone density, cupping or fraying without widening in the metaphysis of humerus, femur, radius and ulna (white arrows) are observed.
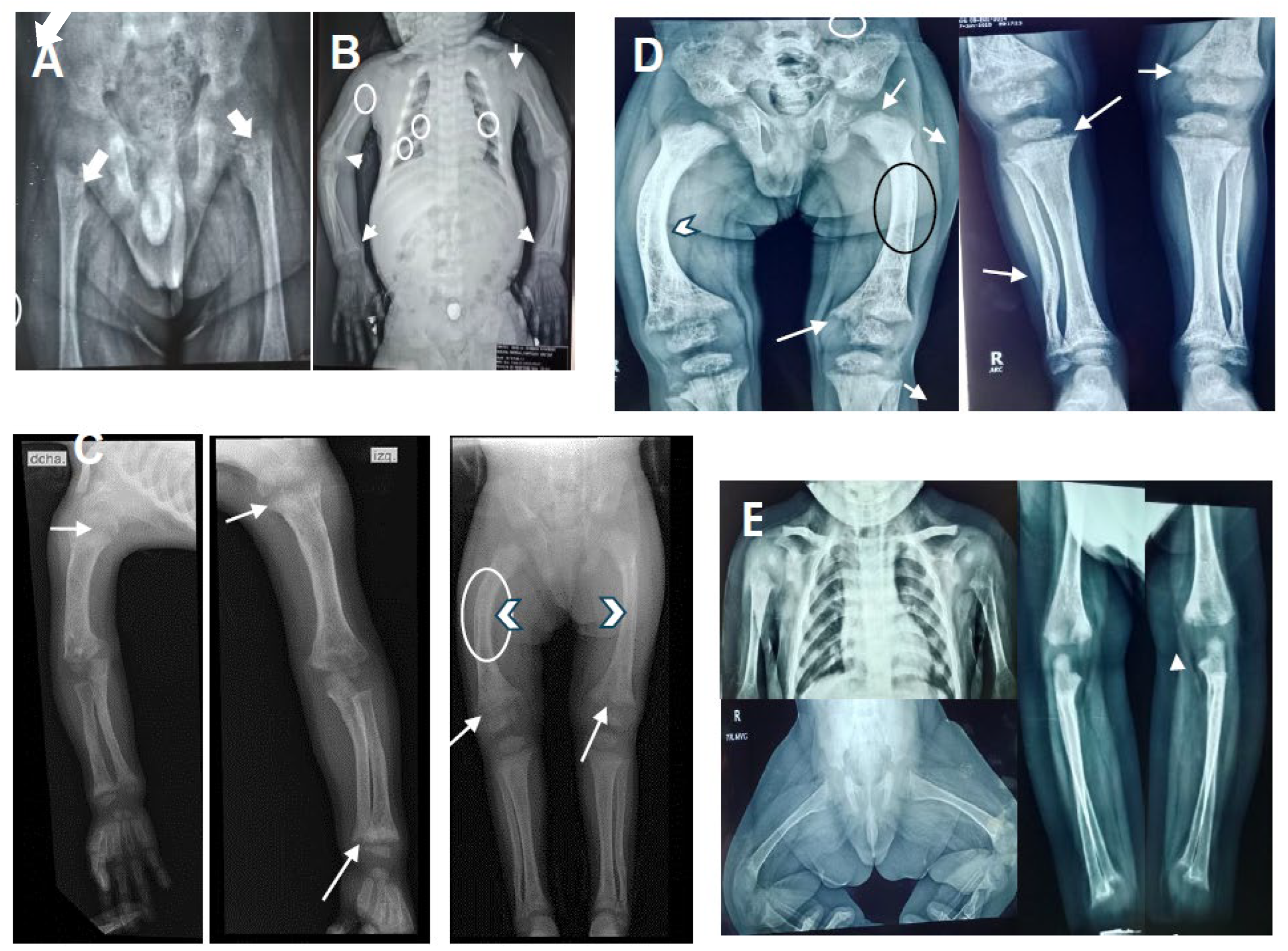
Figure 2.
(A) Shows the pedigree of the family illustrating the carrier status of the parents, the proband (arrow) and his affected younger brother. (B) Shows the blue-gray sclera in our proband. (C) and (D) Shows the partial electropherogram of the recurrent heterozygous variant in exon 8 and the novel heterozygous pathogenic variant in exon 2 in the proband, respectively. (E) Shows the partial electropherogram of the father with the c1319_1325dupCCCACCC (p.phe443Profs*24) variant in the exon 8 and (F) the novel c.227C>A (p.Trp76*) variant in exon 2 of CYP27B1 gene in the mother.
Figure 2.
(A) Shows the pedigree of the family illustrating the carrier status of the parents, the proband (arrow) and his affected younger brother. (B) Shows the blue-gray sclera in our proband. (C) and (D) Shows the partial electropherogram of the recurrent heterozygous variant in exon 8 and the novel heterozygous pathogenic variant in exon 2 in the proband, respectively. (E) Shows the partial electropherogram of the father with the c1319_1325dupCCCACCC (p.phe443Profs*24) variant in the exon 8 and (F) the novel c.227C>A (p.Trp76*) variant in exon 2 of CYP27B1 gene in the mother.
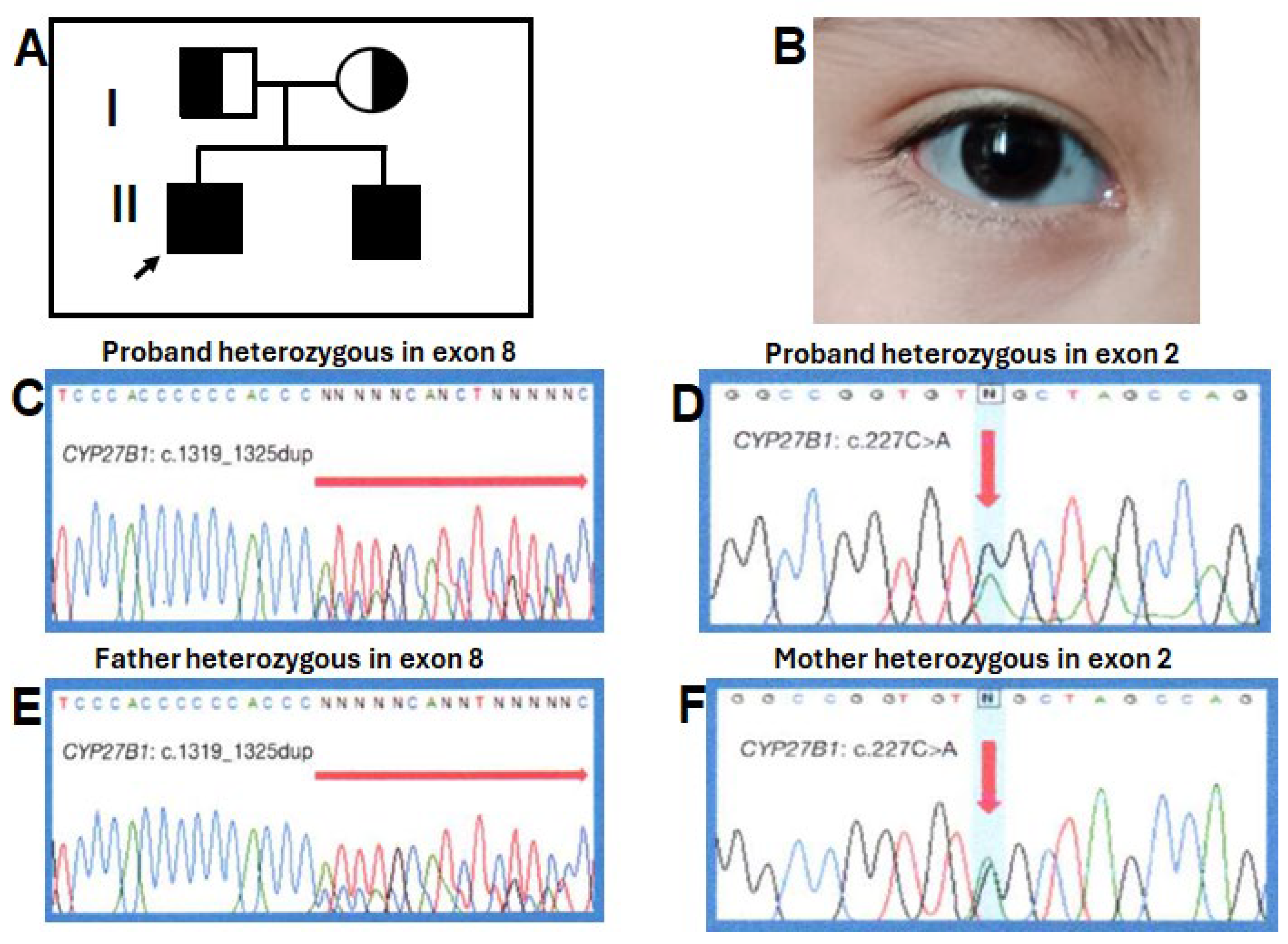

Table 1.
Changes in biochemical parameters before and after calcitriol treatment in our VDDR1A patients.
Table 1.
Changes in biochemical parameters before and after calcitriol treatment in our VDDR1A patients.
| Proband (patient 1) | Brother (patient 2) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Features | Reference ranges | level pre-treatment |
After 2 at 18 months of treatment | After 18 at 53 months of treatment | After 53 months of treatment | F of ANOVA test, P value |
Pre-treatment | 16 mo post-treatment | T Student test, P value |
| Age, y (year), mo (months) | 13 mo | 16 at 32 mo | 32 at 67 mo | 67 to 82 mo | < 3 mo | 3 to 19 mo | |||
| Calcium, mg/dl, mean+ SD | 8.3-10.6 | 8.3 | 8.14 + 0.6 | 8.6.0+ 0.2 | 9.5+ 0.4 | <0.001* | 8.0 | 9.0 | 0.02** |
| Phosphorus, mg/dl, mean+ SD | 2.4-5.1 | 2.1 | 3.1+ 1.1 | 3.2+ 0.5 | 4.8+ 0.7 | <0.006* | 3.8 | 4.2 | 0.59 |
| AP, UI/L, mean+ SD | 45-129 | 9200 | 5098+ 2108 | 2635+ 300 | 171+ 62 | 0.02* | 1336 | 682 | 0.05 |
| PTH, pg/ml, mean+ SD | 10-88 | 1350 | 702 + 185 | 289 + 137 | 60+ 27 | <0.001* | 314 | 67 | < 0.001** |
| 25(OH)D3, ng/ml, mean+ SD | 30-100 | 32.9 | 73.9 + 13 | 51.9 + 12 | 62+ 15 | 0.06 | 81 | 91 | 0.62 |
| 1,25(OH)2D3 pg/ml, mean+ SD | 19.6-54.3 | 6 | 36.7 + 14 | 34.8 + 12 | 45.3+ 4 | 0.12 (<0.001**) | 9 | 43 | 0.01** |
| Height, cm | 67 | 67 at 77 | 77 at 92 | 92 at 103 | NA | 59 | 79 | ||
| Height, SD | -3.2 | -4.3 to -5.3 | -5.3 to -6.6 | -6.6 to -5.6 | NA | -0.6 | -1.3 | ||
| Calcitriol dose, μg /day | NA | 0.5 | 1.5 | 1.5 | NA | NA | 1.0 | ||
| Average of biochemical parameters were analyzed with ANOVA of repeated measures, (*) P statistically significant; SD: standard deviation; NA, not applicable; (**) P statistically significant in the pre- and post-treatment with paired T Student test. | |||||||||
Table 2.
Genetic and Clinical response to calcitriol treatment in VDDR1A cases involving the c.1319_1325dupCCCACCC homozygous or compound with others variants in the CYP27B1 gene.
Table 2.
Genetic and Clinical response to calcitriol treatment in VDDR1A cases involving the c.1319_1325dupCCCACCC homozygous or compound with others variants in the CYP27B1 gene.
| Cases/ Captation age | DNA mutation | Exon | Amino acid change |
Phenotype | Clinical response at calcitriol treatment | Author |
|---|---|---|---|---|---|---|
| 2 cases (4 mo). 2 cases (18 and 19 mo). 1 case (INR) |
c.1319_1325dupCCCACCC. c.1319_1325dupCCCACC. c.1166G>A; c.1079 C>A |
8 8 7 6 |
p.Phe443Profs*24. p.Phe443Profs*24. p.Arg389His; p.Ser360* |
Severe hypocalcemic seizure. Delay in walking, mild hypocalcemia. Bowed legs. |
INR | (16) |
| 1 case (13 mo) | c.1510C > T. | 8 9 | p.Q504*. | Multiple fractures, bossing frontal, and classic VDDR1A | Good biochemical response at 10 mo posttreatment. Growth and deformity NR. | (7) |
| Case 8 (INR) Case 7 (INR) |
c.574A>G; c.1319_1325dupCCCACCC. c.1319_1325dupCCCACCC |
3 8 |
p.K192E; p.Phe443Profs*24. p.Phe443Profs*24. |
Mild phenotype Severe phenotype |
INR | (12) |
| Case 5 (14 mo) Case 8 (24 mo) |
c.1319_1325dupCCCACCC. c.1319_1325dupCCCACCC |
8 8 |
p.Phe443Profs*24. p.Phe443Profs*24. |
Growth retardation, hypocalcemia. Inability to walk and mild hypocalcemia. |
INR | (10) |
| 2 cases (INR) |
c.1319_1325dupCCCACCC. | 8 | p.Phe443Profs*24. |
Hypocalcemic seizure, severe growth retardation, walking difficulty and skeletal deformities | INR | (15) |
| 3 cases (4-18 mo) | c.1319_1325dupCCCACCC. | 88 8 | p.Phe443Profs*24. |
Hypocalcemic seizures in infancy, had rickets, dental anomaly, fractures. | Good biochemical response, two patients > 12 years persisted deformity. | (3) |
| 12 cases | c.1319_1325dupCCCACCC (Eight cases involving this variant) |
8 8 | p.Phe443Profs*24. |
Delayed walking, severe growth retardation. | Good rickets and biochemical response (6 mo to 15.6y of follow-up, 58% patients remained with short stature | (21) |
| Case 1 (proband; 13 mo) Case 2 (Brother; 3 mo) |
c.227G>A; c.1319_1325dupCCCACCC. c.227G>A; c.1319_1325dupCCCACCC. |
2 8 8 2 8 |
p.Trp76*; p.Phe443Profs*24. p.Trp76*; p.Phe443Profs*24. |
Low-normal calcemia, no seizures, fractures, severe growth retardation, sclera gray, Cafe-au-lait spots, frontal bossing, mild medial facial hypoplasia, pectum carinatum. Sclera gray. |
Good biochemical response, partial to rickets and bad to grown o deformity prevention. Good biochemical, rickets, growth response and to prevention of deformities. |
This study. |
OD: Onset of disease, CH: Compound heterozygous mutation, INR: Information not reported. DAH: Attention deficit-hyperactivity disorder, y: year old, mo: months.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



