
Submitted:
17 September 2024
Posted:
18 September 2024
You are already at the latest version
Abstract
SOCS2, an E3 ubiquitin ligase, regulates the JAK/STAT signaling pathway, essential for cytokine signaling and immune responses. Its dysregulation contributes to cardiovascular diseases (CVDs) by promoting abnormal cell growth, inflammation, and resistance to cell death. This study aimed to elucidate the molecular mechanisms underlying the interactions between Lumbricus-derived proteins and peptides and SOCS2, with a focus on identifying potential therapeutic candidates for CVDs. Utilizing a multifaceted approach, advanced computational methodologies, including 3D structure modeling, protein-protein docking, 100 ns molecular dynamics (MD) simulations, and MM/PBSA calculations, were employed to assess the binding affinities and functional implications of Lumbricus-derived proteins on SOCS2 activity. The findings revealed that certain proteins, such as Lumbricin, Chemoattractive glycoprotein ES20, and Lumbrokinase-7T1, exhibited similar activities to standard antagonists in modulating SOCS2 activity. Furthermore, MM/PBSA calculations were employed to assess the binding free energies of these proteins with SOCS2. Specifically, Lumbricin exhibited an average ΔGbinding of -59.25 kcal/mol, Chemoattractive glycoprotein ES20 showed -55.02 kcal/mol, and Lumbrokinase-7T1 displayed -69.28 kcal/mol. These values suggest strong binding affinities between these proteins and SOCS2, reinforcing their potential therapeutic efficacy in cardiovascular diseases. Further in vitro and animal studies are recommended to validate these findings and explore broader applications of Lumbricus-derived proteins.
Keywords:
cardiovascular disease
; earthworm
; lumbrokinase
; molecular dynamics simulation
; SOCS2
; protein-protein interactions
1. Introduction
Cardiovascular diseases (CVDs) represent a major global health issue, profoundly affecting both health outcomes and healthcare systems. According to the World Health Organization (WHO), CVDs cause an alarming 17.9 million deaths each year, positioning them as the primary cause of mortality worldwide [1]. The range of CVDs includes a variety of conditions affecting the heart and blood vessels, each characterized by distinct pathophysiological mechanisms and clinical presentations [2,3]. Some of the most common CVDs include coronary artery disease, hypertension, stroke, heart failure, and peripheral artery disease. These conditions result from complex pathological mechanisms such as endothelial dysfunction, atherosclerosis, inflammation, thrombosis, and vascular remodeling, which collectively disturb cardiovascular homeostasis [4,5]. Despite advancements in the field of medical science and healthcare delivery, conventional treatment methods for cardiovascular diseases predominantly concentrate on addressing risk factors, relieving symptoms, and modifying the progression of the disease [6]. Medications like statins, antiplatelet agents, and drugs for controlling blood pressure have proven effective in lowering cardiovascular morbidity and mortality. Yet, these treatments have drawbacks, including potential side effects, interactions with other drugs, and variability in effectiveness among different groups of patients [7,8]. Additionally, the rise of drug resistance presents a substantial obstacle to the sustained efficacy of current therapies, underscoring the necessity for alternative treatment strategies [9,10].
Suppressor of cytokine signaling 2 (SOCS2), an E3 ubiquitin ligase, has emerged as a critical regulator within the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway, a key mediator of cytokine signaling involved in inflammation and immune responses [11,12]. Dysregulated SOCS2 expression has been implicated in various pathogenic processes underlying CVDs, including aberrant cellular proliferation, inflammatory cytokine production, and apoptotic resistance in cardiac and vascular tissues [13,14]. Research has highlighted SOCS2's multifaceted roles in cardiovascular pathophysiology. For instance, in experimental models of myocardial infarction, elevated SOCS2 levels correlate with increased myocardial fibrosis and impaired cardiac function, suggesting a contributory role in adverse remodeling post-infarction [15,16]. Moreover, SOCS2 influences vascular smooth muscle cell proliferation and migration, which are critical processes in atherosclerotic plaque development and stability. Targeting SOCS2 presents a promising therapeutic strategy for mitigating cardiovascular pathology. By modulating SOCS2 activity, either through direct or indirect inhibition, researchers aim to temper excessive inflammation, promote vascular health, and potentially attenuate the progression of atherosclerosis and other CVDs [17,18].
Besides focusing on SOCS2, natural products have attracted growing interest as reservoirs of bioactive compounds with therapeutic promise against cardiovascular diseases. Earthworms, specifically from the Lumbricus genus, have become particularly fascinating due to their abundant biochemical composition and pharmacological properties [19]. Earthworms have been utilized across different cultures and over centuries in traditional medicine for treating a range of conditions such as inflammation, wounds, and gastrointestinal disorders [20]. This historical background highlights the promise of bioactive compounds derived from earthworms in tackling modern healthcare issues like CVDs. Specifically, earthworms contain Lumbrokinase, a bioactive component known for its potent fibrinolytic effects. Lumbrokinase has demonstrated thrombolytic properties, aiding in the dissolution of fibrin clots, which could potentially mitigate or reverse cardiac fibrosis, a characteristic feature of various cardiovascular disorders [21,22]. Bioactive compounds sourced from earthworms contribute positively to cardiovascular health by employing diverse mechanisms, encompassing anti-inflammatory, antioxidant, and vasodilatory activities [23,24]. Through regulation of these pathways, bioactive compounds from earthworms show promise in attenuating the fundamental pathological mechanisms behind CVDs, thereby enhancing patient outcomes. Moreover, investigating these compounds from earthworms presents a fresh and hopeful avenue in drug research and advancement, holding the potential for discovering novel therapeutic agents that boast enhanced effectiveness and safety profiles in combating CVDs. Molecular simulation techniques such as molecular docking and molecular dynamics simulations are pivotal in modern drug discovery. They are treasured for uncovering intricate protein-protein interactions and aiding in the discovery of promising therapeutic candidates [25,26]. In the context of CVDs, where the molecular mechanisms underlying pathogenesis are often intricate and multifactorial [27,28], molecular simulation techniques offer valuable insights into the interactions between bioactive molecules and their target proteins.
The goals of this study encompassed a thorough exploration of the potential therapeutic implications of targeting SOCS2 in CVDs using bioactive compounds derived from earthworms of the Lumbricus genus. Initially, our objective was to investigate the intricate protein-protein interactions between earthworm-derived bioactive compounds and SOCS2 through molecular docking techniques. By simulating how these compounds bind to specific regions within the SOCS2 protein structure, we aimed to identify lead molecules with strong binding affinity and favorable interactions, guiding the selection and optimization of potential therapeutic candidates. Subsequently, our study aimed to predict the binding affinities and interaction modes between the identified earthworm-derived compounds and distinct binding sites on the SOCS2 protein. Through molecular docking simulations, we aimed to characterize the molecular events responsible for the formation of stable protein-protein complexes, providing insights into the structural basis of compound-target interactions. Moreover, we sought to explore the dynamic behavior and structural integrity of protein-protein complexes formed between earthworm-derived compounds and SOCS2 using molecular dynamics simulations. By modeling the movements and interactions of atoms within these complexes over time, we aimed to elucidate the conformational dynamics and energetic principles governing the binding process, thereby deepening our understanding of the underlying mechanisms of action. Furthermore, our study aimed to assess the therapeutic potential of the identified earthworm-derived compounds in CVDs by targeting SOCS2. Through computational analyses and in silico modeling, we aimed to evaluate the effectiveness and specificity of these compounds in modulating SOCS2 function and alleviating the pathological mechanisms associated with cardiovascular diseases. This research lays the groundwork for future preclinical and clinical investigations into potential therapeutic interventions.
2. Results
2.1.3. D Structure Modeling
In the initial phase of 3D structure modeling in this study, we systematically gathered peptide and protein sequences sourced from earthworms of the Lumbricus genus using the UniProt database. This process yielded a substantial dataset comprising 979 records. To maintain the dataset's quality and reliability for subsequent analyses, stringent criteria were applied during the selection of peptide and protein sequences from Lumbricus earthworms. Several key criteria were employed to filter and refine the dataset obtained from UniProt. Initially, sequences were assessed for completeness to ensure only those with sufficient information for accurate 3D modeling were included, thereby reducing the risk of generating incomplete or erroneous structural models [29]. Furthermore, rigorous quality control protocols were implemented to identify and rectify sequencing errors, ambiguous residues, or any irregularities that might undermine the integrity of subsequent analyses. Sequences that did not meet these quality standards were excluded to ensure the overall accuracy and reliability of the dataset [30]. Additionally, duplicate entries were identified and eliminated from the dataset to avoid redundancy and potential biases in subsequent analyses. A thorough comparison and validation process of sequence data ensured that each unique protein or peptide appeared only once in the final dataset. Moreover, maintaining the taxonomic accuracy of the earthworm species represented in the dataset was essential to ensure specificity and relevance to the Lumbricus genus. Taxonomic verification procedures [31] were utilized to confirm the origin of all sequences from earthworms belonging to the Lumbricus genus, thereby preventing the inclusion of irrelevant or misclassified data. The database of protein sequences sourced from earthworms (Lumbricus genus) is available in Supplementary Data 1. This database includes detailed information on protein/peptide names, UniProt IDs, sequences, and identified protein binding sites (number of residues).
Next, the 3D structures of these proteins and peptides were generated using specialized computational tools designed for their specific structural attributes. For proteins and peptides that had suitable templates in the PDB, the I-TASSER algorithm was utilized. I-TASSER is recognized for its capability to predict precise 3D structures by utilizing template structures from the PDB. This method ensures accurate predictions of tertiary structures through analysis of sequence similarity and structural homology [32]. For proteins and peptides without existing templates in the PDB or newly introduced sequences in UniProt, AlphaFold was employed. AlphaFold represents a cutting-edge deep learning technique for predicting protein structures, revolutionizing this field with its high accuracy. It is particularly adept at predicting 3D structures for sequences lacking homologous structures in the PDB. By leveraging sophisticated machine learning algorithms trained on extensive protein databases, AlphaFold can predict precise 3D structures based solely on the amino acid sequences of proteins [33]. The 3D structure modeling phase of the research employed advanced computational techniques to create precise and dependable structural models for the proteins and peptides sourced from Lumbricus earthworms (Figure 1). These models form the basis for subsequent analyses, such as protein-protein docking simulations and molecular dynamics simulations, facilitating a thorough exploration of the interactions between bioactive compounds from earthworms and the target protein SOCS2. This investigation aims to identify potential therapeutic strategies for treating CVDs.
2.2. Protein-Protein Docking Simulation
The protein-protein docking simulations conducted in this study aimed to uncover the complex interactions between SOCS2 and proteins or peptides derived from earthworms of the Lumbricus genus, focusing specifically on identifying potential therapeutic candidates for cardiovascular diseases. Figure 2 illustrates the optimal binding poses of a standard agonist, a standard antagonist, and the top-performing Lumbricus-derived protein. The molecular docking results provide a comprehensive overview of the binding affinity, structural parameters, and clustering characteristics of the various SOCS2 complexes formed with proteins and peptides from earthworms. Specifically, comparisons were made with standard agonist (SOCS2: EpoR peptide) and antagonist (SOCS2: N4BP1) complexes, which served as benchmarks for assessing the effectiveness of the Lumbricus-derived compounds. A critical measure of the interaction strength between molecules is the binding affinity, quantified by the ΔG (Gibbs free energy) value. Lower ΔG values indicate stronger binding, suggesting more stable and favorable interactions between the proteins or peptides and SOCS2. The comparison between the top-performing proteins and peptides derived from Lumbricus and the standard agonist (SOCS2: EpoR peptide) and antagonist (SOCS2:N4BP1) complexes based on binding affinity provides valuable insights into the potential efficacy of Lumbricus-derived bioactive compounds in modulating SOCS2 activity compared to established proteins.
Firstly, the standard agonist, SOCS2: EpoR peptide, demonstrated a binding affinity with a ΔG value of -8.80 kcal/mol and a dissociation constant (Kd) of 6.50e-7 M. This indicates a strong interaction between SOCS2 and EpoR peptide, highlighting its efficacy as an agonist for SOCS2 activity modulation. In contrast, the standard antagonist, SOCS2:N4BP1, exhibited a slightly lower binding affinity, with a ΔG value of -8.30 kcal/mol and a Kd of 1.50e-6 M. Despite its lower binding affinity compared to the agonist, SOCS2:N4BP1 still displayed a notable interaction with SOCS2, indicative of its efficacy as an antagonist for SOCS2 activity inhibition. Among the Lumbricus-derived complexes, Cytochrome b, SCBP3 protein, Lumbricin, Chemoattractive glycoprotein ES20, Histone H3, and Lumbrokinase-7T1 emerged as top-performing candidates based on their low values of binding affinity ΔG (kcal/mol). Cytochrome b exhibited an ΔG value of -15.1 kcal/mol, surpassing the binding affinity of the standard agonist, SOCS2: EpoR peptide. This suggests that Cytochrome b may possess a stronger interaction with SOCS2, potentially surpassing the efficacy of EpoR peptide and N4BP1 in modulating SOCS2 activity. Similarly, SCBP3 protein, Lumbricin, and Chemoattractive glycoprotein ES20 displayed notable binding affinities with ΔG values of -12.6 kcal/mol, -12.3 kcal/mol, and -12.2 kcal/mol, respectively (Table 1 and Figure 3c). These values compare favorably with the binding affinities of the standard agonist and antagonist, underscoring the potential of these Lumbricus-derived compounds as effective modulators of SOCS2 activity for addressing cardiovascular diseases. Complete docking results are available in Supplementary Data 2. This dataset includes complex identifiers, HADDOCK scores, binding affinities (ΔG), dissociation constants (Kd), cluster sizes, RMSD values, van der Waals energies, electrostatic energies, desolvation energies, restraints violation energies, buried surface areas, and Z-scores.
Furthermore, the cluster size and RMSD values provide crucial structural insights into the protein-protein complexes. Cluster size refers to the number of distinct conformations or poses observed within the ensemble of generated protein-protein complexes [34]. A larger cluster size indicates greater structural diversity, suggesting multiple binding modes or orientations between the proteins and peptides from Lumbricus and SOCS2. This diversity within the complexes reflects potential variations in interaction configurations, influencing their functional properties and biological impacts [35]. For instance, the standard agonist SOCS2: EpoR peptide complex exhibits a notably large cluster size of 22, indicating the presence of diverse conformations or binding modes between SOCS2 and EpoR peptide. This diversity suggests the potential for versatile interactions, which could play crucial roles in various cellular processes regulated by ubiquitination. Conversely, the standard antagonist SOCS2:N4BP1 complex has a smaller cluster size of 19, implying fewer distinct binding configurations compared to the agonist complex. This difference in cluster size may reflect the specific nature of the antagonist interaction and its regulatory role in modulating SOCS2 activity. Analyzing the Lumbricus-derived protein/peptide complexes, it's evident that the cluster sizes vary across different interactions. For instance, complexes involving proteins like SCBP3 protein and Lumbricin exhibit relatively larger cluster sizes (41 and 43, respectively), suggesting structural diversity and potential functional versatility in their interactions with SOCS2. On the other hand, complexes such as SOCS2:Lysosomal membrane glycoprotein and SOCS2:Extracellular globin-4 have smaller cluster sizes (5 and 6, respectively), indicating a more limited range of binding configurations.
Conversely, RMSD values measure the variations or discrepancies in structure among distinct conformations within a given cluster. Reduced RMSD values indicate limited deviation or greater structural similarity among various conformations, implying heightened stability and consistency in the binding interactions observed throughout the simulation period [36]. This stability demonstrates the resilience of the protein-protein complexes, ensuring they maintain their specific structural arrangements despite variations or disturbances in their environment [37]. For instance, the standard agonist SOCS2:EpoR peptide complex exhibits an acceptable RMSD value of 1.7 Å, indicating minimal structural deviation among its conformations and suggesting a stable and well-defined binding mode. Similarly, Lumbricus-derived complexes like SOCS2:Cytochrome b (1.0 Å) and SOCS2:SCBP3 protein show low RMSD values (1.2 Å), indicating stable binding interactions and consistent structural configurations. In contrast, complexes with higher RMSD values, such as SOCS2:Intermediate filament protein (RMSD = 4.3 Å), suggest greater structural variability among their conformations, potentially reflecting dynamic binding interactions with SOCS2. This higher RMSD value suggests potential conformational flexibility or transient interactions, which could impact the functional significance of these complexes in cellular processes [38].
The analysis of intermolecular contacts (ICs) and non-interacting surfaces (NIS) for the protein-protein complexes provides valuable insights into the interaction dynamics between SOCS2 and various earthworm-derived proteins and peptides. The comparison between these complexes and the standard agonist (SOCS2: EpoR peptide) and antagonist (SOCS2: N4BP1) reveals significant differences in their interaction profiles, which can be linked to their potential efficacy in modulating SOCS2 activity (Table 2). The SOCS2: EpoR peptide complex, serving as the standard agonist, exhibited 3 charged-charged, 8 charged-polar, 12 charged-apolar, 2 polar-polar, 11 polar-apolar, and 12 apolar-apolar contacts. The NIS values for charged and apolar interactions were 25.55 and 38.69, respectively. In comparison, the SOCS2: N4BP1 complex, which functions as the standard antagonist, displayed a higher number of charged-charged (5), charged-polar (11), charged-apolar (20), and polar-polar (6) contacts, with slightly lower polar-apolar (11) and apolar-apolar (8) contacts. The NIS values were 29.71 for charged and 39.49 for apolar interactions.
Several proteins and peptides derived from Lumbricus showed distinct interaction profiles with SOCS2, suggesting varying efficacy in modulating its activity. The SOCS2: Cytochrome b complex, for instance, displayed high charged-apolar (25), polar-apolar (38), and apolar-apolar (33) contacts, with non-interacting surface (NIS) values of 15.13 for charged and 52.85 for apolar interactions. This indicates a potentially strong binding affinity. Cytochrome c oxidase subunit 3 showed balanced interactions, with significant polar-apolar and apolar-apolar contacts and NIS values of 14.71 (charged) and 49.41 (apolar), suggesting strong stability and efficacy. The SCBP3 protein complex, with 27.83 NIS (charged) and 41.74 (apolar), also displayed robust binding capability. Lumbricin exhibited higher numbers of charged interactions, indicating strong potential as a SOCS2 modulator. Chemoattractive glycoprotein ES20, with high numbers of charged and polar interactions and NIS values of 22.98 (charged) and 42.39 (apolar), also showed promise.
The statistical analysis aimed at exploring the relationship between the HADDOCK score and Root Mean Square Deviation (RMSD) unveiled a Pearson correlation coefficient (r) of 0.687. This coefficient quantifies the strength and direction of the linear relationship between these two variables. A value of 0.687 signifies a medium positive correlation, suggesting that there is a tendency for the HADDOCK score to decrease as the RMSD decreases. In the context of molecular docking, a lower HADDOCK score indicates a better docking quality and a lower RMSD reflects a closer match to the reference structure. Thus, the positive correlation implies that improvements in docking quality, as indicated by lower HADDOCK scores, are associated with a more accurate docking pose, as represented by lower RMSD values. This relationship underscores the consistency between these two metrics in evaluating the quality of protein-protein docking simulations. Further analysis revealed that approximately 46.75% of the proteins and peptides derived from Lumbricus demonstrated favorable RMSD values, defined as RMSD values equal to or less than 2.00 Å (Figure 3a). The favorable RMSD values indicate that many of the predicted protein-protein complexes have structural conformations closely matching experimental or reference structures, highlighting the overall success of the docking simulations in accurately predicting these arrangements. However, the analysis also revealed three outliers with very large RMSD values. These outliers suggest instances where the predicted structures significantly deviate from the experimental or reference structures. Such deviations could be due to inaccuracies in the docking algorithm, limitations in the input experimental data, or the inherent complexities of the protein-protein interactions being studied [39].
The statistical analysis to examine the relationship between the HADDOCK score and binding affinity (ΔG) revealed a Pearson correlation coefficient (r) of 0.491 (Figure 3b). This coefficient reflects the strength and direction of the linear relationship between these two variables. A value of 0.491 suggests a moderate positive correlation, indicating that as the HADDOCK score decreases (indicating better docking quality), the binding affinity also tends to decrease. This means that improvements in docking quality, as indicated by lower HADDOCK scores, are associated with stronger binding between the proteins, as shown by lower binding affinity values. Conversely, higher HADDOCK scores correlate with weaker binding affinity, underscoring a discernible trend between docking quality and binding strength.
The HADDOCK scoring system effectively captures key aspects of protein-protein interactions that influence binding affinity, including molecular surface complementarity, electrostatic interactions, and van der Waals forces. This indicates that HADDOCK's scoring metrics align well with the physical principles governing these interactions, enhancing the reliability of its predictions. The correlation matrix depicted in Figure 3d provides deeper insights into the relationship between binding energy (kcal/mol) and its individual energy components. Specifically, the positive correlation scores between binding affinity and both van der Waals energy (correlation score: 0.63) and desolvation energy (correlation score: 0.39) are particularly noteworthy. Van der Waals forces are crucial in stabilizing the complex by facilitating close contact between the protein surfaces, while desolvation energy reflects the energetic cost of removing water molecules from the binding interface. A positive correlation with van der Waals energy implies that as these interactions become stronger, the overall binding affinity increases, indicating a more stable protein-protein complex [40,41]. Similarly, the positive correlation with desolvation energy suggests that as the system pays the energetic cost to remove water molecules, the resulting interactions between the proteins are more favorable, thus increasing the binding affinity [42]. Conversely, the correlation between binding affinity and electrostatic energy revealed a negative value (-0.22), signifying an inverse relationship between these factors. This implies that as electrostatic energy increases, the binding affinity tends to decrease. Electrostatic interactions involve the attraction or repulsion between charged residues on the protein surfaces, influencing the stability of protein-protein complexes [43]. However, interpreting this correlation requires caution due to the multifaceted nature of protein-protein interactions. While electrostatic energy plays a significant role, other variables beyond the HADDOCK score can also impact binding affinity. Factors such as specific amino acid residues at the binding interface, which may facilitate or hinder interactions, post-translational modifications that alter protein structure and function, and environmental conditions such as pH and ion concentrations, can all influence the overall stability and strength of protein-protein complexes [44,45].
The thorough examination of hydrogen bond interactions between SOCS2 and the highest-performing proteins sourced from the earthworm (Lumbricus genus) provides insights into the molecular mechanisms that govern their binding interactions (Table 3). These interactions are pivotal for stabilizing protein-protein complexes and facilitating precise recognition between the involved proteins [46]. The interactions predominantly involve specific residues and atoms crucial for stabilizing the protein-protein complexes, focusing particularly on the ligand binding sites of SOCS2, namely Arg73, Ser75, Ser76, and Arg96. The interactions observed between SOCS2 and its standard agonist (EpoR peptide) and antagonist (N4BP1) molecules served as benchmarks for evaluating the binding efficacy of Lumbricus-derived proteins and peptides. The EpoR peptide exhibited multiple hydrogen bonds with key residues such as Val55, Ser76, Arg96, Lys113, and others, with interaction distances ranging from 2.69 Å to 3.33 Å. Conversely, N4BP1 demonstrated interactions involving Gln32, Arg41, Tyr49, Asp74, and others, emphasizing its distinct binding profile characterized by shorter interaction distances (2.57 Å to 2.78 Å). These results underscore the specificity and strength of interactions necessary for SOCS2 modulation by both agonist and antagonist molecules. The proteins and peptides from the Lumbricus genus, including Cytochrome b, SCBP3 protein, Lumbricin, Lumbrokinase-7T1, and others, displayed diverse binding interactions with SOCS2, highlighting their potential as novel modulators of SOCS2 activity. Cytochrome b, for instance, engaged in hydrogen bonds with His77 and Arg96, indicating a stable binding interface critical for functional interaction. Similarly, SCBP3 protein exhibited interactions involving Lys59 and Arg96, demonstrating specificity in binding residues essential for complex stability. Lumbricin and Lumbrokinase-7T1 revealed extensive hydrogen bonding networks with residues such as Arg41, Tyr49, Asp74, and Arg96, underscoring their potential to competitively bind with SOCS2 and influence its regulatory functions. These interactions were characterized by moderate to strong interaction distances, indicative of robust binding affinity crucial for therapeutic efficacy.
2.3. Molecular Dynamics (MD) Simulation
The MD simulation results offer a comprehensive understanding of the behavior of protein-protein complexes formed between Lumbricus-derived proteins and SOCS2. Throughout the 100 ns simulation, SOCS2 maintained a relatively stable conformation, as evidenced by the average RMSD values ranging from 2.413 to 2.599 Å without significant spikes (Figure 4a). This stability suggests that SOCS2 interactions with both standard agonist and antagonist, as well as Lumbricus-derived proteins, were dynamically consistent over the simulation period. When analyzing the RMSD values, which indicate the deviation of protein structures from their initial conformations, notable differences emerge among the complexes. For instance, the SOCS2:EpoR peptide (Standard Agonist) complex displays a slightly higher average RMSD (2.423 Å) compared to apo-protein SOCS2 (2.417 Å). This observation suggests a moderate increase in structural flexibility upon the binding of the standard agonist, indicating potential conformational adjustments required for effective binding. Conversely, the RMSD value for the SOCS2:N4BP1 (Standard Antagonist) complex (2.496 Å) remains similar to that of the apo-protein, indicating that the binding of the antagonist may not significantly perturb the structural stability of SOCS2. Furthermore, the Lumbricus-derived protein complexes, including Cytochrome b, SCBP3 protein, Lumbricin, Chemoattractive glycoprotein ES20, and Lumbrokinase-7T1, exhibit slightly higher average RMSD values ranging from 2.467 to 2.587 Å compared to the standard complexes. These differences suggest potential variations in the dynamic behavior and conformational changes induced by the binding of Lumbricus-derived proteins. The higher RMSD values imply that these Lumbricus-derived proteins may interact with SOCS2 in a manner that elicits different structural adjustments or conformational dynamics compared to the standard agonist and antagonist.
During MD simulations, RMSF analysis was performed to assess the flexibility of individual amino acid residues within SOCS2. The average RMSF values obtained ranged from 0.876 to 1.397 Å, reflecting moderate flexibility in various regions of the protein. This analysis provides valuable insights into the mobility and flexibility of specific residues, enhancing our understanding of their functional roles within the protein structure [47]. Upon examining the interaction between the top-performing Lumbricus-derived protein complex and SOCS2, it was apparent that this interaction disrupted hydrogen bonds with specific residues, especially within the regions encompassing amino acid residues Arg73 to Thr83 and Arg96 to Phe104 (Figure 4b). These regions are notably critical as they constitute the active binding site of SOCS2 as a target receptor [48]. The disruption of hydrogen bonds in critical areas led to greater residue fluctuations compared to the SOCS2 agonist and apo-protein complex. This increase in residue fluctuations suggests enhanced mobility and flexibility of these residues when the top-performing Lumbricus-derived protein binds to SOCS2. Notably, this pattern of increased flexibility resembles that of N4BP1, a known standard antagonist. The similarity in flexibility patterns at these specific residues indicates that the top-performing proteins from Lumbricus have the potential to act as inhibitors, much like standard antagonists. This finding is significant as it suggests that Lumbricus-derived proteins could modulate SOCS2 activity by acting as inhibitors, similar to known antagonists. By disrupting hydrogen bonds and increasing the flexibility of key binding site residues, these proteins may interfere with SOCS2's functional interactions, presenting promising avenues for developing therapeutic interventions targeting SOCS2-mediated cellular processes. Further investigation into the structural and functional implications of these interactions is necessary to fully understand their therapeutic potential and to develop new treatments for SOCS2-related diseases.
The radius of gyration (RoG) offered insights into the compactness or degree of expansion of protein structures during MD simulations [49]. For the SOCS2 complexes, the average RoG values ranged from 1.674 to 2.678 Å. The RoG values for the standard SOCS2 agonist (EpoR peptide) and antagonist (N4BP1) complexes were 2.101 Å and 2.162 Å, respectively. For the Lumbricus-derived protein complexes, RoG values ranged from 2.176 to 2.678 Å, slightly higher than the standard agonist but comparable to the antagonist. The number of hydrogen bonds formed between the two interacting proteins was indicative of the strength and specificity of their interaction. For the standard complexes, the SOCS2: EpoR peptide agonist complex formed 11 hydrogen bonds, while the SOCS2:N4BP1 antagonist complex formed 24 hydrogen bonds. Comparatively, the Lumbricus-derived protein complexes displayed similar or slightly higher numbers of hydrogen bonds, with values ranging from 16 to 39. Notably, the SOCS2:Cytochrome b complex formed the highest number of hydrogen bonds (39), followed closely by the SOCS2: Peroxidasin complex (35) (Table 4). The findings indicated that the interactions between SOCS2 and Lumbricus-derived proteins featured a comparable or slightly higher number of hydrogen bonds relative to the standard interactions. This suggests robust and specific binding between these proteins, which could enhance their functional relevance. Furthermore, the RoG values indicated that the Lumbricus-derived protein complexes displayed similar levels of compactness or dispersion compared to the standard complexes, suggesting they maintained stable structural conformations throughout the simulation period.
2.4. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Calculations
The MM/PBSA calculations provided valuable insights into the thermodynamic stability and binding affinity of the SOCS2 protein-protein complexes, offering a robust method to assess their molecular interactions [50]. We assessed the strength of the protein-protein interactions by calculating the average ΔGbinding values, which indicate the binding free energy, for each complex. In the case of the standard complexes, SOCS2 bound to EpoR peptide (Standard Agonist) demonstrated a mean ΔGbinding of -42.60 kcal/mol, indicating a substantial level of stability in the interaction. Conversely, when bound to N4BP1 (Standard Antagonist), SOCS2 exhibited a higher mean ΔGbinding of -42.51 kcal/mol, suggesting a comparatively weaker interaction. However, the Lumbricus-derived protein complexes presented intriguing findings, showcasing similar or even more negative mean ΔGbinding values compared to the standard complexes. Notably, SOCS2 complexed with Lumbrokinase-7T1 displayed a notably high mean ΔGbinding of -69.28 kcal/mol, indicative of a robust and energetically favorable interaction. This suggests that the Lumbricus-derived protein has a strong affinity for SOCS2, potentially surpassing the binding strength observed with the standard agonist and antagonist. Furthermore, the complexes formed between SOCS2 and Lumbricin and Chemoattractive glycoprotein ES20 exhibited mean ΔGbinding values of -59.25 kcal/mol and -55.02 kcal/mol, respectively (Table 5). These values suggest strong binding affinities comparable to or even exceeding those observed in the standard complexes. This implies that the Lumbricus-derived proteins possess significant potential in modulating SOCS2 activity, potentially rivaling or surpassing the efficacy of standard proteins.
Furthermore, a detailed examination of the binding free energy of individual amino acid residues within SOCS2 provided profound insights into the molecular mechanisms governing binding specificity and affinity. Notably, Arg96 and Ser78 were identified as crucial contributors to the observed antagonistic activity within the complexes. In interactions between SOCS2 and the top-performing protein from Lumbricus, these specific residues demonstrated significantly higher binding affinity values compared to the standard agonist complex. The elevated binding affinity of Arg96 and Ser78 underscores their pivotal roles in mediating the observed antagonistic effects (Figure 5). These residues are likely involved in essential interactions that govern complex stability and specificity, influencing overall functional outcomes. The heightened affinity observed in the complexes involving Lumbricus-derived proteins highlights the importance of these interactions in modulating SOCS2 activity and emphasizes their potential as key factors in therapeutic efficacy. By elucidating the roles of individual amino acid residues in binding energetics, this analysis offers critical insights into the structural basis of protein-protein interactions. The identification of Arg96 and Ser78 as significant contributors to antagonistic activity enhances our understanding of the molecular determinants underlying the intricate interplay between SOCS2 and its interacting partners. These findings pave the way for targeted manipulation of specific residues to effectively modulate SOCS2 function, presenting promising opportunities for developing therapeutic interventions aimed at SOCS2-associated pathways.
3. Discussion
The research employed a comprehensive approach to explore the interactions between SOCS2 and proteins and peptides sourced from Lumbricus earthworms, focusing particularly on their potential therapeutic implications in cardiovascular diseases. Emphasizing meticulous data collection and stringent quality control measures, the study utilized advanced computational techniques and methodologies for tasks such as 3D structure modeling, protein-protein docking simulations, molecular dynamics (MD) simulations, and binding free energy calculations. During the 3D structure modeling phase, the study utilized bioinformatics and computational biology tools to generate precise and dependable structural models of the Lumbricus-derived proteins and peptides. By systematically gathering sequences from the UniProt database and applying rigorous selection criteria, the study ensured the dataset's integrity and relevance. This approach aligns with previous research efforts that have successfully predicted protein structures with high precision using bioinformatics tools [51]. Furthermore, employing both I-TASSER and AlphaFold algorithms enabled thorough modeling of proteins and peptides, regardless of whether homologous structures were available in the PDB. This approach significantly expanded the breadth and depth of our structural modeling endeavors [33].
The research explored the intricate interactions between SOCS2 and proteins derived from Lumbricus earthworms using protein-protein docking simulations, aiming to identify potential therapeutic strategies for cardiovascular diseases. By meticulously comparing the binding affinities of these Lumbricus-derived proteins with those of standard agonist and antagonist complexes, the study identified promising candidates that exhibited comparable or enhanced effectiveness in modulating SOCS2 activity. These findings align with earlier studies emphasizing the therapeutic potential of natural compounds in managing cardiovascular diseases. Previous research has particularly highlighted the medicinal properties of Lumbricus earthworms, including their primary component, Lumbrokinase, which has shown promise in treating cardiovascular conditions [52]. By elucidating the molecular intricacies of Lumbrokinase's interaction with the SOCS2 pathway, this study contributes novel insights into the therapeutic potential of Lumbrokinase for cardiovascular treatment. Additionally, the analysis of cluster size and RMSD values provided insights into the structural diversity and stability of protein-protein complexes, offering valuable information for rational drug design and optimization [53]. MD simulations further elucidated the dynamic behavior of protein-protein complexes over time, uncovering potential mechanisms for modulating SOCS2 activity. The study provided detailed insights into the structural dynamics and functional implications of SOCS2-therapeutic protein interactions by examining residue flexibility and hydrogen bond interactions. This aligned with previous studies that had used MD simulations to investigate protein-protein interactions and elucidate their dynamic behavior [54]. The study observed that Lumbricin, Chemoattractive glycoprotein ES20, and Lumbrokinase-7T1 demonstrated sustained interactions with the SOCS2 protein throughout the simulation period, akin to the behavior exhibited by the standard antagonist complex. However, it's important to note that while these proteins showed promising interactions with SOCS2, specific experimental studies directly linking them to SOCS2's E3 ligase activity are currently lacking in the literature. Furthermore, the study discussed the structural features and key interaction residues within these protein-protein complexes, elucidating the molecular basis underlying their binding affinity and activity. By analyzing the conformational changes and intermolecular forces at play during the MD simulations, the study highlighted the structural motifs and binding pockets crucial for stabilizing the interactions between the Lumbricus-derived proteins and SOCS2. The resemblance in activity between these Lumbricus-derived proteins and the standard antagonist suggests that they may function through similar mechanisms or binding modes, warranting further investigation into their therapeutic potential.
4. Limitations, Clinical Implications, And Future Works
While the study provides valuable insights into the potential therapeutic interactions between SOCS2 and proteins derived from Lumbricus earthworms, several limitations should be acknowledged. Firstly, the findings are based on computational protein-protein docking simulations, which, while robust, rely on predictive models and may not fully capture the complexities of real biological systems. Experimental validation is essential to confirm the observed interactions and their functional implications in vitro and in vivo. Secondly, the study primarily focused on binding affinities and structural dynamics without directly measuring biological outcomes such as enzymatic inhibition or cellular responses. Future research should incorporate functional assays to validate the therapeutic effects of these interactions on SOCS2 activity and explore their broader implications in disease models. Thirdly, the specific mechanisms through which Lumbricus-derived proteins modulate SOCS2 function, particularly regarding E3 ligase activity, remain speculative. Experimental studies investigating these mechanisms are crucial to elucidate their precise therapeutic potential and optimize their application in clinical settings.
The study's findings have significant clinical implications for cardiovascular disease management and potentially other SOCS2-related conditions. Identifying natural compounds from Lumbricus earthworms that exhibit comparable efficacy to standard agonists and antagonists highlights their potential as alternative or adjunctive therapies. These compounds may offer novel strategies for modulating SOCS2 activity, potentially leading to more targeted and effective treatments with fewer side effects compared to conventional therapies. Furthermore, understanding the molecular interactions between Lumbricus-derived proteins and SOCS2 can pave the way for the development of tailored therapeutic approaches. This knowledge could inform the design of novel medications that specifically target SOCS2-related pathways, potentially improving patient outcomes and quality of life in various disease contexts.
Future research directions should aim to address the identified limitations and expand upon the current findings. Experimental studies are crucial to validate the computational predictions and confirm the biological relevance of the observed interactions. Specifically, employing biochemical assays and cellular models will provide deeper insights into how Lumbricus-derived proteins affect SOCS2 function and E3 ligase activity. Moreover, investigating the therapeutic efficacy of these proteins in preclinical disease models will be essential to evaluate their potential clinical translation. This includes assessing their pharmacokinetics, bioavailability, and safety profiles to establish their feasibility as therapeutic agents. Furthermore, exploring the synergy between Lumbricus-derived proteins and existing therapies could uncover combination strategies that enhance therapeutic outcomes. This approach could potentially lead to personalized treatment regimens tailored to individual patient needs, maximizing therapeutic efficacy while minimizing adverse effects.
5. Materials and Methods
5.1. Materials
Protein and peptide sequences derived from earthworms of the Lumbricus genus were obtained through a systematic search approach using UniProt, a comprehensive database housing protein sequences and functional information. The search strategy utilized the MeSH term “Lumbricus” within UniProtKB to specifically target proteins and peptides associated with this genus. Subsequently, retrieved data underwent meticulous curation to remove duplicate entries, ensuring the dataset's integrity and accuracy for subsequent analyses. This curation process was essential to minimize redundancy and potential biases, thereby enhancing the reliability and validity of the study findings. By rigorously retrieving and curating data, a comprehensive and high-quality dataset was established, forming the basis for investigating the bioactive constituents derived from earthworms and their potential therapeutic roles in cardiovascular diseases.
5.2. Computing Power
For this study, computational analyses were conducted using a workstation equipped with the following specifications: an Intel® Core™ i9-12900KF CPU running at 3.90GHz with 16 cores, an NVIDIA GeForce RTX 4090 graphics card featuring 24GB GDDR6X, 64GB DDR5 RAM, an 8TB hard disk drive, and an Ubuntu operating system.
5.3. 3D Structure Modeling
Several computational tools were utilized to generate the three-dimensional structures of the protein and peptide sequences acquired. Initially, I-TASSER (Iterative Threading ASSEmbly Refinement) was applied to proteins and peptides that had corresponding templates in the Protein Data Bank (PDB). I-TASSER excels in constructing 3D structures using available templates from the PDB, utilizing these templates to predict the tertiary structure of the queried sequences [55]. Proteins and peptides without templates in the PDB were modeled using AlphaFold. AlphaFold is a cutting-edge deep learning approach for protein structure prediction, renowned for its ability to accurately forecast the 3D structures of proteins, including cases where homologous structures are unavailable [33]. Next, the active sites of proteins and peptides sourced from Lumbricus were examined using CASTp 3.0. CASTp is a tool designed to detect and analyze active sites and binding pockets within protein structures, offering crucial insights into their functional characteristics and potential sites for ligand binding [56]. Additionally, the SOCS2 protein, which was selected as the target for subsequent docking studies, was obtained from the PDB under the accession code 6I5N with a resolution of 1.98 Å [57]. This step ensured that the target receptor structure was readily available for the following molecular docking simulations, enabling the study of protein-protein interactions between the bioactive compounds derived from Lumbricus earthworm and SOCS2.
5.4. Protein-Protein Docking Simulation
In this section, we thoroughly investigated the interactions between the proteins and peptides obtained from Lumbricus and the SOCS2 target receptor. Employing protein-protein docking simulations, we aimed to uncover critical aspects of the binding mechanism. This included identifying pivotal residues crucial for forming protein-protein complexes, understanding the types of intermolecular interactions at play, assessing binding affinities, examining binding modes, and analyzing orientations. To delineate the binding sites of the target receptor, we utilized PDBSum, a computational tool tailored for detailed summaries of protein structures and their interactions [58]. This examination provided us with insights into how essential residues are spatially arranged within SOCS2's binding site and their potential interactions with Lumbricus-derived proteins and peptides. To maintain the precision of our analysis, we refined the receptor using Swiss-PdbViewer v4.1.1 [59] before proceeding with protein-protein docking analysis, ensuring a reliable foundation for our subsequent investigations. To delve deeper into understanding whether the proteins and peptides derived from Lumbricus earthworms exhibit agonistic or antagonistic effects towards SOCS2, we utilized well-established molecules for comparison. EpoR peptide, identified by its PDB ID: 6I5N [57], serves as a standard agonist known to bind to SOCS2 within the PDB complex. In contrast, N4BP1, with a PDB ID: 8T48 [60], acts as a potent suppressor of E3 ligase activity. By comparing Lumbricus-derived compounds to these known standards, we aimed to evaluate their interactions with SOCS2, particularly focusing on binding pocket residues (Arg73, Ser75, Ser76, and Arg96) [61]. These residues are pivotal in determining the specificity and strength of interactions between SOCS2 and its binding partners. Following this, protein-protein docking calculations were conducted using the advanced interface option within the standalone version of High Ambiguity Driven Protein-Protein Docking (HADDOCK). HADDOCK is a well-established computational tool for modeling protein-protein interactions, utilizing ambiguous interaction restraints derived from experimental or computational sources [62,63]. Using this method, we evaluated the interactions between Lumbricus proteins and peptides and SOCS2, predicting potential binding modes and affinities. Optimal protein-protein docking results for each complex were selected based on two key criteria: the highest number of observed clusters or populations, indicating the reliability of predicted interactions, and the highest docking score (HADDOCK score), which measures the strength of binding affinity between Lumbricus proteins and peptides and SOCS2 within the protein-protein complex.
5.5. Molecular Dynamics (MD) Simulation
Molecular dynamics (MD) simulations were used to study the dynamics and stability of the protein-protein complexes involving Lumbricus proteins and peptides with SOCS2. These simulations were performed using GROMACS 2022.5, a well-established molecular dynamics simulation software renowned for its efficiency and accuracy in modeling biomolecular systems [64]. For the simulations, we utilized the Optimized Potentials for Liquid Simulations (OPLS-AA/L) force field, which accurately models molecular interactions within the system [65]. The dimensions of the simulation box were set using default cubic box parameters to accommodate the biomolecular complexes effectively. Standard protocols were followed to prepare simulation input files, which included adding water molecules using the Single Point Charge Extended (SPCE) model and incorporating counterions to maintain system neutrality [66]. Energy minimization was performed using the steepest-descent method to eliminate steric clashes and achieve system relaxation to a stable state. Following this, a two-phase equilibration process was executed. Initially, in phase 1, the system underwent equilibration in the NVT ensemble to regulate temperature fluctuations and stabilize conditions. Subsequently, in phase 2, equilibration was conducted in the NPT ensemble to maintain constant pressure and temperature. Once equilibrated, production MD simulations were conducted over 100 nanoseconds to observe the dynamics of the protein-protein complexes over an extended period. Throughout the simulations, parameters such as Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (RoG), potential energies, and intermolecular hydrogen bonding interactions were monitored and analyzed to assess complex stability and conformational dynamics. Visualization of critical residues and intermolecular interactions within the predicted protein-protein complexes was conducted through manual inspection using molecular visualization software like PyMOL [67] and UCSF Chimera [68]. These tools enabled the examination and interpretation of essential structural characteristics and interactions within the simulated complexes, offering valuable insights into the mechanisms that govern the binding and stability of the proteins and peptides from Lumbricus with SOCS2.
5.6. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Calculations
The Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) method, employing MD simulations, was employed to investigate the protein-protein interactions involving proteins and peptides derived from Lumbricus and SOCS2. MD simulations produced a range of protein conformations, and representative snapshots were chosen for detailed analysis [69]. Each snapshot was subjected to thorough energy computations, encompassing calculations of gas-phase energy, estimation of solvation energy using a continuum solvent model, and entropy calculations. These energy components were integrated to determine the binding free energy of the protein-protein complex [70,71]. To conduct these computations, we utilized the gmx_MMPBSA module available within the GROMACS simulation package [72,73]. This module facilitates the precise and efficient calculation of binding free energies for biomolecular complexes. The MM/PBSA method is renowned for its ability to predict the binding free energy of protein-protein interactions, establishing it as a valuable tool for understanding the energetic aspects of biomolecular interactions [74]. The MM/PBSA binding free energy calculation is derived from the following equation:
where:
ΔG_binding = ΔG_complex - ΔG_proteinX1 - ΔG_proteinX2
- ΔG_binding: the binding free energy associated with the formation of the protein-protein complex.
- ΔG_complex: the free energy of the fully solvated protein-protein complex.
- ΔG_proteinX1: the free energy of protein 1 in its solvated state when unbound.
- ΔG_proteinX2: the free energy of protein 2 in its solvated state when unbound.
The binding free energy was determined by calculating the difference between the free energy of the complex and the combined free energies of the unbound proteins. This calculation provided insights into the energetic alterations that occurred during the formation of the protein-protein complex, thereby elucidating the strength and stability of the interaction.
5.7. Statistical Analysis
In this study, statistical analysis was performed to investigate the relationships among all parameters derived from both molecular docking and molecular dynamics simulations. The data from these computational experiments underwent thorough statistical analysis and interpretation using SPSS 25 statistical software [75] and OriginLab Pro 2022 [76]. This analytical approach allowed for exploring correlations, trends, and patterns within the dataset, offering valuable insights into the relationships between various variables and parameters. Moreover, statistical analysis helped validate computational outcomes and identify significant findings, enhancing the understanding of the molecular interactions under scrutiny.
6. Conclusions
In conclusion, this study conducted a thorough investigation into the interactions between Lumbricus-derived proteins and SOCS2, revealing their potential therapeutic implications for cardiovascular diseases. Employing a comprehensive approach integrating bioinformatics, computational modeling, and molecular dynamics simulations, the research yielded valuable insights into the structural characteristics and binding affinities of Lumbricus-derived proteins and peptides with SOCS2. The findings indicated that certain proteins such as Lumbricin, Chemoattractive glycoprotein ES20, and Lumbrokinase-7T1 demonstrated activity comparable to standard antagonists in regulating SOCS2 function, suggesting their candidacy for therapeutic applications in cardiovascular diseases. Moreover, the study underscored the significance of computational methodologies in drug discovery, particularly for natural products possessing diverse chemical structures and biological activities. By leveraging advanced computational tools, the study efficiently screened a wide array of candidate compounds for their potential interactions with SOCS2, overcoming the challenges associated with traditional drug discovery methods. This approach not only accelerated the discovery process but also provided mechanistic insights into how these compounds operate at the molecular level. However, it is essential to recognize the study's limitations, primarily its reliance on computational predictions that necessitate validation through experimental assays. Future research efforts should focus on validating these findings using biochemical and structural biology techniques, as well as conducting preclinical studies in relevant disease models. Furthermore, exploring the broader therapeutic potential of Lumbricus-derived proteins beyond cardiovascular diseases and investigating their synergistic effects with existing pharmacological agents could open new avenues for drug development and personalized medicine initiatives.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Data S1: Database of protein sequences sourced from earthworms (Lumbricus genus), including detailed information on protein/peptide names, UniProt IDs, sequences, and identified protein binding sites (number of residues); Supplementary Data S2: Complete docking results, including complex identifiers, HADDOCK scores, binding affinities (ΔG), dissociation constants (Kd), cluster sizes, RMSD values, van der Waals energies, electrostatic energies, desolvation energies, restraints violation energies, buried surface areas, and Z-scores.
Author Contributions
Conceptualization, Nasser Alotaiq and Doni Dermawan; Data curation, Nasser Alotaiq; Formal analysis, Doni Dermawan; Funding acquisition, Nasser Alotaiq; Investigation, Doni Dermawan; Methodology, Nasser Alotaiq and Doni Dermawan; Project administration, Nasser Alotaiq; Resources, Nasser Alotaiq; Software, Doni Dermawan; Supervision, Nasser Alotaiq; Validation, Nasser Alotaiq and Doni Dermawan; Visualization, Doni Dermawan; Writing—original draft, Nasser Alotaiq and Doni Dermawan; Writing—review & editing, Nasser Alotaiq and Doni Dermawan. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-RG23154).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- WHO. Cardiovascular diseases (CVDs): World Health Organization; 2021 [cited 2024 01th April]. Available from: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
- Roth Gregory A, Mensah George A, Johnson Catherine O, Addolorato G, Ammirati E, Baddour Larry M, et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J Am Coll Cardiol. 2020;76(25):2982-3021. [CrossRef]
- Del Buono MG, Montone RA, Camilli M, Carbone S, Narula J, Lavie CJ, et al. Coronary Microvascular Dysfunction Across the Spectrum of Cardiovascular Diseases: JACC State-of-the-Art Review. J Am Coll Cardiol. 2021;78(13):1352-71. PubMed PMID: 34556322; PubMed Central PMCID: PMC8528638. [CrossRef]
- Gimbrone MA, García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016;118(4):620-36. [CrossRef]
- Marc T. Cardiovascular Disease: An Introduction. 2018. p. 1-90.
- Ullah A, Kumar M, Sayyar M, Sapna F, John C, Memon S, et al. Revolutionizing Cardiac Care: A Comprehensive Narrative Review of Cardiac Rehabilitation and the Evolution of Cardiovascular Medicine. Cureus. 2023;15(10):1-11. Epub 20231004. PubMed PMID: 37927717; PubMed Central PMCID: PMC10624210. [CrossRef]
- Jansen-Chaparro S, López-Carmona MD, Cobos-Palacios L, Sanz-Cánovas J, Bernal-López MR, Gómez-Huelgas R. Statins and Peripheral Arterial Disease: A Narrative Review. Front Cardiovasc Med. 2021;8:1-18. Epub 20211122. PubMed PMID: 34881314; PubMed Central PMCID: PMC8645843. [CrossRef]
- Rousan TA, Thadani U. Stable Angina Medical Therapy Management Guidelines: A Critical Review of Guidelines from the European Society of Cardiology and National Institute for Health and Care Excellence. Eur Cardiol. 2019;14(1):18-22. PubMed PMID: 31131033; PubMed Central PMCID: PMC6523058. [CrossRef]
- Kario K, Hoshide S, Narita K, Okawara Y, Kanegae H, Aoki K, et al. Cardiovascular Prognosis in Drug-Resistant Hypertension Stratified by 24-Hour Ambulatory Blood Pressure: The JAMP Study. Hypertension. 2021;78(6):1781-90. [CrossRef]
- Wijkman MO, Malachias MVB, Claggett BL, Cheng S, Matsushita K, Shah AM, et al. Resistance to antihypertensive treatment and long-term risk: The Atherosclerosis Risk in Communities study. J Clin Hypertens (Greenwich). 2021;23(10):1887-96. Epub 20210921. PubMed PMID: 34547175; PubMed Central PMCID: PMC8678845. [CrossRef]
- Hu X, li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduction and Targeted Therapy. 2021;6(1):402. [CrossRef]
- Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984-2009. PubMed PMID: 30267440; PubMed Central PMCID: PMC6237706. [CrossRef]
- Liang C, Zhang L, Lian X, Zhu T, Zhang Y, Gu N. Circulating Exosomal SOCS2-AS1 Acts as a Novel Biomarker in Predicting the Diagnosis of Coronary Artery Disease. BioMed Research International. 2020;2020:1-10. [CrossRef]
- Zhang H, Dhalla NS. The Role of Pro-Inflammatory Cytokines in the Pathogenesis of Cardiovascular Disease. International Journal of Molecular Sciences. 2024;25(2):1082. PubMed PMID:. [CrossRef]
- Shook PL, Singh M, Singh K. Macrophages in the Inflammatory Phase following Myocardial Infarction: Role of Exogenous Ubiquitin. Biology (Basel). 2023;12(9). Epub 20230920. PubMed PMID: 37759657; PubMed Central PMCID: PMC10526096. [CrossRef]
- Klaeske K, Dix M, Adams V, Jawad K, Eifert S, Etz C, et al. Differential Regulation of Myocardial E3 Ligases and Deubiquitinases in Ischemic Heart Failure. Life (Basel). 2021;11(12). Epub 20211218. PubMed PMID: 34947961; PubMed Central PMCID: PMC8708923. [CrossRef]
- Rico-Bautista E, Flores-Morales A, Fernández-Pérez L. Suppressor of cytokine signaling (SOCS) 2, a protein with multiple functions. Cytokine Growth Factor Rev. 2006;17(6):431-9. Epub 20061027. PubMed PMID: 17070092. [CrossRef]
- Durham G, Williams J, Nasim T, Palmer T. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends in Pharmacological Sciences. 2019;40. [CrossRef]
- Mustafa R, Saiqa A, Domínguez J, Jamil M, Manzoor S, Wazir S, et al. Therapeutic Values of Earthworm Species Extract from Azad Kashmir as Anticoagulant, Antibacterial, and Antioxidant Agents. Can J Infect Dis Med Microbiol. 2022;2022(1):1-20. [CrossRef]
- Cooper EL, Balamurugan M, Huang CY, Tsao CR, Heredia J, Tommaseo-Ponzetta M, Paoletti MG. Earthworms dilong: ancient, inexpensive, noncontroversial models may help clarify approaches to integrated medicine emphasizing neuroimmune systems. Evid Based Complement Alternat Med. 2012;2012(1):1-11. Epub 20120725. PubMed PMID: 22888362; PubMed Central PMCID: PMC3410320. [CrossRef]
- Dong Y, Woo YM, Lee YH, Ahn MY, Lee DG, Lee SH, et al. Data on the potent fibrinolytic effects of the Lumbricus rubellus earthworm and the Perinereis linea lugworm. Data Brief. 2019;26(1):1-7. Epub 20190905. PubMed PMID: 31667249; PubMed Central PMCID: PMC6811935. [CrossRef]
- Lai CH, Han CK, Shibu MA, Pai PY, Ho TJ, Day CH, et al. Lumbrokinase from earthworm extract ameliorates second-hand smoke-induced cardiac fibrosis. Environ Toxicol. 2015;30(10):1216-25. Epub 20140405. PubMed PMID: 24706507. [CrossRef]
- Chen M, Wang L, Zheng C, Ma A, Hu K, Xiang A, et al. Novel ACE inhibitory peptides derived from bighead carp (Aristichthys nobilis) hydrolysates: Screening, inhibition mechanisms and the bioconjugation effect with graphene oxide. Food Biosci. 2023;52:1-14.
- Prakash M, Balamurugan M, Parthasarathi K, Gunasekaran G, Cooper E, Ranganathan L. Anti-ulceral and anti-oxidative properties of “earthworm paste” of Lampito mauritii (Kinberg) on Rattus Norvegicus. Eur Rev Med Pharmacol Sci. 2006;11:9-15.
- Dermawan D, Prabowo BA, Rakhmadina CA. In silico study of medicinal plants with cyclodextrin inclusion complex as the potential inhibitors against SARS-CoV-2 main protease (Mpro) and spike (S) receptor. Inform Med Unlocked. 2021;25:1-18.
- Challapa-Mamani MR, Tomás-Alvarado E, Espinoza-Baigorria A, León-Figueroa DA, Sah R, Rodriguez-Morales AJ, Barboza JJ. Molecular Docking and Molecular Dynamics Simulations in Related to Leishmania donovani: An Update and Literature Review. Trop Med Infect Dis. 2023;8(10):1-13. Epub 20230926. PubMed PMID: 37888585; PubMed Central PMCID: PMC10610989. [CrossRef]
- Sánchez-Gloria JL, Arellano-Buendía AS, Juárez-Rojas JG, García-Arroyo FE, Argüello-García R, Sánchez-Muñoz F, et al. Cellular Mechanisms Underlying the Cardioprotective Role of Allicin on Cardiovascular Diseases. Int J Mol Sci. 2022;23(16):1-21. Epub 20220813. PubMed PMID: 36012349; PubMed Central PMCID: PMC9409331. [CrossRef]
- Choudhury TZ, Garg V. Molecular genetic mechanisms of congenital heart disease. Curr Opin Genet Dev. 2022;75:1-13.
- Chen S, Fan G, Li J. Improving completeness and accuracy of 3D point clouds by using deep learning for applications of digital twins to civil structures. Adv Eng Inform. 2023;58:1-13. [CrossRef]
- Davis EM, Sun Y, Liu Y, Kolekar P, Shao Y, Szlachta K, et al. SequencErr: measuring and suppressing sequencer errors in next-generation sequencing data. Genome Biol. 2021;22(1):1-18. Epub 20210125. PubMed PMID: 33487172; PubMed Central PMCID: PMC7829059. [CrossRef]
- Faits T, Odom A, Castro-Nallar E, Crandall K, Johnson W. Metagenomic profiling pipelines improve taxonomic classification for 16S amplicon sequencing data2022.
- Zhang Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 2009;77 Suppl 9:100-13. [CrossRef]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583-9. [CrossRef]
- Mohammadi S, Narimani Z, Ashouri M, Firouzi R, Karimi-Jafari MH. Ensemble learning from ensemble docking: revisiting the optimum ensemble size problem. Sci Rep. 2022;12(410):1-15. Epub 20220110. PubMed PMID: 35013496; PubMed Central PMCID: PMC8748946. [CrossRef]
- Saha A, Panja A, Shyamal M, Mandal. Influence of anionic co-ligands on the structural diversity and catecholase activity of copper(II) complexes with 2-methoxy-6-(8-iminoquinolinylmethyl)phenol. RSC Adv. 2014;4. [CrossRef]
- Kufareva I, Abagyan R. Methods of Protein Structure Comparison. Methods in molecular biology (Clifton, NJ). 2012;857:231-57. [CrossRef]
- Russell R, Alber F, Aloy P, Davis F, Korkin D, Pichaud M, et al. A structural perspective on protein-protein interactions. Curr Opin Struct Biol. 2004;14:313-24. [CrossRef]
- Dagliyan O, Proctor EA, D'Auria KM, Ding F, Dokholyan NV. Structural and dynamic determinants of protein-peptide recognition. Structure. 2011;19(12):1837-45. PubMed PMID: 22153506; PubMed Central PMCID: PMC3240807. [CrossRef]
- Guedes IA, Pereira FSS, Dardenne LE. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front Pharmacol. 2018;9:1-18. Epub 20180924. PubMed PMID: 30319422; PubMed Central PMCID: PMC6165880. [CrossRef]
- Desantis F, Miotto M, Di Rienzo L, Milanetti E, Ruocco G. Spatial organization of hydrophobic and charged residues affects protein thermal stability and binding affinity. Sci Rep. 2022;12(1):12087. Epub 20220715. PubMed PMID: 35840609; PubMed Central PMCID: PMC9287411. [CrossRef]
- Bitencourt-Ferreira G, Veit-Acosta M, De Azevedo Jr W. Van der Waals Potential in Protein Complexes. 20532019. p. 79-91.
- Cramer J, Krimmer S, Heine A, Klebe G. Paying the Price of Desolvation in Solvent-Exposed Protein Pockets: Impact of Distal Solubilizing Groups on Affinity and Binding Thermodynamics in a Series of Thermolysin Inhibitors. Journal of Medicinal Chemistry. 2017;60. [CrossRef]
- Zhou H-X, Pang X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chemical Reviews. 2018;118. [CrossRef]
- Kastritis PL, Rodrigues JPGLM, Bonvin AMJJ. HADDOCK2P2I: A Biophysical Model for Predicting the Binding Affinity of Protein–Protein Interaction Inhibitors. J Chem Inf Model. 2014;54(3):826-36. [CrossRef]
- Kastritis PL, Bonvin AM. On the binding affinity of macromolecular interactions: daring to ask why proteins interact. J R Soc Interface. 2013;10(79):1-27. Epub 20121212. PubMed PMID: 23235262; PubMed Central PMCID: PMC3565702. [CrossRef]
- Dermawan D, Sumirtanurdin R, Dewantisari D. Simulasi dinamika molekular reseptor estrogen alfa dengan andrografolid sebagai anti kanker payudara. Indones J Pharm Sci Technol. 2019;6(2):65-76.
- Craveur P, Joseph AP, Esque J, Narwani TJ, Noël F, Shinada N, et al. Protein flexibility in the light of structural alphabets. Front Mol Biosci. 2015;2:1-20. Epub 20150527. PubMed PMID: 26075209; PubMed Central PMCID: PMC4445325. [CrossRef]
- Maspero E, Mari S, Valentini E, Musacchio A, Fish A, Pasqualato S, Polo S. Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep. 2011;12(4):342-9. Epub 20110311. PubMed PMID: 21399620; PubMed Central PMCID: PMC3077247. [CrossRef]
- Sanusi ZK, Lobb KA. Insights into the Dynamics and Binding of Two Polyprotein Substrate Cleavage Points in the Context of the SARS-CoV-2 Main and Papain-like Proteases. Molecules. 2022;27(23):1-16. Epub 20221126. PubMed PMID: 36500348; PubMed Central PMCID: PMC9740519. [CrossRef]
- Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51(1):69-82. Epub 20101130. PubMed PMID: 21117705; PubMed Central PMCID: PMC3029230. [CrossRef]
- Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins. 2019;87(12):1011-20. Epub 20191023. PubMed PMID: 31589781; PubMed Central PMCID: PMC6927249. [CrossRef]
- Wang YH, Chen KM, Chiu PS, Lai SC, Su HH, Jan MS, et al. Lumbrokinase attenuates myocardial ischemia-reperfusion injury by inhibiting TLR4 signaling. J Mol Cell Cardiol. 2016;99:113-22. Epub 20160805. PubMed PMID: 27503317. [CrossRef]
- Lenselink EB, Louvel J, Forti AF, van Veldhoven JPD, de Vries H, Mulder-Krieger T, et al. Predicting Binding Affinities for GPCR Ligands Using Free-Energy Perturbation. ACS Omega. 2016;1(2):293-304. Epub 20160830. PubMed PMID: 30023478; PubMed Central PMCID: PMC6044636. [CrossRef]
- Salsbury FR, Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol. 2010;10(6):738-44. PubMed PMID: 20971684; PubMed Central PMCID: PMC2981647. [CrossRef]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9(1):40. [CrossRef]
- Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46(1):363-7. [CrossRef]
- Kung W-W, Ramachandran S, Makukhin N, Bruno E, Ciulli A. Structural insights into substrate recognition by the SOCS2 E3 ubiquitin ligase. Nature Communications. 2019;10(1):2534. [CrossRef]
- Laskowski RA, Jabłońska J, Pravda L, Vařeková RS, Thornton JM. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018;27(1):129-34. Epub 20171027. PubMed PMID: 28875543; PubMed Central PMCID: PMC5734310. [CrossRef]
- Johansson MU, Zoete V, Michielin O, Guex N. Defining and searching for structural motifs using DeepView/Swiss-PdbViewer. BMC Bioinformatics. 2012;13(173):1-10. Epub 20120723. PubMed PMID: 22823337; PubMed Central PMCID: PMC3436773. [CrossRef]
- Gitlin AD, Maltzman A, Kanno Y, Heger K, Reja R, Schubert AF, et al. N4BP1 coordinates ubiquitin-dependent crosstalk within the IκB kinase family to limit Toll-like receptor signaling and inflammation. Immunity. 2024;57(5):973-86.
- Ramachandran S, Makukhin N, Haubrich K, Nagala M, Forrester B, Lynch DM, et al. Structure-based design of a phosphotyrosine-masked covalent ligand targeting the E3 ligase SOCS2. Nature Communications. 2023;14(1):6345. [CrossRef]
- Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. Journal of the American Chemical Society. 2003;125(7):1731-7. [CrossRef]
- van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol. 2016;428(4):720-5. Epub 20150926. PubMed PMID: 26410586. [CrossRef]
- Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845-54. [CrossRef]
- Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J Chem Theory Comput. 2015;11(7):3499-509. Epub 2015/07/21. PubMed PMID: 26190950; PubMed Central PMCID: PMC4504185. [CrossRef]
- Yuet P, Blankschtein D. Molecular Dynamics Simulation Study of Water Surfaces: Comparison of Flexible Water Models. The journal of physical chemistry B. 2010;114:13786-95. [CrossRef]
- Schrödinger. The PyMOL Molecular Graphics System. 2.4 ed2020.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-12. PubMed PMID: 15264254. [CrossRef]
- Tian S, Sun H, Pan P, Li D, Zhen X, Li Y, Hou T. Assessing an ensemble docking-based virtual screening strategy for kinase targets by considering protein flexibility. J Chem Inf Model. 2014;54(10):2664-79. Epub 20140929. PubMed PMID: 25233367. [CrossRef]
- Yuan Z, Chen X, Fan S, Chang L, Chu L, Zhang Y, et al. Binding Free Energy Calculation Based on the Fragment Molecular Orbital Method and Its Application in Designing Novel SHP-2 Allosteric Inhibitors. Int J Mol Sci [Internet]. 2024; 25(1):[1-24 pp.].
- Rifai EA, Ferrario V, Pleiss J, Geerke DP. Combined Linear Interaction Energy and Alchemical Solvation Free-Energy Approach for Protein-Binding Affinity Computation. J Chem Theory Comput. 2020;16(2):1300-10. [CrossRef]
- Valdés-Tresanco MS, Valdés-Tresanco ME, Valiente PA, Moreno E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. Journal of Chemical Theory and Computation. 2021;17(10):6281-91. [CrossRef]
- Miller BR, 3rd, McGee TD, Jr., Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of chemical theory and computation. 2012;8(9):3314-21. [CrossRef]
- Panday SK, Alexov E. Protein-Protein Binding Free Energy Predictions with the MM/PBSA Approach Complemented with the Gaussian-Based Method for Entropy Estimation. ACS Omega. 2022;7(13):11057-67. Epub 2022/04/14. PubMed PMID: 35415339; PubMed Central PMCID: PMC8991903. [CrossRef]
- IBM. IBM SPSS Statistics for Windows. Version 25.0 ed. New York: IBM Corp; 2017.
- OriginLab. Origin(Pro). 2022 ed. Northampton, MA, USA: OriginLab Corporation; 2022.
Figure 1.
3D structural modeling results illustrating proteins and peptides derived from earthworms of the Lumbricus genus. (a) Cytochrome b. (b) SCBP3 protein. (c) Lumbricin. (d) Chemoattractive glycoprotein ES20. (e) Histone H3. (f) Lumbrokinase-7T1.
Figure 1.
3D structural modeling results illustrating proteins and peptides derived from earthworms of the Lumbricus genus. (a) Cytochrome b. (b) SCBP3 protein. (c) Lumbricin. (d) Chemoattractive glycoprotein ES20. (e) Histone H3. (f) Lumbrokinase-7T1.
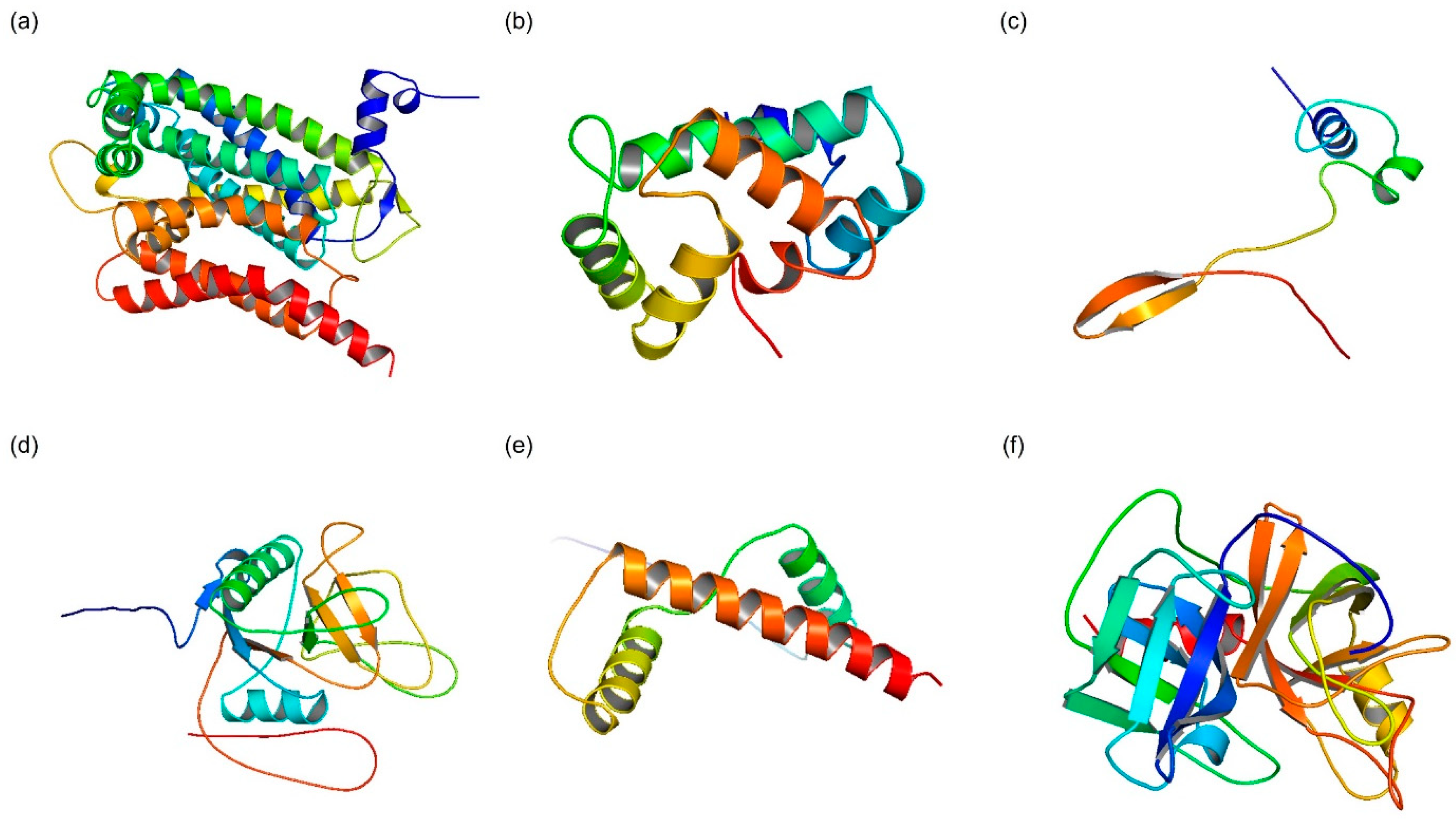
Figure 2.
Molecular docking simulations illustrate the optimal binding orientations for interactions between proteins derived from the earthworm (Lumbricus genus) and SOCS2. (a) SOCS2: EpoR peptide (agonist) complex. (b) SOCS2:N4BP1 (antagonist) complex. (c) SOCS2: Cytochrome b complex. (d) SOCS2:SCBP3 protein complex. (e) SOCS2:Lumbricin complex. (f) SOCS2: Lumbrokinase-7T1 complex.
Figure 2.
Molecular docking simulations illustrate the optimal binding orientations for interactions between proteins derived from the earthworm (Lumbricus genus) and SOCS2. (a) SOCS2: EpoR peptide (agonist) complex. (b) SOCS2:N4BP1 (antagonist) complex. (c) SOCS2: Cytochrome b complex. (d) SOCS2:SCBP3 protein complex. (e) SOCS2:Lumbricin complex. (f) SOCS2: Lumbrokinase-7T1 complex.
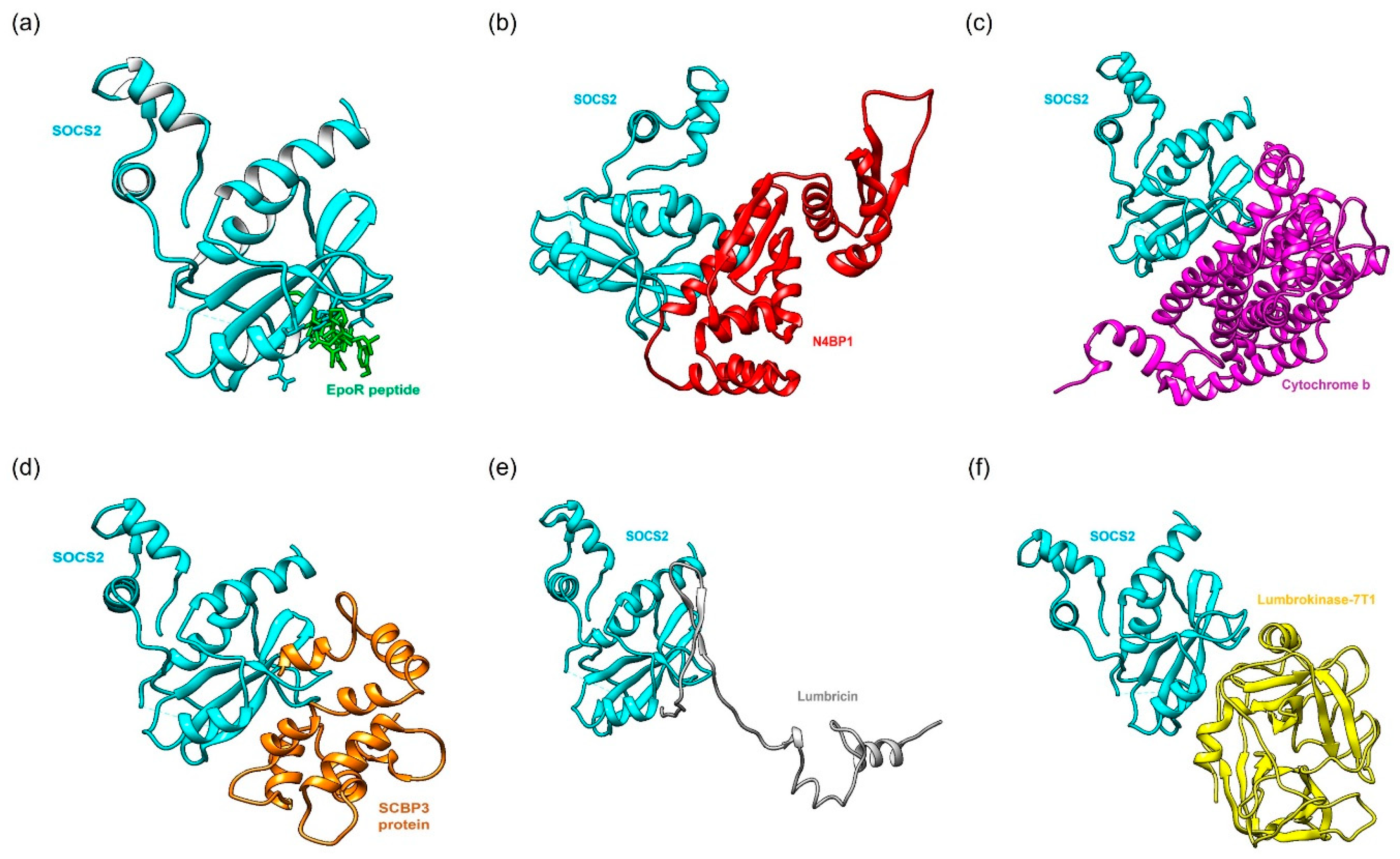
Figure 3.
Summary of molecular docking outcomes. (a) Correlation between HADDOCK score and RMSD. (b) Correlation between HADDOCK score and binding affinity. (c) Binding affinity values of the top-performing proteins derived from Lumbricus earthworms, with a threshold of -11.0 kcal/mol. (d) Correlation matrix illustrating the relationship between binding energy (kcal/mol) and individual energy components.
Figure 3.
Summary of molecular docking outcomes. (a) Correlation between HADDOCK score and RMSD. (b) Correlation between HADDOCK score and binding affinity. (c) Binding affinity values of the top-performing proteins derived from Lumbricus earthworms, with a threshold of -11.0 kcal/mol. (d) Correlation matrix illustrating the relationship between binding energy (kcal/mol) and individual energy components.
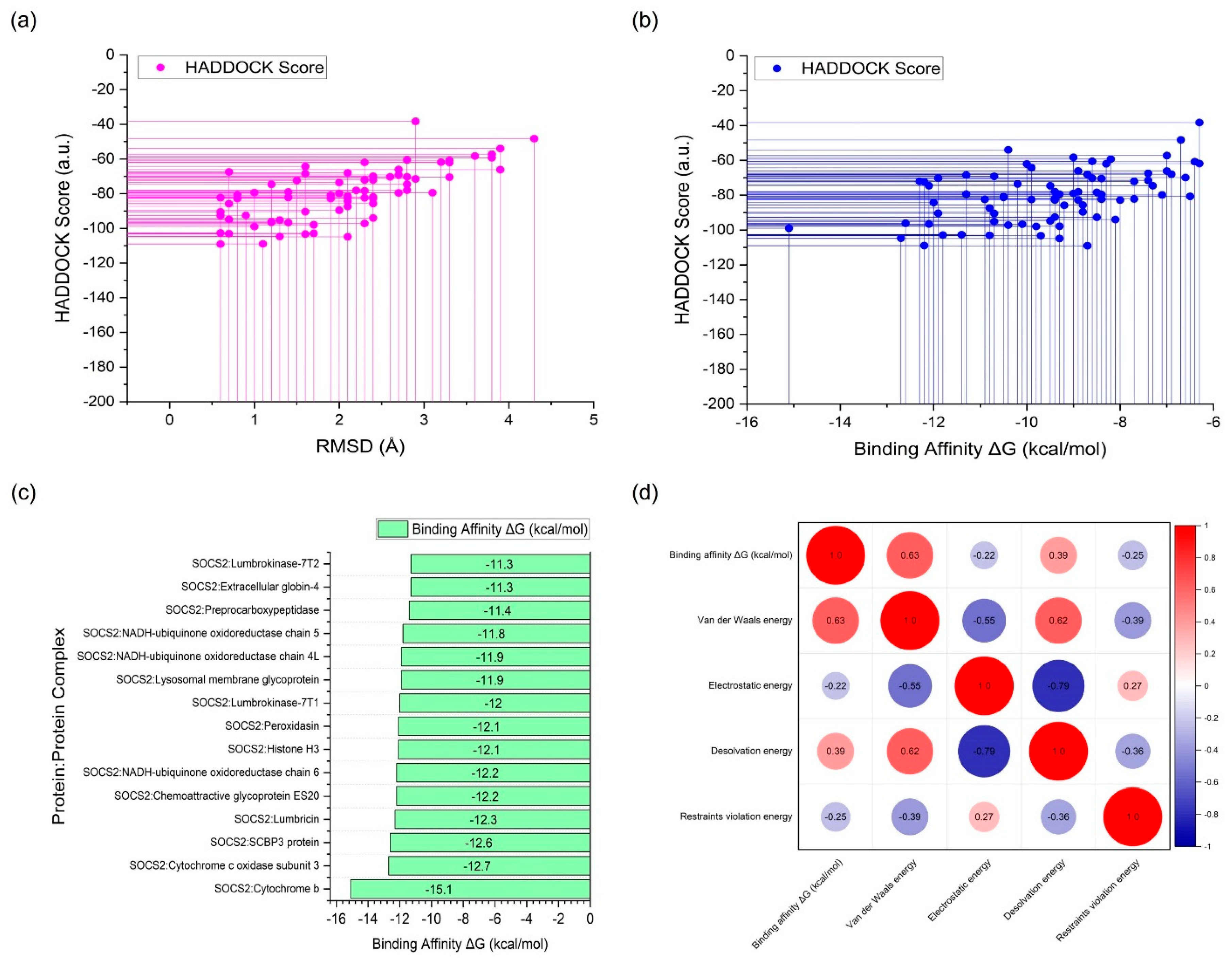
Figure 4.
Analysis of molecular dynamics (MD) simulations for complexes formed between proteins sourced from the Lumbricus genus earthworm and SOCS2 included several key parameters: (a) Root mean square deviation (RMSD) assessed structural stability, (b) Root mean square fluctuation (RMSF) depicted residue flexibility, (c) Radius of gyration (RoG) illustrated structural compactness, and (d) Number of hydrogen bonds highlighted intermolecular interactions.
Figure 4.
Analysis of molecular dynamics (MD) simulations for complexes formed between proteins sourced from the Lumbricus genus earthworm and SOCS2 included several key parameters: (a) Root mean square deviation (RMSD) assessed structural stability, (b) Root mean square fluctuation (RMSF) depicted residue flexibility, (c) Radius of gyration (RoG) illustrated structural compactness, and (d) Number of hydrogen bonds highlighted intermolecular interactions.
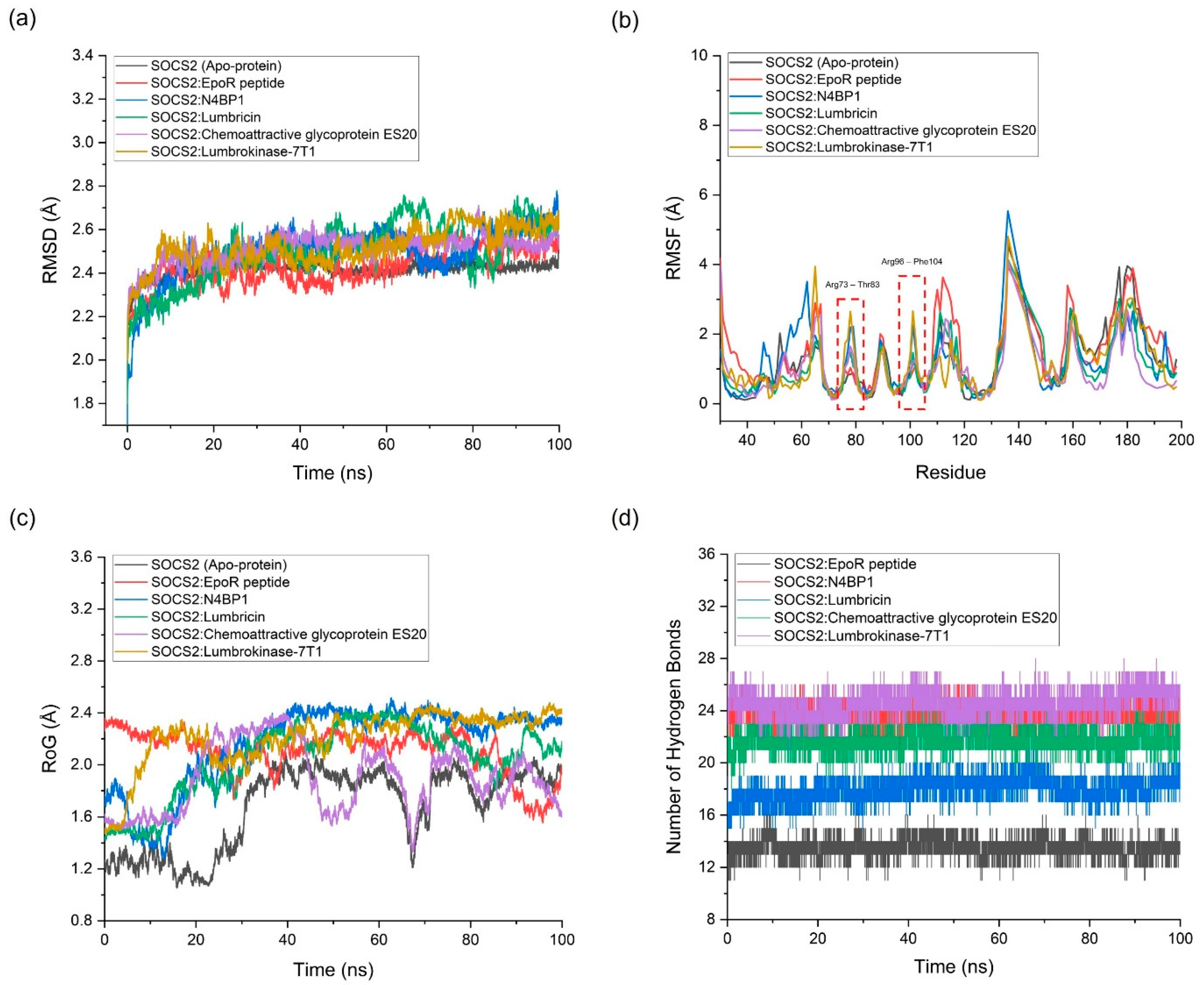
Figure 5.
Analysis of binding free energy for individual amino acids in SOCS2 interactions with the EpoR peptide (standard agonist), N4BP1 (standard antagonist), and the top three proteins derived from earthworms (Lumbricus genus) using MM/PBSA calculations.
Figure 5.
Analysis of binding free energy for individual amino acids in SOCS2 interactions with the EpoR peptide (standard agonist), N4BP1 (standard antagonist), and the top three proteins derived from earthworms (Lumbricus genus) using MM/PBSA calculations.
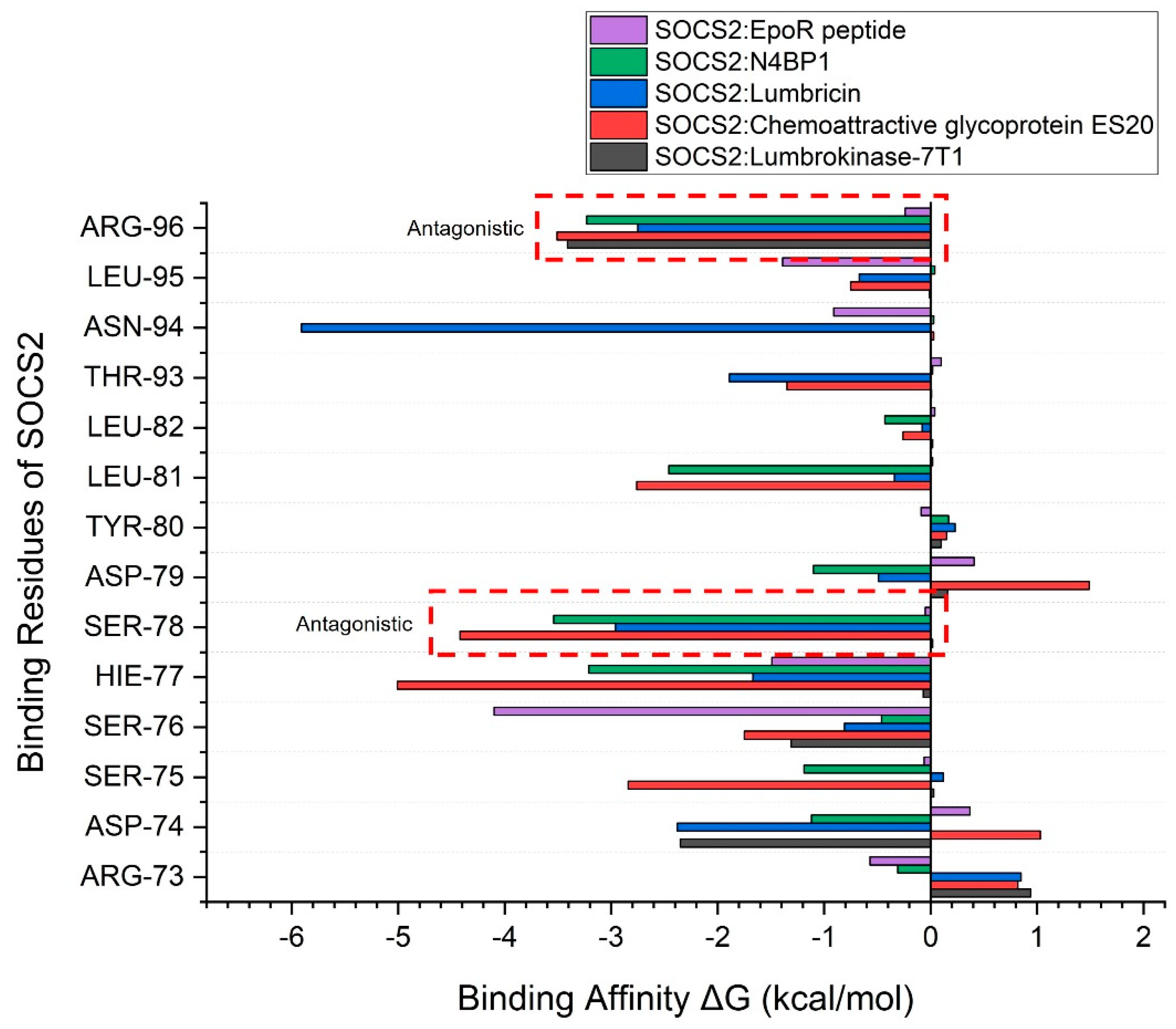

Table 1.
Molecular docking outcomes: Binding affinities and structural attributes of SOCS2 interactions with proteins and peptides obtained from Lumbricus earthworms.
Table 1.
Molecular docking outcomes: Binding affinities and structural attributes of SOCS2 interactions with proteins and peptides obtained from Lumbricus earthworms.
| Complex | HADDOCK Score (a.u.) |
Binding Affinity ΔG (kcal/mol) | Kd (M) |
Cluster size | RMSD (Å) |
|---|---|---|---|---|---|
| Standard | |||||
| SOCS2: EpoR peptide (Standard Agonist) | -81.0 +/- 3.5 | -8.8 | 6.50e-7 | 22 | 1.7 +/- 0.1 |
| SOCS2:N4BP1 (Standard Antagonist) |
-94.3 +/- 11.4 | -8.3 | 1.50e-6 | 17 | 1.2 +/- 0.3 |
| Protein and peptide derived from earthworm (Lumbricus genus) | |||||
| SOCS2:Cytochrome b | -99.0 +/- 8.2 | -15.1 | 2.30e-11 | 27 | 1.0 +/- 0.6 |
| SOCS2:Cytochrome c oxidase subunit 3 | -104.7 +/- 6.3 | -12.7 | 1.10e-09 | 15 | 1.3 +/- 0.5 |
| SOCS2:SCBP3 protein | -96.2 +/- 6.2 | -12.6 | 1.30e-09 | 41 | 1.2 +/- 0.9 |
| SOCS2:Lumbricin | -72.1 +/- 3.7 | -12.3 | 2.10e-09 | 43 | 2.3 +/- 0.3 |
| SOCS2:Chemoattractive glycoprotein ES20 | -72.4 +/- 5.3 | -12.2 | 2.30e-09 | 20 | 1.5 +/- 1.0 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 6 | -109.0 +/- 7.1 | -12.2 | 2.30e-09 | 14 | 1.1 +/- 0.7 |
| SOCS2:Histone H3 | -96.6 +/- 4.0 | -12.1 | 3.10e-09 | 8 | 1.2 +/- 0.2 |
| SOCS2:Peroxidasin | -74.6 +/- 7.6 | -12.1 | 2.70e-09 | 8 | 2.8 +/- 0.0 |
| SOCS2:Lumbrokinase-7T1 | -84.3 +/- 2.8 | -12.0 | 3.70e-09 | 43 | 2.1 +/- 0.2 |
| SOCS2:Lysosomal membrane glycoprotein | -70.3 +/- 2.3 | -11.9 | 4.30e-09 | 5 | 2.6 +/- 0.9 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 4L | -90.4 +/- 5.4 | -11.9 | 4.10e-09 | 12 | 1.6 +/- 1.0 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 5 | -102.9 +/- 11.6 | -11.8 | 4.70e-09 | 7 | 1.7 +/- 0.3 |
| SOCS2:Preprocarboxypeptidase | -102.7 +/- 9.1 | -11.4 | 9.70e-09 | 19 | 0.6 +/- 0.4 |
| SOCS2:Extracellular globin-4 | -79.4 +/- 10.8 | -11.3 | 1.20e-08 | 6 | 1.0 +/- 0.6 |
| SOCS2:Lumbrokinase-7T2 | -68.5 +/- 2.5 | -11.3 | 1.10e-08 | 35 | 1.6 +/- 0.5 |
Table 2.
Intermolecular contacts and non-interacting surface areas for SOCS2 complexes with standard agonist, antagonist, and earthworm-derived proteins and peptides.
Table 2.
Intermolecular contacts and non-interacting surface areas for SOCS2 complexes with standard agonist, antagonist, and earthworm-derived proteins and peptides.
| Complex |
ICs charged-charged | ICs charged-polar | ICs charged-apolar | ICs polar-polar | ICs polar-apolar | ICs apolar-apolar | NIS charged | NIS apolar |
|---|---|---|---|---|---|---|---|---|
| Standard | ||||||||
| SOCS2: EpoR peptide (Standard Agonist) | 3 | 8 | 12 | 2 | 11 | 12 | 25.55 | 38.69 |
| SOCS2:N4BP1 (Standard Antagonist) |
5 | 11 | 20 | 6 | 11 | 8 | 29.71 | 39.49 |
| Protein and peptide derived from earthworm (Lumbricus genus) | ||||||||
| SOCS2:Cytochrome b | 0 | 0 | 25 | 0 | 38 | 33 | 15.13 | 52.85 |
| SOCS2:Cytochrome c oxidase subunit 3 | 2 | 7 | 22 | 6 | 30 | 26 | 14.71 | 49.41 |
| SOCS2:SCBP3 protein | 2 | 3 | 24 | 2 | 27 | 16 | 27.83 | 41.74 |
| SOCS2:Lumbricin | 7 | 9 | 20 | 0 | 22 | 12 | 26.34 | 40.98 |
| SOCS2:Chemoattractive glycoprotein ES20 | 8 | 15 | 24 | 5 | 23 | 16 | 22.98 | 42.39 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 6 | 0 | 3 | 21 | 7 | 32 | 29 | 14.29 | 52.01 |
| SOCS2:Histone H3 | 7 | 6 | 27 | 0 | 20 | 14 | 27.56 | 42.67 |
| SOCS2:Peroxidasin | 13 | 20 | 24 | 5 | 22 | 6 | 26.23 | 41.86 |
| SOCS2:Lumbrokinase-7T1 | 3 | 15 | 12 | 4 | 24 | 10 | 17.72 | 41.14 |
| SOCS2:Lysosomal membrane glycoprotein | 12 | 17 | 18 | 5 | 18 | 11 | 20.49 | 39.02 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 4L | 0 | 8 | 21 | 2 | 24 | 14 | 17.73 | 46.82 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 5 | 0 | 3 | 17 | 1 | 25 | 29 | 14.12 | 50.10 |
| SOCS2:Preprocarboxypeptidase | 7 | 17 | 23 | 5 | 20 | 28 | 26.18 | 40.05 |
| SOCS2:Extracellular globin-4 | 15 | 18 | 28 | 2 | 12 | 6 | 31.28 | 37.04 |
| SOCS2:Lumbrokinase-7T2 | 5 | 10 | 16 | 9 | 24 | 11 | 19.03 | 41.69 |
Note:
- ICs: Number of intermolecular contacts
- NIS: Non-interacting surface
Table 3.
Detailed examination of hydrogen bond interactions occurring between SOCS2 at its binding sites and the most effective proteins originating from earthworms (Lumbricus genus), focusing on identifying specific residues and atoms crucial for the binding mechanism.
Table 3.
Detailed examination of hydrogen bond interactions occurring between SOCS2 at its binding sites and the most effective proteins originating from earthworms (Lumbricus genus), focusing on identifying specific residues and atoms crucial for the binding mechanism.
| Complex | Residue (Receptor) | Protein Atom (Receptor) |
Residue (Interacting Protein/Peptide) |
Protein Atom (Interacting Protein/Peptide) |
Interaction Distance (Å) |
|---|---|---|---|---|---|
| SOCS2: EpoR peptide (Standard Agonist) | Val55 | N | Asp8 | OD1 | 2.76 |
| Ser76 | OG | Asp8 | OD1 | 3.09 | |
| Arg96 | NH1 | Glu3 | O | 2.69 | |
| Arg96 | NH2 | Glu3 | O | 3.33 | |
| Lys113 | NZ | Ser1 | OG | 2.73 | |
| SOCS2:N4BP1 (Standard Antagonist) | Gln32 | NE2 | Glu178 | OE1 | 2.77 |
| Arg41 | NH1 | Glu118 | OE1 | 2.65 | |
| Arg41 | NH2 | Glu118 | OE2 | 2.57 | |
| Tyr49 | OH | Lys132 | NZ | 2.78 | |
| Asp74 | OD2 | Lys132 | NZ | 2.55 | |
| Ser75 | O | Asn136 | ND2 | 3.04 | |
| Ser78 | O | Ser174 | OG | 2.63 | |
| Asp79 | OD2 | Lys145 | NZ | 2.62 | |
| Arg96 | NH2 | Glu138 | OE1 | 2.67 | |
| SOCS2:Cytochrome b | His77 | NE2 | Leu122 | O | 2.74 |
| Arg96 | NH2 | Ile119 | O | 2.72 | |
| SOCS2:SCBP3 protein | Lys59 | NZ | Asp50 | O | 2.58 |
| Arg96 | NH1 | Leu68 | O | 2.66 | |
| SOCS2:Lumbricin | Arg41 | NH1 | Glu56 | O | 3.08 |
| Arg41 | NH1 | Glu56 | OE1 | 2.60 | |
| Arg41 | NH2 | Glu56 | O | 2.98 | |
| Tyr49 | OH | Lys53 | NZ | 2.83 | |
| Ser52 | OG | Glu63 | OE1 | 2.59 | |
| Asp74 | OD2 | Lys53 | NZ | 2.65 | |
| Ser78 | OG | Ile46 | O | 2.67 | |
| Asp79 | OD1 | Arg45 | NH1 | 2.65 | |
| Thr93 | OG1 | Glu72 | OE2 | 2.70 | |
| Asn94 | N | Glu72 | OE2 | 2.73 | |
| Asn94 | ND2 | Glu72 | OE1 | 2.64 | |
| Arg96 | NE | Glu72 | O | 2.74 | |
| Arg96 | NH2 | Glu72 | O | 2.87 | |
| SOCS2:Lumbrokinase-7T1 | Glu57 | OE2 | Lys20 | NZ | 2.55 |
| Asp74 | OD2 | Arg17 | NH1 | 2.69 | |
| Asp74 | OD2 | Arg17 | NH2 | 2.72 | |
| Ser76 | O | Gln19 | NE2 | 3.20 | |
| Asp101 | OD1 | Asn249 | ND2 | 2.66 | |
| Asp101 | OD2 | Ser170 | OG | 2.66 | |
| Cys111 | O | Lys168 | NZ | 2.91 | |
| Lys113 | NZ | Gly32 | O | 2.87 | |
| Lys113 | NZ | Gln35 | OE1 | 2.66 | |
| Leu116 | O | Asn214 | ND2 | 2.98 |
Table 4.
Time-averaged structural properties obtained from the MD simulations of SOCS2 protein-protein complexes.
Table 4.
Time-averaged structural properties obtained from the MD simulations of SOCS2 protein-protein complexes.
| Complex | Average RMSD (Å) | Average RMSF (Å) | Average RoG (Å) |
Number of Hydrogen Bonds Between the Two Proteins | Potential Energy (kcal/mol) |
|---|---|---|---|---|---|
| Standard | |||||
| SOCS2 (Apo-protein) | 2.417 | 1.089 | 1.674 | N/A | -158,603.87 |
| SOCS2:EpoR peptide (Standard Agonist) | 2.423 | 1.397 | 2.101 | 11 | -159,708.72 |
| SOCS2:N4BP1 (Standard Antagonist) | 2.496 | 1.179 | 2.162 | 24 | -459,214.66 |
| Protein and peptide derived from earthworm (Lumbricus genus) | |||||
| SOCS2:Cytochrome b | 2.467 | 0.876 | 2.182 | 39 | -471,304.36 |
| SOCS2:Cytochrome c oxidase subunit 3 | 2.504 | 1.122 | 2.176 | 30 | -450,869.63 |
| SOCS2:SCBP3 protein | 2.587 | 1.301 | 2.198 | 22 | -316,369.57 |
| SOCS2:Lumbricin | 2.495 | 1.031 | 2.148 | 18 | -694,628.89 |
| SOCS2:Chemoattractive glycoprotein ES20 | 2.512 | 0.941 | 2.176 | 22 | -621,068.14 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 6 | 2.487 | 1.001 | 2.178 | 20 | -466,577.25 |
| SOCS2:Histone H3 | 2.413 | 0.917 | 2.287 | 16 | -629,402.13 |
| SOCS2:Peroxidasin | 2.599 | 1.106 | 2.678 | 35 | -2,208,424.10 |
| SOCS2:Lumbrokinase-7T1 | 2.523 | 1.123 | 2.213 | 27 | -1,065,029.39 |
| SOCS2:Lysosomal membrane glycoprotein | 2.511 | 1.115 | 2.298 | 31 | -1,645,741.31 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 4L | 2.498 | 1.178 | 2.199 | 26 | -406,049.87 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 5 | 2.487 | 1.101 | 2.190 | 21 | -1,187,770.16 |
| SOCS2:Preprocarboxypeptidase | 2.596 | 1.259 | 2.188 | 30 | -560,689.83 |
| SOCS2:Extracellular globin-4 | 2.543 | 1.028 | 2.287 | 23 | -473,808.36 |
| SOCS2:Lumbrokinase-7T2 | 2.524 | 1.060 | 2.214 | 24 | -764,436.84 |
Table 5.
The average binding free energy (ΔGbinding) for the SOCS2 protein-protein complexes is reported with standard deviation in kcal/mol units, as determined by MM/PBSA calculations.
Table 5.
The average binding free energy (ΔGbinding) for the SOCS2 protein-protein complexes is reported with standard deviation in kcal/mol units, as determined by MM/PBSA calculations.
| Complex | MM/PBSA Calculation Results ΔGbinding (kcal/mol) | Average (kcal/mol) | ||
|---|---|---|---|---|
| I | II | III | ||
| Standard | ||||
| SOCS2:EpoR peptide (Standard Agonist) | -42.84 | -43.48 | -41.48 | -42.60 |
| SOCS2:N4BP1 (Standard Antagonist) | -42.34 | -42.34 | -42.85 | -42.51 |
| Protein and peptide derived from earthworm (Lumbricus genus) | ||||
| SOCS2:Cytochrome b | -50.57 | -49.93 | -49.82 | -50.11 |
| SOCS2:Cytochrome c oxidase subunit 3 | -44.93 | -44.93 | -44.87 | -44.91 |
| SOCS2:SCBP3 protein | -29.33 | -29.45 | -29.45 | -29.41 |
| SOCS2:Lumbricin | -59.22 | -59.26 | -59.26 | -59.25 |
| SOCS2:Chemoattractive glycoprotein ES20 | -53.69 | -57.76 | -53.60 | -55.02 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 6 | -52.81 | -51.48 | -53.15 | -52.48 |
| SOCS2:Histone H3 | -44.05 | -44.84 | -45.82 | -44.90 |
| SOCS2:Peroxidasin | -42.91 | -42.91 | -42.91 | -42.91 |
| SOCS2:Lumbrokinase-7T1 | -69.22 | -69.35 | -69.27 | -69.28 |
| SOCS2:Lysosomal membrane glycoprotein | -34.42 | -34.89 | -34.75 | -34.69 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 4L | -48.14 | -48.35 | -48.35 | -48.28 |
| SOCS2:NADH-ubiquinone oxidoreductase chain 5 | -37.74 | -36.11 | -36.11 | -36.65 |
| SOCS2:Preprocarboxypeptidase | -39.55 | -38.03 | -39.58 | -39.05 |
| SOCS2:Extracellular globin-4 | -50.09 | -49.68 | -50.30 | -50.02 |
| SOCS2:Lumbrokinase-7T2 | -46.18 | -45.80 | -47.11 | -46.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



