Introduction
ME/CFS is a complex disorder characterized by fatigue, cognitive impairment, autonomic instability, and immune dysfunction[
1]. The pathophysiology remains poorly understood, but emerging evidence suggests metabolic and neurotransmitter dysregulation play crucial roles[
2,
3]. This hypothesis proposes that ME/CFS results from dysfunction in noradrenergic neurons due to excessive insulin receptor sensitivity.
Phosphatidylcholine, a vital phospholipid, is integral to membrane integrity and receptor function. Deficiency in this molecule may contribute to increased insulin receptor sensitivity, leading to dysregulated norepinephrine signaling. Additionally, phosphatidylcholine depletion may impair red blood cell deformability, liver metabolism, and inflammatory balance. This model provides a comprehensive framework for understanding ME/CFS pathogenesis and its diverse symptom presentation.
The Sympathetic Nervous System and Noradrenergic Neurons
The synthesis of norepinephrine begins with the amino acid phenylalanine, which is first converted into tyrosine by the enzyme phenylalanine hydroxylase. Tyrosine is then processed by tyrosine hydroxylase to form L-DOPA, which subsequently undergoes decarboxylation to become dopamine. Finally, dopamine is converted into norepinephrine by the enzyme dopamine beta-hydroxylase.
Once synthesised, norepinephrine is stored in synaptic vesicles and released into the synaptic cleft upon neuronal stimulation. It then acts on postsynaptic noradrenergic receptors to modulate synaptic activity. The reuptake of norepinephrine into the neuron via the norepinephrine transporter (NET) is a critical process that regulates neurotransmitter levels and neuronal signaling. In the brain, norepinephrine is predominantly released from the locus coeruleus and has widespread effects on various neural circuits. Norepinephrine exerts its effects through adrenergic receptors, including alpha and beta receptors, which modulate synaptic transmission and neuronal excitability.
The sympathetic nervous system (SNS) is a critical component of the autonomic nervous system, responsible for the ‘fight or flight’ response. It orchestrates physiological changes in response to stress and maintains homeostasis by modulating cardiovascular, respiratory, and metabolic functions. The SNS is made up of noradrenergic neurons which release norepinephrine as its primary neurotransmitter, which is released from sympathetic nerve terminals to exert its effects on target tissues throughout the body.
Reduced Norepinephrine Transporters and Beta-2 Adrenergic Receptor Down-Regulation
This hypothesis proposes that reduced NET expression on noradrenergic neurons leads to elevated extracellular norepinephrine. Reduced expression or dysfunction of these transporters can lead to increased extracellular norepinephrine levels, while depleting intracellular norepinephrine stores. Research has found high plasma norepinephrine levels in ME patients and one particular study found there were subgroups of high and low norepinephrine which indicates subtypes of different noradrenergic dysfunction(3).
As well as causing a deficiency of norepinephrine in the synaptic vesicle of the neuron, prolonged elevation of extracellular norepinephrine can trigger down-regulation of the β2-adrenergic receptor (β2-AR) on target cells, which may diminish noradrenergic signaling. β2-ARs are critical in mediating physiological responses such as vasodilation, bronchodilation, and metabolic regulation, and their down-regulation can contribute to problems such as increased blood pressure, dysregulated glucose metabolism, reduced lipolysis, reduced vasodilation of blood vessels, altered neurotransmission, reduced gastrointestinal motility and a dysregulated immune response. Research has found ME patient immune cell β2-ARs to be less responsive to β2-AR agonists[
4].
The reduced expression of NET would reduce norepinephrine reuptake into the neuron and could result in a reduction of norepinephrine levels within the neuron. This would explain the low levels of DHPG found in ME patients in a recent NIH Intramural ME Study[
2].
Overtraining Syndrome
Overtraining syndrome (OTS) is a condition resulting from excessive exercise without adequate rest, leading to a persistent decline in performance, fatigue, and autonomic dysfunction. One of the key findings in OTS is a reduced density of β2-ARs on cell surfaces, which impairs SNS function, reducing the body’s ability to respond to stress and physical exertion[
5]. This downregulation of β2-ARs is hypothesised to contribute to the chronic fatigue, impaired exercise tolerance, and autonomic symptoms seen in OTS. Interestingly, there is a striking overlap between the symptoms of OTS and ME, including profound fatigue, post-exertional malaise, and autonomic dysregulation, such as orthostatic intolerance. Furthermore, a significant number of ME patients report being avid exercisers or athletes prior to the onset of illness, suggesting that excessive physical activity or overtraining may be a predisposing factor for the development of ME in some individuals. The similarities in symptoms and underlying mechanisms, such as adrenergic receptor downregulation, highlight potential shared pathways in the pathophysiology of OTS and ME/CFS, although the precise triggers and disease course differ between the two conditions.
Chronotropic incompetence (CI) is the inability to increase heart rate commensurate with metabolic demand and can contribute to exertion intolerance. Research has shown that when undertaking a cardiopulmonary exercise test (CPET) test on two concurrent days, ME patients don’t perform as well on the second day as healthy controls, and appear to have increased CI on day two. Research shows that CI involves cardiac β-adrenergic receptor down-regulation, and so this down-regulation of beta adrenergic receptors on the second day CPET test could be due to excessive and prolonged norepinephrine release during the CPET test on day one causing down-regulation of adrenergic receptors[
6]. However it should also be considered that the exercise on day one could worsen a norepinephrine deficiency, affecting the performance on day two.
Negative Feedback Mechanism of Norepinephrine Regulation
The alpha 2 adrenergic autoreceptor feedback mechanism plays a critical role in maintaining norepinephrine homeostasis. High extracellular norepinephrine levels activate alpha 2 adrenergic autoreceptors on presynaptic neurons, which inhibits further norepinephrine synthesis and release. Reduced NET expression can lead to sustained high extracellular norepinephrine levels, disrupting this feedback mechanism and causing a cyclical pattern of excessive norepinephrine release followed by excessive activation of autoreceptors and inhibition of norepinephrine synthesis, which would worsen the neuronal norepinephrine deficiency. This may explain the boom and bust pattern of symptom severity that many ME patients experience, although it is also possible that the boom and bust pattern of symptoms could actually be due to excessive norepinephrine release down-regulating postsynaptic receptors, or possibly a combination of both.
Insulin’s Role in Regulating Norepinephrine Transporters
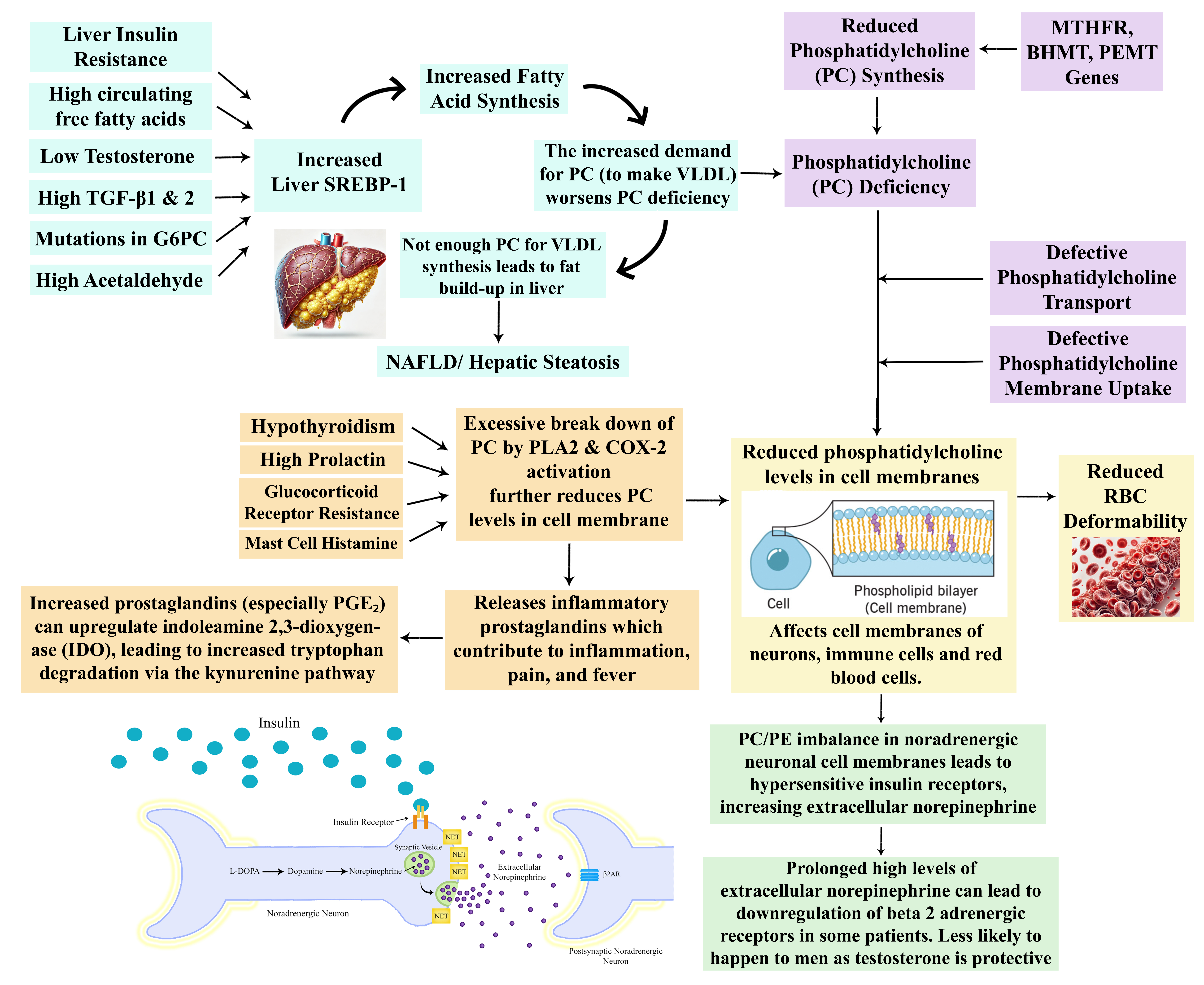
Insulin plays a key role in the regulation of NETS. Research has shown that insulin reduces norepinephrine reuptake by reducing NET expression on noradrenergic neurons[
7]. In ME patients, it is possible that dysregulated insulin signaling exacerbates NET dysfunction. Insulin reduces the expression of NETS on noradrenergic neurons, and in the setting of prolonged hyperinsulinemia, this could lead to increased extracellular norepinephrine levels.
Recent studies have highlighted insulin dysregulation in ME patients, with genetic research identifying variants associated with insulin signaling and secretion. In particular gene variants for the ATP9A, KCNB1 and CLOCK genes have been found to be risk factors for ME, and these genes are involved in insulin secretion from the pancreas[
8]. The insulin receptor INSR gene has also been found to be a risk factor for both ME and Long COVID[
9]. Hypersensitivity of the neuronal insulin receptor could also cause a reduction in NETS.
Whilst this hypothesis is focusing on dysregulated noradrenergic neuron signaling as the main cause of most symptoms in ME, it should be noted that high insulin levels have been shown to increase the expression of dopamine transporters in dopamine neurons of the striatum[
10]. This leads to increased re-uptake of dopamine from the synaptic cleft, resulting in reduced extracellular dopamine levels. This could contribute to the depletion of extracellular dopamine, particularly in brain regions involved in reward and motivation, such as the striatum.
Glucocorticoid Receptor Resistance
Glucocorticoid receptor (GR) resistance refers to a condition in which tissues become less responsive to the effects of glucocorticoids, despite normal or elevated levels of circulating hormones such as cortisol. The human GR is encoded by the NR3C1 gene, which produces two main isoforms, GR-α and GR-β, via alternative splicing. GR-α is the classic isoform responsible for mediating the anti-inflammatory and metabolic effects of glucocorticoids, while GR-β acts as a dominant-negative inhibitor of GR-α, reducing its transcriptional activity. Dysregulation in the balance between these two isoforms can lead to GR resistance.
Evidence suggests that ME patients may exhibit GR resistance. A study by Kavelaars et al. (2000) demonstrated the maximal effect of dexamethasone on T-cell proliferation is significantly reduced in ME patients as compared with controls, which may indicate a reduced responsiveness to cortisol’s regulatory effects on inflammation and metabolic homeostasis[
4]. It could be possible that some ME patients have genetic variants that make their glucocorticoid receptors more vulnerable to developing resistance.
GR resistance also has significant implications for insulin signaling. Cortisol normally promotes insulin resistance by antagonising insulin receptor signaling, reducing glucose uptake in peripheral tissues. However, in the context of GR resistance, tissues may exhibit heightened sensitivity to insulin due to diminished cortisol-mediated insulin antagonism. This effect could be particularly pronounced in noradrenergic neurons. Reduced glucocorticoid signaling may lead to increased insulin receptor sensitivity on these neurons, amplifying the effects of insulin and leading to disruptions in norepinephrine homeostasis. Research shows that corticosterone up-regulates the expression and function of the NET[
11].
How Cell Membrane Phosphatidylcholine Deficiency Affects Insulin Signaling
Phosphatidylcholine (PC) is the most abundant phospholipid in cell membranes, constituting approximately 40–50% of total phospholipid content. It plays a fundamental role in maintaining membrane structure, fluidity, and the optimal function of transmembrane proteins, including receptors. The insulin receptor undergoes dynamic conformational changes upon insulin binding, a process that is influenced by membrane lipid composition. PC deficiency can alter the biophysical properties of the membrane, reducing fluidity and disrupting lipid rafts—microdomains essential for receptor clustering and signal transduction. Furthermore, PC is crucial for the proper endocytosis and recycling of the insulin receptor. Upon insulin binding, the receptor-ligand complex undergoes clathrin-mediated endocytosis, which modulates receptor availability at the cell surface. PC-deficient membranes may impair this process, reducing receptor internalization and degradation, leading to prolonged receptor presence on the membrane and an increased sensitivity to circulating insulin. This effect could contribute to pathological insulin hypersensitivity, particularly in neurons where insulin signaling is tightly regulated.
Phosphatidylcholine is synthesised through two primary pathways: the Kennedy pathway and the phosphatidylethanolamine N-methyltransferase (PEMT) pathway. The Kennedy pathway utilises dietary choline to generate PC. The PEMT pathway, primarily active in hepatocytes, involves the sequential methylation of phosphatidylethanolamine (PE) to PC, a process requiring S-adenosylmethionine (SAMe) as a methyl donor. Once synthesised, PC is transported to cellular membranes via lipid transfer proteins or vesicular trafficking mechanisms, ensuring the appropriate distribution of PC across different organelle and plasma membrane compartments.
The PEMT pathway is particularly sensitive to disruptions in methylation metabolism, and gene variants in MTHFR, BHMT, and PEMT can impair its function, leading to reduced PC synthesis. MTHFR and BHMT gene variants can lower methionine synthesis and subsequently reduce the availability of SAMe, the key methyl donor for PEMT-catalyzed reactions. A reduction in SAMe levels directly impairs PEMT enzyme activity, limiting the conversion of PE to PC. Additionally, PEMT gene variants can further exacerbate this dysfunction by directly decreasing the catalytic efficiency of the enzyme, compounding the impact of reduced methyl donor availability. These deficiencies in PC synthesis could disproportionately affect tissues reliant on PEMT activity for maintaining membrane phospholipid composition, including neurons, where altered PC levels may contribute to dysregulated insulin receptor signaling.
Defects in Phosphatidylcholine Transport and Membrane Uptake
Even when PC synthesis is sufficient, defects in PC transport and membrane uptake can contribute to functional phosphatidylcholine deficiency. Proper PC distribution depends on lipid transport proteins such as ATP-binding cassette transporters and phospholipid transfer proteins, which facilitate the movement of PC between intracellular compartments and the plasma membrane. Dysfunction in these transport processes could lead to regional PC depletion, particularly in cell membranes where receptor signaling is highly dependent on lipid microenvironments. Furthermore, disruptions in lipid raft composition due to altered PC-to-cholesterol ratios can impair receptor mobility and clustering, affecting insulin receptor signaling efficiency. Such deficiencies may play a role in insulin dysregulation by altering the receptor’s responsiveness to extracellular insulin concentrations, contributing to metabolic dysfunction.
Lipoproteins, particularly high-density lipoprotein (HDL) and low-density lipoprotein (LDL), also play a critical role in the systemic transport of phosphatidylcholine. HDL facilitates the redistribution of PC between tissues through the phospholipid transfer protein (PLTP)-mediated exchange of phospholipids, while LDL delivers PC to cells via receptor-mediated endocytosis. Impaired lipoprotein metabolism, as seen in conditions of dyslipidemia, hepatic dysfunction, or genetic variants affecting lipid transport proteins, can disrupt PC availability to peripheral tissues. A deficiency in circulating PC-rich lipoproteins may further contribute to reduced membrane PC content, exacerbating the effects of impaired synthesis or intracellular transport.
Phosphatidylcholine Depletion Due to Phospholipase A2 and COX-2 Activation
Phosphatidylcholine (PC) deficiency in cell membranes can arise from excessive activation of phospholipase A2 (PLA2), a key enzyme that hydrolyzes the sn-2 position of glycerophospholipids, releasing arachidonic acid (AA) from PC and other phospholipids. This process serves as the initial step in the biosynthesis of eicosanoids, including prostaglandins, thromboxanes, and leukotrienes. Under normal physiological conditions, PLA2 activity is tightly regulated to balance membrane phospholipid composition with eicosanoid production. However, chronic inflammatory signaling, oxidative stress, and hormonal dysregulation can lead to excessive PLA2 activation, resulting in accelerated PC degradation. Furthermore, increased phospholipase activity is often accompanied by upregulation of cyclooxygenase-2 (COX-2), an inducible enzyme that converts free arachidonic acid into pro-inflammatory prostaglandins. Sustained COX-2 activation exacerbates phospholipid depletion by perpetuating inflammatory signaling cascades, further increasing PLA2 activity in a feed-forward loop. This cycle can lead to progressive depletion of PC within cellular membranes, impairing membrane-dependent processes, including receptor signaling, vesicular trafficking, and cellular homeostasis.
Elevated Prostaglandin Levels and Their Pathophysiological Consequences
The excessive liberation of arachidonic acid due to PLA2 activation and its subsequent conversion by COX-2 leads to increased levels of pro-inflammatory prostaglandins, particularly prostaglandin E2 (PGE2) and prostaglandin D2 (PGD2). These bioactive lipid mediators exert widespread effects on pain perception, inflammation, neurotransmitter metabolism, and immune regulation. Elevated PGE2 is a well-established contributor to hyperalgesia and chronic pain, acting via EP receptor activation to sensitise nociceptors and lower pain thresholds.
Additionally, PGE2 and PGD2 modulate tryptophan metabolism by enhancing indoleamine 2,3-dioxygenase (IDO) activity, shifting tryptophan catabolism towards the production of kynurenine and its downstream metabolites. This shift not only depletes tryptophan availability for serotonin synthesis, potentially contributing to mood disturbances and fatigue, but also generates neurotoxic metabolites such as quinolinic acid, which can exacerbate neuroinflammation and oxidative stress.
Furthermore, prostaglandins influence immune cell function, with PGE2 exhibiting immunosuppressive effects by inhibiting T-cell proliferation and skewing immune responses toward a Th2-dominant profile. In contrast, PGD2 has been implicated in mast cell activation and eosinophil recruitment, further perpetuating inflammatory and allergic responses. The combined effects of prostaglandin elevation can thus contribute to systemic inflammation, altered neurotransmission, and immune dysregulation, all of which may play a role in conditions associated with phospholipid abnormalities. It should be noted that some ME patients who have very low synthesis of PC may actually have such low membrane PC levels that they produce less prostaglandins than normal.
Mechanisms Driving Phospholipase A2 and COX-2 Activation
Multiple physiological factors can contribute to the excessive activation of PLA2 and COX-2, leading to phosphatidylcholine depletion. One significant regulatory factor is prolactin, which has been shown to enhance PLA2 activity in immune cells and other tissues. Elevated prolactin levels, as seen in stress responses, pregnancy, and prolactinomas, may therefore contribute to increased phospholipid hydrolysis and subsequent prostaglandin production.
Glucocorticoid receptor resistance is another potential driver of PLA2 and COX-2 upregulation. Under normal conditions, glucocorticoids exert anti-inflammatory effects by inhibiting COX-2 transcription and downregulating PLA2 activity through annexin-1 induction. However, in states of glucocorticoid resistance—such as chronic stress, inflammation, or metabolic dysfunction—this inhibitory feedback is weakened, leading to unchecked activation of these lipid-metabolizing enzymes.
Additionally, mast cell activation represents a potent trigger for COX-2 upregulation and phospholipid breakdown. Upon degranulation, mast cells release histamine and tryptase, both of which have been shown to induce COX-2 expression in endothelial cells, fibroblasts, and immune cells. Histamine, acting through H1 and H2 receptors, can enhance inflammatory cytokine production, while tryptase—a serine protease—activates protease-activated receptors (PARs), leading to NF-κB-mediated COX-2 transcription. This mast cell-driven inflammatory response creates a local microenvironment of elevated prostaglandins and chronic phospholipid turnover, further depleting PC stores.
Taken together, these mechanisms illustrate how hormonal imbalances, immune dysregulation, and chronic inflammation can converge to drive excessive membrane phospholipid metabolism, leading to a state of persistent PC depletion and altered cellular signaling dynamics.
Phosphatidylcholine Requirement for VLDL Synthesis and Its Role in Hepatic Lipid Homeostasis
Phosphatidylcholine (PC) is an essential phospholipid required for the proper assembly and secretion of very low-density lipoproteins (VLDL) in the liver. VLDL serves as the primary vehicle for exporting triglycerides (TGs) from hepatocytes into circulation, preventing intracellular lipid accumulation. The process of VLDL formation involves the incorporation of triglycerides and cholesterol into lipid droplets, which are then enveloped by an amphipathic monolayer containing apolipoprotein B (ApoB) and phospholipids, primarily PC. A deficiency of PC disrupts this process, impairing the maturation and secretion of VLDL. Consequently, triglycerides accumulate within hepatocytes, leading to hepatic steatosis, commonly referred to as non-alcoholic fatty liver disease (NAFLD). Experimental models have demonstrated that hepatic PC depletion via genetic disruption of phosphatidylethanolamine N-methyltransferase (PEMT)—a key enzyme in PC biosynthesis—results in triglyceride accumulation and liver dysfunction. Furthermore, impaired VLDL secretion due to inadequate PC levels can exacerbate systemic metabolic disturbances by altering lipid distribution and plasma lipoprotein profiles.
SREBP-1 Activation and the Redistribution of Phosphatidylcholine to VLDL
Sterol regulatory element-binding protein 1 (SREBP-1) is a transcription factor that regulates hepatic lipogenesis, including the synthesis of fatty acids and triglycerides. Under normal conditions, SREBP-1 activation is tightly controlled to balance lipid production with metabolic demands. However, chronic up-regulation of SREBP-1, as seen in insulin resistance and metabolic syndrome, drives excessive lipogenesis, increasing the need for phosphatidylcholine to accommodate the heightened demand for VLDL secretion. This creates a metabolic trade-off in which hepatic PC stores are preferentially diverted toward lipoprotein synthesis at the expense of maintaining adequate PC availability for cellular membranes throughout the body. As a result, systemic phosphatidylcholine depletion may impair the structural integrity and function of cell membranes in multiple tissues, including neurons and red blood cells. In this context, conditions that strongly activate SREBP-1 can act as metabolic stressors that push phosphatidylcholine levels below a critical threshold, precipitating widespread cellular dysfunction and systemic metabolic disturbances.
Factors Contributing to SREBP-1 Activation and Potential Triggers of Phosphatidylcholine Deficiency
-
1.
Hyperinsulinemia and Hepatic Insulin Resistance
Elevated insulin levels are one of the most potent activators of SREBP-1. Insulin stimulates SREBP-1c transcription through the activation of PI3K/Akt signaling, promoting lipogenesis even in states of metabolic dysfunction. Paradoxically, insulin resistance—a condition characterised by impaired insulin signaling in peripheral tissues—often coexists with hepatic insulin sensitivity in the SREBP-1 pathway. This selective insulin resistance leads to persistent SREBP-1 activation, fueling triglyceride accumulation and increasing the demand for phosphatidylcholine to support VLDL synthesis. In this scenario, phosphatidylcholine depletion may be exacerbated as hepatocytes prioritise VLDL secretion to compensate for the increased lipid burden.
The gene variants for the ATP9A, KCNB1 and CLOCK genes have been found to be risk factors for ME, and these genes are involved in insulin secretion from the pancreas[
8]. Dysregulated pancreatic insulin production could contribute to the development of hepatic insulin resistance and increased SREBP-1 activation.
-
2.
Elevated Circulating Free Fatty Acids (FFAs)
Increased FFAs, particularly saturated fatty acids, can also drive hepatic lipogenesis and SREBP-1 activation. Elevated plasma FFAs are commonly observed in obesity, metabolic syndrome, and prolonged fasting states where adipose tissue undergoes excessive lipolysis. The influx of FFAs into the liver provides substrate for triglyceride synthesis, further upregulating SREBP-1 to manage lipid storage and export. This increased hepatic lipid processing imposes a greater demand on PC availability, leading to its preferential utilization for lipoprotein assembly rather than maintaining membrane integrity.
-
3.
Testosterone Dysregulation
Testosterone influences lipid metabolism, and alterations in androgen levels can modulate SREBP-1 activity. In men, low testosterone levels are associated with increased hepatic SREBP-1 expression and heightened triglyceride synthesis, whereas in women with polycystic ovary syndrome (PCOS), elevated testosterone has been linked to hepatic lipid accumulation and NAFLD. Testosterone-driven SREBP-1 activation can therefore contribute to hepatic PC depletion, particularly in individuals with androgen imbalances.
-
4.
High Acetaldehyde Exposure
Acetaldehyde, a toxic intermediate of ethanol metabolism, disrupts lipid homeostasis by modifying key regulatory proteins and impairing mitochondrial function. Chronic alcohol consumption elevates acetaldehyde levels, which in turn stimulates SREBP-1 and enhances triglyceride synthesis. The resulting lipid accumulation exacerbates the need for phosphatidylcholine in VLDL formation, further depleting hepatic PC stores. This mechanism is particularly relevant in alcoholic liver disease but may also apply to non-alcoholic conditions where endogenous acetaldehyde production is increased due to gut dysbiosis or impaired aldehyde dehydrogenase (ALDH) activity.
-
5.
TGF-β Signaling and Fibrotic Progression
Transforming growth factor-beta (TGF-β) is a key regulator of hepatic fibrosis and extracellular matrix remodeling. Chronic activation of TGF-β signaling has been shown to enhance SREBP-1 expression, leading to excessive lipid accumulation in hepatocytes. This pathway is particularly relevant in conditions such as non-alcoholic steatohepatitis (NASH) and liver fibrosis, where chronic inflammation and fibrogenesis further increase metabolic stress on hepatic PC homeostasis.
-
6.
Iron Overload
Iron overload, as seen in hereditary hemochromatosis, promotes oxidative stress through the Fenton reaction, generating reactive oxygen species (ROS) that disrupt metabolic homeostasis. ROS-induced stress upregulates SREBP-1 via the PERK-eIF2α-ATF4 pathway and enhances lipogenic gene transcription. Additionally, iron accumulation increases insulin signaling and inflammatory cytokine release (TNF-α, IL-6), further stimulating SREBP-1 activation through NF-κB and JAK/STAT pathways. This drives hepatic triglyceride accumulation and diverts phosphatidylcholine toward VLDL synthesis, potentially depleting systemic PC availability and contributing to metabolic dysfunction.
Accuracy of the Hemoglobin A1c Test
Altered red blood cell (RBC) deformability in ME/CFS patients may have implications beyond impaired oxygen transport, potentially affecting the accuracy of glycemic biomarkers such as hemoglobin A1c (HbA1c). The HbA1c test relies on the assumption that erythrocytes have a uniform lifespan of approximately 120 days, during which glucose non-enzymatically glycosylates hemoglobin in a time-dependent manner. However, reduced RBC deformability, as observed in ME/CFS, has been associated with shortened erythrocyte lifespan and altered glucose diffusion dynamics within erythrocytes. These factors may lead to an underestimation of glycation levels, thereby yielding falsely lower HbA1c values in individuals with underlying insulin resistance. Consequently, reliance on HbA1c alone may fail to detect dysglycemia in ME/CFS patients, necessitating the use of alternative biomarkers such as fructosamine, fasting insulin, or continuous glucose monitoring to accurately assess metabolic status.
Is There A Prodrome Phase?
In some patients there may be two stages to the disease. There may be a prodrome phase which precedes the onset of full-blown ME. The evidence suggests that prolonged exposure to high extracellular norepinephrine and SNS hyperactivity could lead to gradual down-regulation of the β2ARs.
During the prodrome phase, some ME patients may experience a prolonged period of sympathetic dominance. This state of over-activation has been documented in several chronic conditions, including Long COVID, where patients show increased SNS tone, as evidenced by reduced heart rate variability and other autonomic markers[
13]. In such a state, symptoms such as tachycardia, anxiety, sleep disturbances, and heightened stress responses might predominate, signaling an overactive SNS. This hyperactive phase could last for months, with the body trying to maintain homeostasis in the face of continuous high extracellular norepinephrine.
Prolonged norepinephrine stimulation leads to β2-AR down-regulation, a process that takes time and repeated exposure to high levels of norepinephrine. It is plausible that this process of receptor down-regulation is gradual and may take many months to happen. Patients in the prodrome phase may not yet exhibit full ME symptoms including PEM but may experience signs of SNS over-activation such as palpitations, gastrointestinal dysregulation and cognitive dysfunction.
Some patients with Long COVID manage to recover within 6-12 months, potentially because their autonomic dysregulation resolves before β2-ARs undergo significant down-regulation. Early treatment interventions aimed at rebalancing autonomic function or reducing norepinephrine levels may help prevent progression to receptor down-regulation and, consequently, ME. However, for individuals who suffer a second immune challenge—such as a viral infection, vaccination, or physical injury—this may hasten the β2-AR down-regulation process. The introduction of another stressor could amplify norepinephrine release, leading to an accelerated decline in receptor sensitivity and the rapid onset of ME. A return to exercise before correcting this dysregulation could also be enough to cause β2-AR down-regulation, as exercise releases norepinephrine.
Norepinephrine Dysregulation in the Brain and Its Impact on Astrocyte Function
Norepinephrine is a key neurotransmitter involved in a variety of brain functions, including arousal, attention, cognition, and the regulation of the sleep-wake cycle. However, dysregulation of norepinephrine signaling, particularly chronic elevations in extracellular norepinephrine, can have detrimental effects on brain function and may contribute to the pathophysiology of ME. One critical aspect of norepinephrine dysregulation is it’s impact on astrocytes, the glial cells that play a crucial role in regulating brain energy metabolism, neurotransmitter balance, cerebral blood flow, and waste clearance through the glymphatic system. In ME, norepinephrine dysregulation could lead to astrocytic dysfunction, resulting in a cascade of metabolic and neuronal disturbances.
Norepinephrine Dysregulation of Astrocytes and Glucose Uptake
Astrocytes are vital for maintaining brain energy homeostasis, in part by regulating glucose uptake from the bloodstream. Norepinephrine acts on astrocytic β2ARs, which are known to stimulate glucose uptake via increased expression of glucose transporters, such as GLUT1. While this process is essential for meeting the energy demands of the brain during periods of heightened activity, chronic norepinephrine stimulation, as may occur in ME, could lead to excessive glucose uptake by astrocytes, particularly in the hypothalamus and cortex. This overactivity of astrocytes, driven by continuous norepinephrine signaling, has been proposed to lead to an overproduction of lactate.
Excessive astrocytic lactate production may contribute to the elevated brain lactate levels observed in ME patients, as demonstrated by magnetic resonance spectroscopy studies[
14]. Elevated lactate is a marker of metabolic dysfunction and is associated with increased fatigue, cognitive impairment, and altered sleep patterns, all of which are characteristic symptoms of ME.
Impact of High Lactate Levels on Sleep
The accumulation of lactate in the brain has significant implications for sleep regulation. Lactate is thought to act as a signaling molecule that modulates sleep-wake cycles, particularly slow-wave sleep (SWS), which is the deepest and most restorative phase of sleep. Under normal conditions, the brain’s lactate levels decrease during sleep, particularly during SWS, as the brain clears out metabolic byproducts. However, elevated lactate levels, driven by chronic norepinephrine activation of astrocytes, may interfere with this process, leading to disrupted sleep architecture and unrefreshing sleep, which are hallmark features of ME.
Lactate as a Preferential Fuel
Continuous norepinephrine stimulation of astrocytes may also shift the brain’s neuronal fuel preference from glucose to lactate. This metabolic adaptation is similar to what has been observed in patients with diabetes who experience repetitive hypoglycaemic episodes. In these patients, the brain adapts to rely more heavily on lactate as an energy source, rather than glucose, during periods of hypoglycaemia. In ME, chronic norepinephrine activation may cause a similar shift in metabolic substrate utilisation, particularly in astrocytes of the hypothalamus, which plays a crucial role in energy balance and homeostasis. Moreover, prolonged reliance on lactate as a fuel may impair neuronal function, as neurons require a delicate balance between glucose and lactate metabolism to maintain optimal function.
Sex Differences in Astrocytic Response to Norepinephrine Dysregulation
Emerging evidence suggests that astrocytes may respond differently to chronic norepinephrine dysregulation in males and females, with distinct implications for metabolic and neuronal function. Research has shown there are sex-specific differences in astrocytic metabolism and signaling in response to elevated norepinephrine levels[
15]. In male rats, chronic norepinephrine stimulation led to reduced astrocyte β2-AR expression, which could result in decreased glucose uptake and lactate production in astrocytes. This suggests that males may have a protective down-regulation of the β2-AR pathway in response to high norepinephrine, which helps to moderate the metabolic impact.
In contrast, female rats exhibited increased β2-AR expression in response to high norepinephrine, which could amplify glucose uptake and lactate production by astrocytes. The up-regulation of β2-ARs in females means that they may be more vulnerable to the effects of high extracellular norepinephrine in the brain, as the overstimulation of astrocytes can lead to excessive lactate accumulation and more pronounced metabolic disturbances.
Norepinephrine, Astrocytes and the Glymphatic System
Astrocytes play a central role in regulating the glymphatic system, a waste clearance pathway in the brain that is responsible for removing metabolic byproducts and toxins, particularly during sleep. Norepinephrine is a key regulator of the glymphatic system, as it modulates astrocytic aquaporin-4 (AQP4) channels, which facilitate the movement of cerebrospinal fluid through the brain parenchyma. Under normal conditions, norepinephrine levels decrease during sleep, allowing the glymphatic system to function optimally and clear out waste products such as amyloid-beta and tau proteins. However, in ME, chronic high extracellular norepinephrine may impair glymphatic function by interfering with AQP4 channel regulation, leading to the accumulation of toxic metabolic byproducts in the brain. This could further exacerbate cognitive dysfunction and contribute to the development of neuroinflammatory processes in ME.
Norepinephrine, Astrocytes and Neurotransmitter Regulation
Astrocytes play a key role in regulating neurotransmitter levels, particularly glutamate and gamma-aminobutyric acid (GABA), both of which are crucial for maintaining proper excitatory and inhibitory balance in the brain. The neurotransmitter norepinephrine has a significant impact on astrocyte-mediated glutamate clearance, and its dysregulation could contribute to neurotransmitter imbalances in ME patients. In healthy states, norepinephrine enhances astrocytic glutamate uptake by increasing the expression of glutamate transporters, thereby reducing extracellular glutamate and preventing excitotoxicity.
An increased glutamate uptake by astrocytes could lead to lower extracellular glutamate. Although this might protect against excitotoxicity, a persistent reduction in glutamate levels may disrupt normal synaptic transmission, contributing to cognitive dysfunction and fatigue. Glutamate is critical for excitatory signaling and cognitive processes, so chronically reduced levels could impair memory formation, learning, and overall brain function. This mechanism could help explain the cognitive impairments often reported in ME patients.
Astrocytic Regulation of Cerebral Blood Flow
Astrocytes are key mediators of cerebral blood flow, as they respond to neuronal activity by releasing vasoactive substances that dilate or constrict blood vessels. Norepinephrine plays a role in modulating this astrocytic response through its actions on adrenergic receptors. In ME, chronic norepinephrine dysregulation may impair astrocytic regulation of blood flow, leading to reduced oxygen and nutrient delivery to neurons. This could contribute to the cognitive deficits and fatigue commonly experienced by ME patients, as impaired cerebral blood flow can compromise brain function and exacerbate metabolic dysfunction.
The Effect of Testosterone on Symptom Profile and Illness Risk
Testosterone’s role in increasing the expression of the β2AR may provide protective effects in males[
16]. This may explain the lower prevalence of ME in males as compared to females.
Moreover, testosterone enhances the activity of catechol-O-methyltransferase (COMT), an enzyme that breaks down extracellular norepinephrine into normetanephrine, thereby reducing norepinephrine levels in synaptic clefts[
17]. Enhanced COMT activity, by reducing norepinephrine accumulation, may prevent over-activation of adrenergic receptors, particularly in the sympathetic nervous system, further decreasing the risk of receptor down-regulation. These factors may contribute to a lower risk of illness onset in males (they are more likely to recover from the prodrome phase mentioned earlier).
Potential Links to Endometriosis
Endometriosis is a chronic gynaecological condition characterised by the presence of endometrial-like tissue outside the uterus, primarily on the pelvic organs. This condition affects approximately 10% of women of reproductive age and is associated with debilitating symptoms such as chronic pelvic pain, heavy menstrual bleeding, and infertility. Emerging evidence suggests a potential overlap between endometriosis and ME, with one research paper indicating that approximately one-third of female ME patients are also diagnosed with endometriosis[
18]. This co-occurrence suggests a possible shared pathophysiology between the two conditions.
One notable feature of endometriosis is the presence of reactive hypoglycaemia in affected individuals. Reactive hypoglycaemia refers to episodes of low blood sugar following a meal, typically a result of excessive insulin release. In a study published by Mathias et al. (2001), it was demonstrated that patients with endometriosis exhibit abnormal insulin responses, characterised by heightened insulin sensitivity in peripheral tissues, including nerves[
19]. The authors found that the cellular membranes of endometriosis patients’ peripheral nerves were hyper-responsive to insulin. This finding is particularly relevant to the hypothesis presented in this paper, which posits that the pathophysiology of ME involves increased sensitivity of noradrenergic neuron insulin receptors.
Another connection to endometriosis is the fact that prostaglandins have been found to be increased in endometriosis patients. Prolactin has also been found to be increased in endometriosis patients and this connection may be explained by prolactin’s action on phospholipase A2. Research has shown that the prolactin inhibitor Bromocriptine can improve endometriosis symptoms. As dopamine also controls prolactin levels, medications which increase dopamine (for example low-dose aripiprazole) may also help to reduce high prolactin levels in ME and endometriosis patients with high prolactin levels.
Mast Cell Activation as Both a Root Cause and a Downstream Effect in ME/CFS
Mast cell activation syndrome (MCAS) has been implicated in the pathophysiology of ME/CFS, with evidence suggesting it may act as both a primary driver of disease and a secondary consequence of other dysregulated pathways. In certain patients, MCAS could serve as a root cause by promoting metabolic and inflammatory disturbances that contribute to disease onset. One proposed mechanism involves the protease tryptase, a mast cell-derived enzyme that has been shown to influence hepatic lipid metabolism. Tryptase can activate protease-activated receptors (PARs) in hepatocytes, leading to up-regulation of sterol regulatory element-binding protein 1 (SREBP-1) and increased lipogenesis. Additionally, mast cell-derived histamine and tryptase can activate phospholipase A2 (PLA2) and cyclooxygenase-2 (COX-2), increasing the breakdown of cell membrane phospholipids, including PC.
Conversely, in other patients, MCAS may emerge as a downstream effect of autonomic and immune dysregulation. In this paper it has been suggested that sustained high levels of norepinephrine can lead to the downregulation of β₂-adrenergic receptors (β₂-ARs). If this downregulation happens to mast cell β₂-ARs, this could lead to increased mast cell reactivity. This effect may be further exacerbated by glucocorticoid receptor resistance in mast cells. Since functional glucocorticoid receptors are required for mast cell stabilisation, resistance to glucocorticoid signaling could result in an uninhibited mast cell response, perpetuating a hyperinflammatory state. This creates a vicious cycle in which MCAS exacerbates phospholipid depletion, inflammation, and metabolic dysfunction, feeding back into the pathological mechanisms of ME/CFS.
Regardless of whether MCAS serves as a primary trigger or a secondary consequence, its contribution to systemic inflammation, membrane lipid dysregulation, and microcirculatory impairment suggests that targeting mast cell activation may be a critical therapeutic strategy. Even in cases where MCAS is a downstream effect, its ability to amplify disease pathology warrants clinical intervention to break the cycle of inflammation and metabolic dysfunction.
A Component-Based Approach to Understanding and Treating ME/CFS
Rather than classifying patients into rigid subtypes, a more effective approach to ME/CFS management may involve identifying and addressing key dysfunctional components on an individual basis. Given the complex interplay of metabolic, immune, and neurological abnormalities observed in ME/CFS, each patient may present with a unique combination of pathological factors. A targeted, component-based model allows for more precise intervention and may prevent unintended exacerbation of symptoms that can arise when only one aspect of dysfunction is treated in isolation. The primary components to assess include phosphatidylcholine dysregulation, liver dysfunction, excessive prostaglandin production, mast cell activation, and norepinephrine dysregulation.
The first component is phosphatidylcholine (PC) dysregulation, which may arise from impairments in PC synthesis, transport, or membrane uptake. Identifying the underlying cause is essential, as treatment strategies vary. For individuals with reduced PC synthesis due to genetic variants affecting the phosphatidylethanolamine N-methyltransferase (PEMT) or folate pathways, supplementation with choline, 5-methyltetrahydrofolate, or phosphatidylcholine may be beneficial. However, indiscriminate PC supplementation without addressing concurrent metabolic dysfunctions may lead to unintended consequences, such as excessive VLDL production and triglyceride accumulation.
The second component involves liver dysfunction, particularly in relation to insulin resistance and lipid metabolism. Dysregulation of hepatic function can alter PC availability, impair bile acid metabolism, and exacerbate systemic metabolic disturbances. Clinical assessment should determine whether liver dysfunction is linked to insulin resistance, inflammation, or phospholipid imbalances. Potential therapeutic interventions include TUDCA (tauroursodeoxycholic acid) for bile acid support, berberine for modulating lipid and glucose metabolism, intermittent fasting to improve insulin sensitivity, and dietary modifications to reduce hepatic stress. However, long-term management should prioritise identifying and addressing the root cause of hepatic dysfunction to achieve sustainable improvements.
The third component, excessive prostaglandin production, is a key driver of inflammation and pain in many ME/CFS patients. Prostaglandin levels can be directly measured, and elevated levels may indicate increased phospholipase A2 (PLA2) and cyclooxygenase-2 (COX-2) activity, leading to excessive membrane phospholipid breakdown. Treatment strategies may include COX-2 inhibitors such as celecoxib, antihistamines to reduce mast cell-derived inflammatory mediators, omega-3 supplementation to shift the balance toward anti-inflammatory eicosanoids, and dietary modifications to lower omega-6 intake. Additional interventions such as resveratrol, quercetin, and L-theanine supplementation may further modulate inflammatory pathways. Crucially, identifying the underlying cause of prostaglandin excess—whether due to immune activation, hormonal imbalances, or metabolic dysfunction—is necessary to implement an effective long-term strategy.
The fourth component, mast cell activation can act as both a primary driver and a secondary consequence of ME/CFS pathology. Persistent mast cell activation contributes to systemic inflammation, histamine-mediated symptoms, and increased breakdown of membrane phospholipids via PLA2 activation. If mast cell activation is identified, targeted interventions such as antihistamines, dietary avoidance of high-histamine foods, and diamine oxidase (DAO) supplementation may help mitigate symptoms. However, mast cell activation must be understood in the broader context of the patient’s pathology. For example, if β₂-adrenergic receptor downregulation due to chronic norepinephrine elevation is driving mast cell activation, addressing autonomic dysfunction may be necessary to restore proper immune regulation.
The fifth component, norepinephrine dysregulation, is often secondary to the other imbalances but can persist in a subset of patients. In most individuals, norepinephrine abnormalities should resolve once phospholipid metabolism, liver function, inflammation, and mast cell activity are corrected. However, if prolonged downregulation of β₂-adrenergic receptors has occurred due to chronic exposure to high extracellular norepinephrine, receptor recovery may take 8–12 weeks following resolution of the underlying stressors.
A minority of patients may continue to experience symptoms of mild norepinephrine or dopamine dysregulation, which could be influenced by genetic variations in catechol-O-methyltransferase (COMT) affecting neurotransmitter breakdown rates. In these cases, COMT gene testing may help guide treatment. Individuals with fast COMT variants may exhibit low dopamine and norepinephrine levels, whereas those with slow COMT variants may have excessive catecholamine activity. If neurotransmitter dysregulation persists after addressing all other components, targeted pharmacological interventions may be required. For patients with high extracellular norepinephrine, guanfacine (a selective α₂A-adrenergic receptor agonist) can help reduce sympathetic overactivity. In cases of low norepinephrine, duloxetine (a norepinephrine reuptake inhibitor) may be beneficial. If dopamine deficiency remains an issue, low-dose aripiprazole can provide partial agonist activity at dopamine receptors, improving neurotransmitter balance.
A key principle of this component-based approach is that treating one dysfunction in isolation may worsen other aspects of the disease. For example, supplementing with phosphatidylcholine without addressing underlying liver dysfunction could lead to excessive triglyceride production, increasing the risk of metabolic syndrome and insulin resistance. Likewise, increasing PC availability without correcting excessive PLA2 activation may inadvertently fuel prostaglandin overproduction, worsening inflammation and pain. Furthermore, attempts to correct norepinephrine dysregulation without addressing the root metabolic and inflammatory disturbances may lead to incomplete or short-lived symptom relief. Therefore, a comprehensive strategy that assesses and corrects all relevant components is critical to achieving meaningful clinical improvements. Future research should focus on refining this model through patient stratification and biomarker-driven treatment approaches, ensuring that interventions are tailored to individual pathophysiology.
References
- Arron HE, Marsh BD, Kell DB, Khan MA, Jaeger BR, Pretorius E. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: The Biology of a Neglected Disease. Front Immunol. 2024;15:1386607. [CrossRef]
- Walitt B, Singh K, LaMunion SR, Hallett M, Jacobson S, Chen K, et al. Deep Phenotyping of Post-Infectious Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Nat Commun. 2023; 15(1):907. [CrossRef]
- Nguyen CC, Kumar S, Zucknick M, Kristensen VN, Gjerstad J, Nilsen H, Wyller VB. Associations Between Clinical Symptoms, Plasma Norepinephrine, and Deregulated Immune Gene Networks in Subgroups of Adolescents with Chronic Fatigue Syndrome. Brain Behav Immun. 2019;76:82-96. [CrossRef]
- Kavelaars A, Kuis W, Knook LME, Sinnema G, Heijnen CJ. Disturbed Neuroendocrine-Immune Interactions in Chronic Fatigue Syndrome. J Clin Endocrinol Metab. 2000;85(2):692-696. [CrossRef]
- Fry AC, Schilling BK, Weiss LW, Chiu LZF. β2-Adrenergic Receptor Downregulation and Performance Decrements During High-Intensity Resistance Exercise Overtraining. J Appl Physiol. 2006;101(6):1664-72. [CrossRef]
- Goto T, Kikuchi S, Mori K, Nakayama T, Fukuta H, Seo Y, et al. Cardiac β-Adrenergic Receptor Downregulation, Evaluated by Cardiac PET, in Chronotropic Incompetence. J Nucl Med. 2021;62(7): 996–998. [CrossRef]
- Robertson SD, Matthies HJG, Owens WA, Sathananthan V, Christianson NSB, Kennedy JP, et al. Insulin Reveals Akt Signaling as a Novel Regulator of Norepinephrine Transporter Trafficking and Norepinephrine Homeostasis. J Neurosci. 2010;30(34):11305-16. [CrossRef]
- Das S, Taylor K, Kozubek J, Sardell J, Gardner S. Genetic Risk Factors for ME/CFS Identified Using Combinatorial Analysis. J Transl Med. 2022;20(1):598. [CrossRef]
- Taylor K, Pearson M, Das S, Sardell J, Chocian K, Gardner S. Genetic risk factors for severe and fatigue dominant long COVID and commonalities with ME/CFS identified by combinatorial analysis. J Transl Med. 2023;21(1):775. 2023. [CrossRef]
- Figlewicz DP, Szot P, Chavez M, Woods SC, Veith RC. Intraventricular insulin increases dopamine transporter mRNA in rat VTA/substantia nigra. Brain Res. 1994;644(2):331-4. [CrossRef]
- Sun Z, Fan Y, Zha Q, Zhu MY. Corticosterone up-regulates expression and function of norepinephrine transporter in SK-N-BE(2)C cells. J Neurochem. 2010;113(1): 105–116. [CrossRef]
- Saha AK, Schmidt BR, Wilhelmy J, Nguyen V, Abugherir A, Lechuga TJ, et al. Red blood cell deformability is diminished in patients with Chronic Fatigue Syndrome. Clin Hemorheol Microcirc. 2019;71(1):113–6. [CrossRef]
- da Silva ALG, Vieira LP, Dias LS, Prestes CV, Back GD, Goulart CL, et al. Impact of long COVID on heart rate variability at rest and during deep breathing maneuver. Sci Rep. 2023;13:22695. [CrossRef]
- Lee JS, Sato W, Son CG. Brain-regional characteristics and neuroinflammation in ME/CFS patients from neuroimaging: A systematic review and meta-analysis. Autoimmun. Rev. 2024;23:2. [CrossRef]
- Ibrahim MMH, Bheemanapally K, Briski KP. Norepinephrine regulation of adrenergic receptor expression, 5′ AMP-activated protein kinase activity, and glycogen metabolism and mass in male versus female hypothalamic primary astrocyte cultures. ASN Neuro. 2020;12:17. [CrossRef]
- Carbajal-García A, Reyes-García J, Casas-Hernández MF, Flores-Soto E, Díaz-Hernández V, Solís-Chagoyán H, Sommer B, Montaño LM. Testosterone augments β2 adrenergic receptor genomic transcription increasing salbutamol relaxation in airway smooth muscle. Mol. Cell. Endocrinol. 2020;510:110801. [CrossRef]
- Scardapane L, Cardinali DP. Effect of estradiol and testosterone on catechol-O-methyl transferase activity of rat superior cervical ganglion, pineal gland, anterior hypophysis and hypothalamus. J Neural Transm. 1977;40(1):81-6. [CrossRef]
- Boneva RS, Lin JM, Wieser F, Nater UM, Ditzen B, Taylor RN, Unger ER. Endometriosis as a comorbid condition in chronic fatigue syndrome (CFS): secondary analysis of data from a CFS case-control study. Front. Pediatr. 2019; 7: 195. [CrossRef]
- Mathias JR, Franklin RR. Neural dysfunction of the gastrointestinal tract associated with endometriosis: a disease of insulin sensitivity. Fertil Steril. 2002;77(25). [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).