
Submitted:
19 September 2024
Posted:
23 September 2024
You are already at the latest version
Abstract
Recently, the availability of many omics data source has given the rise of modelling biological networks for each individual or patient. Such networks are able to represent individual-specific characteristics, providing insights into the condition of each person. Given a set of networks of individuals, a network representing a particular condition (e.g., an individual with a specific disease) may be seen as an anomaly network. Consequently, the use of Graph Anomaly Detection techniques may support such analysis. Among the others, Generative Adversarial Networks present optimal per- formances in anomaly detection. This paper presents ADIN (Anomaly Detection in Individual Networks), a framework based on Generative Adversarial Attributed Networks (GAANs) for anomaly detection in convergence/divergence patients at- tributed networks. Preliminary results on networks generated from computational biology gene expression data demonstrate the effectiveness of our approach in detecting and explaining bladder cancer patients.
Keywords:
ISN
; GANN
; Patient
1. Introduction
Anomaly detection in network data is pivotal for numerous applications, ranging from security surveillance to system health monitoring [1,2]. The challenge intensifies when dealing with individual-specific networks where each network’s structure and pattern vary significantly from others. Traditional anomaly detection techniques, which typically rely on static thresholds or predefined metrics, need to catch up in such scenarios due to their inability to adapt to the unique characteristics of each network [1,3]. Recent advances in artificial intelligence have led to the development of Generative Adversarial Networks (GANs), which have shown promising results in learning complex data distributions. Leveraging GANs for anomaly detection involves training these models to generate network data that mimics the normal operational state of a specific network [4,5]. Once trained, these models can identify deviations from the learned normal behaviour as anomalies. This paper proposes the use of GANs to address the challenge of anomaly detection in convergence/divergence patients attributed networks. To address the challenge of graph anomaly detection using GANs we modified the known Generative Adversarial Attributed Network anomaly detection (GAAN) to extract explanations.
Our approach adapts the adversarial training methodology to tailor the model to the nuanced differences of each network, thereby enabling more precise detection of anomalies that are otherwise overlooked by conventional methods. The main contributions of this paper are as follows:
- We are the first to use a GAAN model to perform anomalous node (patients) detection on attributed convergence/divergence networks obtained from patients gene expression data;
- We compared results obtained with GAAN against a baseline of commonly used machine learning (ML) binary classifiers algorithms trained and tested using raw gene expression data;
- We used explainability to explain the predictions of both ML and GAAN models. Those are useful to understand the principal genes related with a given pathology;
- We developed an open-source freely available graphical user interface to reproduce our case studies results and to produce new ones using appropriate gene expression data formats;
- We demonstrate the effectiveness of our algorithm through experiments on real-world gene expression profiling datasets, comparing the obtained results with other machine learning (ML) methods.
The remainder of the paper is organized as follows: Section 2 discusses main related work; Section 3 presents the proposed framework and discusses the main characteristics; Section 4 shows the effectiveness of the framework; finally, Section 5 concludes the paper.
Section 2 introduces Material and Methods used for this experiment, for the Material point of view we report information on the gene expression dataset used and for the Methods we introduce the GAAN architecture and the convergence/divergence paradigm. Section 3 describes the experimental setup in detail, providing the model hyperparameters and results obtained. Finally, Section 4 concludes the paper and discusses potential directions for future research.
2. Related Work
3. The Proposed Framework
We developed an integrated program called ADIN (Anomaly Detection in Individual Networks) with the main objective of detecting anomalies in gene expression data using both deep and machine learning techniques and comparing the results obtained by the GAAN model. The program is designed to identify significant deviations in gene expression patterns by using both structural and attribute information from individualized coexpression networks [6]. This method allows for the detection of potential anomalies that could indicate underlying biological abnormalities.
The program is built upon a modular framework that incorporates data preprocessing, network construction, and anomaly detection components. The primary components are as follows:
- Data upload and preprocessing module: Starting from a GEO dataset, some preprocessing steps need to be applied to clean the dataset: remove duplicates, handle null gene expression values;
- ML binary classification module: Evaluates the performances of a baseline of commonly used algorithms for binary classification from gene expression data;
- Network Constructor: Generates an attributed network from gene expression data using the convergence/divergence network paradigm [7]. This is a submodule, integrated inside the DL graph based module;
- DL Anomaly Detection module: Evaluates the performance of the trained GAAN in the anomaly detection task and compares them against ML results. After training the GAAN model on "Normal" nodes, new nodes are tested, and those that significantly deviate from the learned normal patterns are flagged as "Anomalous".
Figure 1 depicts the framework of our program.
The workflow of the proposed experiment is divided into distinct stages and is depicted in Figure 2:
- Data preprocessing to handle null values;
- Training and evaluation of Machine Learning classifiers to build a baseline of classifiers algorithms used to confront our results. Those classifiers work directly on raw gene expression data and can classify each patient with two labels: normal or anomalous. Explainability of this proposed ML baseline is also made available to the user by using SHAP values;
- Convergence/Divergence network creation: Starting from the raw data we build a network of patients where each node is a patient, nodes attributes are their gene expression values while edges between two nodes are computed using their gene expression profile similarity;
- Training, evaluation, comparison and explainability of the deep learning GAAN model: We developed, trained and tested an Explainable GAAN model that takes in input this convergence/divergence network and performs a node-level binary classification by performing anomaly detection using all the information available in the graph structure: edges and node attributes.
To detect anomalies within an attributed convergence/divergence network, we used the Generative Adversarial Attributed Network (GAAN) [8] available in the PyGOD python library [9,10] for attributed network anomaly detection. This architecture consists of two key components: a Generator and a Discriminator, each designed to leverage both the network structure and the attributes of the nodes (gene expression values in our case).
The Generator produces synthetic node attributes that closely resemble the real data. It takes random noise as input, along with the observed network structure, and generates new node attribute vectors that mimic the distribution of real gene expression data. The goal of the Generator is to produce outputs that are indistinguishable from real ones, thereby attempting to "fool" the Discriminator.
The network structure input to the Generator is preserved, while the node attributes are modified in a way that reflects realistic gene expression profiles. The generated attributes are then reintegrated with the existing network structure to form a synthetic attributed network.
The Discriminator serves as a classifier that differentiates between real and synthetic networks. It takes as input both the node attributes and the network structure. The Discriminator is trained to assign a higher probability to real network and a lower probability to the synthetic one generated by the Generator.
The architecture of the Discriminator is designed to handle both the topological information (edges) and the attributed information (node features) simultaneously. By analyzing the correlation between the structure and attributes, the Discriminator can effectively distinguish between normal and anomalous nodes, as anomalies often present irregularities in either the structure or the node attributes.
The Generator and Discriminator are trained in an adversarial manner. During the training process, the Generator continually improves its ability to produce realistic networks by learning from the feedback provided by the Discriminator. Conversely, the Discriminator improves its accuracy in identifying real versus synthetic networks. The interplay between these two networks enhances the system’s ability to detect anomalies, as the Discriminator becomes more adept at spotting irregularities in both the gene expression data and the network structure.
After training, the Discriminator is utilized to detect anomalies in the convergence/divergence patient attributed network. It does this by evaluating the likelihood that a given networks (or its components) is real or synthetic. A network, or a component (node, node attributes, edges), that the Discriminator classifies as having a low probability of being real are flagged as anomalous. This approach allows for the identification of genes or interactions that deviate significantly from normal patterns, providing insight into potential biological abnormalities or disease mechanisms.
The general GAAN architecture is depicted in Figure 3.
3.1. Machine Learning Algorithms in ADIN
To evaluate the effectiveness of the deep learning graph-based GAAN model, we compared its performance against several traditional machine learning algorithms commonly used for binary classification. These algorithms were chosen for their relevance to anomaly detection in high-dimensional datasets such as gene expression profiles.
- Logistic Regression (LR), is a linear model, simple to implement and interpret, used for binary classification tasks. It predicts the probability that a given instance belongs to one of two classes by applying a logistic function to a linear combination of the input features. Effective for problems where classes are linearly separable;
- Naive Bayes (NB), is a probabilistic classifier that applies Bayes’ theorem with the assumption that features, in our case gene expressions, are conditionally independent given the class label. NB is computationally efficient and works well with high-dimensional data. Performs well when the independence assumption holds approximately true;
- Random Forest (RF) is an ensemble learning method that constructs a multitude of decision trees during training and outputs the class that is the mode of the classes predicted by individual trees. Can handle large datasets with high dimensionality, reduce over-fitting through ensemble averaging, and provide feature importance scores;
- Decision Tree (DT) is a non-parametric model that splits the dataset into subsets based on the most significant attribute at each node, forming a tree-like structure. It’s easy to visualize and interpret, works well on both categorical and continuous data, and requires only little data preprocessing;
- k-Nearest Neighbors (KNN) is a simple, instance-based learning algorithm that classifies a data point based on the majority class among its k nearest neighbors in the feature space. It’s easy to implement and doesn’t require a training phase. Effective for problems where the decision boundary is irregular;
- Support Vector Machine (SVM) is a powerful classifier that finds the hyperplane which best separates the classes in the feature space, often using a kernel trick to handle non-linear relationships. It’s effective in high-dimensional spaces and with clear margin of separation. Particularly useful for binary classification tasks;
- Linear Discriminant Analysis (LDA) is a linear classification technique that finds a linear combination of features that best separates two or more classes by maximizing the ratio of between-class variance to within-class variance. It’s well-suited for situations where classes are linearly separable. It also provides dimensionality reduction as part of the classification process.
3.2. Metrics for Model Evaluation
Let TP and TN be the number of patients (or nodes) correctly identified as normal or abnormal, respectively, and FP and FN the number of normal or abnormal patients (or nodes) misclassified, each ML or DL model has been evaluated in terms of the following performance metrics:
- ROC AUC is calculated as the Area Under the - curve, also known as the ROC curve.
4. Case Studies
4.1. Using ADIN to Detect Bladder Cancer from Gene Expression Data
4.1.1. Dataset
We used the Gene Expression Omnibus (GEO) data repository to access the freely available dataset from with accession code GSE37815. This dataset contains gene expression profiling microarray data obtained using GPL6102 Illumina human-6 v2.0 expression beadchip. Profiling was performed to differentiate between normal cells and from Non-Muscle-Invasive Bladder Cancer (NMIBC) and predict patient prognosis: Normal or Tumour (Anomalous). The dataset contains gene expression values of 27552 genes for 24 patients. Table 1 reports the distribution of the two diagnosis in the dataset, as we can see, this distribution is unbalanced: number of patients with cancer is three times higher than that of healthy control patients.
Gene expression values and clinical diagnosis information were first extracted from the GEO series matrix file. To address missing values, a class-dependent mode imputation strategy was applied. Specifically, for each gene, missing values were replaced by the mode expression value calculated separately for cancer patients and healthy controls, ensuring that imputation preserved the inherent class structure. Gene identifiers, such as those in the format "cg00000292" from platforms like GPL6102 (Illumina human-6 v2.0), were mapped to their corresponding gene names for better interpretability and to enable a more useful model explainability.
4.1.2. Results using Machine Learning on Raw Data
To train and test our machine learning baseline, the dataset was splitted into training and test sets using a 30/70 random split. More information on the train-test split are reported in Table 2.
The train set is then used to compare the performance of the previously mentioned machine learning models in the binary classification task. We used a 2-fold stratified cross validation technique to assess the performances. The trained models are then validated using the test set obtaining the binary classification performance metrics reported in Table 3.
Our framework uses the shapley values technique [11] available in the Shap python library [12] to obtain interpretable explainations from the predictions obtained by each machine learning algorithm. Using this approach, we can assess, for each patient, the contribution of individual genes to the model’s prediction by quantifying how much each gene expression value influenced the model prediction. This allows us to identify key genetic markers that drive the classification decision and provides a patient-specific breakdown of feature importance, facilitating a deeper understanding of the biological factors associated with the diagnosis. The Shapley values show the average contribution of each gene across all test samples, with genes ranked by their overall impact on the model’s predictions. Positive Shapley values indicate genes that consistently push the model towards a cancer diagnosis, while negative values indicate genes that drive the prediction towards a non-cancer classification. Figure 4 depicts three subfigures obtained by applying shapley values on the Random Forest predictions. Subfigure (A) displays a force plot illustrating the classification of a healthy patient, where the contribution of genes aligns with a non-cancer outcome. In contrast, subfigure (B) provides a force plot for the classification of an NMIBC patient, showing how specific genes strongly influence the model’s prediction towards a cancer diagnosis. Finally, subfigure (C) show the top 10 genes contributing to model predictions of the entire test set. This summary plot highlights the key genes that play a significant role in the model’s performance across the test cohort.
4.1.3. Results Using GAAN
We began by analyzing gene expression profiling data to construct convergence/divergence attributed network, where each node represents an individual patients. The attributes of these nodes correspond to the gene expression levels observed in the dataset. Each node was labelled as either "Normal" or "Anomalous" based on the patient diagnosis the dataset provides. To determine the connections (edges) between the nodes, we employed a correlation-based method, which calculates the pairwise correlation coefficients between the gene expression profiles of these patients. We used to
As we reported in Table 4, we splitted the dataset in two disjoint sets for model training and evaluation using a test set percentage of 80%. Different splitting in the train-test is required since GAAN is an anomaly detection tool, that needs to be trained using only "Normal" instances (graphs in our case) so it can be able to detect abnormalities during test.
5. Conclusions
In this study, we have shown the effectiveness of Generative Adversarial Attributed Networks (GAANs) for detecting anomalies in individual-specific networks (ISNs), especially in the context of identifying diseases using gene expression data. Our framework, ADIN (Anomaly Detection in Individual Networks), uses GAANs to identify subtle irregularities in biological network patterns that traditional methods might miss.
Our experimental results demonstrate that GAANs outperform conventional machine learning classifiers in identifying anomalous nodes in networks created from gene expression data. The success of the model is due to its ability to generate realistic network data and identify deviations from the norm, providing a robust method for anomaly detection.
Integrating explainability into the anomaly detection process improves the interpretability of our model, allowing us to pinpoint specific genetic markers associated with diseases. This capability is particularly valuable in medical research and diagnostics, as understanding the genetic basis of abnormalities can lead to more effective treatments.
Looking ahead, the development of an open-source graphical user interface for ADIN will provide a valuable tool for researchers and clinicians, promoting wider adoption and continuous improvement of anomaly detection techniques in genomics. Future work will focus on enhancing the model’s accuracy and computational efficiency, exploring its applicability to other types of omic data, and improving its scalability for larger datasets, which is critical for advancing personalized medicine and patient-specific treatments.
Code Availability
The software developed for this study is available at ... Gene expression data from the Gene Expression Omnibus GSE37815 series is used as a case study to validate and test this software. The full dataset and documentation can be downloaded from https://www.ncbi.nlm.nih.gov/geo/query /acc.cgi?acc=GSE37815.
Acknowledgments
UL Ph.D. fellow is partially funded by Relatech S.p.A.
PHG has been partially supported by the Next Generation EU - Italian NRRP, Mission 4, Component 2, Investment 1.5, call for the creation and strengthening of ’Innovation Ecosystems’, building ’Territorial R&D Leaders’ (Directorial Decree n. 2021/3277) - project Tech4You - Technologies for climate change adaptation and quality of life improvement, n. ECS0000009.
PV has been partially supported by project "SERICS"(PE00000014) under the MUR National Recovery and Resilience Plan funded by the European Union - NextGenerationEU.
References
- Zitnik, M.; Li, M.M.; Wells, A.; Glass, K.; Morselli Gysi, D.; Krishnan, A.; Murali, T.M.; Radivojac, P.; Roy, S.; Baudot, A.; Bozdag, S.; Chen, D.Z.; Cowen, L.; Devkota, K.; Gitter, A.; Gosline, S.J.C.; Gu, P.; Guzzi, P.H.; Huang, H.; Jiang, M.; Kesimoglu, Z.N.; Koyuturk, M.; Ma, J.; Pico, A.R.; Pržulj, N.; Przytycka, T.M.; Raphael, B.J.; Ritz, A.; Sharan, R.; Shen, Y.; Singh, M.; Slonim, D.K.; Tong, H.; Yang, X.H.; Yoon, B.J.; Yu, H.; Milenković, T. Current and future directions in network biology. Bioinformatics Advances 2024, 4, vbae099, [https://academic.oup.com/bioinformaticsadvances/article-pdf/4/1/vbae099/58811290/vbae099.pdf]. [CrossRef] [PubMed]
- Guzzi, P.H.; Lomoio, U.; Veltri, P. GTExVisualizer: a web platform for supporting ageing studies. Bioinformatics 2023, 39, btad303. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Milenković, T. Survey of local and global biological network alignment: the need to reconcile the two sides of the same coin. Briefings in bioinformatics 2018, 19, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Cannataro, M. μ-CS: An extension of the TM4 platform to manage Affymetrix binary data. BMC bioinformatics 2010, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Milano, M.; Guzzi, P.H.; Tymofieva, O.; Xu, D.; Hess, C.; Veltri, P.; Cannataro, M. An extensive assessment of network alignment algorithms for comparison of brain connectomes. BMC bioinformatics 2017, 18, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Latapiat, V.; Saez, M.; Pedroso, I.; Martin, A.J.M. Unraveling patient heterogeneity in complex diseases through individualized co-expression networks: a perspective. Frontiers in Genetics 2023, 14. [Google Scholar] [CrossRef]
- Zanin, M.; Tuñas, J.M.; Menasalvas, E. Understanding diseases as increased heterogeneity: a complex network computational framework. Journal of The Royal Society Interface 2018, 15, 20180405, [https://royalsocietypublishing.org/doi/pdf/10.1098/rsif.2018.0405]. [CrossRef] [PubMed]
- Chen, Z.; Liu, B.; Wang, M.; Dai, P.; Lv, J.; Bo, L. Generative Adversarial Attributed Network Anomaly Detection. Proceedings of the 29thACMInternational Conference on Information&Knowledge Management; Association for Computing Machinery: New York, NY, USA, 2020; CIKM ’20, p. 1989–1992. [CrossRef]
- Liu, K.; Dou, Y.; Ding, X.; Hu, X.; Zhang, R.; Peng, H.; Sun, L.; Yu, P.S. PyGOD: A Python Library for Graph Outlier Detection. Journal of Machine Learning Research 2024, 25, 1–9. [Google Scholar]
- Liu, K.; Dou, Y.; Zhao, Y.; Ding, X.; Hu, X.; Zhang, R.; Ding, K.; Chen, C.; Peng, H.; Shu, K.; Sun, L.; Li, J.; Chen, G.H.; Jia, Z.; Yu, P.S. BOND: Benchmarking Unsupervised Outlier Node Detection on Static Attributed Graphs. Advances in Neural Information Processing Systems; Koyejo, S.; Mohamed, S.; Agarwal, A.; Belgrave, D.; Cho, K.; Oh, A., Eds. Curran Associates, Inc., 2022, Vol. 35, pp. 27021–27035.
- Rozemberczki, B.; Watson, L.; Bayer, P.; Yang, H.T.; Kiss, O.; Nilsson, S.; Sarkar, R. The Shapley Value in Machine Learning, 2022. [CrossRef]
- Lundberg, S.M.; Lee, S.I. A Unified Approach to Interpreting Model Predictions. In Advances in Neural Information Processing Systems 30; Guyon, I.; Luxburg, U.V.; Bengio, S.; Wallach, H.; Fergus, R.; Vishwanathan, S.; Garnett, R., Eds.; Curran Associates, Inc., 2017; pp. 4765–4774.
Figure 1.
The Figure summarises the main software module sof the ADIN framework. A Gene Data Processing module is responsible for reading and the gene expression data for further processing in Network Data Processing and Data Analysis. The former is responsible for creating the convergence/divergence attributed network from gene expression data, while the latter implements all the analysis. The explainability module implements provides explanation of the artificial intelligence algorithms contained in data analysis, while visualization offer visualization capabilities.
Figure 1.
The Figure summarises the main software module sof the ADIN framework. A Gene Data Processing module is responsible for reading and the gene expression data for further processing in Network Data Processing and Data Analysis. The former is responsible for creating the convergence/divergence attributed network from gene expression data, while the latter implements all the analysis. The explainability module implements provides explanation of the artificial intelligence algorithms contained in data analysis, while visualization offer visualization capabilities.
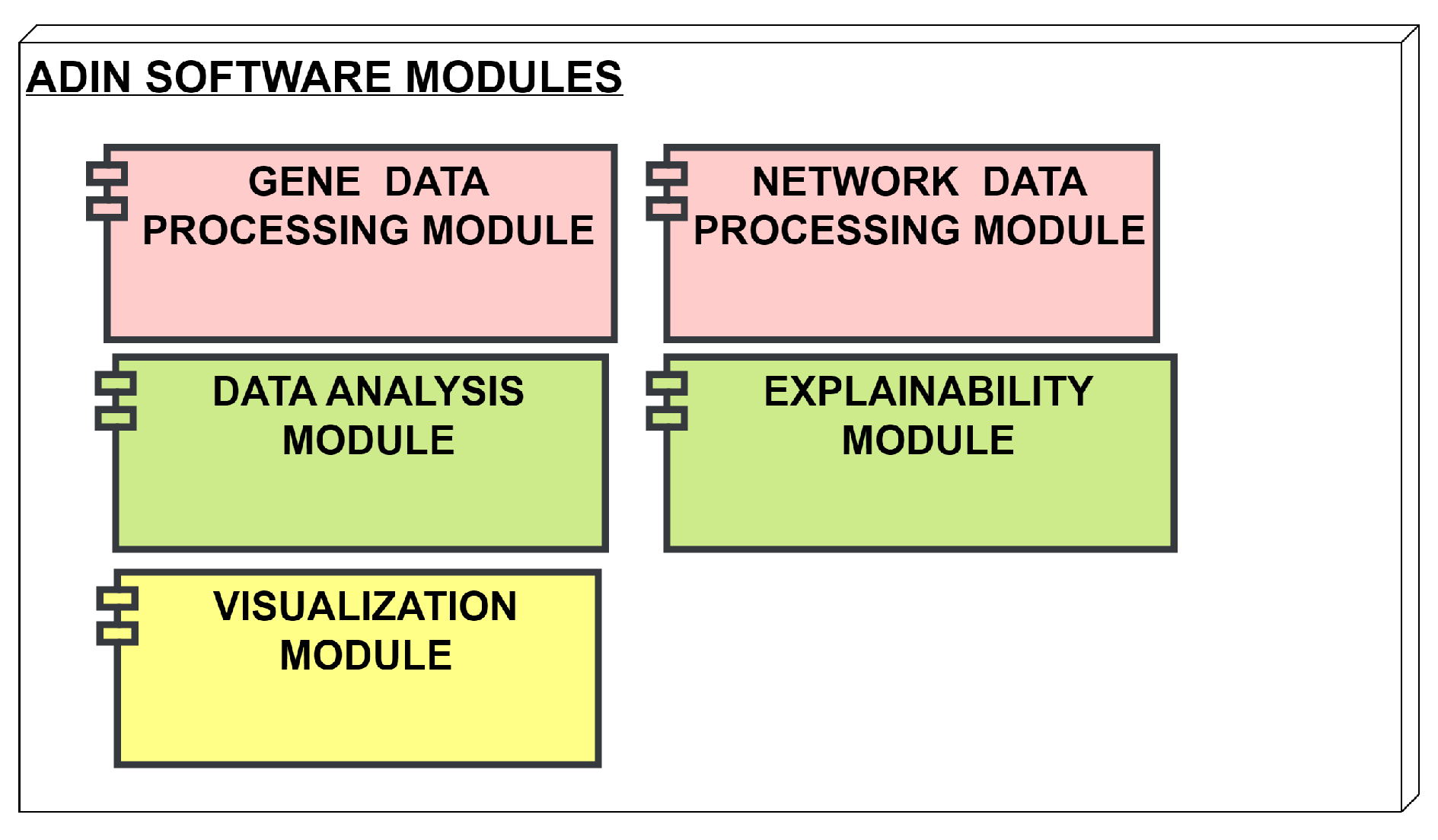
Figure 2.
The Figure depicts a general ADIN experiment workflow: we start with a gene expression file from the GEO platform, then we apply preprocessing. We can choose between an ML or DL based analysis and then compare the results and extract model explanations.
Figure 2.
The Figure depicts a general ADIN experiment workflow: we start with a gene expression file from the GEO platform, then we apply preprocessing. We can choose between an ML or DL based analysis and then compare the results and extract model explanations.
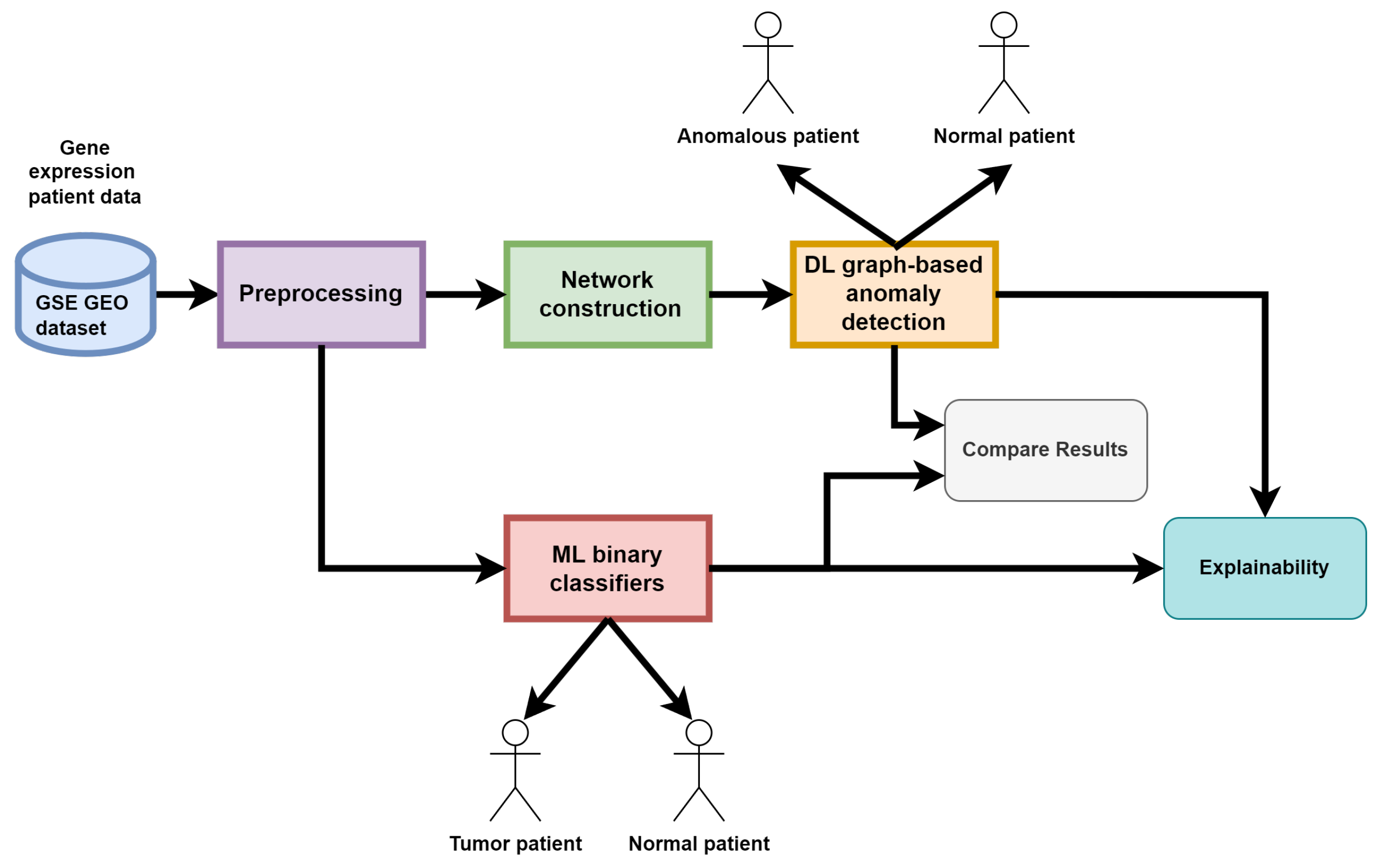
Figure 3.
GAAN architecture.
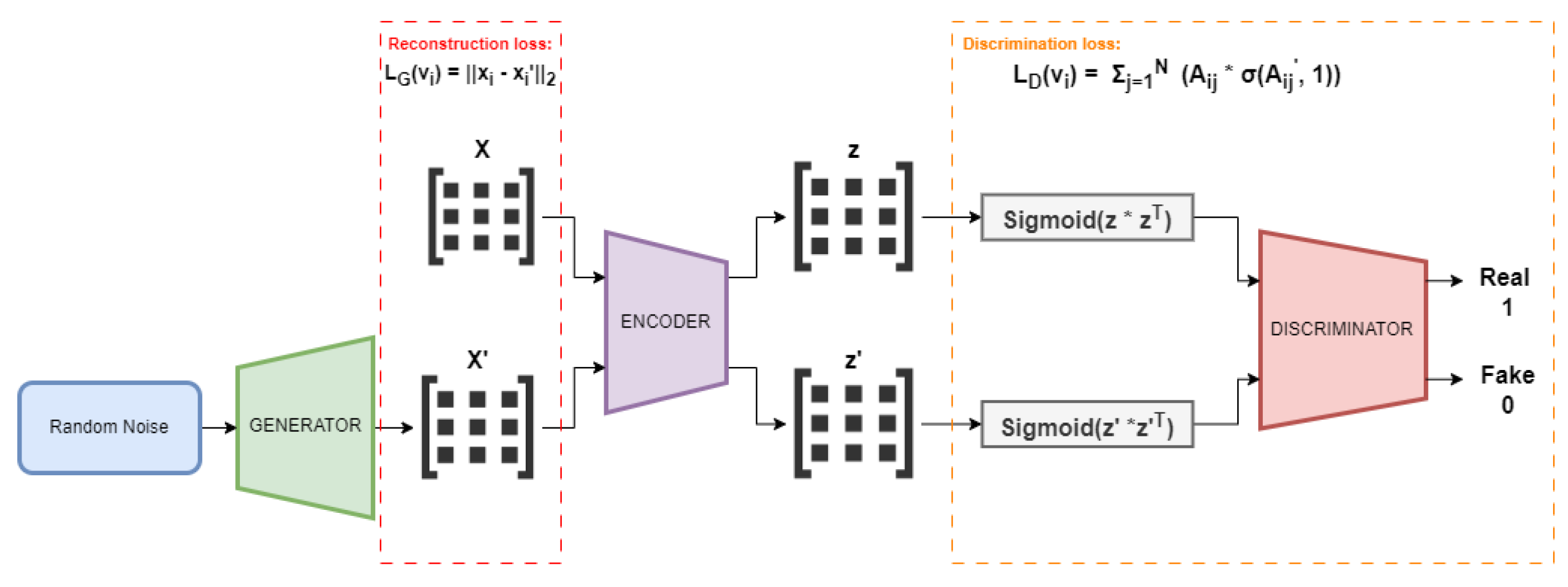
Figure 4.
Shap plots obtained using the random forest model: (A) force plot for the classification of an Healthy Patient; (B) force plot for the classification of an NMIBC Patient; (C) Top 10 genes ranked by their shapley value for the classification of the whole test set.
Figure 4.
Shap plots obtained using the random forest model: (A) force plot for the classification of an Healthy Patient; (B) force plot for the classification of an NMIBC Patient; (C) Top 10 genes ranked by their shapley value for the classification of the whole test set.


Table 1.
Dataset label distribution.
| Label | Count |
|---|---|
| Normal patient | 6 |
| Tumour NMIBC patient | 18 |
Table 2.
Dataset Train and Test split for the machine learning binary classification problem.
| Set | Count Normal | Count Anomalous |
|---|---|---|
| Train | 2 | 5 |
| Test | 4 | 11 |
Table 3.
Machine Learning algorithms performance on the test set
| Model | Acc | f1 | Sens. | Spec. | AUC | Prec |
| LR | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| KNN | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| LDA | 0.882 | 0.929 | 1.00 | 0.50 | 0.75 | 0.867 |
| DT | 0.882 | 0.929 | 1.00 | 0.50 | 0.75 | 0.867 |
| RF | 0.882 | 0.929 | 1.00 | 0.50 | 0.75 | 0.867 |
| NB | 0.765 | 0.867 | 1.00 | 0.00 | 0.50 | 0.765 |
| SVM | 0.765 | 0.867 | 1.00 | 0.00 | 0.50 | 0.765 |
Table 4.
Machine Learning algorithms performance on the test set
| Set | Count Normal | Count Anomalous |
|---|---|---|
| Train | 4 | 0 |
| Test | 2 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



