Submitted:
20 September 2024
Posted:
20 September 2024
You are already at the latest version
Abstract
Chronic Myeloid Leukemia (CML) is one of the most commonly found types of myeloproliferative neoplasms, characterized by increased proliferation of granulocytic cells without losing their differentiation ability. Imatinib, a Tyrosine Kinase Inhibitor (TKI), can be effectively used as therapy for CML. However, imatinib can affect bone turnover, thus having clinical implications on the bones of CML patients undergoing long-term imatinib therapy. However, parameters that can accurately describe the bone condition in CML patients receiving imatinib still need further study. A combination of imaging techniques such as Bone Mineral Density (BMD) and bone turnover activity markers such as C-terminal telopeptide of type I collagen (CTX-1) and Osteocalcin has the potential to be used as monitoring parameters for bone density abnormalities in CML patients receiving imatinib. This article explains the rationale for using BMD, CTX-1, and Osteocalcin as monitoring parameters of bone remodeling in CML patients receiving imatinib. First, the physiological process of bone turnover will be explained. Then, we describe the role of tyrosine kinase in bone metabolism. Next, the impact of imatinib on BMD, CTX-1, and Osteocalcin will be explained. In conclusion, the assessment of bone health of CML patients on Imatinib should include both BMD tests and bone turnover marker assays such as CTX-1 and Osteocalcin.
Keywords:
Chronic Myeloid Leukemia
; Bone Mineral Density
; Bone Turnover
; Tyrosine Kinase Inhibitor
; CTX-1
; Osteocalcin
; Imatinib
1. Introduction
Chronic myelogenous leukemia (CML) is linked to the Philadelphia chromosome, which results from a reciprocal translocation between chromosomes 9 and 22, producing the BCR-ABL chimeric protein with uncontrolled tyrosine kinase activity. This myeloproliferative disorder is characterized by the excessive accumulation of seemingly normal myeloid cells in the bone marrow and peripheral blood. In addition to this myeloid cell buildup, the bone marrow microenvironment in CML also experiences significant changes, such as increased levels of type III collagen (reticulin fibrosis) and enhanced angiogenesis [1].
Protein kinase activity is dysregulated across all cancer types. Imatinib, a tyrosine kinase inhibitor (TKI) that targets ABL, BCR-ABL, PDGFRA, and c-KIT, is widely used in treating chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST). While Imatinib is typically well-tolerated, its side effects, particularly its impact on bone health, require careful consideration [2]. In a study by Berman et al., it was found that osteocalcin (OCN) levels, an indicator of bone formation by osteoblasts, were reduced in 95% of patients receiving long-term Imatinib therapy, with 37% showing undetectable OCN levels, suggesting diminished bone formation in these individuals. Despite this, only 47% of patients exhibited a decrease in bone mineral density (BMD), while 21% showed an increase, and 32% remained unchanged. The study also identified hypophosphatemia and hyperphosphaturia in 26% of patients, a reduction in serum calcium in 84%, and elevated parathyroid hormone (PTH) levels in 42% of patients. These measurements, including OCN, phosphate, calcium, and PTH levels, were taken during the first two years of Imatinib therapy [3].
Bone density changes in CML patients are typically assessed by measuring bone mineral density (BMD). However, BMD is limited in that it only detects bone disturbances after a significant reduction in bone volume has occurred. In contrast, bone turnover markers (BTMs) can identify abnormalities in bone turnover before changes in BMD become apparent and even independently of BMD variations [4]. A systematic review by Shetty et al. highlighted that BTM measurements are reliable as complementary tools for evaluating microarchitectural changes affecting bone quality, detecting fracture risk, and managing bone density disorders [4]. BTMs are broadly categorized into markers of bone formation—such as osteocalcin (OCN), procollagen type I N-terminal propeptide (PINP), and serum alkaline phosphatase (ALP)—and markers of bone resorption, which include collagen degradation products like collagen type I telopeptides (CTX-1, NTX-1), bone sialoprotein, osteoclastic enzymes, and osteocyte activity markers[5].
Imatinib can influence bone turnover, resulting in clinical effects on the bone health of CML patients undergoing long-term therapy. Detecting changes in bone turnover requires non-invasive assessments, including biochemical markers like CTX-1 and Osteocalcin, as well as radiological evaluations of bone mineral density (BMD). This article aims to elucidate the rationale for utilizing BMD, CTX-1, and Osteocalcin as monitoring parameters for bone remodeling in CML patients treated with Imatinib. It will first outline the physiological processes of bone turnover, followed by a discussion on the role of tyrosine kinase in bone metabolism and its effect on serum and phosphate levels in CML patients. Subsequently, the article will explore the impact of Imatinib on BMD, CTX-1, and Osteocalcin.
2. Physiology of Bone Turnover
Bone is a metabolically active structure that undergoes continuous remodeling throughout life. After reaching peak bone mass, bones are consistently remodeled through a process involving bone resorption followed by bone formation within basic multicellular units, known as "bone remodeling units." Under optimal physiological conditions, bone resorption takes approximately 10 days, while bone formation requires around 3 months. Each year, up to 20% of the entire skeleton is replaced through this remodeling process. [4,6,7].
The continuous repair process preserves the functional integrity and strength of the adult skeleton through cycles of bone remodeling. Four cell types are involved in bone modeling and remodeling: chondrocytes, osteoblasts, osteocytes, and osteoclasts. Chondrocytes are the initial cell type to emerge during development. In early embryogenesis, mesenchymal precursors create a mold and differentiate into chondrocytes, which proliferate and excrete a matrix containing elastin and type II collagen to form a cartilaginous analog of the bone [6,7].
Osteoblasts constitute 4-6% of bone cells. They originate from multipotent mesenchymal stem cells, which have the potential to differentiate into chondrocytes, osteoblasts, or adipocytes. Osteoblasts typically have a lifespan of about two weeks, except for those that become bone-lining cells or osteocytes. Under normal physiological conditions, osteoblasts produce type I collagen fibers, which make up 90% of the organic matrix of bone. Additionally, osteoblasts secrete proteins that regulate the assembly of collagen fibers and facilitate mineralization.[8] Osteocytes make up 90% to 95% of bone cells and originate from osteoblasts that become embedded within the bone matrix. Serving as mechanosensors, osteocytes regulate bone modeling and remodeling by controlling osteoclasts through the RANKL/RANK pathway and influencing osteoblasts via Wnt signaling modulation. Additionally, osteocytes play a critical role in regulating phosphate homeostasis [9].
Osteoclasts comprise only 1% to 2% of bone cells, responsible for resorbing bone matrix and minerals. They attach to the bone surface requiring resorption and form a sealing zone where the osteoclast polarizes into a ruffled border and a basolateral membrane. Carbonic anhydrase II (a specific osteoclast pump) and chloride channels create an acidic environment within the resorption lacunae, leading to the dissolution of hydroxyapatite to release calcium and phosphate ions. Simultaneously, secreted cysteine proteases (such as cathepsin K) digest the organic components of the bone[10].
The bone remodeling cycle, influenced by the hormone parathyroid hormone (PTH), consists of a catabolic phase (bone resorption) and an anabolic phase (new bone formation). This ongoing repair process maintains the functional integrity and strength of the adult skeleton through the bone remodeling cycle. The basic multicellular unit involved in bone remodeling comprises osteoclasts and osteoblasts, whose activities are regulated by osteocytes. This dynamic interaction ensures the continuous renewal and maintenance of bone tissue, critical for skeletal health and function[11].
Under basal conditions, osteocytes secrete TGF-β and sclerostin, which inhibit osteoclastogenesis and the formation of bone by Wnt-activated osteoblasts[12]. Increased mechanical loading or local micro-damage results in a decrease in local TGF-β levels and activates bone cells to recruit osteoclast progenitors[13]. Osteocytes and bone-lining cells express macrophage colony-stimulating factor (M-CSF) and RANKL, both essential for osteoclastogenesis[14]. RANKL induces changes in osteoclast migration, survival, attachment to the bone surface, and the formation of the sealing zone. Additionally, osteoblasts and bone marrow stromal cells express osteoprotegerin (OPG), which inhibits the RANK-RANKL signaling pathway[15]. The ratio of RANKL to OPG is crucial in determining the differentiation and activity of osteoclasts. Local cytokines and systemic hormones that regulate bone remodeling include TNF-α, IL-1, prostaglandin E2, estrogen, parathyroid hormone (PTH), and glucocorticoids. The resorption phase lasts for 30-40 days. Paracrine signals released from the degraded matrix recruit osteoblasts, who initiate bone formation over the subsequent 150 days. Osteoblasts secrete and mineralize new bone matrix (osteoid) to fill the resorption cavities. During the bone formation process, some osteoblasts become embedded in the newly formed bone and undergo terminal differentiation into osteocytes. The secretion of sclerostin and other inhibitors by these osteocytes halts bone formation, returning to a quiescent state where osteoblasts become bone-lining cells. [6,7]
3. Interaction of Bone Mineral Density, CTX-1 and Osteocalcin
3.1. Bone Mineral Density
BMD (Bone Mineral Density) testing is crucial for diagnosing osteoporosis, assessing future fracture risk, and monitoring treatment effectiveness[16]. Trabecular bone exhibits a higher metabolic rate than cortical bone, making regions with a high trabecular-to-cortical ratio the ideal sites for BMD measurements. These areas, known as regions of interest, typically include the axial skeleton, which is preferred for screening due to significant changes in trabecular density. However, other sites like the forearm, tibia, and calcaneus are also suitable for BMD analysis [17].
DEXA (Dual-Energy X-ray Absorptiometry) is the preferred method for assessing BMD due to its efficiency, characterized by high precision, quick execution, and minimal radiation exposure. It is commonly used to measure BMD at key sites like the lumbar spine and femur and provides absolute aBMD measurements (g/cm2) which are essential for consistent tracking and comparison across studies[18]. Despite these advantages, DEXA's major limitation is the potential for measurement inconsistencies, with variability that can reach up to 20% between scans. [17]
A BMD T-score of ≤–2.5 at the hip defines osteoporosis, while a T-score between -1 and -2.5 defines osteopenia. Additionally, patients with fragility fractures are classified as having osteoporosis regardless of their T-score[19]. Factors that can affect DEXA BMD results include significant weight changes, absolute body size, and measurement variances specific to the DEXA machine used. The variability inherent in DEXA machines means that significant changes in BMD must exceed 0.055 and 0.045 g/cm^2 at the spine and hip, respectively, corresponding to about a 4–7% change in serial BMD measurements, depending on the initial BMD values[16].
3.2. The C-Terminal Telopeptide of Type I Collagen
The C-terminal telopeptide of type I collagen (CTX-1) is a degradation product of Type 1 collagen generated by the enzymatic activity of cathepsin K. CTX exists in two forms: the α and β isomerized forms. These isomerized forms undergo further isomerization to form the D and L forms. Spontaneous β isomerization of the α isoform occurs with protein aging. Therefore, changes in the ratio of the α and β isomer forms occur with new bone formation in physiological conditions such as childhood growth and pathological conditions such as malignant bone diseases [20].
The International Osteoporosis Foundation and the International Federation of Clinical Chemistry and Laboratory Medicine have proposed serum CTX-1 as a reference marker for bone resorption to assess fracture risk and monitor therapy[4]. Serum CTX-1 levels (a marker of osteoclastic activity) have been shown to inversely correlate with the efficacy of drugs in suppressing bone resorption. CTX-1 levels in patients with fragility fractures were found to be higher in those with persistent treatment, supporting the use of CTX-1 for monitoring bone resorption suppression[21]. Another study by Bjerre-Bastos et al. successfully demonstrated the usefulness of CTX-1 and CTX-2 in predicting the need for total joint replacement in cases of osteoarthritis [22].The main issue with measuring CTX is its circadian variation, with a peak in the early morning (around 5:00 AM) and a nadir in the afternoon (around 2:00 PM). Additionally, food intake affects CTX levels, with postprandial levels being 20% lower than fasting levels. Therefore, CTX-1 sampling is recommended in the morning after an overnight fast[4].
3.3. Osteocalcin
Osteocalcin (OCN) is a hydroxyapatite-binding protein synthesized exclusively by osteoblasts, odontoblasts, and hypertrophic chondrocytes, serving as a marker of osteoblastic activity and bone formation. OCN constitutes 15% of the non-collagenous bone matrix and is indicative of bone formation. OCN undergoes post-translational modifications, one of which involves the carboxylation of specific amino acid residues. The carboxylated form of OCN is known as carboxylated OCN (cOCN) and is found in bone, while the uncarboxylated form is referred to as undercarboxylated OCN (ucOCN or n-mid osteocalcin) and is present in circulation. N-mid osteocalcin specifically refers to the N-terminal or amino-terminal portion of the OCN molecule. This region is prone to cleavage during the carboxylation process, and the resulting fragments, including N-mid osteocalcin, can be used as markers to assess bone turnover[23,24].
The use of OCN as a biomarker for bone formation offers advantages such as tissue specificity, wide availability, and low variability. Serum OCN as a bone remodeling biomarker can be useful in assessing osteoporosis and predicting fracture risk in elderly individuals, particularly in women [25]. However, the utility of OCN is limited by its short half-life, instability of the intact molecule, and the influence of vitamin K status, renal function, and circadian rhythm[26]. Additionally, samples for OCN measurement have specific collection and transportation requirements due to OCN's instability. It is recommended that samples be stored at approximately 4°C and processed within 4 hours of collection. Consistent hemolysis of samples can reduce OCN levels, likely by increasing OCN degradation. Due to this instability, largely caused by the labile C-terminal sequence of 6 amino acids, the measurement of the N-Mid-OCN fragment (amino acids 1–43) has shown good clinical utility [23,27].
3.4 Interplay of BMD, CTX-1 and OCN
The relationship between BMD, CTX-1, and OCN involves complex interactions within the bone remodeling process (Figure 1). Osteoblasts and osteoclasts are key cells in bone remodeling, with osteoblasts responsible for bone formation and osteoclasts for bone resorption. Bone Mineral Density (BMD) is a crucial measure that reflects the balance between bone formation and resorption, processes primarily regulated by osteoblasts and osteoclasts. BMD increases when bone formation by osteoblasts surpasses bone resorption by osteoclasts, leading to stronger bones. Conversely, when resorption outpaces formation, BMD decreases, indicating bone loss[28].
Furthermore, CTX-1 is a well-established marker of bone resorption. It is released into the bloodstream during the process of bone remodeling when osteoclasts break down the C-terminal region of type I collagen, which is the most abundant protein in bone. CTX-1 levels in the serum are indicative of osteoclastic activity and can be used to monitor the effectiveness of treatments. The specificity of CTX-1 as a marker comes from its release during the cathepsin K-mediated degradation of bone collagen, distinguishing it from other resorption markers that may be influenced by different metabolic pathways[29,30,31].
On the other hand, OCN is a protein produced by osteoblasts during bone formation and is widely recognized as a marker of bone turnover. Specifically, the undercarboxylated form of osteocalcin, known as N-mid OCN, is released into the bloodstream and plays a crucial role in reflecting the rate of bone turnover. This marker is significant because it not only provides insights into bone metabolism but also interacts with various signaling pathways, including those related to energy metabolism and insulin sensitivity[32]. The endocrine functions of OCN have been linked to metabolic processes such as glucose regulation and fat metabolism, highlighting its broader role beyond bone health[33,34,35]. The measurement of N-mid OCN in clinical settings can therefore provide valuable information about both bone health and metabolic functions, making it a useful biomarker in the assessment of conditions like osteoporosis and metabolic syndromes.
4. Role of Tyrosine Kinase in Bone Metabolism
All types of malignancies can cause disruptions in kinase signaling. Tyrosine-protein kinases catalyze the phosphorylation of specific tyrosine residues and are key regulators of signaling pathways that involve cell proliferation, differentiation, and apoptosis. These proteins exhibit constitutive tyrosine kinase activity, which stimulates hematopoietic transformation and myeloproliferative. The dominant isoform, BCR-Abl, is a protein found in over 90% of patients with Chronic Myelogenous Leukemia (CML). This protein's unregulated kinase activity is central to the pathogenesis of CML, driving the aberrant growth and survival of leukemic cells [36].BCR-Abl is identified in the Philadelphia chromosome, which is a hallmark of Chronic Myelogenous Leukemia (CML). The chromosomal translocation between chromosomes 9 and 22 results in the formation of the BCR-Abl tyrosine kinase, which constitutively activates Abl kinase through the BCR promoter. This leads to continuous proliferative signaling. This aberrant kinase activity disrupts normal cellular control mechanisms, driving the unchecked proliferation of myeloid cells characteristic of CML. [37].
Recent research has shown that tyrosine kinases play a very important role in the regulation of bone metabolism, specifically osteoblasts and osteoclasts that are responsible for bone formation as well as bone resorption respectively. Among these kinase families, Src family is particularly prominent. The function of osteoclasts is dependent on the activity of Src kinase while it also inhibits bone synthesis by osteoblasts. Osteoporosis develops in mice that lack Src due to impaired function of osteoclasts and increased bone formation indicating dual effect of Src on resorption promotion and formation inhibition[38]. Another crucial one is MerTK kinase which was found to negatively regulate β-catenin and Smad signaling pathways during the early stages of bone development, whereas also suppressing osteoclastogenesis through an elevated OPG/RANKL ratio. This results to increase in bone volume but reduction in bone resorption in Mertk knockouts hence suggesting therapeutic potential for targeting MerTK for treatment of such lytic conditions like osteoporosis [39]. Moreover, non-receptor tyrosine kinase Matk, which inhibits Src, modulates osteoclast and osteoblast activities independently and through c-Src,[40]. Therefore, tyrosine kinases can be considered as regulators or controllers of bones mass.
5. Impact of Tyrosine Kinase Inhibitors to Bone Turnover
Since tyrosine kinase plays a significant role as regulators of bone mass, tirosine kinase inhibitors (TKI) consequently affact bone remodelling process. Research found that Imatinib inhibits MSC proliferation from which osteoblasts are originated[41]. Dasatinib appears to direct differentiation away from the adipocyte lineage and toward the osteoblast lineage[42]. VEGF inhibitors (sunitinib and cabozantinib) and RET inhibitors (vandetanib) have been shown to interfere with the osteoblast differentiation process[43,44]. Fully differentiated osteoblasts are known as osteocytes and, along with immature osteoblasts, secrete RANKL, the primary driver of osteoclast differentiation and activity[45]. However, the effects of TKIs on osteocytes are largely unknown[46].
TKIs can cause an increase in parathyroid hormone (PTH) levels. Several studies have reported that TKIs, particularly those like imatinib, dasatinib, and sunitinib, may lead to secondary hyperparathyroidism[46]. This condition occurs because TKIs can reduce calcium levels by inhibiting osteoclast activity, which decreases bone resorption and subsequently lowers serum calcium. The body responds to this drop in calcium by increasing the secretion of PTH to maintain calcium homeostasis[47]. A tonic increase in PTH leads to increased RANKL production, which is expected to result in increased osteoclast differentiation and activity[48]. However, despite hyperparathyroidism, some TKIs have been found to inhibit osteoclast activity[49]. Osteoclasts originate from hematopoietic stem cells in the bone marrow and are stimulated by factors including RANKL to differentiate into mature multinucleated cells. Src kinase has been shown to enhance osteoclast differentiation and activity; dasatinib is a potent Src kinase inhibitor[50]. M-CSF-induced pre-osteoclast differentiation into mature osteoclasts has been shown to be inhibited by sunitinib and imatinib[51,52].
6. Impact of Imatinib to BMD, CTX-1 and Osteocalcin
Recent studies have indicated changes in bone and mineral metabolism in patients receiving imatinib, with secondary hyperparathyroidism and decreased bone remodeling observed during long-term imatinib treatment. In vitro studies have revealed that imatinib enhances bone formation in CML patients [36,53,54]. A longitudinal study on 17 CML patients found that serum parathyroid hormone levels increased over 4 years, with 7 out of 17 patients developing secondary hyperparathyroidism. However, mean areal and volumetric BMD remained stable and even higher in the cortical compartment compared to controls[55].
Imatinib is thought to affect bone mineral metabolism through the inhibition of other tyrosine kinases present in bone cells, including PDGFRa and PDGFRb, C-kit, and the C-fms receptor on monocytes and macrophages[37]. Dib et al. suggested that imatinib may influence mature osteoclasts through c-fms inhibition, and imatinib may have clinical value in treating diseases where bone destruction occurs due to excessive M-CSF production, such as osteoporosis, inflammation-induced osteolysis, and tumor-induced osteolysis[52]. A study by Dewar et al. in mice also found that imatinib might be an effective antiosteolytic agent. Imatinib inhibits osteoclast formation and activity in vivo. The inhibition of osteoclast differentiation is likely due to the inhibition of c-fms signaling, while the inhibition of osteoclast function occurs indirectly through decreased RANK expression[56].
On the other hand, studies in mice have shown narrowing of the growth plate in the proximal tibia of animals treated with imatinib. Imatinib has antiresorptive effects on osteoclasts that interfere with the length of tubular bones, particularly in prepubertal animals [36]. A study by O'Sullivan found that imatinib does not affect BMD in the long term. This aligns with recent studies in healthy mice where imatinib administration did not increase BMD and did not alter biochemical markers of bone resorption. Although imatinib promotes early osteoblast differentiation, it also reduces mineralization, and this effect is more pronounced at low imatinib concentrations. The effect of imatinib on osteoblast differentiation also depends on the cell's maturation stage. Therefore, it appears that CML patients initially experience increased bone formation with imatinib therapy, which does not persist after the first 1 or 2 years of treatment [57,58]. This was evidenced in a study by Choeyprasert et al. on 6 CML children, which found a correlation with low BMD without affecting bone parameters, with a high prevalence of vitamin D deficiency [59].
Several studies have found a direct correlation between imatinib use and the bone turnover markers CTX-1 and N-mid OCN. Jaeger et al. found that in 17 children with CML undergoing prolonged imatinib therapy, serum CTX-I levels were above the normal range in 57% of patients, and N-mid OCN levels showed a significant linear decrease of -0.30 µg per week (p=0.04)[60]. Tauer also observed a significant decrease in N-mid OCN in mice treated with imatinib for 10 weeks, although no difference was found in CTX-1 levels [61]. We summarize the results of the studies in Table 1.
7. Rationale for Combination of BMD, Osteocalcin and CTX-1 to Monitor Bone Remodelling
BMD examination with DEXA is the gold standard for diagnosing osteoporosis and monitoring treatment. However, changes in BMD are slow and require at least ≥1 year to assess treatment efficacy. Although BMD measured by DEXA is used to diagnose osteoporosis according to WHO recommendations, bone quality also determines bone strength and fracture risk. Furthermore, some clinical trials have shown that the reduction in fracture risk associated with anti-resorptive therapy can occur independently of changes in BMD. Therefore, for a complete assessment of bone strength, BMD should be combined with an evaluation of bone quality. One important contributor to bone strength is the rate of bone remodeling, which can be assessed by measuring bone turnover markers (BTM) [62].
Bone turnover markers (BTM) are a series of proteins or protein-derived biomarkers released during bone remodeling by osteoblasts or osteoclasts. BTM can provide prognostic information regarding fracture risk that complements radiographic measurements of bone mass. However, testing using BTM must consider a large number of preanalytical factors and comorbid clinical conditions that affect BTM levels. BTM responds quickly to changes in bone physiology, making it useful for determining patient response and adherence to osteoporosis therapy[27].
Osteoclasts secrete TRAP and Cathepsin K, which metabolize collagen I into DPD, PYR, NTX, and CTX metabolites. Meanwhile, osteoblasts produce BAP, pro-collagen, and OCN. Therefore, both can be used as markers of bone formation and resorption, or what is known as bone turnover markers. Wu et al. stated that in relation to the low adherence rates to osteoporosis treatment, experts reached a consensus on the use of BTM to increase awareness and short-term monitoring of osteoporosis treatment in the Asia-Pacific region [62]. The experts support the use of BTM, particularly CTX-1 and PINP, as short-term monitoring tools to help physicians assess responses to osteoporosis therapy and make early adjustments to treatment regimens before BMD examinations. The absolute value or rate of change from baseline BTM can be used to monitor the efficacy of osteoporosis therapy [62]. A systematic review by Hong et al. also found that changes in OCN are useful in evaluating long-term changes in BMD after anti-osteoporosis drug interventions [63].
8. Conclusions
In the case of CML patients on Imatinib the assessment of bone health should include both BMD tests and bone turnover marker assays such as CTX-1 and Osteocalcin as they help in determining bone modeled changes earlier than BMD. Imatinib and other TKIs changes in the activity of both osteoclast and osteoblast cells and therefore the risk for bone side effects should be managed properly. All in all, a thorough strategy towards the follow-up of bone health is important so as to improve the management of CML patients under long term treatment with Imatinib.
Author Contributions
NI did formal analysis, writing, and editing of the manuscript. MA, AS, SUYB, MND, PNAA, and PZR contributed to conception, editing, and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data is available upon request.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pandey, N.; Yadav, G.; Kushwaha, R.; Verma, S.P.; Singh, U.S.; Kumar, A.; Mishra, P. Effect of Imatinib on Bone Marrow Morphology and Angiogenesis in Chronic Myeloid Leukemia. Adv. Hematol. 2019, 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother Res Pract. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Berman, E.; Girotra, M.; Cheng, C.; Chanel, S.; Maki, R.; Shelat, M.; Strauss, H.W.; Fleisher, M.; Heller, G.; Farooki, A. Effect of long term imatinib on bone in adults with chronic myelogenous leukemia and gastrointestinal stromal tumors. Leuk. Res. 2013, 37, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Paul, T.V.; Shetty, S.; Kapoor, N.; Bondu, J.D.; Thomas, N. Bone turnover markers: Emerging tool in the management of osteoporosis. Indian J. Endocrinol. Metab. 2016, 20, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.P.; Don-Wauchope, A.; Douville, P.; Albert, C.; Vasikaran, S.D. Current use of bone turnover markers in the management of osteoporosis. Clin. Biochem. 2022, 109-110, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nandiraju, D.; Ahmed, I. Human skeletal physiology and factors affecting its modeling and remodeling. Fertil. Steril. 2019, 112, 775–781. [Google Scholar] [CrossRef]
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Chi, G.; Xu, J.; Tan, Y.; Xu, J.; Lv, S.; Xu, Z.; Xia, Y.; Li, L.; Li, Y. Extracellular matrix stiffness controls osteogenic differentiation of mesenchymal stem cells mediated by integrin α5. Stem Cell Res. Ther. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, C.; Ferretti, M. The Osteocyte: From “Prisoner” to “Orchestrator”. J. Funct. Morphol. Kinesiol. 2021, 6, 28. [Google Scholar] [CrossRef]
- Veis, D.J.; O’Brien, C.A. Osteoclasts, Master Sculptors of Bone. Annu. Rev. Pathol. : Mech. Dis. 2023, 18, 257–281. [Google Scholar] [CrossRef]
- Martin, T.J.; Seeman, E. Bone Remodeling and Modeling: Cellular Targets for Antiresorptive and Anabolic Treatments, Including Approaches Through the Parathyroid Hormone (PTH)/PTH-Related Protein Pathway. Neurospine 2023, 20, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ruan, X.; Li, J.; Wang, B.; Chen, J.; Wang, X.; Wang, P.; Tu, X. The Osteocyte Stimulated by Wnt Agonist SKL2001 Is a Safe Osteogenic Niche Improving Bioactivities in a Polycaprolactone and Cell Integrated 3D Module. Cells 2022, 11, 831. [Google Scholar] [CrossRef] [PubMed]
- Jackson, E.; Lara-Castillo, N.; Akhter, M.P.; Dallas, M.; Scott, J.M.; Ganesh, T.; Johnson, M.L. Osteocyte Wnt/β-catenin pathway activation upon mechanical loading is altered in ovariectomized mice. Bone Rep. 2021, 15, 101129. [Google Scholar] [CrossRef] [PubMed]
- Vuoti, E.; Lehenkari, P.; Tuukkanen, J.; Glumoff, V.; Kylmäoja, E. Osteoclastogenesis of human peripheral blood, bone marrow, and cord blood monocytes. Sci. Rep. 2023, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Leon-Oliva, D.; Barrena-Blázquez, S.; Jiménez-Álvarez, L.; Fraile-Martinez, O.; García-Montero, C.; López-González, L. , et al. The RANK–RANKL–OPG System: A Multifaceted Regulator of Homeostasis, Immunity, and Cancer. Medicina 2023, 59, 1752. [Google Scholar] [CrossRef]
- Phillips, P.J.; Phillipov, G. Bone mineral density - frequently asked questions. . 2006, 35, 341–4. [Google Scholar]
- Kranioti, E.F.; Bonicelli, A.; García-Donas, J.G.; Bonicelli, R. Bone-mineral density: clinical significance, methods of quantification and forensic applications. Res. Rep. Forensic Med Sci. 2019, ume 9, 9–21. [Google Scholar] [CrossRef]
- Francisco, I.; Nunes, C.; Pereira, F.; Travassos, R.; Ribeiro, M.P.; Marques, F.; McEvoy, M.; Santos, M.; Oliveira, C.; Marto, C.M.; et al. Bone Mineral Density through DEXA and CBCT: A Systematic Review with Meta-Analysis. Appl. Sci. 2023, 13, 5962. [Google Scholar] [CrossRef]
- Chen, Y.-P.; Chan, W.P.; Zhang, H.-W.; Tsai, Z.-R.; Peng, H.-C.; Huang, S.-W.; Jang, Y.-C.; Kuo, Y.-J. Automated osteoporosis classification and T-score prediction using hip radiographs via deep learning algorithm. Ther. Adv. Musculoskelet. Dis. 2024, 16. [Google Scholar] [CrossRef]
- Demeuse, J.; Determe, W.; Grifnée, E.; Massonnet, P.; Schoumacher, M.; Huyghebaert, L.; Dubrowski, T.; Peeters, S.; Le Goff, C.; Cavalier, E. CHARACTERIZATION OF C-TERMINAL TELOPEPTIDE OF TYPE I COLLAGEN (CTX) SPECIES IN HUMAN PLASMA AND SERUM USING HIGH RESOLUTION MASS SPECTROMETRY. Osteoarthr. Cartil. 2024, 32, 759. [Google Scholar] [CrossRef]
- Bellemare, M.; Senay, A.; Delisle, J.; Banica, A.; Beaumont, P.; Giroux, M.; Jodoin, A.; Laflamme, Y.; Leduc, S.; MacThiong, J.; et al. Assessment of CTX-1 Bone Biomarker As An Indicator of Antiresorptive Therapy Efficacy And of Persistence In A Fracture Liaison Service. Value Heal. 2016, 19, A531. [Google Scholar] [CrossRef]
- Bjerre-Bastos, J.; Bay-Jensen, A.-C.; Karsdal, M.; Byrjalsen, I.; Andersen, J.; Riis, B.; Christiansen, C.; Bihlet, A. Biomarkers of bone and cartilage turnover CTX-I and CTX-II predict total joint replacements in osteoarthritis. Osteoarthr. Cartil. 2019, 27, S31–S32. [Google Scholar] [CrossRef]
- Takahashi, M.; Kushida, K.; Nagano, A.; Inoue, T. Comparison of the analytical and clinical performance characteristics of an N-MID versus an intact osteocalcin immunoradiometric assay. Clin. Chim. Acta 2000, 294, 67–76. [Google Scholar] [CrossRef]
- Patti, A.; Gennari, L.; Merlotti, D.; Dotta, F.; Nuti, R. Endocrine Actions of Osteocalcin. Int. J. Endocrinol. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Kuo, T.-R.; Chen, C.-H. Bone biomarker for the clinical assessment of osteoporosis: recent developments and future perspectives. Biomark. Res. 2017, 5, 1–9. [Google Scholar] [CrossRef]
- Brennan-Speranza, T.C.; Conigrave, A.D. Osteocalcin: An Osteoblast-Derived Polypeptide Hormone that Modulates Whole Body Energy Metabolism. Calcif. Tissue Int. 2014, 96, 1–10. [Google Scholar] [CrossRef]
- Greenblatt, M.B.; Tsai, J.N.; Wein, M.N. Bone Turnover Markers in the Diagnosis and Monitoring of Metabolic Bone Disease. Clin. Chem. 2017, 63, 464–474. [Google Scholar] [CrossRef]
- Wang, L.; You, X.; Zhang, L.; Zhang, C.; Zou, W. Mechanical regulation of bone remodeling. Bone Res. 2022, 10, 1–15. [Google Scholar] [CrossRef]
- Kuo, T.-R.; Chen, C.-H. Bone biomarker for the clinical assessment of osteoporosis: recent developments and future perspectives. Biomark. Res. 2017, 5, 1–9. [Google Scholar] [CrossRef]
- Greenblatt, M.B.; Tsai, J.N.; Wein, M.N. Bone Turnover Markers in the Diagnosis and Monitoring of Metabolic Bone Disease. Clin. Chem. 2017, 63, 464–474. [Google Scholar] [CrossRef]
- Rosen, H.N.; Moses, A.C.; Garber, J.; Iloputaife, I.D.; Ross, D.S.; Lee, S.L.; Greenspan, S.L. Serum CTX: A New Marker of Bone Resorption That Shows Treatment Effect More Often Than Other Markers Because of Low Coefficient of Variability and Large Changes with Bisphosphonate Therapy. Calcif. Tissue Int. 2000, 66, 100–103. [Google Scholar] [CrossRef]
- Li, J.; Zhang, H.; Yang, C.; Li, Y.; Dai, Z. An overview of osteocalcin progress. J. Bone Miner. Metab. 2016, 34, 367–379. [Google Scholar] [CrossRef]
- Ferron, M.; McKee, M.D.; Levine, R.L.; Ducy, P.; Karsenty, G. Intermittent injections of osteocalcin improve glucose metabolism and prevent type 2 diabetes in mice. Bone 2012, 50, 568–575. [Google Scholar] [CrossRef]
- Ferron, M.; Hinoi, E.; Karsenty, G.; Ducy, P. Osteocalcin differentially regulates β cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl. Acad. Sci. 2008, 105, 5266–5270. [Google Scholar] [CrossRef]
- Zhou, B.; Li, H.; Liu, J.; Xu, L.; Zang, W.; Wu, S. , et al. Intermittent injections of osteocalcin reverse autophagic dysfunction and endoplasmic reticulum stress resulting from diet-induced obesity in the vascular tissue via the NFκB-p65-dependent mechanism. Cell Cycle. 2013, 12, 1901–1913. [Google Scholar] [CrossRef]
- Lodish, M.B.; A Stratakis, C. Endocrine side effects of broad-acting kinase inhibitors. Endocrine-Related Cancer 2010, 17, R233–R244. [Google Scholar] [CrossRef]
- Alemán, J.; Farooki, A.; Girotra, M. Effects of tyrosine kinase inhibition on bone metabolism: untargeted consequences of targeted therapies. Endocrine-Related Cancer 2014, 21, R247–R259. [Google Scholar] [CrossRef]
- Matsubara, T.; Yasuda, K.; Mizuta, K.; Kawaue, H.; Kokabu, S. Tyrosine Kinase Src Is a Regulatory Factor of Bone Homeostasis. Int. J. Mol. Sci. 2022, 23, 5508. [Google Scholar] [CrossRef]
- Ryu, K.-Y.; Pokhrel, N.K.; Jung, H.-J.; Kim, H.J.; Seok, J.; Kim, T.-Y.; Kim, H.J.; Lee, J.H.; Kim, J.-Y.; Kim, Y.-G.; et al. Mer tyrosine kinase regulates bone metabolism, and its deficiency partially ameliorates periodontitis- and ovariectomy-induced bone loss in mice. JBMR Plus 2024, 8, ziad014. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Kim, N. c-Src-Dependent and -Independent Functions of Matk in Osteoclasts and Osteoblasts. J. Immunol. 2018, 200, 2455–2463. [Google Scholar] [CrossRef]
- Vandyke, K.; Fitter, S.; Drew, J.; Fukumoto, S.; Schultz, C.G.; Sims, N.A.; Yeung, D.T.; Hughes, T.P.; Zannettino, A.C.W. Prospective Histomorphometric and DXA Evaluation of Bone Remodeling in Imatinib-Treated CML Patients: Evidence for Site-Specific Skeletal Effects. J. Clin. Endocrinol. Metab. 2013, 98, 67–76. [Google Scholar] [CrossRef]
- Boufker, H.I.; Lagneaux, L.; Najar, M.; Piccart, M.; Ghanem, G.; Body, J.-J.; Journé, F. The Src inhibitor dasatinib accelerates the differentiation of human bone marrow-derived mesenchymal stromal cells into osteoblasts. BMC Cancer 2010, 10, 298–298. [Google Scholar] [CrossRef]
- Grüllich, C. Cabozantinib: Multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [CrossRef]
- Brassard, M.; Neraud, B.; Trabado, S.; Salenave, S.; Brailly-Tabard, S.; Borget, I.; Baudin, E.; Leboulleux, S.; Chanson, P.; Schlumberger, M.; et al. Endocrine Effects of the Tyrosine Kinase Inhibitor Vandetanib in Patients Treated for Thyroid Cancer. J. Clin. Endocrinol. Metab. 2011, 96, 2741–2749. [Google Scholar] [CrossRef]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’brien, C.A. Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLOS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef]
- Lodish, M.B. Kinase Inhibitors: Adverse Effects Related to the Endocrine System. J. Clin. Endocrinol. Metab. 2013, 98, 1333–1342. [Google Scholar] [CrossRef]
- Brown, R.L. Tyrosine kinase inhibitor-induced hypothyroidism: incidence, etiology, and management. Target. Oncol. 2011, 6, 217–226. [Google Scholar] [CrossRef]
- Lossdörfer, S.; Götz, W.; Jäger, A. PTH(1-34)-induced changes in RANKL and OPG expression by human PDL cells modify osteoclast biology in a co-culture model with RAW 264.7 cells. Clin. Oral Investig. 2010, 15, 941–952. [Google Scholar] [CrossRef]
- Alemán, J.; Farooki, A.; Girotra, M. Effects of tyrosine kinase inhibition on bone metabolism: untargeted consequences of targeted therapies. Endocrine-Related Cancer 2014, 21, R247–R259. [Google Scholar] [CrossRef]
- Miyazaki, T.; Tanaka, S.; Sanjay, A.; Baron, R. The role of c-Src kinase in the regulation of osteoclast function. Mod Rheumatol. 2006, 16, 68–74. [Google Scholar] [CrossRef]
- Maita, S.; Yuasa, T.; Tsuchiya, N.; Mitobe, Y.; Narita, S.; Horikawa, Y.; Hatake, K.; Fukui, I.; Kimura, S.; Maekawa, T.; et al. Antitumor effect of sunitinib against skeletal metastatic renal cell carcinoma through inhibition of osteoclast function. Int. J. Cancer 2011, 130, 677–684. [Google Scholar] [CrossRef]
- Dib, I.E.H.; Gallet, M.; Mentaverri, R.; Sévenet, N.; Brazier, M.; Kamel, S. Imatinib mesylate (Gleevec®) enhances mature osteoclast apoptosis and suppresses osteoclast bone resorbing activity. Eur. J. Pharmacol. 2006, 551, 27–33. [Google Scholar] [CrossRef]
- Fitter, S.; Dewar, A.L.; Kostakis, P.; To, L.B.; Hughes, T.P.; Roberts, M.M.; Lynch, K.; Vernon-Roberts, B.; Zannettino, A.C.W. Long-term imatinib therapy promotes bone formation in CML patients. Blood 2008, 111, 2538–2547. [Google Scholar] [CrossRef]
- Berman, E.; Nicolaides, M.; Maki, R.G.; Fleisher, M.; Chanel, S.; Scheu, K.; Wilson, B.-A.; Heller, G.; Sauter, N.P. Altered Bone and Mineral Metabolism in Patients Receiving Imatinib Mesylate. New Engl. J. Med. 2006, 354, 2006–2013. [Google Scholar] [CrossRef]
- Jönsson, S.; Standal, T.; Olsson, B.; Mellström, D.; Wadenvik, H. Secondary hyperparathyroidism but stable bone-mineral density in patients with chronic myeloid leukemia treated with imatinib. Am. J. Hematol. 2012, 87, 550–552. [Google Scholar] [CrossRef]
- Dewar, A.L.; Farrugia, A.N.; Condina, M.R.; To, L.B.; Hughes, T.P.; Vernon-Roberts, B.; Zannettino, A.C.W. Imatinib as a potential antiresorptive therapy for bone disease. Blood 2006, 107, 4334–4337. [Google Scholar] [CrossRef]
- O’sullivan, S.; Horne, A.; Wattie, D.; Porteous, F.; Gamble, G.; Browett, P.; Grey, A. Bone metabolism during long-term treatment with imatinib. Leuk. Lymphoma 2013, 54, 1783–1785. [Google Scholar] [CrossRef]
- Terpos, E.; Apperley, J.; Milojkovic, D. Imatinib and chronic myeloid leukemia: close to the bone. Leuk. Lymphoma 2013, 54, 1581–1582. [Google Scholar] [CrossRef]
- Choeyprasert, W.; Yansomdet, T.; Natesirinilkul, R.; Wejaphikul, K.; Charoenkwan, P. Adverse effects of imatinib in children with chronic myelogenous leukemia. Pediatr. Int. 2016, 59, 286–292. [Google Scholar] [CrossRef]
- Jaeger, B.A.S.; Tauer, J.T.; Ulmer, A.; Kuhlisch, E.; Roth, H.J.; Suttorp, M. Changes in bone metabolic parameters in children with chronic myeloid leukemia on imatinib treatment. Med Sci. Monit. 2012, 18, CR721–CR728. [Google Scholar] [CrossRef]
- Tauer, J.T.; Hofbauer, L.C.; Jung, R.; Gerdes, S.; Glauche, I.; Erben, R.G.; Suttorp, M. Impact of Long-Term Exposure to the Tyrosine Kinase Inhibitor Imatinib on the Skeleton of Growing Rats. PLOS ONE 2015, 10, e0131192. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Chang, Y.F.; Chen, C.H.; Lewiecki, E.M.; Wüster, C.; Reid, I.; Tsai, K.S.; Matsumoto, T.; Mercado-Asis, L.B.; Chan, D.C.; et al. Consensus Statement on the Use of Bone Turnover Markers for Short-Term Monitoring of Osteoporosis Treatment in the Asia-Pacific Region. J. Clin. Densitom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Liu, D.; Wu, F.; Wang, M.; Cen, Y.; Ma, L. Correlation between Bone Turnover Markers and Bone Mineral Density in Patients Undergoing Long-Term Anti-Osteoporosis Treatment: A Systematic Review and Meta-Analysis. Appl. Sci. 2020, 10, 832. [Google Scholar] [CrossRef]
Figure 1.
Interaction of CTX-1, Osteocalcin and Bone Mineral Density. Osteoclasts produce CTX-1 which is a marker of bone resorption. Osteoblasts produce Osteocalcin which is a marker of bone formation. When bone formation rate surpasses the bone resorption rate, Bone Mineral density (BMD) increases. On the other hand, when bone resorption rate surpasses the bone formation rate, BMD decreases. CTX-1: C-terminal telopeptide of type I collagen.
Figure 1.
Interaction of CTX-1, Osteocalcin and Bone Mineral Density. Osteoclasts produce CTX-1 which is a marker of bone resorption. Osteoblasts produce Osteocalcin which is a marker of bone formation. When bone formation rate surpasses the bone resorption rate, Bone Mineral density (BMD) increases. On the other hand, when bone resorption rate surpasses the bone formation rate, BMD decreases. CTX-1: C-terminal telopeptide of type I collagen.
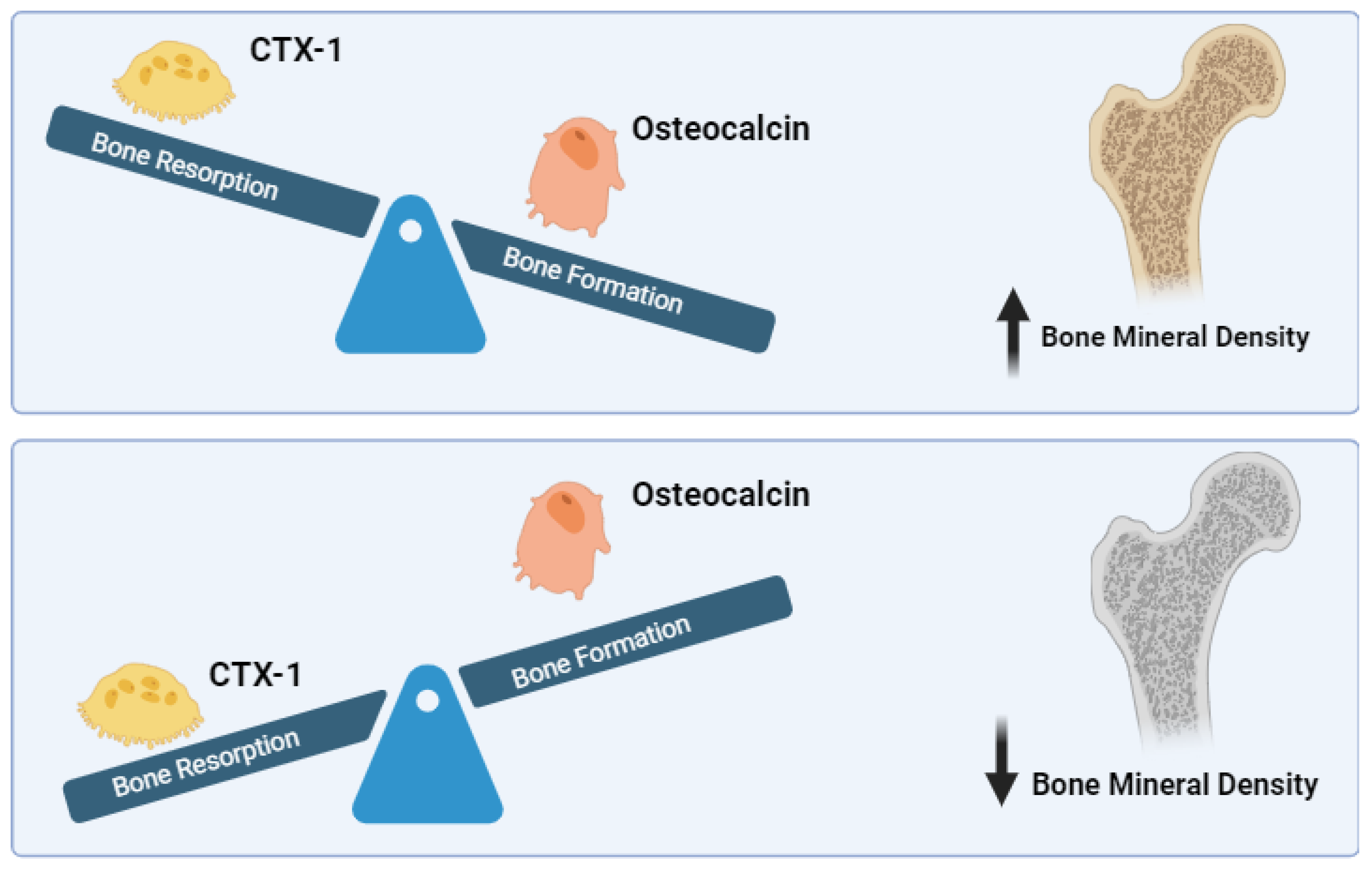

Table 1.
Summary of Study About Impact of Imatinib to BMD, CTX-1 and Osteocalcin.
| Study/Authors | Study Type | Participants/Subjects | Key Observations |
|---|---|---|---|
| Jönsson et al. [55] | Longitudinal Study | 17 CML patients | 7/17 developed secondary hyperparathyroidism; Increased serum parathyroid hormone levels over 4 years; Mean areal and volumetric BMD remained stable; cortical BMD higher than controls. |
| Dib et al. 45 | Clinical/Preclinical | Not specified | Imatinib influences mature osteoclasts through c-fms inhibition; Potential clinical value in treating osteoporosis and osteolysis. |
| Dewar et al. [56] | Animal Study | Mice | Imatinib inhibits osteoclast formation and activity in vivo ; Effective antiosteolytic agent; affects c-fms signaling and RANK expression. |
| O'Sullivan50 | Animal Study | Healthy mice | Early osteoblast differentiation promoted, reduced mineralization at low concentrations; No long-term effect on BMD; does not alter bone resorption markers |
| Choeyprasert et al. [59] | Clinical Study | 44 CML children | Correlation with low BMD without affecting bone parameters; high prevalence of vitamin D deficiency; Imatinib linked to low BMD and vitamin D deficiency |
| Jaeger et al [60] | Clinical Study | 17 children with CML on prolonged imatinib | Decrease in N-mid OCN, CTX-I levels high in a majority of patients. |
| Tauer et al. [61] | Animal Study | Mice | Similar findings as Jaeger et al. in terms of N-mid OCN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



