Submitted:
20 September 2024
Posted:
23 September 2024
You are already at the latest version
Abstract
Programmed cell death 4 (Pdcd4) is a tumor suppressor, which has been demonstrated to efficiently suppress tumorigenesis. Biochemically, Pdcd4 binds with translation initiation factor 4A and represses protein translation. Beyond its role in tumor suppression, increasing evidence indicates that Pdcd4 enhances the chemosensitivity of several chemotherapeutic drugs. To date, several translational targets of Pdcd4 have been identified. These targets govern important signal transduction pathways, and their attenuation may enhance chemosensitivity or overcome drug resistance. This review will discuss the signal transduction pathways regulated by Pdcd4 and the potential mechanisms through which Pdcd4 enhances chemosensitivity or counteracts drug resistance.
Keywords:
eIF4A
; mTORC2-Akt pathway
; β-catenin pathway
; JNK pathway
; paclitaxel
; doxorubicin
; fluorouracil
; platinum-containing drug
; IGF1R/IR inhibitor
1. Introduction
Although many types of cancer cells are initially susceptible to chemotherapy, over time, they can develop resistance to the treatment, leading to relapse. Resistance to chemotherapeutics can be categorized as either intrinsic or acquired. Intrinsic resistance occurs when resistance-mediating factors are present in cancer cells before chemotherapy, making the treatment ineffective from the beginning. Acquired resistance develops during the treatment of cancers that were initially responsive, resulting from DNA mutations or activation of alternative cellular signaling pathways during treatment. Programmed cell death 4 (Pdcd4) is a tumor suppressor, which has shown to efficiently inhibit cell proliferation, survival, migration/invasion in various cancer cells [1,2]. Analyses of the Pdcd4 expression levels in the panel of NCI-60 cancer cell lines suggested that Pdcd4 protein level contributes to cellular sensitivity to tamoxifen and geldanamycin [3]. In addition, the Pdcd4 protein levels also found to be down-regulated in the recurrent colorectal cancer patients compared to non-recurrent colorectal patients [4]. These findings suggest that Pdcd4 plays a critical role in drug resistance. In this review, we explore the mechanism by which the loss of Pdcd4 contributes to drug resistance through the regulation of protein translation by Pdcd4.
2. Pdcd4 as a Protein Translation Inhibitor
Pdcd4 was initially recognized as a tumor suppressor due to its downregulation in promotion-resistant mouse epidermal JB6 cells, which results in the acquisition of a promotion-sensitive phenotype [5]. Conversely, ectopic expression of Pdcd4 cDNA in promotion-sensitive JB6 cells leads to the development of promotion-resistance and suppression of tumor phenotype [6,7]. Subsequently, Pdcd4 was found to exert multifaceted effects, including inhibiting tumor cell invasion and metastasis [8,9,10,11,12], reducing proliferation [13,14,15], and promoting apoptosis [16] (Figure 1).
Beyond suppressing tumorigenesis, Pdcd4 has been shown to inhibit protein translation as well. Using yeast two-hybrid screens, Pdcd4 was found to bind with translation initiation factor (eIF) 4A [17]. Crystallography and point mutation analyses further confirmed that Pdcd4 binds with eIF4A through two MA-3 domains that consists with 8 or 9 α-helices [18,19,20] (Figure 2). Apart from the MA-3 domains, Pdcd4 protein possesses several intriguing features. First, the Ser residue at position 67 of Pdcd4 can be phosphorylated by Akt (also called protein kinase B) or Ribosomal protein S6 kinase (p70S6K) resulting in proteasomal degradation of Pdcd4 [21,22]. Second, the Ser residue at position 457 also can be phosphorylated by Akt for nuclear localization [22]. Third, the Arg residue at position 110 can be methylated by protein arginine methyltransferase 5 (PRMT5). The methylation at the Arg residue impairs the Pdcd4 tumor suppressor function [23,24]. Last, Pdcd4 contains two clusters of positively charged amino acids that have been reported to interact with RNA [25,26] (Figure 2).
By binding with eIF4A, Pdcd4 inhibits the eIF4A’s helicase activity [17]. eIF4A is an ATP-dependent RNA helicase. The role of eIF4A in protein translation initiation is to unwind the secondary structure of mRNA at 5’ untranslated region (5′UTR), enabling the translation initiation complex to scan the mRNA [27]. Therefore, inhibition of the eIF4A’s helicase activity by Pdcd4 is expected to suppress protein translation, especially the mRNAs with a secondary structure at 5’UTR. This idea was supported by the observations that Pdcd4 represses the translation of luciferase reporter with an artificial stem-loop structure, which has a free energy of -44.8 kcal/mol, greater than the one without the structure [18]. In addition, cells treated with the eIF4A inhibitor, silvestrol, showed decreased luciferase activity in the reporter with the structured Sin1 5’UTR (stress-activated-protein kinase interacting protein 1), but not in the one lacking the Sin1 5’UTR [28]. These studies indicate that Pdcd4 preferentially inhibits translation of mRNAs with structured 5’UTR. Besides inhibiting translation through the eIF4A-dependent mechanism, Pdcd4 also inhibits translation via an eIF4A-independent mechanism. It has been reported that Pdcd4 directly binds to the c-myb mRNA interacting with poly A binding protein and possibly blocks translation elongation [29]. Liwak et al. reported that Pdcd4 directly binds to mRNA of the X-linked inhibitor of apoptosis (XIAP) and Bcl-xL at internal ribosome entry site (IRES) elements and blocks the translational initiation complex formation [30]. A recent study using cryo-electron microscopy suggests that Pdcd4 may hamper translation through the binding to the 40S ribosome, interfering with the formation of the translation initiation complex [31]. Up-to date, several translational targets of Pdcd4 have been identified including Sin1 [28], p70S6K1 [32], p53 [33], c-myb [34], XIAP [30], and Bcl-xL [30]. These Pdcd4 translational targets play crucial roles in governing multiple signaling transduction pathways that regulate proliferation, survival, apoptosis, and drug resistance.
3. Pdcd4 Regulates Signaling Pathway to Control Tumorigenesis
3.1. mTORC2-Akt Pathway
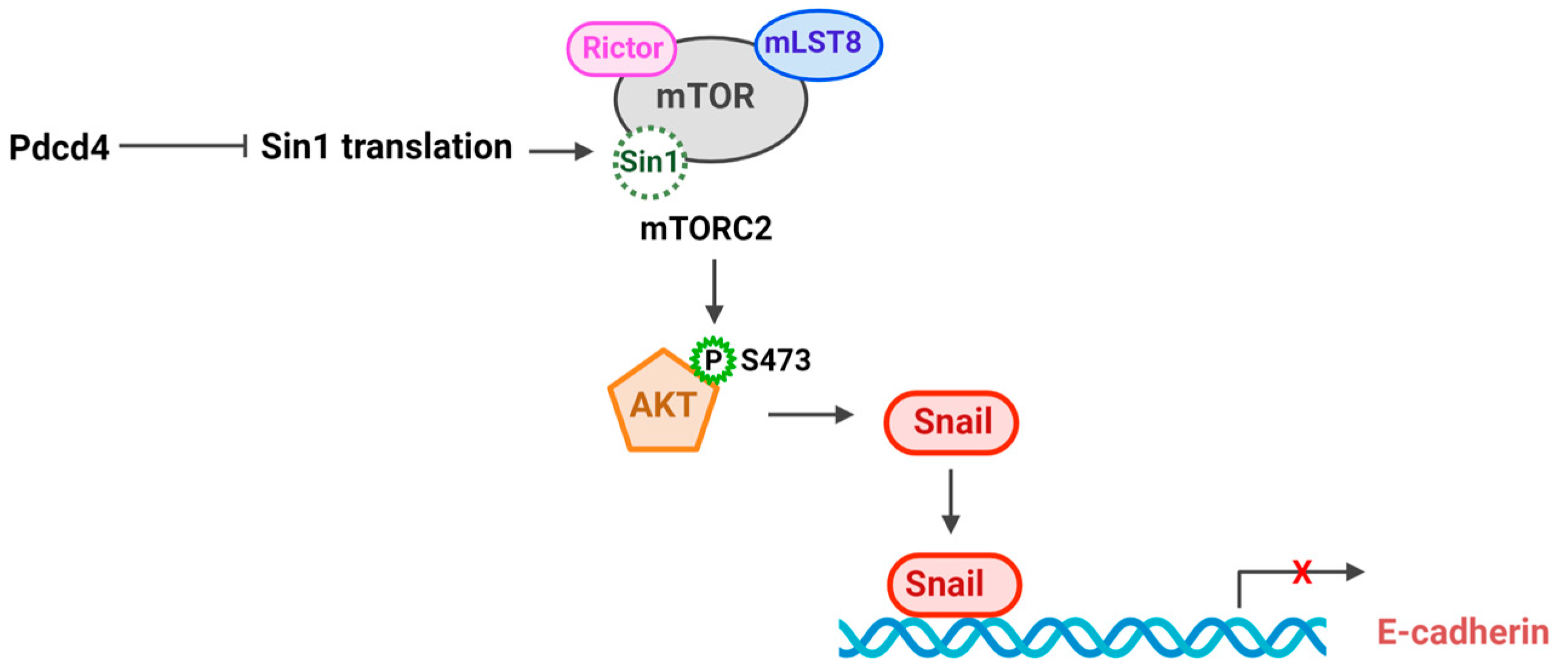
Akt is a serine/threonine kinase that regulates key cellular processes including survival, proliferation, growth, apoptosis, and metabolism [35]. Akt is frequently activated in various types of cancers via phosphorylation at Thr308 and Ser473. Phosphorylation of Akt at Thr308 is primarily controlled by phosphoinositide 3-kinase (PI3K)- phosphoinositide-dependent kinase 1 (PDK1) signaling pathway, which drives its basal kinase activity. Additionally, Akt can be phosphorylated at Ser 473 by Mammalian target of rapamycin complex 2 (mTORC2). Phosphorylation of Akt at both Thr308 and Ser473 leads to the maximal activation of Akt kinase activity [36]. The mTORC2 complex, composed of the core components mTOR, Rictor, Sin1, and mLST8, regulates a range of biological functions, including survival, actin organization, and metabolism, by phosphorylating Akt, protein kinase Cα (PKCα), and serum- and glucocorticoid-regulated kinase 1 (SGK1) [37]. Studies have indicated that Pdcd4 knockdown leads to an increase in Akt phosphorylation at Ser473, whereas the overexpression of Pdcd4 results in a reduction in phosphorylation [38,39]. The Akt phosphorylation at Ser473 and mTORC2 kinase activity are also elevated in Pdcd4 nude MEF cells [28]. The increased mTORC2 activity in Pdcd4 knockdown cells is attributed to the enhanced Sin1 protein translation, which in turn activates mTORC2 kinase activity and promotes cell invasion [28]. Regulation of Sin1 translation by Pdcd4 is mediated by eIF4A since the Pdcd4 mutant defective in binding with eIF4A barely inhibits mTORC2 activity and cell invasion [28] (Fig. 3). In addition to inhibiting Sin1 translation, Pdcd4 has been reported to bind with Rictor, resulting in suppression of mTORC2 activity [40]. It is possible that Pdcd4, by binding with Rictor, prevents the formation of the mTORC2 complex. However, the precise mechanism underlying this interaction requires further investigation.
3.2. E-cadherin-β catenin Pathway
Knockdown or knockout of Pdcd4 activates mTORC2-Akt signaling and concomitantly up-regulates Snail expression and down-regulates E-cadherin expression [12]. Conversely, knockdown of Akt reverses the Pdcd4 knockdown induced-increase in Snail expression and restores E-cadherin level [11,28](Figure 3). These results suggest that mTORC2-Akt signaling crosstalks with E-cadherin-β-catenin signaling pathway. E-cadherin is a transmembrane protein whose cytoplasmic domain is associated with β-catenin. Under normal physiological condition, β-catenin released from E-cadherin undergoes is rapidly phosphorylation by glycogen synthase kinase 3 (GSK3) and subsequently degraded via the proteasome pathway. When GSK3 activity is inhibited, β-catenin remains unphosphorylated. This unphosphorylated form translocates to the nucleus and binding withanother transcription factor, Tcf4. The β-catenine/Tcf4 complex, then, activates the transcription of target genes involved in β-catenin signaling [41](Figure 4). E-cadherin expression can be regulated by zinc finger transcription repressors of Snail family, including Snail and Slug [42]. Snail binds to a specific E-box (CAGGTG) at proximal E-cadherin promoter, recruiting HDAC to modify the local chromatin structure, thereby repressing E-cadherin transcription [43].
The possible mechanism is that knockdown of Pdcd4 up-regulates Snail expression resulting in down-regulation of E-cadherin to release β-catenin from inner membrane to cytoplasm. The free β-catenin may then evade proteasomal degradation due to the inactivation of GSK-3 by Akt [14]. Subsequently, the free β-catenin translocates into the nucleus, where it activates β-catenin-dependent transcription, leading to the stimulation of oncogene expression, including u-PAR and c-Myc (Figure 4). The mechanism by which Pdcd4 regulates Snail expression remains unknown and warrants further investigation.
3.3. JNK-AP-1 Pathway
Pdcd4 knockdown has been reported to up-regulate c-Myc transcript factor, which then binds to the promoter of MAP4K1 (mitogen-activated protein kinase kinase kinase kinase 1) and stimulates its transcription [10,44]. MAP4K1 is an upstream kinase of c-jun terminal kinase (JNK) pathway. The increase in MAP4K1 expression by Pdcd4 knockdown leads to JNK activation and translocation from cytosol to nucleus, where it phosphorylates and activates c-Jun [6,7,45,46]. c-Jun, a member of the Jun protein family, can form heterodimers with other Jun family members (JunB and JunD) and with the Fos protein family protein (c-Fos, FosB, Fra-1, and Fra-2). These interactions result in the formation of the AP-1 transcription factor, which is enssential for regulating cell proliferation, transformation, survival, and invasion [47] (Figure 5).
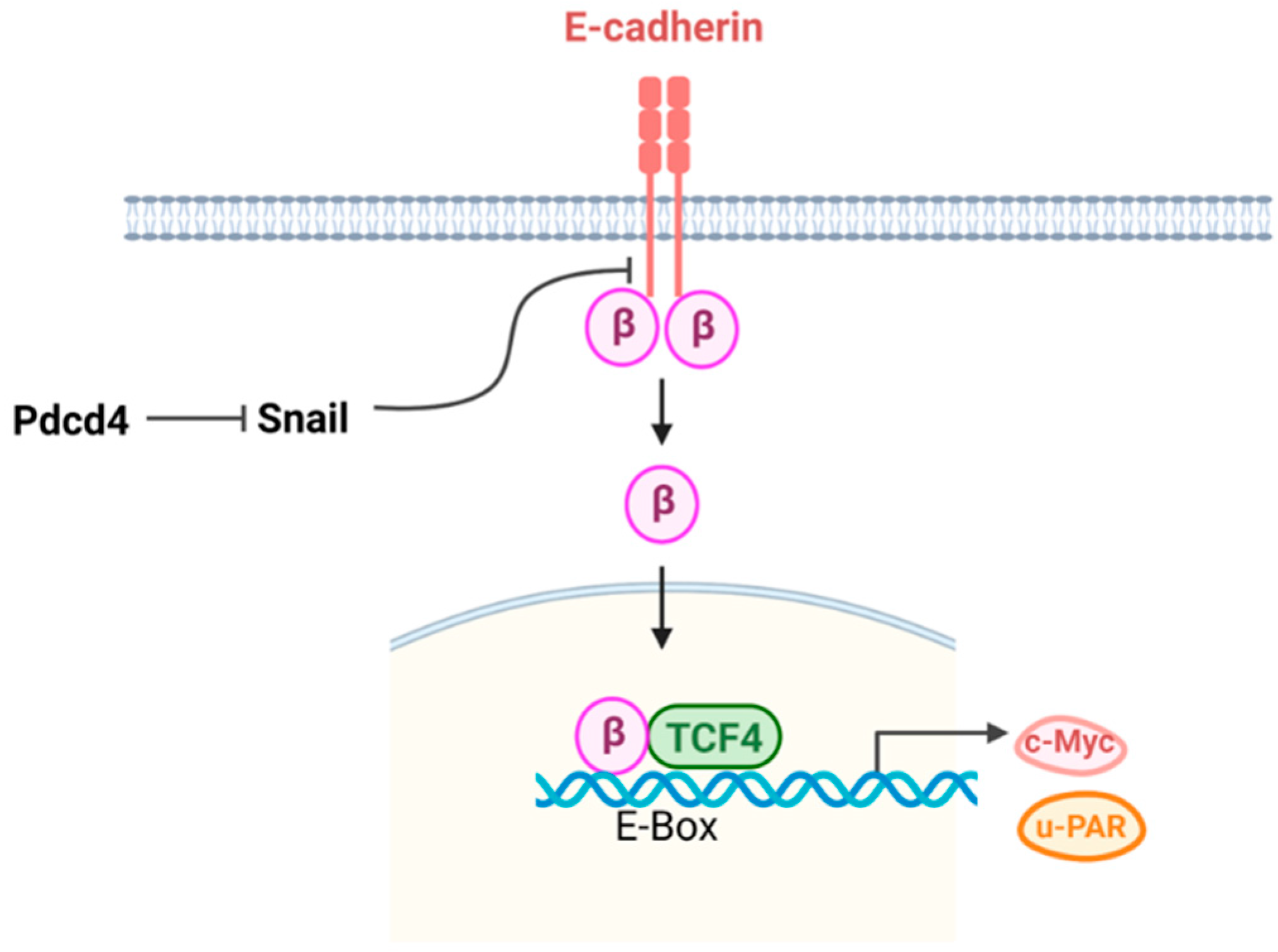
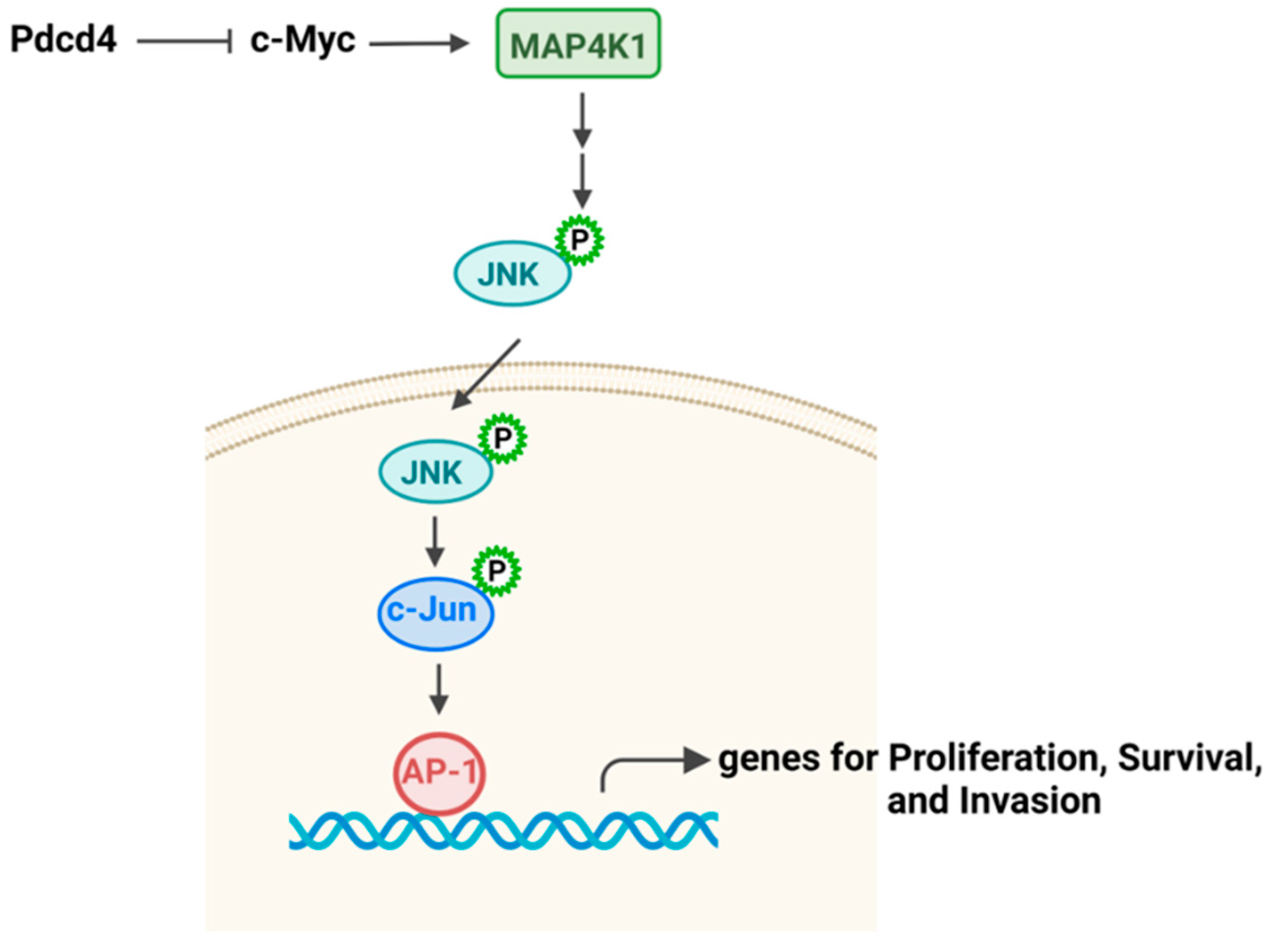
Figure 4.
Pdcd4 suppresses E-cadherin-β catenin pathway. The transcription repressor Snail suppresses E-cadherin expression, leading to β-catenin nuclear translocation and binding with Tcf4 to stimulate c-Myc and u-PAR expression.
Figure 4.
Pdcd4 suppresses E-cadherin-β catenin pathway. The transcription repressor Snail suppresses E-cadherin expression, leading to β-catenin nuclear translocation and binding with Tcf4 to stimulate c-Myc and u-PAR expression.
Figure 5.
Pdcd4 suppresses JNK-AP-1 pathway. Pdcd4 inhibits MAP4K1 expression resulting in suppression of JNK-AP-1 pathway.
Figure 5.
Pdcd4 suppresses JNK-AP-1 pathway. Pdcd4 inhibits MAP4K1 expression resulting in suppression of JNK-AP-1 pathway.
4. Loss of Pdcd4 Enhances Chemotherapy Resistance
Growing evidence links the loss of Pdcd4 to chemotherapy resistance in various cancer types, though the exact mechanism remains unclear. In this section, we will explore the potential mechanisms by which Pdcd4 overcomes resistance to paclitaxel, doxorubicin, fluorouracil, platinum compounds, and IGF1R-IR inhibitors, where Pdcd4 is downregulated.
4.1. Paclitaxel
Paclitaxel, originally isolated from Pacific yew tree, Taxus brevifolia, has been approved by FDA for treatment of ovarian, cervical, breast, esophageal, lung, pancreatic cancers, and Kaposi's sarcoma. Paclitaxel binds to the microtubule polymers, stabilizing them and preventing disassembly. This inhibits proper metaphase spindle segregation, leading to prolonged activation of the mitotic checkpoint and ultimately triggering apoptosis [48]. Although paclitaxel effectively inhibits tumor growth in various approved cancer types, many patients eventually develop resistance after an initial positive response to the treatment.
Pdcd4 has been shown to enhance the chemosensitivity and overcome the resistance to paclitaxel. Overexpression of Pdcd4 cDNA has been demonstrated to enhance the chemosensitivity of paclitaxel in colon [49] and prostate cancer cells [50]. Conversely, down-regulation of Pdcd4 by Pdcd4 siRNA promotes the resistance to paclitaxel treatment in breast cancer cells [51,52]. Mechanically, Moustafa-Kamal et al. [53] found that Pdcd4 knockdown significantly prolongs mitotic survival in paclitaxel-treated HeLa cells. In contrast, HeLa cells treated with the S6K inhibitor, PF-4708671, exhibit increased Pdcd4 protein level, consequently improving the survival in response to paclitaxel treatment. Since Pdcd4 acts as an inhibitor of eIF4A, using the eIF4A inhibitor hippuristanol to mimic the effect of Pdcd4 showed that hippuristanol induces cell death in a dose-dependent manner when combined with paclitaxel. These findings collectively suggest that Pdcd4 plays a crucial role in enhancing paclitaxel chemosensitivity. Combination of paclitaxel and an eIF4A inhibitor, such as hippuristanol, appears to be a promising strategy for overcoming paclitaxel resistance. As mentioned in section 2, inhibiting eIF4A is expected to suppress translation of a set of mRNAs involved in critical cellular functions. However, the specific downstream targets of eIF4A involved in paclitaxel resistance remain unclear and require further study.
4.2. Doxorubicin
Doxorubicin, also known as Adriamycin, is an FDA approved drug used alone or in combination of other therapies to treat various blood cancers and solid tumors [54,55]. Doxorubicin functions as a topoisomerase II inhibitor. Topoisomerase II is an essential enzyme for DNA replication and chromosome segregation, and its inhibition causes a delay in mitosis, leading to cell death when prolonged [56]. Studies have shown that Pdcd4 overexpression enhances the sensitivity to doxorubicin treatment in multiple myeloma cells [57]. Additionally, Gonzalez-Ortiz et al. [58] demonstrated that Pdcd4 expression is down-regulated in the doxorubicin-resistant MDA-MB-231 breast cancer cells, which subsequently reduces the interaction of Pdcd4 and eIF4A, implying that eIF4A downstream targets are involved in doxorubicin resistance. Knockdown of eIF4A in these resistant cells decreases focal adhesion kinase (FAK) expression, suggesting that FAK is one of the eIF4A downstream targets [58]. FAK, a protein tyrosine kinase, regulates cellular migration, proliferation, and survival [59], playing a critical role in doxorubicin resistance. Inhibition of FAK activity has been shown to overcome resistance in doxorubicin-resistant lung and breast cancer cells [60,61].
4.3. Fluorouracil
Fluorouracil (5-FU) is an uracil analog, where the hydrogen atom at the C-5 position is replaced by a fluorine atom. It has been widely used to treat gastrointestinal, breast, gynecological, and head and neck cancers [62]. 5-FU inhibits thymidylate synthase activity, and thereby suppresses deoxythymidine mono-phosphate production required for DNA replication and repair [63]. Additionally, 5-FU can be misincorporated into RNA and DNA in place of uracil or thymine, disrupting normal nucleic acid function [63]. As a result, 5-FU treatment impairs cell cycle progression and ultimately induces cell death. However, resistance to 5-FU frequently develops, reducing the efficacy of the therapy and leading to poor prognosis.
Several reports have shown that Pdcd4 expression is significantly down-regulated in 5-FU resistant colorectal or pancreatic cancer cells [4,64,65], highlighting its importance in 5-FU resistance. The loss of Pdcd4 expression in these resistant cells is mediated by microRNA or long non-coding RNA (LncRNA) inhibition. Conversely, ectopic expression of Pdcd4 in 5-FU-resistant HCT116 and SW480 cells restores their sensitivity to 5-FU treatment. Over-expression of Pdcd4 in 5-FU-resistant colorectal cells induces apoptosis and inhibits invasion by up-regulating the pro-apoptotic Bax and down-regulating the invasion-promoting MMP-2 and MMP-9 [4].
One potential mechanism by which Pdcd4 regulates Bax, MMP-2, and MMP-9 expression is through the inhibition of NF-κB to suppress their transcription [66]. NF-κB consists of the subunits such as p105/p50, p100/p52, p65, c-Rel, and RelB, which form homo- or heterodimers [67]. In the cytosol, IκB binds with NF-κB to prevent NF-κB nuclear translocation. Upon phosphorylation by IKKα/IKKβ, IκB is degraded, allowing NF-κB to translocate into nucleus and activate its target gene transcription [68]. Pdcd4 is known to inhibit Akt activation, and Akt knockdown suppresses IKKα/IKKβ activation [14]. Therefore, Pdcd4 likely suppresses MMP-2 and MMP-9 expression through the Akt- IKKα/IKKβ-NF-κB axis.
4.4. Platinum-Containing Drug
Platinum-containing drugs, such as cisplatin, carboplatin, oxaliplatin, ormaplatin, and enloplatin, are commonly used to treat various cancers, including lung, ovarian, head and neck, breast, and brain cancers. However, resistance to these drugs often patients develops, limiting their effectiveness. Platinum-containing drugs work by interfering with the DNA repair mechanisms, causing DNA damage through the formation of crosslinks with the purine bases, primarily guanine and adenine [69], ultimately leading to cancer cell death. Pdcd4 has been reported to enhance the chemosensitivity of cisplatin. In a comparison of Pdcd4 expression level across various ovarian cancer cell lines, Zhang et al. found that the cells with higher Pdcd4 level are more sensitive to cisplatin [70]. Over-expression of Pdcd4 further increases the cisplatin chemosensitivity, promoting cell death via elevated level of cleaved caspase-3 and cleaved caspase-8 in both cultured cells and mice [70]. On the contrary, down-regulation of Pdcd4 reduces the cisplatin sensitivity [70,71,72].
Liu et al. reported that loss of Pdcd4 expression leads to the upregulation of multidrug resistance mutation 1 (MDR1) in cisplatin-resistant HeLa cells [73]. MDR1, also known as ABCB1, is an efflux pump that transports anticancer drugs from the inside to the outside of the cell membrane, contributing drug resistance [74]. MDR1 is frequently overexpressed in cancer cells, leading to the development of resistance to various anticancer drugs [75]. Liu et al. also found that Pdcd4 associates with YB1 (Y-box binding protein 1), a transcription factor, and subsequently binds to the MDR1promoter region to repress its transcription [73]. In addition to transcriptional regulation, Pdcd4 may also regulate MDR1 translation through Akt inhibition. As mentioned earlier, Pdcd4 inhibits Akt activation by suppressing the translation of Sin1 [28]. Non-phosphorylated YB1 is able to bind with the MDR1 mRNA at capped 5’-end, which blocks the translation initiation factors assembly and thereby suppresses translation of MDR1. Upon phosphorylation by Akt, YB1 reduces its affinity for the MDR1 mRNA at capped 5’-end, relieving translational repression and allowing MDR1 expression to increase [76].
4.5. IGF1R/IR Inhibitors
Insulin-like growth factor 1 receptor (IGF-1R) and insulin receptor (IR) are receptor tyrosine kinase that, upon binding with IGF1, IGF2, or insulin, activates downstream signaling pathways, such as PI3K-Akt and Ras-ERK pathway. These pathways primarily regulate cancer cell proliferation, survival, and apoptosis [77]. Therefore, inhibiting IGF1R/IR activation is expected to efficiently suppress tumor growth. Two types of IGF1R/IR inhibitors have been developed, i.e. monoclonal antibody and small molecule inhibitors [78]. The monoclonal antibodies block the binding of ligands to IGF1R/IR, while the small molecule inhibitors compete for the ATP binding site of IGF1R/IR. Although the in vitro and animal studies have shown promising results with IGF1R/IR inhibitors in suppressing tumorigenesis, clinical outcomes have shown that patients treated with these inhibitors experience a lack of survival benefit and an increase in adverse events [79]. The inefficacy of IGF1R/IR inhibitors is likely due to the developed resistance.
In an analysis of 27 colorectal cancer cell lines, Pitt et al. classified 16 cell lines, including HCT116 and SW480 cells, as resistant, and six cell lines, including HT29 and GEO cells, as sensitive to the IGF1R/IR inhibitor, OSI-906 [80]. Interestingly, the resistant cells exhibit relatively low levels of Pdcd4 protein, while sensitive cell show high Pdcd4 levels, suggesting that Pdcd4 expression correlates with the chemosensitivity to OSI-906 in colorectal cancer cells [81]. Subsequently, overexpression of Pdcd4 in IGF1R/IR inhibitor-resistant cells restores sensitivity to the inhibitor, while Pdcd4 knockdown induces a resistant phenotype [81].
When resistant colorectal or breast cancer cells are treated with IGF1R/IR inhibitors, such as OSI-906, BMS-754807, or GSK1838705A, it induces phosphorylation of p70S6K [32,81,82]. However, Pdcd4 overexpression inhibits the IGF1R/IR inhibition-induced p70S6K phosphorylation. Activation of p70S6K1 has been shown to promote cell survival by phosphorylation of Mouse Double Minute 2 [83], an E3 ubiquitin ligase. Phosphorylation of MDM2 by p70S6K1 leads to polyubiquitination and degradation of p53, thereby suppressing p53-dependent apoptosis [84]. Further investigation revealed that Pdcd4 specifically inhibits translation of p70S6K1, but not p70S6K2, to control the level of p70S6K1 phosphorylation [32]. Subsequently, a combination treatment of OSI-906 (an IGF1R/IR inhibitor) and PF-4708671 (a p70S6K inhibitor) in mice significantly reduces colorectal tumor growth [81]. Further studies are needed to evaluate the efficacy of this combination in overcoming IGF1R/IR resistance.
5. Conclusions and Perspectives
Pdcd4 is increasingly recognized as a critical player in combating drug resistance, primarily due to its role as an inhibitor of protein translation. The loss or downregulation of Pdcd4 is associated with heightened drug resistance, while its upregulation enhances cancer cells sensitivity to chemotherapy. Therefore, Pdcd4 holds potential as both a biomarker for predicting treatment responses and a therapeutic target for re-sensitizing drug-resistant cancer cells. Further exploration of the molecular mechanisms by which Pdcd4 re-sensitizes drug-resistant cancer cells could unveil new therapeutic targets, offering the dual benefits of suppressing tumor progression and mitigating drug resistance. Additionally, the identification of new agents that effectively restore Pdcd4 expression or function may eventually lead to clinical applications that boosts the efficacy of existing therapies in overcoming resistance.
Author Contributions
Writing-original draft and preparation, Q.W.; writing-review and editing, H.-S. Y.; visualization, Q.W. and H.-S. Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by National Cancer Institute grant CA272483 and CA279455.
Acknowledgments
The grammar was edited by ChatGPT and the figures were created using BioRender.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Matsuhashi, S.; Manirujjaman, M.; Hamajima, H.; Ozaki, I. Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yang, H.S. The role of Pdcd4 in tumour suppression and protein translation. Biol Cell 2018, 110, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.P.; Camalier, C.E.; Stark, C.; Colburn, N.H. Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity. Mol Cancer Ther 2004, 3, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, L.; Hou, Z.; Liu, W.; Wang, H.; Zhou, T.; Li, Y.; Chen, S. LncRNA HAND2-AS1 inhibits 5-fluorouracil resistance by modulating miR-20a/PDCD4 axis in colorectal cancer. Cell Signal 2020, 66, 109483. [Google Scholar] [CrossRef]
- Cmarik, J.L.; Min, H.; Hegamyer, G.; Zhan, S.; Kulesz-Martin, M.; Yoshinaga, H.; Matsuhashi, S.; Colburn, N.H. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci U S A 1999, 96, 14037–14042. [Google Scholar] [CrossRef]
- Yang, H.S.; Jansen, A.P.; Nair, R.; Shibahara, K.; Verma, A.K.; Cmarik, J.L.; Colburn, N.H. A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation. Oncogene 2001, 20, 669–676. [Google Scholar] [CrossRef]
- Yang, H.S.; Knies, J.L.; Stark, C.; Colburn, N.H. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene 2003, 22, 3712–3720. [Google Scholar] [CrossRef]
- Nieves-Alicea, R.; Colburn, N.H.; Simeone, A.M.; Tari, A.M. Programmed cell death 4 inhibits breast cancer cell invasion by increasing tissue inhibitor of metalloproteinases-2 expression. Breast Cancer Res Treat 2009, 114, 203–209. [Google Scholar] [CrossRef]
- Santhanam, A.N.; Baker, A.R.; Hegamyer, G.; Kirschmann, D.A.; Colburn, N.H. Pdcd4 repression of lysyl oxidase inhibits hypoxia-induced breast cancer cell invasion. Oncogene 2010, 29, 3921–3932. [Google Scholar] [CrossRef]
- Yang, H.S.; Matthews, C.P.; Clair, T.; Wang, Q.; Baker, A.R.; Li, C.C.; Tan, T.H.; Colburn, N.H. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol 2006, 26, 1297–1306. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Z.; Yang, H.S. Downregulation of tumor suppressor Pdcd4 promotes invasion and activates both beta-catenin/Tcf and AP-1-dependent transcription in colon carcinoma cells. Oncogene 2008, 27, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Sun, Z.X.; Allgayer, H.; Yang, H.S. Downregulation of E-cadherin is an essential event in activating beta-catenin/Tcf-dependent transcription and expression of its target genes in Pdcd4 knockdown cells. Oncogene 2010, 29, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Ozaki, I.; Xia, J.; Kuwashiro, T.; Kojima, M.; Takahashi, H.; Ashida, K.; Anzai, K.; Matsuhashi, S. PDCD4 Knockdown Induces Senescence in Hepatoma Cells by Up-Regulating the p21 Expression. Front Oncol 2018, 8, 661. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, W.; Wang, Q.; Yang, H.S. AKT Activation by Pdcd4 Knockdown Up-Regulates Cyclin D1 Expression and Promotes Cell Proliferation. Genes Cancer 2011, 2, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Ahn, H.S.; Lee, J.Y.; Matsuhashi, S.; Park, W.Y. Up-regulation of PDCD4 in senescent human diploid fibroblasts. Biochem Biophys Res Commun 2002, 293, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ozaki, I.; Mizuta, T.; Hamajima, H.; Yasutake, T.; Eguchi, Y.; Ideguchi, H.; Yamamoto, K.; Matsuhashi, S. Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 2006, 25, 6101–6112. [Google Scholar] [CrossRef]
- Yang, H.S.; Jansen, A.P.; Komar, A.A.; Zheng, X.; Merrick, W.C.; Costes, S.; Lockett, S.J.; Sonenberg, N.; Colburn, N.H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol 2003, 23, 26–37. [Google Scholar] [CrossRef]
- Yang, H.S.; Cho, M.H.; Zakowicz, H.; Hegamyer, G.; Sonenberg, N.; Colburn, N.H. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol 2004, 24, 3894–3906. [Google Scholar] [CrossRef]
- Loh, P.G.; Yang, H.S.; Walsh, M.A.; Wang, Q.; Wang, X.; Cheng, Z.; Liu, D.; Song, H. Structural basis for translational inhibition by the tumour suppressor Pdcd4. EMBO J 2009, 28, 274–285. [Google Scholar] [CrossRef]
- Chang, J.H.; Cho, Y.H.; Sohn, S.Y.; Choi, J.M.; Kim, A.; Kim, Y.C.; Jang, S.K.; Cho, Y. Crystal structure of the eIF4A-PDCD4 complex. Proc Natl Acad Sci U S A 2009, 106, 3148–3153. [Google Scholar] [CrossRef]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Palamarchuk, A.; Efanov, A.; Maximov, V.; Aqeilan, R.I.; Croce, C.M.; Pekarsky, Y. Akt phosphorylates and regulates Pdcd4 tumor suppressor protein. Cancer Res 2005, 65, 11282–11286. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.M.; Clegg, J.M.; Uchida, K.A.; Powers, M.A.; Ullman, K.S. Enhanced arginine methylation of programmed cell death 4 protein during nutrient deprivation promotes tumor cell viability. J Biol Chem 2014, 289, 17541–17552. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.A.; Fay, M.M.; Factor, R.E.; Welm, A.L.; Ullman, K.S. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res 2011, 71, 5579–5587. [Google Scholar] [CrossRef]
- Bohm, M.; Sawicka, K.; Siebrasse, J.P.; Brehmer-Fastnacht, A.; Peters, R.; Klempnauer, K.H. The transformation suppressor protein Pdcd4 shuttles between nucleus and cytoplasm and binds RNA. Oncogene 2003, 22, 4905–4910. [Google Scholar] [CrossRef]
- Wedeken, L.; Ohnheiser, J.; Hirschi, B.; Wethkamp, N.; Klempnauer, K.H. Association of Tumor Suppressor Protein Pdcd4 With Ribosomes Is Mediated by Protein-Protein and Protein-RNA Interactions. Genes Cancer 2010, 1, 293–301. [Google Scholar] [CrossRef]
- Xue, C.; Gu, X.; Li, G.; Bao, Z.; Li, L. Expression and Functional Roles of Eukaryotic Initiation Factor 4A Family Proteins in Human Cancers. Front Cell Dev Biol 2021, 9, 711965. [Google Scholar] [CrossRef]
- Wang, Q.; Zhu, J.; Wang, Y.W.; Dai, Y.; Wang, Y.L.; Wang, C.; Liu, J.; Baker, A.; Colburn, N.H.; Yang, H.S. Tumor suppressor Pdcd4 attenuates Sin1 translation to inhibit invasion in colon carcinoma. Oncogene 2017, 36, 6225–6234. [Google Scholar] [CrossRef]
- Fehler, O.; Singh, P.; Haas, A.; Ulrich, D.; Muller, J.P.; Ohnheiser, J.; Klempnauer, K.H. An evolutionarily conserved interaction of tumor suppressor protein Pdcd4 with the poly(A)-binding protein contributes to translation suppression by Pdcd4. Nucleic Acids Res 2014, 42, 11107–11118. [Google Scholar] [CrossRef]
- Liwak, U.; Thakor, N.; Jordan, L.E.; Roy, R.; Lewis, S.M.; Pardo, O.E.; Seckl, M.; Holcik, M. Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol Cell Biol 2012, 32, 1818–1829. [Google Scholar] [CrossRef]
- Brito Querido, J.; Sokabe, M.; Diaz-Lopez, I.; Gordiyenko, Y.; Zuber, P.; Du, Y.; Albacete-Albacete, L.; Ramakrishnan, V.; Fraser, C.S. Human tumor suppressor protein Pdcd4 binds at the mRNA entry channel in the 40S small ribosomal subunit. Nat Commun 2024, 15, 6633. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Zhu, J.; Zheng, H.; Chen, S.; Chen, L.; Yang, H.S. IGF-1R inhibition induces MEK phosphorylation to promote survival in colon carcinomas. Signal Transduct Target Ther 2020, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Wedeken, L.; Singh, P.; Klempnauer, K.H. Tumor suppressor protein Pdcd4 inhibits translation of p53 mRNA. J Biol Chem 2011, 286, 42855–42862. [Google Scholar] [CrossRef]
- Singh, P.; Wedeken, L.; Waters, L.C.; Carr, M.D.; Klempnauer, K.H. Pdcd4 directly binds the coding region of c-myb mRNA and suppresses its translation. Oncogene 2011, 30, 4864–4873. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Kearney, A.L.; Cooke, K.C.; Norris, D.M.; Zadoorian, A.; Krycer, J.R.; Fazakerley, D.J.; Burchfield, J.G.; James, D.E. Serine 474 phosphorylation is essential for maximal Akt2 kinase activity in adipocytes. J Biol Chem 2019, 294, 16729–16739. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes (Basel) 2020, 11. [Google Scholar] [CrossRef]
- Wei, N.A.; Liu, S.S.; Leung, T.H.; Tam, K.F.; Liao, X.Y.; Cheung, A.N.; Chan, K.K.; Ngan, H.Y. Loss of Programmed cell death 4 (Pdcd4) associates with the progression of ovarian cancer. Mol Cancer 2009, 8, 70. [Google Scholar] [CrossRef]
- Zhen, Y.; Li, D.; Li, W.; Yao, W.; Wu, A.; Huang, J.; Gu, H.; Huang, Y.; Wang, Y.; Wu, J.; et al. Reduced PDCD4 Expression Promotes Cell Growth Through PI3K/Akt Signaling in Non-Small Cell Lung Cancer. Oncol Res 2016, 23, 61–68. [Google Scholar] [CrossRef]
- Bera, A.; Das, F.; Ghosh-Choudhury, N.; Kasinath, B.S.; Abboud, H.E.; Choudhury, G.G. microRNA-21-induced dissociation of PDCD4 from rictor contributes to Akt-IKKbeta-mTORC1 axis to regulate renal cancer cell invasion. Exp Cell Res 2014, 328, 99–117. [Google Scholar] [CrossRef]
- Fuchs, S.Y.; Ougolkov, A.V.; Spiegelman, V.S.; Minamoto, T. Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle 2005, 4, 1522–1539. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Yang, H.S. Pdcd4 knockdown up-regulates MAP4K1 expression and activation of AP-1 dependent transcription through c-Myc. Biochim Biophys Acta 2012, 1823, 1807–1814. [Google Scholar] [CrossRef]
- Liu, J.; Zhai, R.; Zhao, J.; Kong, F.; Wang, J.; Jiang, W.; Xin, Q.; Xue, X.; Luan, Y. Programmed cell death 4 overexpression enhances sensitivity to cisplatin via the JNK/c-Jun signaling pathway in bladder cancer. Int J Oncol 2018, 52, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Bitomsky, N.; Bohm, M.; Klempnauer, K.H. Transformation suppressor protein Pdcd4 interferes with JNK-mediated phosphorylation of c-Jun and recruitment of the coactivator p300 by c-Jun. Oncogene 2004, 23, 7484–7493. [Google Scholar] [CrossRef]
- Ozanne, B.W.; McGarry, L.; Spence, H.J.; Johnston, I.; Winnie, J.; Meagher, L.; Stapleton, G. Transcriptional regulation of cell invasion: AP-1 regulation of a multigenic invasion programme. Eur J Cancer 2000, 36, 1640–1648. [Google Scholar] [CrossRef]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef]
- Wang, D.; Hou, Q.; Zhao, L.; Gao, J.; Xiao, Y.; Wang, A. Programmed cell death factor 4 enhances the chemosensitivity of colorectal cancer cells to Taxol. Oncol Lett 2019, 18, 1402–1408. [Google Scholar] [CrossRef]
- Shiota, M.; Izumi, H.; Tanimoto, A.; Takahashi, M.; Miyamoto, N.; Kashiwagi, E.; Kidani, A.; Hirano, G.; Masubuchi, D.; Fukunaka, Y.; et al. Programmed cell death protein 4 down-regulates Y-box binding protein-1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res 2009, 69, 3148–3156. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Bottai, G.; Nuciforo, P.G.; Di Tommaso, L.; Giovannetti, E.; Peg, V.; Losurdo, A.; Perez-Garcia, J.; Masci, G.; Corsi, F.; et al. MicroRNA-21 links epithelial-to-mesenchymal transition and inflammatory signals to confer resistance to neoadjuvant trastuzumab and chemotherapy in HER2-positive breast cancer patients. Oncotarget 2015, 6, 37269–37280. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wu, Y.Q.; Zhang, S.P. MiR-21-5p enhances the progression and paclitaxel resistance in drug-resistant breast cancer cell lines by targeting PDCD4. Neoplasma 2019, 66, 746–755. [Google Scholar] [CrossRef] [PubMed]
- Moustafa-Kamal, M.; Kucharski, T.J.; El-Assaad, W.; Abbas, Y.M.; Gandin, V.; Nagar, B.; Pelletier, J.; Topisirovic, I.; Teodoro, J.G. The mTORC1/S6K/PDCD4/eIF4A Axis Determines Outcome of Mitotic Arrest. Cell Rep 2020, 33, 108230. [Google Scholar] [CrossRef] [PubMed]
- Ajzashokouhi, A.H.; Bostan, H.B.; Jomezadeh, V.; Hayes, A.W.; Karimi, G. A review on the cardioprotective mechanisms of metformin against doxorubicin. Hum Exp Toxicol 2020, 39, 237–248. [Google Scholar] [CrossRef]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J Oncol Pharm Pract 2020, 26, 434–444. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Wu, Y.; Zhu, X.; Shen, R.; Huang, J.; Xu, X.; He, S. miR-182 contributes to cell adhesion-mediated drug resistance in multiple myeloma via targeting PDCD4. Pathol Res Pract 2019, 215, 152603. [Google Scholar] [CrossRef]
- Gonzalez-Ortiz, A.; Pulido-Capiz, A.; Castaneda-Sanchez, C.Y.; Ibarra-Lopez, E.; Galindo-Hernandez, O.; Calderon-Fernandez, M.A.; Lopez-Cossio, L.Y.; Diaz-Molina, R.; Chimal-Vega, B.; Serafin-Higuera, N.; et al. eIF4A/PDCD4 Pathway, a Factor for Doxorubicin Chemoresistance in a Triple-Negative Breast Cancer Cell Model. Cells 2022, 11. [Google Scholar] [CrossRef]
- Schaller, M.D. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Dragoj, M.; Milosevic, Z.; Bankovic, J.; Tanic, N.; Pesic, M.; Stankovic, T. Targeting CXCR4 and FAK reverses doxorubicin resistance and suppresses invasion in non-small cell lung carcinoma. Cell Oncol (Dordr) 2017, 40, 47–62. [Google Scholar] [CrossRef]
- Datta, A.; Bhasin, N.; Kim, H.; Ranjan, M.; Rider, B.; Abd Elmageed, Z.Y.; Mondal, D.; Agrawal, K.C.; Abdel-Mageed, A.B. Selective targeting of FAK-Pyk2 axis by alpha-naphthoflavone abrogates doxorubicin resistance in breast cancer cells. Cancer Lett 2015, 362, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Grem, J.L. Mechanisms of Action and Modulation of Fluorouracil. Semin Radiat Oncol 1997, 7, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, W.; Wang, L.; Zhang, Y.; Zhang, X.; Chen, M.; Wang, F.; Yu, J.; Ma, Y.; Sun, G. MicroRNA-21 induces 5-fluorouracil resistance in human pancreatic cancer cells by regulating PTEN and PDCD4. Cancer Med 2016, 5, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liang, Y.; Wu, K.; Wang, C.; Zhang, T.; Peng, R.; Zou, F. Repressing PDCD4 activates JNK/ABCG2 pathway to induce chemoresistance to fluorouracil in colorectal cancer cells. Ann Transl Med 2021, 9, 114. [Google Scholar] [CrossRef]
- Westermarck, J.; Kahari, V.M. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J 1999, 13, 781–792. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci STKE 2006, 2006, re13. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Song, X.; Liu, C.; Shi, Y.; Wang, Y.; Afonja, O.; Ma, C.; Chen, Y.H.; Zhang, L. Programmed cell death 4 enhances chemosensitivity of ovarian cancer cells by activating death receptor pathway in vitro and in vivo. Cancer Sci 2010, 101, 2163–2170. [Google Scholar] [CrossRef]
- Ren, W.; Wang, X.; Gao, L.; Li, S.; Yan, X.; Zhang, J.; Huang, C.; Zhang, Y.; Zhi, K. MiR-21 modulates chemosensitivity of tongue squamous cell carcinoma cells to cisplatin by targeting PDCD4. Mol Cell Biochem 2014, 390, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, H.; Shen, H.; Li, H. microRNA-106a modulates cisplatin sensitivity by targeting PDCD4 in human ovarian cancer cells. Oncol Lett 2014, 7, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ke, J.; Liu, Y.; Rao, H.; Tang, Z.; Liu, Y.; Zhang, Z.; You, L.; Luo, X.; Sun, Z.; et al. The interaction between PDCD4 and YB1 is critical for cervical cancer stemness and cisplatin resistance. Mol Carcinog 2021, 60, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Evdokimova, V.; Ruzanov, P.; Anglesio, M.S.; Sorokin, A.V.; Ovchinnikov, L.P.; Buckley, J.; Triche, T.J.; Sonenberg, N.; Sorensen, P.H. Akt-mediated YB-1 phosphorylation activates translation of silent mRNA species. Mol Cell Biol 2006, 26, 277–292. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Wang, P.; Mak, V.C.; Cheung, L.W. Drugging IGF-1R in cancer: New insights and emerging opportunities. Genes Dis 2023, 10, 199–211. [Google Scholar] [CrossRef]
- Beckwith, H.; Yee, D. Minireview: Were the IGF Signaling Inhibitors All Bad? Mol Endocrinol 2015, 29, 1549–1557. [Google Scholar] [CrossRef]
- Pitts, T.M.; Tan, A.C.; Kulikowski, G.N.; Tentler, J.J.; Brown, A.M.; Flanigan, S.A.; Leong, S.; Coldren, C.D.; Hirsch, F.R.; Varella-Garcia, M.; et al. Development of an integrated genomic classifier for a novel agent in colorectal cancer: approach to individualized therapy in early development. Clin Cancer Res 2010, 16, 3193–3204. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Q.; Chen, L.; Yang, H.S. Inhibition of p70S6K1 activation by Pdcd4 overcomes the resistance to an IGF-1R/IR inhibitor in colon carcinoma cells. Mol Cancer Ther 2015, 14, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Ekyalongo, R.C.; Mukohara, T.; Kataoka, Y.; Funakoshi, Y.; Tomioka, H.; Kiyota, N.; Fujiwara, Y.; Minami, H. Mechanisms of acquired resistance to insulin-like growth factor 1 receptor inhibitor in MCF-7 breast cancer cell line. Invest New Drugs 2013, 31, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Meng, Q.; Vogt, P.K.; Zhang, R.; Jiang, B.H. A downstream kinase of the mammalian target of rapamycin, p70S6K1, regulates human double minute 2 protein phosphorylation and stability. J Cell Physiol 2006, 209, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.D.; Donner, D.B. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci 2002, 27, 462–467. [Google Scholar] [CrossRef]
Figure 1.
Pdcd4 is a tumor suppressor. Experimental evidence has shown that Pdcd4 suppresses various cancer cell characteristics including inflammation, proliferation, survival, invasion, metastasis. Pdcd4 also promotes cell apoptosis and overcomes drug resistance.
Figure 1.
Pdcd4 is a tumor suppressor. Experimental evidence has shown that Pdcd4 suppresses various cancer cell characteristics including inflammation, proliferation, survival, invasion, metastasis. Pdcd4 also promotes cell apoptosis and overcomes drug resistance.
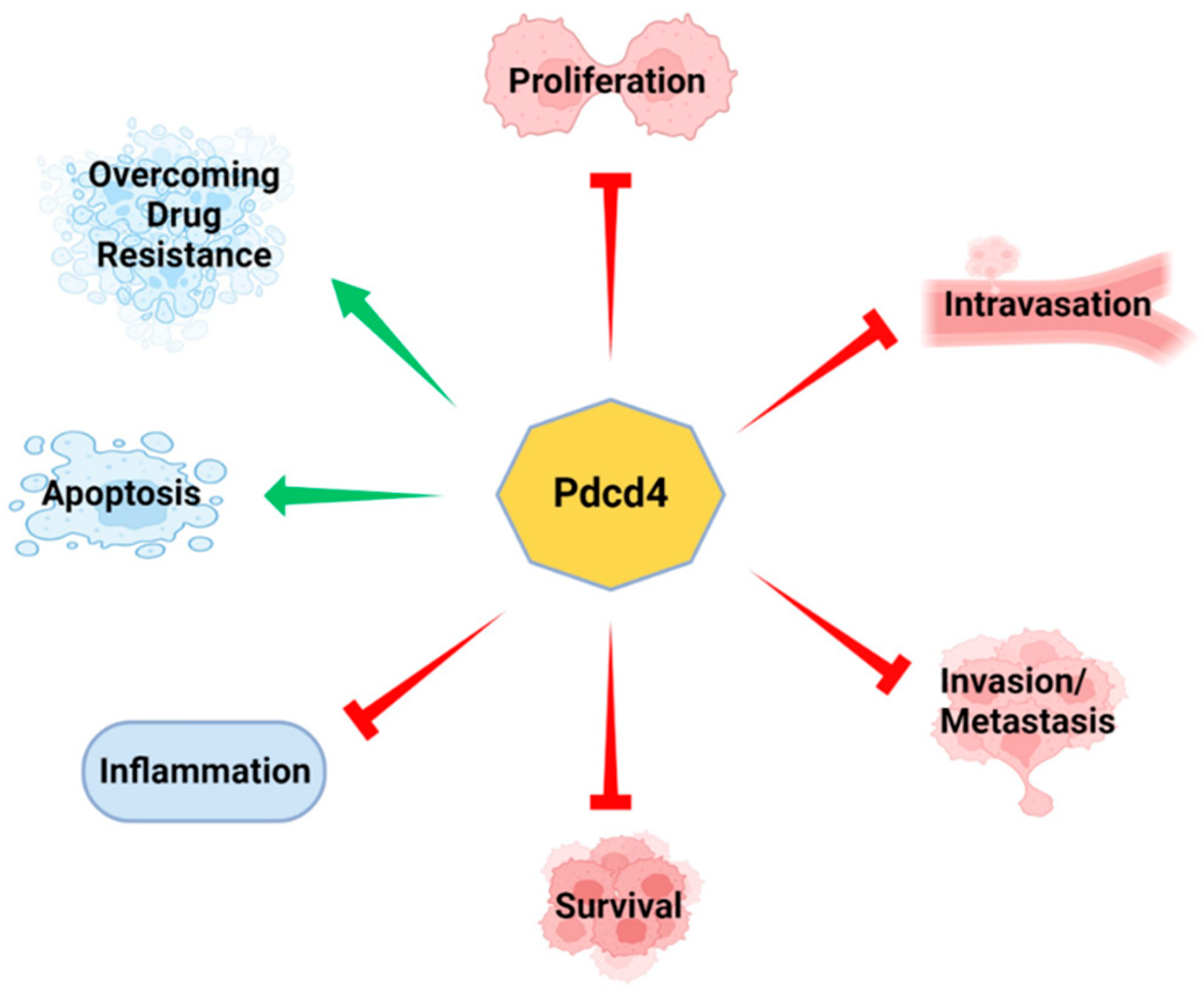
Figure 2.
Schematic diagram of the functional motifs of Pdcd4. Pdcd4 features: (1) Pdcd4 use two MA-3 domains to bind with eIF4A to inhibit protein translation; (2) The Ser67 can be phosphorylated by Akt or p70S6K for proteasome degradation; (3) The Ser457 can be phosphorylated by Akt for nuclear localization; (4) The Arg110 can be methylated by PRMT% to attenuate the tumor suppression function; (5) The positive amino acid cluster (+++) has been suggested to bind with RNAs.
Figure 2.
Schematic diagram of the functional motifs of Pdcd4. Pdcd4 features: (1) Pdcd4 use two MA-3 domains to bind with eIF4A to inhibit protein translation; (2) The Ser67 can be phosphorylated by Akt or p70S6K for proteasome degradation; (3) The Ser457 can be phosphorylated by Akt for nuclear localization; (4) The Arg110 can be methylated by PRMT% to attenuate the tumor suppression function; (5) The positive amino acid cluster (+++) has been suggested to bind with RNAs.
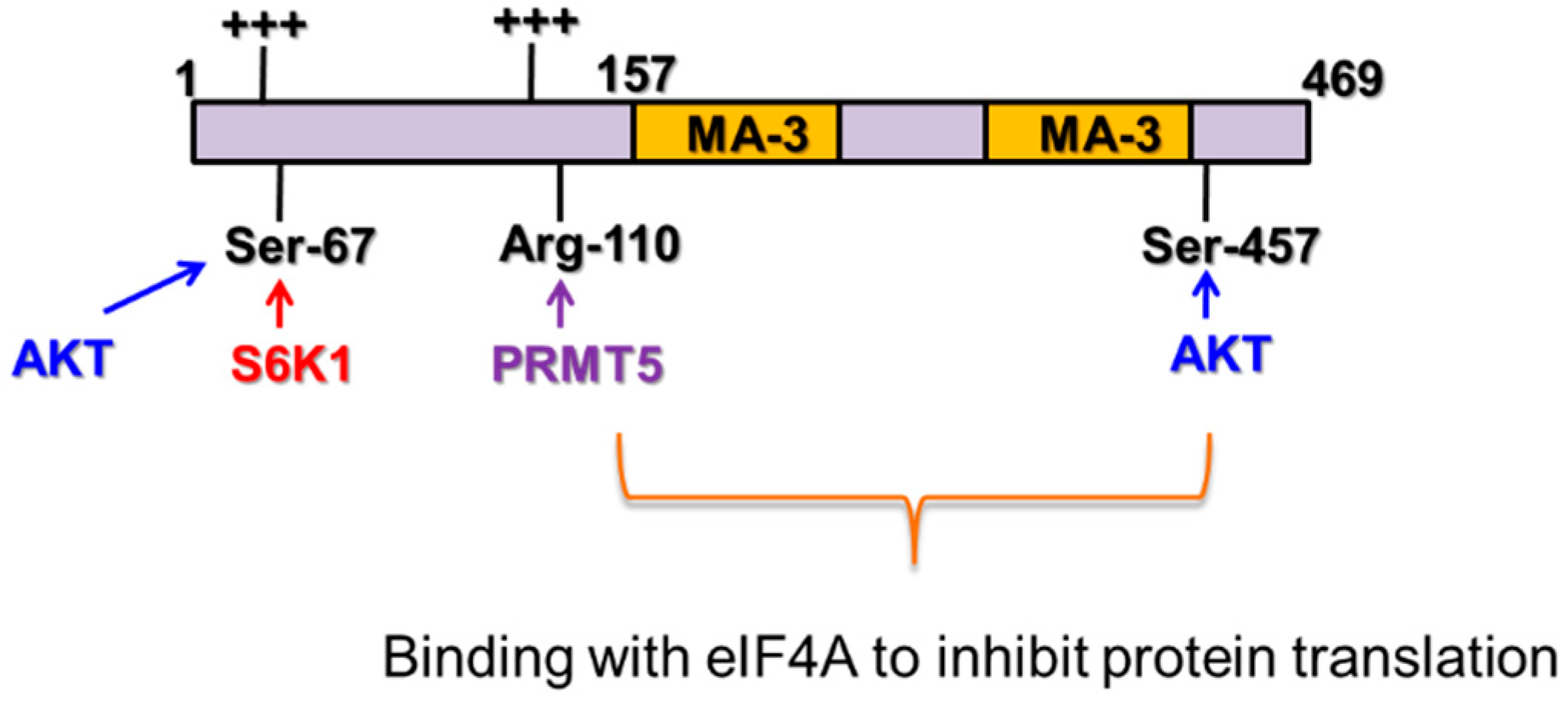
Figure 3.
Pdcd4 suppresses mTORC2-Akt pathway.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



