
Submitted:
21 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
Alzheimer's disease (AD) is the most prevalent form of dementia. With the global aging population, the incidence of AD and related dementias is projected to rise dramatically, posing significant public health challenges. Traditionally, the diagnosis of AD has relied on the detection of amyloid-beta (Aβ) plaques and neurofibrillary tangles of hyperphosphorylated tau in the brain, assessed through invasive cerebrospinal fluid (CSF) analysis and costly positron emission tomography scans. However, these methods are not feasible for widespread screening or use in low-resource settings. Recent advances in blood-based biomarkers offer a promising alternative for the early detection and monitoring of AD. These biomarkers provide a minimally invasive, cost-effective means of tracking the pathological progression of AD before the onset of clinical symptoms, potentially enabling earlier and more accurate diagnosis. Blood-based biomarkers, including those related to Aβ, phosphorylated tau, and neurofilament light polypeptide, have demonstrated significant potential in reflecting the underlying neuropathology of AD and correlating with traditional CSF and imaging biomarkers. This review synthesizes the latest research on blood-based biomarkers in AD, highlighting their role in the disease's etiopathogenesis, diagnostic accuracy, and prognostic value. We discuss the emerging evidence on proteinopathies, neuroinflammation, vascular pathology, and bioenergetic profiles, emphasizing the clinical applicability of these biomarkers. Furthermore, we explore the integration of blood-based biomarkers with advanced neuroimaging techniques and their potential to revolutionize the diagnostic landscape of AD, particularly in resource-limited settings. We have integrated protein-protein interaction (PPI) and miRNA network analyses to enhance the analysis. By incorporating these, we can explore the interactions and relationships between different proteomic, genomic, and epigenomic biomarkers, potentially unveiling new insights into the underlying biological mechanisms of AD. The continued development and validation of these biomarkers could lead to more accessible and equitable AD diagnostics, ultimately improving patient outcomes.
Keywords:
Alzheimer's disease (AD)
; blood-based biomarkers
; amyloid-beta (Aβ)
; phosphorylated tau (p-Tau)
; neurofilament light polypeptide (NfL)
; non-invasive diagnostics
; neurodegeneration
; proteinopathy
; neuroinflammation
; vascular pathology
; early detection
; prognostics
; advanced neuroimaging
; dementia
1. Background
Neurodegeneration in Alzheimer's disease (AD) is associated with toxic amyloid-beta oligomers, protein aggregates, intra-neuronal neurofibrillary tangles consisting of hyperphosphorylated microtubule-associated tau protein, synaptic dysfunction, reduced cerebral glucose metabolism, and mitochondrial dysfunction [1]. Undoubtedly, AD accounts for more than 50-70% of cases among all neurodegenerative dementias, and it is estimated that approximately 44 million people worldwide are living with AD dementia, a number that could triple by the year 2050 [2]. Notably, only a tiny percentage (1%) of AD is inherited, known as early-onset AD (EOAD). Most cases are sporadic and generally appear after age 65, also known as late-onset AD (LOAD) [3]. The age of onset for LOAD can vary between countries. It is typically 65 in the USA, whereas in India, it generally develops after 60 years of age [3]. In addition to genetic factors, other factors, including reduced physical activity, poor diet, diabetes, cerebrovascular disease, and stress, are major risk factors responsible for the disease's progression.
According to the 2018 National Institute on Aging-Alzheimer's Association (NIA-AA) framework, AD should be considered in a biological context rather than a syndromal context using an A/T/N classification system. In this system, "A" represents the concentration of Aβ biomarkers, "T" means the level of tau, and "N" reflects biomarkers of neurodegeneration [4]. Such categorization prioritizes the classification of AD biomarkers according to the pathological mechanism. However, this classification framework also assumes the equivalence of cerebrospinal fluid (CSF) and imaging biomarkers within each AT(N) category [5]. Ample evidence reveals that this is not always the case. The AD diagnosis can be strengthened by including other biomarkers that reflect parameters like brain vascularity changes, Lewy body pathology markers, and neuroinflammation [5].
Notably, in recent years, the understanding of AD has moved from diagnosing and characterizing AD based on clinical presentation alone to diagnosing the disease biologically. Biologically based diagnosis and disease staging is now transitioning from priorities dominated by research alone to priorities required for research and clinical care. The high cost of positron emission tomography (PET) and the invasiveness of CSF sampling are significant obstacles to population screening to detect potentially manageable pre-clinical AD [6]. From this purview, minimally invasive sampling from blood and fluid-based components holds great promise to revolutionize AD's diagnostic and prognostic work-up in day-to-day clinical practice. This approach is particularly timely considering the recent advent of anti-amyloid-β immunotherapies. Using and standardizing plasma and fluid-based assays and other non-invasive neuroimaging techniques would reduce the need for more costly and invasive investigations involving CSF samples or PET scans [7].
This review summarizes evidence on blood and fluid-based biomarkers that can be used in clinical practice to diagnose AD due to their minimal invasiveness. Along with recent developments in noninvasive neuroimaging techniques, we aim to integrate an understanding of various aspects of this heterogeneous disease that can facilitate the development of new diagnostic tools.
2. Methodology
We conducted a comprehensive search of databases, including PubMed, Embase, and Google Scholar, using specific search terms: "Alzheimer's disease," "Alzheimer's disease and related dementia," AND "plasma," OR "blood," OR "serum," AND "biomarkers,"
We also reviewed reference lists to locate relevant articles that were not captured in the initial search. Our inclusion criteria prioritized articles published in English and focused on plasma biomarkers for AD in the last ten years.
We successfully created our network model using the STRING database 12.0 (https://string-db.org/). The query proteins were uploaded with their specific gene symbol as obtained from the National Center for Biotechnology Information (NCBI) database. We then created our network using the following functional settings: a complete STRING network showing functional and physical interactors of the query proteins, with network edges based on evidence from text-mining, co-expression, curated databases, gene neighborhood, gene fusions, gene co-occurrence, and protein homology. The minimum interaction score required was kept at a high confidence level (≥ 0.700)
The interaction network of the predicted AD miRNA biomarkers with validated target genes was achieved and visualized in the miRNeT 2.0 database (https://www.mirnet.ca/) to prioritize the selective miRNA biomarkers and their roles in gene regulation in both peripheral blood and brain tissue. To cluster the final enlisted miRNA biomarkers, we selected the parameters like organism: H. sapiens (human), ID type: miRBase ID, Tissue types: Peripheral blood and brain, Targets: miRTarBase v9.0 and used two network topology features, degree and betweenness centrality on the miRNA-miRNA network.
3. Results/Discussion
3.1. Current Insights into Different Biomarker Categorizations.
The development of in-vivo biomarkers has shifted the diagnosis of AD from the late or advanced dementia stages of the disease to earlier stages. It has introduced the potential for pre-symptomatic diagnosis. Categorization of biomarkers refers to grouping biomarkers into categories that reflect a common proteinopathy pathway or pathogenic process. According to the recent recommendation by the Alzheimer's Association workgroup, AD biomarkers can be broadly categorized into:
- Core biomarkers of AD neuropathological changes
- Non-specific biomarkers that are important in AD pathogenesis but are also involved in other brain diseases
- Biomarkers of common non-AD pathologies
Such broad categorization and sub-categorization rely on specific proteinopathy pathways or a pathogenic sequence. Importantly, imaging biomarkers reveal the cumulative effects and capture topographic data based on the established neuropathic construct. In contrast, blood or fluid-based biomarkers generally represent the dynamics of analytes' production/clearance ratio at a given time.
The recent updates, as per the international working group, incorporate recently developed blood-based markers (BBM) of "A," "T," and (N) [8,9]. Core AD biomarkers, according to recent updates, are those in "A" (Aβ) and "T" (tau) categories, where the A category represents Aβ proteinopathy pathway-related biomarkers; it is well known that soluble aggregation-prone Aβ peptides are the critical building units of insoluble Aβ fibrillary aggregates in plaques, and it represents the temporal relation of different biochemical pools of the proteinopathy pathway through fluid-based assay and imaging findings [9] [Figure-1(A)].
Figure 1.
Figure 1(A): From ATN to AT1T2NIVS biomarker categorization of fluid analyte: The proposed new criteria by the NIA-AA 2024 working group solicits 'A' and 'T' as the core biomarkers for diagnosis and staging of AD. Besides, the revised scheme also recognizes an expanded suite of additional markers that detect non-specific biomarkers involved in AD pathophysiology ('N' and 'I') and non-AD co-pathological biomarkers ('V' and 'S'). The core category also distinguishes between core 1 and core 2 biomarkers. Figure 1(B) depicts the biomarker profile and respective categorization based on the "A," "T," and "N" systems. Binarizing the three AT(N) biomarker types leads to eight biomarker profiles. Based on the biomarker profile, each individual can be placed into one of the three general biomarker categories: standard AD biomarkers, the Alzheimer's continuum, and those with non-AD pathological changes.
Figure 1.
Figure 1(A): From ATN to AT1T2NIVS biomarker categorization of fluid analyte: The proposed new criteria by the NIA-AA 2024 working group solicits 'A' and 'T' as the core biomarkers for diagnosis and staging of AD. Besides, the revised scheme also recognizes an expanded suite of additional markers that detect non-specific biomarkers involved in AD pathophysiology ('N' and 'I') and non-AD co-pathological biomarkers ('V' and 'S'). The core category also distinguishes between core 1 and core 2 biomarkers. Figure 1(B) depicts the biomarker profile and respective categorization based on the "A," "T," and "N" systems. Binarizing the three AT(N) biomarker types leads to eight biomarker profiles. Based on the biomarker profile, each individual can be placed into one of the three general biomarker categories: standard AD biomarkers, the Alzheimer's continuum, and those with non-AD pathological changes.
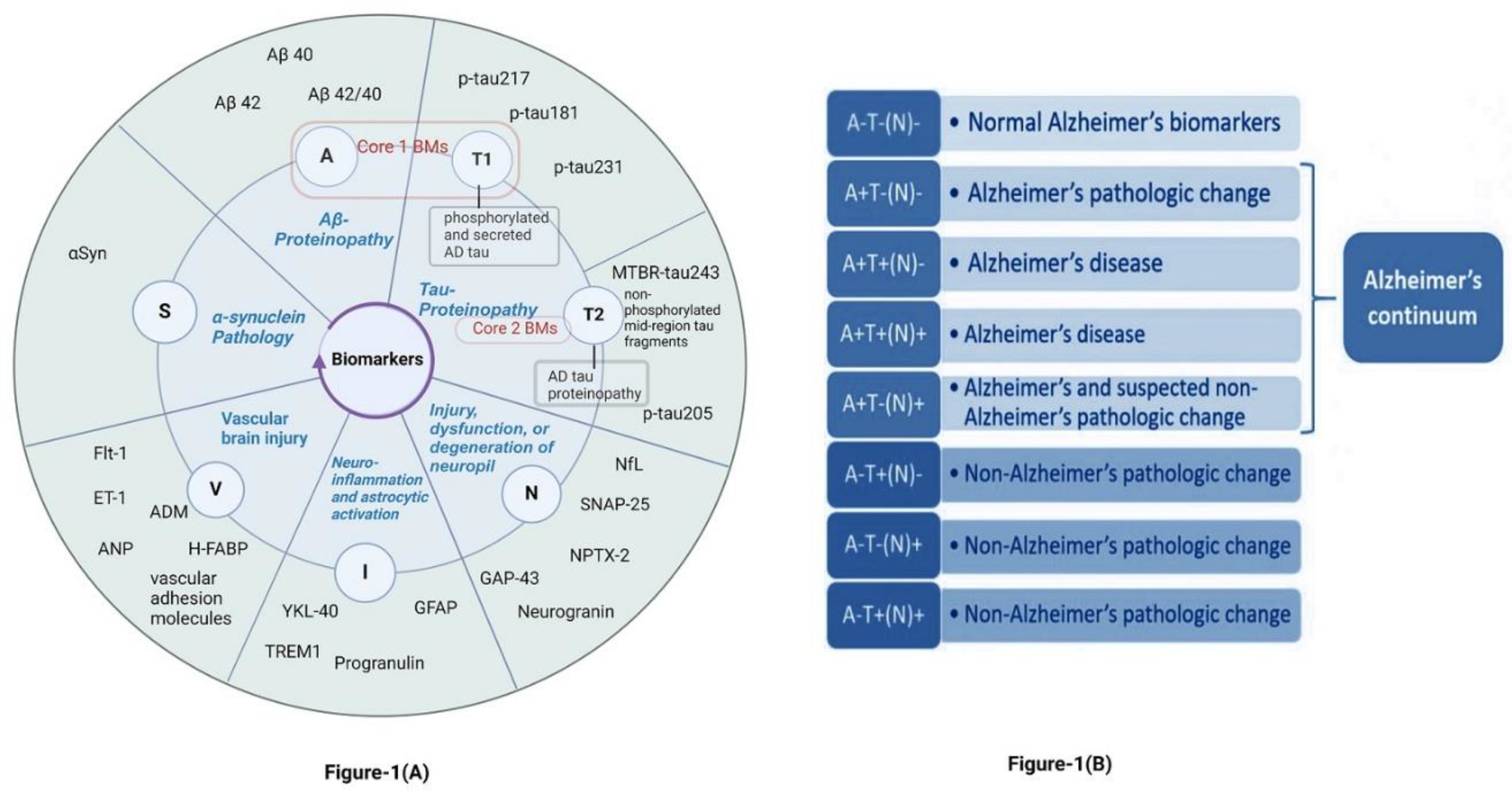
Accordingly, "T" biomarkers also exhibit varying temporal relationships across the spectrum. These differences can be categorized into subcategories: T1 and T2. T1 represents tau PET imaging or biofluid analytes of soluble tau fragments that react to amyloid plaques or soluble Aβ species in the plaque penumbra. T2 refers to tau PET imaging or biofluid analytes that indicate the presence of AD tau aggregates. Consequently, core biomarkers (A and T1) define the initial stage of AD as detectable in vivo and capable of identifying AD in symptomatic and asymptomatic individuals.
On the contrary, Core 2 biomarkers (T2 category) include tau-PET and specific soluble tau fragmented residues related to tau-proteinopathy and generally reflect significant evolution of AD pathogenesis. Hence, Core 2 biomarkers do not detect the initial presence of the disease and are highly associated with Aβ pathology. Therefore, Core 2 biomarkers, combined with Core 1, may be used to stage the biological disease severity [9].
In general, AD biomarkers can be classified into three major categories:
[A] Diagnostic markers (PET imaging and CSF analysis for Aβ and Tau)
[B] Blood-based markers: Protein and microRNA analyzed from whole blood or serum
[C] Fluid-based biomarkers: saliva and urine
[D] Non-invasive neuroimaging techniques: functional near-infrared spectroscopy (fNIRS), magnetoencephalography, structural MRI, diffusion tensor imaging, single-photon emission computed tomography, magnetic resonance spectroscopy, and functional MRI.
Notably, in neuroimaging with MRI, different morphometric methods for data processing have significantly improved clinical understanding. In this review, we will discuss the recent advancements in blood-related biomarkers as novel approaches.
3.2. The In-Hitherto Context for Developing Blood-Based Biomarkers in AD
The development of blood-based AD biomarkers is increasingly integral to research and clinical evaluation, from understanding the disease etiopathogenesis to disease monitoring and therapeutic efficacy. They are pivotal in swift clinical decisions and optimizing resource allocation and healthcare efficiency. Furthermore, they are crucial in population screening for early detection of AD and participant screening to determine eligibility for AD-related clinical trials [10]. Hence, adopting this strategy can positively contribute to public health scenarios, especially in middle and low-income countries. The versatile application of blood-based biomarkers can effectively harness the potential of ongoing research and innovation to enhance better outcomes for individuals diagnosed with AD.
Furthermore, blood-based biomarker tests, offering a non-invasive alternative to CSF study or PET scans, can aid in diagnosing AD in symptomatic patients with cognitive impairment. More importantly, they can guide therapeutic decisions, paving the way for personalized treatment and disease management. In resource-limited clinical facilities, where performing CSF analysis or PET scans is challenging as a routine procedure, blood-based screening of patients with dementia can be a practical solution [11]. Looking ahead, the potential of blood-based screening to effectively identify AD in asymptomatic individuals is a promising prospect. This could empower early interventions, potentially slowing down the onset and progression of dementia, thereby reducing the disease burden and improving long-term outcomes. This approach can foster early detection, accurate diagnosis, and personalized therapeutic intervention in AD management, transforming the field into a state-of-the-art [Figure 2]. Ultimately, large-scale validation studies can help establish the reliability, sensitivity, and specificity of blood-based biomarkers across different populations, aiding in AD diagnosis and patient stratification based on disease severity and progression in clinical settings.
3.3. BBBMs Related to Proteinopathy in Early Detection of AD
Research on AD has shown that proteins expressed in brain tissues can be found in peripheral circulation. Blood-based biomarkers related to underlying proteinopathy are emerging as crucial tools in identifying AD-related pathology. Several studies have linked proteinopathy-related plasma biomarkers with corresponding PET imaging, CSF biomarkers, and cognitive staging [12] [Figure 3], highlighting the growing significance of blood-based biomarkers in the field.
3.3.1. Plasma Proteinopathy-Related Biomarkers of Core AD Pathology:
Core AD-related biomarkers in plasma are those in the A (Aβ) and T (Tau) categories. The A category denotes biomarkers of Aβ proteinopathy pathways and reflects on the "stage of the biochemical pool" of different Aβ aggregates. T represents “the timing relationship” across the spectrum of differentially modified forms of Tau proteins.
3.3.1.1. Aβ and Its Variations in Plasma:
Identifying and validating accurate and reliable blood-based markers of amyloid proteinopathy has been tremendously challenging. This is because plasma or serum Aβ42 levels are 10-100 times lower than their CSF counterpart, and Aβ structural epitopes can be masked by their binding affinity to plasma proteins [13]. Additionally, the variable peripheral source of Aβ and these factors present reliable and consistent measures of Aβ in peripheral circulation across different laboratories and study cohorts of the various populations using the conventional enzyme-linked immunoassay [14]. However, recent advances in peripheral Aβ measurement using cutting-edge techniques, including an immunomagnetic reduction, single-molecule array (SIMOA), immunoprecipitation (IP), and liquid chromatography-mass spectroscopy (LC-MS), are adding significant advantages for accurate quantification and standardization of peripheral Aβ level in AD across different laboratories [15,16,17]. In a head-to-head study including ten different assays, the LC-MS method displayed the best diagnostic performance among all tested assays [17].
Notably, the advancement of SIMOA has allowed the measurement of Aβ with high precision. It has already demonstrated the ability to measure and demonstrate the plasma Aβ40/Aβ42 levels with precision to accurately predict amyloid-positive PET scans in both cognitively normal and impaired individuals [13]. However, the study by Blennow and Zetterberg et al. [18] revealed that AD patients with pathological CSF signatures showed significant differences in plasma Aβ42 from control, implying a limited potential of plasma Aβ for distinguishing pre-clinical AD with CSF pathologies. Interestingly, a study by Guo et al. revealed different dynamic trends throughout the AD disease continuum. The plasma Aβ42/40 levels were significantly reduced in the cognitively unimpaired (CU) A+T+ compared with the CU A-T- group. Similar trends were observed between the mild cognitive impairment (MCI) or AD dementia group and the CU A-T- group. Additionally, the AD dementia group depicted reduced plasma Aβ 42/40 level relative to CU A+T- and MCI+ groups. However, individual plasma levels of Aβ42 and Aβ40 were unchanged except for increased AB40 levels in the AD dementia group compared to the CU A-T- group [19].
Plasma composite biomarkers (normalized scores for APP669–711/Aβ1-42 and Aβ1-40/Aβ1-42) demonstrated a strong relationship with 80.4% accuracy between plasma and CSF levels among patients with AD. Such association performs comparably to CSF Aβ42 in determining brain Aβ burden. Using IP-MS, Nakamura et al. [20] showed that plasma Aβ better-predicted brain Aβ burden than when subjects were classified using Aβ PET. Furthermore, Schindler SE et al. [21] showed that the plasma Aβ 42/40 ratio, combined with age and Apolipoprotein E status, can reflect good accuracy in diagnosing brain amyloidosis with an LC-MS technique. In 2017, a study by Ovod et al. [22] showed that the plasma Aβ42/40 ratio measured by LC-MS had good accuracy with an AUC of 0.88 to differentiate amyloid positivity in Aβ-PET or CSF. The relationship between Aβ biomarker abnormality and AD progression has been depicted in Figure 3.
However, the challenges to the widespread use of plasma Aβ as a surrogate measure of brain amyloid pathology are significant. On this note, it is essential to mention that the differences in plasma Aβ levels between Aβ-PET (+) and Aβ-PET (-) are only around 10-15%, whereas it is around 40%-60% when measured in CSF [23]. Henceforth, using a combination of biomarkers usually improves the overall accuracy of Aβ measurement. Understanding the impact of cohort differences, processing procedures in different analytical methods, and the role of other AD risk factors has improved plasma Aβ assays' diagnostic properties. Using the clinically available PrecivityADTM test, an LC-MS-based method, a plasma Aβ42/40 ratio cut-off value of 0.0975 had an AUC of 0.81 and an accuracy of 75% when adjusting to cohort differences, the diagnostic performance improved the AUC to 0.86 and the accuracy to 81%. The AUC increased to 0.90 and the accuracy to 86% with additional adjustments for age and Apolipoprotein E status. The diagnostic accuracy of this method was not significantly affected by potential confounding variables like differences in plasma sample collection in different cohorts [24].
3.3.1.2. Plasma p-Tau:
In the recent updates by the NIA-AA working group, different subtypes of Tau have been placed in the T1 (Core-1) and T2 (Core-2) sub-categories within the core AD-related pathological markers [9]. The fundamental concepts of Core-1 and Core-2 AD biomarkers in these recent updates are differentiated by the timing of cognitive abnormality onset, where the Core-1 category represents the initial stage of AD neuropathological changes in-vivo in both symptomatic and asymptomatic individuals, whereas Core-2 biomarkers reflect strongly on Aβ pathology. Hence, Tau accumulation is a more precise biomarker for cognitive decline and is strongly associated with underlying AD pathology. It also predicts the risk of future dementia among individuals with MCI [25]. So far, the CSF total tau is a non-specific marker as it is found to be elevated in traumatic brain injury and acute stroke. Hence, it is more of a rational marker for neuronal injury.
Tau's physiological role is to stabilize microtubules in neurons' axons, and the degeneration of neuraxial structures results in increased Tau release from neuronal structures [26]. Additionally, tau undergoes truncation and subsequent phosphorylation, leading to NFT aggregation in the proximal axoplasm. Abnormal phosphorylation and truncation of the Tau protein are the main etiologies for NFT formation in AD and other tauopathies.
The tau protein bears multiple phosphorylation sites. T1 represents the ‘phosphorylated and secreted tau’ (p-tau217, p-tau181, p-tau 231), and T2 represents ‘AD tau proteinopathy' (microtubular-binding region-tau243, p-tau205, non-phosphorylated mid-region tau fragments) [9]. Like plasma Aβ, the challenge for developing plasma-based Tau is the significantly lower concentration of Tau in the blood than in the CSF. The CSF level of Tau is about 2–300 pg/ml, while the plasma concentration is approximately 100-fold lower, about 5 pg/ml.
Significant progress has been made in developing highly sensitive assays, especially MS-based techniques, which have greatly improved plasma p-tau identification and quantification. Plasma p-tau181 is strongly associated with the Aβ-PET and CSF P-tau181 measures and portrays high specificity to discriminate AD from other tauopathies [27,28]. Additionally, plasma p-tau 181 has been found to distinguish between Aβ-PET (+) and Aβ-PET (-) individuals along with the disease progression to dementia and tau-burdened brain areas with AD-related atrophic changes [29]. In this context, T1-related plasma-tau variations, i.e., p-tau217, p-tau181, p-tau231, are significantly higher in AD patients than cognitively unimpaired ones. Interestingly, a study by Guo et al. [19] revealed the dynamic trend of plasma p-tau181 throughout the disease continuum [Figure 1(B)]; plasma p-tau181 level increases in CU A+T+ compared to CU A-T-. A similar trend was observed in the MCI+ or AD dementia group in comparison to CU A-T-. At the same time, only p-tau181 levels were higher in CU A+T+ and MCI+ groups in contrast to the CU A+T- group.
Notably, plasma p-tau217 has been found to show more precision in diagnostic accuracy than p-tau 181 in CSF and plasma and also accurately predicted the progression of individuals from subjective cognitive decline (SCD) and MCI to dementia when combined with other risk factors [30]. Ashton et al., using the Simoa-based assay, revealed that plasma p-tau231, like CSF p-tau217, could discriminate patients with and without AD-pathology during post-mortem assessment with an AUC of 0.99 [31].
Additionally, a study using the BioFINDER cohort reported that the plasma p-tau217 could accurately predict (AUC=0.83) the progression from MCI to AD within four years, and the diagnostic accuracy further increased (AUC=0.91) when plasma p-tau217 was added with Apolipoprotein E genotype [32]. Janelidze et al. [33] also revealed that plasma p-tau217 was significantly elevated before tau-PET became positive in cognitively unimpaired Aβ-PET+ older individuals.
Interestingly, studies by Gonzalez Ortiz et al. [34] on brain-derived tau (BD-Tau) from the plasma of AD individuals have shown that it outperforms plasma and total tau. Unlike Nfl, BD-tau has shown better specificity towards AD-related neurodegeneration. Hence, it demonstrates a potential to complete the blood AT(N) scheme as a valuable biomarker for AD-dependent neurodegeneration evaluation. Notably, the plasma core 2 biomarkers, mainly those in the T2 category, including specific soluble tau fragments associated with tau proteinopathy and non-phosphorylated tau fragments, are still in the pre-clinical development phase [35].
Based on these recent advancements, it is pretty appalling that plasma p-tau estimation can precisely diagnose AD based on its clinical and pathological criteria. Additionally, it can identify individuals in the early stages of AD and monitor the pathological continuum for individuals at higher risk of cognitive decline. However, it should be remembered that the large variability in plasma p-tau measurement across the different analytical platforms and the lack of universally accepted biomarkers' cutoff value precludes its rational widespread usage, most notably in clinical settings.
3.3.2. Proteinopathy-Related Biomarkers of Non-Core AD Pathology
Apart from the proteinopathy-associated core AD plasma biomarkers, other plasma-based molecules due to underlying proteinopathy of non-core AD pathogenesis have also been extensively studied.
3.3.2.1. Biomarker of TAR-DNA Binding Proteinopathy:
TAR DNA-binding protein 43 (TDP-43) is a nuclear protein encoded by the TARDBP gene and is involved in various aspects of RNA processing, including transcription, splicing, and transport. It is a 43-kDa protein initially identified as a binding protein to the TAR (Trans-Activation Response) element of the HIV-1 virus [36,37]. TDP-43 pathology has also been observed in a subset of AD cases, particularly in association with hippocampal sclerosis, suggesting its involvement in the broader spectrum of neurodegenerative disorders [38]. Hippocampal Sclerosis and TDP-43 are presumably part of the later neuropathological changes in AD. Given its role in neurodegenerative diseases, there has been interest in using blood TDP-43 levels as a potential biomarker for conditions like Amyotrophic Lateral Sclerosis (ALS), Frontotemporal Dementia (FTD), and AD. Elevated levels of TDP-43 in blood, particularly in plasma or serum, have been observed in some studies of ALS and FTD patients, suggesting its potential utility as a non-invasive biomarker [39].
One of the many challenges with measuring TDP-43 in blood is the sensitivity and specificity of the assays used. TDP-43 exists in multiple forms (e.g., full-length, truncated, phosphorylated), and distinguishing these forms can be technically demanding [40,41]. Additionally, because TDP-43 is ubiquitously expressed in various tissues, its presence in blood may not always correlate directly with neurodegenerative disease [42].
TDP-43 and its phosphorylated derivative form can be measured in a platelet lysate. A-Phospho (S409/410-2) TDP-43 is identified as a selective antibody for AD patients that discriminates AD from non-demented control and amyotrophic lateral sclerosis (based on platelet analysis of phospho-TDP43. This AD-selective antibody may be a screening tool to strengthen AD diagnosis and cognitive tests [43]. Patient populations with mild cognitive impairment, mild dementia, and frontal lobe dementia need to be assessed for the phosphorylated TDP-43 profile.
3.3.2.2. Blood-Based Biomarkers of Synuclein Pathology:
The brain afflicted by AD is neuropathologically defined by extracellular amyloid-β plaques and neurofibrillary tangles composed of hyperphosphorylated tau proteins accumulating intraneuronally [44]. However, accumulating evidence suggests that the presynaptic protein α-synuclein (αSyn), well known to be associated with Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy (MSA), is involved in the pathophysiology of AD [45]. Lewy-related pathology, primarily comprised of αSyn, is present in a majority of autopsied AD brains, and higher levels of αSyn in the cerebrospinal fluid (CSF) of patients with MCI and AD have been linked to cognitive decline [44]. Recent studies also suggest that the asymptomatic accumulation of Aβ plaques is associated with higher CSF αSyn levels in subjects at risk of sporadic AD and in individuals with autosomal dominant AD [44]. Experimental evidence has further linked αSyn mainly to tau hyperphosphorylation and the pathological actions of Aβ and the Apolipoprotein E ε4, the latter being a major genetic risk factor for AD and dementia with Lewy bodies [46].
Therefore, the need for measuring αSyn in body fluids, especially blood, becomes the need of the hour to detect early AD pathology. However, the major hindrance in this is the non-neurological sources of αSyn like RBC, which have them in abundance. Interestingly, 99% of the total blood αSyn is found in blood components; among those 99 %, the majority lies within RBCs [47]. Now, due to their abundance and fragility, the lysed RBCs can release αSyn in various fluid compartments of the body like blood (where they usually remain), CSF (where they can accidentally enter), thus leading to elevation of αSyn in the plasma or CSF[47].
Studies have shown that while total α-synuclein levels in the blood may not differ significantly between patients with neurodegenerative diseases and healthy controls, the levels of oligomeric or phosphorylated forms of α-synuclein might be more closely associated with disease states [47]. Thus, Assays with high specificity, which can detect even the slightest of variations in blood αSyn and can also differentiate effectively between the various forms of αSyn, are the need of the hour. However, in a study, it was shown that the fall of αSyn in CSF in Lewy body dementia (LBD) patients was more than that compared to AD patients or healthy controls, indicating that the αSyn was accumulating more in brain matter than in CSF in LBD patients. However, for AD, we can see that the fall of αSyn in CSF was not very pronounced and was comparable to that in healthy adults [47,48].
Regarding blood levels, a study conducted by Daniele et al. [49] showed a significant difference in levels of αSyn /tau and αSyn /Aβ 42 heterodimers in the centrifuged RBCs of healthy controls and those with AD. Another study by Laske et al. showed that the serum levels of αSyn in AD and healthy controls were not much different. Still, both significantly differed from the serum αSyn levels in LBD patients. Thus, the correlation between serum αSyn levels of healthy controls and AD is still in question and has not been adequately established [50].
Several studies suggest that Aβ 42, tau, and α-syn interact in vivo to promote aggregation and accumulation of each other and accelerate cognitive dysfunction [51]. Interestingly, their expression levels and aggregation processes are not restricted to the brain but reach peripheral tissues by the bloodstream, possibly configuring a systemic disease [52].
3.3.2.3. Serum Dickkopf-1(DKK1) as Candidate Blood-Based Biomarker in AD:
DKK-1 or Dickkopf-1 is a critical member of the DKK protein family. It is a secretory glycoprotein and has its most significant role in determining the fate of cells in vertebrates. Besides, it has been found lately to be linked to neurodegeneration as well as regeneration, and its role in AD has been the latest topic of research [53]. It is an endogenous indirect inhibitor of the WNT/beta-catenin pathway, which has its role established in embryogenesis and adult homeostasis. Some studies have shown that the occurrence of AD and the patient’s cognitive decline was associated with dysregulation of the DKK1 and Wnt-signalling, including familial or early-onset AD and sporadic or late-onset AD [54]. DKK1 expression level increased significantly in the cerebrospinal fluid, plasma, and brain tissue of AD patients and AD transgenic mice [53].
Studies performed by Caricasole et al. [54] suggested that DKK-1 induction has a role in initiating the pathological cascade reaction of Aβ and increasing the phosphorus action of Tau protein. Other studies have indicated that the role of DKK1 was played by inhibiting the endogenous WNT ligands that were important for the maintenance of neural synapses [55]. Knockdown of the expression of DKK1 by siRNA in the hippocampus increased the regeneration of hippocampal neurons. It enhanced the spatial working memory and consolidation of memory, thereby reversing the age-related memory integration impairment [56].
DKK1, due to its capacity to cross the blood-brain barrier (BBB) at elevated serum concentrations, may accelerate AD progression. This highlights the urgency and importance of investigating DKK1 as a potential therapeutic target for AD. The therapeutic effect of such intervention may be linked to synaptic protection and hippocampal circuit reconstruction. Notably, animal behavior tests have shown that inhibiting DKK1 improves spatial memory tasks [57], a finding supported by electrophysiological data on DKK1's role in long-term potentiation (LTP) [57].
Elevated serum levels of DKK-1 have been reported in AD patients compared to healthy controls. This elevation correlates with disease severity, particularly cognitive decline and synaptic loss. Thus, DKK-1 level elevation in blood can help us identify subjects at risk of developing AD before manifesting the disease symptoms [58]. Also, tracking the trends in serum DKK-1 can help us understand the treatment prognosis and identify disease progression. Lastly, given its specific association with the Wnt signaling pathway and Aβ pathology, serum DKK-1 levels might help differentiate AD from other neurodegenerative conditions that do not exhibit this pathway disruption.
3.3.2.4. Plasma Visinin-Like Protein-1 (VILIP-1):
VILIP-1 is a new emerging biomarker reflecting different aspects of the heterogeneous pathophysiology of AD. VILIP-1 is a calcium-binding protein of neuronal calcium sensor (NCS), expressed in neuronal pericaria, dendrites, and some axons, and is involved in maintaining neuronal growth, survival, and synaptic plasticity [59]. In AD, disturbed calcium homeostasis followed by neuronal degeneration causes the release of VILIP-1 in the extracellular space. A case-control study by Halbgebauer et al. [60] indicates that the Simoa assay of CSF VILIP-1 and serum VILIP-1 could be highly sensitive and reliable for AD diagnosis. Additionally, they found a significant increase in CSF VILIP-1 levels in AD compared to control groups, as well as in Parkinson's disease, behavioral variant frontotemporal dementia ALS, and (LBD). Serum VILIP-1 was elevated in AD patients in comparison to the control groups, but surprisingly, no such significant difference in concentrations was observed with AD-MCI patients and other neurodegenerative groups [61]. From these findings, we can hypothesize that serum VILIP -1 alone cannot be a potential biomarker for early AD diagnosis. Nevertheless, monitoring the ratio of CSF-VILIP-1 to serum VILIP-1 can be crucial.
3.4. Blood-Based Biomarkers of Neuronal and Synaptic Injury
In 2018, NIA-AA guidelines incorporated neurodegeneration as the third biomarker (N) to define AD pathology. Notably, in their 2024 revised update, recently developed BBBMs of A, T, and (N) have been included [9]. Additionally, as mentioned earlier, the group of biomarkers representing neuronal injury, dysfunction, or degeneration of neuropil has been placed under the broad category of "Biomarkers of non-specific processes involved in AD pathophysiology." However, neurodegeneration or neuronal dysfunction alone could not be significant enough to function as a diagnostic marker as its dynamic alteration could precisely predict the progression of AD-continuum. For that matter, biomarkers of synaptic dysfunction due to the loss of synaptic plasticity and integrity represent the very early pathological alterations in AD. Synaptic dysfunction is directly triggered by Aβ and tau pathology and indirectly by the consequences of neuroinflammatory responses.
3.4.1. Plasma Neurofilaments (NfL) as AD Diagnostic and Disease Progression Biomarker:
Neurofilaments are one of the main proteins expressed within neuronal cells. They are found in the axons of all neuronal cells and play a crucial role in maintaining nerve impulses' structural integrity and conduction velocity, thereby keeping the axonal caliber [62]. Degeneration of large-caliber axons is an essential feature of AD neurodegeneration.
Post neuroaxonal injury, there is a surge in neurofilament proteins in the blood and cerebrospinal fluid (CSF). Recent breakthroughs have unveiled plasma NfL's potential in monitoring various neurodegeneration elements, such as glucose metabolism, cognitive function, structural imaging, and future atrophy [63]. In a study conducted by Mattsson et al. [64], it was found that patients with MCI and AD dementia, as well as those with preclinical AD, prodromal AD, and AD dementia, all exhibited higher NfL rates at baseline compared to controls. This study also supported the idea of tracking NfL levels longitudinally as a marker of neurodegeneration across the preclinical stage and different clinical stages of AD.
Unlike the traditional method of using CSF samples to quantify NfL, recent strides have been made in measuring Neurofilament levels in the blood. The advent of 3rd/4th generation enzyme-linked immunosorbent assay (ELISA) [65], and the more sensitive electrochemiluminescence assay technology [65] have revolutionized this process. Interestingly, Gou et al. [19] revealed that plasma NfL is not significantly elevated until the MCI+ stage. Interestingly, plasma NfL level was relatively higher in the MCI+ group than in the CU A-T- and CU A+T- groups. Plasma NfL also showed the same trend in the AD dementia group relative to CU A+T+ and CU A+T- and MCI+ groups. Moreover, SIMOA has made it feasible to accurately identify the slightest disease-induced changes at all doses, including in healthy individuals [65]. NfL also shows promising results as a treatment response biomarker for protopathic lesion-induced neurodegeneration [66].
3.4.2. BBBMs Related to Pre-Synaptic Dysfunction:
Synaptosome-associated protein 25 (SNAP-25) is a crucial protein primarily residing in the presynaptic vesicles and is associated with synaptic degradation. Studies have shown an increasing trend in CSF SNAP-25 in the AD population, with a decreasing index in the cerebral cortex, indicating the extent of synaptic dysfunction [67]. Notably, CSF SNAP-25 can differentiate AD from Parkinson’s disease and ALS, and an increased concentration of CSF SNAP-25 is also found in Creutzfeldt-Jakob disease, highlighting its potential to distinguish between various neurodegenerative diseases [68]. In contrast, very few studies have documented the possible association between plasma SNAP-25 and AD progression. Interestingly, a survey by Agliardi et al. [69] has shown a decreasing trend in neuron-derived exosomes containing SNAP-25 derived from plasma and associated with cognitive status evaluated by MMSE.
Neuronal pentraxin 2 (NPTX-2), a protein associated with inhibitory circuit dysfunction, has shown a remarkable trend as a biomarker related to synaptic dysfunction. A longitudinal study by Libiger et al. based on CSF proteomics has shown a correlation between the changing rate of NPTX-2 level and the rate of cognitive decline [70]. However, the fundamental understanding of the role of NPTX-2 in AD is still unclear. A recent study has revealed a reduced NPTX-2 level in the plasma NDEs of AD patients, which can show such alteration that can be detected a decade before the onset of AD-associated dementia, suggesting its potential as a biomarker for early detection of AD [71]. Additionally, CSF growth-associated protein (GAP-43), another potential biomarker related to pre-synaptic dysfunction, has been found to display an increasing trend in AD dementia and is correlated with Aβ burden and NFT formation in the hippocampus, amygdala, and cerebral cortex [72]. However, the plasma-based association of GAP-43 with AD is questionable. On that note, a recent study by Jia et al. has revealed that neuro-exosomal GAP-43 could predict MCI-AD in 4-7 years advance [73].
3.4.3. BBBMs Related to Post-Synaptic Protein Dysfunction:
Neurogranin (NG), a 78 AAs long post-synaptic protein, is associated with synaptic dysfunction and neuronal injury [74]. Previous findings have revealed the importance of NG in maintaining synaptic plasticity, long-term potentiation (LTP), and long-term depression (LTD) [75]. A high level of CSF NG is positively correlated with brain AB burden and the extent of Tau pathology, and a specific type of NG, i.e., NG 48-76, has been found to increase significantly during the brain neurodegenerative process [76]. However, the CSF NG is poorly specific for AD-related pathological changes, and plasma NG did not reveal any associative trend between AD and healthy controls [77]. However, a decreasing trend in plasma-derived neuron-derived exosomes (NDE) containing NG has shown a positive correlation with cognitive decline [78].
3.5. Blood-Based AD-Related Biomarkers of Vascular Pathology.
The relationship between vascular pathology and blood-based biomarkers linked to AD provides essential new information on the illness's processes, especially the interplay between neurodegeneration and vascular dysfunction.
3.5.1. Fms-Like Tyrosine Kinase-1 (Flt-1) in AD-Related Vascular Changes:
VEGFR-1, or Flt-1, plays a crucial role in AD. It is a receptor for VEGFs and is essential for inflammation, vascular permeability, and angiogenesis. The disruption of the BBB in AD is linked to Flt-1 dysregulation, which affects angiogenesis and increases vascular permeability. This disintegration facilitates the entry of hazardous chemicals into the brain, promoting neuroinflammation and neurodegeneration [79,80]. A major pathogenic characteristic of AD is brain microvascular dysfunction, which has been associated with elevated Flt-1 levels. Recent research by Lau et al. [81] suggests a connection between enhanced angiogenesis and immunological activation and the endothelial overexpression of Flt-1 in AD. Measuring Flt-1 levels in the blood not only helps assess vascular involvement in AD but also opens up a new avenue for early detection of vascular changes associated with the disease, sparking further interest in this area of research.
3.5.2. Role of Endothelin 1 (ET-1) in AD-Associated Vascular Pathology:
Vascular endothelial cells primarily produce ET-1, a potent vasoconstrictor. Blood flow and vascular tone are significantly regulated by it. Elevated levels of ET-1 are linked to vascular dysfunction and decreased cerebral blood flow in AD. ET-1 can exacerbate neuronal damage through tau-pathology and Aβ-deposition. Chronic vasoconstriction brought on by ET-1 may cause ischemia and hypoperfusion in the brain, which would further exacerbate AD pathogenesis [82] .ET-1 levels may indicate vascular impairment in AD. A study done by Palmer et al. [83] showed that ET-1 protein was significantly higher in AD than in control tissue and also provided evidence of overactivity of the endothelin system in AD, further supporting the suggestion that endothelin receptor antagonists may be of value for the treatment of this disease.
3.5.3. Alteration of Adrenomedullin (ADM) in AD
ADM regulates blood pressure, promotes vasodilation, and preserves the integrity of the endothelium barrier [84]. Vascular dysregulation and endothelial dysfunction have been associated with altered levels of ADM in AD. Inhibiting oxidative stress and inflammation, two factors crucial to AD pathogenesis, is one of the neuroprotective effects of ADM. The breakdown of BBB caused by dysregulated ADM levels may allow Aβ and other neurotoxic substances to enter the brain more efficiently [85]. A study by Ferrero et al. [86] compared the ADM levels between the cortex of AD patients and controls, and ADM was found to be significantly higher in the cortex of AD patients.
3.5.4. Role of Atrial Natriuretic Peptide (ANP) in AD-Related Vascular Alterations
The cardiac hormone ANP, which controls salt homeostasis, fluid balance, and blood pressure, may offer hope in treating AD. It contributes to cardiovascular homeostasis and has vasodilatory effects [87]. Given That individuals with AD often have circulatory dysfunctions, ANP levels may be affected by the condition. It has been demonstrated that ANP affects cerebral ischemia risk and cerebrovascular tone [88]. The pathophysiology of AD may worsen due to reduced cerebral blood flow caused by dysregulation of ANP signaling.
Furthermore, ANP might be involved in controlling the brain's clearance of Aβ [89]. Tracking ANP levels in AD patients may help understand the disease's cerebrovascular and cardiovascular aspects. A study by Mahinrad et al. found an increased number of ANP receptors in AD brains compared to non-AD brains [90]. Pathways connected to ANP may present treatment options to enhance vascular function and lessen AD.
3.5.5. Vascular Immune Interaction and Gamma Interferon/Chemokine (C-X-C) Ligand 9 (MIG/CXCL-9) in AD
MIG/CXCL-9, a chemokine, may hold significant diagnostic value in AD. Its primary function is to attract immune cells, especially T lymphocytes, to areas of inflammation [91]. It is involved in immunological surveillance and inflammatory reactions and is produced in response to interferon-gamma [92]. Elevated MIG/CXCL-9 levels are linked to neuroinflammation and the migration of immune cells to the brain in AD. This chemokine aggravates amyloid pathology and neuronal impairment by contributing to the chronic inflammatory state seen in AD [93]. The blood's concentration of MIG/CXCL-9 may indicate ongoing neuroinflammatory processes and how the vascular and immune systems interact in AD.MIG/CXCL-9 may be a biomarker for vascular-immune interactions and neuroinflammation in AD. Reducing MIG/CXCL-9 or its signaling pathways could potentially assist.
3.5.6. Role of Heart-Type Fatty Acid Binding Protein (H-FABP) in AD-Related Vascular Pathology:
AD can arise and evolve as a result of chronic vascular disease and impaired cerebral blood flow regulation. Heart-type fatty acid binding protein (H-FABP) plays a role in fatty acid metabolism and lipid transport, and elevated levels of H-FABP indicate oxidative damage and systemic inflammation. Its levels could potentially reflect underlying vascular pathology that can be elevated in CSF of patients suffering from various forms of neurodegenerative diseases such as AD, Parkinson's disease with dementia, Lewy body dementia (LBD), vascular dementia (VaD), and Creutzfeldt-Jakob disease [94]. Several studies have found a positive correlation between H-FABP and its ability to act as a diagnostic and prognostic factor in AD. In a study done by Desikan et al. to determine the role of fatty acid binding protein in neurodegeneration, H-FABP, in the earliest stages of AD, was found to be associated with atrophy of entorhinal cortex and other AD-vulnerable regions of interest in the brain [95]. A significant association between H-FABP and p-tau and several apolipoproteins, including Apolipoprotein E and ApoCIII, suggests a strong relationship between neuronal lipid biology and neurodegeneration [95]. An interesting finding of this study points to the association of H-FABP and increased Aβ aggregation, highlighting the role of phospholipids, cholesterol, and protein transporters in Aβ dyshomeostasis and its possible use as a therapeutic target.
3.5.7. Alteration of Vascular Adhesion Molecule (AM) Expression and Endothelial Dysfunction in AD
Endothelial dysfunction has been linked to cerebrovascular disease, with elevated levels of AMs associated with the presence or progression of small and large vessel disease and white matter hyperintensities. [96,97]. Studies have implicated adhesive proteins in multiple pathological linkages of MCI and AD, including amyloid plaque degradation, diffusion, and inflammation [98]. Soluble vascular cell adhesion molecule-1 (sVCAM-1), as opposed to soluble intercellular adhesion molecule-1, is more likely associated with atherosclerotic load determined by angiography or echocardiography [99]. Thus, elevated levels might indicate the burden of atherosclerosis in AD and vascular dementia. Free β-amyloid inhibits endothelial nitric oxide synthase (NOS) activity, causing endothelial dysfunction and increasing AM [100]. Several investigations that have measured CSF and blood levels of intercellular adhesion molecule-1, vascular cell adhesion molecule-1 (VCAM-1), and interleukin 15 in AD have produced conflicting results mainly due to differences in sample size and cognitive status of controls and the presence of confounding factors [101]. A study by Zuliani, G. et al. [102] concluded that sVCAM-1 was elevated in vascular dementia and late-onset AD, without any cerebrovascular disease, with no significant changes in E-selectin level. Drake et al. [103] also showed a similar positive association between sVCAM-1 and cognitive decline in AD but no such correlation with I-CAM1 and E-selectin levels. A study by Chen et al. [98] demonstrated VCAM-1 and ALCAM as strong predictors of AD with a significant correlation with age and the severity of cognitive decline, with no significance on intercellular adhesion molecule levels. In comparison, a study by Janelidze et al. [101] found a substantial rise of intercellular adhesion molecule and VCAM-1, YKl-40, interleukin 15, and Flt-1 in AD's preclinical and prodromal stages. It was associated with cognitive decline and increased risk of subsequent development of AD.
3.6. Blood-Based Biomarkers of Oxidative Stress and Bioenergetics
3.6.1. BBBMs Related to Oxidative Stress:
Oxidative stress can accompany AD and MCI-related pathological changes and is considered to be a crucial upstream factor in disease progression. The resultants of free radical damage, such as aldehydes and lipid hydroperoxides, can readily diffuse into the peripheral circulation. Studies have revealed that BBB permeability and integrity are affected significantly in both AD and vascular dementia, and products of oxidative stress represent potential biomarkers for AD diagnosis [104,105]. However, oxidative stress markers in the blood in AD are consistent as they can be influenced by underlying co-morbidities such as diabetes and metabolic syndrome or cardiovascular diseases.
Notably, AD-related oxidative stress is due to the Aβ misfolding, which activates the resting microglia. The NADPH oxidase inside the microglia is activated, leading to free radical generation in AD patients [106]. Additionally, the Aβ peptide is an essential source of free radicals in AD, and it has been found that Aβ directly produces free radicals for which methionine at the 35th position is responsible [107]. Besides, Aβ binds with redox-active metals, which function as a catalytic factor for free radical production. In that context, it has been found that Fe+2 concentration is increased in the AD brain. Furthermore, the oxidative stress-related burden precedes the senile plaques and tangle formation stage [108,109].
Metabolic products secondary to lipid peroxidation (LPO) accumulate in neurons without any AD-related pathological changes, and these intermediates formed in the brain may traverse through BBB easily, considering they are small and lipophilic [110]. Several studies have revealed the importance of malondialdehyde (MDA), a product mainly arising from polyunsaturated fatty acid (PUFA), and 4-hydroxynonenal (4-HNE), another essential product during the peroxidation of linoleic and arachidonic acid in plasma as potential biomarkers of brain oxidative stress in AD [111]. In plasma, both the levels of MDA and 4-HNE are increased in MCI-AD compared to controls [112,113]. Importantly, isoprostane represents the best available biomarkers of lipid peroxidation these days, and several studies have revealed that increased F2-isoprostanes (F2-IsoPs) in body fluids, including plasma, CSF, and urine, a potential marker of oxidative stress during MCI phase of AD [114]. Furthermore, its concentration correlated with the disease continuum from SCD to MCI to AD. Although within MCI and AD groups, F2-IsoPs did not correlate with memory impairment duration or cognitive test scores [115]. Interestingly, it has been found that fibroblasts and lymphoblasts from patients with familial AD, in contrast to sporadic AD, carry APP and presenilin-1 gene mutations and showed an increase in MDA and 4-HNE [116].
Free radicals may further impair essential proteins' structural and functional properties directly or secondarily by attacking them as end-products of lipid peroxidation [117]. The reaction of various ROS and RNS species can lead to the formation of 3-nitrotyrosine and dinitrotyrosine, and their concentration is increased in CSF and plasma of AD individuals [118]. Additionally, mini-mental state examination (MMSE) correlated negatively with 3-nitrotyrosine concentration in CSF, and the total protein nitration measure in brain samples differed significantly in MCI compared to healthy controls in the inferior parietal lobule and hippocampus [118,119]. Studies with a plasma proteomic approach have revealed that specific oxidation products in AD, identified as isoforms of human transferrin, hemopexin, alpha-1-antitrypsin, and fibrinogen gamma-chain precursor proteins, are significantly increased [120]. Furthermore, the increased carbonyl protein and tyrosine in IgG have also been documented in AD. However, only one study revealed increased 3-nitrotyrosine and dinitrotyrosine levels in the plasma of AD patients [121].
Notably, oxidative damage of DNA or RNA in chemical modulation of DNA bases or deoxyribose has been measured in AD individuals. The most notable finding to determine the gradient of DNA-oxidative damage in AD has been the nucleoside 8-hydroxyguanosine (8-OHG) level, which is significantly higher in the lymphocytes of AD patients than in controls [122]. Other studies have revealed elevated oxidized pyrimidines and purines in the peripheral blood of AD patients compared to age-matched control individuals. The oxidative damage to DNA in plasma is a far earlier process in the pathogenesis of AD [123].
Importantly, studies on the antioxidant level in blood have also been crucial as their levels are decreased due to increased oxidative stress during the earlier stages of AD. Several studies have shown reduced plasma levels of vitamin E, vitamin C, or Vitamin A in the plasma of AD individuals under normal dietary circumstances without any supplementation [124]. However, several other studies did not find significant differences in the levels of plasma of AD individuals in comparison to controls. Overall, changes in the total antioxidant capacity of the plasma have been significantly decreased in AD patients and negatively correlated with the disease duration [125].
3.6.2. Blood-Based Bioenergetic Profiling
Blood-based bioenergetic profiling has emerged as a reliable and minimally invasive approach to assessing mitochondrial function. Circulating blood cells, including platelets and peripheral blood mononuclear cells (PBMCs), exhibit high rates of electron transport chain (ETC) activity and metabolic flexibility. The mitochondrial cascade hypothesis proposes mitochondrial dysfunction in the form of lower mitochondrial respiration as one of the earliest hallmarks of AD and one of the main causative factors leading to senile plaque and NFT formation, mainly in sporadic or late-onset AD (LOAD) individuals [126]. Concomitantly, alterations in mitochondrial quality processes, i.e., fusion, fission, and autophagy, can trigger neurodegeneration. Mitochondrial DNA (mt-DNA) mutations correlate with cognitive decline risk and AD pathogenesis [127]. Aβ deposition has also been negatively associated with mitochondrial bioenergetics, including ETC uncoupling, reduced ATP production, and increased ROS generation [128].
Notably, PBMC from early AD patients revealed decreased expression of mitochondrial respiratory complex I-V genes (OxPHOS genes) and mitochondrial ribosomal complex subunits compared to the control group [129]. These changes lead to accelerated mitochondrial dysfunction and enhanced oxidative damage. Studies have revealed that PBMC from sporadic AD patients had decreased basal oxygen consumption rate (OCR) and proton leak without changing maximum respiratory capacity compared to age-matched controls [130] At the same time, a reduction in basal OCR and maximum respiratory capacity was evident in lymphocyte mitochondria [131]. Interestingly, platelets have also been found to have abnormalities in the ETC of AD patients, and it is considered an emerging biomarker in peripheral blood in AD patients [132]. Several studies have reported decreased activity and expression of the complex-IV enzyme and its subunits in the platelets of AD individuals [129].
Additionally, fibroblasts from both sporadic and familial AD cases have revealed functional abnormality and an increase in their numbers in peripheral circulation. The ETC function in fibroblasts from AD patients reflects higher variability than in other blood cells [133]. Studies have also reported impaired glucose uptake by the fibroblasts from AD individuals [134]. However, it remains a grey area whether the functional and structural changes of the ETC seen in AD individuals are due to primary mitochondrial changes (mitochondrial hypothesis of AD) or the Aβ deposition-mediated alterations.
Mitochondrial dynamics has also been found to be significantly impaired in AD individuals and can be a potential approach to study these changes as biomarkers of early AD dementia. In this context, the formation of a complex formed by S-nitrosothiol and dynamin-related protein 1 (Drp1), i.e., SNO-Drp1, can result in increased mitochondrial fission, loss of synapses and neuronal damage in mice model and primary neuronal culture as well as in post-mortem tissue. SNO-Drp1 is increased in peripheral blood lymphocytes in AD patients, and preventing nitrosylation has been found to reduce neuronal loss [134]. There are also contradictory findings that SNO-Drp1 does not differ significantly in AD compared to controls [135].
Current literature has also suggested the importance of studying mitochondrial calcium signaling as a promising biomarker. Interestingly, AD's abnormal mitochondrial Ca+2 concentration and signaling are directly associated with Aβ and tau protein or by the mutated PSEN1 and PSEN2 genes in familial AD [136].
Recent studies on blood-based indices of mitochondrial DNA (mt-DNA) copy number and cell-free mtDNA (cf-mt-DNA) have revealed that mt-DNA copy number was significantly associated with cognitive impairment, and cf-mtDNA was also found to be higher in these cases when compared to controls [137]. Hence, cellular mt-DNA copy number can be used as a potential biomarker of mitochondrial biogenesis and cellular energetics to reflect upon mitochondrial health in AD. In this context, a study by Reid et al. has revealed that assessment of 8-oxo-7,8-dihydro guanine (8oxoG) somatic single nucleotide variants (sSNVs) (8oxoG sSNVs) can serve as a better mitochondrial dysfunction-related biomarker [138]. In contrast, due to its inflammatory endophenotype, the circulating cf-mtDNA (ccf-mtDNA) 8oxoG variant can be used as an improved biomarker [139].
The most critical challenge of using mitochondrial function as a biomarker is the versatility of the measurement methods used. Functional assessment of mitochondria requires several weeks of processing biological samples from patients. This would be expensive and time-consuming, making the development of a practical, widely available mitochondria-based biomarker difficult. Besides, a handful of studies have investigated mitochondrial abnormalities in AD by recruiting large patient cohorts. Therefore, further investigations on a larger scale are needed to identify more precise and accurate mitochondrial function-related abnormalities that will be considered standard biomarkers in the coming days.
In Table 1, the oxidative stress and bioenergetic dysfunction-related biomarkers in AD pathology are highlighted. The STRING network model indicates interactions between proteins associated with oxidative stress and mitochondrial dynamics, suggesting their co-expression or co-occurrence in AD, as depicted in [Figure 4].
3.7. Blood-Based Biomarkers of Neuroinflammation and Immune Dysregulation
One of the most relevant factors in AD pathogenesis is chronic neuroinflammation and the involvement of microglia in this process [140]. These inflammatory markers can be potential supplements to the AD diagnostic panel. Most of these inflammatory biomarkers can be analyzed from blood or its derivatives. Their concentration can be quantified by ELISA or other immunoassays, such as electrochemiluminescence immunoassay and mesoscale discovery immunoassay V-PLEX.
AD can be characterized by prominent astrogliosis, mostly seen as surrounding amyloid plaques with activated astrocyte processes participating in neuritic plaque formation [141]. The intermediate filament glial fibrillary acidic protein (GFAP) is highly upregulated in reactive astrocytes. These reactive astrocytes contribute to neuroinflammation by releasing pro-inflammatory cytokines and chemokines. This chronic inflammation contributes to the progression of AD [142]. Preliminary data on GFAP suggests that the plasma biomarker performs better than its CSF counterpart in identifying AD pathology [143]. Several studies show the marked elevation of GFAP in AD and MCI, without significant change in frontotemporal dementia (FTD) and progressive supranuclear palsy (PSP) and its association with longitudinal decline in revised Addenbrooke’s cognitive examination scores in MCI and AD, thus adding a prognostic value [144]. In a study done by Benedet et al. to measure GFAP levels across the whole AD continuum, plasma GFAP levels, unlike its CSF levels, were found to be higher among individuals with preclinical AD and reached significantly greater levels at symptomatic stages of AD, along with a positive correlation with Aβ pathology [145]. It is noteworthy that Gou et al. [19], as mentioned earlier, revealed the dynamic trends through the AD continuum [Figure 1(B)] where plasma GFAP level was significantly increased in the CU A+T- compared to the CU A-T- group. The same trend was found in CU A+T+ compared to CU A-T- group. Plasma GFAP also showed the same trend in the AD dementia group relative to CU A+T+ and CU A+T- and MCI+ groups.
Several pro-inflammatory cytokines, including IL-α, IL-β, interleukin 6, and chemokines, including monocyte chemoattractant protein CCL2, are altered in serum and CSF of AD individuals compared to controls [146]. However, such alterations may not be directly associated with AD and can be related to aging and other systemic diseases. White blood cell CX3CL1, also called fractaline, is significantly elevated in the plasma of MCI and AD [147]. CCL23 plasma concentration has also been found to have a predictive value towards MCI-to-AD progression. C-C chemokine ligands or RANTES plasma concentrations are elevated in AD and correlate with the neuroinflammatory burden [148].
Progranulin, a growth factor expressed in neurons and microglia, has been associated with neuroinflammatory modulation, such as microgliosis and astrogliosis. Studies have revealed that the increased progranulin-expressing gene, GRN, is in the blood of MCI and AD patients [149,150]. Interestingly, YKL-40, a chitinase-3-like protein (encoded by the CHI3L1 gene), was found to be increasingly expressed in astrocytes during neuroinflammatory changes. A longitudinal study has revealed that plasma YKL-40 level is negatively correlated with cognitively healthy individuals at risk of developing AD and showed a positive correlation with the result of sensitive free and cued selective reminding test or FCSRT [151].
Additionally, interleukin 33 and the soluble form of its receptors ST2 (sST2) are associated with neuroinflammation. Still interestingly, in AD, it plays a protective role and is found to be downregulated in brain tissues of MCI and AD individuals. However, its plasma concentration is higher in MCI and AD than controls. Hence, a higher plasma concentration of interleukin 33 is associated with better cognitive function [152], but the question remains: why is this cytokine elevated in AD and MCI? A possible answer could be that AD patients have a higher concentration of sST2, attenuating the effective concentration of interleukin 33, and it can contribute to cognitive function in the AD disease continuum [153].
Recently, increased levels of triggering receptor expressed on myeloid cells 2 (TREM2) mRNA levels in peripheral mononuclear cells have been found to have the distinguishing ability between aMCI, AD, and healthy control individuals and to be dependent on the Apolipoprotein E genotype [154]. A study by Hu et al. [155] showed TREM2 expression in monocytes to be consistent with RNA-based observation in circulating monocytes. Like TREM2, TREM1 has also been found to be a possible biomarker [156]. Interestingly, the soluble form of secreted TREM2 or sTREM2 concentration is lower in plasma and associated with Aβ accumulation and CSF p-Tau level in AD [157]. However, such alteration of sTREM2 has also been found in vascular dementia, questioning the specificity of this biomarker.
3.8. Blood-Based Epigenetic Biomarkers Related to Early Detection and Prognosis of AD
Epigenetics has proven to be a valuable tool in gaining a deeper understanding of the pathogenesis of AD. In the search for more relevant blood biomarkers for AD, the role of epigenetic mechanisms that mediate the interaction between the genome and the environment is becoming increasingly significant. These mechanisms are emerging as key contributors to the pathogenesis of AD.
3.8.1. DNA Methylation-Based Markers:
DNA methylation, an epigenetic marker influenced by genetic inheritance and environmental factors, shows significant potential for predicting AD. The methylation patterns in blood may offer valuable insights into the biological mechanisms contributing to AD. For instance, specific changes in methylation have been linked to neurodegenerative processes, suggesting that these epigenetic alterations could serve as early indicators of AD risk. The ability to identify these methylation changes years before the onset of clinical symptoms underscores DNA methylation as a promising approach for early diagnosis and intervention in AD. This is particularly relevant given that the pathological characteristics of AD can manifest long before cognitive decline becomes noticeable [158].
Studies have investigated the role of DNA methylation in creating epigenetic clocks, which connect biological age to disease mechanisms. Changes in DNA methylation in blood have been linked to CSF biomarkers of AD, including Aβ and tau proteins. A thorough analysis that included more than 111,000 cases of AD and almost 678,000 controls revealed 1,168 cytosine-phosphate guanine (CpG) sites significantly associated with the risk of AD. Among them, 52 CpG sites related to 32 genes demonstrated consistent associations with AD risk, including genes such as CNIH4 and THUMPD3, which were not previously recognized as risk genes for AD [159].
3.8.2. Potential Blood-Based microRNA Biomarkers for AD
MicroRNAs (miRNAs) have a complex role in the development of AD. They influence various facets of the condition, including amyloid beta (Aβ) metabolism, tau phosphorylation, neuroinflammation, and synaptic function.
A literature review revealed that among the 137 miRNAs identified as altered in the blood of AD patients, 36 have been confirmed in at least one independent study [160]. Some of the most frequently reported miRNAs are hsa-miR-146a, hsa-miR-125b, and hsa-miR-135a. These three miRNAs have been consistently found to be altered in the blood, CSF, and brain tissue of individuals with AD [160]. One study discovered three miRNA signatures (miR-92a-3p, miR-486-5p, miR-29a-3p) that may differentiate between preclinical, MCI due to AD, and healthy controls [161]. Another study identified a panel of 12 miRNAs that could predict AD progression in patients with MCI, with eight validated through qPCR [162]. The dysregulated miRNAs found in the blood of AD patients have been linked to various pathways associated with AD pathogenesis, including the regulation of amyloid precursor protein cleavage, expression of presenilin-1 and BACE1, oxidative stress, neuroinflammation, cell cycle regulation, synaptic transmission, cell signaling, and metabolism [161,163,164].
After comparing the networks of enlisted miRNAs [Table 2] between peripheral blood and brain tissue, it was found that the miRNA miR-93-5p has a more significant interaction with the validated genes in both [Figure 5]. This may indicate that it could serve as a critical blood biomarker for future AD diagnosis.
3.8.3. Potential Blood-Based Long Non-Coding RNA (lncRNA) Biomarkers for AD
Recent studies have discovered multiple lnc-RNAs in the blood that may serve as promising biomarkers for the detection and progression of AD. Among them, the plasma levels of NEAT1 and BC200 were significantly elevated in AD patients compared to healthy controls. They can effectively differentiate AD patients from controls with high sensitivity and specificity. Additionally, NEAT1 could distinguish between MCI and advanced AD compared to controls. Furthermore, plasma levels of BC200 showed a positive correlation with the age of the patients [165].
Additionally, plasma levels of BACE1 lncRNA were significantly elevated in patients with AD compared to non-AD controls. Receiver Operating Characteristic (ROC) analysis demonstrated that BACE1 exhibited high specificity (88%) for AD [166]. Other lncRNAs, such as NDM29, FAS-AS1, and GAS5-AS1, were also assessed but did not reveal significant differences between AD patients and controls [165]. In-silico analysis of RNA-sequencing data identified 33 lncRNAs that were upregulated and 13 that were downregulated in AD patients compared to controls [166]. These studies indicate that plasma levels of NEAT1, BC200, and BACE1 lncRNAs are promising blood-based biomarkers that could assist in early AD detection and monitoring disease progression. Still, more extensive validation studies are necessary to confirm their clinical applicability. Additionally, integrating these lncRNA biomarkers with other blood-based markers, such as amyloid-beta and tau proteins, may enhance diagnostic accuracy further.
3.8.4. Markers Related to Histone Modification and DNA Alteration
Histone modifications, such as acetylation, methylation, and phosphorylation, are essential for regulating gene expression and have been shown to affect cognitive functions, learning, and memory. In the context of AD, research suggests that there are both losses and gains of specific histone marks, indicating a complex interaction of epigenetic changes that contribute to the disease's progression and pathology [167,168]. For example, histone acetylation is linked to active gene expression, whereas methylation can activate or repress genes based on the specific context. In AD, changes in these modifications are associated with cognitive decline and neurodegeneration, underscoring their potential as biomarkers for the disease [167]. DNA methylation age, which reflects biological aging, has also been correlated with AD pathology, indicating that accelerated epigenetic aging may contribute to the disease [169,170]. Similarly, alterations in DNA methylation patterns have been connected to AD. Research has demonstrated that differential methylation occurs in genomic regions associated with AD susceptibility, suggesting that these changes may precede clinical symptoms and act as early disease indicators [167,168].
The dynamic nature of histone and DNA modifications creates opportunities to develop diagnostic and prognostic biomarkers for AD. These epigenetic markers could assist in early detection, tracking disease progression, and assessing treatment responses. Moreover, targeting these modifications with epigenetic drugs, such as histone deacetylase inhibitors, is being investigated as a therapeutic approach to alter disease outcomes [167,168,169,170]. Histone modifications and DNA alterations are increasingly recognized as significant biomarkers in AD. These epigenetic changes influence gene expression without altering the fundamental DNA sequence, playing a vital role in the development of the disease.
3.8.5. Circular RNA (circRNA) Related Biomarkers
Research has identified circRNAs as potential biomarkers for AD, presenting an encouraging pathway for early diagnosis and differentiation from other forms of dementia. One study established a panel of six circRNAs that effectively distinguish AD patients from cognitively healthy individuals and those with different types of dementia, such as vascular dementia and Parkinson’s disease dementia. This panel was validated using three independent datasets, demonstrating its specificity and potential as a dependable biomarker for AD [171].
It has revealed distinct expression profiles of circRNAs in AD, with certain circRNAs being either upregulated or downregulated in the blood of affected patients. For example, hsa_circ_0003391 has been identified as significantly downregulated in AD patients compared to those with other types of dementia, indicating its potential as a diagnostic marker [172].
The capability of circRNAs to traverse the blood-brain barrier, combined with their specific expression profiles in various tissues, enhances their potential as biomarkers. They could aid in creating non-invasive diagnostic tests for AD, enabling earlier intervention and improved disease management [173,174]. CircRNAs have been shown to play a significant role in the pathophysiology of AD. They can function as ‘sponges’ for microRNAs, thereby influencing gene regulation and potentially contributing to the progression of the disease. Their stability and abundance in the nervous system make them appealing candidates for biomarker development.
3.9. Plasma Exosome-Based AD-Related Biomarkers
Exosomes derived from CSF, blood, and neural cells can potentially emerge as biomarkers for diagnosing AD. Proteomic analysis of CSF-derived exosomal vesicles (EV) revealed that more than 400 unique proteins are involved in AD pathogenesis [175]. According to Muraoka et al. [176], some proteins, such as HSPA1A, NPEPPS, and PTGFRN, are essential to monitor the progression of MCI converted to AD. Apart from that, the total Tau and p-181-tau levels in CSF-derived EVs were higher in patients with AD compared to the healthy control [177]. The encapsulating lipid bilayer of exosomes makes it efficient to cross the blood-brain barrier without losing any biomarker, reaching many biological fluids such as blood, urine, saliva, and synovial fluid [178].
Fiandaca et al. revealed that the levels of P-S396-tau and p-tau 181 in NDE could predict the development of AD up to 10 years before the clinical onset of sporadic AD [179]. Moreover, higher levels of α-globin, β-globin, and δ-globin in NDE were found in patients with AD compared to those in controls. In contrast, in plasma NDEs, the levels of presynaptic proteins such as synaptotagmin and synaptophysin and post-synaptic proteins, including synaptopodin and neurogranin, were reported to be markedly lower [180]. Metabolism-based blood NDE includes P-S312-IRS-1, which showed higher levels in AD than control subjects without any changes in T-IRS-1. However, down-regulation of P-panY-IRS-1 and N-(1-carboxymethyl)-L-lysine has been reported in AD patients compared to controls. Additionally, other NDE biomarkers, such as Ser/Tyr phosphorylation of insulin receptor substrate 1, lysosomal enzymes, and ubiquitin, show marked differences between AD patients and healthy controls [181].
Studies have suggested that astrocyte-derived exosomes (ADEs) could be emerging biomarkers for AD diagnosis. The levels of complement proteins such as C1q, C3b, and cytokines, including interleukin 6, TNF-α, and IL-1β in ADEs significantly differed between AD individuals and controls. Additionally, elevated levels of C1q, C4b, factor D, fragments Bb, C5b, C3b, and C5b-C9 in plasma ADEs could serve as predictive biomarkers for the progression of MCI to AD [182].
Several scientific studies have indicated that microglia can engulf neurons or synapses containing tau and transfer tau to other neurons through phagocytosis and exosomes. This suggests a link between microglia, microglia-derived exosomes, and tau pathology, and reducing their presence significantly slows down the spread of tau. When neurons absorb microglia-derived exosomes containing tau, it triggers further abnormal tau aggregation. Additionally, activated microglia can release exosomes containing inflammatory markers such as iNOS, IL-1β, TNF-α, MHC class II, interleukin 6, as well as miR-155, miR-146a, and miR-124, along with pro-resolving genes like interleukin 10 and arginase 1. This results in a more damaging pro-inflammatory state throughout the brain [179,183]. However, it has been reported that microglia and neighboring neurons could collaborate to clear Aβ peptides through exosomes.
A summary of various exosome-based BBB models related to AD is provided in Table 1.
4. Future Directions and Conclusion:
Despite the recent advancements in identifying and implementing blood-based biomarkers as a diagnostic test for AD, several avenues for future research and development still exist. A significant aspect of this challenge lies in refining assay techniques, longitudinal validation of studies, and multi-modal approaches. Observational studies and clinical trials should be conducted with better representative cohorts. Almost all observational studies in underrepresented groups have been undertaken with the convenience of samples with definitive selection biases. Hence, proper epidemiological diversification and real-world data on biomarker studies are needed to ascertain valid relationships at the population level.
Recent upliftment in the sensitivity of measuring techniques and assays, including IP and LC-MS, has allowed blood tests to detect subtle pathology. The most robust strategy will be to combine a cluster of blood-based biomarkers to maximize diagnostic accuracy and specificity. Improved understanding of the role of immune and inflammatory processes, microglia, and astroglial biology in AD pathogenesis has a more prominent role in the biological characterization and prognosis, especially if brain tissue-specific changes can be detected in the peripheral circulation.
Apart from individual proteins as potential biomarkers, an array of network proteins linked to each other might suggest a different picture. As individual protein changes may give false positive results in multifactorial disease, a hub of proteins tightly linked with physical and functional interaction might provide a broader perspective of the disease pathology. Hence, we suggest a cumulative protein network from the STRING database based on our candidate, plasma-based biomarker proteins that will indicate a molecular outlook of the related proteins linked with functional and physical interactome. Similarly, the predicted miRNA biomarkers' interaction network with validated target genes can also reflect on the broader perspective of the AD continuum. These networks are based upon the relative individual biomarkers that might be done as cumulative tests to get a positive result in determining the disease stage and progression.
Most importantly, there is a significant need for a more profound understanding of the temporal relationship between the biomarker types and the pathophysiology of AD, including cognitive impairment. To date, the reliability and accuracy of blood-based investigations of AD are under scrutiny, and there is a variation in performance levels across different tests. Besides many important variables such as age, sex, genetics, and related co-morbidities, race and ethnicity can significantly influence the blood-based biomarker level. Additionally, in many countries, including the United States, insurance companies do not cover or reimburse the costs of blood-based AD tests. On this note, the lack of population diversity in blood-related AD biomarker development research prevents this approach from widespread adoption and integration with clinical practice. The increasing availability of BBM will undoubtedly make the complete “AT1T2NIVS criteria” for AD diagnosis more widely deployable within World Bank–designated low—to middle-income countries where PET and CSF-based biomarkers may not be readily available, and an acceleration in the disease burden is evident.
While complex challenges remain, ongoing efforts to harness blood tests' diagnostic and prognostic potential hold promise for transforming the landscape of AD/ADRD diagnosis and treatment.
Author Contributions
Mrinmay Dhauria (mrinmay.dhauria@gmail,com) collaborated on the conception, organization, and execution of the research project; Ritwick Mondal (ritwickraw@gmail.com) collaborated on the conception, organization, and execution of the research project, the writing of the first draft of the manuscript; Shramana Deb (shramandeb1039@gmail.com) collaborated on the conception, organization, and execution of the research project, the writing of the first draft of the manuscript; Gourav Shome (gshome007@gmail.com) collaborated on the conception, organization, and execution of the research project; Dipanjan Chowdhury (dipanjan.1729@gmail.com) collaborated on data organization, and coordination of literature searches from different databases; Shramana Sarkar (shramana99.sarkar@gmail.com) collaborated on data organization, and coordination of literature searches from different databases; Julián Benito-León (jbenitol67@gmail.com) collaborated on the conception, organization, and execution of the research project and the writing of the first draft of the manuscript.
Data Availability Statement
Anonymized data will be made available upon reasonable request to qualified researchers. Requests should be directed to Dr. Julián Benito-León at jbenitol67@gmail.com.
Acknowledgments
Julián Benito-León is supported by the National Institutes of Health, Bethesda, MD, USA (NINDS #R01 NS39422) and by the Recovery, Transformation, and Resilience Plan at the Ministry of Science and Innovation (grant TED2021-130174B-C33, NETremor).
Conflicts of Interest
None.
Abbreviations
Aβ, Amyloid-beta; ADM, Adrenomedullin; AD, Alzheimer's disease; ADAD, Autosomal Dominant Alzheimer's Disease; ADNPC, Alzheimer's disease neuropathological changes; AGER, Advanced Glycosylation End-Product Specific Receptor; ALS, Amyotrophic Lateral Sclerosis; AM, Adhesion Molecule; ANP, Atrial Natriuretic Peptide; APOCIII, Apolipoprotein CIII; APP, Amyloid Precursor Protein; AT(N), Amyloid/Tau/Neurodegeneration; BACE1, Beta-Secretase 1; BACE2, Beta-Secretase 2; BBB, Blood-brain barrier; BBBM, Blood-based biomarkers; BD-Tau, Brain-derived tau; BV-FTD, Behavioral variant frontotemporal dementia; CCL, C-C motif chemokine ligand; CF-mt-DNA, Cell-free mitochondrial DNA; CircRNA, Circular RNA; COX20, Cytochrome C Oxidase Subunit 20; COX5A, Cytochrome C Oxidase Subunit 5A; COX6A2, Cytochrome C Oxidase Subunit 6A2; COX8A, Cytochrome C Oxidase Subunit 8A; CSF, Cerebrospinal fluid; CXCL9, Chemokine (C-X-C Motif) Ligand 9; DCFDA, Dichloro-fluorescein Diacetate Assay; DKK1, Dickkopf-related protein 1; DLS, Dynamic Light Scattering; DNM1L, Dynamin 1 Like; ELISA, Enzyme-Linked Immunosorbent Assay; EOAD, Early-onset Alzheimer's disease; ET-1, Endothelin 1; EV, Exosomal vesicles; FABP3, Fatty Acid Binding Protein 3; FDG, Fluorodeoxyglucose; Flt-1, Fms-like tyrosine kinase-1; FTD, Frontotemporal dementia; GAP-43, Growth-Associated Protein 43; GC-MS, Gas Chromatography-Mass Spectrometry; GFAP, Glial fibrillary acidic protein; H-FABP, Heart-type fatty acid binding protein; HPLC, High-Performance Liquid Chromatography; IFN-γ, Interferon-gamma; IL, Interleukin; IHC, Immunohistochemistry; Immuno-PCR, Immuno-polymerized Chain Reaction; IP-MS, Immunoprecipitation-Mass Spectroscopy; LC-MS, Liquid Chromatography-Mass Spectroscopy; LTD, Long-term depression; LOAD, Late-onset Alzheimer's disease; lncRNA, Long Non-coding RNA; MAPT, Microtubule Associated Protein Tau; MCI, Mild cognitive impairment; MIG/CXCL-9, Monokine induced by gamma interferon; MMSE, Mini-mental state examination; MT-ND1, Mitochondrially Encoded NADH Oxidoreductase Core Subunit 1; MT-ND2, Mitochondrially Encoded NADH Oxidoreductase Core Subunit 2; MT-ND3, Mitochondrially Encoded NADH Oxidoreductase Core Subunit 3; MT-ND4, Mitochondrially Encoded NADH Oxidoreductase Core Subunit 4; MT-ND5, Mitochondrially Encoded NADH Oxidoreductase Core Subunit 5; NDE, Neuron-derived exosomes; NDUFA4, NADH Oxidoreductase Subunit A4; NDUFS3, NADH Oxidoreductase Iron-Sulfur Protein 3; NEFL, Neurofilament Light Chain; NG, Neurogranin; NfL, Neurofilament light polypeptide; NOX1, NADPH Oxidase 1; NOX3, NADPH Oxidase 3; NOX4, NADPH Oxidase 4; NPTX-2, Neuronal pentraxin 2; PET, Positron emission tomography; PRKN, Parkin RBR E3 Ubiquitin Protein Ligase; PSEN1, Presenilin 1; PSEN2, Presenilin 2; RBC, Red blood cells; SCD, Subjective cognitive decline; SIMOA, Single Molecular Array; SNAP25, Synaptosome-associated protein 25; SNCA, Synuclein Alpha; sVCAM-1, Soluble vascular cell adhesion molecule-1; SPR, Surface Plasmon Resonance; sST2, Soluble form of the interleukin 33 receptor; TDP-43, TAR DNA-binding protein 43; TREM1, Triggering Receptor Expressed on Myeloid Cells 1; TREM2, Triggering Receptor Expressed on Myeloid Cells 2; VCAM1, Vascular Cell Adhesion Molecule; VILIP-1, Visinin-like protein 1; V-PLEX, Validated Plasma exchange; YKL-40, Chitinase 3 Like 1.
References
- Vanessa de Jesus, R.; Guimarães, F.M.; Diniz, B.S.; Forlenza, O.V. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, TAU protein or both? Dementia neuropsychologia 2009, 3, 188. [Google Scholar]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: the challenge of the second century. Science translational medicine 2011, 3, sr1–sr77. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Early-onset Alzheimer disease. Neurologic clinics 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Jack Jr, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimer's dementia 2018, 14, 535–562. [Google Scholar] [PubMed]
- Klyucherev, T.O.; Olszewski, P.; Shalimova, A.A.; Chubarev, V.N.; Tarasov, V.V.; Attwood, M.M.; Syvänen, S.; Schiöth, H.B. Advances in the development of new biomarkers for Alzheimer’s disease. Translational neurodegeneration 2022, 11, 25. [Google Scholar]
- Angioni, D.; Delrieu, J.; Hansson, O.; Fillit, H.; Aisen, P.; Cummings, J.; Sims, J.; Braunstein, J.; Sabbagh, M.; Bittner, T. Blood biomarkers from research use to clinical practice: What must be done? A report from the EU/US CTAD Task Force. The journal of prevention of Alzheimer's disease 2022, 9, 569–579. [Google Scholar] [CrossRef]
- Schreiner, T.G.; Croitoru, C.G.; Hodorog, D.N.; Cuciureanu, D.I. Passive Anti-Amyloid Beta Immunotherapies in Alzheimer’s Disease: From Mechanisms to Therapeutic Impact. Biomedicines 2024, 12, 1096. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's dementia 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Jack Jr, C.R.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; Hansson, O.; Ho, C.; Jagust, W.; McDade, E. Revised criteria for diagnosis and staging of Alzheimer's disease: Alzheimer's Association Workgroup. Alzheimer's Dementia 2024. [Google Scholar] [CrossRef]
- Arslan, B.; Zetterberg, H.; Ashton, N.J. Blood-based biomarkers in Alzheimer’s disease–moving towards a new era of diagnostics. Clinical Chemistry Laboratory Medicine 2024, 62, 1063–1069. [Google Scholar] [CrossRef]
- Assfaw, A.D.; Schindler, S.E.; Morris, J.C. Advances in blood biomarkers for Alzheimer disease (AD): A review. The Kaohsiung Journal of Medical Sciences 2024, 40, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.C.; Kinney, J.W.; Ritter, A.; Andrews, R.D.; Toledano Strom, E.N.; Lukic, A.S.; Koenig, L.N.; Revta, C.; Fillit, H.M.; Zhong, K. Relationships between plasma biomarkers, tau PET, FDG PET, and volumetric MRI in mild to moderate Alzheimer's disease patients. Alzheimer's & Dementia: Translational Research & Clinical interventions 2024, 10, e12490. [Google Scholar]
- Brand, A.L.; Lawler, P.E.; Bollinger, J.G.; Li, Y.; Schindler, S.E.; Li, M.; Lopez, S.; Ovod, V.; Nakamura, A.; Shaw, L.M. The performance of plasma amyloid beta measurements in identifying amyloid plaques in Alzheimer’s disease: a literature review. Alzheimer's research therapy 2022, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Yi, S.; Han, J.-y.; Park, S.Y.; Jang, J.-W.; Chun, I.K.; Kim, S.E.; Lee, B.S.; Kim, G.J.; Yu, J.S.J.A. s. r. ; therapy, Oligomeric forms of amyloid-β protein in plasma as a potential blood-based biomarker for Alzheimer’s disease. Alzheimer's research therapy 2017, 9, 1–10. [Google Scholar] [CrossRef]
- Li, D.; Mielke, M.M. An update on blood-based markers of Alzheimer’s disease using the SiMoA platform. Neurology therapy 2019, 8 (Suppl 2), 73–82. [Google Scholar] [CrossRef]
- Wang, R.; Sweeney, D.; Gandy, S.E.; Sisodia, S.S. The Profile of Soluble Amyloid β Protein in Cultured Cell Media: detection and quantification of amyloid β protein and variants by immunoprecipitation-mass spectrometry. Journal of Biological Chemistry 1996, 271, 31894–31902. [Google Scholar] [CrossRef]
- Korecka, M.; Shaw, L.M. Mass spectrometry-based methods for robust measurement of Alzheimer's disease biomarkers in biological fluids. Journal of neurochemistry 2021, 159, 211–233. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Cerebrospinal fluid biomarkers for Alzheimer's disease. Journal of Alzheimer's Disease 2009, 18, 413–417. [Google Scholar] [CrossRef]
- Guo, Y.; Shen, X.-N.; Wang, H.-F.; Chen, S.-D.; Zhang, Y.-R.; Chen, S.-F.; Cui, M.; Cheng, W.; Dong, Q.; Ma, T. The dynamics of plasma biomarkers across the Alzheimer’s continuum. Alzheimer's Research Therapy 2023, 15, 31. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.; Xiong, C. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [Google Scholar] [CrossRef] [PubMed]
- Ovod, V.; Ramsey, K.N.; Mawuenyega, K.G.; Bollinger, J.G.; Hicks, T.; Schneider, T.; Sullivan, M.; Paumier, K.; Holtzman, D.M.; Morris, J.C. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer's Dementia 2017, 13, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Li, W.; Chen, Y.; Lin, Y.; Wang, B.; Guo, Q.; Miao, Y. Plasma Aβ as a biomarker for predicting Aβ-PET status in Alzheimer’s disease: A systematic review with meta-analysis. Journal of Neurology, Neurosurgery Psychiatry 2022, 93, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Kirmess, K.M.; Meyer, M.R.; Holubasch, M.S.; Knapik, S.S.; Hu, Y.; Jackson, E.N.; Harpstrite, S.E.; Verghese, P.B.; West, T.; Fogelman, I. The PrecivityAD™ test: Accurate and reliable LC-MS/MS assays for quantifying plasma amyloid beta 40 and 42 and apolipoprotein E proteotype for the assessment of brain amyloidosis. Clinica Chimica Acta 2021, 519, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Qiao, Y.; Choi, J.; Raman, R.; Ringman, J.M.; Shi, Y.; Initiative, A. s. D. N. Enhanced association of tau pathology and cognitive impairment in mild cognitive impairment subjects with behavior symptoms. Journal of Alzheimer's Disease 2022, 87, 557–568. [Google Scholar] [CrossRef]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. The physiological roles of tau and Aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta neuropathologica 2020, 140, 417–447. [Google Scholar] [CrossRef]
- Brickman, A.M.; Manly, J.J.; Honig, L.S.; Sanchez, D.; Reyes-Dumeyer, D.; Lantigua, R.A.; Lao, P.J.; Stern, Y.; Vonsattel, J.P.; Teich, A.F. Plasma p-tau181, p-tau217, and other blood-based Alzheimer's disease biomarkers in a multi-ethnic, community study. Alzheimer's Dementia 2021, 17, 1353–1364. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. Jama 2020, 324, 772–781. [Google Scholar] [CrossRef]
- McGrath, E.R.; Beiser, A.S.; O’Donnell, A.; Yang, Q.; Ghosh, S.; Gonzales, M.M.; Himali, J.J.; Satizabal, C.L.; Johnson, K.A.; Tracy, R.P. Blood phosphorylated tau 181 as a biomarker for amyloid burden on brain PET in cognitively healthy adults. Journal of Alzheimer's Disease 2022, 87, 1517–1526. [Google Scholar] [CrossRef]
- Gonzalez-Ortiz, F.; Ferreira, P.C.; González-Escalante, A.; Montoliu-Gaya, L.; Ortiz-Romero, P.; Kac, P.R.; Turton, M.; Kvartsberg, H.; Ashton, N.J.; Zetterberg, H. A novel ultrasensitive assay for plasma p-tau217: performance in individuals with subjective cognitive decline and early Alzheimer's disease. Alzheimer's Dementia 2024, 20, 1239–1249. [Google Scholar] [CrossRef]
- Ashton, N.J.; Brum, W.S.; Di Molfetta, G.; Benedet, A.L.; Arslan, B.; Jonatis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M. Diagnostic accuracy of the plasma ALZpath pTau217 immunoassay to identify Alzheimer’s disease pathology. medRxiv 2023. [Google Scholar]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Initiative, A. s. D. N.; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nature medicine 2021, 27, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Berron, D.; Smith, R.; Strandberg, O.; Proctor, N.K.; Dage, J.L.; Stomrud, E.; Palmqvist, S.; Mattsson-Carlgren, N.; Hansson, O. Associations of plasma phospho-tau217 levels with tau positron emission tomography in early Alzheimer disease. JAMA neurology 2021, 78, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ortiz, F.; Turton, M.; Kac, P.R.; Smirnov, D.; Premi, E.; Ghidoni, R.; Benussi, L.; Cantoni, V.; Saraceno, C.; Rivolta, J. Brain-derived tau: a novel blood-based biomarker for Alzheimer’s disease-type neurodegeneration. Brain 2023, 146, 1152–1165. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; van der Kant, R.; Hansson, O. Tau biomarkers in Alzheimer's disease: towards implementation in clinical practice and trials. The Lancet Neurology 2022, 21, 726–734. [Google Scholar] [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Montelongo, R.; Lorenzo-Salazar, J.M.; Estévez-Herrera, J.; García-Luis, J.; Íñigo-Campos, A.; Rubio-Rodríguez, L.A.; Muñoz-Barrera, A.; Trujillo-González, R. Transactive response DNA-binding protein (TARDBP/TDP-43) regulates cell permissivity to HIV-1 infection by acting on HDAC6. International journal of molecular sciences 2022, 23, 6180. [Google Scholar] [CrossRef]
- Gatignol, A.; Duarte, M.; Daviet, L.; Chang, Y.-N.; Jeang, K.-T. Sequential steps in Tat trans-activation of HIV-1 mediated through cellular DNA, RNA, and protein binding factors. Gene expression 1996, 5, 217. [Google Scholar]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 pathology in Alzheimer’s disease. Molecular neurodegeneration 2021, 16, 1–15. [Google Scholar] [CrossRef]
- Lopez, O.L.; Kofler, J.; Chang, Y.; Berman, S.B.; Becker, J.T.; Sweet, R.A.; Nadkarni, N.; Patira, R.; Kamboh, M.I.; Cohen, A.D. Hippocampal sclerosis, TDP-43, and the duration of the symptoms of dementia of AD patients. Annals of clinical translational neurology 2020, 7, 1546–1556. [Google Scholar] [CrossRef]
- Katisko, K.; Huber, N.; Kokkola, T.; Hartikainen, P.; Krüger, J.; Heikkinen, A.-L.; Paananen, V.; Leinonen, V.; Korhonen, V.E.; Helisalmi, S. Serum total TDP-43 levels are decreased in frontotemporal dementia patients with C9orf72 repeat expansion or concomitant motoneuron disease phenotype. Alzheimer's research therapy 2022, 14, 151. [Google Scholar] [CrossRef]
- Cordts, I.; Wachinger, A.; Scialo, C.; Lingor, P.; Polymenidou, M.; Buratti, E.; Feneberg, E. TDP-43 proteinopathy specific biomarker development. Cells 2023, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Cenik, B.; Cenik, B.K.; Herz, J.; Yu, G. TDP-43 in central nervous system development and function: clues to TDP-43-associated neurodegeneration. Biological chemistry 2012, 393, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Wilhite, R.; Sage, J.M.; Bouzid, A.; Primavera, T.; Agbas, A. Platelet phosphorylated TDP-43: an exploratory study for a peripheral surrogate biomarker development for Alzheimer's disease. Future Science OA 2017, 3, FSO238. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Molecular neurodegeneration 2019, 14, 23. [Google Scholar] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson's disease. Cold Spring Harbor perspectives in medicine 2012, 2, a009399. [Google Scholar] [CrossRef]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P. Red blood cells are the major source of alpha-synuclein in blood. Neurodegenerative diseases 2008, 5, 55–59. [Google Scholar] [CrossRef]
- Kasuga, K.; Tokutake, T.; Ishikawa, A.; Uchiyama, T.; Tokuda, T.; Onodera, O.; Nishizawa, M.; Ikeuchi, T. Differential levels of α-synuclein, β-amyloid42 and tau in CSF between patients with dementia with Lewy bodies and Alzheimer's disease. Journal of Neurology, Neurosurgery Psychiatry 2010, 81, 608–610. [Google Scholar] [CrossRef]
- Kasuga, K.; Nishizawa, M.; Ikeuchi, T. α-Synuclein as CSF and Blood Biomarker of Dementia with Lewy Bodies. International Journal of Alzheimer’s Disease 2012, 2012, 437025. [Google Scholar] [CrossRef]
- Daniele, S.; Baldacci, F.; Piccarducci, R.; Palermo, G.; Giampietri, L.; Manca, M.L.; Pietrobono, D.; Frosini, D.; Nicoletti, V.; Tognoni, G. α-Synuclein heteromers in red blood cells of Alzheimer’s disease and Lewy body dementia patients. Journal of Alzheimer's Disease 2021, 80, 885–893. [Google Scholar] [CrossRef]
- Laske, C.; Fallgatter, A.J.; Stransky, E.; Hagen, K.; Berg, D.; Maetzler, W. Decreased α-synuclein serum levels in patients with Lewy body dementia compared to Alzheimer’s disease patients and control subjects. Dementia geriatric cognitive disorders 2011, 31, 413–416. [Google Scholar] [CrossRef]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic interactions between Aβ, tau, and α-synuclein: acceleration of neuropathology and cognitive decline. Journal of Neuroscience 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Baldacci, F.; Daniele, S.; Piccarducci, R.; Giampietri, L.; Pietrobono, D.; Giorgi, F.S.; Nicoletti, V.; Frosini, D.; Libertini, P.; Lo Gerfo, A. Potential diagnostic value of red blood cells α-synuclein heteroaggregates in Alzheimer’s disease. Molecular Neurobiology 2019, 56, 6451–6459. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Gu, X.; Li, H.; Lei, S.; Wang, Z.; Wang, J.; Yin, P.; Zhang, C.; Wang, F.; Liu, C. The role of DKK1 in Alzheimer’s disease: a potential intervention point of brain damage prevention? Pharmacological Research 2019, 144, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer's brain. Journal of Neuroscience 2004, 24, 6021–6027. [Google Scholar] [CrossRef] [PubMed]
- Purro, S.A.; Galli, S.; Salinas, P.C. Dysfunction of Wnt signaling and synaptic disassembly in neurodegenerative diseases. Journal of molecular cell biology 2014, 6, 75–80. [Google Scholar] [CrossRef]
- Seib, D.R.; Corsini, N.S.; Ellwanger, K.; Plaas, C.; Mateos, A.; Pitzer, C.; Niehrs, C.; Celikel, T.; Martin-Villalba, A. Loss of Dickkopf-1 restores neurogenesis in old age and counteracts cognitive decline. Cell stem cell 2013, 12, 204–214. [Google Scholar] [CrossRef]
- Marzo, A.; Galli, S.; Lopes, D.; McLeod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A. Reversal of synapse degeneration by restoring Wnt signaling in the adult hippocampus. Current Biology 2016, 26, 2551–2561. [Google Scholar] [CrossRef]
- Tay, L.; Leung, B.; Yeo, A.; Chan, M.; Lim, W.S. Elevations in Serum Dickkopf-1 and disease progression in community-dwelling older adults with mild cognitive impairment and mild-to-moderate Alzheimer’s disease. Frontiers in Aging Neuroscience 2019, 11, 278. [Google Scholar] [CrossRef]
- Tarawneh, R.; D'Angelo, G.; Macy, E.; Xiong, C.; Carter, D.; Cairns, N.J.; Fagan, A.M.; Head, D.; Mintun, M.A.; Ladenson, J.H. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Annals of neurology 2011, 70, 274–285. [Google Scholar] [CrossRef]
- Halbgebauer, S.; Steinacker, P.; Riedel, D.; Oeckl, P.; Anderl-Straub, S.; Lombardi, J.; von Arnim, C.A.; Nagl, M.; Giese, A.; Ludolph, A.C. Visinin-like protein 1 levels in blood and CSF as emerging markers for Alzheimer’s and other neurodegenerative diseases. Alzheimer's research therapy 2022, 14, 175. [Google Scholar] [CrossRef]
- Mavroudis, I.A.; Petridis, F.; Chatzikonstantinou, S.; Karantali, E.; Kazis, D. A meta-analysis on the levels of VILIP-1 in the CSF of Alzheimer’s disease compared to normal controls and other neurodegenerative conditions. Aging Clinical Experimental Research 2021, 33, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Rao, M.V.; Nixon, R.A. Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harbor perspectives in biology 2017, 9, a018309. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Shi, M.; Li, Y.; Zhao, X. Elevated plasma neurofilament light was associated with multi-modal neuroimaging features in Alzheimer’s Disease signature regions and predicted future tau deposition. Research Square 2024. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Andreasson, U.; Zetterberg, H.; Blennow, K.; neurology, A.s.D.N.I.J.J. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. 2017, 74, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Barro, C.; Andreasson, U.; Derfuss, T.; Lindberg, R.; Sandelius, Å.; Liman, V.; Norgren, N.; Blennow, K.; Zetterberg, H. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clinical Chemistry Laboratory Medicine 2016, 54, 1655–1661. [Google Scholar] [CrossRef]
- Bäckström, D.; Linder, J.; Jakobson Mo, S.; Riklund, K.; Zetterberg, H.; Blennow, K.; Forsgren, L.; Lenfeldt, N. NfL as a biomarker for neurodegeneration and survival in Parkinson disease. Neurology 2020, 95, e827–e838. [Google Scholar] [CrossRef]
- Kivisäkk, P.; Carlyle, B.C.; Sweeney, T.; Quinn, J.P.; Ramirez, C.E.; Trombetta, B.A.; Mendes, M.; Brock, M.; Rubel, C.; Czerkowicz, J. Increased levels of the synaptic proteins PSD-95, SNAP-25, and neurogranin in the cerebrospinal fluid of patients with Alzheimer’s disease. Alzheimer's research therapy 2022, 14, 58. [Google Scholar] [CrossRef]
- Halbgebauer, S.; Steinacker, P.; Hengge, S.; Oeckl, P.; Rumeileh, S.A.; Anderl-Straub, S.; Lombardi, J.; Von Arnim, C.A.; Giese, A.; Ludolph, A.C. CSF levels of SNAP-25 are increased early in Creutzfeldt-Jakob and Alzheimer’s disease. Journal of Neurology, Neurosurgery Psychiatry 2022, 93, 1059–1065. [Google Scholar] [CrossRef]
- Agliardi, C.; Guerini, F.R.; Zanzottera, M.; Bianchi, A.; Nemni, R.; Clerici, M. SNAP-25 in serum is carried by exosomes of neuronal origin and is a potential biomarker of Alzheimer’s disease. Molecular neurobiology 2019, 56, 5792–5798. [Google Scholar] [CrossRef]
- Libiger, O.; Shaw, L.M.; Watson, M.H.; Nairn, A.C.; Umaña, K.L.; Biarnes, M.C.; Canet-Avilés, R.M.; Jack Jr, C.R.; Breton, Y.A.; Cortes, L. Longitudinal CSF proteomics identifies NPTX2 as a prognostic biomarker of Alzheimer's disease. Alzheimer's Dementia 2021, 17, 1976–1987. [Google Scholar] [CrossRef]
- Saunders, T.S.; Gadd, D.A.; Spires-Jones, T.L.; King, D.; Ritchie, C.; Muniz-Terrera, G. Associations between cerebrospinal fluid markers and cognition in ageing and dementia: A systematic review. European Journal of Neuroscience 2022, 56, 5650–5713. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Q.; Skudder-Hill, L.; Toyota, T.; Wei, W.; Adachi, H. CSF GAP-43 as a biomarker of synaptic dysfunction is associated with tau pathology in Alzheimer’s disease. Scientific Reports 2022, 12, 17392. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Zhu, M.; Kong, C.; Pang, Y.; Zhang, H.; Qiu, Q.; Wei, C.; Tang, Y.; Wang, Q.; Li, Y. Blood neuro-exosomal synaptic proteins predict Alzheimer's disease at the asymptomatic stage. Alzheimer's Dementia 2021, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, T.; Blandino, V.; Maniscalco, L.; Matranga, D.; Graziano, F.; Guajana, F.; Agnello, L.; Lo Sasso, B.; Gambino, C.M.; Giglio, R.V. Biomarkers related to synaptic dysfunction to discriminate alzheimer’s disease from other neurological disorders. International Journal of Molecular Sciences 2022, 23, 10831. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Gerges, N.Z. Neurogranin and synaptic plasticity balance. Communicative integrative biology 2010, 3, 340–342. [Google Scholar] [CrossRef]
- Kvartsberg, H.; Portelius, E.; Andreasson, U.; Brinkmalm, G.; Hellwig, K.; Lelental, N.; Kornhuber, J.; Hansson, O.; Minthon, L.; Spitzer, P. Characterization of the postsynaptic protein neurogranin in paired cerebrospinal fluid and plasma samples from Alzheimer’s disease patients and healthy controls. Alzheimer's research therapy 2015, 7, 1–9. [Google Scholar] [CrossRef]
- Wellington, H.; Paterson, R.W.; Portelius, E.; Törnqvist, U.; Magdalinou, N.; Fox, N.C.; Blennow, K.; Schott, J.M.; Zetterberg, H. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology 2016, 86, 829–835. [Google Scholar] [CrossRef]
- He, M.; Sun, L.; Cao, W.; Yin, C.; Sun, W.; Liu, P.; Tan, L.; Xu, Z.; Zhao, W. Association between plasma exosome neurogranin and brain structure in patients with Alzheimer’s disease: A protocol study. BMJ Open 2020, 10, e036990. [Google Scholar] [CrossRef]
- Shibuya, M.J.G. cancer, Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti-and pro-angiogenic therapies. 2011, 2, 1097–1105. [Google Scholar]
- Ceci, C.; Lacal, P.M.; Barbaccia, M.L.; Mercuri, N.B.; Graziani, G.; Ledonne, A. The VEGFs/VEGFRs system in Alzheimer’s and Parkinson’s diseases: Pathophysiological roles and therapeutic implications. J Pharmacological Research 2024, 107101. [Google Scholar]
- Lau, S.-F.; Cao, H.; Fu, A.K.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proceedings of the National Academy of Sciences 2020, 117, 25800–25809. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.; Ashby, E.L.; Wellington, D.; Barrow, V.M.; Palmer, J.C.; Kehoe, P.G.; Esiri, M.M.; Love, S. Pathophysiology of white matter perfusion in Alzheimer’s disease and vascular dementia. Brain 2014, 137, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.C.; Tayler, H.M.; Love, S. Endothelin-converting enzyme-1 activity, endothelin-1 production, and free radical-dependent vasoconstriction in Alzheimer's disease. Journal of Alzheimer's Disease 2013, 36, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Geven, C.; Kox, M.; Pickkers, P. Adrenomedullin and adrenomedullin-targeted therapy as treatment strategies relevant for sepsis. Frontiers in Immunology 2018, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Inflammation and white matter damage in vascular cognitive impairment. Stroke 2009, 40 (3_suppl_1), S20–S23. [Google Scholar] [CrossRef]
- Ferrero, H.; Larrayoz, I.M.; Martisova, E.; Solas, M.; Howlett, D.R.; Francis, P.T.; Gil-Bea, F.J.; Martínez, A.; Ramírez, M. Increased levels of brain adrenomedullin in the neuropathology of Alzheimer’s disease. J Molecular Neurobiology 2018, 55, 5177–5183. [Google Scholar] [CrossRef]
- Noda, M.; Matsuda, T. Central regulation of body fluid homeostasis. Proceedings of the Japan Academy, Series B 2022, 98, 283–324. [Google Scholar] [CrossRef]
- Mahinrad, S.; Sabayan, B.; Garner, C.R.; Lloyd-Jones, D.M.; Sorond, F.A. N-terminal pro brain, N-terminal pro atrial natriuretic peptides, and dynamic cerebral autoregulation. Journal of the American Heart Association 2020, 9, e018203. [Google Scholar] [CrossRef]
- Qi, X.-m.; Ma, J.-f. The role of amyloid beta clearance in cerebral amyloid angiopathy: more potential therapeutic targets. Translational neurodegeneration 2017, 6, 1–12. [Google Scholar] [CrossRef]
- Mahinrad, S.; Bulk, M.; Van Der Velpen, I.; Mahfouz, A.; van Roon-Mom, W.; Fedarko, N.; Yasar, S.; Sabayan, B.; Van Heemst, D.; Van Der Weerd, L. Natriuretic peptides in post-mortem brain tissue and cerebrospinal fluid of non-demented humans and Alzheimer’s disease patients. Frontiers in Neuroscience 2018, 12, 864. [Google Scholar] [CrossRef]
- Hong, J.; Cheng, H.; Wang, P.; Wu, Y.; Lu, S.; Zhou, Y.; Wang, X.B.; Zhu, X. CXCL9 may serve as a potential biomarker for primary Sjögren’s syndrome with extra-glandular manifestations. Arthritis Research Therapy 2024, 26, 26. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, T.K.; Dunachie, S.J.; Todryk, S.; Hill, A.V.; Fletcher, H.A. MIG (CXCL9) is a more sensitive measure than IFN-γ of vaccine induced T-cell responses in volunteers receiving investigated malaria vaccines. Journal of immunological methods 2009, 340, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Sun, Y.; Xie, X.; Zhao, Y. Blood and CSF chemokines in Alzheimer’s disease and mild cognitive impairment: a systematic review and meta-analysis. Alzheimer's research therapy 2023, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Hertze, J.; Ohlsson, M.; Nägga, K.; Höglund, K.; Basun, H.; Annas, P.; Lannfelt, L.; Andreasen, N.; Minthon, L. Cerebrospinal fluid levels of heart fatty acid binding protein are elevated prodromally in Alzheimer's disease and vascular dementia. Journal of Alzheimer's Disease 2013, 34, 673–679. [Google Scholar] [CrossRef]
- Desikan, R.S.; Thompson, W.K.; Holland, D.; Hess, C.P.; Brewer, J.B.; Zetterberg, H.; Blennow, K.; Andreassen, O.A.; McEvoy, L.K.; Hyman, B.T. Heart fatty acid binding protein and Aβ-associated Alzheimer’s neurodegeneration. Molecular neurodegeneration 2013, 8, 1–9. [Google Scholar] [CrossRef]
- Sashindranath, M.; Nandurkar, H.H. Endothelial dysfunction in the brain: setting the stage for stroke and other cerebrovascular complications of COVID-19. Stroke 2021, 52, 1895–1904. [Google Scholar] [CrossRef]
- Jickling, G.C.; Ander, B.P.; Zhan, X.; Stamova, B.; Hull, H.; DeCarli, C.; Sharp, F.R. Progression of cerebral white matter hyperintensities is related to leucocyte gene expression. Brain 2022, 145, 3179–3186. [Google Scholar] [CrossRef]
- Chen, J.; Dai, A.-X.; Tang, H.-L.; Lu, C.-H.; Liu, H.-X.; Hou, T.; Lu, Z.-J.; Kong, N.; Peng, X.-Y.; Lin, K.-X. Increase of ALCAM and VCAM-1 in the Plasma Predicts the Alzheimer’s Disease. Frontiers in Immunology 2023, 13, 1097409. [Google Scholar] [CrossRef]
- Papasavvas, E.; Azzoni, L.; Pistilli, M.; Hancock, A.; Reynolds, G.; Gallo, C.; Ondercin, J.; Kostman, J.R.; Mounzer, K.; Shull, J. Increased soluble vascular cell adhesion molecule-1 plasma levels and soluble intercellular adhesion molecule-1 during antiretroviral therapy interruption and retention of elevated soluble vascular cellular adhesion molecule-1 levels following resumption of antiretroviral therapy. Aids 2008, 22, 1153–1161. [Google Scholar]
- Austin, S.A.; Katusic, Z.S. Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. Journal of Cerebral Blood Flow Metabolism 2020, 40, 392–403. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Stomrud, E.; Lindberg, O.; Palmqvist, S.; Zetterberg, H.; Blennow, K.; Hansson, O. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology 2018, 91, e867–e877. [Google Scholar] [CrossRef]
- Zuliani, G.; Cavalieri, M.; Galvani, M.; Passaro, A.; Munari, M.; Bosi, C.; Zurlo, A.; Fellin, R. Markers of endothelial dysfunction in older subjects with late onset Alzheimer's disease or vascular dementia. Journal of the neurological sciences 2008, 272, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.D.; Chambers, A.B.; Ott, B.R.; Daiello, L.A.; Initiative, A. s. D. N. Peripheral markers of vascular endothelial dysfunction show independent but additive relationships with brain-based biomarkers in association with functional impairment in Alzheimer’s disease. Journal of Alzheimer's Disease 2021, 80, 1553–1565. [Google Scholar] [CrossRef] [PubMed]
- Farrall, A.J.; Wardlaw, J.M. Blood–brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiology of aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.O.; Toescu, E.C.; Popescu, L.M.; Bajenaru, O.; Muresanu, D.F.; Schultzberg, M.; Bogdanovic, N. Blood-brain barrier alterations in ageing and dementia. Journal of the neurological sciences 2009, 283, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Fan, L.M.; Liu, F.; Smith, C.; Li, J.-M. Nox2 dependent redox-regulation of microglial response to amyloid-β stimulation and microgliosis in aging. Scientific reports 2020, 10, 1582. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Kanski, J. Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer’s amyloid β-peptide 1–42. Peptides 2002, 23, 1299–1309. [Google Scholar] [CrossRef]
- Lovell, M.; Robertson, J.; Teesdale, W.; Campbell, J.; Markesbery, W. Copper, iron and zinc in Alzheimer's disease senile plaques. Journal of the neurological sciences 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Bartzokis, G.; Sultzer, D.; Cummings, J.; Holt, L.E.; Hance, D.B.; Henderson, V.W.; Mintz, J. In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Archives of general psychiatry 2000, 57, 47–53. [Google Scholar] [CrossRef]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 1991, 26, 421–425. [Google Scholar] [CrossRef]
- Markesbery, W.; Lovell, M. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiology of aging 1998, 19, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.; Close, D.; Stern, S. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer’s disease. Neurobiology of aging 2002, 23, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Greilberger, J.; Koidl, C.; Greilberger, M.; Lamprecht, M.; Schroecksnadel, K.; Leblhuber, F.; Fuchs, D.; Oettl, K. Malondialdehyde, carbonyl proteins and albumin-disulphide as useful oxidative markers in mild cognitive impairment and Alzheimer's disease. Free radical research 2008, 42, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D.; Clark, C.M.; Lee, V.M.Y.; Trojanowski, J.Q.; Rokach, J.; FitzGerald, G.A. Increased 8, 12-iso-iPF2α-VI in Alzheimer's disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Annals of neurology 2000, 48, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.; Yao, Y.; Hyman, B.; Growdon, J.; Pratico, D. Plasma F2A isoprostane levels in Alzheimer’s and Parkinson’s disease. Neurodegenerative Diseases 2007, 4, 403–405. [Google Scholar] [CrossRef]
- Cecchi, C.; Fiorillo, C.; Sorbi, S.; Latorraca, S.; Nacmias, B.; Bagnoli, S.; Nassi, P.; Liguri, G. Oxidative stress and reduced antioxidant defenses in peripheral cells from familial Alzheimer’s patients. Free Radical Biology Medicine 2002, 33, 1372–1379. [Google Scholar] [CrossRef]
- Rao, A.; Bharani, M.; Pallavi, V. Role of antioxidants and free radicals in health and disease. Adv Pharmacol Toxicol 2006, 7, 29–38. [Google Scholar]
- Butterfield, D.A.; Reed, T.T.; Perluigi, M.; De Marco, C.; Coccia, R.; Keller, J.N.; Markesbery, W.R.; Sultana, R. Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer's disease. Brain research 2007, 1148, 243–248. [Google Scholar] [CrossRef]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer's disease and link to cognitive impairment. Journal of neurochemistry 2005, 92, 255–263. [Google Scholar] [CrossRef]
- Yu, H.L.; Chertkow, H.M.; Bergman, H.; Schipper, H.M. Aberrant profiles of native and oxidized glycoproteins in Alzheimer plasma. Proteomics 2003, 3, 2240–2248. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mattioli, P.; Aldred, S.; Cecchetti, R.; Stahl, W.; Griffiths, H.; Senin, U.; Sies, H.; Mecocci, P. Plasma antioxidant status, immunoglobulin g oxidation and lipid peroxidation in demented patients: relevance to Alzheimer disease and vascular dementia. Dementia geriatric cognitive disorders 2004, 18, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Mecocci, P.; Polidori, M.C.; Cherubini, A.; Ingegni, T.; Mattioli, P.; Catani, M.; Rinaldi, P.; Cecchetti, R.; Stahl, W.; Senin, U. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Archives of neurology 2002, 59, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, E.; Sardas, S.; Aslan, S.; Isik, E.; Karakaya, A.E. Detection of oxidative DNA damage in lymphocytes of patients with Alzheimer's disease. Biomarkers 2004, 9, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rivière, S.; Birlouez-Aragon, I.; Nourhashémi, F.; Vellas, B. Low plasma vitamin C in Alzheimer patients despite an adequate diet. International journal of geriatric psychiatry 1998, 13, 749–754. [Google Scholar] [CrossRef]
- Sinclair, A.J.; Bayer, A.J.; Johnston, J.; Warner, C.; Maxwell, S.R. Altered plasma antioxidant status in subjects with Alzheimer's disease and vascular dementia. International journal of geriatric psychiatry 1998, 13, 840–845. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer's disease: Role in pathogenesis and novel therapeutic opportunities. British journal of pharmacology 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Gao, R.; Ma, S.L. Is mitochondria DNA variation a biomarker for AD? Genes 2022, 13, 1789. [Google Scholar] [CrossRef]
- Mahapatra, G.; Gao, Z.; Bateman III, J.R.; Lockhart, S.N.; Bergstrom, J.; DeWitt, A.R.; Piloso, J.E.; Kramer, P.A.; Gonzalez-Armenta, J.L.; Amick, K.A. Blood-based bioenergetic profiling reveals differences in mitochondrial function associated with cognitive performance and Alzheimer's disease. Alzheimer's Dementia 2023, 19, 1466–1478. [Google Scholar] [CrossRef]
- Bhatia, S.; Rawal, R.; Sharma, P.; Singh, T.; Singh, M.; Singh, V. Mitochondrial dysfunction in Alzheimer’s disease: opportunities for drug development. Current Neuropharmacology 2022, 20, 675. [Google Scholar] [CrossRef]
- Maynard, S.; Hejl, A.-M.; Dinh, T.-S. T.; Keijzers, G.; Hansen, Å.M.; Desler, C.; Moreno-Villanueva, M.; Bürkle, A.; Rasmussen, L.J.; Waldemar, G. Defective mitochondrial respiration, altered dNTP pools and reduced AP endonuclease 1 activity in peripheral blood mononuclear cells of Alzheimer's disease patients. Aging 2015, 7, 793. [Google Scholar] [CrossRef]
- Coskun, P.; Helguera, P.; Nemati, Z.; Bohannan, R.C.; Thomas, J.; Samuel, S.E.; Argueta, J.; Doran, E.; Wallace, D.C.; Lott, I.T. Metabolic and growth rate alterations in lymphoblastic cell lines discriminate between Down syndrome and Alzheimer’s disease. Journal of Alzheimer's Disease 2017, 55, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Veitinger, M.; Varga, B.; Guterres, S.B.; Zellner, M. Platelets, a reliable source for peripheral Alzheimer’s disease biomarkers? Acta neuropathologica communications 2014, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Toledo, G.; Silva-Lucero, M.-d.-C.; Herrera-Díaz, J.; García, D.-E.; Arias-Montaño, J.-A.; Cardenas-Aguayo, M.-d.-C. Patient-derived fibroblasts with presenilin-1 mutations, that model aspects of Alzheimer’s disease pathology, constitute a potential object for early diagnosis. Frontiers in Aging Neuroscience 2022, 14, 921573. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral glycolysis in neurodegenerative diseases. International journal of molecular sciences 2020, 21, 8924. [Google Scholar] [CrossRef]
- Bossy, B.; Petrilli, A.; Klinglmayr, E.; Chen, J.; Lütz-Meindl, U.; Knott, A.B.; Masliah, E.; Schwarzenbacher, R.; Bossy-Wetzel, E. S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer's disease. Journal of Alzheimer's disease 2010, 20 (s2), S513–S526. [Google Scholar] [CrossRef]
- Huang, D.-X.; Yu, X.; Yu, W.-J.; Zhang, X.-M.; Liu, C.; Liu, H.-P.; Sun, Y.; Jiang, Z.-P. Calcium signaling regulated by cellular membrane systems and calcium homeostasis perturbed in Alzheimer’s disease. Frontiers in Cell Developmental Biology 2022, 10, 834962. [Google Scholar] [CrossRef]
- Trumpff, C.; Michelson, J.; Lagranha, C.J.; Taleon, V.; Karan, K.R.; Sturm, G.; Lindqvist, D.; Fernström, J.; Moser, D.; Kaufman, B.A. Stress and circulating cell-free mitochondrial DNA: A systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion 2021, 59, 225–245. [Google Scholar] [CrossRef]
- Reid, D.M.; Barber, R.C.; Jones, H.P.; Thorpe Jr, R.J.; Sun, J.; Zhou, Z.; Phillips, N.R. Integrative blood-based characterization of oxidative mitochondrial DNA damage variants implicates Mexican American’s metabolic risk for developing Alzheimer’s disease. Scientific reports 2023, 13, 14765. [Google Scholar] [CrossRef]
- Moya, G.E.; Rivera, P.D.; Dittenhafer-Reed, K.E. Evidence for the role of mitochondrial DNA release in the inflammatory response in neurological disorders. International journal of molecular sciences 2021, 22, 7030. [Google Scholar]
- Miao, J.; Ma, H.; Yang, Y.; Liao, Y.; Lin, C.; Zheng, J.; Yu, M.; Lan, J. Microglia in Alzheimer’s disease: Pathogenesis, mechanisms, and therapeutic potentials. Frontiers in aging neuroscience 2023, 15, 1201982. [Google Scholar]
- Frost, G.R.; Li, Y.-M. The role of astrocytes in amyloid production and Alzheimer's disease. Open biology 2017, 7, 170228. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of microglia and astrocytes in Alzheimer’s disease: from neuroinflammation to Ca2+ homeostasis dysregulation. Cells 2022, 11, 2728. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Shin, K.Y.; Chang, K.-A. GFAP as a potential biomarker for Alzheimer’s disease: a systematic review and meta-analysis. Cells 2023, 12, 1309. [Google Scholar] [CrossRef] [PubMed]
- Parvizi, T.; König, T.; Wurm, R.; Silvaieh, S.; Altmann, P.; Klotz, S.; Rommer, P.S.; Furtner, J.; Regelsberger, G.; Lehrner, J. Real-world applicability of glial fibrillary acidic protein and neurofilament light chain in Alzheimer’s disease. Frontiers in Aging Neuroscience 2022, 14, 887498. [Google Scholar] [CrossRef] [PubMed]
- Benedet, A.L.; Milà-Alomà, M.; Vrillon, A.; Ashton, N.J.; Pascoal, T.A.; Lussier, F.; Karikari, T.K.; Hourregue, C.; Cognat, E.; Dumurgier, J. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA neurology 2021, 78, 1471–1483. [Google Scholar] [CrossRef]
- Domingues, C.; AB da Cruz e Silva, O.; Henriques, A. Impact of cytokines and chemokines on Alzheimer's disease neuropathological hallmarks. Current Alzheimer Research 2017, 14, 870–882. [Google Scholar] [CrossRef]
- Perea, J.R.; Lleó, A.; Alcolea, D.; Fortea, J.; Ávila, J.; Bolós, M. Decreased CX3CL1 levels in the cerebrospinal fluid of patients with Alzheimer’s disease. Frontiers in Neuroscience 2018, 12, 609. [Google Scholar] [CrossRef]
- Vacínová, G.; Vejražkova, D.; Rusina, R.; Holmerová, I.; Vaňková, H.; Jarolímová, E.; Včelák, J.; Bendlová, B.; Vaňková, M. Regulated upon activation, normal T cell expressed and secreted (RANTES) levels in the peripheral blood of patients with Alzheimer’s disease. Neural Regeneration Research 2021, 16, 796–800. [Google Scholar] [CrossRef]
- Martens, L.H.; Zhang, J.; Barmada, S.J.; Zhou, P.; Kamiya, S.; Sun, B.; Min, S.-W.; Gan, L.; Finkbeiner, S.; Huang, E.J. Progranulin deficiency promotes neuroinflammation and neuron loss following toxin-induced injury. The Journal of clinical investigation 2012, 122, 3955–3959. [Google Scholar] [CrossRef]
- Mendsaikhan, A.; Tooyama, I.; Walker, D.G. Microglial progranulin: involvement in Alzheimer’s disease and neurodegenerative diseases. Cells 2019, 8, 230. [Google Scholar] [CrossRef]
- Vergallo, A.; Lista, S.; Lemercier, P.; Chiesa, P.A.; Zetterberg, H.; Blennow, K.; Potier, M.-C.; Habert, M.-O.; Baldacci, F.; Cavedo, E. Association of plasma YKL-40 with brain amyloid-β levels, memory performance, and sex in subjective memory complainers. Neurobiology of aging 2020, 96, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Hua, F.; Zhang, L.; Lin, Y.; Fang, P.; Chen, S.; Ying, J.; Wang, X. Dual roles of interleukin-33 in cognitive function by regulating central nervous system inflammation. Journal of Translational Medicine 2022, 20, 369. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Piancone, F.; La Rosa, F.; Galimberti, D.; Fenoglio, C.; Scarpini, E.; Clerici, M. IL-33 and its decoy sST2 in patients with Alzheimer’s disease and mild cognitive impairment. Journal of neuroinflammation 2020, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Casati, M.; Ferri, E.; Gussago, C.; Mazzola, P.; Abbate, C.; Bellelli, G.; Mari, D.; Cesari, M.; Arosio, B. Increased expression of TREM 2 in peripheral cells from mild cognitive impairment patients who progress into Alzheimer's disease. European journal of neurolog 2018, 25, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Tan, M.-S.; Yu, J.-T.; Sun, L.; Tan, L.; Wang, Y.-L.; Jiang, T.; Tan, L. Increased expression of TREM2 in peripheral blood of Alzheimer's disease patients. Journal of Alzheimer's Disease 2014, 38, 497–501. [Google Scholar] [CrossRef]
- Lu, Q.; Xie, Y.; Qi, X.; Yang, S. TREM1 as a novel prognostic biomarker and tumor immune microenvironment evaluator in glioma. Medicine (Baltimore) 2023, 102, e36410. [Google Scholar] [CrossRef]
- Španić Popovački, E.; Babić Leko, M.; Langer Horvat, L.; Brgić, K.; Vogrinc, Ž.; Boban, M.; Klepac, N.; Borovečki, F.; Šimić, G. Soluble TREM2 concentrations in the cerebrospinal fluid correlate with the severity of neurofibrillary degeneration, cognitive impairment, and inflammasome activation in Alzheimer’s disease. Neurology international 2023, 15, 842–856. [Google Scholar] [CrossRef]
- AlMansoori, M.E.; Jemimah, S.; Abuhantash, F.; AlShehhi, A. Predicting early Alzheimer’s with blood biomarkers and clinical features. Scientific Reports 2024, 14, 6039. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, J.; Yang, Y.; Zhang, Z.; Zhong, H.; Zeng, G.; Zhou, D.; Nowakowski, R.S.; Long, J.; Wu, C. Identification of candidate DNA methylation biomarkers related to Alzheimer’s disease risk by integrating genome and blood methylome data. Translational psychiatry 2023, 13, 387. [Google Scholar] [CrossRef]
- Nagaraj, S.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. microRNA diagnostic panel for Alzheimer’s disease and epigenetic trade-off between neurodegeneration and cancer. Ageing research reviews 2019, 49, 125–143. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Tarazona-Sánchez, A.; Braza-Boils, A.; Balaguer, A.; Ferré-González, L.; Cañada-Martínez, A.J.; Baquero, M.; Cháfer-Pericás, C. Plasma microRNAs as potential biomarkers in early Alzheimer disease expression. Scientific Reports 2022, 12, 15589. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Tordera, L.; Papandreou, C.; Novau-Ferré, N.; García-González, P.; Rojas, M.; Marquié, M.; Chapado, L.A.; Papagiannopoulos, C.; Fernàndez-Castillo, N.; Valero, S. Exploring small non-coding RNAs as blood-based biomarkers to predict Alzheimer’s disease. Cell Bioscience 2024, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Sadlon, A.; Takousis, P.; Evangelou, E.; Prokopenko, I.; Alexopoulos, P.; Udeh-Momoh, C.-M.; Price, G.; Middleton, L.; Perneczky, R.; Initiative, A. s. D. N. Association of Blood MicroRNA Expression and Polymorphisms with Cognitive and Biomarker Changes in Older Adults. The Journal of Prevention of Alzheimer's Disease 2024, 11, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Yuen, S.C.; Liang, X.; Zhu, H.; Jia, Y.; Leung, S.-w. Prediction of differentially expressed microRNAs in blood as potential biomarkers for Alzheimer’s disease by meta-analysis and adaptive boosting ensemble learning. Alzheimer's research therapy 2021, 13, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Khodayi, M.; Khalaj-Kondori, M.; Feizi, M.A.H.; Bonyadi, M.J.; Talebi, M. Plasma lncRNA profiling identified BC200 and NEAT1 lncRNAs as potential blood-based biomarkers for late-onset Alzheimer’s disease. EXCLI journal 2022, 21, 772. [Google Scholar]
- Feng, L.; Liao, Y.-T.; He, J.-C.; Xie, C.-L.; Chen, S.-Y.; Fan, H.-H.; Su, Z.-P.; Wang, Z. Plasma long non-coding RNA BACE1 as a novel biomarker for diagnosis of Alzheimer disease. BMC neurology 2018, 18, 1–8. [Google Scholar] [CrossRef]
- Santana, D.A.; Smith, M. d. A. C.; Chen, E.S. Histone modifications in Alzheimer’s disease. Genes 2023, 14, 347. [Google Scholar] [CrossRef]
- De Plano, L.M.; Saitta, A.; Oddo, S.; Caccamo, A. Epigenetic Changes in Alzheimer’s Disease: DNA Methylation and Histone Modification. Cells 2024, 13, 719. [Google Scholar] [CrossRef]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA methylation biomarkers in aging and age-related diseases. Frontiers in Genetics 2020, 11, 480672. [Google Scholar] [CrossRef]
- Thrush, K.L.; Bennett, D.A.; Gaiteri, C.; Horvath, S.; van Dyck, C.H.; Higgins-Chen, A.T.; Levine, M.E. Aging the brain: multi-region methylation principal component based clock in the context of Alzheimer’s disease. Aging 2022, 14, 5641. [Google Scholar] [CrossRef]
- Ren, Z.; Chu, C.; Pang, Y.; Cai, H.; Jia, L. A circular RNA blood panel that differentiates Alzheimer’s disease from other dementia types. Biomarker Research 2022, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, X.; Chen, Y.-H.; Zhang, K. Identification of circular RNA hsa_Circ_0003391 in peripheral blood is potentially associated with Alzheimer's disease. Frontiers in Aging Neuroscience 2020, 12, 601965. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Morikawa, S.; Nakashima, M.; Yoshikawa, S.; Taniguchi, K.; Sawamura, H.; Suga, N.; Tsuji, A.; Matsuda, S. CircRNAs and RNA-binding proteins involved in the pathogenesis of cancers or central nervous system disorders. Non-coding RNA 2023, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, F.; He, A.T.; Yang, B.B. Circular RNAs: expression, localization, and therapeutic potentials. Molecular therapy 2021, 29, 1683–1702. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, S.; Jedrychowski, M.P.; Tatebe, H.; DeLeo, A.M.; Ikezu, S.; Tokuda, T.; Gygi, S.P.; Stern, R.A.; Ikezu, T. Proteomic profiling of extracellular vesicles isolated from cerebrospinal fluid of former national football league players at risk for chronic traumatic encephalopathy. Frontiers in neuroscience 2019, 13, 1059. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P.; Yanamandra, K.; Ikezu, S.; Gygi, S.P.; Ikezu, T. Proteomic profiling of extracellular vesicles derived from cerebrospinal fluid of Alzheimer’s disease patients: a pilot study. Cells 2020, 9, 1959. [Google Scholar] [CrossRef]
- Guo, M.; Yin, Z.; Chen, F.; Lei, P. Mesenchymal stem cell-derived exosome: a promising alternative in the therapy of Alzheimer’s disease. Alzheimer's Research Therapy 2020, 12, 1–14. [Google Scholar]
- Colombo, E.; Borgiani, B.; Verderio, C.; Furlan, R. Microvesicles: novel biomarkers for neurological disorders. Frontiers in physiology 2012, 3, 63. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L. Identification of preclinical Alzheimer's disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimer's Dementia 2015, 11, 600–607. e1. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Nogueras-Ortiz, C.; Mustapic, M.; Mullins, R.J.; Abner, E.L.; Schwartz, J.B.; Kapogiannis, D. Deficient neurotrophic factors of CSPG4-type neural cell exosomes in Alzheimer disease. The FASEB Journal 2019, 33, 231. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. The FASEB Journal 2015, 29, 589. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J. Advancing medicine for Alzheimer’s disease: A plasma neural exosome platform. The FASEB Journal 2020, 34, 13079–13084. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.-A. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte–neuron communication. PLoS biology 2013, 11, e1001604. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Timeline of patients' journey integrated with blood-based biomarkers: The figure depicts a patient's journey starting with visits to primary care clinical set-up and subsequently to a specialist neurologist or dementia specialist. Validated blood-based biomarkers (BBBMs) can be used as a diagnostic parameter in addition to clinical assessment. Additionally, BBBMs may be utilized to screen healthy populations at risk for AD and monitor therapeutic response and disease advancement.
Figure 2.
Timeline of patients' journey integrated with blood-based biomarkers: The figure depicts a patient's journey starting with visits to primary care clinical set-up and subsequently to a specialist neurologist or dementia specialist. Validated blood-based biomarkers (BBBMs) can be used as a diagnostic parameter in addition to clinical assessment. Additionally, BBBMs may be utilized to screen healthy populations at risk for AD and monitor therapeutic response and disease advancement.
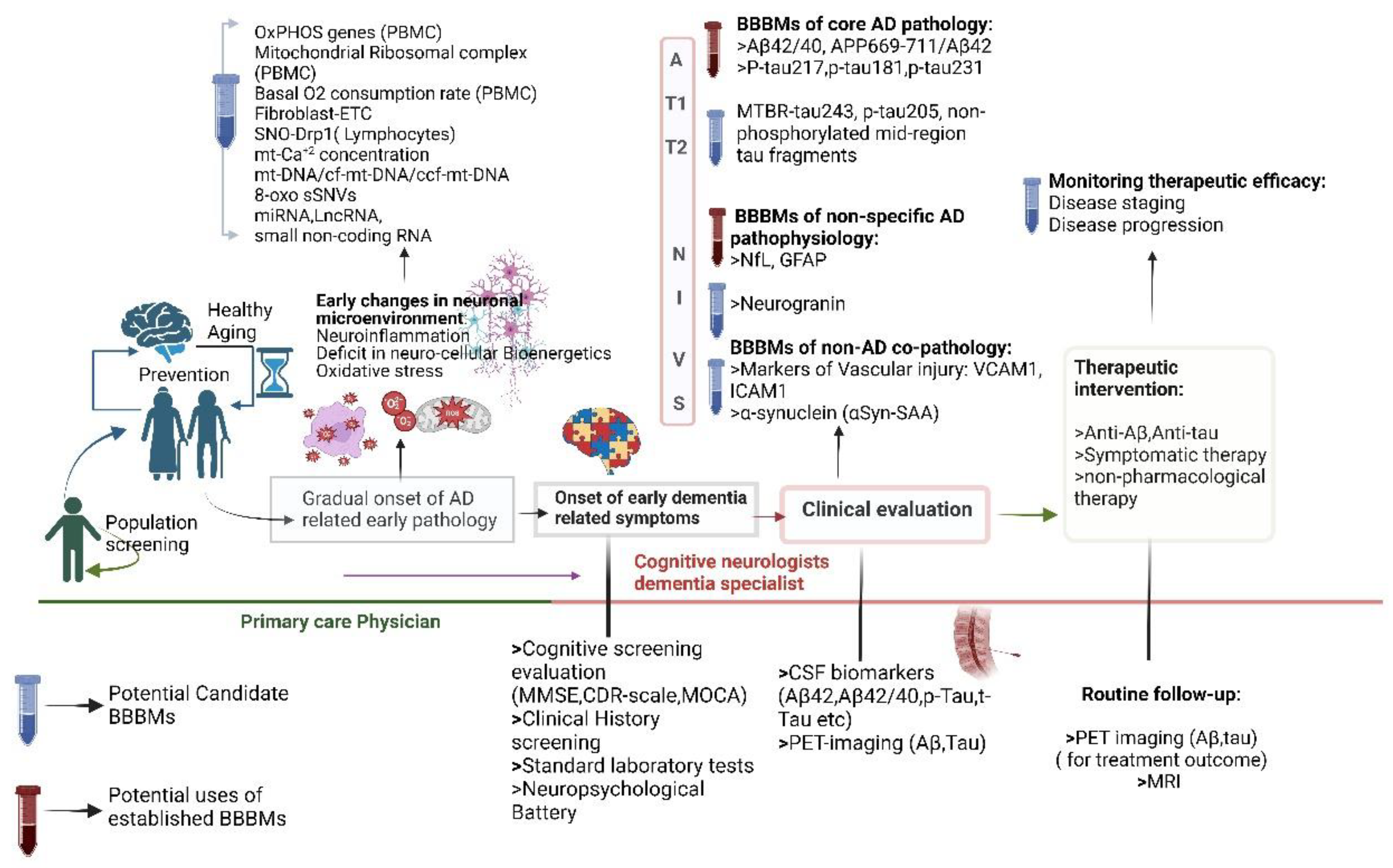
Figure 3.
An approximate relationship between biomarker abnormality and AD disease continuum (pre-clinical, prodromal, and AD dementia).
Figure 3.
An approximate relationship between biomarker abnormality and AD disease continuum (pre-clinical, prodromal, and AD dementia).
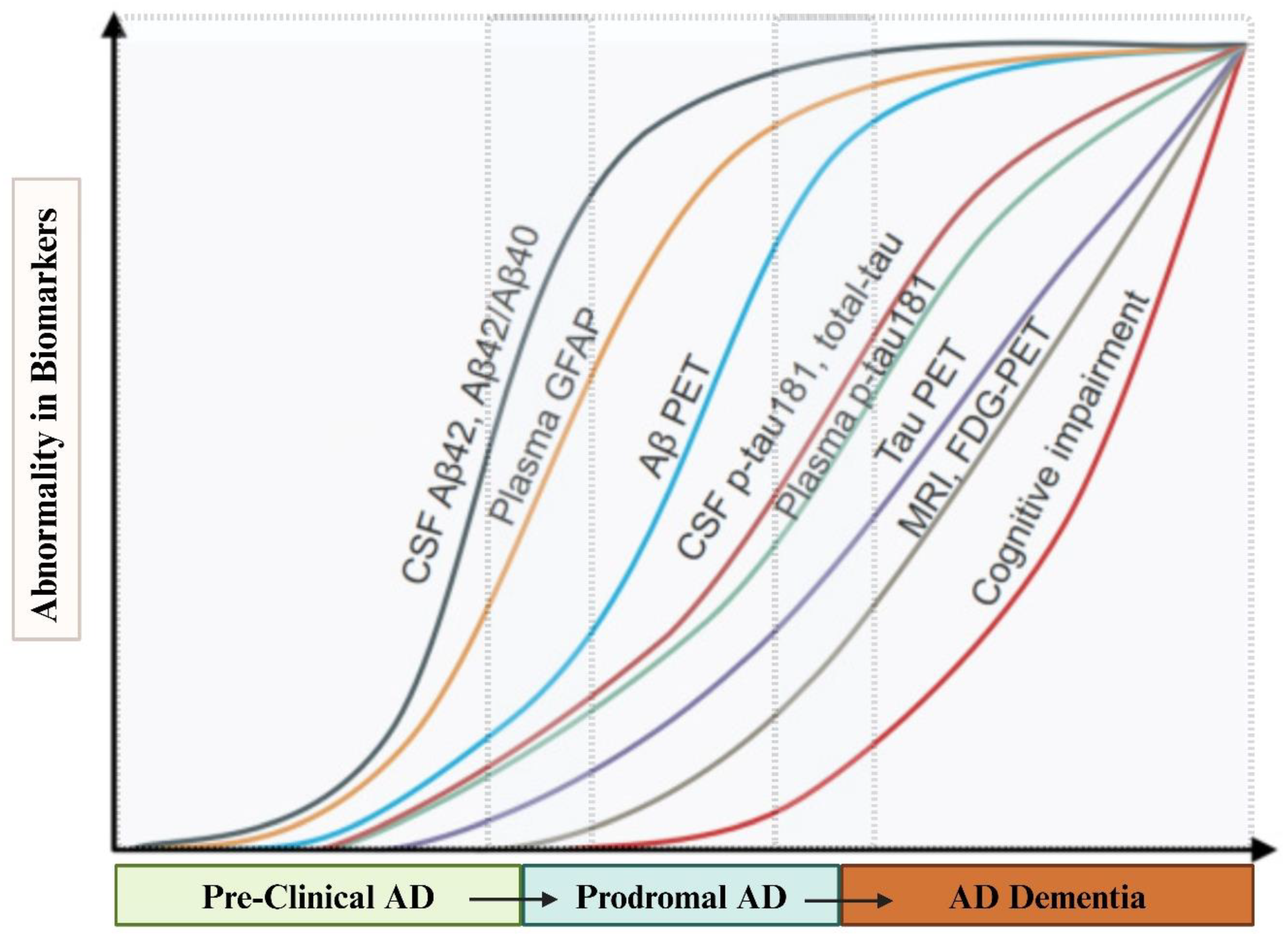
Figure 4.
STRING Protein-Protein Interaction Network. Nodes: Colored nodes: Query proteins and first shell of interactors; White nodes: Second shell of interactors; Empty nodes: Proteins of unknown 3D structure; Filled nodes: Proteins with known or predicted 3D structure. Edges: Edges represent protein-protein associations, indicating proteins that jointly contribute to a shared function. Colors indicate the types of evidence for the association: Blue: Known Interactions from curated databases. Pink: Experimentally determined interactions. Green: Predicted interactions from gene neighborhood. Red: Predicted interactions from gene fusions. Dark Blue: Predicted interactions from gene co-occurrence. Yellow: Interactions from text-mining. Black: Co-expression. Light Blue: Protein homology.
Figure 4.
STRING Protein-Protein Interaction Network. Nodes: Colored nodes: Query proteins and first shell of interactors; White nodes: Second shell of interactors; Empty nodes: Proteins of unknown 3D structure; Filled nodes: Proteins with known or predicted 3D structure. Edges: Edges represent protein-protein associations, indicating proteins that jointly contribute to a shared function. Colors indicate the types of evidence for the association: Blue: Known Interactions from curated databases. Pink: Experimentally determined interactions. Green: Predicted interactions from gene neighborhood. Red: Predicted interactions from gene fusions. Dark Blue: Predicted interactions from gene co-occurrence. Yellow: Interactions from text-mining. Black: Co-expression. Light Blue: Protein homology.
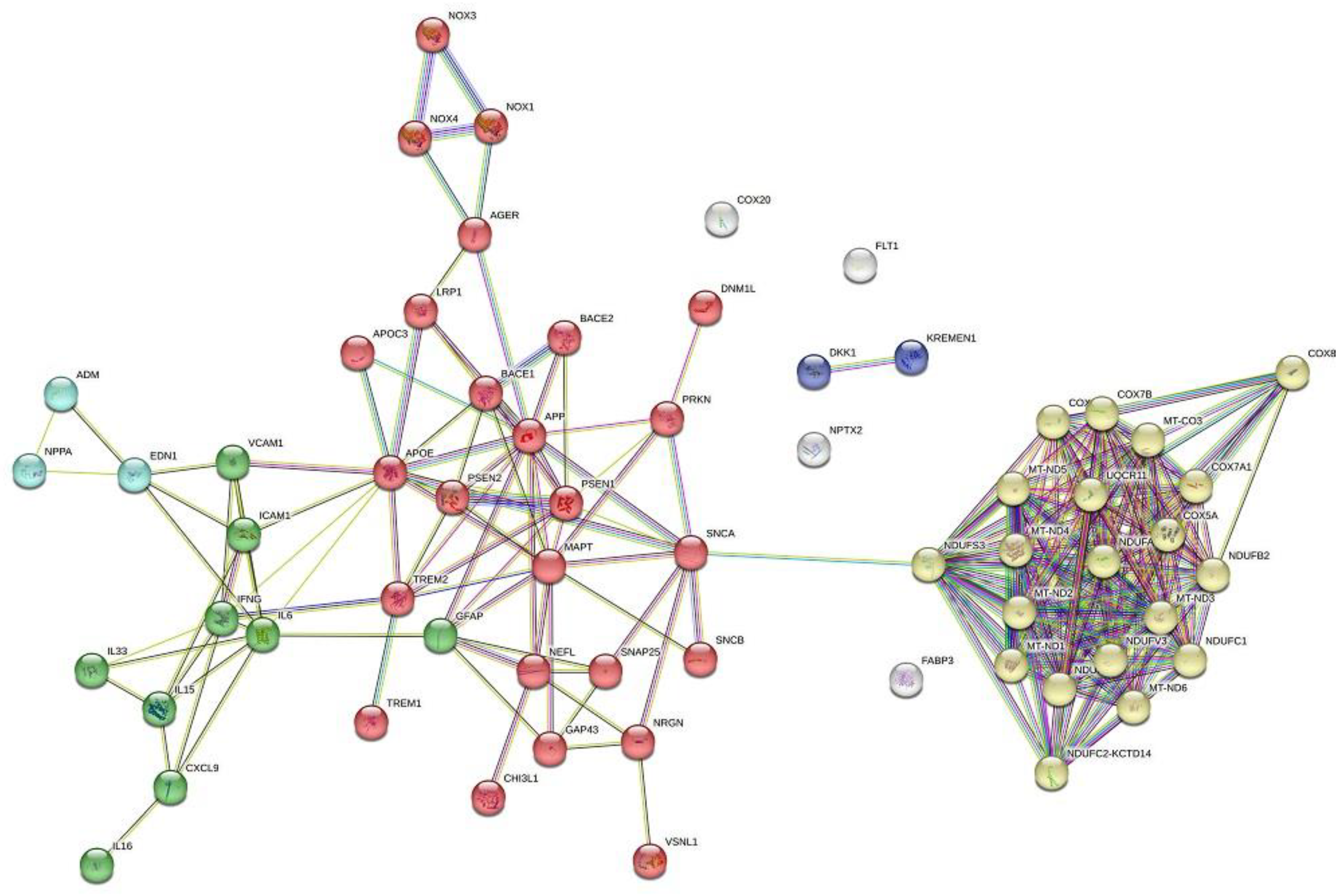
Figure 5.
The miRNA interaction network of the (A) 4 predicted miRNA markers of AD in peripheral blood, and (B) 8 predicted miRNA markers of AD in brain tissue. Nodes: Square nodes: Predicted miRNAs, the color intensity and the size of the square denotes the number of interactions with target genes (increase the size of the square as the number of interactions increases). Circular nodes: Target genes. The deep color denotes a gene without interaction with other genes, and the light color depicts the interaction between two or more genes. Edges: Edges represent miRNA-genes interactions or gene-gene interactions within the network. The miRNA interaction network is one of AD's three predicted miRNA markers.
Figure 5.
The miRNA interaction network of the (A) 4 predicted miRNA markers of AD in peripheral blood, and (B) 8 predicted miRNA markers of AD in brain tissue. Nodes: Square nodes: Predicted miRNAs, the color intensity and the size of the square denotes the number of interactions with target genes (increase the size of the square as the number of interactions increases). Circular nodes: Target genes. The deep color denotes a gene without interaction with other genes, and the light color depicts the interaction between two or more genes. Edges: Edges represent miRNA-genes interactions or gene-gene interactions within the network. The miRNA interaction network is one of AD's three predicted miRNA markers.
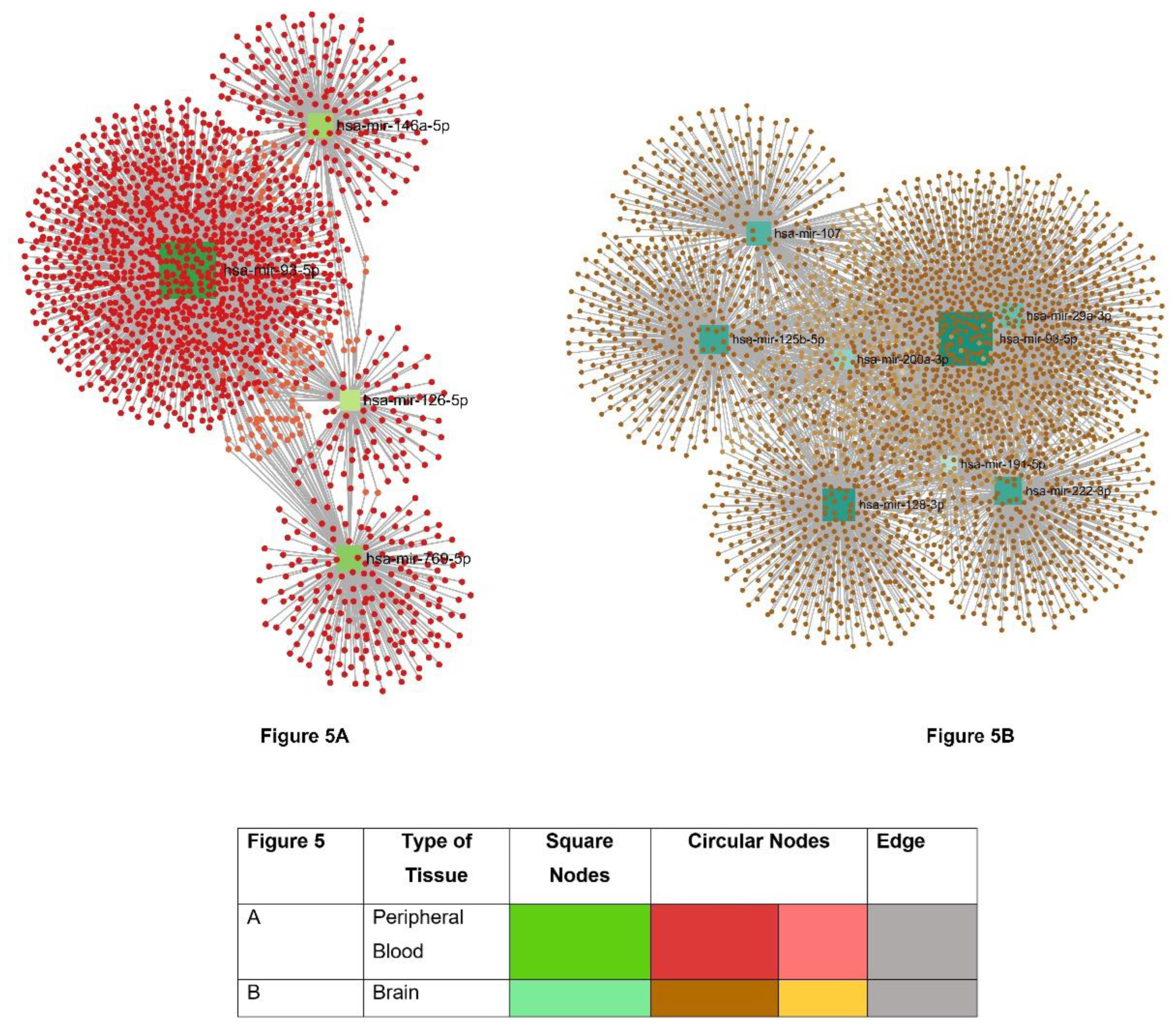

Table 1.
Overview of Blood-Based Biomarkers in Alzheimer's Disease: Assay Techniques, Pathomechanisms, and Their Role in Diagnostics and Prognostics.
Table 1.
Overview of Blood-Based Biomarkers in Alzheimer's Disease: Assay Techniques, Pathomechanisms, and Their Role in Diagnostics and Prognostics.
| Name of the BBMs | Underlying Pathophysiology |
Current Categorization of BBBMs (NIA-AA'2024) | Relevance in AD | Trend of the biomarker in Plasma | Assessment techniques | |
|---|---|---|---|---|---|---|
| Amyloid β | Aβ42 | Plasma proteinopathy- Amyloidogenic | Core 1 biomarker (A) |
Early detection of AD in asymptomatic individuals, with progression from normal cognition to MCI or AD | Decreased in AD and MCI in comparison to controls | 1: ELISA 2: Luminex xMAP Technology 3: SIMOA 4: LCMS 5: Immunoprecipitation Mass Spectrometry |
| Aβ40 | Plasma proteinopathy- Amyloidogenic | Core 1 biomarker (A) |
Early detection of AD in asymptomatic individuals, with progression from normal conition to MCI or AD | Decreased in AD and MCI in comparison to controls | ||
| Aβ42/40 | Plasma proteinopathy- Amyloidogenic | Core 1 biomarker (A) |
Identify early stages of AD and predict cognitive decline in concordance with CSF and neuroimaging biomarkers. | Decreased Aβ42/Aβ40 Ratio in AD and MCI compared to control | ||
| Tau | p-tau217 | Plasma proteinopathy-Tauopathy phosphorylated and secreted AD tau | Core 1 biomarker (T1) |
Early detection of AD in people without symptoms -Accurately predict the progression of individuals from subjective cognitive decline (SCD) and MCI to dementia when combined with other risk factors. |
Increased in AD and MCI in compared to controls | 1: ELISA 2: Luminex xMAP Technology 3: SIMOA 4: LCMS 4: Immunoprecipitation Mass Spectrometry |
| p-tau181 | Plasma proteinopathy-Tauopathy phosphorylated and secreted AD tau | Core 1 biomarker (T1) |
Early detection of AD in people without symptoms -Distinguishes between Aβ-PET (+) and Aβ-PET (-) individuals, along with the disease progression to dementia and tau-burdened brain areas with AD-related atrophic changes. |
Increased in AD and MCI compared to controls | ||
| p-tau 231 | Plasma proteinopathy-Tauopathy phosphorylated and secreted AD tau | Core 1 biomarker (T1) |
-Early detection of AD in people without symptom -Discriminates patients with and without AD pathology during post-mortem assessment. |
Increased in AD and MCI compared to controls | ||
| MTBR-tau243 | Plasma proteinopathy-Tauopathy AD tau proteinopathy |
Core 2 biomarker (T2) |
Elevated in later stages of AD Staging of biological disease severity along with Core 1 biomarker -strongly associated with tau-PET and disease progression. |
Increased in AD and MCI compared to controls | ||
| Non-phosphorylated mid-region tau fragments |
Plasma proteinopathy-Tauopathy AD tau proteinopathy |
Core 2 biomarker (T2) |
Elevated in later stages of AD -staging of biological disease severity along with Core 1 biomarker. |
Increased in AD and MCI compared to controls | ||
| α-synuclein (αSyn) | αSyn/tau | Proteinopathy-related biomarkers of non-core AD pathology Synuclein pathology | Biomarkers of non-AD co-pathology (S) |
Total α-synuclein levels in the blood may not differ significantly between patients with neurodegenerative diseases. The oligomeric or phosphorylated form of α-synuclein accelerates cognitive dysfunction. |
Decreased in AD and MCI compared to controls | 1: Seed Amplification Assays: -Protein Misfolding Cyclic Amplification (PMCA) -Real-Time Quaking-Induced Conversion (RT-QuIC) 2: ELISA and Western blotting 3: Quantitative Mass Spectrometry 4: Luminex xMAP Technology 5: SPR- DLS 6: Immuno-PCR |
| αSyn /Aβ 42 | Biomarkers of non-AD co-pathology | Increased in AD and MCI compared to controls | ||||
| DKK-1 or Dickkopf-1 | Proteinopathy-related biomarkers of non-core AD pathology. | Research Biomarker | Elevated levels correlate with disease severity, particularly cognitive decline and synaptic loss and help differentiate AD from other neurodegenerative conditions |
Increased in AD | 1: ELISA and Western blotting 2: Luminex xMAP Technology 3: Immuno-PCR 4: Mass Spectrometry |
|
| VILIP-1 | Proteinopathy-related biomarkers of non-core AD pathology. | Research Biomarker | Increased levels are seen in AD, but there is no significant difference in concentrations with AD-MCI patients and other neurodegenerative groups. |
Increased in AD | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3. Immuno-PCR 4. Mass Spectrometry |
|
| Plasma Neurofilaments (NfL) | Injury, dysfunction, or degeneration of neuropil | Biomarkers of non-specific processes involved in AD pathophysiology (N) |
Increased levels in Aβ-positive patients with AD and MCI are associated with the degree of cognitive impairment as well as used as monitoring biomarkers to indicate the severity of neurodegeneration. | Increased in AD and MCI vs controls | 1. ELISA (Enzyme-Linked Immunosorbent Assay) 2. Luminex xMAP Technology 3. ECLIA 4. Mass Spectrometry 5. SIMOA |
|
| SNAP-25 | Neuronal and synaptic injury-pre synaptic dysfunction | Biomarkers of non-specific processes involved in AD pathophysiology (N) |
CSF concentrations can distinguish between various neurodegenerative diseases like AD, Parkinson’s dieases, and ALS. | Decreased in AD compared to controls | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| Neuronal pentraxin 2 (NPTX-2) | Neuronal and synaptic injury-pre synaptic dysfunction | Biomarkers of non-specific processes involved in AD pathophysiology (N) |
Potential as a probable biomarker for early detection of AD. | Decreased in AD compared to control | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| Growth-associated protein (GAP-43) | Neuronal and synaptic injury-pre synaptic dysfunction | Biomarkers of non-specific processes involved in AD pathophysiology (N) |
Potential as a probable biomarker for early detection of AD. | Increased in AD compared to control | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| Neurogranin (NG) | Neuronal and synaptic injury- post-synaptic protein dysfunction | Biomarkers of non-specific processes involved in AD pathophysiology (N) |
Potential as a probable biomarker for early detection of AD. | Decreased in AD compared to control | 1. ELISA 2. Luminex xMAP Technology 3. ECLIA 4. Mass Spectrometry 5. SIMOA |
|
| Fms-like tyrosine kinase-1 (Flt-1) | Vascular Damages related to AD | Research Biomarker (V) |
Assess total vascular involvement and early detection of vascular changes associated with AD. |
Increased in AD compared to control | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| Endothelin 1 (ET-1) | Vascular Damages related to AD | Research Biomarker (V) |
Indicates vascular impairment in AD. | Increased in AD compared to AD | 1. ELISA 2. Luminex xMAP Technology 3. Immuno-PCR 4. Mass Spectrometry |
|
| Atrial natriuretic peptide (ANP) | Vascular Damage related to AD | Research Biomarker (V) |
Causes reduced cerebral blood flow and impairment of neurovascular health. . |
Increased in AD compared to control | 1. ELISA 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| MIG/CXCL-9 | Vascular Damage related to AD | Research Biomarker (V) |
Indicate ongoing chronic neuroinflammatory processes. | Increased in AD compared to control | 1. ELISA 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| heart-type fatty acid binding protein (H-FABP) | Vascular Damage related to AD | Research Biomarker (V) |
Potential as a probable biomarker for early detection of AD as it was found to be elevated in the pre-clinical phase of AD dementia. | Increased in AD compared to control | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| Vascular adhesion molecular | sVCAM-1 | Vascular Damage related to AD | Research Biomarker (V) |
Indicate the burden of atherosclerosis in AD with elevated sVCAM, indicating a significant correlation between age and the severity of cognitive decline. |
Increased in AD compared to control | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
| Soluble intercellular adhesion molecule-1 | Vascular Damage related to AD | Research Biomarker (V) |
Indicate the burden of atherosclerosis in AD with elevated Soluble intercellu-lar adhesion molecule-1. | Increased in AD compared to control | 1. ELISA and Western blotting 2. Luminex xMAP Technology 3.Immuno-PCR 4. Mass Spectrometry |
|
| Metabolic products secondary to lipid peroxidation (LPO) | malondialdehyde (MDA) | Oxidative Stress | Research Biomarker | Increased levels in familial AD that carry APP and presenilin-1 gene mutations. | Increased in AD compared to control | 1. HPLC 2. LC-MS 3. ELISA 4. GC-MS |
| 4-hydroxynonenal (4-HNE) | Oxidative Stress | Research Biomarker | Increased levels in familial AD that carry APP and presenilin-1 gene mutations. | Increased in AD compared to control | ||
| increased F2-isoprostanes (F2-IsoPs) | Oxidative Stress | Research Biomarker | potential marker of oxidative stress during the MCI phase of AD, and its quantity correlates with the disease continuum from SCD to MCI to AD. | Increased in AD compared to control | ||
| Free radicals | ROS | Oxidative Damage | Research Biomarker | ROS modifies neuronal macromolecules and induces τ protein hyperphosphorylation during prodromal AD phases. | Increased in AD | 1. DCFDA 2. Electron Spin Resonance (ESR) Spectroscopy 3.Nitroblue Tetrazolium (NBT) Assay 4. Flow Cytometry with ROS-sensitive dyes |
| RNS | Oxidative damage | Research Biomarker | Nitrosylation of critical proteins in neurons impairs their function, promoting neurodegenerative processes. | Increased in AD | 1. Nitrotyrosine ELISA Electron Spin Resonance (ESR) Spectroscopy Western Blot for 3-Nitrotyrosine-modified Proteins |
|
| Nucleoside 8-hydroxyguanosine (8-OHG) | Oxidative Damage | Research Biomarker | It is notable for determining the gradient of DNA oxidative damage in AD patients. It allows us to determine oxidative damage to plasma DNA early in AD. | Increased in lymphocytes of AD patients compared to control | 1. ELISA 2. HPLC and Electrochemical detection 3. LC-MS 4. Western Blot using specific anti-5.8-OHG antibodies 6. Immunoprecipitation 7. GC-MS |
|
| Mitochondrial respiratory complex I-V genes (OxPHOS genes) | Bioenergetic abnormality | Research Biomarker | An imbalance in nuclear and mitochondrial genome-encoded OXPHOS transcripts may cause a negative feedback loop that lowers mitochondrial translation and compromises OXPHOS efficiency. This would likely result in the generation of harmful reactive oxygen species. |
Reduced expression in early AD patients | 1. qPCR Western Blot IHC Blue Native Gel Electrophoresis |
|
| SNO-Drp1 | Bioenergetic abnormality | Research Biomarker | SNO-Drp1 can result in increased mitochondrial fission, loss of synapses, and neuronal damage in mice models and primary neuronal culture, as well as in post-mortem tissue. | Increased in peripheral blood lymphocytes in AD patients. There are also contradictory findings that SNO-Drp1 does not differ significantly in AD compared to controls. | 1. Biotin Switch Assay Mass Spectrometry Nitroso-Proteome Profiling Immunoprecipitation and Western blot |
|
| mitochondrial DNA (mt-DNA) | Bioenergetic abnormality | Research Biomarker | mtDNA copy number acts as an indirect indicator for several functioning mitochondria and thus provides info regarding bioenergetics as a factor for AD progression. | Decreased in patients with AD | 1. qPCR Digital droplet PCR Southern Blotting |
|
| 8oxoG sSNVs | Bioenergetic abnormality | Research Biomarker | due to their inflammatory endophenotype, the circulating cf-mtDNA (ccf-mtDNA) 8oxoG variant can be used as an improved biomarker. | Increased in AD patients | 1. 8-oxoG DNA Glycosylase (OGG1) Assay Comet Assay with Fpg (Formamidopyrimidine-DNA Glycosylase) ELISA HPLC with electrochemical detection |
|
| circulating cf-mtDNA (ccf-mtDNA) | Bioenergetic abnormality | Research Biomarker | cellular mt-DNA copy number can be used as a potential biomarker of mitochondrial biogenesis and cellular energetics to reflect upon mitochondrial health in AD. | Increased in AD patients | qPCR Digital droplet PCR Southern Blotting |
|
| The intermediate filament glial fibrillary acidic protein (GFAP) | Neuroinflammation and Immune Dysregulation | Research Biomarker (I) |
The marker of astrogliosis can be seen in chronic inflammatory processes like progressing AD. | Increases in AD patients | 1. ELISA ECLIA mesoscale discovery immunoassay V-PLEX |
|
| CX3CL1 (Fractalkine) | Neuroinflammation and Immune Dysregulation | Research Biomarker (I) |
significantly elevated in the plasma of MCI and AD compared to other neuroinflammatory disease processes. | Increases in AD and MCI | 1. ELISA Western blot IHC Flow Cytometry Luminex |
|
| CCL23 | Neuroinflammation and Immune Dysregulation | Research Biomarker (I) |
Their plasma concentration has also been found to have a predictive value toward MCI-to-AD progression. | Increases in AD | 1. ELISA Western blot IHC Flow Cytometry Luminex |
|
| C-C chemokine ligands or RANTES | Neuroinflammation and Immune Dysregulation | Research Biomarker (I) |
Elevated in AD and correlate with the neuroinflammatory burden. | Increases in AD | ELISA Western blot IHC Flow Cytometry 5. Luminex |
|
| YKL-40 | Neuroinflammation and Immune Dysregulation | Research Biomarker (I) |
increasingly expressed in astrocytes during neuroinflammatory changes. Plasma YKL-40 level shown to have a positive correlation with the result of sensitive free and cued selective reminding test | Increases in AD | 1. ELISA 2. Western blot 3. IHC 4. Flow Cytometry 5. Luminex |
|
| Progranulin | Neuroinflammation and Immune Dysregulation | Research Biomarker (I) |
Studies have revealed that the increased progranulin-expressing gene GRN is in the blood of MCI and AD patients. | Increases in AD | 1. ELISA 2. Western blot 3. IHC 4. Flow Cytometry 5. Luminex |
|
| Triggering receptor expressed on myeloid cells 2 (TREM2) | Neuroinflammation and Immune Dysregulation | Research Biomarker (I) |
mRNA levels in peripheral mononuclear cells have been found to have the distinguishing ability between aMCI, AD, and healthy control individuals and to be dependent on the Apolipoprotein E genotype. | Increases in AD | 1. ELISA 2. Western blot 3. IHC 4. Flow Cytometry 5. Luminex |
|
| NDE | P-S396-tau | Tauopathy | Research Biomarker | Can predict the development of AD up to 10 years before the clinical onset of sporadic AD. | Increased in AD | Proteomic Analysis of the EV’s like ELISA |
| p-tau 181 | Tauopathy | Research Biomarker | Can predict the development of AD up to 10 years before the clinical onset of sporadic AD | Increased in AD and MCI vs healthy controls | 1 ELISA 2. Ultra-sensitive inhouse SIMOA |
|
| Synaptotagmin | Synaptopathy | Research Biomarker | Its impairment leads to decreased neurotransmission, neuroplasticity, and long-term potentiation, thus hampering memory formation. | Reduced in AD | 1. ELISA LC-MS SIMOA |
|
| synaptophysin | Synaptic loss and dysfunction | Research Biomarker | Loss of proper functioning synapse leads to impaired signal transmission and, thus, cognitive impairment. | Reduced in AD | 1. ELISA LC-MS SIMOA |
|
| P-S312-IRS-1 | Neuroinflammation and Insulin Resistance | Research Biomarker | Its increment promotes insulin resistance, leading to progressive neurodegeneration. | Increased in AD vs. controls | 1. ELISA LC-MS SIMOA |
|
| P-panY-IRS-1 | Insulin resistance and Synaptic dysfunction | Research Biomarker | Its reduction promotes insulin resistance, leading to progressive neurodegeneration. | Downregulated in AD | 1. ELISA LC-MS SIMOA |
|
| N-(1-carboxymethyl)-L-lysine | ROS mediated damage | Research Biomarker | can differentiate early to moderate AD. | Downregulated in AD | 1. ELISA LC-MS SIMOA |
|
| MDE | Tauopathy | Research Biomarker | When neurons absorb microglia-derived exosomes containing tau, it triggers further abnormal tau aggregation | Increases in AD | ELISA LC-MS |
|
| ADE | Neuroinflammation | Research Biomarker | Plasma levels of various complement components, such as C1q, C3b, and factor D, could serve as predictive biomarkers for the progression of MCI to AD | Increases in AD | 1. ELISA LC-MS |
|
Table 2.
List of miRNAs based on extensive literature search found in whole blood, plasma, and serum as different sources.
Table 2.
List of miRNAs based on extensive literature search found in whole blood, plasma, and serum as different sources.
| Source of the miRNA | Names of miRNA | Reference |
|---|---|---|
| Whole Blood | hsa-miR-107 | [160,161,162,164] |
| Plasma | hsa-miR-92a-3p, hsa-miR-486-5p, hsa-miR-29a-3p, hsa-miR-107, hsa-miR-128-3p, hsa-miR-132-3p, hsa-miR-34c-5p, hsa-let-7d-5p, hsa-miR-191-5p, hsa-miR-200a-3p, hsa-miR-483-5p, hsa-miR-486-5p, hsa-miR-502-3p, hsa-miR-548k, hsa-miR-339-5p, hsa-miR-221-5p, hsa-miR-144-5p, hsa-miR-382-5p, hsa-miR-146b-5p, hsa-miR-224-5p, hsa-miR-625-5p, hsa-miR-769-5p, hsa-miR-454-5p, hsa-miR-548d-5p, hsa-miR-877-5p, hsa-miR-146a, hsa-miR-125b, | |
| Serum | hsa-miR-106-b-3p, hsa-miR-22-3p, hsa-miR-126-5p, hsa-miR148b-5p, hsa-miR-181c-3p, hsa-miR-93-5p, hsa-miR-29c-3p, hsa-miR-132-3p, hsa-miR-222-3p, hsa-let-7d-5p, hsa-miR-191-5p, hsa-miR-146a, hsa-miR-125b, hsa-miR-135a | |
| PBMC | hsa-miR-128-3p, hsa-miR-34c-5p, |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



