
Submitted:
23 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
Despite improvements in participation in population-based screening programs, colorectal cancer remains a major cause of cancer-related mortality worldwide. Targeted interventions are desirable to reduce the health and economic burden of this disease. Two-dimensional monolayers of colorectal cancer cell lines represent the traditional in vitro models for disease and are often used for diverse purposes, including delineation of molecular pathways associated with disease aetiology or to gauge drug efficacy. The lack of complexity in such models, chiefly the limited epithelial cell diversity and differentiation, attenuated mucus production, lack of microbial interactions and mechanical stresses, has driven interest in development of more holistic and physiologically relevant in vitro model systems. In particular, established ex vivo patient-derived explant and patient-derived tumour xenograft models have been supplemented by progress in organoid and microfluidic organ-on-a-chip cultures. Here we discuss the applicability of advanced culturing technologies, such as organoid systems, as models for colorectal cancer and for testing chemotherapeutic drug sensitivity and efficacy. We highlight current challenges associated with organoid technologies and discuss their future for more accurate disease modelling and personalized medicine.
Keywords:
intestinal organoid
; 3D culture
; colorectal cancer
; organ-on-a-chip
; disease modelling
; therapeutic screening
; ex vivo models
1. Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide [1]. Despite the establishment of screening programmes in several countries and improved management of CRC, approximately 2 million new diagnoses and 1 million deaths occurred in 2020, and this is projected to increase to 3.2 million new cases in 2040 [1]. The increasing prevalence of CRC is mirrored by the rise in associated healthcare costs where studies have pointed to rising costs and healthcare burden related to management of the disease in countries with high incidences such as the UK, US and NZ [2,3,4,5]. Improved efficiencies in the clinical management of CRC, particularly in the prevention, detection, and treatment, hold the potential to reduce the growing burden of this disease.
Treatment options for advanced CRC in particular have grown, with newer targeted chemotherapies and immunotherapies increasing survival for patients with metastatic disease [6,7]. Examples include monoclonal antibodies such as cetuximab (Erbitux) and panitumumab (Vectibix) that target epidermal growth factor receptor (EGFR), immune checkpoint inhibitors nivolumab (Opdivo) and pembrolizumab (Keytruda) [8,9], and agents targeting vascular endothelial growth factor (VEGFR) signalling to disrupt tumour vascularisation such as bevacizumab (Avastin) and ramucirumab (Cyramza) [10]. Despite the proliferation of novel therapeutics, the clinical uptake of these specific therapies is limited by the dearth of biomarkers to direct their use in CRC. Accepted markers are few: KRAS/BRAF mutation contra-indicating the use of anti-EGFR immunotherapy and MSI-H status indicating a higher probability of response to checkpoint inhibitor therapy. The majority of patients with CRC therefore benefit little from the emergence of new immunotherapies.
Identifying clinically beneficial therapeutics that can be directed against CRC remains challenging, with a high number of drug candidates never reaching the market. Study of the drug approval process suggests that even of those drugs reaching Phase 2 or 3 clinical trials, up to 79% fail to meet the necessary safety and efficacy profile for approval [11,12]. The likelihood of obtaining US FDA drug approval is especially low for anti-cancer therapeutics, with an overall success rate as low as 3% [13]. Contemporary strategies offered to address this lack of success relate to “fast-fail” approaches to improve selection of promising candidates, including novel computational (AI/ML) approaches to drug design, identifying better surrogate markers to measure physiological endpoints, and improving the predictivity of model systems by applying ex vivo models with direct relevance to humans.
Since a landmark paper in 2009 which demonstrated the ability of intestinal stem cells to self-organise into viable intestinal organoids in vitro [14], organoid models have gained traction as viable preclinical models for drug screening, with the potential to augment or replace traditional 2D monolayer cultures in this setting. Organoids are 3D stem cell-derived multicellular systems that can represent a specific organ, including its cellular diversity, morphology and tissue architecture. To date, organoids have been derived to represent many organ systems including the intestine, liver, kidney, and brain. Organoids can be derived from pluripotent stem cells or directly from tissue biopsy material. When obtained from patients directly, these patient-derived organoid models have been shown to retain the genetic characteristics of the original tissue and are amenable to long term culture and cryopreservation [15], representing significant technological advances for development of personalised or precision therapies.
Incorporating phenotypic screening using ex vivo patient-derived model systems, such as patient-derived organoid (PDO), xenograft (PDX), or explant (PDE) models, into the initial stages of discovery is one approach that can potentially improve the success rate and efficiency of therapeutic drug discovery. Choice of model system requires some consideration and varies in their fidelity to intestinal pathophysiology, with each model system naturally presenting their own unique set of limitations, e.g. heterogeneity, complexity, scalability, amenability to automation and throughput, and is summarised in Table 1. Organoid models would fit best into three “domains” in the discovery and pre-clinical phases: early-phase target/drug discovery and validation, mechanistic studies/pre-clinical refinement, and pre-clinical efficacy/toxicity (Figure 1) and also presents opportunity for capturing patient diversity at each phase [16]. While there are many publications describing in vitro models for CRC drug screening [17,18,19], this review discusses the current status and challenges associated with culturing and screening patient-derived 3D organoids as a preclinical tool for CRC disease modelling, drug evaluation and prediction of clinical outcome.
2. Ex Vivo Epithelial Organoids
Colonic epithelial organoids are recognised as a promising model system for studying colonic carcinogenesis and offer a number of advantages to facilitate the identification of underlying molecular pathways and novel predictive markers of therapeutic response. When used as an adjunct to existing pre-clinical models of tumourigenesis, together these can form critical points for decision-making in the research and development of new diagnostics and pharmacological therapies. Organoids can be generated from whole isolated crypts or from isolated crypt-base stem cells from the primary tissue [15]. Epithelial tissue is obtained relatively easily from mucosal biopsy during colonoscopy, from both normal and tumour tissue and organoids derived from these tissues can be perpetually expandable while retaining characteristics of the donor tissue [26]. This feature is of critical importance --- while we can induce mutations in epithelial stem cells to replicate carcinogenesis, the transformation does not guarantee carcinogenesis. This is obvious when comparing precursor lesions which may eventually give rise to cancer from disparate molecular and functional pathways. The ability to scale organoid cultures to become a source of various tumour precursor tissue through to advanced carcinoma for in vitro large-scale high-throughput screening and follow-up experimentation[15,27]. Preservation of the genomic and transcriptomic heterogeneity present in different subtypes of colonic tumours make these organoid models highly appealing [28]. For example, it is difficult to replicate models representing the different molecular subtypes of CRC with high fidelity, such as models that have high levels of microsatellite instability are thought to arise outside the conventional adenoma pathway and are associated with BRAF V600E mutations, or those with defective DNA mismatch repair mechanisms, which are associated with resistance to 5-fluorouracil chemotherapy [29]. Besides differences in tumour subtypes, considerable genetic variability exists within a single tumour mass, attributable to the progressive local expansion of divergent subclones which are themselves the product of an underlying global genetic instability. Truly representative organoid models which incorporate the heterogeneity present within tumours overcome known potential confounders for biomarker and drug discovery efforts which utilize 2D in vitro cultures derived from clonally restricted cell lines.
Assembly of a “living biobank” of matching normal, precancerous and cancerous, patient-derived epithelial organoids potentially provides an immensely valuable source of tissue to support organoids for drug discovery, in particular for personalised treatments and when utilising precancerous adenomas, to test drugs at an early stage of disease to prevent progression [25,30]. These patient-derived organoids recapitulate the heterogeneity and genetic mutations of the individuals, creating a biobank representative of the clinical heterogeneity. For example, Luo et al (2023)[31] recently reported on the establishment of patient derived adenoma organoid biobank consisting of 37 organoid lines from 33 patients which was then used to screen a 139 compound library for drugs that could inhibit adenoma growth. Similarly, there are a number of reports detailing successful establishment colorectal tumour organoid biobanks for modelling disease subtypes and for predicting patient response to therapy [25,28,32,33,34,35,36,37,38]. Table 2 lists registered clinical trials involving patient-derived organoids to guide therapy.
Establishment of patient-derived colorectal organoids can be complicated by technical issues such as microbial contamination and unwanted mixed normal or stromal cell populations if tumour margins are not well defined. The intestinal tract contains bacteria, viruses and yeasts that may be present within mucosal tissues or the adherent mucus at the time of surgery. For example, unwanted overgrowth of microbial contamination was the cause for failure of the establishment of 21% of primary oesophageal organoid cultures in one study [39]. Bacterial and fungi may be suppressed through treatment of the starting material and initial cultures with a cocktail of antibiotics (e.g. penicillin, streptomycin, gentamycin and antifungals such as amphotericin B or nystatin) [40]. The use of metronidazole has been reported for controlling anaerobic bacteria associated with primary colonic tissues [41]. However, prolonged use of such antibiotics should be avoided since antibiotics may both inhibit cell growth and conceal low-level infection from organisms such as mycoplasma. Instead, surveillance for infection through microscopic observation, biochemical or molecular testing (e.g. qPCR, 16S sequencing), and the use of aseptic culture technique and sterile reagents/consumables are recommended [40]. Maintaining sterile technique and adequate biological containment is also important given the possibility of pathogenic viral infection.
Establishing organoid models from primary tissues presents the risk of culturing undesired cell populations from admixed tissues. The difficulty of obtaining organoids from epithelial tumours that are composed purely of neoplastic cells in long-term culture is a problem now highlighted by several studies of prostate and lung cancer-derived organoids [42,43,44]. Where clearly abnormal morphologies exist, morphology alone may serve as a convenient marker for manual separation of tumour and normal organoids in conjunction with mutational analysis and copy number profiling [42,45]. Selection media deficient in selected components such as stem cell niche growth factors normally supplied by the stroma, may provide the means to eliminate contaminating normal epithelial cells from admixed cultures. Since the majority of colorectal tumours carry mutations in the Wnt signalling pathway, removal of Wnt and R-spondin from culture medium may reliably produce pure cultures of tumour organoids [46]. Use of selection media however may result in a diminished number of successfully established organoid lines, perhaps concordant with the success rate for patient-derived cell line generation [42,47]. Given the genetic heterogeneity within the neoplasms it is unclear how many subclones may be retained through selection more precise selection through addition of chemical inhibitors targeting wild-type signalling pathways such as TP53, may better preserve an organoid culture success rate suitable for large-scale screening [42].
3. Developing Model Complexity
3.1. Replicating the Tumour Microenvironment
To date, the majority of patient derived tumour organoid models described in literature are composed of colonic epithelial cells and do not replicate the tumour microenvironment. This includes representation of the surrounding stromal compartment such as the vasculature required to sustain the growth of tumours in vivo or the immune microenvironment that influences patient response to therapy and drug resistance. Due to their ability to directly affect tumour cell death, tumour-infiltrating cytotoxic T lymphocytes are of particular interest in anti-tumour responses. Frequency and activity of cytotoxic T lymphocytes isolated from the blood of patients with CRC are not ideal prognostic markers [48]. Accurate prediction of lymphocyte activity in the in vivo tumour micro-environment may be frustrated by the methods used to isolate, culture and stimulate them in vitro. Re-incorporation of infiltrating or resident immune effector cells into in vitro tumour models can assist in identifying markers of effective anti-tumour immune responses [49], studying the on-target off-tumour toxicities of immunotherapy treatments, capturing clinical toxicities not predicted by conventional tissue-based models as well as inter-patient variabilities in drug and immunotherapy responses [50]. Collection of peripheral blood mononuclear cells (PBMC) from donors is an ideal source of patient-matched T lymphocytes. In one study, two-week co-culture of autologous T lymphocytes with IFN-γ-stimulated tumour organoids enabled the identification of patients whose organoids stimulated cytotoxic T lymphocytes, i.e. separation into responders and non-responders based on MHC-I status and cytolytic efficiency [49].
T lymphocytes with modified chimeric antigen receptors (CAR) have been used in haematological malignancies, however, there has been varying success in their use for other cancers. Co-culture of matched patient-derived normal and tumor organoids with CAR-engineered NK-92 cells has been reported as a sensitive in vitro platform to evaluate CAR efficacy and tumor specificity [51]. The authors noted that killing efficiency varied between organoids of different sizes with slower kinetics observed for larger organoids, which might reflect the CAR response against solid tumor masses in vivo more closely.
Lymphocyte co-culture with healthy, non-cancerous colonic organoids has also been used to identify potential cross-reactivity in responder T cells. One consideration raised by the tumour-lymphocyte co-culture is the possibility of animal proteins in the supporting matrix, in this case Geltrex™, an EHS sarcoma-derived hydrogel, generating cross-reactive T lymphocytes against host epithelial cells [49].
An alternative method for generating a more realistic in vitro colorectal tumour model employed a common organoid culture medium in an air-liquid interface (ALI) culture system [52]. The described ALI system preserved the stromal fibroblast and a diverse array of functional immune cells from myeloid and lymphoid lineages. Demonstrating the utility of a complex tumour model, TIL responses to PD-1/PD-1L immunotherapy could be quantified and co-cultured, though a progressive decline with repeated passage was apparent in colonic cultures.
Drug responses to anti-cancer chemotherapy are also influenced by the permeability and biochemistry of endothelium. Methods available to replicate angiogenesis in organoid models are reviewed in Grebenyuk & Ranga (2019)[53]. At present, many patient-derived organoid models are cultured in multi-well plates under static conditions with establishment of tumour-vascular interactions involving co-culture of the organoid above a monolayer of endothelial cells. One challenge associated with vascularising patient-derived organoids is their differing growth requirements and supporting ECM requirements in vitro, as recently highlighted in Rajasekar et al. (2020) [54]. The authors determined that self-assembly of endothelial cells into microvascular structures was impeded by collagen/laminin-rich gels, such as the Matrigel favoured for colonic organoids. To compensate, a perfusion model was engineered to include a hydrogel matrix consisting of fibrin, Matrigel and culture media compatible with both microvasculature and colonic organoids growth.
3.2. Host-Microbial Interactions
A diverse community of trillions of microorganisms resides in the mucosal surfaces of the gastrointestinal tract (GIT) from the mouth to the rectum and play a key role in mucosal health and homeostasis. The number of microbes varies along the different segments of GIT with the greatest number (1010 – 1014 CFU/ml) of these microbes residing in the colonic mucosa [55,56]. The gut microbiota in particular plays a major role in maintaining intestinal physiological and immune homeostasis, however, microbiota dysbiosis and enrichment of certain pathobionts have been associated with the development of intestinal and extra-intestinal disorders, including CRC [55].
The majority of evidence on the role of the microbiota on CRC is derived from association studies and the causal link is still largely unexplained. As such, intestinal organoids provide a valuable tool to address the association-causation gap between microbes and CRC through mechanistic studies to investigate how these microorganisms could contribute to the disease. For example, multiple studies have exposed organoids to live bacteria, bacterial lysates or bacterial-derived toxins or metabolites to determine their role in CRC development. Table 3 summarises studies that have utilised intestinal organoids and bacterial co-culture including live bacteria, bacterial-derived toxins, conditioned media, extracellular vesicles, and metabolites.
Overall, the current evidence indicates that organoids offer a promising tool for understanding the role of the microbiota in both cancer development and treatment outcomes. The most effective way to understand the full spectrum of the role of microbes is by coculturing a community of live microbes with organoids. However, the key limitation of the current organoid systems is that it does not offer the growth conditions present in the human intestine including the oxygen gradient that supports the growth of both aerobic and anaerobic bacteria, or mechanical forces such as luminal flow or peristalsis. Currently, there are multiple attempts to overcome these limitations. Using microinjection offers a better option to coculture anaerobic microorganisms as the organoid lumen has lower oxygen concentration. The key challenge is that the microinjection process is time-consuming and labour-intensive and generally requires specialised equipment, which is not practical where higher throughput is desired. To automate the microinjection process, Williamson et al. established a high-throughput semi-automated microinjection system which was able to microinject around 90 organoids/h. While this offers a promising step for automating microinjection, further optimisation is needed to create a fully automated and affordable microinjection system [75]. Another way to overcome the inaccessibility of the lumen in 3D organoids is by dissociating and growing them as monolayers which offer easier access to apical and basal surfaces, but this still requires a way to deplete oxygen on the luminal/apical side of the epithelium. Currently, there are multiple attempts to create a culture system that interfaces normoxic and hypoxic environments using culture devices of varying degrees of sophistication. One recently described method uses a colonic epithelial organoid-derived monolayer cultured on a semi-permeable insert and the anaerobic bacteria are introduced to the apical compartment. Non-porous rubber is used to seal the apical chamber generating a hypoxic environment while permitting oxygen perfusion into the basal chamber to maintain epithelial respiration. In this device, epithelium showed intact polarity, mucus layer and stem cell hierarchy [76]. To introduce the luminal flow, Sunuwar et al. cultured human enteroid-derived monolayer in fluidic-based intestine-chips and exposed it to heat-stable enterotoxin A derived from enterotoxigenic E. coli under three conditions i) static fluid, ii) apical and basolateral flow and iii) flow and repetitive stretch. As compared to the static fluid, introducing fluid flow increases epithelial height and the section of cyclic GMP at baseline and in response to enterotoxin [77]. Developing engineering-based approaches to improving the growth conditions within the organoid systems will further allow for a better understanding of how these microbes collectively contribute to CRC carcinogenesis.
3.3. Intestine-on-a-Chip
Advances in tissue engineering, microfluidics and microfabrication technology has permitted the culture of human cells in an environment mimicking important aspects of anatomy and complex, dynamic physiological functions. Still in its infancy, intestinal organ-on-a-chip models are being designed to mimic the native environment by compartmentalising the organ specific functional components and by incorporating dynamic fluid flow to simulate blood circulation and perfusion of the intestinal lumen to support co-culture with immune cells, endothelial cells, microbes, and stromal cells [78]. These models have the potential to overcome some of the limitations of organoids which predominantly contains the epithelial layer and an enclosed lumen which limits access for nutrients or drugs, and does not permit mechanical stimuli to be applied [79]. Currently, the majority of organ-on-a-chip models utilises commercially available cell lines or pluripotent stem cells differentiated into multiple cell types of the intestine for proof of concept studies [79,80,81]. While representing a significant improvement over standard cell lines, models derived from iPSCs typically display a foetal phenotype, failing to reach the maturity of an adult organ. Adult stem cells (ASCs) derived from tissue biopsy are more difficult to obtain than iPSCs and are limited in their ability to differentiate to non-epithelial cells, and hence require coculture conditions with multiple cell types to create a physiologically relevant platform. However, ASCs allow for more accurate modelling of tumour processes and provide opportunity for personalised medicine and therapies to be explored.
Although microfluidic systems provide an ideal platform for development of an intestine-on-a-chip that can be used for cancer modelling or drug screening application, technical challenges still need to be overcome before they are able to be widely adopted. For application as a fast-fail approach to drug discovery, i.e. deploying models with high physiological relevance earlier in the drug discovery phase, the low-throughput microfluidic devices typically used to answer research questions will need to be adapted to high-throughput screening. Scaling these low throughput systems involves surmounting key biological and engineering challenges summarised in Probst et al. (2018) [82]. Limitations such as the requirement for specialised skills for consistent and reproducible microfabrication and use of the device and challenges associated with working with the microscale size currently limit broader adoption outside of the research community. Additionally, intestine-on-a-chip models are still limited in their ability to recreate the layers of the intestinal walls such as the smooth muscle layer required for peristalsis or the enteric nervous system which stimulates production of signalling molecules [79]. Technical challenges also exist for inclusion of the microbial community into an intestine-on-a-chip platform which is critical to recreate the true microenvironment and recapitulate the in vivo response [81]. Sanchez-Salazar et al (2023) attempts to address three recognised limitations of the intestine-on-a-chip technology as it applies to CRC. The authors designed a versatile platform with a rhomboid chamber to enable microtissues to be continuously perfused with culture medium [83]. In this instance, the well characterised Caco2 cell line was used to demonstrate extended culture of the cells and proof of concept drug assays with 5-FU. Overall, the continuous improvement of intestine-on-a-chip systems will widen the spectrum of organoid applications to address different aspects of CRC development, treatment and prevention.
4. Conclusions
The applications for three-dimensional culture is expanding throughout academic and industrial settings, indicating that physiological relevance is desirable despite the complexity and cost of these advanced cellular models. It should be emphasised that the field is still evolving, with fundamental challenges associated with faithfully recapitulating the intestinal organ still to be addressed, e.g. retaining the in vivo diversity of cellular types and the response to stress or biological insult. Standardisation and control over variability and development of reproducible and validated assays applicable to 3D culture is another aspect undergoing continued development, including the use of AI for microscopy and high content imaging. Additionally, microfluidic devices promise to provide enhanced monitoring and control over culture conditions, offering real-time, continuous and non-destructive measurement of biomarkers through arrays of biosensors. Increased development of miniaturised microfluidic systems within a SBS plate format, incorporating gravity-driven perfusion, presents an interesting compromise between complex, high-fidelity culture systems and the convenience of fitting into existing higher throughput workflows. Furthermore, research to reproducibly manufacture microfluidic devices to scale for applications beyond the academic research is ongoing, and examples of commercial availability of these devices continue to grow.
As with all model systems, a balance between complexity and practicality must be struck. The use of banked, patient-derived organoids together with new “-omic” technologies to guide assays measuring biological function may significantly improve the efficiency of biomarker discovery and drug development studies. By achieving a higher quality of putative biomarkers and drug responses from organoid-based screening, clinical validation can be initiated with greater confidence, resulting in higher success rates in human studies. Moreover, refinement of preclinical research and development using these models will enable novel chemoprevention strategies and screening of new drugs, leading to improved patient outcomes for colorectal cancer and other intestinal disease.
5. Future Directions
Ex vivo technologies in cancer research, including for those being developed for CRC research, encompasses complex cellular models that include multiple cell types and the intestinal microbiome that are more physiologically relevant than current 2D immortalised cell lines currently in use today. While primary cell lines, PDX, and PDE are regarded as closely representative of the in vivo response, they lack key features that make 3D culture systems, such as organoid models, attractive for disease modelling and therapeutic testing. However, before organoid technology is more widely adopted for clinical use, important factors also need to be considered. These include standardisation of procedures for culturing and storage, development of robust workflows and protocols that are transferable and transparent, ethical considerations (e.g., patient informed consent), and manufacturing to scale of reagents and component parts. For research and development to progress toward real world clinical application, regulatory frameworks need to be developed and deployed consistently across multiple jurisdictions.
In the context of CRC, intestinal organoids have the potential to improve outcomes for colorectal cancer patients. In particular, the development of organoid biobanks with samples derived from patients has the ability to represent the heterogeneity of the disease, including the different molecular subtypes and clinical course of the disease. When compared with organoids derived from matched adjacent normal mucosa, both efficacy and toxicity of potential therapeutics can be assessed. While single organ-on-a-chip models have been reported in the literature, interconnecting these individual organs into multi-organ systems remains challenging. When multi-organ systems are combined, this replication of the “body-on-a-chip” has the potential to provide a platform to test the effect of drugs on different organs, which is an important advancement in drug discovery. Patient specific information, such as tumour specific microenvironment, immune responses, drug metabolism and pharmacokinetics, and drug resistance mechanisms can be more accurately assessed. Traditionally, drug efficacy and toxicity are tested in vivo using animal models which is costly, time and resource intensive, often fails to provide accurate data for human studies and presents ethical arguments. As the technology develops and evolves, the organ-on-a-chip and body-on-a-chip concepts have the potential to revolutionise our understanding of the disease and shape clinical studies to the benefit of the patients.
Author Contributions
Conceptualization, S.R.D and K.Y.C.F.; writing—original draft preparation, S.R.D and K.Y.C.F; writing—review and editing, G.A., A.R., C.M., I.K.P., M.H., R.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Morgan, E.; et al. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef]
- Blakely, T.; et al. Patterns of cancer care costs in a country with detailed individual data. Med Care 2015, 53, 302–9. [Google Scholar] [CrossRef]
- Laudicella, M.; et al. Cost of care for cancer patients in England: evidence from population-based patient-level data. Br J Cancer 2016, 114, 1286–92. [Google Scholar] [CrossRef]
- Mariotto, A.B.; et al. Projections of the cost of cancer care in the United States: 2010-2020. J Natl Cancer Inst 2011, 103, 117–28. [Google Scholar] [CrossRef]
- Mishra, J.; et al. Prospective of colon cancer treatments and scope for combinatorial approach to enhanced cancer cell apoptosis. Crit Rev Oncol Hematol 2013, 86, 232–50. [Google Scholar] [CrossRef]
- Ciombor, K.K., C. Wu, and R.M. Goldberg, Recent therapeutic advances in the treatment of colorectal cancer. Annu Rev Med 2015, 66, 83–95. [Google Scholar] [CrossRef]
- Xie, Y.-H., Y. -X. Chen, and J.-Y. Fang, Comprehensive review of targeted therapy for colorectal cancer. Signal Transduction and Targeted Therapy 2020, 5, 22. [Google Scholar] [CrossRef]
- Jonker, D.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. New England Journal of Medicine 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E. , Integration of the anti-EGFR agent panitumumab into clinical practice in metastatic colorectal cancer. Clin Adv Hematol Oncol 2007, 5, 611–3. [Google Scholar]
- Chen, M.-H.; et al. How May Ramucirumab Help Improve Treatment Outcome for Patients with Gastrointestinal Cancers? Cancers 2021, 13, 3536. [Google Scholar] [CrossRef]
- Hwang, T.J.; et al. Failure of Investigational Drugs in Late-Stage Clinical Development and Publication of Trial Results. JAMA Intern Med 2016, 176, 1826–1833. [Google Scholar]
- Dowden, H. and J. Munro, Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov 2019, 18, 495–496. [Google Scholar] [CrossRef]
- Wong, C.H., K. W. Siah, and A.W. Lo, Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Sato, T.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–5. [Google Scholar] [CrossRef]
- Sato, T.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 2011, 141, 1762–72. [Google Scholar] [CrossRef]
- Co, J.Y.; et al. Toward Inclusivity in Preclinical Drug Development: A Proposition to Start with Intestinal Organoids. Advanced Biology 2023, 7, 2200333. [Google Scholar] [CrossRef]
- Du, Y.; et al. Development of a miniaturized 3D organoid culture platform for ultra-high-throughput screening. J Mol Cell Biol 2020, 12, 630–643. [Google Scholar] [CrossRef]
- Gilazieva, Z.; et al. Promising Applications of Tumor Spheroids and Organoids for Personalized Medicine. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Kaushik, G., M. P. Ponnusamy, and S.K. Batra, Concise Review: Current Status of Three-Dimensional Organoids as Preclinical Models. Stem Cells 2018, 36, 1329–1340. [Google Scholar] [CrossRef]
- Li, X., A. Ootani, and C. Kuo, An Air-Liquid Interface Culture System for 3D Organoid Culture of Diverse Primary Gastrointestinal Tissues. Methods Mol Biol 2016, 1422, 33–40. [Google Scholar]
- Wang, X.; et al. Cloning and variation of ground state intestinal stem cells. Nature 2015, 522, 173–178. [Google Scholar] [CrossRef] [PubMed]
- de Sousa, E.M.F.; et al. Modeling Colorectal Cancer Progression Through Orthotopic Implantation of Organoids. Methods Mol Biol 2020, 2171, 331–346. [Google Scholar]
- Co, J.Y.; et al. Controlling Epithelial Polarity: A Human Enteroid Model for Host-Pathogen Interactions. Cell Rep 2019, 26, 2509–2520. [Google Scholar] [CrossRef]
- Duleba, M.; et al. An Efficient Method for Cloning Gastrointestinal Stem Cells From Patients via Endoscopic Biopsies. Gastroenterology 2019, 156, 20–23. [Google Scholar] [CrossRef]
- van de Wetering, M.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–45. [Google Scholar] [CrossRef]
- Pauli, C.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov 2017, 7, 462–477. [Google Scholar] [CrossRef]
- Kondo, J.; et al. High-throughput screening in colorectal cancer tissue-originated spheroids. Cancer Sci 2019, 110, 345–355. [Google Scholar] [CrossRef]
- Yan, H.H.N.; et al. Organoid cultures of early-onset colorectal cancers reveal distinct and rare genetic profiles. Gut 2020, 69, 2165–2179. [Google Scholar] [CrossRef]
- Sargent, D.J.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol 2010, 28, 3219–26. [Google Scholar] [CrossRef]
- Bolck, H.A.; et al. Cancer Sample Biobanking at the Next Level: Combining Tissue With Living Cell Repositories to Promote Precision Medicine. Front Cell Dev Biol 2019, 7, 246. [Google Scholar] [CrossRef]
- Luo, Z.; et al. Establishment of a large-scale patient-derived high-risk colorectal adenoma organoid biobank for high-throughput and high-content drug screening. BMC Medicine 2023, 21, 336. [Google Scholar] [CrossRef]
- Fujii, M.; et al. Human Intestinal Organoids Maintain Self-Renewal Capacity and Cellular Diversity in Niche-Inspired Culture Condition. Cell Stem Cell 2018, 23, 787–793. [Google Scholar] [CrossRef]
- Fujii, M.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Roerink, S.F.; et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 2018, 556, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; et al. Colorectal Cancer Organoid-Stroma Biobank Allows Subtype-Specific Assessment of Individualized Therapy Responses. Cancer Discov 2023, 13, 2192–2211. [Google Scholar] [CrossRef]
- Engel, R.M.; et al. Modeling colorectal cancer: A bio-resource of 50 patient-derived organoid lines. Journal of Gastroenterology and Hepatology 2022, 37, 898–907. [Google Scholar] [CrossRef]
- He, X.; et al. Patient-derived organoids as a platform for drug screening in metastatic colorectal cancer. Frontiers in Bioengineering and Biotechnology 2023, 11. [Google Scholar] [CrossRef]
- Mo, S.; et al. Patient-Derived Organoids from Colorectal Cancer with Paired Liver Metastasis Reveal Tumor Heterogeneity and Predict Response to Chemotherapy. Advanced Science 2022, 9, 2204097. [Google Scholar] [CrossRef]
- Derouet, M.F.; et al. Towards personalized induction therapy for esophageal adenocarcinoma: organoids derived from endoscopic biopsy recapitulate the pre-treatment tumor. Sci Rep 2020, 10, 14514. [Google Scholar] [CrossRef]
- Nims, R.W. and P. J. Price, Best practices for detecting and mitigating the risk of cell culture contaminants. In Vitro Cell Dev Biol Anim 2017, 53, 872–879. [Google Scholar]
- Aref, A.R.; et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018, 18, 3129–3143. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; et al. Challenges in Establishing Pure Lung Cancer Organoids Limit Their Utility for Personalized Medicine. Cell Rep 2020, 31, 107588. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Karthaus, W.R.; et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 2014, 159, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Wallaschek, N.; et al. Establishing Pure Cancer Organoid Cultures: Identification, Selection and Verification of Cancer Phenotypes and Genotypes. J Mol Biol 2019, 431, 2884–2893. [Google Scholar] [CrossRef]
- Lannagan, T.R.M.; et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 2019, 68, 684–692. [Google Scholar] [CrossRef]
- Wilding, J.L. and W.F. Bodmer, Cancer cell lines for drug discovery and development. Cancer Res 2014, 74, 2377–84. [Google Scholar] [CrossRef]
- Berzins, S.P.; et al. A Role for MAIT Cells in Colorectal Cancer. Front Immunol 2020, 11, 949. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598. [Google Scholar] [CrossRef]
- Harter, M.F.; et al. Analysis of off-tumour toxicities of T-cell-engaging bispecific antibodies via donor-matched intestinal organoids and tumouroids. Nature Biomedical Engineering 2024, 8, 345–360. [Google Scholar] [CrossRef]
- Schnalzger, T.E.; et al. 3D model for CAR‐mediated cytotoxicity using patient‐derived colorectal cancer organoids. The EMBO Journal 2019, 38, e100928. [Google Scholar]
- Neal, J.T.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef]
- Grebenyuk, S. and A. Ranga, Engineering Organoid Vascularization. Front Bioeng Biotechnol 2019, 7, 39. [Google Scholar] [CrossRef]
- Rajasekar, S.; et al. IFlowPlate-A Customized 384-Well Plate for the Culture of Perfusable Vascularized Colon Organoids. Adv Mater 2020, 32, e2002974. [Google Scholar] [CrossRef]
- de Vos, W.M.; et al. Gut microbiome and health: mechanistic insights. Gut 2022, 71, 1020–1032. [Google Scholar] [CrossRef]
- O'Hara, A.M. and F. Shanahan, The gut flora as a forgotten organ. EMBO Rep 2006, 7, 688–93. [Google Scholar] [CrossRef]
- Sogari, A.; et al. Tolerance to colibactin correlates with homologous recombination proficiency and resistance to irinotecan in colorectal cancer cells. Cell Rep Med 2024, 5, 101376. [Google Scholar] [CrossRef]
- Iftekhar, A.; et al. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat Commun 2021, 12, 1003. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; et al. Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Allen, J.; et al. Colon Tumors in Enterotoxigenic Bacteroides fragilis (ETBF)-Colonized Mice Do Not Display a Unique Mutational Signature but Instead Possess Host-Dependent Alterations in the APC Gene. Microbiol Spectr 2022, 10, e0105522. [Google Scholar] [CrossRef]
- Sayed, I.M.; et al. The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef]
- Engevik, M.A.; et al. Fusobacterium nucleatum Secretes Outer Membrane Vesicles and Promotes Intestinal Inflammation. mBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, W.; et al. Cytolethal Distending Toxin Promotes Replicative Stress Leading to Genetic Instability Transmitted to Daughter Cells. Front Cell Dev Biol 2021, 9, 656795. [Google Scholar] [CrossRef]
- Miyakawa, Y.; et al. Gut Bacteria-derived Membrane Vesicles Induce Colonic Dysplasia by Inducing DNA Damage in Colon Epithelial Cells. Cell Mol Gastroenterol Hepatol 2024, 17, 745–767. [Google Scholar] [CrossRef]
- Zhang, L.; et al. Enterococcus faecalis promotes the progression of colorectal cancer via its metabolite: biliverdin. J Transl Med 2023, 21, 72. [Google Scholar] [CrossRef]
- Holst, L.M.; et al. Fecal Luminal Factors from Patients with Gastrointestinal Diseases Alter Gene Expression Profiles in Caco-2 Cells and Colonoids. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Zhang, Y.G.; et al. Vitamin D Receptor Protects Against Dysbiosis and Tumorigenesis via the JAK/STAT Pathway in Intestine. Cell Mol Gastroenterol Hepatol 2020, 10, 729–746. [Google Scholar] [CrossRef]
- Tang, Q.; et al. Endogenous Coriobacteriaceae enriched by a high-fat diet promotes colorectal tumorigenesis through the CPT1A-ERK axis. NPJ Biofilms Microbiomes 2024, 10, 5. [Google Scholar] [CrossRef]
- Mowat, C.; et al. Short chain fatty acids prime colorectal cancer cells to activate antitumor immunity. Front Immunol 2023, 14, 1190810. [Google Scholar] [CrossRef]
- Sugimura, N.; et al. Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut 2021, 71, 2011–21. [Google Scholar] [CrossRef]
- Iwama, T.; et al. Bacteria-derived ferrichrome inhibits tumor progression in sporadic colorectal neoplasms and colitis-associated cancer. Cancer Cell Int 2021, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Mackie, G.M.; et al. Bacterial cancer therapy in autochthonous colorectal cancer affects tumor growth and metabolic landscape. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Gao, Y.; et al. Fusobacterium nucleatum enhances the efficacy of PD-L1 blockade in colorectal cancer. Signal Transduct Target Ther 2021, 6, 398. [Google Scholar] [CrossRef]
- Williamson, I.A.; et al. A High-Throughput Organoid Microinjection Platform to Study Gastrointestinal Microbiota and Luminal Physiology. Cell Mol Gastroenterol Hepatol 2018, 6, 301–319. [Google Scholar] [CrossRef]
- Sasaki, N.; et al. Development of a Scalable Coculture System for Gut Anaerobes and Human Colon Epithelium. Gastroenterology 2020, 159, 388–390. [Google Scholar] [CrossRef]
- Sunuwar, L.; et al. Mechanical Stimuli Affect Escherichia coli Heat-Stable Enterotoxin-Cyclic GMP Signaling in a Human Enteroid Intestine-Chip Model. Infect Immun 2020, 88. [Google Scholar] [CrossRef]
- Mitrofanova, O.; et al. Bioengineered human colon organoids with in vivo-like cellular complexity and function. Cell Stem Cell 2024. [Google Scholar] [CrossRef]
- Bein, A.; et al. Microfluidic Organ-on-a-Chip Models of Human Intestine. Cellular and Molecular Gastroenterology and Hepatology 2018, 5, 659–668. [Google Scholar] [CrossRef]
- Gonçalves, I.M.; et al. Organ-on-a-Chip Platforms for Drug Screening and Delivery in Tumor Cells: A Systematic Review. Cancers 2022, 14, 935. [Google Scholar] [CrossRef]
- Workman, M.J.; et al. Enhanced Utilization of Induced Pluripotent Stem Cell–Derived Human Intestinal Organoids Using Microengineered Chips. Cellular and Molecular Gastroenterology and Hepatology 2018, 5, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Probst, C., S. Schneider, and P. Loskill, High-throughput organ-on-a-chip systems: Current status and remaining challenges. Current Opinion in Biomedical Engineering 2018, 6, 33–41. [Google Scholar] [CrossRef]
- Sánchez-Salazar, M.G.; et al. 3D-Printed Tumor-on-Chip for the Culture of Colorectal Cancer Microspheres: Mass Transport Characterization and Anti-Cancer Drug Assays. Bioengineering 2023, 10, 554. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Patient-derived organoids can be applied to both disease modelling and the preclinical phases of therapeutic testing.
Figure 1.
Patient-derived organoids can be applied to both disease modelling and the preclinical phases of therapeutic testing.
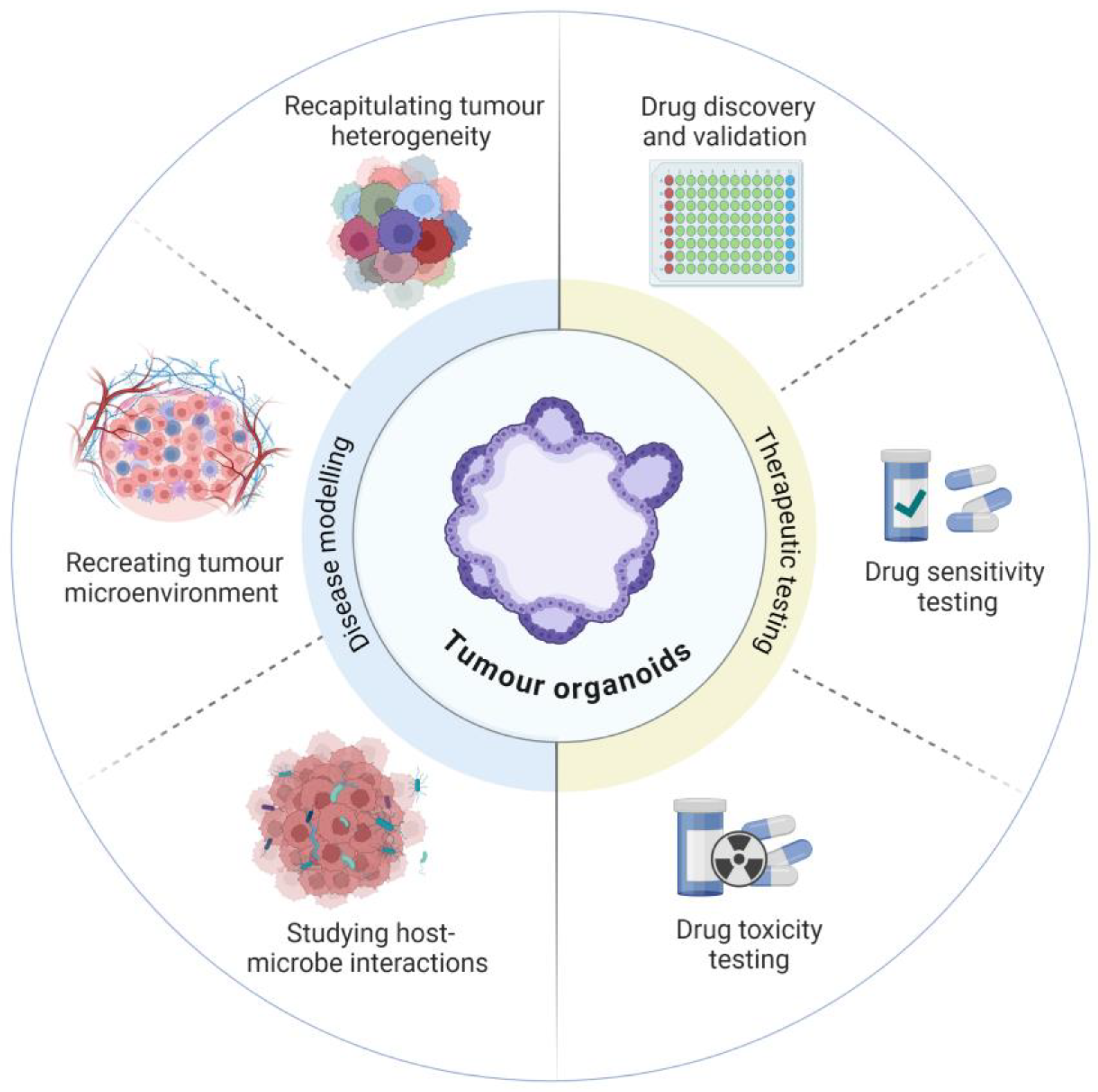

Table 1.
Design considerations in modelling colorectal cancer for drug discovery using patient-derived tissues.
Table 1.
Design considerations in modelling colorectal cancer for drug discovery using patient-derived tissues.
| Primary intestinal cells (Transwell support) | Patient-derived organoids | Organ-on-a-Chip | Patient derived xenografts | Patient derived explants | |
|---|---|---|---|---|---|
| Represents in vivo system — Native organ structures? | Crypt-like formation in collagen-based gels with ALI [20,21] | “Spherical“ (3D), with crypt-like formation, differentiated cell types present [15] | Organoid, Tubular (3D) & planar (2D) | “Spherical” (3D) [22] | High fidelity, overall architecture retained, mixed mucosal cell types |
| Preservation of Intra-tumoural heterogeneity | Low | Medium | Variable | Medium | High |
| Clonogenicity | High, >70% in stem cell culture, low in ALI [21] | Low, possibly <2% under differentiating conditions [21] | NA | NA | Low, differentiation & maturation close to normal |
| Accessible lumen | No | Yes, [23] | Organoids: no Planar/tubular cultures: yes | No | Yes |
| Long-term culture | Stem cells repeat passages | Yes | Possible, likely depends on ECM stability | Viability diminished after 3–5 passages | Static culture: Viability declines after 7 days medium perfusion: Bioreactor: 30 days |
| Throughput | Low-throughput format | Scalable, grown in multiwell format, up to 1536-well plates | Generally low to medium | Difficult to achieve — Labour and costs prohibitive even for organoid grafts | Non-scalable — limited by size of starting material |
| Biobanking | Stem cell banking [21,24] | High success rate, existing CRC organoid banks [25] | NA | Tumours can be banked | Can be cryopreserved but not expandable |
| Genetic manipulation | Yes | Yes | Yes | Direct from tumour: No Organoid grafts: Yes | No |
Table 2.
Current registered clinical trials involving patient-derived organoids. References: https://anzctr.org.au/TrialSearch.aspx; https://clinicaltrials.gov/. Last accessed Sept 5, 2024.
Table 2.
Current registered clinical trials involving patient-derived organoids. References: https://anzctr.org.au/TrialSearch.aspx; https://clinicaltrials.gov/. Last accessed Sept 5, 2024.
| Trial number | Study Title | Study status | Conditions | Interventions | Country |
|---|---|---|---|---|---|
| NCT05669586 | Organoids Predict Therapeutic Response in Patients With Multi-line Drug-resistant Lung Cancer | Recruiting | Lung Cancer|Organoid | phase 2 | China |
| NCT04768270 | The Culture of Ovarian Cancer Organoids and Drug Screening | Recruiting | Ovarian Cancer | observational, patient registry | China |
| NCT05092009 | Lung Cancer Organoids and Patient Derived Tumor Xenografts | Recruiting | Lung Cancer | observational | Netherlands |
| NCT05290961 | The Culture of Advanced or Recurrent Ovarian Cancer Organoids and Drug Screening | Recruiting | Ovarian Neoplasms | observational, patient registry | China |
| NCT06064682 | An Organoid-based Functional Precision Medicine Trial in Osteosarcoma | Recruiting | Osteosarcoma | observational, standard of care biopsy | USA |
| NCT05577689 | Novel Therapy Target in Metastatic Prostate Cancer | Not yet recruiting | Prostate Neoplasms | observational | China |
| NCT05832398 | Precision Chemotherapy Based on Organoid Drug Sensitivity for Colorectal Cancer | Recruiting | Colorectal Cancer | interventional | China |
| NCT04931394 | Organoid-Guided Adjuvant Chemotherapy for Pancreatic Cancer | Recruiting | Pancreatic Cancer | interventional, phase 3 | China |
| NCT04931381 | Organoid-Guided Chemotherapy for Advanced Pancreatic Cancer | Recruiting | Advanced Pancreatic Cancer | interventional, phase 3 | China |
| NCT06268652 | Patient Derived Organoid-guided Personalized Treatment Versus Treatment of Physician's Choice in Breast Cancer | Recruiting | Breast Cancer|Refractory Breast Carcinoma | interventional, phase 3 | China |
| NCT05024734 | Guiding Instillation in Non Muscle-invasive Bladder Cancer Based on Drug Screens in Patient Derived Organoids | Recruiting | Bladder Cancer|Non-muscle Invasive | interventional, phase 2 | Switzerland |
| NCT05725200 | Study to Investigate Outcome of Individualized Treatment in Patients With Metastatic Colorectal Cancer | Recruiting | Metastatic Colorectal Cancer | interventional, phase 2 | Norway |
| NCT06468527 | Clinical Trial to Evaluate the Efficacy and Safety of Dirocaftor/Posenacaftor/Nesolicaftor in Adults With CF | Recruiting | Cystic Fibrosis | interventional, phase 2 | Netherlands |
| NCT06102824 | Organoid-based Functional Precision Therapy for Advanced Breast Cancer | Recruiting | HER2-negative Breast Cancer|Advanced Breast Cancer | interventional, phase 2 | China |
| NCT05352165 | The Clinical Efficacy of Drug Sensitive Neoadjuvant Chemotherapy Based on Organoid Versus Traditional Neoadjuvant Chemotherapy in Advanced Rectal Cancer | Not yet recruiting | Neoadjuvant Therapy | interventional | China |
| NCT06227065 | Precise Neoadjuvant Chemoresection of Low Grade NMIBC | Not yet recruiting | Bladder Cancer, Non-muscle Invasive Bladder Cancer | interventional, phase 2 | Switzerland |
| NCT03979170 | Patient-derived Organoids of Lung Cancer to Test Drug Response | Recruiting | Lung cancer | observational, patient registry | Switzerland |
| NCT03283527 | Chemoradioresistance in Prospectively Isolated Cancer Stem Cells in Esophageal Cancer-Organoid: RARE STEM-Organoid | Recruiting | Esophageal cancer | observational | Netherlands |
| 387579 (ACTRN12624000684527p) | FORECAST-II Feasibility of using Organoid Response to inform treatments for patients with Colorectal cancer staring first-line therapy | Not yet recruiting | Colorectal Cancer | Diagnosis / Prognosis |
Australia |
| 386544 (ACTRN12623001136695) | ORganoId GuIded N-of-1 (ORIGIN-1) Trial: A phase 4 study to investigate whether people with cystic fibrosis (CF) with rare cystic fibrosis transmembrane regulator (CFTR) mutations who have an in vitro response to Trikafta will also have a clinically meaningful response to Trikafta versus placebo | Not yet recruiting | Cystic Fibrosis | interventional, phase 4 | Australia |
| 380279 (ACTRN12620001353987) | FORECAST 1. Feasibility of using Organoid Response to find Effective Treatments for patients with Colorectal cancer After failure of Standard Therapy | Recruitment closed | Metastatic Colorectal Cancer | interventional | Australia |
| NCT03544255 | Drug Screening of Pancreatic Cancer Organoids Developed From EUS-FNA Guided Biopsy Tissues | Unknown status | Pancreatic Cancer | observational | China |
| NCT03544047 | Clinical Study on Drug Sensitivity Verification or Prediction of Therapy for Breast Cancer by Patient-Derived Organoid Model | Unknown status | Breast cancer | interventional | China |
Table 3.
Intestinal co-culture with bacterial species and application to colorectal cancer.
| Organoid co-culture models: pro-tumorigenesis mechanisms | |||
|---|---|---|---|
| Organoid type and species | Bacterial species | Effect shown | Reference |
| Human CRC organoids | Colibactin-producing E. coli DH10B | DNA damage (double-strand break (DSB)) | [57] |
| Murine colon organoids | pks+ E. coli | DSB, genomic instability, chromosomal aberrations andgenetic mutations | [58] |
| Human intestinal organoids | pks+ E. coli | DNA damage and oncogenic mutational signatures | [59] |
| Human intestinal organoids | Enterotoxigenic B. fragilis | Did not induce a unique mutational pattern | [60] |
| Mouse and human colon organoids | F. nucleatum, E. coli K12 strain DH10B, E. coli strain LF82 and Helicobacter pylori | F. nucleatum downregulated expression of DNA repair protein (NEIL2), increased the accumulation of DNA damage and production of the IL-8 | [61] |
| Murine intestinal organoids | Bacterial lysates of wild-type C. jejuni (WT) or C. jejuni mutcdtB | DNA damage | [62] |
| Human colon organoids | F. nucleatum conditioned media | Increased inflammatory responses characterised by increased secretion of TNF and activation of NF-κB, p-ERK, p-CREB signalling pathways | [63] |
| Human intestinal organoids | E. coli-derived cytolethal distending toxin | DNA damage | [64] |
| Human intestinal organoids | Actinomyces odontolyticus -derived lipoteichoic acid-rich membrane vesicles | DSB | [65] |
| Human CRC organoids | Biliverdin, a key metabolite produced by CRC-associated E. faecalis | Increased the expression of cell proliferation marker Ki67 | [66] |
| Human colon organoids | Faecal supernatant from colon cancer patients | Alterations in gene expression | [67] |
| Human and murine colon organoids | Faecal supernatant from a cancer mouse model lacking intestinal vitamin D receptor | Activation of JAK/STAT3 signalling and increase in PCNA and β-catenin expression | [68] |
| Organoid co-culture models: protective mechanisms | |||
| Murine colon organoids | Coriobacteriaceae (Cori.ST1911) and Lactobacillus murinus (La.mu730) | Upregulated expression of carnitine palmitoyltransferase 1A (CPT1A), and downregulated MUC2 protein. Lactobacillus murinus (La.mu730) reversed negative effect of Cori.ST1911 |
[69] |
| Human and murine CRC organoids | Short chain fatty acids | Upregulated expression of Type I IFN Stimulated Genes (CXCL10 and ISG15) which are important for anti-tumour immune response | [70] |
| Human CRC organoids | Lactobacillus gallinarum supernatant | Induction of apoptosis | [71] |
| Human adenoma and CRC organoids | Lactobacillus casei- derived ferrichrome | Tumour suppression response by upregulating the expression of DNA damage-inducible transcript 3 | [72] |
| Organoid co-culture models: mechanisms related to treatment response | |||
| Murine tumour organoids | Salmonella enterica serovar Typhimurium (aromatase A–deficient Salmonella Typhimurium (STmΔaroA) | Altered gene expression analysis including reduced expression of stem cell and EMT markers, increased expression of innate immunity proteins | [73] |
| Human CRC organoid | F. nucleatum | Enhanced efficacy of anti-PD-L1 immunotherapy | [74] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



