
Submitted:
23 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
Oncolytic measles virus (MeV) is a promising anti-cancer treatment. However, the production of high titers of infectious MeV (typically 107–109 TCID50 per dose) is challenging because the virus is unstable under typical production conditions. The objective of this study was to investigate how the multiplicity of infection (MOI) and different media – serum-containing (SCM), serum-free (SFM) and chemically-defined (CDM) – affect MeV production. We infected Vero cells at MOIs of 0.02, 0.2 or 2 TCID50 mL-1 and the lowest MOI resulted in the largest number of infected cells towards the end of the production period. However, this did not equate to higher maximum MeV titers, which were similar for all MOIs. The medium had a moderate effect, generating maximum titers of 0.89–2.17 × 106, 1.08–1.25 × 106 and 4.58–9.90 × 105 TCID50 mL‑1 for SCM, SFM and CDM, respectively. Infection at a low MOI often required longer process times to reach maximum yields. Our findings show that SCM, SFM and CDM are equally suitable for MeV production in terms of yield and process time. This will allow MeV production in serum-free conditions, addressing the safety risks and ethical concerns associated with the use of serum.
Keywords:
multiplicity of infection (MOI)
; media adaption
; chemically-defined medium (CDM)
; serum-free medium (SFM)
; viral vaccines
; vectors
; virus-like particles (VLPs)
; cell culture research
; process development
1. Introduction
Cancer is the second leading cause of death worldwide [1]. An emerging cancer treatment approach is the use of oncolytic viruses, such as the Measles morbillivirus (MeV) vaccine strains [2], which are being tested in the clinic for the treatment of ovarian cancer and glioma [3]. As well as its ability to infect and lyse tumor cells [4], MeV also induces systemic anti-tumor immune responses [5]. The high affinity of MeV for tumors reflects the uptake of the virus via surface receptors such as Nectin-4, CD150/SLAM and the ubiquitous receptor CD46, which are commonly overexpressed on cancer cells [6,7,8]. Infected cells change morphologically as they fuse with adjacent cells, form multi-nuclear giant cells (syncytia) and finally lyse. This virus-mediated cell damage, also known as the cytopathic effect, is one of the mechanisms of action of oncolytic virotherapy [9].
A high dose of infectious MeV is necessary for successful treatment: typically 107–109 TCID50 [10], and in one exceptional case a systemic application of 1011 TCID50 [11]. This requires highly efficient production processes, whereby MeV is produced under optimal conditions with the highest possible yield. Potent host cells (e.g., Vero or MRC-5 cells) enable the production of high titers of MeV up to bioreactor scale [12,13,14,15], but a major challenge is the physicochemical instability of the virus. MeV is rapidly inactivated at the optimum temperature for the cultivation of mammalian cells (37 °C) [16] and is also highly unstable at pH < 7, which is problematic for MeV production in systems without pH regulation [16]. It is therefore necessary to determine the optimal time of harvest (TOH) with precision, because missing this optimal time can result in MeV losses of several log units within hours [17]. In addition to the TOH, the multiplicity of infection (MOI) is a critical process parameter that must be optimized to maximize virus yields. The MOI is defined as the ratio of infectious virus particles (or their relative units) to host cells at the time of infection (TOI). Theoretically, high MOIs allow the simultaneous infection of all host cells whereas low MOIs result in a proportion of cells being infected at the beginning of the process. The non-infected cells continue to proliferate and are then infected by newly produced viruses during secondary infections. This initially leads to populations of infected and non-infected cells but it is likely that all cells are infected later in the process. A high MOI is beneficial for short process times but requires a larger amount of virus stock for inoculation. The low MOI strategy allows the growth of non-infected cells, which results in a greater number of cells being infected at the end [18]. Given that MeV infection is a statistical process, it can be reasonably assumed that an increase in the number of virus particles will inevitably lead to an increase in the number of infected (and thus virus-producing) cells [17]. Previous studies have only considered the cell concentration at the time of infection without analyzing the number of cells actually infected. [13,14]. Monitoring the number of infected cells would help a priori to determine the most appropriate MOI for an optimal space–time yield of infectious MeV. However, it must be noted that a successful infection with MeV does not necessarily result in high virus titers. This is because the virus must replicate within the host cell and be released in order to achieve high yields [19].
A critical element for an effective MeV production process is the choice of the cell culture medium, which must be optimized for both the infection process and virus replication and release. High MeV yields have already been achieved in serum-containing medium (SCM) using Vero cells in a microcarrier-based stirred-tank reactor (STR). However, the use of serum in cell culture media has several disadvantages including high costs, ethical concerns, and (especially in immunocompromised cancer patients) contamination with pathogens and the risk of serum-induced allergic reactions. In addition, the undefined nature of serum leads to batch-to-batch variation, which directly affects MeV quality and raises safety concerns. The use of serum-free medium (SFM) and chemically defined medium (CDM) is therefore preferred in the biopharmaceutical industry [20]. SFM was previously tested for MeV production and showed promising results in terms of yield and process efficiency [13,15]. However, little information is available about the production of MeV in CDM.
Here we investigated the effect of MOI and different media on MeV production. We adapted Vero cells for growth in one SFM and two CDMs to determine the suitability of these media for MeV production in static cultivation systems. The media were compared to standard Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). To evaluate the influence of the MOI on static MeV production, we used a model MeV strain (Section 2.3) allowing the ratio changes between total cells and MeV-infected cells to be monitored by flow cytometry. In addition, we used fluorescence microscopy to investigate the course of infection and its spread during static cultivation, and determined the concentrations of relevant substrates and metabolites in the culture supernatant.
2. Materials and Methods
2.1. Cultivation of Vero Cells
The adherent cell line Vero-B4 was purchased from DSMZ (#ACC 33). For general maintenance, the cells were seeded into T-75 or T-175 tissue culture flasks with venting caps (Sarstedt, #83.3911.002 or #83.3912.002) at 5—10 × 103 cells cm-2 with 0.28 mL cm-2 culture medium and were incubated at 37 °C in an 8% CO2 atmosphere. The cells were passaged after reaching 90–100% confluence.
For harvesting, the cell layer was washed twice with phosphate-buffered saline (PBS; without Ca2+/Mg2+; Sigma-Aldrich, #D8537) and covered with at least 0.08 mL cm-2 trypsin/EDTA (prepared from a 10× stock in PBS; Biochrom, #L2153) or 1× TrypZean solution (Sigma-Aldrich; #T3449). Trypsinization was carried out at 37 °C in an 8% CO2 atmosphere until the cells detached from the surface.
The enzymatic reaction was stopped by adding three times the trypsin volume of SCM or, in the case of SFM/CDM, by adding trypsin inhibitor (stock solution 1 mg mL-1 in PBS; Sigma-Aldrich, #T6522) using the same volume as TrypZean. The detached cells were centrifuged (250 × g, 5 min, room temperature) and resuspended in fresh medium before seeding into new T-flasks or use for MeV production. When cultured in SCM, the centrifugation step was omitted and the cell suspension was used directly. Cell concentrations were determined using a Scepter 2.0 cell counter (Merck, #PHCC20060) and the corresponding Scepter 60 µm sensors (Merck, #PHCC60500).
2.2. Adaption of Vero Cells to SFM and CDM
Vero cells were adapted from standard DMEM (Gibco, Thermo Fisher Scientific, USA; #41965062), supplemented with 10% FBS (Capricorn Scientific, #FBS-11A), to one SFM (VP-SFM; Gibco, #11681020), supplemented with 4 mM l-glutamine (200 mM stock; Sigma-Aldrich, #G7513), and two non-commercial research CDMs (73614C and 73835C) kindly provided by Merck (Germany) and supplemented according to their instructions.
After thawing Vero cells in standard medium (DMEM with 10% FBS) and the first passaging, the cells were adapted to new media during the next passage by successive media exchanges over 3 days. We started with a 75/25 ratio of standard/new medium from 0.33 days post seeding (dps), then switched to 37.5/62.5 from 1 dps, then to 7.5/92.5 from 2 dps, and finally to 100% VP-SFM or CDM from 3 dps. The cells were then cultured and expanded for three further passages before master cell banks (MCBs) were generated. For this purpose, we used a chemically-defined freezing medium without components of animal origin (10 mg mL-1 human serum albumin; Sigma-Aldrich, #A1653-5G), 9 mg mL-1 NaCl, 10% (v/v) dimethyl sulfoxide (Sigma-Aldrich, #D2650) in demineralized water). Working cell banks (WCBs) were generated after thawing a MCB vial and three subsequent passages.
2.3. Measles Virus Strain
The infectious model strain MeV NSe ld-EGFP (derived from the Edmonston B-derived MeV vaccine strain NSe) was kindly provided by Prof. Guy Ungerechts, National Center for Tumor Diseases (Heidelberg, Germany). The virus genome carries an eGFP transgene, and enhanced GFP is therefore expressed by host cells after infection.
2.4. MeV Production in Six-Well Plates
Vero cells (from the WCB) were seeded at 10,000 cells cm-2 in six-well tissue culture plates (Sarstedt, #83.3920) with 0.28 mL cm-2 of the appropriate medium and were incubated at 37 °C in an 8% CO2 atmosphere. The cells were infected 0.83 dps at MOIs of 0.02, 0.2 and 2 TCID50 cell-1 by adding the required MeV stock volume. We assumed that the changes in cell number from seeding to the TOI were negligible (in relation to MOI with log10-level differences) due to the lag phase. The infectious virus titers of the MeV stocks were determined using a TCID50 assay in eight technical replicates. Mock-infected Vero cell cultures were used as controls. To increase the stability of the virus, the temperature was reduced to 32 °C immediately after infection. Because the volume of MeV stock we added was dependent on the culture medium and MOI, the medium was exchanged in all cultures at 1.25 days post-infection (dpi) to ensure the same media conditions and filling volumes. Based on our experience, the number of viruses released at this time point is negligible.
For each medium, culture supernatant samples were taken from one well per day for further analysis. TCID50 assay samples were stored at –80 °C and cells were detached with trypsin for immediate analysis by flow cytometry. The cell densities of total and infected (GFP+) cells were recalculated from the cell concentration results. As the resuspension of trypsinized MeV-infected cells had to be comparatively rough, especially in the later course of infection (due to sticky cell aggregates), it is likely that a certain degree of mechanical cell lysis was anticipated. This intrinsic error during cell quantification should be considered regarding total cell densities.In addition, one well containing cells growing in each medium was prepared for fluorescence microscopy at 3 and 5 dpi.
2.5. Flow Cytometry
Harvested cells were, if necessary, diluted to a concentration of 1–5 × 105 cells mL-1. We transferred 200 μL of the cell suspension to a 96-well plate with a flat bottom for analysis using a Guava easyCyte flow cytometer (Cytek Biosciences, USA) and guavaSoft Incyte software. The cell population was separated from debris by creating a gate in the sideward scatter (SSH-HLin)-forward scatter (FSC-HLin) dot plot. In the selected gate, 1300–5000 events were recorded per sample at an average flow rate of 0.59 μL s-1. The wells were mixed for 6 s between individual measurements. The fluorescence signal was detected after passing a 583/26 nm bandpass filter with a photomultiplier as Yel-B-H log and logarithmically amplified (five decades). The fluorescence signal of the gated cell population was exported as an abundance histogram (counts vs Yel-B-H log). The autofluorescence of non-infected cells was amplified to be close to the ordinate. The fluorescence intensity threshold for GFP+ (infected) cells was set by filtering out > 98% of the non-infected control cells. Cells with a fluorescence signal exceeding the measurement range were registered as the maximum signal value. The experiments were carried out in technical duplicates (data points represent the mean value).
2.6. Fluorescence Microscopy
The infection status of the adherent Vero cell cultures was also checked by fluorescence microscopy 3 and 5 dpi. The GFP signal was used to determine the number of MeV NSe ld-EGFP infected cells, whereas cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; ITW Reagents, #A1001,0010) to determine the total number of cells. Vero cell cultures were washed twice with 0.1 mL cm-2 PBS and then mixed with 0.1 mL cm-2 DAPI staining solution (0.1 µg mL-1 DAPI in 5% (v/v) methanol in PBS) and incubated for 30–40 min at room temperature in the dark. The cells were not fixed beforehand in order to preserve their native state. After washing with PBS, fluorescence images were captured automatically using the Cytation 3 (Agilent BioTek, USA) and associated Gen 5 software (Agilent BioTek). We used 2.5× or 10× objectives, GFP and DAPI, and four (2×2) or nine (3×3) images per well, stitched.
2.7. Determination of the Infectious MeV Titer
The virus titer was determined using a TCID50 (median tissue culture infectious dose) assay. A Vero cell suspension with 50,000 cells mL-1 in DMEM (10% FCS) was prepared and 0.2 mL of the suspension was pipetted into all wells of 96-well tissue culture plates (Sarstedt, #83.3924). The plates were incubated at 37 °C in an 8% CO2 atmosphere for least 4 h. MeV samples were serially diluted in rows of separate 96-well plates in 1:10 dilution steps (100 to 10-11) with DMEM (10% FCS). The pipette tips were changed at each dilution step. We transferred 30 μL of each sample dilution to eight wells containing Vero cells and incubated the plates for 6–7 days at 32 °C in an 8% CO2 atmosphere. After incubation, we used the Cytation 3 and Gen 5 software to capture automatic images of GFP expression with a 2.5× objective (six per well (2×3), stitched) to confirm infection with MeV NSe ld-EGFP. The TCID50 titer was determined based on Spearman-Karber calculations of the 50% endpoint dilution [21,22].
2.8. Substrate and Metabolite Concentrations
Samples from the cultures were centrifuged (2000 × g, 2 min, room temperature) and the glucose and lactate concentrations in the supernatant were determined using the Biosen C-line analyzer (EKF Diagnostics, Germany). Ammonia, glutamine, glutamate and pyruvate were measured semi-automatically using the Cedex Bio Analyzer (Roche, Switzerland). Lactate dehydrogenase (LDH) enzyme activity in the supernatant was monitored as a marker of cell lysis using the same device. All measurements were carried out according to the manufacturers’ instructions.
3. Results and Discussion
3.1. Growth Kinetics of Vero Cells Adapted to SFM and CDM
We compared the growth kinetics of Vero cells adapted to SFM and CDM to cells in standard DMEM during static cultivation (Figure 1).
The highest cell densities ~6 days after seeding were achieved in DMEM (10% FBS) with 180,000 ± 10,000 cells cm-2. The two CDMs (73614C and 73835C) and VP-SFM resulted in cell densities of 120,000–155,000 cells cm-2. The CDMs and SFM therefore reached sufficient cell densities, with growth kinetics largely comparable to SCM. Direct comparison indicated the slightly weaker overall performance of VP-SFM.
3.2. Influence of MOI on Virus Yields and TOH
We compared the influence of MOI (0.02, 0.2 and 2 TCID50 cell-1) on static MeV production in the Vero cells adapted to SFM and CDM and those in SCM (Figure 2). We analyzed the course of MeV production by the number of infected cells (GFP+ cells) per growth area and the infectious MeV titer in the culture supernatant.
3.2.1. MOI-Dependent Growth Kinetics
We observed MOI-dependent cell growth in all media during MeV production. Cultures infected at an MOI of 2 or 0.2 experienced a short cell growth phase and reached a maximum cell density of 12,000–24,000 cells cm-2 1.5–2.2 dpi. The only exception was observed in VP-SFM at an MOI of 0.2, where the maximum cell density was reached at a later point in time (26,010 cells cm-2 3.2 dpi). After reaching the maximum, the cytopathic effect of MeV reduced the total cell number, so that fewer than 6400 cells cm-2 were left 5–6 dpi. This probably reflected a combination of cell lysis and the fusion of infected cells with adjacent cells, leading to the formation of polynucleated giant cells (syncytia) [9]. The detachment of apoptotic cells from the growth surface may also have contributed because only adherent cells were quantified after washing and detaching the cell layer. Moreover, virus-induced alterations in cell metabolism and cell cycle progression also limit cell proliferation. In contrast, the lowest MOI (0.02) prolonged cell proliferation up to 4.2 dpi, regardless of the culture medium. The peak cell density was therefore up to 0.74 times higher in DMEM/73614C, and even up to 2.31 times higher in VP-SFM/73835C, compared to MOIs of 0.2 and 2. Even 7 dpi, more than 12,570 cells cm-2 remained in cultures infected at an MOI of 0.02.
Substrate uptake, lactate and ammonia accumulation, and LDH release are summarized in n Figure 3. During MeV amplification, the glucose and glutamine concentrations remained above the critical values (glucose > 5 mM, glutamine > 0.5 mM), which were already defined for Vero cell STR cultivations with bolus feeds [23]. Ammonia accumulated to a maximum of 2.5 mM, which is only half the IC50 (50% reduction in growth) of 5 mM for Vero cells [24]. Lactate, which indicates exponential growth in uninfected Vero cell cultures, did not exceed the critical concentration of 19 mM in any of the cultures infected at MOIs of 0.2 or 2. At an MOI of 0.02, the lactate concentration 7 dpi was 14.6–23.2 mM and thus above or near to the critical value. The LDH activity in the culture supernatant increased in all MeV-infected cultures (from 5 dpi at the latest), which can be explained by the lysis of the cells during the course of MeV infection. Pyruvate (a component of VP-SFM and the CDMs) was consumed, remaining at the highest levels in the mock-infected cultures and lowest in the heavily MeV-infected (MOI 2) cultures. However, the Vero cells were not dependent on pyruvate given their ability to grown in DMEM, which lacks this compound. The slow and MOI-dependent consumption of substrates and formation of metabolites in the MeV-infected cultures is associated with the lower cell densities compared to the mock-infected cultures.
3.2.2. MOI-Dependent Infection Kinetics
Infected cells were detected by flow cytometry based on the presence of GFP, providing evidence of successful MeV propagation at each tested MOI (Figure 2). The infection ratio increased over time and ultimately exceeded 95%. The higher the MOI, the less time was required to reach 50% infected cells (1.36 ± 0.23 dpi at MOI = 2, 2.75 ± 0.27 dpi at MOI = 0.2 and 4.14 ± 0.36 dpi at MOI = 0.02) and subsequently ~100% infection. This relationship can be explained by the variable number of completed infection cycles and the subsequent secondary infections.
Compared to an MOI of 2, it is evident that the lower MOIs lead to a multi-stage infection process in which newly-formed viruses infect the remaining uninfected host cells. The longer expansion phase of Vero cells observed when using an MOI of 0.02 may therefore reflect the delayed spread of infection through the cell population. As expected, the highest MOI resulted in a greater number of infected cells at the beginning of the process. Although the density of infected Vero cells increased somewhat later when using the medium MOI, the trends were similar, mirroring the observations regarding total cell density. The medium and high MOIs resulted in a maximum density of 7500–19,000 infected cells cm-2 3 dpi at the latest. By 5 dpi, fewer than 6000 infected cells cm-2 were left. At the low MOI, we observed a sharp increase in the number of infected cells between 3 and 5 dpi, which then stayed more or less stable for 2–3 days in DMEM and CDM but decreased immediately in VP-SFM. Importantly, the maximum number of infected cells was up to 4.16 times higher compared to the MOIs of 0.2 and 2 (with the exception of DMEM, where the difference was minimal).
The hill slopes of the sigmoidal curves of GFP+ cells over time (especially noticeable in DMEM, with hill slopes of 1.11 ± 0.02) were mostly similar, indicating that the propagation of infection is MOI-independent and its course is only temporally shifted. Deviating hill slopes (e.g., in VP-SFM with MOI = 2) probably reflected measurement inaccuracies combined with a low number of data points. For future studies, we would therefore recommend more measurements per day. Because the time intervals between “reaching 50% infected cultures” depending on the MOI were very similar in all media, with ∆t = 1.39 ± 0.16 (MOI 2 vs 0.2) and ∆t = 1.39 ± 0.11 (MOI 0.2 vs 0.02), the replication cycle of MeV NSe ld-EGFP appears to last ≤ 1.4 d.
Theoretically, an MOI of 2 TCID50 cell-1 should be sufficient for simultaneous infection of the whole cell population. However, at the highest MOI, the proportion of infected cells at the beginning of the process (~1.6 dpi) was in some cases below 95%. Notably, the identification of infected cells was delayed because sufficient GFP accumulation is needed to distinguish infected from non-infected cells. Furthermore, different cell cycle phases in the cell population and the uneven distribution of infectious virus particles on host cells (Poisson distribution [25]) could explain the reduced infection efficiency at the TOI.
3.2.3. MOI-Dependent MeV Yield
The influence of MOI on the infection process and cell growth was reflected in the virus release profile. Consistent with the presence of more virus-infected cells at the beginning of the process with higher MOIs, the virus titer also increased more rapidly. The optimal TOH was therefore achieved 1 day sooner. On the other hand, there was little difference in the virus release profile when comparing the medium and high MOIs in the CDMs (optimum TOH = 4–5 dpi). Even so, a change in MOI from 2 to 0.2 did not substantially affect the maximum virus yield, which was 0.89–2.17 × 106 TCID50 mL-1 in DMEM (10% FCS), 1.08–1.25 × 106 TCID50 mL-1 in VP-SFM, and 4.58–9.90 × 105 TCID50 mL-1 in the CDMs. These titers are similar to those reported for MeV produced in Vero cells in VP-SFM using unregulated systems [26]. It is notable that the previously reported results differed from our findings regarding MOI-dependent TOH. Although lower MOIs were used, the maximum MeV titers were reached earlier. However, this discrepancy may reflect the use of a different MeV strain, suggesting a re-evaluation of the data is needed for each individual strain. In both cases there is an MOI range that differs by one log level (MOI = 2 and 0.2 in our case, MOI = 0.1 and 0.01 previously [26]) but still shows a very similar MeV production profile over process time. Presumably, this reflects the mainly small differences in the time course of infected cells as well as host cell availability, as described above.
The subsequent loss of infectivity can be attributed to the virus inactivation rate, which is higher than its release rate. In addition to thermal inactivation, proteases released during cell lysis might damage MeV proteins, such as the surface proteins required for adsorption (H protein and F protein). Furthermore, metabolites and debris released during cell lysis accumulate in the supernatant over time, probably affecting the structure and aggregation of MeV, for example by changing the pH [16]. At MOIs of 0.2 and 2, the peak virus titer was accompanied by a drop in cell density (60–93% of Xmax). This suggests that a large quantity of virus particles is released by cell lysis. The titer increased almost continuously throughout the process with an MOI of 0.02. Given the limited timeframe, it remains unclear whether the virus titer would have increased had more time been available. The continued presence of infected cells in the cultures suggests that such an increase is likely. To achieve this, it would be necessary to ensure an adequate supply of nutrients and oxygen for MeV production and host cell survival, while maintaining optimal process conditions (e.g., pH and the concentrations of substrates and inhibitory metabolites).
The expectation that larger numbers of infected cells enhance the virus yield was not confirmed in our static batch cultivation experiment. This has also been shown in a host cell screening for MeV production, in which both adherent and suspension cells were examined. Although MeV infection was successful and nearly complete in most experiments (six of nine host cell types > 95% infected by MeV), the maximum MeV titers varied by up to 4 log levels. Vero cells proved to be the most productive host cells for MeV [19]. However, the extent to which the proportion of MeV-infected Vero cells affects the maximum MeV yield was not investigated here. Even if more infected cells were available for MeV production at an MOI of 0.02, the maximum yield of infectious MeV was either similar to or even 0.5–1.2 log10 lower than an MOI of 0.2 or 2. One potential explanation is that we only assessed GFP+ cells (compared to the mock-infected control). The different GFP intensities of GFP+ cells, which were higher at higher MOIs, were not taken into account because this would have complicated the presentation of the results. Higher GFP intensities could represent a greater viral load per cell and thus lead to higher MeV production rates. However, given that GFP intensity can also be influenced by other transcriptional and translational factors, the relationship to MeV production is not clear.
Our findings align with those of previous studies that have demonstrated a positive effect of higher MOIs on MeV yields [12,27]. For the propagation of MeV in MRC-5 cells, increasing the MOI from 0.001 to 0.01 increased yield from 7.5 × 105 to 7.5 × 106 TCID50 mL-1 [12]. In contrast, other reports suggest that higher MeV yields are achieved by reducing the MOI, but at the expense of longer process times [27,28]. Moreover, MeV production in Vero cells using a microcarrier-based STR process with various MOIs (0.0005, 0.001 and 0.02) resulted in MeV titers reaching almost the same order of magnitude [13].
The selection of an appropriate MOI involves a compromise between production time, virus yield and the required volume of virus stock [20] and also appears to be dependent on the MeV strain. Furthermore, any increase in cell lysis and the accumulation of inactivated MeV particles makes the medium more complex, which directly affects downstream processing. To generate a high yield of infectious MeV in a short process time while reducing the required MeV stock volume, we would recommend an MOI of 0.2 for the production of Edmonston B-derived MeV strains in static cultivation systems among the three MOIs we tested.
3.2.4. Additional Monitoring of the MeV Infection Course by Fluorescence Microscopy
In addition to flow cytometry and TCID50 assays, the influence of the MOI on static MeV production was also monitored by fluorescence microscopy. The MeV-induced cytopathic effect is illustrated in Figure 4. The fusion of the Vero cells into syncytia was evidenced by the aggregation of nuclei and the large-area GFP fluorescence, which was caused by MeV NSe ld-EGFP infection. In the late stage of the culture process, some areas of the original cell layer were completely lysed, as indicated by empty areas without stained cell nuclei.
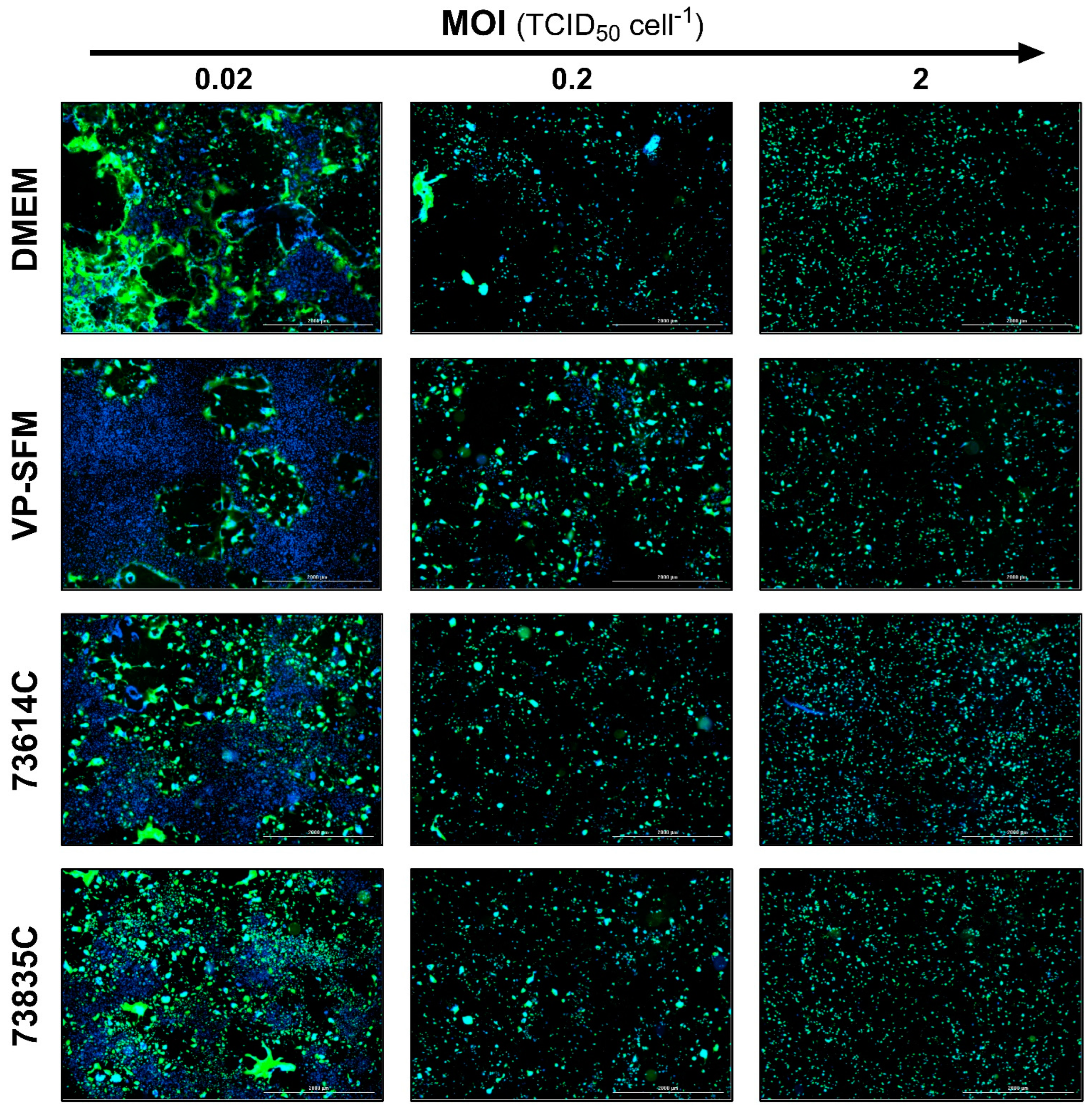
The levels of GFP expression at the three MOIs tested during the infection course are shown for the different culture media at 3 and 5 dpi (Figure 5 and Figure 6). At 3 dpi, the GFP signal was related to the MOI as anticipated (Figure 5).
At 5 dpi (Figure 6), the green fluorescence and cell number declined for all media at the highest MOI (2.0 TCID50 cell-1) compared to 3 dpi, which can be explained by cell lysis due to MeV replication and release. In contrast, the lowest MOI (0.02 TCID50 cell -1) showed a spread of infection, recognizable by larger GFP+ areas. In addition, “infection halos” were visible in the layer where cells were absent, but which was bordered by green fluorescent cells. In contrast, some cell-covered areas showed no green fluorescence.
These data suggest that MeV particles released in static cultivations tend to infect neighboring cells. Furthermore, cell-cell contacts, not only during the formation of syncytia but possibly also through the direct transmission of MeVs at the contact points, could play an important role in the spread of infection in static Vero cell cultures.
3.3. Comparison of MeV Production in SCM, SFM and CDMs
To assess the impact of the culture medium on MeV production in more detail, we compared the total cell density and proportion of infected cells as well as the MeV titer in four different media (Supplement 1).
3.3.1. Medium-Dependent Growth Kinetics of Non-Infected and Infected Vero Cell Cultures
As can be seen from the largely similar growth kinetics of the mock-infected control cells (Figure 2), each medium provided sufficient nutrients and sustained a favorable environment for host cell metabolism and proliferation for at least 6 days.
Infected cultures also showed comparable progress in terms of total cell density in all media at high MOIs. (Supplement 1). However, VP-SFM achieved a higher total cell density (~30% higher than DMEM or 73835C, and ~100% higher than 73614C) at an MOI of 0.02. Nevertheless, the number of GFP+ (MeV-infected) cells in VP-SFM during cultivation did not substantially exceed the numbers observed in the other media at any MOI. Taking into account methodological deviations, such as cell loss during sample preparation, the course of infected cells at MOIs of 0.2 and 2 was very similar in all four media. At an MOI of 0.02, the number of GFP+ cells peaked in all media 4–5 dpi, but the peak density in DMEM was 36.0–62.9% lower compared to the other media. As stated in Section 3.2, the number of infected cells was more stable in DMEM and the CDMs, whereas a sharp decline was immediately observed in VP-SFM.
3.3.2. Medium-Dependent Infection Kinetics
The influence of the medium on the progress of infection in the cultures was assessed by estimating the percentage of GFP+ cells (Supplement 1). The infection spread fastest in CDM 73614C (50% GFP+ cells reached after 1.1, 2.4 and 3.7 dpi at MOIs of 0.02, 0.2 and 2, respectively), closely followed by CDM 73835C (1.2, 2.6 and 3.9 dpi). In DMEM (10% FBS) and VP-SFM, the infection states were reached slightly later, with the biggest difference (+ 0.9 days offset) in VP-SFM with an MOI of 0.02. The slower spread of infection in VP-SFM explains the prolonged proliferation of host cells at an MOI of 0.02 compared to the other media.
The initially slower spread of infection in DMEM compared to the CDMs may reflect the neutralization of viruses by serum proteins, which are added to DMEM in the form of FBS. An interaction between serum proteins (e.g., complement system or acute phase proteins) and MeVs might prevent the virus binding to cell-surface receptors and thus inhibit further infection (especially at lower MOIs). However, given that a different MeV stock was applied in each medium, and the TCID50 assay has an error of ± 0.5 log10 [29,30,31], we anticipate some variation in terms of initially infected cells at the supposedly same MOI. Surprisingly, the proportion of GFP+ cells revealed consistent dynamics of infection spreading in SCM, SFM and the CDMs, as shown by the similar slopes of the sigmoidal fit (Section 3.2). This suggests that the infection course is to a great extent independent of the medium.
3.3.3. Medium-Dependent MeV Yield
Despite the partly different evolution and maximum number of infected cells, there were only small differences between the media in terms of the maximum infectious MeV titer, even when varying the MOI. The maximum titer was 0.61–1.08 × 106 TCID50 mL-1 at an MOI of 0.2 and 0.46–2.17 × 106 TCID50 mL-1 at an MOI of 2. However, the titer range was somewhat wider (0.09–1.08 × 106 TCID50 mL-1) at an MOI of 0.02. In VP-SFM, the maximum MeV yield was 0.5–1.0 log10 lower than in DMEM and the CDMs. Therefore, the high density of infected cells in VP-SFM and the subsequent sharp drop in cell count did not lead to a virus boost. Depending on the culture medium, the maximum MeV yield was observed after different process times. For example, at an MOI of 0.2, the optimum TOH in the CDMs was reached 1–2 days earlier (4 dpi) than in VP-SFM and DMEM. Interestingly, this correlated with the earlier spread of infection in the CDMs. MeV stability in all tested media can thus be considered very similar. Although the exact composition of the media is not known, the stability of the MeV titer in VP-SFM and the CDMs over time may reflect the presence of additives. Ideally, cell culture medium for the production of MeV should contain additives that protect virus particles against thermal and mechanical inactivation, and prevent virus aggregation [32,33]. The amino acid composition of the medium also influences MeV synthesis in HeLa cells [34].
Serum is an important source of vitamins, growth factors, hormones, amino acids, adhesion factors and cytokines in cell culture media [9]. The addition of 10% FBS resulted in a 0.5–1.5 log10 increase in the titer of the Edmonston MeV strain in Vero cells compared to medium without serum [28]. However, the extent to which serum has a positive effect on the MeV yield appears to depend on the culture medium. We observed no major detrimental effects on the MeV yield or process time when using VP-SFM or Merck CDMs compared to SCM. Even in a STR, MeV production in VP-SFM at low to moderate shear stress (below 0.25 N m-2) resulted in yields similar to production in SCM [15]. Furthermore, no significant difference in MeV production was observed in the CDMs Hektor and InVitrus (both Cell Culture Technologies, Switzerland) compared to VP-SFM [32].
4. Conclusion
We found that the combination of MOI and growth medium influences both MeV infection and release. The CDMs were as suitable as the SCM for MeV production, which overcomes all the negative effects of FCS during production (e.g., batch-to-batch variation and ethical and safety concerns surrounding the use of animal serum). The time course of MeV release was faster in the CDMs, especially in the low and medium MOI range, which was in agreement with the faster spreading of infection. Nevertheless, further investigation is required to assess the performance of the CDMs in dynamic bioreactors, specifically in flow conditions with higher shear forces, because serum proteins have been shown to protect the shear-sensitive MeV in this environment [15]. The potential for flow cytometry to detect Vero cells infected with the MeV NSe ld-EGFP would provide a deeper understanding of the production process. The ratio of infected cells can be monitored to determine the most appropriate MOI for an optimal space-time yield of infectious MeV. However, the use of this method to monitor MeV production in STRs may prove challenging, given that adherent Vero cells growing on microcarriers such as Cytodex 1 can be difficult to detach. The aforementioned method would be more applicable to Vero cells adapted for growth in suspension [35,36,37] (Eckhardt et al., data to be published). Fluorescence microscopy offered further insights in the process of MeV spreading and is recommended in carrier-based STR processes to monitor MeV infection and propagation efficiency during production. Our results confirmed that it is possible to transfer a static MeV production process from SCM to SFM or even CDM, resulting in comparable high MeV titers.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, D.E. and J.M.; investigation, J.M., D.E. and J.F., supported by J.P.K., I.S., L.E.W. and S.F.; resources, D.S. and P.C.; writing—original draft preparation, J.M.; writing—review and editing, D.E., D.S. and P.C.; visualization, J.M. and D.E.; supervision, D.S. and D.E; project administration, D.S. and P.C.; funding acquisition, D.S. and P.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Merck (Darmstadt, Germany) and by the research fund (“Sonderforschungsfond”) of the University of Applied Sciences THM.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank Merck (Darmstadt, Germany) for providing the chemically-defined cell culture media and Prof. Guy Ungerechts (National Center for Tumor Diseases, Heidelberg, Germany) for the MeV NSe ld-EGFP strain. We thank Richard M. Twyman for professional editing of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef]
- Loewe, D.; Dieken, H.; Grein, T.A.; Weidner, T.; Salzig, D.; Czermak, P. Opportunities to debottleneck the downstream processing of the oncolytic measles virus. Crit. Rev. Biotechnol. 2020, 40, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Galanis, E. Potential and clinical translation of oncolytic measles viruses. Expert Opin. Biol. Ther. 2017, 17, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving oncolytic viruses into the clinic: clinical-grade production, purification, and characterization of diverse oncolytic viruses. Mol. Ther. Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W.; Fisher, K.D. Oncolytic viruses: finally delivering. Br. J. Cancer 2016, 114, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Aref, S.; Bailey, K.; Fielding, A. Measles to the Rescue: A Review of Oncolytic Measles Virus. Viruses 2016, 8. [Google Scholar] [CrossRef]
- Noyce, R.S.; Richardson, C.D. Nectin 4 is the epithelial cell receptor for measles virus. Trends Microbiol. 2012, 20, 429–439. [Google Scholar] [CrossRef]
- Hsu, E.C.; Sarangi, F.; Iorio, C.; Sidhu, M.S.; Udem, S.A.; Dillehay, D.L.; Xu, W.; Rota, P.A.; Bellini, W.J.; Richardson, C.D. A single amino acid change in the hemagglutinin protein of measles virus determines its ability to bind CD46 and reveals another receptor on marmoset B cells. J. Virol. 1998, 72, 2905–2916. [Google Scholar] [CrossRef]
- Weiss, K.; Salzig, D.; Mühlebach, M.D.; Cichutek, K.; Pörtner, R.; Czermak, P. Key Parameters of Measles Virus Production for Oncolytic Virotherapy. American Journal of Biochemistry and Biotechnology 2012, 8, 81–98. [Google Scholar] [CrossRef]
- Msaouel, P.; Opyrchal, M.; Dispenzieri, A.; Peng, K.W.; Federspiel, M.J.; Russell, S.J.; Galanis, E. Clinical Trials with Oncolytic Measles Virus: Current Status and Future Prospects. Curr. Cancer Drug Targets 2018, 18, 177–187. [Google Scholar] [CrossRef]
- Russell, S.J.; Federspiel, M.J.; Peng, K.-W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O'Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin. Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef]
- Trabelsi, K.; Majoul, S.; Rourou, S.; Kallel, H. Development of a measles vaccine production process in MRC-5 cells grown on Cytodex1 microcarriers and in a stirred bioreactor. Appl. Microbiol. Biotechnol. 2012, 93, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Gerstenberger, J.; Salzig, D.; Mühlebach, M.D.; Cichutek, K.; Pörtner, R.; Czermak, P. Oncolytic measles viruses produced at different scales under serum-free conditions. Eng. Life Sci. 2015, 15, 425–436. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Salzig, D.; Weidner, T.; Czermak, P. High titer oncolytic measles virus production process by integration of dielectric spectroscopy as online monitoring system. Biotechnol. Bioeng. 2018, 115, 1186–1194. [Google Scholar] [CrossRef]
- Grein, T.A.; Loewe, D.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Aeration and Shear Stress Are Critical Process Parameters for the Production of Oncolytic Measles Virus. Front. Bioeng. Biotechnol. 2019, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Loewe, D.; Häussler, J.; Grein, T.A.; Dieken, H.; Weidner, T.; Salzig, D.; Czermak, P. Forced Degradation Studies to Identify Critical Process Parameters for the Purification of Infectious Measles Virus. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Grein, T.A.; Weidner, T.; Czermak, P. Concepts for the Production of Viruses and Viral Vectors in Cell Cultures. In New Insights into Cell Culture Technology; Gowder, S.J.T., Ed.; InTech, 2017; ISBN 978-953-51-3133-5. [Google Scholar]
- Grein, T.A.; Loewe, D.; Dieken, H.; Salzig, D.; Czermak, P. High titer oncolytic measles virus production process by integration of dielectric spectroscopy as online monitoring system. Biotechnol. Bioeng. 2017. [Google Scholar] [CrossRef]
- Grein, T.A.; Schwebel, F.; Kress, M.; Loewe, D.; Dieken, H.; Salzig, D.; Weidner, T.; Czermak, P. Screening different host cell lines for the dynamic production of measles virus. Biotechnol. Prog. 2017, 33, 989–997. [Google Scholar] [CrossRef]
- Kiesslich, S.; Kamen, A.A. Vero cell upstream bioprocess development for the production of viral vectors and vaccines. Biotechnol. Adv. 2020, 44, 107608. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Archiv f. experiment. Pathol. u. Pharmakol 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Ramakrishnan, M.A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 2016, 5, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, Y.E.; Rubingh, O.; Wijffels, R.H.; van der Pol, L.A.; Bakker, W.A.M. Improved poliovirus D-antigen yields by application of different Vero cell cultivation methods. Vaccine 2014, 32, 2782–2788. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yi, X.; Zhang, Y. Improvement of Vero cell growth in glutamate-based culture by supplementing ammoniagenic compounds. Process Biochemistry 2006, 41, 2386–2392. [Google Scholar] [CrossRef]
- Ellis, E.L.; Delbrück, M. The Growth of Bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Bacher, J.; Lali, N.; Steiner, F.; Jungbauer, A. Cytokines as fast indicator of infectious virus titer during process development. J. Biotechnol. 2024, 383, 55–63. [Google Scholar] [CrossRef]
- Baczko, K.; Lazzarini, R.A. Efficient propagation of measles virus in suspension cultures. J. Virol. 1979, 31, 854–855. [Google Scholar] [CrossRef]
- Scott, J.V.; Choppin, P.W. Enhanced yields of measles virus from cultured cells. J. Virol. Methods 1982, 5, 173–179. [Google Scholar] [CrossRef]
- The European Agency for the Evaluation of Medicinal Products. Note for guidance on virus validation studies: The design, contribution and interpretation of studies validating the inactiviation and removal of viruses. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/note-guidance-virus-validation-studies-design-contribution-and-interpretation-studies-validating-inactivation-and-removal-viruses_en.pdf (accessed on 23 April 2024).
- Mayer, V.; Frank, A.-C.; Preinsperger, S.; Csar, P.; Steppert, P.; Jungbauer, A.; Pereira Aguilar, P. Removal of chromatin by salt-tolerant endonucleases for production of recombinant measles virus. Biotechnol. Prog. 2023, 39, e3342. [Google Scholar] [CrossRef]
- Steppert, P.; Mosor, M.; Stanek, L.; Burgstaller, D.; Palmberger, D.; Preinsperger, S.; Pereira Aguilar, P.; Müllner, M.; Csar, P.; Jungbauer, A. A scalable, integrated downstream process for production of a recombinant measles virus-vectored vaccine. Vaccine 2022, 40, 1323–1333. [Google Scholar] [CrossRef]
- Eckhardt, D.; Bossow, S.; Klee, J.-P.; Boshof, B.; Ungerechts, G.; Czermak, P.; Salzig, D. Improved Production Strategies for Oncolytic Measles Viruses as a Therapeutic Cancer Treatment. In Bioprocess and Analytics Development for Virus-based Advanced Therapeutics and Medicinal Products (ATMPs); Gautam, S., Chiramel, A.I., Pach, R., Eds.; Springer International Publishing: Cham, 2023; pp. 375–405. ISBN 978-3-031-28488-5. [Google Scholar]
- Schlehuber, L.D.; McFadyen, I.J.; Shu, Y.; Carignan, J.; Duprex, W.P.; Forsyth, W.R.; Ho, J.H.; Kitsos, C.M.; Lee, G.Y.; Levinson, D.A.; et al. Towards ambient temperature-stable vaccines: the identification of thermally stabilizing liquid formulations for measles virus using an innovative high-throughput infectivity assay. Vaccine 2011, 29, 5031–5039. [Google Scholar] [CrossRef]
- Romano, N.; Scarlata, G. Amino acids requirements of measles virus in HeLa cells. Arch. Gesamte Virusforsch. 1973, 43, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Paillet, C.; Forno, G.; Kratje, R.; Etcheverrigaray, M. Suspension-Vero cell cultures as a platform for viral vaccine production. Vaccine 2009, 27, 6464–6467. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.F.; Guilbault, C.; Li, X.; Elahi, S.M.; Ansorge, S.; Kamen, A.; Gilbert, R. Development of suspension adapted Vero cell culture process technology for production of viral vaccines. Vaccine 2019, 37, 6996–7002. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-K.; Park, J.; Seo, D.-W. Suspension culture of Vero cells for the production of adenovirus type 5. Clin. Exp. Vaccine Res. 2020, 9, 48–55. [Google Scholar] [CrossRef]
Figure 1.
Growth kinetics of adapted Vero cells in static batch cultivation. Vero cells were seeded in six-well plates (5000 cells cm-2) and incubated at 37 °C in an 8% CO2 atmosphere. Cells from three wells per medium were detached using trypsin every day and counted using a Scepter 2.0 cell counter (n = 3, mean ± SD).
Figure 1.
Growth kinetics of adapted Vero cells in static batch cultivation. Vero cells were seeded in six-well plates (5000 cells cm-2) and incubated at 37 °C in an 8% CO2 atmosphere. Cells from three wells per medium were detached using trypsin every day and counted using a Scepter 2.0 cell counter (n = 3, mean ± SD).
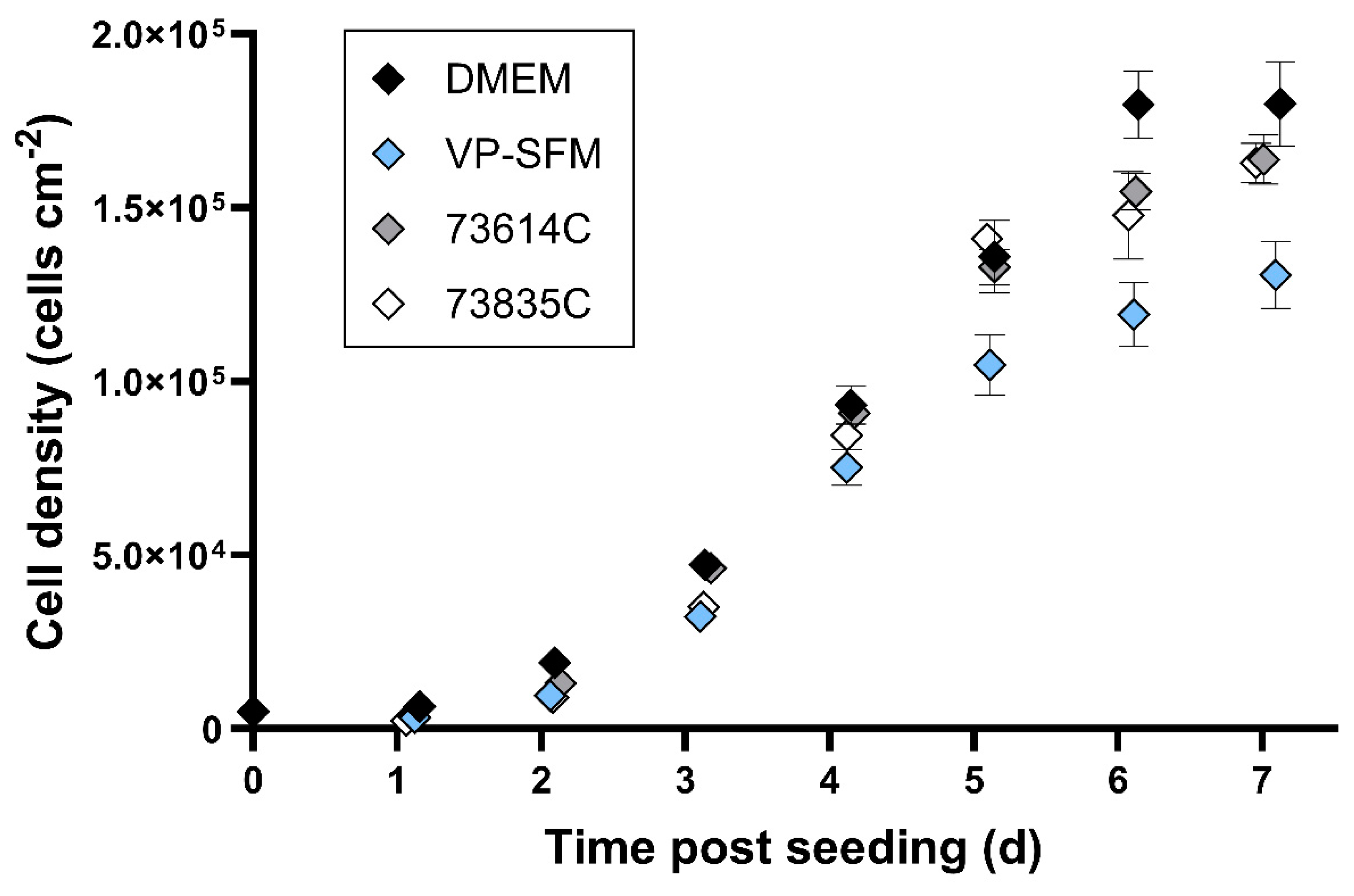
Figure 2.
Measles virus production in adherent Vero cells at different MOIs in six-well plates. After the infection of Vero cells with MeV, the cultivation temperature was reduced from 37 to 32 °C and a complete media exchange was carried out ~1 day later. The concentration of total cells and the ratio of infected cells (%GFP+ cells) in four media were determined by flow cytometry, allowing the recalculation of cell densities. Criterion GFP+: fluorescence intensity greater than the mock-infected control. Sigmoidal curve fitting of %GFP+ cells over time, forced through points [–0.82;0] and [0;0] (time points of cell seeding and MeV infection). MeV titers were determined using the cell-based 50% endpoint dilution method (TCID50 assay). LOD: limit of detection.
Figure 2.
Measles virus production in adherent Vero cells at different MOIs in six-well plates. After the infection of Vero cells with MeV, the cultivation temperature was reduced from 37 to 32 °C and a complete media exchange was carried out ~1 day later. The concentration of total cells and the ratio of infected cells (%GFP+ cells) in four media were determined by flow cytometry, allowing the recalculation of cell densities. Criterion GFP+: fluorescence intensity greater than the mock-infected control. Sigmoidal curve fitting of %GFP+ cells over time, forced through points [–0.82;0] and [0;0] (time points of cell seeding and MeV infection). MeV titers were determined using the cell-based 50% endpoint dilution method (TCID50 assay). LOD: limit of detection.
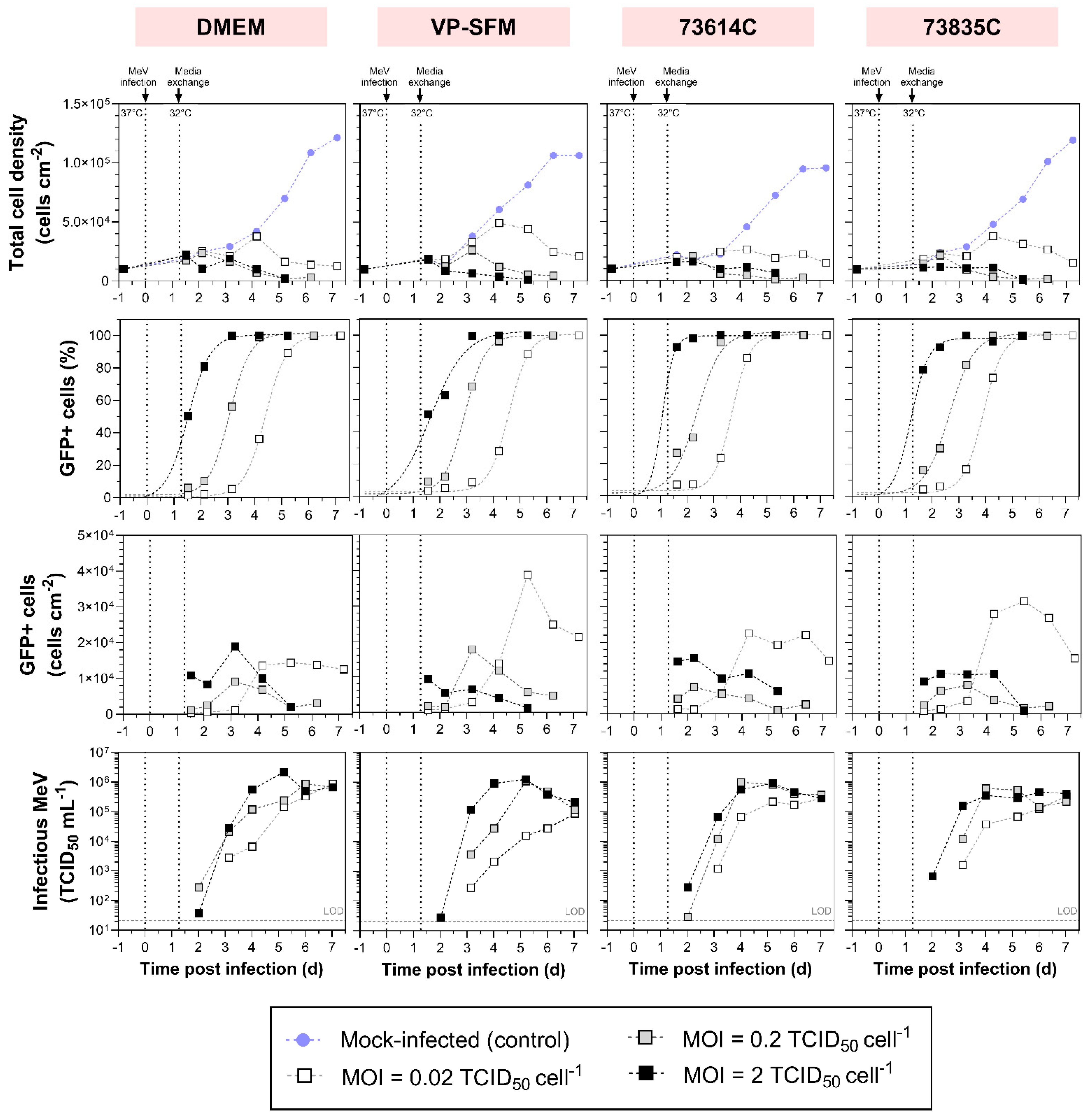
Figure 3.
Substrate and metabolite concentrations and LDH activity during the static production of MeV in DMEM (10% FBS), VP-SFM (4 mM Gln), CDM 73614C and CDM 73835C (both according Merck’s instructions). Following the infection of Vero cells with MeV, the cultivation temperature was reduced from 37 to 32 °C and a complete media exchange was carried out ~1 day later. Data points after medium exchange were not measured, but only copied from the origin data (–0.82 dpi). Solid lines (data plotted left): glucose, glutamine, pyruvate, glutamate. Dashed lines (data plotted right): lactate, ammonia, LDH activity.
Figure 3.
Substrate and metabolite concentrations and LDH activity during the static production of MeV in DMEM (10% FBS), VP-SFM (4 mM Gln), CDM 73614C and CDM 73835C (both according Merck’s instructions). Following the infection of Vero cells with MeV, the cultivation temperature was reduced from 37 to 32 °C and a complete media exchange was carried out ~1 day later. Data points after medium exchange were not measured, but only copied from the origin data (–0.82 dpi). Solid lines (data plotted left): glucose, glutamine, pyruvate, glutamate. Dashed lines (data plotted right): lactate, ammonia, LDH activity.
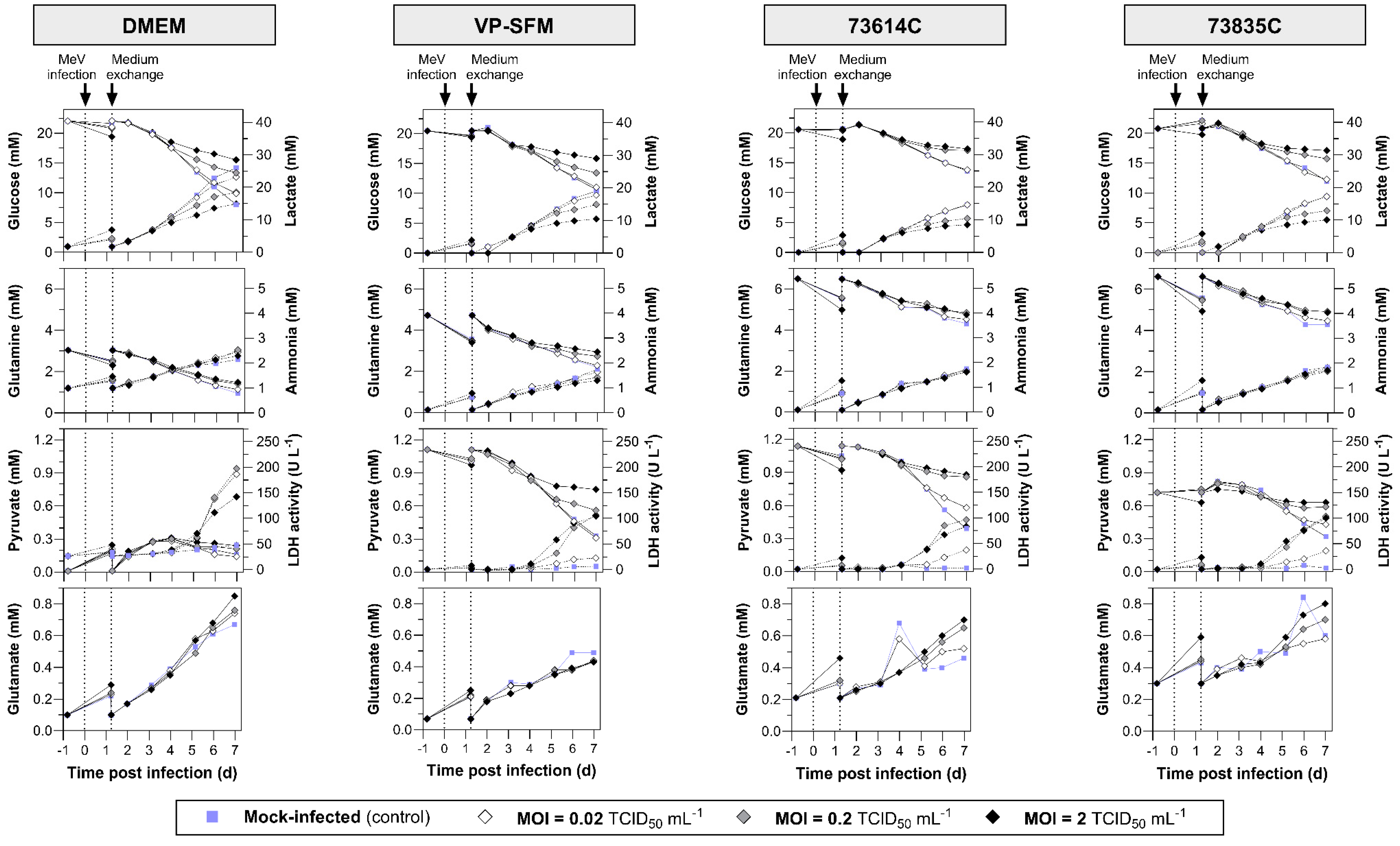
Figure 4.
Fluorescence image of a MeV-infected static Vero cell culture in CDM 73835C (MOI = 0.02 TCID50 cell-1, 5 dpi). The expression of eGFP due to MeV infection is shown in green and cell nuclei are counterstained with DAPI (blue) in this merged image captured using the Cytation 3 (objective magnification: 10×).
Figure 4.
Fluorescence image of a MeV-infected static Vero cell culture in CDM 73835C (MOI = 0.02 TCID50 cell-1, 5 dpi). The expression of eGFP due to MeV infection is shown in green and cell nuclei are counterstained with DAPI (blue) in this merged image captured using the Cytation 3 (objective magnification: 10×).
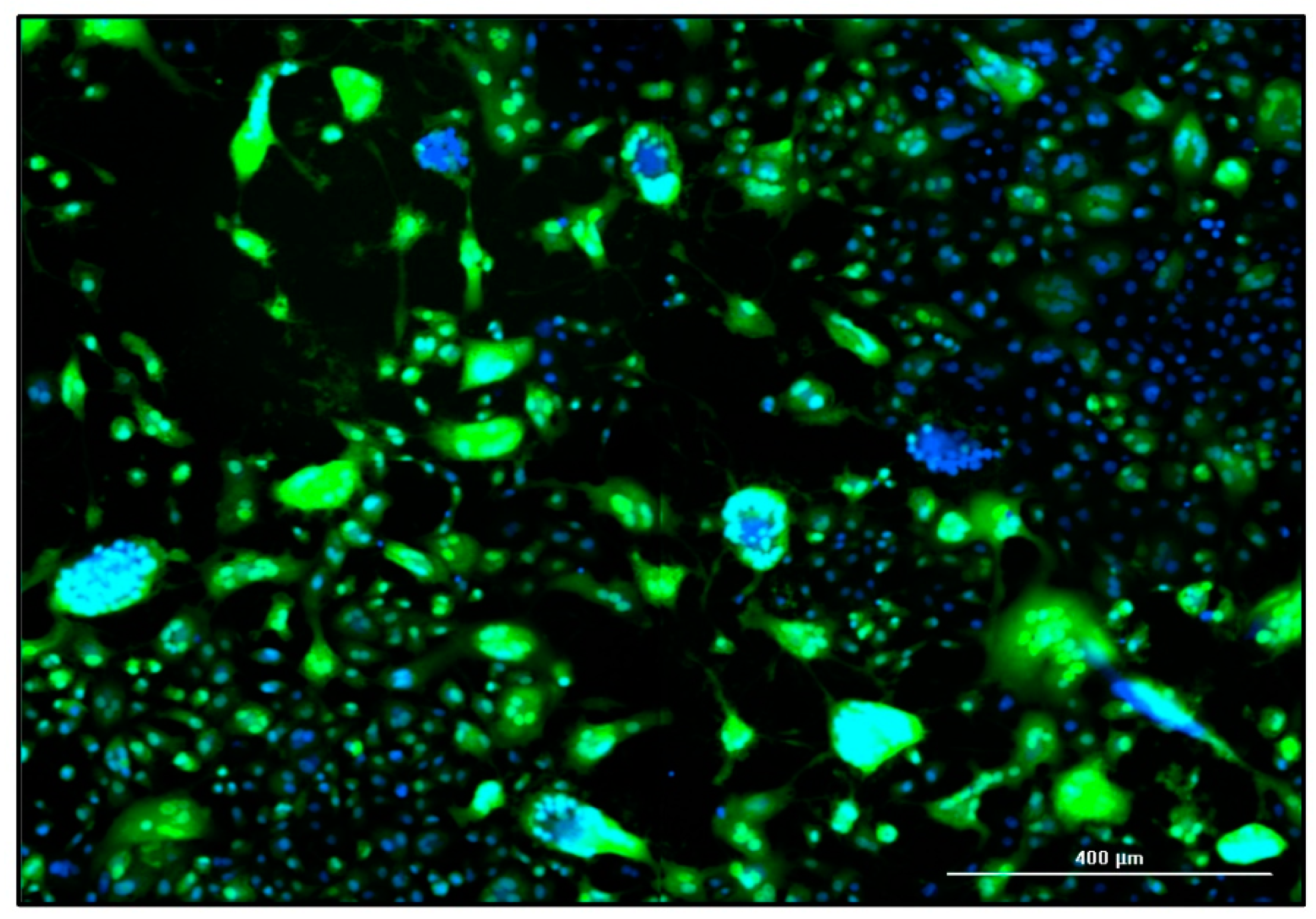
Figure 5.
Fluorescence images of MeV-infected static Vero cell cultures 3 dpi in SCM, SFM and CDMs at different MOIs. The expression of eGFP due to MeV infection is shown in green and cell nuclei are counterstained with DAPI (blue) in this merged image captured using the Cytation 3 (objective magnification: 2.5×, four (2×2) images per well, stitched).
Figure 5.
Fluorescence images of MeV-infected static Vero cell cultures 3 dpi in SCM, SFM and CDMs at different MOIs. The expression of eGFP due to MeV infection is shown in green and cell nuclei are counterstained with DAPI (blue) in this merged image captured using the Cytation 3 (objective magnification: 2.5×, four (2×2) images per well, stitched).

Figure 6.
Figure 6. Fluorescence images of MeV-infected static Vero cell cultures 5 dpi in SCM, SFM and CDMs at different MOIs. The expression of eGFP due to MeV infection is shown in green and cell nuclei are counterstained with DAPI (blue) in this merged image captured using the Cytation 3 (objective magnification: 2.5×, four (2×2) images per well, stitched).
Figure 6.
Figure 6. Fluorescence images of MeV-infected static Vero cell cultures 5 dpi in SCM, SFM and CDMs at different MOIs. The expression of eGFP due to MeV infection is shown in green and cell nuclei are counterstained with DAPI (blue) in this merged image captured using the Cytation 3 (objective magnification: 2.5×, four (2×2) images per well, stitched).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



