
Submitted:
27 September 2024
Posted:
30 September 2024
You are already at the latest version
Abstract
Obviously, the discussion of different factors that could have contributed to the origin of life and evolution represents a clear speculation, since there is no way to check the validity of the most of the related hypotheses on practice, as the corresponding events are not only already happened, but took place in a very distant past. However, there are a few undisputable facts obviously present at the moment, such as the existence of a broad variety of living forms and the abundant presence of intrinsically disordered proteins (IDPs) or hybrid proteins containing ordered domains and intrinsically disordered regions (IDRs) in all those living forms. Since it seems that the currently existing living forms have originated from a common ancestor, their variety represents a result of evolution. Therefore, one could ask a logical question of what role(s) those structure-less and highly dynamic but vastly abundant and multifunctional IDPs/IDRs might have in evolution. This study represents an attempt to consider various ideas pertaining to the potential roles of protein intrinsic disorder in origin of life and evolution.
Keywords:
intrinsically disordered proteins
; protein-protein interactions
; posttranslational modifications
; alternative splicing
; structural heterogeneity
; multifunctionality
; membrane-less organelles
; liquid-liquid phase separation
; origin of life
; evolution
1. Introduction: Who Are You, Mr. IDP?
For the most time of its fruitful existence, the protein science was ruled by the famous “lock-and-key” model proposed in 1894 by a German chemist Hermann Emil Louis Fischer (1852-1919) to describe the molecular mechanisms of enzymatic activity [1]. Here, the unique complementarity of the rigid structures of a substrate and an enzyme were suggested to define the efficiency of catalysis. Therefore, the specific functionality of a given protein was believed to be predetermined by the precise spatial positioning of its amino acid side chains and prosthetic groups, which, in its turn, was predestinated through a defined 3-D structure of this protein (the so-called structure-function paradigm). Despite its numerous limitations, this structure-function paradigm assuming that the protein functionality is directly linked to its unique rigid 3-D structure, acted as a ‘Big Bang’ that gave rise to the universe of modern protein science [2,3], a universe, where ordered proteins with well-defined structures conduct well-defined functions in a “unique sequence – unique structure – unique function” manner.
However, even the most structured proteins, instead of being rigid crystal-like entities, represent dynamic systems with different degree of conformational flexibility [3]. In fact, the 3-D structures of ordered proteins determined by X-ray crystallography and many other ensemble-based techniques represent averaged pictures [4]. This is because proteins are constantly involved in structural rearrangements originating from the fact that the conformational forces stabilizing the protein structure are weak and can be broken even at the ambient temperatures due to the thermal fluctuations [3,5], providing protein groups involved in such interactions with the ability to form new weak interactions with comparable energy [5]. Therefore, ordered proteins exist as dynamic ensembles of interchanging conformations, where structural rearrangements, being of relatively small scale, happen relatively fast (they occur typically in a time scale that is faster than the time required for structure determination by X-ray crystallography and many other physical techniques) [4].
It was also pointed out that not all structures deposited to Protein Data Bank (PDB) [6] are defined throughout the entire protein lengths and instead contain regions of missing electron density (i.e., portions of protein sequences missing from the determined structures) [7,8]. These regions of missing electron density, being flexible or disordered in nature, are incapable of the coherent scattering of X-rays. They are very common in the PDB, as less that 30% of PDB protein structures do not have them [9]. In addition to ordered proteins possessing different degrees of conformational flexibility and ordered proteins containing malleable/disordered regions of varying length, many biologically active proteins are characterized by a complete or almost complete lack of ordered structure under physiological conditions and exist as highly dynamic and heterogeneous conformational ensembles [5,10,11,12,13,14,15]. These IDPs and hybrid proteins containing ordered domains and various IDRs [16], are characterized by remarkable conformational heterogeneity and constitute a significant part of the protein kingdom [17,18,19,20].
Since IDPs/IDRs cannot spontaneously fold under the “physiological” conditions promoting folding of ordered proteins/domains, it was not surprising to find that the universe of protein amino acid sequences can be divided into at least two very different categories: sequences that naturally fold into ordered proteins or domains, and sequences that yield IDPs/IDRs [3,21]. Furthermore, the removal of the restrictions posed by the need to spontaneously fold into ordered structure to become functional dramatically increased the sequence space available to IDPs/IDRs in comparison with the sequence space available to foldable proteins and domains [3,22]. Therefore, the amino acid sequences of the structure-less and ordered proteins are dramatically different [10,12,13,23,24,25]. For example, IDPs with extended disorder (so-called native coils and native pre-molten globules) were shown to be characterized by a low content of hydrophobic residues combined with a high content of similarly charged residues [12]. At the more grained level, the IDPs/IDRs were documented to be significantly depleted in the so-called order-promoting amino acids (Cys, Trp, Tyr, Ile, Phe, Val, Leu, His, Thr, and Asn) and enriched in the disorder-promoting Ala, Gly, Asp, Met, Lys, Arg, Ser, Gln, Pro, and Glu residues [10,13,24,25,26,27,28]. These and other disorder-specific peculiarities of the amino acid sequences were used to design numerous computational tools for the reliable prediction of intrinsic disorder in proteins [10,13,17,29,30,31,32,33,34,35]. The use of those tools has opened a way to evaluate the natural prevalence of protein disorder, revealing that many proteins are expected to contain long IDRs and that the eukaryotic proteomes have a higher fraction of intrinsic disorder than prokaryotic proteomes [17,18,20,36,37,38,39,40]. It was also pointed out that these differences in the disorder distribution within the protein universe can be understood by taking into account the facts that IDPs/IDRs have evolved to have specific functions, being commonly involved in regulation, recognition, and signaling (see below), and that the eukaryotes and especially in muticellular eukaryotic organisms possess complex and well-developed regulation networks that might rely on the capability of IDPs/IDRs to perform the necessary regulatory functions [5,19,41,42]. In fact, being commonly involved in recognition, regulation, and control of various signaling pathways [41,42,43], IDPs/IDRs have unique functional arsenal that is parallel and complementary to the catalytic and transport functions of ordered proteins [24,44,45,46].
2. Roles of Intrinsic Disorder in Origin of Life
2.1. Prebiotic Life on the Earth: Intrinsic Disorder of the Extraterrestrial Peptides
Since glycine was detected among other molecules in comets, meteorites (see [47,48,49]), and in the interstellar medium [50], and since the oligoglycine can be synthesized on the surface of cold solid particles (cosmic dust) [51], one can assume that the extraterrestrial biomolecules contributed to the origin of life on Earth [52]. In fact, CO, C, and NH, which are the three most abundant species in the star-forming interstellar medium were shown to condensate on the surface of cold dust grains and form isomeric glycine monomers in a barrier-less manner, which then can polymerize to produce homo-polymeric peptides of different lengths even at low temperatures under astrophysically relevant conditions in the absence of irradiation or water [51]. Therefore, polypeptides of significant lengths, and not just elementary amino acids such as glycine, may be synthesized in rocky planets in the habitable zone and may have served as an important element when life as we know it, originated ~4 billion years ago (see [53]).
It is unclear if more complex heteropeptides can be synthesized via the mechanism proposed by the for the extraterrestrial polyglycine synthesis [51]. However, meteorites (particularly carbonaceous chondrites) were shown to contain various amino acids. For example, 52 different amino acids were found in the Murchison meteorite, among which 33 were unknown in natural materials, but eight were the amino acids found in terrestrial proteins [54]. Furthermore, a 4641 Da amino acid polymer predominantly containing glycine and some hydroxy-glycine and alanine [55] and a 2320 Da meteoritic protein hemolithin containing two glycine strands, each of 16-residues long, terminated by the iron atoms, and holding additional oxygen and lithium atoms [56] were found in the carbonaceous chondrite CV3 meteorites Acfer 086 and Allende.
Importantly, isomeric polyglycine-based peptides similar to ones of the extraterrestrial origin were strongly predicted to be intrinsically disordered [52]. Therefore, homopolypeptides that can be synthesized extraterrestrially from glycine via the pathway proposed by Krasnokutski et al. [51] or by some other yet unknown mechanisms cannot be ordered. Obviously, this is not a big surprise, as glycine included in such polypeptides, besides being the simplest amino acid, is considered as a disorder-promoting residue. It was also emphasized that such disordered polypeptides of the extraterrestrial origin can be present for long times due to the absence of proteases in the abiotic environment of the primitive Earth [52]. Of course, a peptide bind can be decomposed via the uncatalyzed hydrolysis involving the direct attack of water on the peptide bond. However, the half-time of such uncatalyzed hydrolysis is expected to be as long as 600 years [57]. Furthermore, since the atmosphere of the primordial Earth was reducing and had no molecular oxygen or other reactive oxides, the primordial ocean did not contain molecular oxygen or other reactive oxides as well, which can further slowdown the rate of the spontaneous hydrolysis of the primordial peptides [52]. Concluding this part it is tempting to hypothesize that the extraterrestrial IDPs might have contributed to the prebiotic origin of life on Earth [52]. More detailed description of this important concept will be discussed in the subsequent sections.
2.2. Prebiotic Life on the Earth: Intrinsic Disorder of the Primordial Proteins
The complex 3D structures of modern ordered proteins represent the result of lengthy molecular evolution. What then one can say about structures of the primordial proteins? It is clear that the chances for the first polypeptides that appeared in the primordial soup of the primitive Earth to have unique 3D structures are negligibly slim. Instead, with a very high probability, such polypeptides were intrinsically disordered. We can find indirect clues supporting the validity of this hypothesis while looking at some known facts. Although the Earth formed about 4.5 billion years ago and became cool enough to potentially spawn life around 4.2 billion years ago, the first fossils are dated to 3.85 billion years ago, raising a question of what was happening in those years in between. At the beginning of the 20th century, Alexander I. Oparin (1894-1980) [58] and John Burdon Sanderson Haldane (1892-1964) [59] proposed a model that constitutes a cornerstone of the theory of molecular evolution according to which some organic molecules could have been synthesized spontaneously from the gases of the primitive Earth atmosphere. Such abiotic production of organic molecules would require reducing atmosphere and ample supply of energy in a form of lightning and/or ultraviolet light. The validity of this idea was demonstrated thirty year later, when Stanley Lloyd Miller (1930-2007) and Harold Clayton Urey (1893-1981) conducted elegant experiments deservedly known now as the Miller-Urey experiments and showed that placing the non-organic compounds, such as water vapor, hydrogen, methane, and ammonia, which were believed to represent the major components of the atmosphere of the primordial Earth into a closed system and running a continuous electric current through the system, to simulate lightning storms believed to be common on the early Earth results in the appearance of various organic molecules including some amino acids [60,61]. Importantly, only about half of the modern amino acids was synthesized in these Miller-Urey experiments [60,61] suggesting that the first proteins on Earth may have contained only a few amino acids.
In line with these considerations, the biosynthetic theory of the genetic code evolution suggests that the genetic code evolved from a simpler form encoding fewer amino acids [62], likely in parallel with the invention of biosynthetic pathways for new and chemically more complex amino acids [63]. Peculiarities of the redundancy of the standard genetic code, where 20 amino acids are encoded by 64 codons, provide some support to the validity of this hypothesis. Here, despite the fact that the redundant codons encoding one amino acid may differ in any of their three positions, only the third position of some of such codons may be fourfold degenerate; i.e., represents a position, where all possible nucleotide changes are synonymous as they do not change the amino acid. If these peculiarities of the modern genetic code reflect its evolution, then it is likely that a doublet code preceded the triplet code, indicating that the third position was not used at all in the early genetic code. This means that this early code used 4×4=16 codons, thereby encoding 16 or fewer amino acids, if a termination codon is taken into account [64], indicating that evolutionary old and new amino acids can be potentially discriminated. These and many other observations were used by Edward N. Trifonov to propose the following consensus order of the appearance of the 20 amino acids on the evolutionary scene: G/A, V/D, P, S, E/L, T, R, N, K, Q, I, C, H, F, M, Y, and W [65]. Let’s look at this scale from the view point of protein intrinsic disorder, where residues can be ranged based on their order-promoting (or foldability) potential [10,13,24,25,26,27,28]. In fact, there are three scales that can provide ranking of the tendencies of amino acid residues to promote order or disorder. These are the Top-IDP scale (W, F, Y, I, M, L, V, N, C, T, A, G, R, D, H, Q, K, S, E, and P) [23], the DisProt-based scale (C, W, Y, I, F, V, L, H, T, N, A, G, D, M, K, R, S, Q, E, and P) [66], and the scale based on the average number of contacts per residue in the ordered proteins (W, F, V, I, L, M, V, C, H, R, T, Q, N, S, K, E, D, A, P, and G) [67]. Figure 1 represents comparison of these scales with the amino acid novelty scale proposed by Trifonov and shows that typically, older residues (e.g., G, D, E, P, and S) have a strong tendency to be disorder-promoting, whereas many newer amino acids (e.g., C, W, Y, and F) tend to be order-promoting.
Figure 2 provides another view of these correlations by showing modern genetic code complemented with information on the early and late codons (shown by light red and light blue colors respectively), and on corresponding disorder- and order-promoting residues as evaluated based on the DisProt scale (shown by red and blue colors, respectively). Codons with intermediate age and disorder-neutral residues are shown by light pink and pink colors, respectively. This presentation emphasizes that there is relatively good agreement between the “age” of the residue and its disorder-promoting capacity, with early residues being mostly disorder-promoting, and with the majority of late residues being mostly order-promoting. This conclusion follows from the abundance of the matching colors (light red-red, light blue-blue and light pink-pink). There are only two noticeable exceptions from this rule, V and L, which are early but order-promoting residues.
There are also some other facts that can provide further support to this idea. Since during the early stages of evolution primordial Earth was likely hotter than in our days, more stable codon-anticodon interactions (in the absence additional stabilizing interactions) would be more favorable under these early conditions with presumably higher temperatures [65]. Therefore, thermostability of the codons (measured as melting enthalpies (kcal/M) of the dinucleotide stacks corresponding to the first and second codon positions [68]) should have at least some correlation with the amino acid novelty scale. Figure 3A shows that such correlation is indeed observed, as early amino acids are typically encoded by more thermostable codons. Furthermore, Figure 3B shows that there is also inverse correlation between the codon thermostability and the disorder-promoting capability of amino acids, with disorder-promoting residues being encoded by more thermostable codons. One can also add another angle here and bring into consideration residue buriability, which provides a quantitative measure of the driving force for the burial of an amino acid residue in proteins and thereby contributes to the conformational stability of ordered proteins [69]. Figure 3C shows that the codon thermostability is inversely related to the buriability of the residues encoded by these codons, whereas Figure 3D illustrates the presence of a correlation between the buriability and novelty of residues, where the old residues are expected to be less buriable, whereas high buriability is characteristic to new residues. Finally, Figure 3E shows that the disorder-promoting residues are less buriable than the order-promoting residues.
Taken together, these observations indicate that the primordial polypeptides were intrinsically disordered, as evolutionary old amino acids, being encoded by more thermostable codons, were less buriable and mostly disorder-promoting. Although it is rather unlikely that these disordered primordial polypeptides possessed high catalytic activity [70], undoubtedly they played important roles in the origin of life and were crucial players in early evolution as well. In fact, as per the RNA world theory, the enzymatic activity evolution involved a transfer of catalytic power from catalytic RNAs (known as ribozymes, with an exceptional illustrative example being given by a ribosome, which is an RNA enzyme actually catalyzing the formation of the peptide bonds during protein translation, and which was defined as “a creature with a hundred of waggly tails” since its stability is supported by numerous ribosomal proteins, most of which are disordered in the unbound state and fold at binding to ribosome [71]) to ribonucleoproteins (RNP) and only then to proteins.[72] Based on these premises, in an organism which was the first to invent protein synthesis, the first proteins would be IDPs with some nonspecific RNA chaperone activities rather than specific catalysts [70,73]. However, in the RNA world, where misfolding-prone RNA [74,75] was used for both information storage and catalysis [76], the presence of such disordered RNA chaperones would be highly beneficial to their carriers providing them a significant selective advantage. Furthermore, the transferring of the enzymatic activity from RNAs (ribozymes) to proteins was a logical evolutionary step determined by the higher stability of protein structures than RNA structures and by the dramatic increase in the variability of physicochemical properties of amino acids in comparison with those of nucleotides. Since stable structure represents an important prerequisite for the proper spatial arrangement of catalytic residues, which is needed for the efficient catalysis [77], transferring the catalytic activities to proteins generated strong evolutionary pressure towards proteins with the well-folded structures.
3. Roles of Intrinsic Disorder in Evolution
3.1. Wavy Evolution of Intrinsic Disorder: Back to the Future or Blast from the Past
Figure 4 represents a snapshot of the distribution of intrinsic disorder in the modern proteomes [20] and illustrates the well-known fact that IDPs/IDRs are more prevalent in eukaryotes than in the less complex organisms [17,18,36,37,38,39,40]. As it was already pointed out, this plot representing dependence of the fraction of disordered residues on the proteome size has a well-defined gap between the prokaryotes and eukaryotes, as the majority of the prokaryotic species have 27% or fewer disordered residues, whereas almost all eukaryotes are predicted to have 32% or more disordered residues [20]. This observation indicates the existence of a complex step-wise correlation between the increase in the organism complexity and the increase in the amount of intrinsic disorder and suggests that the “origination” of intrinsic disorder was crucial for moving from the less complex prokaryotic to more complex eukaryotic cells, which contain many intricate innovations that seemingly arose all at once. Therefore, the sharp jump in the levels of proteome disorder parallels a morphological gap between the prokaryotic and eukaryotic cells, indicating that the increased usage of intrinsic disorder paralleled and likely was crucial for the increase in the morphological complexity of the cell [20].
These observations clearly indicate that IDPs/IDRs, with their ability to control various signaling, recognition, and regulation pathways and networks, act as crucial life maintainers in eukaryotic and especially muticellular eukaryotic organisms [5,19,41,42]. They also seem to suggest that the introduction of intrinsic disorder represents a relatively recent evolutionary “invention” that helped moving from prokaryotes to eukaryotes. However, as it was discussed in the previous section, more likely than not, primordial proteins/polypeptides were intrinsically disordered. Therefore, the increased use of intrinsic disorder in eukaryotic organisms clearly represents a blast from the past and can be considered as a “back to the future” event. This is illustrated by Figure 5 schematically showing that the pattern of the global evolution of intrinsic disorder is not straight, but wavy. Here, evolution starts with the highly disordered primordial proteins primarily acting as RNA chaperones. Since the competitive advantage of primitive cells was likely defined by the degree of their independence from the fluctuating environmental conditions linked to the ability to catalyze the production of all the constituents necessary for their independent existence, highly disordered RNA chaperones evolved into the ordered enzymes with well-folded unique 3D structures. At the subsequent evolutionary steps, protein intrinsic disorder was reinvented because IDRs/IDPs have specific features crucial for the regulation of complex processes. This prompted the development of more complex organisms from the last universal ancestor (i.e., the most recent organism from which all organisms now living on Earth descend [78,79]), eventually leading to the advent of the highly elaborated eukaryotic cells.
3.2. Intrinsic Disorder and LLPS: From Prebiotic Life to Origin of Cellular Life and Evolution
The aforementioned Miller-Urey experiments demonstrated that simple building blocks (including amino acids) required for the formation of complex macromolecules could form in environments seemingly mimicking early Earth [60,61]. These amino acids could have naturally assembled into polypeptide chains without the need for the complex biological machinery. The principle possibility of such prebiotic peptide synthesis has been studied for decades, with the researchers investigating different geological settings, such as volcanic geothermal fields, hydrothermal fields, sea-floor sediments, and tidal flats [80,81,82,83,84,85] and also looking at the effects of minerals, salts, ions, and pH [80,81,82,83]. Under highly alkaline conditions, peptide synthesis was favored, and the 20-mer oligopeptides (Gly20 – with no doubts, this was an IDP!) were synthesized [86]. However, such highly alkaline conditions could not support RNA synthesis due to the low stability of this biopolymer. Another attractive possibility was recently demonstrated in the experiments conducted by Yuki Sumie, Keiichiro Sato, Takeshi Kakegawa, and Yoshihiro Furukawa, who have shown that boric acid can catalyze polypeptide synthesis under neutral and acidic conditions leading the to the appearance of 39 residue-long glycine polypeptides (Gly39 – IDP again!) [87]. These observations suggested that in the primordial Earth, polypeptides and proto-proteins could be spontaneously formed from the assembled amino acids in the coastal areas of ancient small continents and islands rich in boric acid [87]. Furthermore, it was indicated that “the same conditions would allow for the formation of RNAs and interactions of primordial proteins and RNAs that could be inherited by RNA-dependent protein synthesis during the evolution of life” [87]. These experiments provided important clues on how early chemistry could have evolved into self-replicating structures. Importantly, the phase separation of primitive macromolecules into liquid coacervates was proposed in the 1920s, by Alexander I. Oparin as the first step in the origin of life [58,88].
Therefore, it is likely that primordial IDPs in general (and polyglycine in particular), liquid-liquid phase separation (LLPS, see below), and membrane-less organelles (MLOs, see blow) played crucial roles in the prebiotic evolution. In fact, it was pointed out that polyglycine with its ability to phase separate, form membrane-less droplets, and amyloid accretions, very likely contributed to organization of the protocell domains, facilitation of the evolution of the genetic code, and the overall transition of the pre-life to the cellular life [89]. IDPs in the form of extraterrestrial polypeptides or the primordial IDPs abiotically synthesize on the early Earth could cause emergence of self-organizing systems that evolved over time following natural selection [90,91,92]. Consistent with this hypothesis, a recent study by Matsuo and Kurihara [93] showed that under appropriate conditions, peptide generation and self-assembly occurs concurrently and can give rise to a proliferating peptide-based droplet through liquid-liquid phase separation in water. Furthermore, it was observed that the droplets experienced a steady growth-division cycle by periodic addition of monomers through autocatalytic self-reproduction [93]. It was also emphasized that LLPS “may represent a primordial mechanism for functional self-assembly of relatively unevolved molecular assemblies in the early stages of the evolution of life” [94].
LLPS-driven primordial coacervate formation did not wane during evolution. Instead, it seems that its fate is similar to that of IDPs. This is reflected in the fact that although different MLOs are found in the cells of all kingdoms of life, the variability of these biomolecular condensates is dramatically increased in eukaryotic cells, as most of the 100+ currently known MLOs/BCs are of eukaryotic origin [95]. A very important aspect related to the functionality of IDPs and IDRs is their crucial role in the regulation and control of LLPS, an important process associated with the biogenesis of various MLOs and biomolecular condensates (BCs) [94,96,97,98,99,100,101,102,103,104,105,106]. In fact, more than a hundred of different MLOs/BCs can be found in the cytoplasm, nucleus, mitochondria (and chloroplasts) of the eukaryotic cells, as well as in the cytoplasm of bacteria and archaea, and, likely, in viruses [95], where they represent “an intricate solution of the cellular need to facilitate and regulate molecular interactions by physically isolating target molecules in specialized compartments in a reversible and controllable way” [102]. IDPs/IDRs are central constituents of all the MLOs investigated so far [98,101,102,107,108,109,110], as their structural plasticity and capability to be involved in multivalent, stochastic, weak, palpation-like interactions are crucial for LLPS leading to the spontaneous separation of a homogeneous solution into two distinct immiscible liquids, or “phases”: a dense phase, and a dilute phase, both characterized by high water content and not separated by the membranes. As a result, MLOs always contain IDPs despite the fact that they differ from each other by the specific sets of their resident proteins [102]. It seems that formation of MLOs/BCs often represent a way of the intracellular compartmentalization of IDPs/IDRs [101,102,108,111,112]. Being liquid in nature, MLOs are characterized by high levels of internal dynamics [94,96,113,114,115,116,117], thereby representing fluid disorder-based ensembles. Since MLOs can be formed on the liner cellular structures, such as chromatin and cytoskeleton, or in/on the membranes, or in the bulk of the nucleoplasm/cytoplasm/matrix/stroma, they are classified as 1D, 2D, or 3D assemblages that can influence each other, thus, representing an important way of the intracellular communication and regulation [118].
It is clear that the protein intrinsic disorder, biological phase separation, and MLO phenomena are interlinked [102,106,107,118,119], since LLPS of specific IDPs is required for the formation of many (if not all) MLOs [98,102,111,120,121,122,123,124,125]. It was pointed out that this IDP/IDR-LLPS-MLO interconnection is redefining the organizational principles of living matter from a rather mechanistic model, where functions of proteins are determined by their rigid globular structures and where intracellular processes occur within the rigid membrane-encapsulated organelles, to a new model, where highly dynamic "biological soft matter" (IDPs and MLOs) positioned at the “edge of chaos” represents a critical foundation of life and defines complexity and evolution of the living things [107].
3.3. Intrinsic Disorder in Nucleic Acid-Binding Proteins
The text book truism defines genetic programing as a classic molecular biology dogma, where genetic information flows from DNA to RNA to protein. However, it is clear now that this straightforward DNA → RNA → protein information flow, being an oversimplification, is mostly applicable to simple organisms. In fact, using it, one can understand how E. coli genome work, as bacterial genomes mostly contain information required for making proteins (typically, ~90% of bacterial genomes are responsible for protein coding). However, the eukaryotic genomes are immensely more complex, as reflected in the facts that genes of higher organisms represents complex mosaics of coding (exons) and noncoding sequences (introns that are removed from the messenger RNA during the process of splicing and can be extraordinarily large, accounting for the majority of the DNA sequence in human genes [126]), all of which are transcribed [127,128,129], with exons covering around the 2.8% of the human genome [126]. Curiously, although most of the non-coding DNA in the eukaryotic proteome was considered non-functional (therefore termed “junk DNA” [130,131]), it was eventually shown that the vast majority (at least 80%) of the human and mouse genomes are in fact transcribed and have assign biochemical functions [132,133]. The majority of the genome sequences conserved between humans and other mammals correspond to the non-coding intergenic and intronic regions, rather than in the protein-coding exons themselves, thereby indicating that these non-coding sequences have critical roles in development and cellular processes. Furthermore, the relative amount of non-coding sequences was shown to increase consistently with the organism complexity [133], indicating that although bacterial genomes are mostly dedicated to making proteins, whereas eukaryotic genomes are mostly dedicated to production of noncoding RNAs with various regulatory functions. Therefore, especially in the complex organisms, RNA does not only acts as a passive mostly linear messenger between DNA and protein but is actively involved in the regulation of genome organization and gene expression [134]. In doing that, RNA can fold into specific 3D structures, which are complex and can be allosterically responsive, and which “can both recruit generic effector proteins and guide the resulting complexes sequence-specifically to other RNAs and DNA” [134].
Obviously, most of the regulatory RNA functions are conducted in close conjunction with the RNA-binding proteins (RBPs), which are intimately involved in regulation of gene expression, post-transcriptional regulations, and protein synthesis, as well as govern the maturation and fate of their target RNA substrates [135,136]. Furthermore, RBPs establish a specific network complementing a network regulating gene activity and differently organizing RNA transcripts in different tissues. The global importance of RBPs is reflected in the fact that human proteome contains at least 1,542 such proteins [135,136], indicating that RBPs represent a third major protein group in human cells in addition to soluble globular proteins and membrane proteins. Based on the comprehensive bioinformatics analysis of ∼548,000 proteins forming nucleiomes (i.e., sets of nucleic acid binding proteins) in 1121 species from Archaea, Bacteria, and Eukaryota it was concluded that the entire nucleiome is enriched in intrinsic disorder, as evidenced by significantly increased intrinsic disorder content in DNA- and RNA-binding proteins relatively to other proteins in corresponding proteomes [137]. This global analysis supported conclusions of earlier studies focused on specific families and classes of DNA- or RNA-binding proteins, with some of the illustrative examples of intrinsically disordered DNA- or RNA-binding proteins being histones [138], ribosomal proteins [71], transcription factors [139,140,141], and proteins involved in the biogenesis and action of yeast [142] and human spliceosomes [143]. Furthermore, focused bioinformatics analysis of the prevalence of intrinsic disorder in human RBPs binding to six common RNA types: messenger RNA (mRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), non-coding RNA (ncRNA), ribosomal RNA (rRNA), and internal ribosome RNA (irRNA) revealed that although RNA-binding proteins are generally enriched in intrinsic disorder, the disorder propensity is unequally distributed across proteins that bind different RNA types [144]. In fact, although the mRNA-, rRNA-, and snRNA-binding proteins were predicted to be significantly enriched in disorder, the proteins that interact with ncRNA and irRNA were not enriched in disorder, and the tRNA-binding proteins were significantly depleted in disorder [144].
4. Intrinsic Disorder as Means for Increasing the Proteome Complexity
4.1. Alternative Splicing
Alternative splicing is an important process by which two or more mature mRNAs are produced from a single mRNA by the inclusion and omission of different segments [145,146], and which therefore serves as an important mechanism for enhancing protein diversity in multicellular eukaryotes [147]. For example, tissue specificity of many proteins is achieved via the alternative splicing. The process is very common especially in higher eukaryotes, with between 35 and 60% of human genes yielding protein isoforms by means of alternatively spliced mRNA [148,149,150]. It was hypothesized that alternative splicing affects diversity of protein functions, such as protein-protein interactions, ligand binding, and enzymatic activity [151,152,153]. In the multicellular organisms such added protein diversity from alternative splicing is important for tissue-specific signaling and regulatory networks.
The aforementioned fact that the spliceosomal RBPs are enriched in intrinsic disorder [142,143] reflects crucial importance of IDPs/IDRs in the splicing of the eukaryotic protein-encoding mRNAs, a process by which a spliceosome removes the non-coding regions (introns) from a pre-messenger RNA (pre-mRNA) transcript and joins the coding regions (exons) to create mature mRNA. Since during splicing, exons from the same gene can be joined in different combinations, leading to different, but related, mRNA transcripts, and since these alternatively spliced mRNAs can be translated into different proteins with distinct structures and functions, IDP-containing spliceosomes play crucial roles in the alternative splicing-driven increase in the proteome complexity. Furthermore, because of their intrinsically disordered nature, many spliceosomal RBPs possess several unrelated functions; i.e., have an ability to moonlight, whereas some spliceosomal RBPs drive LLPS and formation of various MLOs via interaction with RNA. To illustrate disorder status of some of such spliceosomal intrinsically disordered RBPs, Figure 6 represents AlphaFold-generated 3D structural model for one of the moonlighting RBPs involved in the regulation of alternative splicing in nervous system, RNA binding protein fox-1 homolog 2 (RBFOX2; UniProt ID: O43251), which besides regulating the alternative splicing events by binding to 5'-UGCAUGU-3' elements can also act as a negative regulator of the human estrogen receptor (ER) signaling and play a role in some ovarian cancers [154]. Figure 6B represents a per-residue intrinsic disorder profile generated by RIDAO and shows that human RBFOX2 is predicted to have high levels of intrinsic disorder, especially in its N-terminal region preceding the RNA recognition motif (RRM, residues 121-197). Figure 6C shows disorder profile for the spliceosomal RBP serine/arginine repetitive matrix protein 2 (SRRM2, UniProt ID: Q9UQ35), that serves as a component of the minor spliceosome and is thereby required for pre-mRNA splicing but is also involved in the biogenesis of nuclear speckles (NS), which are among the most prominent biomolecular condensates [155]. Figure 6C leaves no doubts that SRRM2 is an extremely disordered protein. Curiously, region comprising residues 197-259, which is sufficient for RNA binding is predicted to be mostly disordered as well.
Importantly, IDPs/IDRs are not only crucial for the control and execution of alternative splicing of precursor pre-mRNAs, but also have vital role on another side of this phenomenon, as protein regions affected by alternative splicing of pre-mRNA are enriched in intrinsic disorder [158]. The fact that alternatively spliced segments of mRNAs mostly encode for IDRs provides important means for avoiding potential conformational catastrophe. This is because in ordered proteins capable of spontaneous folding, most of the amino acid sequence contributes to the folding process and is involved in structural stability, as the specific sequence determines which interactions can form between amino acid residues, ultimately shaping the 3D structure of a protein. In other words, the information containing in a protein amino acid sequence determines its unique 3D structure and thereby acts as a specific protein folding code. Therefore, it is likely that the removal of a piece of an amino acid sequence of a foldable protein containing a part of the said folding code (e.g., as a result of alternative splicing of the corresponding mRNA) would distort the capability of a protein to spontaneously fold in a right structure, causing the aforementioned conformational catastrophe reflected in protein misfolding, aggregation, and associated issues. However, no conformational catastrophe is expected if protein/region is intrinsically disordered, as a removal of a piece with “no structure” would have much of effect on remaining “no structure”. On the other hand, it was proposed that associating alternative splicing with protein disorder enables the time- and tissue-specific modulation of protein function [158]. Since IDRs are frequently utilized in protein binding regions, having alternative splicing of pre-mRNA coupled to IDRs can define tissue-specific signaling and regulatory diversity [158]. Furthermore, since regulatory and signaling elements of IDPs/IDRs can be as short as just a few residues, and since functionally important segments can be located within the IDRs with a high density, functionality of IDPs/IDRs can be completely rewired via the alternative splicing [158]. Therefore, a linkage between alternative splicing and signaling via IDRs represented one of the possible molecular mechanisms leading to the origin of cell differentiation, which ultimately gave rise to multicellular organisms [158].
4.2. Posttranslational Modifications
In addition to the aforementioned alternative splicing complexity of a proteome relative to its encoding genome is known to be dramatically increased via various posttranslational modifications (PTMs) of proteins. These spontaneous or enzymatically catalyzed chemical changes of a polypeptide chain happen after DNA has been transcribed into RNA and translated into protein and can be reversible or irreversible. PTM-related increase in the proteome complexity is determined by the capability of PTMs to extend the range of amino acid structures and physico-chemical properties thereby leading to the diversification of protein structures and functions [159]. It is emphasized that because of various PTMs proteins might contain more than 140 physico-chemically different residues despite the fact that 20 primary amino acids are typically encoded by DNA [159]. It was also indicated that there are as many as 300 physiologically relevant PTMs in higher eukaryotes [160]. Although all amino acid side chains can serve as PTM targets, most commonly protein PTMs are found at side chains that can act as either strong (C, M, S, T, Y, K, H, R, D, and E) or weak (N and Q) nucleophiles, whereas the remaining residues (P, G, L, I, V, A, W, and F) are rarely involved in enzymatically-catalyzed covalent modifications of their side chains [159]. Furthermore, since some commonly observed PTMs (e.g., phosphorylation and glycosylation) are readily reversible by the action of specific demodifying enzymes, the interplay between the corresponding modifying and demodifying enzymes provides important means for rapid and economical control of protein function.
The overall importance of PTMs in various aspects of cellular “life” of proteins is reflected in the fact that as much as 5% of the eukaryotic genomes are expected to encode PTM-related enzymes [160]. In fact, some PTMs are known to regulate the process of protein folding, whereas other PTMs control protein targeting to specific subcellular compartments and interaction with ligands or other proteins, and still other PTMs manage protein functional states affecting catalytic activity of enzymes or the signaling potential of proteins in various signal transduction pathways [161,162]. It is estimated that phosphorylation/dephosphorylation cycles originating from carefully regulated protein kinase and phosphatase activities control functions of one-third of eukaryotic proteins [163]. Not surprisingly, eukaryotic protein kinases constitute one of the largest protein families, where yeast, mouse and human kinomes include 119, 540, and ~520 kinases, human genome contains than 150 genes encoding phosphatases, whereas there are 1019 kinase- and 300 phosphatase-coding genes in Arabidopsis thaliana [163]. Functionality of some proteins is controlled by multiple different PTMs that can act individually or synergistically to fine-tune molecular interactions and modulate overall protein activity and stability [164]. An illustrative example of well-known multi-PTM proteins is given by a family of nuclear IDPs, histones, that are known to undergo acetylation, ADP-ribosyation, methylation, phosphorylation, SUMOylation, and ubiquitylation at different stages of their function [138]. Although for a long time, the N-terminal tails of the core histones containing an extraordinary number of different PTMs were known to play important roles in the nucleosome dynamics and related gene expression and transcription [165], over 30 PTMs have been reported in the core domains of these proteins as well [166].
Importantly, most enzymatically-catalyzed PTMs have intimate connections to protein intrinsic disorder, as PTM sites targeted by modifying enzymes are commonly placed within IDRs. This is illustrated by phosphorylation, for which bioinformatics analysis revealed that many protein phosphorylation sites were located in regions that were structurally characterized as IDRs [167,168]. Furthermore, there is a high correspondence between the prediction of disorder and the occurrence of phosphorylation [169], and amino acid compositions, sequence charge, complexity, and hydrophobicity, as well as many other sequence features of the regions adjacent to phosphorylation sites are very similar to those of IDRs [169]. In addition to phosphorylation, several other PTM types, such as acetylation, fatty acid acylation, methylation, protease digestion, and ubiquitination, have also been observed to preferentially occur within IDRs [45,167,168,170]. These observations indicate that in eukaryotic cells, localization of sites targeted for various PTMs show strong preference for IDRs, making these sites easily accessible to modifying enzymes and explaining the functional promiscuity of those enzymes, where a single enzyme could bind to and modify a wide variety of protein targets.
4.3. Intrinsic Disorder, Structural Heterogeneity, Multifunctionality, and Binding Promiscuity
Importantly, protein intrinsic disorder has multiple flavors, as proteins have different levels and depth of disorder, and different parts of a protein can be (dis)ordered to different degree [42]. This heterogeneity of disorder can be summarized rephrasing the famous opening line of Leo Tolstoy's novel Anna Karenina: “All ordered proteins are alike; each disordered protein is disordered in its own way.” In fact, IDPs/IDRs can exist in the extended (coil- or pre-molten globule-like) or collapsed (molten globule-like) forms [2,5,12,13,15,171,172,173,174], and an IDP/IDR can be more or less compact and possess smaller or larger amounts of flexible secondary/tertiary structure [2,5,12,13,174,175]. Furthermore, a typical IDP/IDR is not structurally homogeneous and instead might contain a multitude of potentially foldable, partially foldable, differently foldable, or not foldable at all structural elements [3,22], indicating that foldability (or structure-coding potential) is non-homogeneously distributed within the amino acid sequences of a protein. One should also keep in mind that this distribution of differently (dis)ordered regions is constantly changing in time, and a given segment of a protein molecule can potentially show different structures or lack of structure at different time points [3,22].
Therefore, protein structure represents a highly dynamic and very heterogeneous entity, where not only the entire protein molecule is expected to be disordered to different degrees, but various protein segments (even rather short ones) can be differently disordered as well [3,22,109,176,177,178]. Such mosaic structural architecture of a protein molecule can be considered as a set of foldons (regions capable of spontaneous folding), non-foldons (segment that do not fold), semi-foldons (regions that are always in a semi-folded state), inducible foldons (segments that can gain structure (at least partially fold) at interaction with binding partners), inducible morphing foldons (regions capable of folding to the different structures at interaction with different binding partners), and unfoldons (important less stable parts of ordered proteins that must unfold (or undergo order-disorder transition, at least partially) in order to make protein active) [3,22,109,176,177,178]. The distribution of these variously (dis)ordered segments (foldons, non-foldons, inducible foldons, inducible morphing foldons, semi-foldons, and unfoldons) is constantly changing in time, and the entire protein has a highly dynamic and morphing structure, which is not rigid or crystal-like [3,22,109,177,178]. Furthermore, many proteins exist as complex structural hybrids possessing ordered and differently disordered domains, thereby defining another level of structural heterogeneity crucial for their functions [16]. Therefore, it is clear that the classification of proteins as ordered and disordered is an obvious oversimplification, as the structure-disorder space of a protein represents a continuum, with no obvious boundary between order and disorder [3,177].
It is clear that such complex, highly dynamic, mosaic-like structural organization of proteins is also reflected in complex disorder-based functionality of proteins, as all the differently (dis)ordered structural segments of proteins (foldons, non-foldons, inducible foldons, inducible morphing foldons, semi-foldons, and unfoldons) might have very different functions. Furthermore, since all those foldons, semi-foldons, non-foldons, inducible foldons, inducible morphing foldons, and unfoldons can be found within one protein molecules, one could clearly see that a protein with such heterogeneous structure is inherently multifunctional. Therefore, the aforementioned protein structural continuum defines protein multifunctionality. These considerations constitute a basis of a “protein structure-function continuum” model, where a functional protein exists as a dynamic conformational ensemble characterized by a broad spectrum of structural features and possessing different functionalities, and which provides a global link between the protein structure and function [179].
Among the important functional features of IDPs/IDRs residing on their lack of stable structure are their ability to serve as hub proteins; i.e., nodes in protein-protein interaction networks that have a very large number of connections to other nodes [180,181,182,183,184,185,186], to bind partners with both high specificity and low affinity [187], to be engaged in promiscuous interactions with unrelated partners such other proteins small molecules, and nucleic acids [188], to contain molecular recognition features (MoRFs), which are short binding regions located within longer disordered regions that can fold at interaction with a partner [180,189,190,191], to adopt different structures upon binding to different partners [10,188,192,193,194,195,196], to form fuzzy complexes, where a significant part of an IDP continues to be disordered even in the bound state outside the binding interface [158,197,198,199,200,201,202], to act as dynamic and sensitive “on-off” switches [199], and to be able to return to their highly dynamic and pliable conformations after the completion of a particular function [3,22].
Disorder-based interactions are commonly of combinatorial and promiscuous in nature, and such combinatorial and promiscuous interactivity define multifunctionality of IDPs/IDRs. An illustrative example of this concept is given by the GPCR-G-protein signaling system, which in humans, includes more than 800 various G-protein-coupled receptors (GPCRs) [203,204,205,206] and a large set of intracellularly located guanine nucleotide-binding proteins (G-proteins), which are heterotrimers composed of α, β, and γ subunits, with their Gα subunit being diversified even further, as there are four major families of the (Gαs, Gαi, Gαq, and Gα12) encoded by 16 human genes [205,207,208]. Furthermore, complexity of this system goes far beyond a multitude of pair-wise ligand-GPCR and GPCR-G-protein interactions, as one GPCR can recognize more than one extracellular signal and interact with more than on G-protein and one ligand can activate more than one GPCR, and multiple GPCRs can couple to the same G-protein [209]. The biological importance of this system cannot be overemphasized, as it recognizes a multitude of extracellular ligands and triggers a variety of intracellular signaling cascades in cellular responses to hormones neurotransmitters, ions, photons, and other environmental stimuli, and are responsible for vision, olfaction, and taste. In fact, more than a 1000 of natural and artificial extracellular ligands, ranging from photons to amines, lipids, nucleotides, organic odorants, peptides, and proteins can interact with and activate GPCRs [206,207], and these signals are used to initiate a wide spectrum of intracellular signaling cascades via interaction of an activated GPCR with a Gα subunit, which is a member of one of the four major Gα families. This results in the activation or modulation of various downstream effector proteins and key secondary messengers [207,210,211]. The combinatorial and promiscuous nature of this system is further reflected in the fact that interactions between the activated GPCRs and Gα proteins are characterized by complex coupling selectivity, where several different GPCRs can pair with the same Gα protein and one GPCR can combine with more than one Gα protein. All these features define the GPCR-G-protein system as a cellular “control panel” capable of detecting an exceptionally diversified set of molecules outside the cell and initiating a broad variety of intracellular signaling cascades in response [212]. This combinatorial promiscuity is further amplified and, in fact, is explained by the presence of intrinsic disorder and associated with it high conformational flexibility of the members of this system. In fact, it was shown that the cytoplasmic and extracellular regions of GPCRs encompass numerous IDRs, multiple disorder-based binding sites, abundant PTM sites, and typically have multiple isoforms generated by alternative splicing [209,213]. Similarly, all human G-proteins contain noticeable levels of functional intrinsic disorder, include numerous sites of various PTMs, include disorder-based interactions sites, and exist as multiple isoforms generated by alternative splicing [209]. Furthermore, both GPCRs and G-proteins often undergo function-associated conformational changes that range from domain motion to binding-induced disorder-to-order transitions. In other words, multifunctionality of these major players of the GPCR-G-protein system is determined by the fact that all these proteins exist as numerous and highly dynamic conformational/basic, inducible/modified, and functioning proteoforms [209].
It is important to note that the combinatorial promiscuity not only can be used to describe the assembly of operating protein systems, but also to define the outputs of action of the corresponding promiscuous reconfigurable signaling networks at the organismal level. This point is illustrated by the action of a family of important chemosensory GPCRs, the olfactory receptors (ORs), which are located in the nasal olfactory epithelium and are responsible for the sense of smell. In humans, ~400 ORs are used to discriminate at least one trillion olfactory stimuli [214]. Obviously, such situation is incompatible with the scenario, where each dedicated OR recognizes one specific odorant molecule. Instead, OR of a particular type can display broad sensitivities to different odorants (i.e., it can recognize multiple odorants), each odorant can promiscuously bind to receptors of many types (i.e., one odorant is recognized by multiple ORs), and different odorants are recognized by different combinations of ORs [215,216]. Therefore, odorants are discriminated in a combinatorial manner [215], where ORs bind odorants promiscuously with different affinities, and the corresponding combinatorial rules define the output signal sent to the brain.
5. Protein Intrinsic Disorder and Evolution of Multicellularity
5.1. Intrinsic Disorder and Proteoforms
It is very likely that IDPs played important roles in at various stages of life origin and evolution, being involved in prebiotic evolution preceding the origin of Tibor Ganti’s Chemoton, a suspected precursor to the first universal common ancestor and, subsequently, to later stages of evolution including early origin of complex multicellularity and the ensuing bilateria during the Cambrian explosion ~571 million years ago [217,218,219,220]. The cornerstone of modern evolutionary theory is the existence of a last universal common ancestor (LUCA), which is a hypothetical common ancestral cell from which the three domains of life, the Bacteria, the Archaea, and the Eukarya have originated [78,79] and which lived roughly 3.5 billion years ago, as it follows from a comprehensive computational analysis using model selection theory without making assumption that sequence similarity indicates a genealogical relationship [79]. The existence of LUCA is supported by multiple observations [79,221,222,223], such as:
- The agreement between phylogeny and biogeography;
- The correspondence between phylogeny and the paleontological record;
- The existence of numerous predicted transitional fossils;
- The hierarchical classification of morphological characteristics;
- The marked similarities of biological structures with different functions (that is, homologies); and
- The congruence of morphological and molecular phylogenies.
Complex multicellularity implies the presence in the organism of multiple differently specialized cells responsible for formation of tissues and organs. Among the molecular mechanisms required for the development of complex multicellularity are means to increase the size of the functional proteome relative to the encoding genome that encodes, which also represents an important phenomenon behind the observation that the complexity of biological systems is mostly determined by their proteome sizes and not by the dimensions of their genomes [224]. This can be illustrated by gene-protein relationship in Homo sapiens [225,226,227,228,229], where the number of protein-coding genes is ranging between 20,000 and 25,000 [132], but the actual number of functionally different proteins is in a range between a few million [230] and several billion [231]. The required structural and functional diversification of a proteomecan be achieved by allelic variations (i.e., single or multiple point mutations (amino acid polymorphisms), indels, single nucleotide polymorphisms (SNPs)), alternative splicing, mRNA editing and other pre-translational mechanisms affecting mRNA, as well as by a wide spectrum of various PTMs of a polypeptide chain. As a result, a single gene encodes a set of distinct protein molecules, known as proteoforms [231]. Since all these aforementioned mechanisms are associated with some changes in the physico-chemical structure of a polypeptide chain, the resulting proteoforms have induced or modified nature. Importantly, protein structural diversity is further enhanced by intrinsic disorder and functionality, giving rise to the conformational or basic proteoforms and functioning proteoforms, respectively [232]. However, since many PTM sites are preferentially located within the IDRs [169,233], since mRNA regions affected by alternative splicing predominantly encode IDRs [158], since IDPs/IDRs act as highly promiscuous binders [5,11,12,14,15,22,24,167,168,175,180,189,199,234,235,236,237,238,239,240,241], and since IDPs/IDRs are characterized by the exceptional spatiotemporal heterogeneity, proteins and protein regions without unique structures represent a very rich source of proteoforms [232].
5.2. Casual Emergence
Since multicellular organisms represent complex systems, their organization and behavior are driven by casual emergence, where the higher scale of a system has stronger causal relationships than its underlying lower scales, allowing macroscales to reduce noise in causal relationships and thereby leading to stronger causes at the higher scale level [242]. Emergence is defined as the appearance of a multi-part, complex system, behavior of which cannot be derived, predicted, or understood by looking at the behavior of its parts. It is one of the characteristic features of complex systems, behavior of which is determined by a set of common rules [243]:
Complex systems contain many heterogeneous components involved in the nonlinear interactions, where a small perturbation may cause a large effect, a proportional effect, or even no effect at all. Therefore, the behavior of a complex system cannot be expressed as a sum of the behaviors of its parts (or of their multiples);
- The constituents of a complex system are interdependent;
- A complex system possesses a structure spanning several scales and may be nested; i.e., the components of a complex system may themselves be complex systems;
- A complex system is capable of emergent behavior, which is unanticipated behavior shown by the system, for example the arising of novel and coherent structures, patterns and properties during the process of self-organization;
- Complexity involves an interplay between chaos (disorder) and order;
- Complexity involves an interplay between cooperation and competition, and complex systems contain both positive (amplifying) and negative (damping) feedbacks;
- Complex systems may have a memory. In other words, the history of a complex system may be important, since due to their dynamic nature, complex systems change over time, and prior states may have an influence on present states (for example, no two genetically identical mice or even two single cells that share the exact same DNA sequence are absolutely identical because of environmental influences, random variations in gene expression, and epigenetic modifications).
It was emphasized that IDPs/IDRs are complex “edge of chaos” systems, as their behavior obeys the aforementioned regulations. “Heterogeneous nature of IDPs is obvious. In fact, IDPs and IDRs are heterogeneous at multiple levels. Globally, they can be compact or extended and their major structural components are heterogeneous too, giving rise to foldons, induced foldons, semi-foldons and non-foldons. These structural components can be independent or interdependent, and they are able to interact nonlinearly. Functional misfolding represents an illustration of the interplay between cooperation and competition. The spatiotemporal complexity of IDPs/IDPRs is further increased by the fact that they and their structural components are always moving between order and disorder. IDPs are able to sense various stimuli and response to these stimuli via corresponding structural changes, where even smallest environmental perturbations might produce large structural and functional outcomes. IDPs/IDPRs possess emergent behavior, since under some conditions they are able to undergo self-organization via stimuli-induced disorder-to-order transitions. Finally, MoRFs, SLiMs and PreSMos represents a memory of the IDP, since they are transiently populated in the non-bound state and may have a profound influence on IDP binding mechanism and on the resulting bound state. All this supports the hypothesis that IDPs/IDPs are positioned at the edge of chaos” [22].
Since in the case of casual emergence groups of features influence the future of a system together, rather than separately, this mechanism is crucial for governing the reliable large-scale responses, such as determining the fate of a single cell, defining intercellular communication and collaboration to form tissues and organs, and even delineating the behavior reaction of an organism in responses to the external stimuli. Although casual emergences was shown to be present in protein-protein interaction (PPI) networks (interactomes) of both prokaryotes and eukaryotes (where a cluster of PPIs can be replaced by a single “macro-node” capable of conducting the same job as the collective), it was more evident in eukaryotes and especially in the complex multicellular eukaryotic organisms [244]. These findings indicated that the more complex organisms tend to more often use higher organization levels of their networks for casual roles, thereby becoming more tolerant to noise and indeterminism of their microscales, as macroscales of interactomes are more resilient than microscales [244]. In this way, their noisy microscales do not serve as primary determinants of the phenotypic outcomes ranging from body structure and body shape to behavior [244]. Importantly, this increase in the casual emergence in complex eukaryotes can explain a rather counter-intuitive observation that the effectiveness of the protein interactomes measured as the effective information which serves as an information-theoretic network quantity based on the entropy of random walker behavior on a network and is reflected in the certainty (or uncertainty) contained in connectivity of analyzed network [245] decreased in moving from prokaryotes to eukaryotes [244]. It is very likely that the observed increase in casual emergence in complex eukaryotes is linked to the higher levels of intrinsic disorder in their proteomes. In fact, although due to the abundant presence of IDPs/IDRs, eukaryotic interactomes at their microscales become noisy, more stochastic, and less effective over evolutionary time [244], the formation of macro-nodes that defines the macroscale structure of the corresponding interactomes is likely to be driven by protein intrinsic disorder.
In attempt to understand what might trigger the transition to multicellularity, the genome and proteome of a single cellular eukaryote, amoeboid holozoan Capsaspora owczarzaki, which is one of the evolutionary closest relative of the first multicellular animals, were investigated [246]. The researchers paid special attention to the genes/proteins involved in the transcriptional regulation as untangling the early evolution of transcription factors (TFs) is critical for understanding of the origin of metazoans and animal development [246]. This analysis revealed that C. owczarzaki contains more transcription factors than any other know single-cellular organism, and that the transcription factors found in this organism are already organized in specific networks that are often found in multicellular animals as well. It was also emphasized that the complexity of the repertoire of transcription factors in C. owczarzaki “is strikingly high, pushing back further the origin of some transcription factors formerly thought to be metazoan specific” [246]. Therefore, it seems that at least some means (in a form of the specific TF-containing networks) required for the animal development were present even before the appearance of the multicellularity, suggesting that the switch to the multicellularity was driven by devising new ways of gene regulation rather than by appearance of more new genes [246]. Figure 7 illustrates the remarkably high level of the global intrinsic disorder content in the C. owczarzaki proteome, which is rather comparable to that of human proteome.
Phenotypic changes on animal lineages are linked to the gain, loss, and modification of gene regulatory elements [247]. Often, such regulation is achieved using cis-regulatory conserved non-exonic elements (CNEEs), which are evolutionarily conserved yet do not overlap with any, coding or noncoding, mature transcript [247], and which show a strong linkage with trait/disease associated single nucleotide polymorphisms [248]. By analyzing genome-wide sets of putative regulatory regions for five vertebrates, including human, to infer the branch on which each CNEE came under selective constraint, it was shown that see it was shown that the evolution of gene regulatory elements is characterized by the presence of three extended periods [247]. It was indicated that instead of the gradual changes in the frequencies of regulatory elements over the past 650 million years, evolution of CNEEs saw three different eras, with early vertebrate evolution lasting from the vertebrate ancestor until about 300 million years ago (when mammals split with birds and reptiles) was characterized by the regulatory gains near the transcription factors and developmental genes. The second period that lasted between 300 and 100 million years ago was characterized by the replacement of the first trend by a high frequency of regulatory innovations near extra-cellular signaling genes, and then, since 100 million years ago, the third period that is affecting at least placental mammals, I characterized by the increase of regulatory innovations for genes involved in post-translational protein modification [247]. Although CNEEs, by default, are non-coding elements, peculiarities of their evolution indicate the crucial roles of regulatory gains of genes mostly encoding for proteins with high levels of intrinsic disorder, such as transcription factors and receptors, or proteins mostly acting by modifying functionality of IDPs/IDRs, such as proteins related to the PTM control. Therefore, this specific CNEE evolution emphasizes the importance of IDPs/IDRs in animal evolution.
5.3. Intrinsic Disorder, Noise/Stochasticity of Transcriptional Regulation, and Development
The examples in the preceding section illustrate the overall complexity of the disorder-based organizing principles of biological networks, which are inherently noisy, and, being promiscuous, rather indiscriminative, and insensitive to the fine details, use combinatorial and fuzzy logics to solve various cellular and organismal queues. Furthermore, all these observations hint to the idea that biological actions are stochastic/noisy, and part of this stochasticity/noisiness is determined by the presence of intrinsic disorder in acting proteins. Importantly, this biological noisiness represents an important driving factor of the development and evolution. This concept can be illustrated by considering the dynamical landscape defining stochastic determination of the cell fate during, for example, differentiation of mouse hematopoietic stem cells into specialized blood cell types via the formation of the multipotent progenitor cells first. One of these multipotent progenitor cells, myeloid progenitor cell, can differentiate either in erythrocytes or the precursors of certain white blood cells, with the choice between these erythroid and myelomonocytic fates being determined by the interplay between the two lineage-determining transcription factors, GATA1 and PU.1 [249]. In this bifurcation, multipotent progenitor cells expressing more of GATA1 will end up in the erythroid state, whereas myeloid state is triggered by higher levels of PU.1 expression. Complexity of this relatively simple system regulated by is determined by the fact that it has sensitive feedbacks, as GATA1 and PU.1, being self-promoting, can inhibit the expression of each other. The dynamics of a resulting binary fate decision system represents an illustration of the phenomenon of "multilineage priming", where a gene-circuit generates stable attractors corresponding to the erythroid and myelomonocytic fates, as well as an uncommitted metastable state characterized by co-expression of both TFs [249]. Here, commitment to a particular cell fate occurs in two stages, where at the first stage, the progenitor state is destabilized in an almost symmetrical bifurcation event, resulting in a poised state at the boundary between the two lineage-specific attractors; second, the cell is driven to the respective, now accessible attractors” [249]. It was also shown that another TF, GATA2, which is antagonistic to PU.1 but boosts the GATA1 expression, plays an important role in differentiation of mouse hematopoietic stem cells by adding to the transcription noise [250]. Here, infrequent, stochastic bursts of transcription lead to the co-expression of these antagonistic TFs in the majority of hematopoietic stem and progenitor cells, thereby opening a possibility for the cells to reach both target lineages more reliably, instead of being stuck on one or another track [250]. In other words, the noisiness of the transcription regulation represents an important way of keeping all the cell-fate option open, where a system maintains a temporally stable probability of cells in every available transcriptional state [250]. Since the major players of this system are transcription factors, it is not surprising that GATA1, GATA2, and PU.1 are highly disordered, as illustrated by Figure 8. It is tempting to assume that this system serves as an illustration of the utilization of protein intrinsic disorder in noisy transcriptional regulation required for cell differentiation.
6. Conclusions
This article analyzes some of the potential implementations of intrinsic disorder in origin of life and evolution. Clearly, views presented here are rather personalized and admittedly subjective. With a very high probability some aspects are incompletely covered, and some other aspects related to this subject are missed. However, one message is absolutely clear: neither origin of life nor evolution would be possible without protein intrinsic disorder. In fact, IDPs, with their highly heterogeneous structural organization and related multifunctionality and enormous interactivity, seem to be perfect life organizers and evolution drivers. Even in a perfect world of highly ordered biological catalysts (enzymes), intrinsic disorder cannot be ridiculed, since the primordial IDPs were the entities that started the molecular evolution of modern enzymes. In fact, the chances that a perfect catalyst with unique 3D structure responsible for a unique catalytic function would spontaneously appear in the primordial Earth are negligible. Instead, one can easily imaging a scenario, where an extremely floppy polypeptide capable of lousy substrate recognition could have a very sloppy catalytic activity. If the rate of the resulting floppy-sloppy “pseudo catalytic” reaction would be even slightly higher than the rate of the corresponding spontaneous, non-catalyzed reaction, one would have an excellent starting point for evolutionary improvement. Obviously, not everything would evolve into highly ordered specialized machines, and numerous modern biological processes are critically dependent on the floppo-sloppiness of IDPs. Life is not something frozen in time and space, and biological processes (especially those in more complex organisms) are not controlled by the precise “chain of command”, being instead stochastic in nature. Acting as crucial constituents of the terrestrial life, IDPs are “edge of the chaos” systems capable of emerging behavior. IDP-driven or IDP-governed, or at least IDP-related emergence is everywhere, and has multiple forms and levels. Evolution is rooted in intrinsic disorder, as IDPs were crucial for origin of life and emergence of protocells, drove the split between prokaryotes and eukaryotes, and orchestrated the emergence of multicellularity.
Author Contributions
Conceptualization, V.N.U.; validation, V.N.U.; formal analysis, V.N.U.; investigation, V.N.U.; writing—original draft preparation, V.N.U.; writing—review and editing, V.N.U.; visualization, V.N.U.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Fischer, E. Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges. 1894, 27, 2985-2993.
- Uversky, V.N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci 2002, 11, 739-756. [CrossRef]
- Uversky, V.N. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci 2013, 22, 693-724. [CrossRef]
- Petsko, G.A.; Ringe, D. Primers in Biology. Protein Structure and Function.; New Science Press Ltd., Sinauer Associates, Inc. Publishers, Blackwell Publishing: London, 2004.
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim Biophys Acta 2010, 1804, 1231-1264. [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol 1977, 112, 535-542.
- Bloomer, A.C.; Champness, J.N.; Bricogne, G.; Staden, R.; Klug, A. Protein disk of tobacco mosaic virus at 2.8 A resolution showing the interactions within and between subunits. Nature 1978, 276, 362-368.
- Bode, W.; Schwager, P.; Huber, R. The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. The refined crystal structures of the bovine trypsinogen-pancreatic trypsin inhibitor complex and of its ternary complex with Ile-Val at 1.9 A resolution. J Mol Biol 1978, 118, 99-112.
- Le Gall, T.; Romero, P.R.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in the Protein Data Bank. J Biomol Struct Dyn 2007, 24, 325-342. [CrossRef]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput 1998, 473-484.
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol 1999, 293, 321-331.
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are "natively unfolded" proteins unstructured under physiologic conditions? Proteins 2000, 41, 415-427.
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J Mol Graph Model 2001, 19, 26-59.
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem Sci 2002, 27, 527-533.
- Daughdrill, G.W.; Pielak, G.J.; Uversky, V.N.; Cortese, M.S.; Dunker, A.K. Natively disordered proteins. In Handbook of Protein Folding, Buchner, J., Kiefhaber, T., Eds.; Wiley-VCH, Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 271-353.
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered. Intrinsically Disordered Proteins 2013, 1, e24157.
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform 2000, 11, 161-171.
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 2004, 337, 635-645. [CrossRef]
- Uversky, V.N. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol 2010, 2010, 568068. [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn 2012, 30, 137-149. [CrossRef]
- Wathen, B.; Jia, Z. Folding by numbers: primary sequence statistics and their use in studying protein folding. Int J Mol Sci 2009, 10, 1567-1589. [CrossRef]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 2013, 1834 932-951. [CrossRef]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett 2008, 15, 956-963.
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys J 2007, 92, 1439-1456. [CrossRef]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38-48.
- Garner, E.; Cannon, P.; Romero, P.; Obradovic, Z.; Dunker, A.K. Predicting Disordered Regions from Amino Acid Sequence: Common Themes Despite Differing Structural Characterization. Genome Inform Ser Workshop Genome Inform 1998, 9, 201-213.
- Williams, R.M.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput 2001, 89-100.
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics 2007, 8, 211. [CrossRef]
- Ferron, F.; Longhi, S.; Canard, B.; Karlin, D. A practical overview of protein disorder prediction methods. Proteins 2006, 65, 1-14. [CrossRef]
- Bourhis, J.M.; Canard, B.; Longhi, S. Predicting protein disorder and induced folding: from theoretical principles to practical applications. Curr Protein Pept Sci 2007, 8, 135-149.
- Dosztanyi, Z.; Sandor, M.; Tompa, P.; Simon, I. Prediction of protein disorder at the domain level. Curr Protein Pept Sci 2007, 8, 161-171.
- Dosztanyi, Z.; Tompa, P. Prediction of protein disorder. Methods Mol Biol 2008, 426, 103-115. [CrossRef]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: an overview. Cell Res 2009, 19, 929-949. [CrossRef]
- Jin, F.; Liu, Z. Inherent Relationships among Different Biophysical Prediction Methods for Intrinsically Disordered Proteins. Biophys J 2013, 104, 488-495. [CrossRef]
- Romero, P.; Obradovic, Z.; Kissinger, C.R.; Villafranca, J.E.; Garner, E.; Guilliot, S.; Dunker, A.K. Thousands of proteins likely to have long disordered regions. Pac Symp Biocomput 1998, 437-448.
- Feng, Z.P.; Zhang, X.; Han, P.; Arora, N.; Anders, R.F.; Norton, R.S. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol Biochem Parasitol 2006, 150, 256-267. [CrossRef]
- Tompa, P.; Dosztanyi, Z.; Simon, I. Prevalent structural disorder in E. coli and S. cerevisiae proteomes. J Proteome Res 2006, 5, 1996-2000. [CrossRef]
- Galea, C.A.; High, A.A.; Obenauer, J.C.; Mishra, A.; Park, C.G.; Punta, M.; Schlessinger, A.; Ma, J.; Rost, B.; Slaughter, C.A.; et al. Large-scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome. Journal of proteome research 2009, 8, 211-226. [CrossRef]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Archaic chaos: intrinsically disordered proteins in Archaea. BMC Syst Biol 2010, 4 Suppl 1, S1. [CrossRef]
- Burra, P.V.; Kalmar, L.; Tompa, P. Reduction in structural disorder and functional complexity in the thermal adaptation of prokaryotes. PLoS One 2010, 5, e12069. [CrossRef]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J 2005, 272, 5129-5148. [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit 2005, 18, 343-384. [CrossRef]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol 2002, 323, 573-584.
- Vucetic, S.; Xie, H.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res 2007, 6, 1899-1916. [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Obradovic, Z.; Uversky, V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res 2007, 6, 1917-1932. [CrossRef]
- Xie, H.; Vucetic, S.; Iakoucheva, L.M.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N.; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res 2007, 6, 1882-1898. [CrossRef]
- Wickramasinghe, N.C.; Wickramasinghe, J.; Napier, W. Comets and the Origin of Life; World Scientific: 2009.
- Nakashima, S.; Kebukawa, Y.; Kitadai, N.; Igisu, M.; Matsuoka, N. Geochemistry and the Origin of Life: From Extraterrestrial Processes, Chemical Evolution on Earth, Fossilized Life's Records, to Natures of the Extant Life. Life (Basel) 2018, 8. [CrossRef]
- Rimola, A.; Balucani, N.; Ceccarelli, C.; Ugliengo, P. Tracing the Primordial Chemical Life of Glycine: A Review from Quantum Chemical Simulations. Int J Mol Sci 2022, 23. [CrossRef]
- Irvine, W.M. Extraterrestrial organic matter: a review. Orig Life Evol Biosph 1998, 28, 365-383. [CrossRef]
- Krasnokutski, S.; Chuang, K.-J.; Jäger, C.; Ueberschaar, N.; Henning, T. A pathway to peptides in space through the condensation of atomic carbon. Nature Astronomy 2022, 6, 381-386.
- Kulkarni, P.; Salgia, R.; Uversky, V.N. Intrinsic disorder, extraterrestrial peptides, and prebiotic life on the earth. J Biomol Struct Dyn 2023, 41, 5481-5485. [CrossRef]
- Rivera-Valentin, E.G.; Filiberto, J.; Lynch, K.L.; Mamajanov, I.; Lyons, T.W.; Schulte, M.; Mendez, A. Introduction-First Billion Years: Habitability. Astrobiology 2021, 21, 893-905. [CrossRef]
- Cronin, J.R.; Pizzarello, S. Amino acids in meteorites. Adv Space Res 1983, 3, 5-18. [CrossRef]
- McGeoch, J.E.; McGeoch, M.W. A 4641Da polymer of amino acids in Acfer 086 and Allende meteorites. arXiv preprint arXiv:1707.09080 2017.
- McGeoch, M.; Dikler, S.; McGeoch, J.E. Hemolithin: a meteoritic protein containing iron and lithium. arXiv preprint arXiv:2002.11688 2020.
- Radzicka, A.; Wolfenden, R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. Journal of the American Chemical Society 1996, 118, 6105-6109.
- Oparin, A.I. The Origin of Life (in Russian); Moscow Worker publisher: Moscow, 1924.
- Haldane, J.B.S. The origin of life. In The Rationalist Annual for the Year 1929, Watts, C.A., Ed.; Watts & Co: London 1929; pp. 3-10.
- Miller, S.L. A production of amino acids under possible primitive earth conditions. Science 1953, 117, 528-529.
- Miller, S.L.; Urey, H.C. Organic compound synthesis on the primitive earth. Science 1959, 130, 245-251.
- Crick, F.H. The origin of the genetic code. J Mol Biol 1968, 38, 367-379, doi:0022-2836(68)90392-6 [pii].
- Wong, J.T. A co-evolution theory of the genetic code. Proc Natl Acad Sci U S A 1975, 72, 1909-1912.
- Jukes, T.H. Possibilities for the evolution of the genetic code from a preceding form. Nature 1973, 246, 22-26.
- Trifonov, E.N. Consensus temporal order of amino acids and evolution of the triplet code. Gene 2000, 261, 139-151, doi:S0378-1119(00)00476-5 [pii].
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: the Database of Disordered Proteins. Nucleic Acids Res 2007, 35, D786-793. [CrossRef]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. To be folded or to be unfolded? Protein Sci 2004, 13, 2871-2877. [CrossRef]
- Xia, T.; SantaLucia, J., Jr.; Burkard, M.E.; Kierzek, R.; Schroeder, S.J.; Jiao, X.; Cox, C.; Turner, D.H. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson-Crick base pairs. Biochemistry 1998, 37, 14719-14735. [CrossRef]
- Zhou, H.; Zhou, Y. Quantifying the effect of burial of amino acid residues on protein stability. Proteins 2004, 54, 315-322. [CrossRef]
- Poole, A.M.; Jeffares, D.C.; Penny, D. The path from the RNA world. J Mol Evol 1998, 46, 1-17.
- Peng, Z.; Oldfield, C.J.; Xue, B.; Mizianty, M.J.; Dunker, A.K.; Kurgan, L.; Uversky, V.N. A creature with a hundred waggly tails: intrinsically disordered proteins in the ribosome. Cell Mol Life Sci 2014, 71, 1477-1504. [CrossRef]
- Jeffares, D.C.; Poole, A.M.; Penny, D. Relics from the RNA world. J Mol Evol 1998, 46, 18-36.
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. Faseb J 2004, 18, 1169-1175. [CrossRef]
- Treiber, D.K.; Williamson, J.R. Beyond kinetic traps in RNA folding. Curr Opin Struct Biol 2001, 11, 309-314, doi:S0959-440X(00)00206-2 [pii].
- Cristofari, G.; Darlix, J.L. The ubiquitous nature of RNA chaperone proteins. Prog Nucleic Acid Res Mol Biol 2002, 72, 223-268.
- Gilbert, W. Origin of life - the RNA world. Nature 1986, 319, 618-618, doi:Doi 10.1038/319618a0.
- Csermely, P. Proteins, RNAs and chaperones in enzyme evolution: A folding perspective. Trends in Biochemical Sciences 1997, 22, 147-149, doi:Doi 10.1016/S0968-0004(97)01026-8.
- Doolittle, W.F. Uprooting the tree of life. Sci Am 2000, 282, 90-95.
- Theobald, D.L. A formal test of the theory of universal common ancestry. Nature 2010, 465, 219-222. [CrossRef]
- Lahav, N.; White, D.; Chang, S. Peptide formation in the prebiotic era: thermal condensation of glycine in fluctuating clay environments. Science 1978, 201, 67-69. [CrossRef]
- Rodriguez-Garcia, M.; Surman, A.J.; Cooper, G.J.T.; Suarez-Marina, I.; Hosni, Z.; Lee, M.P.; Cronin, L. Formation of oligopeptides in high yield under simple programmable conditions. Nat Commun 2015, 6, 8385. [CrossRef]
- Campbell, T.D.; Febrian, R.; McCarthy, J.T.; Kleinschmidt, H.E.; Forsythe, J.G.; Bracher, P.J. Prebiotic condensation through wet-dry cycling regulated by deliquescence. Nat Commun 2019, 10, 4508. [CrossRef]
- Sakata, K.; Kitadai, N.; Yokoyama, T. Effects of pH and temperature on dimerization rate of glycine: evaluation of favorable environmental conditions for chemical evolution of life. Geochimica et Cosmochimica Acta 2010, 74, 6841-6851.
- Imai, E.; Honda, H.; Hatori, K.; Brack, A.; Matsuno, K. Elongation of oligopeptides in a simulated submarine hydrothermal system. Science 1999, 283, 831-833. [CrossRef]
- Ohara, S.; Kakegawa, T.; Nakazawa, H. Pressure effects on the abiotic polymerization of glycine. Orig Life Evol Biosph 2007, 37, 215-223. [CrossRef]
- Muller, F.; Escobar, L.; Xu, F.; Wegrzyn, E.; Nainyte, M.; Amatov, T.; Chan, C.Y.; Pichler, A.; Carell, T. A prebiotically plausible scenario of an RNA-peptide world. Nature 2022, 605, 279-284. [CrossRef]
- Sumie, Y.; Sato, K.; Kakegawa, T.; Furukawa, Y. Boron-assisted abiotic polypeptide synthesis. Commun Chem 2023, 6, 89. [CrossRef]
- Lazcano, A. Historical development of origins research. Cold Spring Harb Perspect Biol 2010, 2, a002089. [CrossRef]
- Lei, L.; Burton, Z.F. Chaos, order and systematics in evolution of the genetic code. 2020.
- Kauffman, S.A. The origins of order: Self-organization and selection in evolution; Oxford University Press, USA: 1993.
- Kacar, B.; Garcia, A.K.; Anbar, A.D. Evolutionary History of Bioessential Elements Can Guide the Search for Life in the Universe. Chembiochem 2021, 22, 114-119. [CrossRef]
- Matveev, V.V. Cell theory, intrinsically disordered proteins, and the physics of the origin of life. Prog Biophys Mol Biol 2019, 149, 114-130. [CrossRef]
- Matsuo, M.; Kurihara, K. Proliferating coacervate droplets as the missing link between chemistry and biology in the origins of life. Nat Commun 2021, 12, 5487. [CrossRef]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Julicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729-1732. [CrossRef]
- Darling, A.L.; Uversky, V.N. Known types of membrane-less organelles and biomolecular condensates. In Droplets of Life: Membrane-Less Organelles, Biomolecular Condensates, and Biological Liquid-Liquid Phase Separation, 1st ed.; Uversky, V.N., Ed.; Elsevier: Amsterdam, Netherlands, 2023; pp. 271-335.
- Brangwynne, C.P. Phase transitions and size scaling of membrane-less organelles. The Journal of cell biology 2013, 203, 875-881. [CrossRef]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat Physics 2015, 11, 899–904. [CrossRef]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett 2015, 589, 15-22. [CrossRef]
- Dundr, M.; Misteli, T. Biogenesis of nuclear bodies. Cold Spring Harb Perspect Biol 2010, 2, a000711. [CrossRef]
- Zhu, L.; Brangwynne, C.P. Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr Opin Cell Biol 2015, 34, 23-30. [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr Opin Struct Biol 2017, 44, 18-30. [CrossRef]
- Uversky, V.N. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv Colloid Interface Sci 2017, 239, 97-114. [CrossRef]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686-1697. [CrossRef]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun Signal 2016, 14, 1. [CrossRef]
- Martin, E.W.; Holehouse, A.S. Intrinsically disordered protein regions and phase separation: sequence determinants of assembly or lack thereof. Emerg Top Life Sci 2020, 4, 307-329. [CrossRef]
- Antifeeva, I.A.; Fonin, A.V.; Fefilova, A.S.; Stepanenko, O.V.; Povarova, O.I.; Silonov, S.A.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. Liquid-liquid phase separation as an organizing principle of intracellular space: overview of the evolution of the cell compartmentalization concept. Cell Mol Life Sci 2022, 79, 251. [CrossRef]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem Sci 2019, 44, 716-728. [CrossRef]
- Darling, A.L.; Liu, Y.; Oldfield, C.J.; Uversky, V.N. Intrinsically Disordered Proteome of Human Membrane-Less Organelles. Proteomics 2018, 18, e1700193. [CrossRef]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog Mol Biol Transl Sci 2019, 166, 1-17. [CrossRef]
- Uversky, V.N. Recent Developments in the Field of Intrinsically Disordered Proteins: Intrinsic Disorder–Based Emergence in Cellular Biology in Light of the Physiological and Pathological Liquid–Liquid Phase Transitions. Annual Review of Biophysics 2021, 50, 135-156. [CrossRef]
- Meng, F.; Na, I.; Kurgan, L.; Uversky, V.N. Compartmentalization and Functionality of Nuclear Disorder: Intrinsic Disorder and Protein-Protein Interactions in Intra-Nuclear Compartments. Int J Mol Sci 2015, 17. [CrossRef]
- Uversky, V.N. The roles of intrinsic disorder-based liquid-liquid phase transitions in the "Dr. Jekyll-Mr. Hyde" behavior of proteins involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Autophagy 2017, 13, 2115-2162. [CrossRef]
- Brangwynne, C.P.; Mitchison, T.J.; Hyman, A.A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc Natl Acad Sci U S A 2011, 108, 4334-4339. [CrossRef]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336-340. [CrossRef]
- Aggarwal, S.; Snaidero, N.; Pahler, G.; Frey, S.; Sanchez, P.; Zweckstetter, M.; Janshoff, A.; Schneider, A.; Weil, M.T.; Schaap, I.A.; et al. Myelin membrane assembly is driven by a phase transition of myelin basic proteins into a cohesive protein meshwork. PLoS biology 2013, 11, e1001577. [CrossRef]
- Feric, M.; Brangwynne, C.P. A nuclear F-actin scaffold stabilizes ribonucleoprotein droplets against gravity in large cells. Nature cell biology 2013, 15, 1253-1259. [CrossRef]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791-805. [CrossRef]
- Nesterov, S.V.; Ilyinsky, N.S.; Uversky, V.N. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim Biophys Acta Mol Cell Res 2021, 1868, 119102. [CrossRef]
- Fonin, A.V.; Antifeeva, I.A.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B.Y.; Kulkarni, P.; Uversky, V.N. Biological soft matter: intrinsically disordered proteins in liquid-liquid phase separation and biomolecular condensates. Essays Biochem 2022, 66, 831-847. [CrossRef]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 2015, 57, 936-947. [CrossRef]
- Mitrea, D.M.; Cika, J.A.; Guy, C.S.; Ban, D.; Banerjee, P.R.; Stanley, C.B.; Nourse, A.; Deniz, A.A.; Kriwacki, R.W. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife 2016, 5. [CrossRef]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci U S A 2015, 112, 7189-7194. [CrossRef]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell 2015, 60, 208-219. [CrossRef]
- Toretsky, J.A.; Wright, P.E. Assemblages: functional units formed by cellular phase separation. The Journal of cell biology 2014, 206, 579-588. [CrossRef]
- Csizmok, V.; Follis, A.V.; Kriwacki, R.W.; Forman-Kay, J.D. Dynamic Protein Interaction Networks and New Structural Paradigms in Signaling. Chem Rev 2016. [CrossRef]
- Rigau, M.; Juan, D.; Valencia, A.; Rico, D. Intronic CNVs and gene expression variation in human populations. PLoS Genet 2019, 15, e1007902. [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5' terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci U S A 1977, 74, 3171-3175. [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5' ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1-8. [CrossRef]
- Williamson, B. DNA insertions and gene structure. Nature 1977, 270, 295-297.
- Eddy, S.R. The C-value paradox, junk DNA and ENCODE. Curr Biol 2012, 22, R898-899. [CrossRef]
- Palazzo, A.F.; Gregory, T.R. The case for junk DNA. PLoS Genet 2014, 10, e1004351. [CrossRef]
- Consortium, T.E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57-74. [CrossRef]
- Taft, R.J.; Pheasant, M.; Mattick, J.S. The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays 2007, 29, 288-299. [CrossRef]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat Rev Genet 2014, 15, 423-437. [CrossRef]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat Rev Genet 2014, 15, 829-845. [CrossRef]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.L.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711-719. [CrossRef]
- Wang, C.; Uversky, V.N.; Kurgan, L. Disordered nucleiome: Abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea. Proteomics 2016, 16, 1486-1498. [CrossRef]
- Peng, Z.; Mizianty, M.J.; Xue, B.; Kurgan, L.; Uversky, V.N. More than just tails: intrinsic disorder in histone proteins. Mol Biosyst 2012, 8, 1886-1901. [CrossRef]
- Bhalla, J.; Storchan, G.B.; MacCarthy, C.M.; Uversky, V.N.; Tcherkasskaya, O. Local flexibility in molecular function paradigm. Mol Cell Proteomics 2006, 5, 1212-1223. [CrossRef]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873-6888. [CrossRef]
- Minezaki, Y.; Homma, K.; Kinjo, A.R.; Nishikawa, K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J Mol Biol 2006, 359, 1137-1149. [CrossRef]
- Coelho Ribeiro Mde, L.; Espinosa, J.; Islam, S.; Martinez, O.; Thanki, J.J.; Mazariegos, S.; Nguyen, T.; Larina, M.; Xue, B.; Uversky, V.N. Malleable ribonucleoprotein machine: protein intrinsic disorder in the Saccharomyces cerevisiae spliceosome. PeerJ 2013, 1, e2. [CrossRef]
- Korneta, I.; Bujnicki, J.M. Intrinsic disorder in the human spliceosomal proteome. PLoS Comput Biol 2012, 8, e1002641. [CrossRef]
- Zhao, B.; Katuwawala, A.; Oldfield, C.J.; Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Intrinsic Disorder in Human RNA-Binding Proteins. J Mol Biol 2021, 433, 167229. [CrossRef]
- Sambrook, J. Adenovirus amazes at Cold Spring Harbor. Nature 1977, 268, 101-104.
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 2003, 72, 291-336. [CrossRef]
- Graveley, B.R. Alternative splicing: increasing diversity in the proteomic world. Trends Genet 2001, 17, 100-107, doi:S0168-9525(00)02176-4 [pii].
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.A.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1-20. [CrossRef]
- Brett, D.; Hanke, J.; Lehmann, G.; Haase, S.; Delbruck, S.; Krueger, S.; Reich, J.; Bork, P. EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett 2000, 474, 83-86, doi:S0014-5793(00)01581-7 [pii].
- Johnson, J.M.; Castle, J.; Garrett-Engele, P.; Kan, Z.; Loerch, P.M.; Armour, C.D.; Santos, R.; Schadt, E.E.; Stoughton, R.; Shoemaker, D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141-2144. [CrossRef]
- Minneman, K.P. Splice variants of G protein-coupled receptors. Molecular interventions 2001, 1, 108-116.
- Thai, T.H.; Kearney, J.F. Distinct and opposite activities of human terminal deoxynucleotidyltransferase splice variants. J Immunol 2004, 173, 4009-4019.
- Scheper, W.; Zwart, R.; Baas, F. Alternative splicing in the N-terminus of Alzheimer's presenilin 1. Neurogenetics 2004, 5, 223-227.
- Norris, J.D.; Fan, D.; Sherk, A.; McDonnell, D.P. A negative coregulator for the human ER. Mol Endocrinol 2002, 16, 459-468. [CrossRef]
- Ilik, I.A.; Malszycki, M.; Lubke, A.K.; Schade, C.; Meierhofer, D.; Aktas, T. SON and SRRM2 are essential for nuclear speckle formation. Elife 2020, 9. [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583-589. [CrossRef]
- Dayhoff, G.W., 2nd; Uversky, V.N. Rapid prediction and analysis of protein intrinsic disorder. Protein Sci 2022, 31, e4496. [CrossRef]
- Romero, P.R.; Zaidi, S.; Fang, Y.Y.; Uversky, V.N.; Radivojac, P.; Oldfield, C.J.; Cortese, M.S.; Sickmeier, M.; LeGall, T.; Obradovic, Z.; et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci U S A 2006, 103, 8390-8395.
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 2005, 44, 7342-7372. [CrossRef]
- Witze, E.S.; Old, W.M.; Resing, K.A.; Ahn, N.G. Mapping protein post-translational modifications with mass spectrometry. Nat Methods 2007, 4, 798-806. [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat Struct Mol Biol 2010, 17, 666-672. [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat Biotechnol 2003, 21, 255-261. [CrossRef]
- Marks, F. Protein Phosphorylation; VCH Weinheim: New York, Basel, Cambridge, Tokyo, 1996.
- Yang, X.J. Multisite protein modification and intramolecular signaling. Oncogene 2005, 24, 1653-1662. [CrossRef]
- Erler, J.; Zhang, R.; Petridis, L.; Cheng, X.; Smith, J.C.; Langowski, J. The role of histone tails in the nucleosome: a computational study. Biophys J 2014, 107, 2911-2922. [CrossRef]
- Mersfelder, E.L.; Parthun, M.R. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res 2006, 34, 2653-2662. [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573-6582.
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv Protein Chem 2002, 62, 25-49.
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O'Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037-1049.
- Radivojac, P.; Vacic, V.; Haynes, C.; Cocklin, R.R.; Mohan, A.; Heyen, J.W.; Goebl, M.G.; Iakoucheva, L.M. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010, 78, 365-380.
- Uversky, V.N. Protein folding revisited. A polypeptide chain at the folding-misfolding-nonfolding cross-roads: which way to go? Cell Mol Life Sci 2003, 60, 1852-1871.
- Turoverov, K.K.; Kuznetsova, I.M.; Uversky, V.N. The protein kingdom extended: ordered and intrinsically disordered proteins, their folding, supramolecular complex formation, and aggregation. Prog Biophys Mol Biol 2010, 102, 73-84. [CrossRef]
- Williams, R.M.; Obradovic, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput 2001, 89-100.
- Uversky, V.N. What does it mean to be natively unfolded? Eur J Biochem 2002, 269, 2-12, doi:2649 [pii].
- Dunker, A.K.; Obradovic, Z. The protein trinity--linking function and disorder. Nat Biotechnol 2001, 19, 805-806.
- Uversky, V.N. Paradoxes and wonders of intrinsic disorder: Complexity of simplicity. Intrinsically Disord Proteins 2016, 4, e1135015. [CrossRef]
- DeForte, S.; Uversky, V.N. Order, Disorder, and Everything in Between. Molecules 2016, 21. [CrossRef]
- Uversky, V.N. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J Biol Chem 2016, 291, 6681-6688. [CrossRef]
- Uversky, V.N. Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J 2015, 282, 1182-1189. [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454-12470. [CrossRef]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett 2006, 580, 2041-2045.
- Ekman, D.; Light, S.; Bjorklund, A.K.; Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol 2006, 7, R45.
- Haynes, C.; Oldfield, C.J.; Ji, F.; Klitgord, N.; Cusick, M.E.; Radivojac, P.; Uversky, V.N.; Vidal, M.; Iakoucheva, L.M. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol 2006, 2, e100.
- Dosztanyi, Z.; Chen, J.; Dunker, A.K.; Simon, I.; Tompa, P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res 2006, 5, 2985-2995.
- Singh, G.P.; Dash, D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins 2007, 68, 602-605.
- Singh, G.P.; Ganapathi, M.; Dash, D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins 2007, 66, 761-765.
- Schulz, G.E. Nucleotide Binding Proteins. In Molecular Mechanism of Biological Recognition, Balaban, M., Ed.; Elsevier/North-Holland Biomedical Press: New York, 1979; pp. 79-94.
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci U S A 1996, 93, 11504-11509.
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J Mol Biol 2006, 362, 1043-1059. [CrossRef]
- Cheng, Y.; Oldfield, C.J.; Meng, J.; Romero, P.; Uversky, V.N.; Dunker, A.K. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468-13477. [CrossRef]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75-83. [CrossRef]
- Landsteiner, K. The Specificity of Serological Reactions; Courier Dover Publications: Mineola, New York, 1936.
- Pauling, L. A theory of the structure and process of formation of antibodies. J Am Chem Soc 1940, 62, 2643-2657.
- Karush, F. Heterogeneity of the binding sites of bovine serum albumin. J Am Chem Soc 1950, 72, 2705-2713.
- Meador, W.E.; Means, A.R.; Quiocho, F.A. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science 1993, 262, 1718-1721.
- Uversky, V.N. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn 2003, 21, 211-234, doi:d=3013&c=4118&p=11847&do=detail [pii].
- Fuxreiter, M.; Tompa, P. Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol 2012, 725, 1-14. [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem Sci 2008, 33, 2-8. [CrossRef]
- Uversky, V.N. Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem Soc Rev 2011, 40, 1623-1634. [CrossRef]
- Permyakov, S.E.; Millett, I.S.; Doniach, S.; Permyakov, E.A.; Uversky, V.N. Natively unfolded C-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins 2003, 53, 855--862.
- Sigalov, A.; Aivazian, D.; Stern, L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 2004, 43, 2049-2061. [CrossRef]
- Sigalov, A.B.; Zhuravleva, A.V.; Orekhov, V.Y. Binding of intrinsically disordered proteins is not necessarily accompanied by a structural transition to a folded form. Biochimie 2007, 89, 419-421. [CrossRef]
- Bjarnadottir, T.K.; Gloriam, D.E.; Hellstrand, S.H.; Kristiansson, H.; Fredriksson, R.; Schioth, H.B. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 2006, 88, 263-273. [CrossRef]
- Anantharaman, V.; Abhiman, S.; de Souza, R.F.; Aravind, L. Comparative genomics uncovers novel structural and functional features of the heterotrimeric GTPase signaling system. Gene 2011, 475, 63-78. [CrossRef]
- Southan, C.; Sharman, J.L.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Alexander, S.P.; Buneman, O.P.; Davenport, A.P.; McGrath, J.C.; Peters, J.A.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016, 44, D1054-1068. [CrossRef]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 2003, 63, 1256-1272. [CrossRef]
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G-protein binding. Nature 2017, 545, 317-322. [CrossRef]
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers - aligning topology maps while minding the gaps. Trends Pharmacol Sci 2015, 36, 22-31. [CrossRef]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Multi-functionality of proteins involved in GPCR and G protein signaling: making sense of structure-function continuum with intrinsic disorder-based proteoforms. Cell Mol Life Sci 2019, 76, 4461-4492. [CrossRef]
- Neves, S.R.; Ram, P.T.; Iyengar, R. G protein pathways. Science 2002, 296, 1636-1639. [CrossRef]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 2001, 22, 368-376. [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem Rev 2017, 117, 139-155. [CrossRef]
- Venkatakrishnan, A.J.; Flock, T.; Prado, D.E.; Oates, M.E.; Gough, J.; Madan Babu, M. Structured and disordered facets of the GPCR fold. Curr Opin Struct Biol 2014, 27, 129-137. [CrossRef]
- Bushdid, C.; Magnasco, M.O.; Vosshall, L.B.; Keller, A. Humans can discriminate more than 1 trillion olfactory stimuli. Science 2014, 343, 1370-1372. [CrossRef]
- Malnic, B.; Hirono, J.; Sato, T.; Buck, L.B. Combinatorial receptor codes for odors. Cell 1999, 96, 713-723. [CrossRef]
- Reddy, G.; Zak, J.D.; Vergassola, M.; Murthy, V.N. Antagonism in olfactory receptor neurons and its implications for the perception of odor mixtures. Elife 2018, 7. [CrossRef]
- Gánti, T. Chemoton theory: theory of living systems; Springer Science & Business Media: 2003.
- Kulkarni, P.; Bhattacharya, S.; Achuthan, S.; Behal, A.; Jolly, M.K.; Kotnala, S.; Mohanty, A.; Rangarajan, G.; Salgia, R.; Uversky, V. Intrinsically Disordered Proteins: Critical Components of the Wetware. Chem Rev 2022, 122, 6614-6633. [CrossRef]
- Katsnelson, A. Did Disordered Proteins Help Launch Life on Earth? ACS Cent Sci 2020, 6, 1854-1857. [CrossRef]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins: The Dark Horse of the Dark Proteome. Proteomics 2018, 18, e1800061. [CrossRef]
- Penny, D.; Foulds, L.R.; Hendy, M.D. Testing the theory of evolution by comparing phylogenetic trees constructed from five different protein sequences. Nature 1982, 297, 197-200. [CrossRef]
- Futuyma, D. Evolutionary biology, 3rd edn Sinauer Associates. Sunderland.[Google Scholar] 1998.
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving genes and proteins; Elsevier: 1965; pp. 97-166.
- Schluter, H.; Apweiler, R.; Holzhutter, H.G.; Jungblut, P.R. Finding one's way in proteomics: a protein species nomenclature. Chem Cent J 2009, 3, 11. [CrossRef]
- Uhlen, M.; Bjorling, E.; Agaton, C.; Szigyarto, C.A.; Amini, B.; Andersen, E.; Andersson, A.C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Molecular & Cellular Proteomics 2005, 4, 1920-1932. [CrossRef]
- Farrah, T.; Deutsch, E.W.; Omenn, G.S.; Sun, Z.; Watts, J.D.; Yamamoto, T.; Shteynberg, D.; Harris, M.M.; Moritz, R.L. State of the Human Proteome in 2013 as Viewed through PeptideAtlas: Comparing the Kidney, Urine, and Plasma Proteomes for the Biology- and Disease-Driven Human Proteome Project. Journal of Proteome Research 2014, 13, 60-75. [CrossRef]
- Farrah, T.; Deutsch, E.W.; Hoopmann, M.R.; Hallows, J.L.; Sun, Z.; Huang, C.Y.; Moritz, R.L. The State of the Human Proteome in 2012 as Viewed through PeptideAtlas. Journal of Proteome Research 2013, 12, 162-171. [CrossRef]
- Reddy, P.J.; Ray, S.; Srivastava, S. The Quest of the Human Proteome and the Missing Proteins: Digging Deeper. Omics-a Journal of Integrative Biology 2015, 19, 276-282, doi:DOI 10.1089/omi.2015.0035.
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575-+. [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int J Anal Chem 2016, 2016, 7436849. [CrossRef]
- Smith, L.M.; Kelleher, N.L.; Consortium for Top Down, P. Proteoform: a single term describing protein complexity. Nat Methods 2013, 10, 186-187. [CrossRef]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int J Mol Sci 2016, 17, 1874. [CrossRef]
- Pejaver, V.; Hsu, W.L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The structural and functional signatures of proteins that undergo multiple events of post-translational modification. Protein Sci 2014, 23, 1077-1093. [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26-59. [CrossRef]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr Opin Struct Biol 2008, 18, 756-764. [CrossRef]
- Dunker, A.K.; Uversky, V.N. Signal transduction via unstructured protein conduits. Nat Chem Biol 2008, 4, 229-230. [CrossRef]
- Uversky, V.N. Disordered competitive recruiter: fast and foldable. J Mol Biol 2012, 418, 267-268. [CrossRef]
- Uversky, V.N.; Dunker, A.K. The case for intrinsically disordered proteins playing contributory roles in molecular recognition without a stable 3D structure. F1000 biology reports 2013, 5, 1. [CrossRef]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol 2002, 12, 54-60. [CrossRef]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 2005, 6, 197-208. [CrossRef]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res 2007, 6, 2351-2366. [CrossRef]
- Comolatti, R.; Hoel, E. Causal emergence is widespread across measures of causation. arXiv preprint arXiv:2202.01854 2022.
- Baranger, M. Chaos, complexity, and entropy - A physics talk for non-physicists. 2001.
- Klein, B.; Hoel, E.; Swain, A.; Griebenow, R.; Levin, M. Evolution and emergence: higher order information structure in protein interactomes across the tree of life. Integr Biol (Camb) 2021, 13, 283-294. [CrossRef]
- Klein, B.; Hoel, E. The emergence of informative higher scales in complex networks. Complexity 2020, 2020, 8932526.
- Sebe-Pedros, A.; de Mendoza, A.; Lang, B.F.; Degnan, B.M.; Ruiz-Trillo, I. Unexpected repertoire of metazoan transcription factors in the unicellular holozoan Capsaspora owczarzaki. Mol Biol Evol 2011, 28, 1241-1254. [CrossRef]
- Lowe, C.B.; Kellis, M.; Siepel, A.; Raney, B.J.; Clamp, M.; Salama, S.R.; Kingsley, D.M.; Lindblad-Toh, K.; Haussler, D. Three periods of regulatory innovation during vertebrate evolution. Science 2011, 333, 1019-1024. [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009, 106, 9362-9367. [CrossRef]
- Huang, S.; Guo, Y.P.; May, G.; Enver, T. Bifurcation dynamics in lineage-commitment in bipotent progenitor cells. Dev Biol 2007, 305, 695-713. [CrossRef]
- Wheat, J.C.; Sella, Y.; Willcockson, M.; Skoultchi, A.I.; Bergman, A.; Singer, R.H.; Steidl, U. Single-molecule imaging of transcription dynamics in somatic stem cells. Nature 2020, 583, 431-436. [CrossRef]
Figure 1.
Correlations between three foldability scales (the scale based on the average number of contacts per residue in the ordered proteins (Galzitskaya) [67], the DisProt-based scale [66], and the Top-IDP scale [23]) and amino acid novelty scale proposed by Trifonov [65]. Red and blue symbols correspond to disorder- and order-promoting residues as defined by DisProt-based scale. Pink and cyan squares with error bars show averaged values. .
Figure 1.
Correlations between three foldability scales (the scale based on the average number of contacts per residue in the ordered proteins (Galzitskaya) [67], the DisProt-based scale [66], and the Top-IDP scale [23]) and amino acid novelty scale proposed by Trifonov [65]. Red and blue symbols correspond to disorder- and order-promoting residues as defined by DisProt-based scale. Pink and cyan squares with error bars show averaged values. .
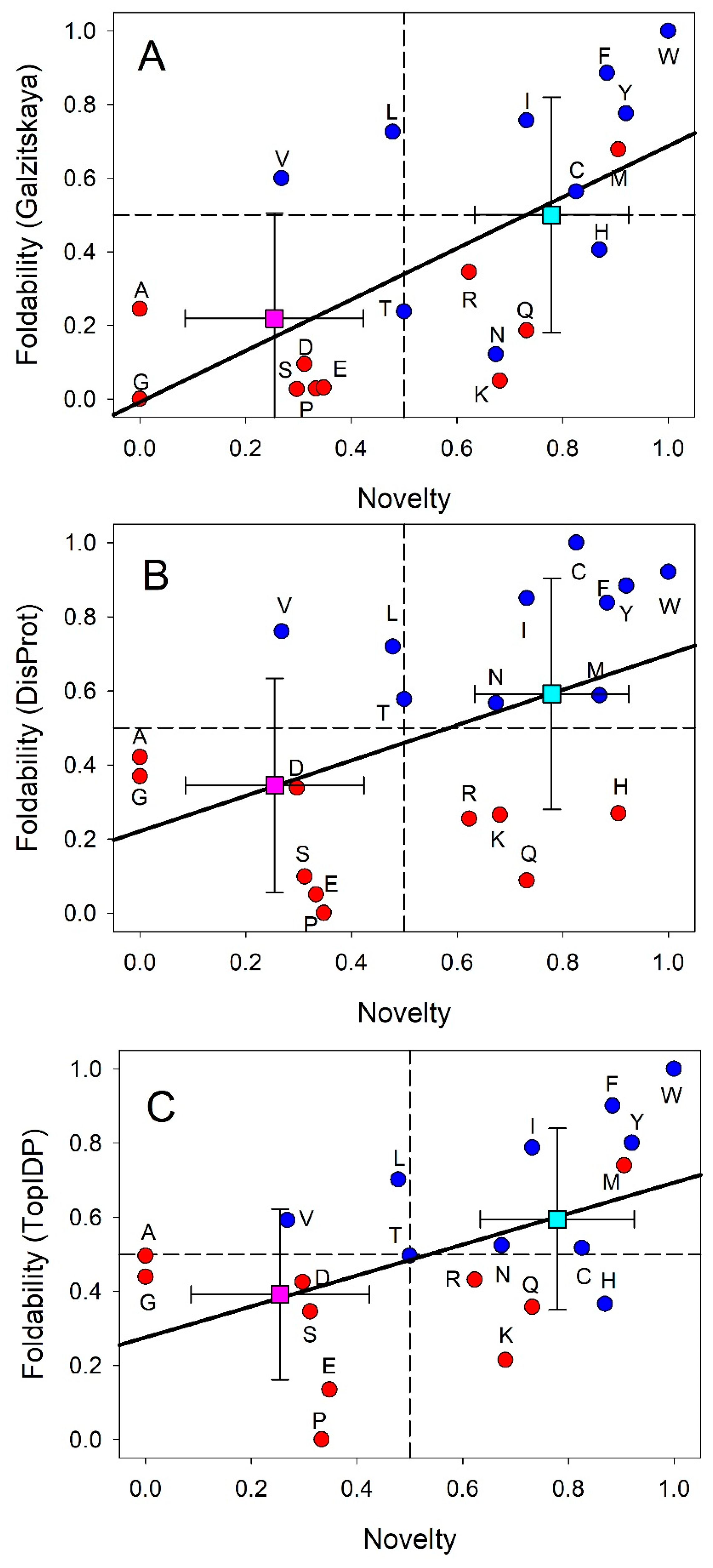
Figure 2.
Modern genetic code with information on the early and late codons (shown by light red and light blue colors, respectively) and disorder- and order-promoting residues (shown by red and blue colors, respectively). Codons with intermediate ages (i.e., those located between early and late codons) are shown by light pink color, whereas disorder-neutral residues are shown by pink color. Reproduced with permission form ref. [3].
Figure 2.
Modern genetic code with information on the early and late codons (shown by light red and light blue colors, respectively) and disorder- and order-promoting residues (shown by red and blue colors, respectively). Codons with intermediate ages (i.e., those located between early and late codons) are shown by light pink color, whereas disorder-neutral residues are shown by pink color. Reproduced with permission form ref. [3].
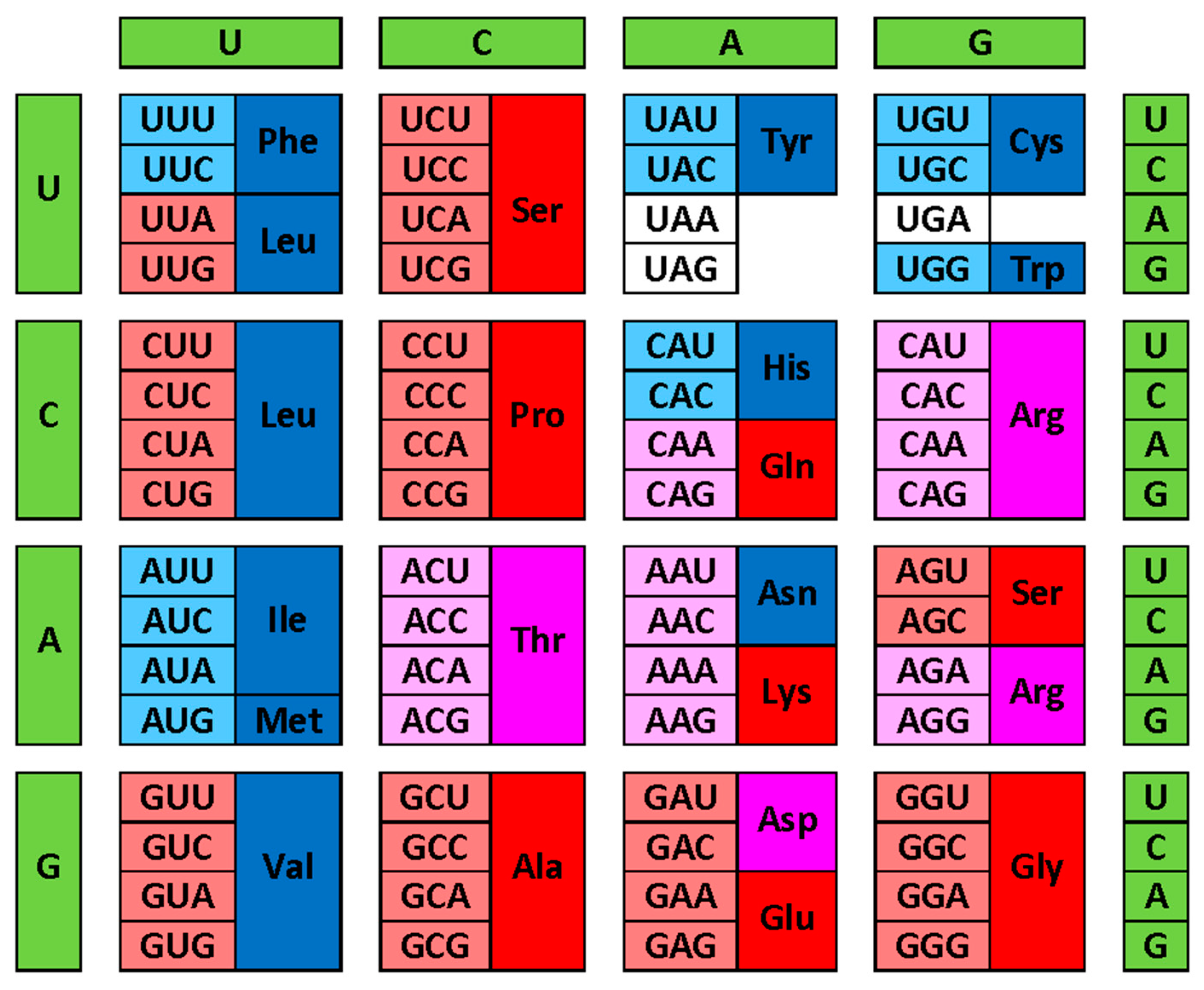
Figure 3.
Correlations between thermostability of the codons (measured as melting enthalpies (kcal/M) of the dinucleotide stacks corresponding to the first and second codon positions [68]) and amino acid novelty of corresponding residue (A), thermostability of codons and DisProt foldability of corresponding residues (B), and thermostability of codons and buriability of corresponding residues (C), buriability of amino acids and their novelty, (D), and DisProt foldability and buriability (E). Red and blue symbols correspond to disorder- and order-promoting residues as defined by DisProt-based scale.
Figure 3.
Correlations between thermostability of the codons (measured as melting enthalpies (kcal/M) of the dinucleotide stacks corresponding to the first and second codon positions [68]) and amino acid novelty of corresponding residue (A), thermostability of codons and DisProt foldability of corresponding residues (B), and thermostability of codons and buriability of corresponding residues (C), buriability of amino acids and their novelty, (D), and DisProt foldability and buriability (E). Red and blue symbols correspond to disorder- and order-promoting residues as defined by DisProt-based scale.

Figure 4.
Correlation between the intrinsic disorder content and proteome size for 3,484 species from viruses, archaea, bacteria, and eukaryotes. Each symbol indicates a species. There are totally six groups of species: viruses expressing one polyprotein precursor (small red circles filled with blue), other viruses (small red circles), bacteria (small green circles), archaea (blue circles), unicellular eukaryotes (brown squares), and multicellular eukaryotes (pink triangles). Each viral polyprotein was analyzed as a single polypeptide chain, without parsing it into the individual proteins before predictions. The proteome size is the number of proteins in the proteome of that species and is shown in log base. The average fraction of disordered residues is calculated by averaging the fraction of disordered residues of each sequence over the all sequences of that species. Disorder prediction is evaluated by PONDR-VSL2B. This figure is reproduced from Reproduced with permission form ref. [3].
Figure 4.
Correlation between the intrinsic disorder content and proteome size for 3,484 species from viruses, archaea, bacteria, and eukaryotes. Each symbol indicates a species. There are totally six groups of species: viruses expressing one polyprotein precursor (small red circles filled with blue), other viruses (small red circles), bacteria (small green circles), archaea (blue circles), unicellular eukaryotes (brown squares), and multicellular eukaryotes (pink triangles). Each viral polyprotein was analyzed as a single polypeptide chain, without parsing it into the individual proteins before predictions. The proteome size is the number of proteins in the proteome of that species and is shown in log base. The average fraction of disordered residues is calculated by averaging the fraction of disordered residues of each sequence over the all sequences of that species. Disorder prediction is evaluated by PONDR-VSL2B. This figure is reproduced from Reproduced with permission form ref. [3].
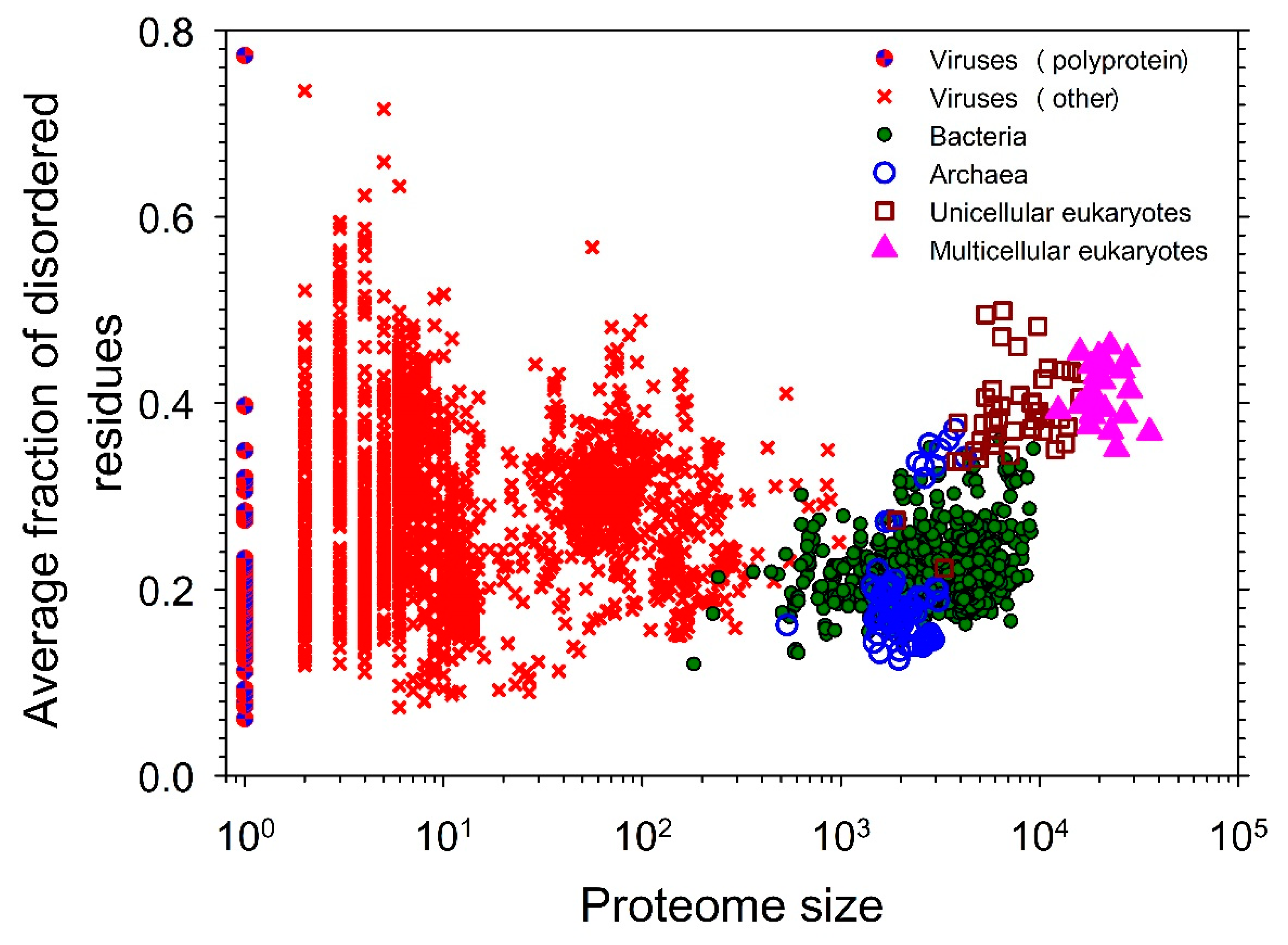
Figure 5.
Wavy pattern of the global evolution of protein intrinsic disorder. X-axis represents evolutionary time and Y-axis shows disorder content in proteins at given evolutionary time point. Here, primordial proteins are expected to be mostly disordered (left-hand side of the plot), proteins in LUA likely are mostly structured (center of the plot), whereas many protein in eukaryotes are either totally disordered or hybrids containing both ordered and disordered regions (right-hand side of the plot). Reproduced with permission form ref. [3].
Figure 5.
Wavy pattern of the global evolution of protein intrinsic disorder. X-axis represents evolutionary time and Y-axis shows disorder content in proteins at given evolutionary time point. Here, primordial proteins are expected to be mostly disordered (left-hand side of the plot), proteins in LUA likely are mostly structured (center of the plot), whereas many protein in eukaryotes are either totally disordered or hybrids containing both ordered and disordered regions (right-hand side of the plot). Reproduced with permission form ref. [3].
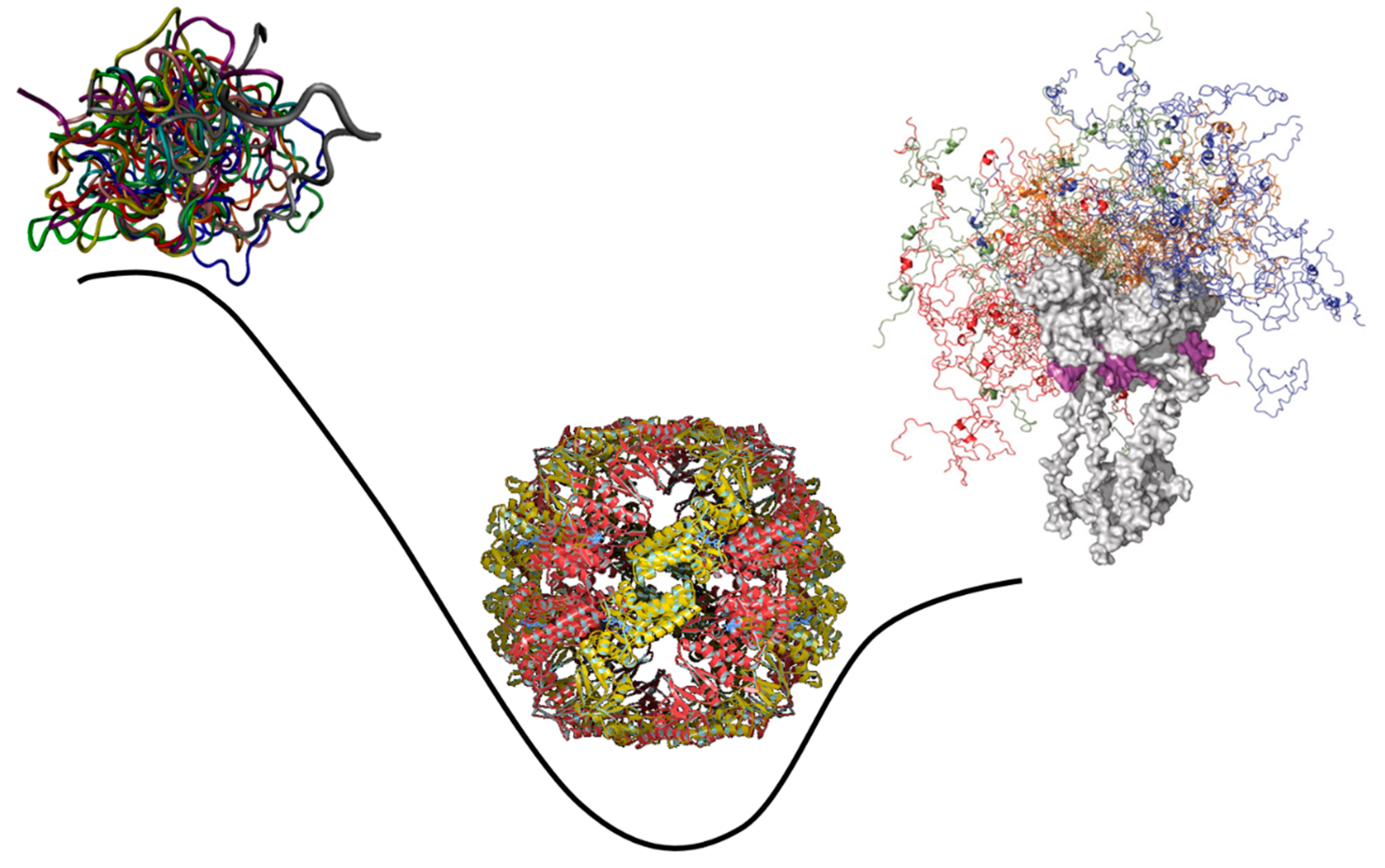
Figure 6.
Intrinsic disorder in spliceosomal proteins. A. 3D structural model generated for one of the moon-lighting spliceosomal proteins RBFOX2 (UniProt ID: O43251) by AlphaFold [156]. Structure is colored according to the model confidence. B. Per-residue intrinsic disorder profile of RBFOX2 generated by RIDAO [157]. C. RIDAO-generated per-residue intrinsic disorder profile of spliceosomal protein SRRM2 (UniProt ID: Q9UQ35) involved in the biogenesis of nuclear speckles.
Figure 6.
Intrinsic disorder in spliceosomal proteins. A. 3D structural model generated for one of the moon-lighting spliceosomal proteins RBFOX2 (UniProt ID: O43251) by AlphaFold [156]. Structure is colored according to the model confidence. B. Per-residue intrinsic disorder profile of RBFOX2 generated by RIDAO [157]. C. RIDAO-generated per-residue intrinsic disorder profile of spliceosomal protein SRRM2 (UniProt ID: Q9UQ35) involved in the biogenesis of nuclear speckles.
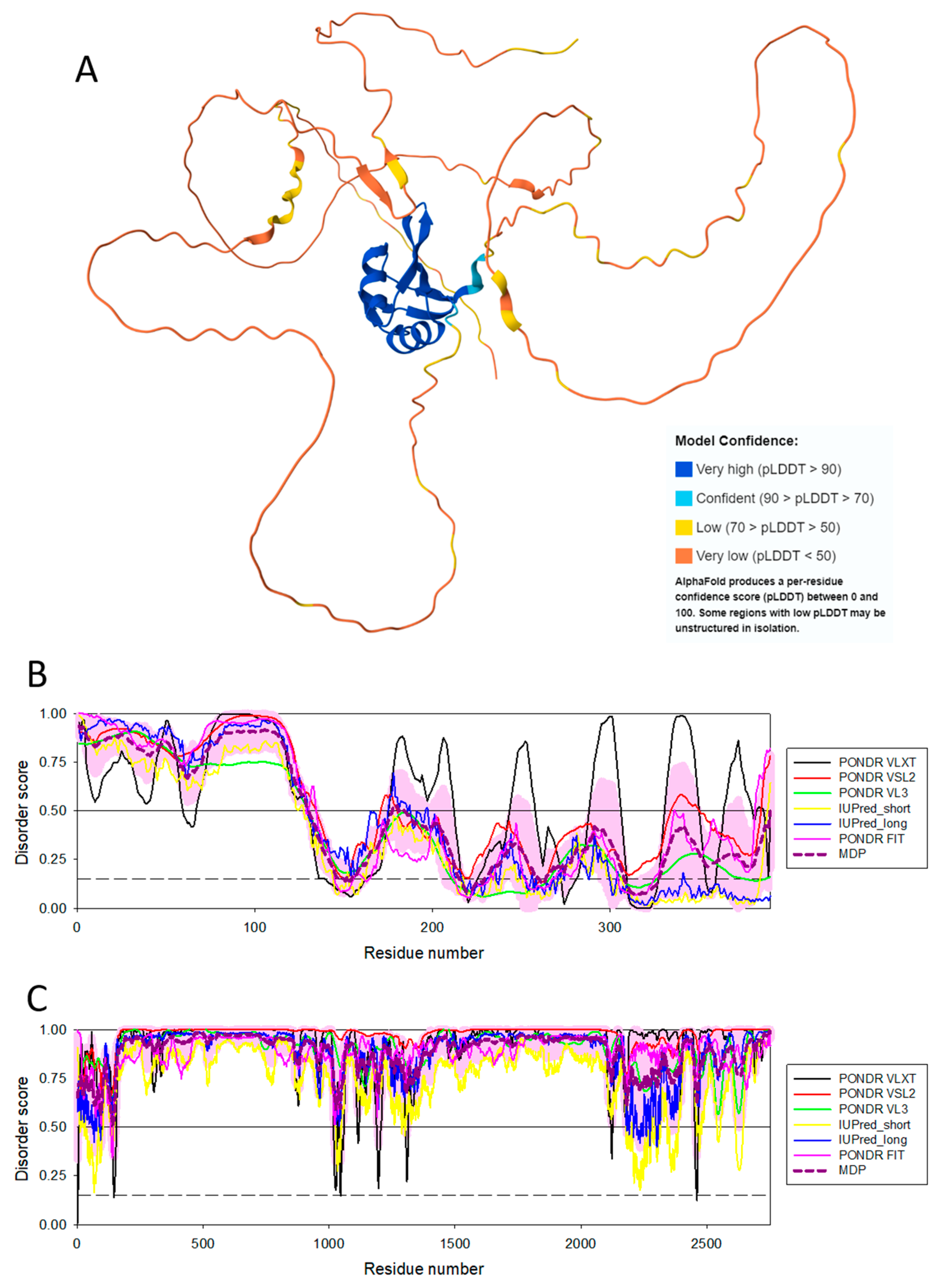
Figure 7.
Multifactorial intrinsic disorder analysis of the entire proteome of amoeboid holozoan Capsaspora owczarzaki containing 9794 proteins. A. PONDR® VSL2 Score vs. VSL2 PONDR® (%) analysis. PONDR® VSL2 (%) is a percent of predicted intrinsically disordered residues (PPIDR), i.e., residues with disorder scores above 0.5. PONDR® VSL2 score is the average disorder score (ADS) for a protein. Based on these parameters, query proteins are classified as ordered (PPIDR < 10%; ADS <0.15), moderately disordered (10% ≤ PPIDR < 30%; 0.15 ≤ ADS < 0.5), and highly disordered (PPIDR ≥ 30%; ADS ≥ 0.5). Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which the predictors disagree with each other. The boundaries of the colored regions represent arbitrary and accepted cutoffs for ADS (y-axis) and the percentage of predicted disordered residues (PPIDR; x-axis). For comparison, in human proteome, 0.4%, 5.1%, 33.7%, 21.0%, and 40.1% proteins are located within blue, light blue, pink, light pink, and red segments, respectively. This distribution observed in human proteome is remarkably close to the distribution reported here for the C. owczarzaki proteins. B. Charge-Hydropathy and Cumulative Distribution Function (CH-CDF) analysis of C. owczarzaki proteins. The CH-CDF plot is a two-dimensional representation that integrates both the CH plot, which correlates a protein's net charge and hydrophobicity with its structural order, and the CDF, which cumulates disorder predictions from the N-terminus to the C-terminus of a protein, offering insight into the distribution of disorder residues. The Y-axis (ΔCH) represents the protein's distance from the CH boundary, indicating the balance between charge and hydrophobicity, while the X-axis (ΔCDF) represents the deviation of a protein's disorder frequency from the CDF boundary. Proteins are then stratified into four quadrants: Quadrant 1 (bottom right) indicates proteins likely to be structured; Quadrant 2 (bottom left) includes proteins that may be in a molten globule state or lack a unique 3D structure; Quadrant 3 (top left) consists of proteins predicted to be highly disordered; Quadrant 4 (top right) captures proteins that present a mixed prediction of being disordered according to CH but ordered according to CDF. For comparison, 59.1%, 25.5%, 12.3% and 3.1% of human proteins are located within the quadrants Q1, Q2, Q3, and Q4, respectively. This indicates that although the C. owczarzaki and human proteomes contain comparable fractions of ordered proteins, but there are noticeably more native molten globules and noticeably less highly disordered proteins in the C. owczarzaki proteome.
Figure 7.
Multifactorial intrinsic disorder analysis of the entire proteome of amoeboid holozoan Capsaspora owczarzaki containing 9794 proteins. A. PONDR® VSL2 Score vs. VSL2 PONDR® (%) analysis. PONDR® VSL2 (%) is a percent of predicted intrinsically disordered residues (PPIDR), i.e., residues with disorder scores above 0.5. PONDR® VSL2 score is the average disorder score (ADS) for a protein. Based on these parameters, query proteins are classified as ordered (PPIDR < 10%; ADS <0.15), moderately disordered (10% ≤ PPIDR < 30%; 0.15 ≤ ADS < 0.5), and highly disordered (PPIDR ≥ 30%; ADS ≥ 0.5). Color blocks indicate regions in which proteins are mostly ordered (blue and light blue), moderately disordered (pink and light pink), or mostly disordered (red). If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect areas in which the predictors disagree with each other. The boundaries of the colored regions represent arbitrary and accepted cutoffs for ADS (y-axis) and the percentage of predicted disordered residues (PPIDR; x-axis). For comparison, in human proteome, 0.4%, 5.1%, 33.7%, 21.0%, and 40.1% proteins are located within blue, light blue, pink, light pink, and red segments, respectively. This distribution observed in human proteome is remarkably close to the distribution reported here for the C. owczarzaki proteins. B. Charge-Hydropathy and Cumulative Distribution Function (CH-CDF) analysis of C. owczarzaki proteins. The CH-CDF plot is a two-dimensional representation that integrates both the CH plot, which correlates a protein's net charge and hydrophobicity with its structural order, and the CDF, which cumulates disorder predictions from the N-terminus to the C-terminus of a protein, offering insight into the distribution of disorder residues. The Y-axis (ΔCH) represents the protein's distance from the CH boundary, indicating the balance between charge and hydrophobicity, while the X-axis (ΔCDF) represents the deviation of a protein's disorder frequency from the CDF boundary. Proteins are then stratified into four quadrants: Quadrant 1 (bottom right) indicates proteins likely to be structured; Quadrant 2 (bottom left) includes proteins that may be in a molten globule state or lack a unique 3D structure; Quadrant 3 (top left) consists of proteins predicted to be highly disordered; Quadrant 4 (top right) captures proteins that present a mixed prediction of being disordered according to CH but ordered according to CDF. For comparison, 59.1%, 25.5%, 12.3% and 3.1% of human proteins are located within the quadrants Q1, Q2, Q3, and Q4, respectively. This indicates that although the C. owczarzaki and human proteomes contain comparable fractions of ordered proteins, but there are noticeably more native molten globules and noticeably less highly disordered proteins in the C. owczarzaki proteome.
Figure 8.
3D structural model generated by AlphaFold [156] for mouse GATA1 (UniProt ID: P17679; A), GATA2 (UniProt ID: O09100; B), and PU.1 (UniProt ID: P17679; C) proteins. Structures are colored according to the model confidence, with blue, cyan, yellow, and orange colors corresponding to the regions with very high, high, low, and very low cinfidence, respectively.
Figure 8.
3D structural model generated by AlphaFold [156] for mouse GATA1 (UniProt ID: P17679; A), GATA2 (UniProt ID: O09100; B), and PU.1 (UniProt ID: P17679; C) proteins. Structures are colored according to the model confidence, with blue, cyan, yellow, and orange colors corresponding to the regions with very high, high, low, and very low cinfidence, respectively.
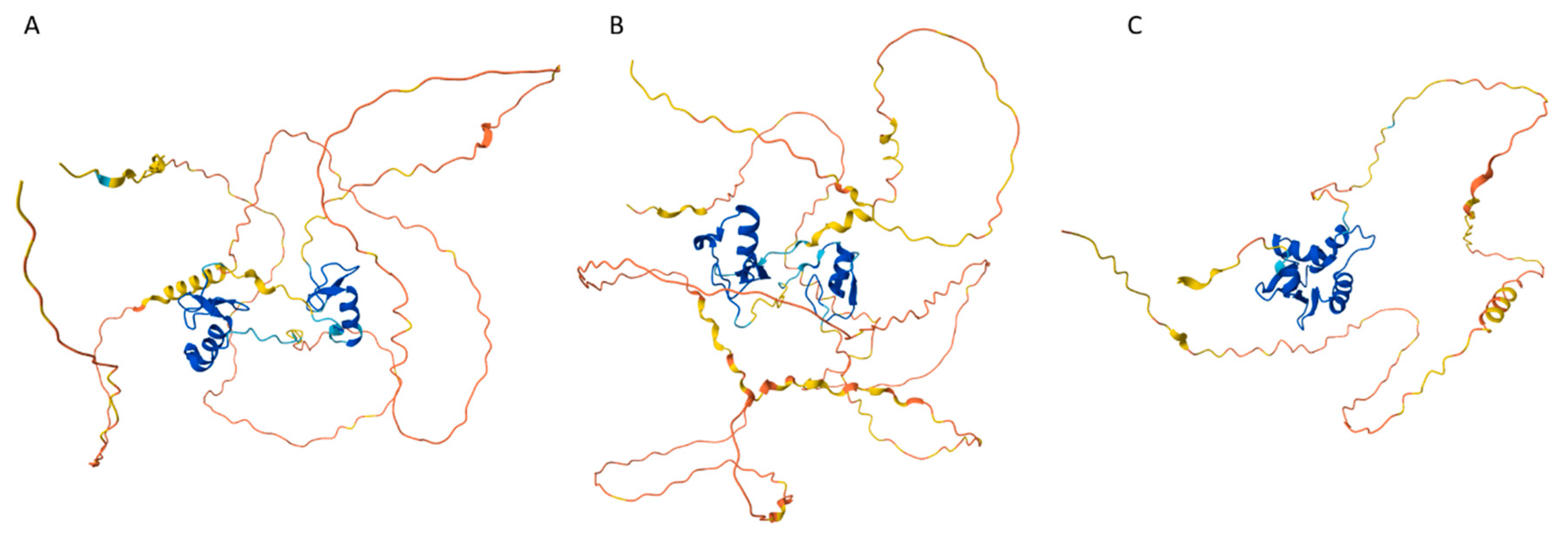
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



