
Preprint
Review
CRISPR-Cas Systems Associated with Electrolyte-Gated Graphene-Based Transistors: How They Work and How to Combine Them
Altmetrics
Downloads
87
Views
48
Comments
0
A peer-reviewed article of this preprint also exists.




Submitted:
29 September 2024
Posted:
30 September 2024
You are already at the latest version
Alerts
Abstract
In this review, recent advances in the combination of CRISPR-Cas systems with graphene-based electrolyte-gated transistors are discussed in detail. In a first part, the functioning of CRISPR-Cas systems is briefly explained, as well as the most common ways to convert their molecular activity into measurable signals. Besides optical means, conventional electrochemical transducers are also developed. However, it seems that the incorporation of CRISPR/Cas systems into transistor devices could be extremely powerful, as the former provides molecular amplification, while the latter provides electrical amplification; combined, the two could help to advance in terms of sensitivity and compete with conventional PCR assays. Today, organic transistors suffer from poor stability in biological media, where graphene materials perform better by being extremely sensitive to their chemical environment while being stable. The need for fast and inexpensive sensors to detect viral RNA arose on the occasion of the COVID-19 crisis, but many other RNA viruses are of interest, such as dengue, hepatitis C, hepatitis E, West Nile fever, Ebola, polio, for which detection means are needed.
Keywords:
Subject: Chemistry and Materials Science - Analytical Chemistry
1. Introduction
Infectious diseases occur when a micro-organism enters the human body, whether in the form of a bacteria, fungi, parasites or viruses. Among the latter, SARS-Cov-2 but also Ebola, Zika, the human immunodeficiency virus (HIV), the various hepatitis viruses and the influenza virus. Despite constant progress in detecting them in the early stages of infection, established and emerging viruses remain major causes of human pathologies, with dramatic consequences: in addition to acute illnesses, viruses are responsible for at least 15-20% of human cancers (liver viruses, papillomaviruses, etc.), and are implicated in numerous neurological disorders and chronic diseases [1].
One of the many challenges in the fight against virus-mediated diseases is the ability to detect viruses in the early stages of infection. It is therefore necessary to find alternatives to conventional laboratory detection methods, moving towards rapid, accurate and easy-to-use detection schemes, otherwise known as Point-Of-Care (POC) devices. These POC devices, whose development has increased sharply in recent years, meet certain criteria defined by the WHO (Word Health Organization) under the REASSURED guideline: Real-time connectivity, Ease of specimen collection, Affordable, Sensitive, Specific, User-friendly, Rapid, Robust, Equipment-free and Deliverable to end users [2].
In recent years, CRISPR-Cas systems have emerged as innovative new tools in molecular diagnostics and have received increasing attention as a powerful tool in the fight against viral infections. These systems are capable of recognizing a specific nucleic acid sequence which activates an enzymatic hydrolysis activity. For example, one CRISPR-Cas13a complex is able to cleave up to 100 RNA strands. This amplification property has the potential to address the major challenges associated with nucleic acid detection and improve the insufficient sensitivity of current alternatives to qPCR (quantitative Polymerase Chain Reaction). The first Cas13- or Cas12-based detection platforms (Specific High-Sensitivity Enzymatic Report UnlOCKing: SHERLOCK), used for the detection of SARS-CoV-2, demonstrated high specificity and sensitivity compared to conventional techniques [3]. However, this approach has certain limitations, notably the presence of multi-step nucleic acid amplification and additional fluorescent labeling, which can affect precise quantification of the RNA target, and increase reaction time and the number of reagents required.
To add another amplification stage to that of the CRISPR-Cas, transistors and more particularly Electrolyte-Gated Field Effect Transistors (EGFETs) offer new possibilities due to their ability to amplify a local electrostatic event into a measurable current or voltage change, in a aqueous environment. In recent years, EGFET biosensors, and in particular those based on graphene (EGGFETs), have received increasing attention because they benefit from the 2D (two-dimensional) nature of graphene's very thin, sensitive layer, the high mobility of its charge carriers and its excellent biocompatibility with biological samples and their stability in biological solutions. As a result, EGGFET biosensors are seen as ideal platforms for enhancing the performance of CRISPR-Cas-mediated devices.
This article reviews the current state of the art in this field. First, the mechanism of a viral infection is explained, along with the various specific biomarkers expressed during such infections and their respective detection techniques. The value of CRISPR-Cas complexes for nucleic acid detection is then presented. Next, the various biosensors, and in particular EGFETs, are introduced, along with their characteristics. Their suitability for the detection of biological species in solution is discussed, and several examples of nucleic acid detection are given. Finally, the various biosensors based on EGFETs in combination with CRISPR-Cas systems are presented, reviewed and discussed.
2. Current Detection Methods of Viral Infections
Viruses are pathogenic organisms that affect human health to varying degrees, ranging from simple discomfort and mild symptoms such as fever and headaches, to respiratory difficulties that can lead to irreversible trauma [4]. What's more, an uncontrolled viral infection can rapidly turn into a pandemic if left unchecked, with health, social and economic consequences. A rapid and robust means of detection is therefore essential to combat the spread of viruses. The mechanisms of infection by a viral organism, as well as current detection methods against these pathogens, are detailed below.
2.1. Current Detection Methods for Viral Infections
2.1.1. General Information on Detecting Viral Infections
During the course of an infectious disease, there are a number of techniques for detecting the presence of an invading virus; these fall into several categories. Direct methods, such as cell culture and microscopy, enable the presence of the virus to be observed visually. Other methods, which can be described as indirect, exploit the presence of various biological compounds associated with a viral infection, known as viral biomarkers. The main biomarkers are antigens and antibodies, detected by immunological methods, as well as genomic RNA and/or DNA, detectable by nucleic acid amplification techniques; the most widespread being PCR (Polymerase Chain Reaction). These indirect methods appear to be the most suitable for the development of sensitive, specific, portable, rapid and easy-to-use diagnostic platforms [5].
Direct Methods
Electron microscopy enables visual observation of viruses, including their structure. It is from this structure that viruses are often named. The coronavirus family was named as such because all the viruses in it have a crown-shaped structure, linked to the presence and specific arrangement of its S (surface) proteins on the membrane [6]. Apart from the visual study of viral particles or certain events in the replication cycle, this technique is little used in diagnosis.
Cell culture is generally the reference test for identifying micro-organisms but results often require several days or weeks. Cell culture isolation is the most sensitive diagnostic tool, since a single viral particle can theoretically result in a positive culture. Although it has the highest sensitivity, this technique is slow, delicate and demanding, making it unsuitable for rapid, sensitive, specific and easy-to-use diagnostic platforms known as Point-of-Care devices (POCs, as defined in the introduction).
Indirect Methods
Immunological detection methods are based on the affinity between viral antigens and their specific antibodies. An antigen is defined as any biological species on the viral particle (a protein, for example), which is capable of triggering an immune response. To do this, the host will create specific antibodies, which will be directed against these antigens, to which they will bind and trigger the immune response with the help of lymphocytes. Among immunological tests, we can cite the case of serological tests, which were used extensively during the SARS-CoV-2 pandemic, using the ELISA (Enzyme Linked ImmunoSorbent Assay) method [7]. The procedure is quite simple: two drops of physiological liquid are brought into contact with a solution containing the specific SARS-CoV-2 antigen, and if the patient has antibodies, an antigen-antibody complex is formed, causing the solution to stain. The main drawback of serological tests is their limited sensitivity at an early stage of infection, when the host has not yet developed specific antibodies. In the case of SARS-CoV-2 infection, it has been shown that antibody production begins in the first week and is generally detectable from the second week onwards [8,9]. In other words, antibodies are only detectable when the infection is on the wane or has already been repelled by the immune system. Therefore, these methods cannot help to contain the spread of a virus.
2.1.2. Nucleic Acid Amplification and Detection Methods
Among the biomarkers that testify to the presence of a viral micro-organism, RNA and DNA are among those most relevant for detecting infectious diseases. Although both carry genetic information, their functions are quite different. DNA constitutes the genetic heritage of a micro-organism, be it a virus, prokaryote or eukaryote. Most of it is contained in the nucleus and is highly stable due to its double-helix structure, making it a kind of genetic trace of the presence of a virus. So, in the event of a viral infection, DNA detection is tantamount to knowing whether the host has been infected by the virus. It does not give precise temporal information on the stage of the disease or its evolution; the DNA will always be present, even once the infection is over. Viral RNA, on the other hand, exists as messenger RNA to code viral proteins, thanks to ribosomes. Its lifespan is very short, from a few minutes to a few days at most, due to the RNA enzymes present in the cytoplasm, which degrade it. Thus, the presence of viral RNA indicates an ongoing infection. Quantifying and monitoring RNA concentration can also help track the evolution of a disease. This is why the detection of nucleic acids, and more specifically of RNAs, is widely used to detect and monitor the evolution of viral infections [10].
Detection methods based on nucleic acids, either DNA or RNA, are generally specific and highly sensitive, and can be used for all categories of infectious micro-organisms. To perform nucleic acid detection, it is first necessary to know the specific sequence of the micro-organism to be detected. This process is called sequencing and involves determining the nucleotide sequence of the DNA or RNA present in a given micro-organism, in order to isolate a unique nucleic acid sequence specific to it. The first species to be sequenced were viruses, as they have small genomes, mostly less than 10 kilobases (or base pairs). The SARS-CoV-2 virus has a genome of 30 kilobases, making it one of the larger viruses. By way of comparison, the genome size of bacteria is of the order of a few million base pairs, and of a few billion in eukaryotic cells. Once sequencing has been completed, it is possible to use the infectious micro-organism's gene sequences to create specific primers, which are then used to amplify the target DNA or RNA. Most nucleic acid detection techniques rely on prior amplification, as DNA and RNA concentrations are very low in biological samples [11].
PCR (Polymerase Chain Reaction) is the most widely used method for amplifying nucleic acids, whether DNA or RNA (in the case of RNA, it's called RT-PCR for Transcription Reverse-PCR). It was the method most widely used during the SAR-CoV-2 pandemic to detect viral RNAs in biological samples and is described in detail in Figure 1.
The PCR process involves several dozen cycles, each divided into 3 steps: denaturation of DNA strands, primer hybridization and polymerase elongation. If the target to be detected is RNA, a preliminary step of transcribing RNA into DNA by a reverse transcriptase enzyme is necessary. DNA denaturation involves heating the sample to around 95°C to separate the two complementary strands. Primers specific to the target DNA are then hybridized to the separated DNA strands. This step is called primer hybridization and takes place between 40 and 65°C, depending on primer size and sequence. These primers serve as a starting point for DNA polymerase enzymes, which synthesize a complementary copy of the DNA strands in a third step taking place at around 70°C. Each PCR cycle doubles the number of starting DNA strands, in less than 10 minutes. These PCR cycles are repeated between 20 and 50 times to obtain sufficient copies of the target sequence. PCR is an amplification method, but not a detection method so that it is not possible to determine the presence or concentration of nucleic acids in a biological sample. To obtain a quantification of the nucleic acid sequence sought, it is necessary to use qPCR (quantitative PCR), also known as real-time PCR.
qPCR is a variant of PCR in which a fluorescent DNA probe is added to the reaction mixture, enabling real-time monitoring of the number of DNA copies produced. These fluorescent probes are of various types and can bind specifically to double-stranded DNA (SYBR technology) or to a precise sequence of replicated DNA (Taqman and Beacon technology) [12]. These probes only fluoresce when they are bound to DNA chains; when they are free in solution, they are extinguished by proximity to a fluorescence quencher. Analysis of fluorescence as a function of the number of PCR cycles enables the initial RNA concentration to be determined, and the evolution of viral infection to be assessed. Although qPCR is a specific and highly sensitive method, the use of a thermocycler and high temperatures (in the range 40-95°C) does not always make it easy to use, and trained laboratory staff are required. These constraints are not compatible with the WHO's ASSURED guideline for sensitive, specific, portable and easy-to-use diagnostic devices. For this reason, isothermal temperature gene amplification methods have been developed, requiring much less instrumentation. Some of these methods use temperatures close to room temperature, such as RPA (Recombinase Polymerase Amplification, between 37 and 42°C) [13], while others require heating devices: LAMP (Loop Mediated Isothermal Amplification, between 60 and 65°C) [14], NASBA (Nucleic Acid Sequence Amplification, between 40 and 55°C) [3,15], or lesser-used ones such as SDA (Strand Displacement Amplification, 60°C) [16], HDA (Helicase-Dependent Amplification, 65°C) [17] and EXPAR (EXponential Amplification Reaction, 55°C) [18]. Like PCR, these isothermal nucleic acid amplification methods use primers, DNA polymerases and other proteins to successfully amplify DNA sequences without the denaturation step. Some of their characteristics are described in Table 1 and will not be detailed here. Although these methods are much less cumbersome from an instrumental point of view, and more suitable for field measurement, their main drawback is their lack of sensitivity. Quantification of viral nucleic acids is therefore not always possible at low concentrations. Specificity isn't always ideal either, with some methods unable to recognize differences of a single base pair in the targeted sequences.
To sum up, nucleic acids, and RNA in particular, are specific biomarkers of infectious diseases, and their quantification makes it possible to monitor the evolution of a viral infection over time. On one hand, PCR is the standard method for detecting nucleic acids, but its limitations make it difficult to use in a POC device. On the other hand, isothermal amplification methods do not yet offer sufficient sensitivity and selectivity to meet POCs needs [19]. Efforts must therefore be concentrated on developing new systems for the quantifiable and sensitive detection of viral RNAs, in diagnostic platforms that are as simple as possible, portable and easy to use. To this end, CRISPR-Cas systems are promising; they are detailed in the remainder of this sub-section.
2.2. The Use of CRISPR-Cas Complexes as a New Means of Nucleic Acid Detection
The discovery of CRISPR dates back to 1987 in Japan, when repeated DNA sequences were observed in the genome of the bacterium Escherichia Coli [21]. This CRISPR (Clustered Regularly Interspaced Spacer with Palindromic Repeats) array is composed of short, repeated nucleotide sequences called repeats, interspersed with short, variable and unique nucleotide sequences called spacers, which originate from the genetic material of viruses or phages involved in previous infections (Figure 2) [22,23]. These spacers are central to the CRISPR defense mechanism, as they provide specific immunity against phages or viruses that possess a complementary DNA sequence. Indeed, adjacent to the CRISPR network are a series of genes encoding Cas proteins (CRISPR associated). The pooling of the CRISPR network and Cas proteins enables the creation of CRISPR-Cas systems, which provide the bacterium with an immune and adaptive defense.
The immune response of the CRISPR-Cas system can be described in two stages: firstly, the acquisition of pathogenic spacers into the CRISPR network, followed by their transcription and use to trigger the immune response. In the first stage, known as gap acquisition or adaptation, viral genome sequences are captured when foreign DNA enters the cell and integrated into the CRISPR network. The CRISPR network is then enriched with a new virus-specific sequence, enabling it to protect itself against the virus - a process akin to bacterial immunization (Figure 2a) [24]. The second stage, during which immunity is implemented, can be divided into two phases. Firstly, in the guide RNA biogenesis phase, the CRISPR array is transcribed and processed to produce short RNAs each containing a sequence with a unique repeat and spacing (Figure 2b). These small RNAs are called crRNAs (CRISPR RNAs) or guide RNAs. Then, in the targeting phase, the crRNAs bind to the Cas proteins already formed, for which they will act as an antisense guide to direct these Cas nucleases towards the foreign genetic material. A catalytic site is created by pairing the crRNA on the Cas enzyme with a crRNA that has the complementary sequence to the virus DNA. When the DNA sequence complementary to the guide RNA sequence is detected by hybridization of the two nucleic acid chains, the CRISPR-Cas complex triggers its nuclease activity, splitting the invading DNA strand in two. The cut DNA strand is then processed and degraded by other enzymes, and the bacterium therefore repels the infection. This property of hydrolyzing DNA strands at the site of regrowth is known as cis-cleavage.
The greatest strength of this technology lies in its specificity: the CRISPR-Cas complex can scan an entire DNA genome in just a few minutes and bind very specifically to the complementary sequence of its guide RNA [25]. This technology is now becoming increasingly widespread in the field of diagnostics and is helping to boost sensor performance. Different types of nucleic acid (RNA or DNA) detection based on CRISPR-Cas are presented below.
2.2.1. Classification of CRISPR-Cas Systems and Complexes of Interest
CRISPR-Cas systems are extremely diverse, distinguished into two classes, six types and 21 subtypes (Figure 3) [25,26,27]. This classification is continually evolving, with new subtypes being discovered every year. The classification of CRISPR-Cas systems is based on the architecture of the signature proteins of each type: class 1 CRISPR-Cas systems are defined by the use of multi-protein vectors, i.e. the complex is composed of multiple proteins. These are the most abundant, accounting for 90% of CRISPR-Cas systems; they are found in bacteria and archaea (prokaryotic single-cell microorganisms) [26]. Class 2 CRISPR-Cas systems account for 10% of CRISPR-Cas systems, and their effector consists of a single multi-domain protein; they are found only in bacteria.
The CRISPR-Cas tools most widely used and simplest to implement are those of class 2 (type II, type V and type VI), since they contain only a single Cas protein. The first to be discovered was CRISPR-Cas9, by Jennifer Doudna and Emmanuelle Charpentier in 2012, revolutionizing genome editing [29]. The main difference with the genome-editing systems used at the time lies in the recognition of the sequence to be edited. Genome-editing systems such as mega-nucleases [30], zinc finger nucleases [31] or TALENs (Transcription Acitvator Like-Effector) [32] use proteins to recognize the DNA sequence to be edited, whereas CRISPR-Cas9 uses a strand of guide RNA [33,34]. Given that it is much easier to produce synthetic RNAs than proteins, CRISPR-Cas9 has quickly become indispensable for genome editing; Jennifer Doudna and Emmanuelle Charpentier won the Nobel Prize in 2020 [35].
Due to its ease of use, CRISPR-Cas9 has been used since 2013 for genetic engineering in human cells or bacteria, whether to regulate gene expression, introduce mutations or also as a genetic marker [36]. The high specificity of CRISPR-Cas9's guide RNA towards the target DNA has enabled it to be used also in the field of biosensors, as Pardee et al. who combined isothermal gene amplification (LAMP) with the nuclease activity of CRISPR-Cas9 to detect the Zika virus [15]. The specificity of CRISPR-Cas technology enabled them to detect and differentiate between American and African strains of the Zika virus, with a resolution of just one nucleotide [15,37].
After 2014, other class 2 CRISPR-Cas systems came to light, with the discovery of the Cas12, Cas13 and Cas14 enzymes [38,39,40,41]. Unlike Cas9, these enzymes have two distinct catalytic sites that grant them non-specific RNAse (Cas13) or DNAse (Cas12 and Cas14) activity [19]. In other words, once activated, Cas13 can non-selectively cut any RNA strand it encounters, leading to the non-specific degradation of all RNAs in solution (Figure 4). Cas12 and Cas14 have a similar nuclease activity towards DNA strands, as they are DNAses. However, their operation remains very similar to that of the other CRISPR-Cas systems described above: a guide RNA binds to a Cas enzyme at the first catalytic site, and searches for a DNA sequence (double-stranded for Cas12, single-stranded for Cas14) or RNA sequence (single-stranded for Cas13) complementary to its guide, referred to as the target RNA (or DNA) sequence. Unlike the process described above in the context of the bacterial immune response, the target RNA hybridizes to the guide RNA, but is not degraded by it, serving as a trigger for the appearance of CRISPR-Cas' second catalytic site. Once the target RNA has hybridized to the guide RNA, the Cas enzyme undergoes a conformational change to activate its nuclease domains and bring out its second catalytic site. The CRISPR-Cas complex is then activated and can cut any strand of RNA (or DNA) it encounters, including other target RNAs. Figure 4 illustrates the mechanism of Cas13 activation, from transcription of the CRISPR network and Cas13-encoding genes, to activation of the complex's RNAse, which hydrolyzes the phosphodiester bridges adjacent to the U (Uracile) nucleotides. Both catalytic sites are present, the first in the REC domain (for Recognition of Target RNA), the second in the NUC domain (for Nuclease).
These discoveries have had a major impact on the nucleic acid detection methods used to date. The "gold standard" method is still PCR, but in the context of creating portable devices for nucleic acid detection, it suffers from certain limitations such as its rather cumbersome instrumentation. By combining the specificity of CRISPR-Cas systems with isothermal nucleic acid amplification methods such as RPA, it becomes possible to create specific, rapid, sensitive and portable devices for DNA or RNA detection [43,44,45]. Some of these new platforms, which use Cas13 such as SHERLOCK, CARMEN or CARVER are detailed below [46,47,48,49].
2.2.2. SHERLOCK: Specific High Sensitivity Reporter Unlocking
The SHERLOCK (Specific High Sensitivity Reporter unLOCKing) method is the first biosensor based on a CRISPR-Cas system to have been implemented. The aim of this method is to detect a DNA or RNA fragment using isothermal RPA gene amplification and the RNAse activity of CRISPR-Cas13.
First, the DNA or RNA fragment to be analyzed must be amplified, as its concentration is initially too low. This preliminary step is an isothermal amplification by a recombinase polymerase (RPA). It takes place at around 37-42°C and enables DNA sequences present in solution to be multiplied in a non-specific way. If the target is RNA, a reverse transcription step is required prior to RPA, in which case the amplification is called RT-RPA (Reverse Transcriptase-RPA). Next, all the amplified DNA fragments are transcribed back into RNA using a T7 RNA Polymerase enzyme, as CRISPR-Cas13 only recognizes RNAs. These target RNAs are brought into contact with the CRISPR-Cas13 complexes, and the enzymatic activity of Cas13 is triggered on recognition of the target RNA complementary to the crRNA it which it has been programmed.
For the detection step, an RNA probe is introduced into the reaction medium, which can be fluorescent as shown in Figure 5. Thanks to the enzymatic activity of Cas13, the RNA probe is split into two parts, resulting in the separation of the fluorescence quencher from the fluorophore, so that the fluorophore signal becomes visible.
Introduced by Gootenberg et al., the SHERLOCK method reduced the detection limit of CRISPR-Cas13 to 10 aM, whereas the detection limit of CRISPR-Cas13 without gene amplification was 50 fM [3,50,51]. The detection of RNA from viruses such as zika virus (ZIKV), dengue virus (DENV), bacterial DNA and cancer mutations in DNA fragments from cells have also been demonstrated using this method [3]. The method was quickly adopted by other laboratories for a variety of applications, such as the detection of Lassa and Ebola viruses [52], malaria [53] and SARS-CoV-2 [54]. The results obtained by SHERLOCK are equivalent or even better than those obtained by qPCR, especially as the instrumental resources are much smaller. A second version of the SHERLOCK platform has been developed by the same group, called SHERLOCKv2, and is even more robust. It enables the simultaneous detection of 3 different RNA targets and 1 DNA target at concentrations as low as 8 zM under certain conditions [55]. In addition, the detection system has evolved from fluorescence-based measurement to colorimetric reading on an immunochromatographic paper strip, which requires no special equipment (as with self-testing for SARS-CoV-2) (Figure 6). For SHERLOCKv2, amplification takes place in a single step: the biological sample is brought into contact with a solution containing crRNA-Cas13 complexes which is amplified by RPA, then this solution is applied directly to the test strip. This was the first portable, simple, rapid and cost-effective CRISPR-Cas-based device offering sensitivity similar to qPCR.
CARVER (Cas13-Assisted Restriction of Viral Expression and Readout) and CARMEN (Combinatorial Arrayed Reactions for Multiplexed Evaluation of Nucleic acids) are nucleic acid detection platforms derived from SHERLOCK [56]. These new platforms use the same amplification and detection process as SHERLOCK but are much more capable of multiplexing. For example, CARVER is inspired by the SHERLOCK method, using CRISPRCas13 to detect the viral RNAs of 3 different viruses simultaneously: influenza A, lymphocytic choriomeningitis and vesicular stomatitis [57]. CARMEN, meanwhile, can simultaneously detect and differentiate 169 human-associated viruses using virus-specific crRNAs [47]. Other platforms also exist for nucleic acid detection with other class 2 CRISPR-Cas systems, such as DETECTR or HOLMES for Cas12 [58,59,60,61,62]. NASBACC or CRISPREXPAR for Cas9 [15,63], or HARRY for Cas14 [64]. Most of these methods are based on the same two-step detection principle: a first step of gene amplification of the target to be analyzed, followed by CRISPR-Cas detection by fluorescence or colorimetric reading on an immunochromatography strip. These methods are designed to detect DNA and will not be discussed in detail here. However, they operate on a very similar principle to SHERLOCK.
2.2.3. Detection of Nucleic Acids by CRISPR-Cas13 without Gene Amplification
Although the methods presented above, such as SHERLOCKv2, are highly sensitive, selective and robust, they require gene amplification, which is not always straightforward. What's more, when the target to be detected is an RNA, reverse transcription into DNA is required prior to gene amplification, followed by further transcription of the amplified DNA so that it can bind to CRISPR-Cas13. All these steps can lead to a loss of measurement specificity, resulting in "false positive" results [65]. Doing away with gene amplification increases specificity and simplifies the detection method. In the case of detection without gene amplification by CRISPR-Cas, Cas13 is the most relevant class 2 enzyme, as it is the only one that recognizes RNA, the others recognizing single- or double-stranded DNA [38]. As described above, detecting RNA rather than DNA is more accurate for quantifying the evolution of an infection, whether viral or bacterial. Currently, RNA detection by CRISPR-Cas13 without gene amplification cannot compete with qPCR, as the limit of detection (LOD) is around 1 fM, which is three orders of magnitude higher than that of PCR [66]. In 2016, the first CRISPR-Cas13-based sensors without gene amplification were developed for the detection of bacteriophage RNA and human cell mRNA for concentrations down to 1 pM [50,51]. This LOD was extended to 50 fM by Gootenberg et al. for the detection of Zika virus and dengue virus RNA [3]. Subsequently, East-Seletsky et al. succeeded in detecting RNA at a limit of 10 fM (6000 copies/μL) [67]. Qin et al. succeeded in detecting Ebola virus RNA up to 50 fM [68]. All these sensors use a fluorophore-extinguisher RNA probe with a fluorescence reader to measure CRISPR-Cas13 activity. Fozouni et al. further lowered the LOD by using two different crRNAs targeting two SARS-CoV-2 genes for detection, achieving an LOD of around 1 fM (270 copies/uL) [69]. The use of different crRNAs enabled them to target more genes present in solution, which tends to lower the LOD. Although this combination of crRNAs improves the sensitivity of the devices, it also increases the possibility of detecting non-targeted RNA and may lead to a false-positive result or an inaccurate measurement when a crRNA does not correspond precisely to the viral sequence of the target RNA [69]. In addition, these authors modified a cell phone with a microscope so that it could be used as a fluorescence reader, again with the aim of obtaining low-cost, portable measuring instruments. In the same vein, Katzmeier et al. in 2019 created a pocket-sized fluorescence reader costing less than $15 that detects RNA by CRISPR-Cas13 with an LOD of 3.7 nM [70].
Other types of CRISPR-Cas13 sensor, such as the one proposed by Johnston et al. emerged during the SARS-Cov-2 pandemic [71]. Their sensor is based on an electrode functionalized by RNA strands that are linked at their other end to glucose oxidase (an enzyme that catalyzes the oxidation of glucose to H2O2). In the presence of CRISPR-Cas13, the RNA strands that bind glucose oxidase to the electrode are hydrolyzed; the current measured at the electrode (due to electrochemical recycling of the enzyme) falls sharply because the enzyme is lifted away. Their device can detect two specific SARS-CoV-2 genes, the envelope gene (E gene) and the gene responsible for viral mRNA transcription (RdRP gene) at concentrations of 4 and 15 fM, respectively. Their sensor also incorporates a microfluidic channel that enables rapid SAR-CoV-2 detection in 30 minutes in biological samples where these genes are concentrated, and at a cost of less than €3 per sensor. All these devices are detailed in Table 2.
To summarize very quickly, diagnostics based on CRISPR-Cas13 technology have evolved from an experimental nucleic acid detection tool to a clinically relevant diagnostic technology for rapid, sensitive and easy RNA detection. The best example being SHERLOCKv2, which uses colorimetric strips that can detect trace amounts of RNA down to 2 aM at a cost of $0.60 per strip [55]. However, several challenges remain to transform CRISPR-Cas technology into a robust diagnostic tool for emerging diseases and pathogens. One of the main shortcomings of current CRISPR-Cas13-based diagnostics is the reliance on an RNA pre-amplification step for targets with sub-femtomolar concentrations [19]. This step adds complexity to the diagnostic, may decrease its specificity, also increases its measurement time as well as its cost [72]. In view of these issues, it is necessary to develop tools that can exploit the RNAse activity of CRISPR-Cas13 and amplify its signal to develop devices that can compete with the sensitivity of qPCR. For this, field-effect transistors appear to be good candidates thanks to their capacitive coupling, which amplifies the phenomena taking place at their interfaces. These will be discussed in greater detail later in this chapter.
3. Electrolytic Gate Field Effect Transistors (EGFETs)
3.1. Introduction
Electrolytic Gate Field-Effect Transistors (EGFETs) are a sub-family of FETs in which the dielectric layer is replaced by an electrolyte, which may be liquid or gel. The electrolyte used acts both as an ionic conductor and as the electrical insulator inherent in the dielectric layer of FETs. EGFETs have rapidly become important elements of advanced bioelectronics, as they are stable in aqueous media, operate at low voltages (generally between -2 V and 2 V) and can translate biological events into electrical signals with good precision [73].
Like FETs, EGFETs are three-electrode devices in which the conductivity of a semiconductor material connected between two electrodes, source and drain, is modulated by a third electrode, the gate. In a conventional EGFET structure, both the semiconductor and the gate are in direct contact with the electrolyte. What's more, the gate need not face the channel, as charge modulation is due to charge accumulation or depletion at the semiconductor/electrolyte interface, which is less sensitive to field line geometry as long as the ionic conductivity of the electrolyte is sufficient. In addition to the classic FET configurations where the gate is located above the semiconductor channel (top gate configuration) or below it (bottom gate configuration), it can also be coplanar (side gate configuration) or extended to an electrolyte other than that of the semiconductor channel (extended gate configuration). The latter two configurations are highly relevant as they are simpler to manufacture, functionalize and integrate into microfluidic systems [74]. The different possible architectures for EGFETs are detailed in Figure 8.
EGFETs are classified into two distinct families, depending on the ion permeability of the semiconductor used. In the case where the semiconductor is impermeable to electrolyte ions, the application of a VG gate potential causes the displacement and accumulation of ions at the gate/electrolyte and semiconductor/electrolyte interfaces (the electrochemical double layers, EDLs). The ions at these interfaces, on the one hand, screen the gate charges and, on the other, cause the accumulation of charge carriers (mirror charges or image charges) in the semiconductor (Figure 9).
Electrochemical double layers (EDLs) can be thought of as nanometer-thick capacitors. The capacitance of each interface can be estimated by the Helmoltz equation, C ≈ kε0/λ where ε0 is the permittivity of vacuum, k is the dielectric constant of the EDL and λ is the Debye length, which can be equated with the thickness of the EDL. With λ of the order of 1 nm and k ≈ 10, a capacitance value of 1-10 µF.cm-2 can be achieved with electrolytes. These capacitance values are at least ten times greater than the capacitance values obtained with conventional solid-state dielectrics (note that the dielectric constant of water is 80 times greater than that of a conventional solid-state dielectric) [75]. This property gives rise to the main advantage of using EGFETs: their operating voltages (VD and VG) are much lower than those of OFETs (below 1 V), making them compatible with the detection of biological species in solution without the appearance of electrolysis [76].
For ion-permeable semiconductors, an EDL is formed only at the interface between the gate (polarizable electrode) and the electrolyte, under the application of a gate potential. The ions present at the semiconductor/electrolyte interface diffuse into the volume of the semiconductor film, modulating the density of free charge carriers in the transistor channel. This process is called electrochemical doping and is well reported in the literature for certain semiconductor polymers in contact with an electrolyte [77]. These transistors are called organic electrochemical transistors (OECTs). The most widely used polymer for the development of OECTs is PEDOT:PSS (poly(3,4-ethylenedioxythiophene):poly(styrene sulfonic acid)) [77]. OECTs exploit the entire volume of the semiconductor film, exhibiting a specific surface area vastly superior to that of an impermeable semiconductor. This is referred to as volume capacitance in the case of an OECT, as opposed to surface capacitance in the case of an EGFET. As a result, the gate/electrolyte interface is the interface of choice for biosensing. In fact, this interface is the one most frequently used in the various works describing OECT-based biosensing, even though some papers use the volume of the semiconductor channel for the detection of biological species (antigens [78], glucose [79], bacteria [80], viruses [81]). None, however, deals with nucleic acid detection in this way. All the work described to date uses a gate electrode (usually gold) functionalized with nucleic acid strands complementary to the targets to be detected [82,83,84,85].
Here, we focused on EGFETs with an impermeable channel. They have the advantage of presenting two functionalizable interfaces for biosensing (channel and gate), which broadens the functionalization possibilities. We will not review organic EGFETs (EGOFETs) but focus of graphene-based ones (EGGFETs).
3.2. Graphene-Based EGFETs
Discovered by Novoselov and Geim in 2004 [86,87] graphene is a material remarkable for its optical, thermal, mechanical and electrical properties. As the first two-dimensional material to be discovered, graphene has been the focus of much research into its fundamental properties and peculiarities. Naturally, researchers have integrated it into electronic devices [88]. Being stable in aqueous media and sensitive to field effect, it is a natural candidate for electrolytic gate transistors.
3.2.1. Graphene and Derivatives
Graphene is a single-layer sheet of two-dimensional carbon atoms structured in a honeycomb lattice. In fact, each carbon atom is linked to three other carbon atoms by single σ bonds. These bonds result from hybridization between the 2s, 2px and 2py orbitals, giving three coplanar sp2 hybridization orbitals (Figure 10a). The presence of non-hybridized 2pz orbitals on each carbon atom enables π-π conjugation across the entire lattice, and thus strong electron delocalization, which gives graphene its excellent electrical conductivity. In reciprocal space, the first Brillouin zone corresponds to the representation of the elementary lattice and is often used to study the electronic properties of a material (Figure 10b). The energy band structure of graphene represented in the first Brillouin zone reveals symmetry points K and K', known as Dirac points [89], or charge neutrality points (CNPs) (Figure 10c). These points are defined as the places where the conduction and valence bands meet, and where they coincide with graphene's Fermi level. Graphene is therefore a zero-bandgap material, and its charge-carrier concentration is zero at the CNP level (Figure 11a) [90].
The most interesting aspect of graphene's energy band structure is that its charge carriers can be described by a Dirac spectrum for massless fermions rather than by the usual Schrödinger equation for non-relativistic particles. Figure 11b shows the mass profile of electrons and holes as a function of their respective concentration n. Close to the Dirac point, both types of charge carrier tend to become massless quasiparticles. These particles are called Dirac fermions and have a velocity only 300 times slower than the speed of light [86]. This result leads to the unique properties of graphene, which is neither a metal nor a semiconductor and is often referred to in the literature as a semi-metal [91].
Although graphene's bandgap is zero, the gate voltage of a transistor can still modulate the density of states in graphene from low-conductivity states near the Dirac point to high-conductivity states elsewhere (Figure 11c). The application of a positive gate voltage induces electrons as charge carriers, while a negative voltage induces holes as charge carriers. This property, known as ambipolarity make graphene a perfectly suitable candidate for electronic devices.
For the realization of printed transistors, some articles report suspensions of graphene sheets in aqueous solvents, but obtaining a stable suspension remains a critical point and limited to low concentrations (< 0.5 mg.mL-1) [92]. In addition, it is necessary to use surfactants to stabilize these suspensions. An alternative to these problems lies in the use of graphene oxide (GO), which is more easily exploitable for making suspensions because its surface energy is much higher and it can be reduced after deposition to give "reduced graphene oxide" (rGO), the properties of which being close to those of graphene.
rGO has a large number of sp2 carbons but differs from pure graphene in the presence of defects such as residual oxygen groups (Figure 13). Despite these defects, rGO has good conductivity and is sensitive to the application of an electric field. In the remainder of this section, all the information provided on graphene-based transistors can also be extended to rGO-based transistors.
3.2.2. Operating Modes of EGGFETs
Figure 14a shows the structure of an EGGFET (Electrolytic Gate Graphene Field Effect Transistor) in top-gate configuration. The transistor's total capacitance is a function of the different capacitances linked to the formation of electrochemical double layers at the interfaces, as explained above. We also note the presence of a third capacitance, denoted Cq, the graphene quantum capacitance (Figure 14b) [95]. The value of this capacitance has already been determined using electrochemical methods [96], but determining its precise value remains difficult, as it is of the same order of magnitude as the other two capacitances of the transistor (it is therefore not negligible).
EGGFETs differ from organic transistors in that the channel is always conductive (transistor always "ON"). The drain current ID can nevertheless be modulated by applying a gate voltage VG but is never zero as long as the drain voltage VD is non-zero. The application of a gate voltage VG leads to the formation of two electrochemical double layers at the gate/electrolyte and graphene/electrolyte interfaces. EGGFETs are characterized by plotting the device's transfer and output curves. The output curves show a linear behavior representative of an ohmic operating regime (Figure 15a). The slope of the various output curves depends on the voltage applied to the gate, i.e. the charge carrier density in the rGO. The transfer curves reflect the ambipolar nature of the rGO. An increase in ID is the consequence of an increase in the density of majority charge carriers, whether electrons (branch "n", for VG above a voltage denoted V*) or holes (branch "p", for VG below V*). Transfer curves thus have the appearance of a V (or U) curve, characterized by a minimum current at potential V*. When VG > V*, the current in the transistor is provided by electrons (in other words, the majority carriers are electrons), and conversely when VG < V* (the majority carriers are holes). In the case of a defect-free graphene sheet, V* is ideally expected to be equal to 0, as the CNP coincides with the Fermi level. However, the Fermi level may differ from the CNP, mainly due to the presence of defects. This is the case of rGO with its residual oxygen groups, which contribute to p-doping and thus to a rightward shift of the transfer curves. A typical transfer curve is shown in Figure 15b. The Dirac cone illustrates the behavior of a p-doped transistor, with a Fermi level below the CNP. Here, V* is around 1.3 V.
3.2.3. Principle of Biosensing with EGGFETs
For EGGFETs, the detection principle is based on the modification of the charge carrier density in the rGO either by a redox process of charge transfer, or by electrostatic interactions (Figure 16). In the case of charge transfer, the target analyte (or any other redox molecule involved in molecular recognition) boosts the channel, thereby altering the Fermi level of the rGO. In the case of an electrostatic interaction, the presence of a charged target analyte at the graphene/electrolyte or grid/electrolyte interface modifies the charge distribution at these interfaces, and thus influences the charge neutrality point. As a consequence, the point V* for which ID is minimal is not then located at the same potential on the transfer curve. Particular attention has to be paid to this principle of electrostatic transduction, which can easily be applied to the detection of biological species, most of which are charged (proteins), or even polyelectrolyte-like (DNA, RNA). The vast majority of these sensors use transistors in which the graphene or rGO channel is functionalized by biological receptors (DNAs, antigens, antibodies).
3.2.4. Using EGGFETs for Biosensing
One of the first applications of EGGFETs was as pH sensors [98]. The operating principle of these sensors is based on the concentration of hydroxide (OH-) and hydronium (H3O+) ions adsorbed on the graphene surface. The concentration of these ions changes with pH, modifying the charge density at the graphene/electrolyte interface, as explained above. More explicitly, as pH decreases, the concentration of H3O+ ions adsorbed to the surface increases, attracting electrons into the channel. This is equivalent to p-type doping (p for positive, like H3O+ cations), which shifts the transfer curves to the left (lower VG). Figure 17 illustrates the operating mechanism of an EGGFET-based pH sensor. In this illustration, the accumulation of anions above the rGO causes a shift of the transfer curves to the right, and the accumulation of cations causes a shift to the left.
For biosensing purpose, it is necessary to functionalize the graphene (or rGO) channel. To achieve this, it is possible to exploit the π-conjugated network, which confers π-stacking possibiities with other π-conjugated molecules. This is the case with RNA or DNA chains that possess conjugated bases and will come to bind non-covalently on graphene. Gao et al. used this property and directly functionalized a graphene channel with DNA strands, complementary to the miRNA to be detected; the sensor can detect miRNAs in 20 minutes with a LOD of 10 fM [99].
This π-stacking property can also be used to graft molecules functionalized with a π-conjugated group such as PBASE (1-pyrenebutanoic acid succinimidyl ester), which serves as a first brick for linking biological compounds. The pyrene group of the PBASE compound interacts with graphene by π-stacking, while the carboxylic acid function is used to graft biological molecules featuring a terminal -NH2 amine group, forming a stable amide bridge linking the two compounds. Seo et al. used this method to functionalize an EGGFET with an antibody specific to the surface protein S of COVID-19 [100]. Tian et al. also used this method to graft DNA strands and detect complementary DNA with an LOD of 0.1 fM; their approach is described in Figure 18 [101].
It has been shown that this method, and more generally the use of an adsorbent spacer group, prevents "useful" nucleic bases from interacting directly with graphene by π-stacking, and leads to improved sensitivity. For example, Chan et al. highlighted that DNA bases that interact with graphene by π-stacking are not available for hybridization with target DNA strands. They therefore use a larger DNA chain, comprising a section of nitrogenous bases for immobilization on graphene and a section for hybridization with the target to be detected. DNA strands hybridized without an immobilization section tend to separate from the graphene under the influence of electrolyte flow, which is not the case for DNA strands immobilized by an ad hoc immobilization segment (Figure 19) [102].
Graphene can also be functionalized covalently. “Formic acid” graphene and “acetic acid” graphene are two compounds of interest for bioreceptor grafting (Figure 20) [103,104]. Both of these compounds have carboxylic acid groups that can be used to covalently link biological targets with an accessible -NH2 amine function. Hensel et al. have demonstrated the grafting of aptamers onto “acetic acid” graphene [103]. These authors reported that due to these functions, the distance between graphene sheets is larger, which avoids problems of steric hindrance and accessibility during amide bridge formation.
Another approach to the covalent functionalization of graphene involves the use of gold nanoparticles (AuNPs), which are spontaneously reduced on graphene (which is a reducing agent, its oxidized form being GO). AuNPs are generally formed by contacting graphene (or rGO) with an aqueous solution of chloroauric salts such as HAuCl4, where gold is in Au(III) oxidation state. On contact with graphene, Au(III) is spontaneously reduced to Au(0) by a sp2 carbon atom, given that the redox potential of Au(III) is 1.0 V and that of graphene 0.22 V (vs. ESH) [105]. AuNPs serve as anchoring points for biological molecules with a thiol group -SH. The main advantage of this functionalization route over those based on π-stacking interactions is that it spaces the probe from the graphene, making it more available. This approach was used by Cai et al. to detect RNA by hybridization with a complementary APN (peptide nucleic acid, uncharged) strand with an LOD of 10 fM [106]. It is described in Figure 21a.
Rather than functionalizing the graphene (or rGO), it is also possible to functionalize the gate electrode. However, the gate of EGGFET transistors is not always functionalizable, as it is often replaced by a non-polarizable Ag/AgCl reference electrode, which makes it possible to control the actual potential applied whatever the electrolyte solution contains, and thus to make measurements more reproducible. Nevertheless, some works such as those by Li et al. describe the use of a gold grid functionalized with DNA strands (Figure 22) [107]. Charged species recognition phenomena at the grid/electrolyte interface modify the charge arrangement and influence the electric field, inducing an evolution of the transistor output current ID. A LOD of 1 fM has been obtained for DNA detection by these authors [107].
4. CRISPR-Cas and Transistors
CRISPR-Cas complexes are relevant systems for biosensing because they involve two sites for recognition and hydrolysis of nucleic acid strands, as detailed in Section 2.2. The first property, called cis-cleavage, is the one mainly used by Cas9, and consists in the recognition of a target DNA by a guide RNA previously bound to the enzyme. The target DNA strand (or RNA) then hybridizes to the guide RNA; this step may be followed by hydrolysis of the target DNA chain, as with Cas9. However, hydrolysis does not always take place, and target DNA/guide RNA hybridization can also serve as a trigger for the appearance of another catalytic site, as in Cas12, Cas13 and Cas14. Once activated by hybridization between target DNA and guide RNA, these enzymes undergo structural reformation to reveal a new catalytic site, triggering non-specific RNAse (or DNAse) activity (i.e. any strand of RNA or DNA in the vicinity of the enzyme is hydrolyzed, without specificity for any particular sequence). This property is known as trans-cleavage.
These two properties of Cas enzymes can be exploited in EGFET transistors, based on specific recognition of a target RNA (cleavage-cis) and amplification of the signal by the RNAse activity of these enzymes (cleavage-trans). The various sensors that use the combination of CRISPR-Cas complexes and EGFETs to detect biological species are discussed below.
4.1. Exploiting Cis-Cleavage
Unlike other Class 2 enzymes, Cas9 has no trans-cleavage activity, but only a cis-cleavage property through recognition of double-stranded DNA at its guide RNA. This property of recognizing a specific sequence in a double-stranded DNA genome is at the heart of CRISPR-Cas technology. The first transistor using CRISPR-Cas complexes and EGFETs was described in 2019 by Hajian et al. who exploit the cis-cleavage property between CRISPR-Cas9 and a double-stranded guide DNA (Figure 23) [108]. The Cas9 enzyme used is first deactivated, to prevent target DNAs from hydrolysis after guide RNA-target DNA hybridization. This deactivated Cas9 enzyme is referred to as dCas9. CRISPR-dCas9 complexes are immobilized on the channel of an EGGFET via a PBA (pyrene butanoic acid) group. An amide bridge is created between the acid function of the PBA and a free -NH2 end of CRISPR-dCas9 to bind the enzyme to the EGGFET. The sensor's detection principle is based on hybridization between the target DNA and CRISPR-dCas9 immobilized on the channel surface.
In the context of a conventional transistor, biorecognition events taking place at a distance greater than the Debye length λ (λ ≈ 1 nm under salinity conditions equivalent to phosphate-buffered saline, PBS) are not efficiently translated because the charges carried by the targets and probes are shielded by free ions in solution. Since Cas enzymes are large riboprotein complexes (> 10 nm), hybridization of the DNA target to the guide RNA therefore takes place outside the Debye length λ; yet it is quantifiable by an evolution of the output current of the ID transistor thanks to the Donnan effect [109].
Indeed, the CRISPR-dCas9 protein layer immobilized on the EGGFET surface can be considered an ion-permeable and selective membrane. Hybridization of the negatively charged DNA target creates an ion-dense layer on the membrane surface, due to the accumulation of cations to maintain charge neutrality. This change in ion concentration between the electrolyte volume and the membrane creates a Donnan potential. This new potential modifies the electric field between the grid and the channel, which in turn modulates the output current. This phenomenon makes it possible to detect biological events at distances greater than the Debye length [110]. It is this effect that also enables the detection of antigen-antibody interactions on the surface of a transistor.
The CRISPR-Chip sensor by Haijian et al. sense target DNA in 15 minutes with a LOD of 1.7 fM.
This sensor was improved Balderston et al. into a device called CRISPR-SNP-Chip, capable of discriminating the mutation of a single base pair in a target DNA [111] (Figure 24). This is made possible by extending the electrical signals analyzed and optimizing the guide RNAs. In addition to the output current I, two other electrical parameters are analyzed: (1) The capacitance C of the graphene/electrolyte interface which changes following hybridization of the target DNA to the CRISPR-dCas9. C influences the slope of the transfer curve at a given potential. (2) The position of the minimum V* of the transfer curve. By exploiting the relative variations of these three parameters (I, C and V*) and optimizing the sequences of the guide RNAs, these authors created a sensor that can discriminate a one-base mismatch within 1 hour, exemplified for the genomic DNA of Amyotrophic Lateral Sclerosis (ALS) and sickle cell disease [111].
Two other works by Li's group report on the use of cis-cleavage of CRISPR-Cas complexes for biosensing [112,113]. In contrast to CRISPR-Chip, these works describe the functionalization of the gate electrode of two EGGFETs. Two sensors are proposed, one based on Cas13 and the other based on Cas12, where these enzymes are grafted onto the gate of an EGGFET via a self-assembled layer and/or a polymer layer. The functionalized gate structures are detailed in Figure 25. The operating principle of these sensors is exactly the same as CRISPR-Chip, with the difference that the gate electrode is functionalized rather than the transistor channel. The pairing of negatively-charged target DNA (or RNA) on CRISPR-Cas12a (or CRISPR-Cas13a) modifies the arrangement of charges at the gate/electrolyte interface, inducing an evolution of the electric field. This results in a shift of the transfer curve towards positive potentials.
Their Cas12a-based sensor is capable of detecting Papillomavirus DNA in 20 minutes with a LOD of 8.3 aM [113]. while the Cas13-based one detects SARS-CoV-2 RNA in 10 minutes with a LOD of 13 aM [112]. The main advantage of the functionalized grid is the possibility of interchanging them according to the target to be detected, as simply dipped in the electrolyte, they are not bound to the rest of the device. Modifying the graphene channel is more difficult and costly.
The various RNA or DNA sensors based on recognition between a target nucleic acid sequence and a Cas enzyme immobilized on one of the two interfaces of an EGGFET transistor are detailed in Table 3.
4.2. Exploiting Trans-Cleavage
The trans-cleavage property is the most interesting for the creation of POC sensors, as it enables a molecular signal amplification. Indeed, a single RNA target activates a CRISPR-Cas13 enzyme capable of hydrolyzing an average of 104 RNA strands [42]. This phenomenon of enzymatic amplification of the signal is very interesting for biosensing, compared with previous generations of sensors based on hybridization of a single RNA probe with a single target, for limited molecular reorganization. The advantage of CRISPR-Cas13 is that the output signal is multiplied by the hydrolysis activity, since many strands of RNA are hydrolyzed, for a much greater (macro)molecular reorganization. It is precisely this trans-cleavage property that enhances the sensitivity of the devices presented above, while specificity is ensured by cis-cleavage of the target RNAs on the CRISPR-Cas enzymes to which they are specific. Clearly, as Cas9 does not exhibit trans-cleavage activity, its use is not possible for this purpose.
Before being used in transistors, CRISPR-Cas13 trans-cleavage was used for electrochemical sensors. This was introduced by the work of Dai et al. in 2019, who proposed an electrochemical sensor named E-CRISPR (Electrochemical CRISPR) for the detection of human papillomavirus and parvovirus B-19, with picomolar sensitivity [114]. Single-stranded DNAs are grafted onto the working electrode and carry a redox probe at its free end. Hydrolysis of these DNA strands by CRISPR-Cas12a releases the redox probes into the electrolyte and reduces the redox current of the probe at the electrode surface. Zhang et al. used the same detection principle with a hairpin RNA probe to bring the redox probe closer to the electrode surface [115]. This sensor is also used for human papillomavirus detection, with a dynamic detection range between 50 pM and 100 fM. Recently, other electrochemical sensors capable of detecting RNA concentrations with an LOD of 10 aM have emerged [116,117]. This is notably the case of the sensor proposed by Lee's group, which is also based on RNA strands bound to an electrode, at the end of which a redox probe is grafted [118]. The sensor's response time is relatively long, around 3h, which gives CRISPR-Cas13 enough time to hydrolyze the RNA strands and obtain an LOD at the attomolar level.
In the following sub-sections are discussed the use of Cas12 and Cas13 in EGGFETs.
4.2.1. DNA Detection by Cas12
As with those presented above, most devices combining CRISPR-Cas12 and EGFETs use graphene-based transistors. In the case of DNA detection, Cas12 and Cas14 enzymes can be used but no paper has yet described the use of Cas14 with EGGFETs, however. With regard to articles employing CRISPR-Cas12, the channel is an interface of choice for functionalization, not least because it allows the use of an Ag/AgCl gate [119].
Within the framework of EGGFETs that exploit the enzymatic activity of Cas12, all employ a non-covalent grafting route through the use of PBASE, detailed above. Although the mode of channel functionalization remains the same for these different works, the DNA probes immobilized on the transistor channel are different. Wang et al. use a 20-nucleotide T DNA probe for the detection of monkeypox (MPXV). Their sensor has an LOD of 1 aM and can differentiate between the different mutations of this disease in 20 minutes at an operating temperature of 52°C [120]. Weng et al. use 20-nucleotides poly-A or poly-C DNA probes to detect human papillomavirus (HPV-16) and the Escherichia coli membrane gene with LODs of 1 aM and 10 aM, respectively [121]. This detection is possible in less than 30 minutes, and incubation of CRISPR-Cas12a on the transistor is performed at room temperature. Their sensor can also differentiate a single mismatch on Escherichia coli plasmid DNA and allows an LOD of 1 pM. For their part, Wang et al. chose to modify the transistor-grafted probe to increase the sensitivity of their device [122], an RNA consisting of 6 T nucleotides and 12 U nucleotides (structure detailed in Figure 26). This structure involves the hydrolysis by CRISPR-Cas12 of only the T part of this DNA-RNA hybrid probe, which is closest to the channel (CRISPR-Cas12 does not hydrolyze polyU sequences), which increases the sensitivity of the device compared with DNA probes made up of a single nucleotide type. The LOD obtained, for African swine fever virus (AFSV), is 0.5 aM, with a 30-minute incubation at room temperature. This is the lowest detection limit described to date for DNA detection using EGGFETs.
4.2.2. DNA Detection by Cas13
Numerous sensors featuring EGGFETs and CRISPR-Cas13 have also been developed in the wake of the COVID-19 pandemic. The first device was developed by Li et al. in 2022 for the detection of the ORF1ab and N genes of SARS-CoV-2 as well as hepatitis C virus (HCV) [123]. The transistor used is an rGO-EGFET, where the rGO channel is functionalized by gold nanoparticles onto which RNA probes are grafted (Figure 27). The RNA probes chosen have a hairpin structure and are oriented horizontally rather than vertically, maximizing the density of negative charges closest to the rGO, within the Debye length. This sensor is capable of detecting RNA after a 20-minute incubation at 37°C and achieves an LOD of 1.56 aM for SARS-CoV-2.
Other EGGFET-based sensors for the detection of SARS-CoV-2 have been developed, such as those proposed by Li [124], Ban [125] and Sun [126]. These three sensors use an EGGFET on which RNA probes are embedded using PBASE. The sensor proposed by Ban et al. targets both SARS-CoV-2 RNA and virus particles, with LODs of 65 aM and 1000 copies.µL-1 respectively. Virus particle detection is made possible by protein-catalyzed capture agents (PCCs) immobilized on the channel, while RNA detection is achieved using CRISPR-Cas13a (Figure 28). The system developed by Li et al. has been validated for the detection of SARS-CoV-2 and respiratory syncytial virus (RSV), with an LOD of 1 aM and an incubation time of 30 minutes at 37°C. The RNA probes used on these two sensors are 20-nucleotide chains (polyU20). Like the polyU-polyT DNA-RNA hybrid probes proposed by Hu et al. for DNA detection using Cas12, Sun et al. have also proposed an RNA-DNA hybrid probe for RNA detection using Cas13 [126]. This probe is composed of 6 U nucleotides followed by 12 T nucleotides, which specifies Cas13 activity towards the RNA part of this probe and accelerates signal detection. With an incubation time of 2h at 37°C, an LOD of 0.25 aM was obtained.
In a view of comparison, Chen et al. have described the use of an EGFET based on the inorganic semiconductor IGZO (Oxide of Zinc, Gallium and Indium), in a device named CAVRED (CRISPR-based Amplification-free Viral RNA Electrical Detection [127]. The IGZO layer is functionalized with APTES ((3-aminopropyl)triethoxysilane) to create amine functions that can bind to RNA probes. In a similar way to EGGFETs, hydrolysis of RNA probes by Cas13 generates a change in charge density at the channel surface that modifies the ID drain current. The CAVRED platform has been validated for the detection of SARS-CoV-2 with a LOD of 1 copy per µL (≈ 1.7 aM).
The various EGGFET-based biosensors exploiting the trans-cleavage property of the CRISPR-Cas systems presented above are grouped together in Table 4 with their different characteristics.
Quite intuitively (and comparing Table 4 with Table 3), it appears that devices using trans-cleavage of CRISPR-Cas complexes are more sensitive than those based on cis-cleavage interaction alone, because the enzymatic activity amplifies the molecular recognition before it is transduced by the transistor. This property positions CRISPR-Cas sensors as credible challengers to PCR (or RT-PCR) techniques.
5. Conclusions
The COVID-19 pandemic highlighted the lack of available alternatives to PCR for rapid, real-time detection of nucleic acids. Discovered in 2007, CRISPR-Cas ribonucleic complexes have recently been able to compete with PCR thanks to their trans-cleavage activity, which amplifies the output signal by four orders of magnitude [42]. This has revolutionized nucleic acid detection, and some devices have been rapidly cleared by the FDA for SARS-CoV-2 detection, such as SHERLOCK. However, these detection platforms still require a pre-amplification step that is difficult to compare with the design of POC devices. The improvements realized these last years prove that CRISPR-Cas complexes and EGGFETs show good synergy, making them plausible candidates for the creation of POC devices for rapid, sensitive and specific detection.
Funding
This research was funded by the Doctoral School ED388 (PhD scholarship of P.G.) and by the French Research Agency for a research grant Project-ANR-21-CE19-0039.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Nichol, S.T.; Arikawa, J.; Kawaoka, Y. Emerging viral diseases. PNAS 2000, 97, 12411–12412. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Iyer, S.S. ASSURED-SQVM diagnostics for COVID-19 : addressing the why, when, where, who, what and how of testing ». Expert Rev. Mol. Diag. 2021, 21, 349–362. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung,J. ; Verdine,V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; Myhrvold, C.; Bhattacharyya, R.P.; Livny, J.; Regev, A.; Koonin, E.V.; Hung, D.T.; Sabeti, P.C.; Collins, J.J.; Zhang, F. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Ñamendys-Silva, S.A.; Rivero-Sigarroa, E.; Dominguez-Cherit, G. Acute Respiratory Distress Syndrome Induced by Pandemic H1N1 2009 Influenza A Virus Infection. Am. J. Resp. Crit. Care Med. 2015, 182, 857–858. [Google Scholar] [CrossRef] [PubMed]
- Cella, L.N.; Blackstock, D.; Yates, M.A.; Mulchandani, A.; Chen, W. Detection of RNA Viruses : Current Technologies and Future Perspectives. Critical Rev. Eukaryotic Gene Expres. 2013, 23, 125–137. [Google Scholar] [CrossRef]
- Kevadiya, B.D.; Machhi, J.; Herskovitz, J.; Oleynikov, M.D.; Blomberg, W.R.; Bajwa, N.; Soni, D.; Das, S.; Hasan, M.; Patel, M.K.; Senan, A.M.; Gorantla, S.; McMillan, J.; Edagwa, B.; Eisenberg, R.; Gurumurthy, C.B.; Reid, S.M.; Punyadeera, C.; Chang, L.; Gendelman, H.E. Diagnostics for SARS-CoV-2 infections. Nature Mater. 2021, 20, 593–605. [Google Scholar] [CrossRef]
- Lequin, R.M. Enzyme Immunoassay (EIA)/Enzyme-Linked Immunosorbent Assay (ELISA). Clinical Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef]
- Li, G.; Chen, X.; Xu, A. Profile of Specific Antibodies to the SARS-Associated Coronavirus. New England J. Med. 2003, 349, 508–509. [Google Scholar] [CrossRef]
- Zhao, J.; Yuan, Q.; Wang, H.; Liu, W.; Liao, X.; Su, Y.; Wang, X.; Yuan, J.; Li, T.; Li, J.; Qian, S.; Hong, C.; Wang, F.; Liu, Y.; Wang, Z.; He, Q.; Li, Z.; He, B.; Zhang, T.; Fu, Y.; Ge, S.; Liu, L.; Zhang, J.; Xia, N.; Zhang, Z. Antibody Responses to SARS-CoV-2 in Patients With Novel Coronavirus Disease 2019. Clinical Infectious Diseases 2020, 71, 2027–2034. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W. Common and unique features of viral RNA-dependent polymerases. Cellular and Molecular Life Sciences 2014, 71, 4403–4420. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, D.; Yang, P.; Poon, L.L.M.; Wang, Q. Viral load of SARS-CoV-2 in clinical samples. The Lancet Infect. Diseases 2020, 20, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lázaro, D.; Hernández, M. Real-time PCR in Food Science : Introduction. Cur. Issues Mol. Biol. 2013, 15, 25–38. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biology 2006, 4, e204. [Google Scholar] [CrossRef]
- Notomi, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, 63e–63. [Google Scholar] [CrossRef]
- Pardee, K.; Green, A.A.; Takahashi, M.K.; Braff, D.; Lambert, G.; Lee, J.W.; Ferrante, T.; Ma, D.; Donghia, N.; Fan, M.; Daringer, N.M.; Bosch, I.; Dudley, D.M.; O’Connor, D.H.; Gehrke, L.; Collins, J.J. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell 2016, 165, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Walker, G.T.; Fraiser, M.S.; Schram, J.L.; Little, M.C.; Nadeau, J.H.; Malinowski, D.P. Strand displacement amplification—an isothermal, in vitro DNA amplification technique. Nucleic Acids Res. 1992, 20, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Xu, Y.; Kong, H. Helicase-dependent isothermal DNA amplification. EMBO reports 2004, 5, 795–800. [Google Scholar] [CrossRef]
- Van Ness, J.; Van Ness, L.K.; Galas, D.J. Isothermal reactions for the amplification of oligonucleotides. PNAS 2003, 100, 4504–4509. [Google Scholar] [CrossRef]
- Kaminski, M.M.; Abudayyeh, O.O.; Gootenberg, J.S. Feng Zhang; Collins, J.J. CRISPR-based diagnostics. Nature Biomedical Engineering 2021, 5, 643–656. [Google Scholar] [CrossRef]
- Yang, S.; Rothman, R.E. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. The Lancet Infec. Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A. CRISPR-Cas immunity in prokaryotes. Nature 2015, 526, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Current Opinion in Microbiology 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Terns, R.M.; Terns, M.P.; White, M.F.; Yakunin, A.F.; Garrett, R.A.; van der Oost, J. , Backofen, R.; Koonin, E.V. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Scott, D.; Shah, S.A.; Siksnys, V.; Terns, M.P.; Venclovas, C.; White, M.F.; Yakunin, A.F.; Yan, W.; Zhang, F.; Garrett, R.A.; Backofen, R.; van der Oost, J.; Barrangou, R.; Koonin, E.V. Evolutionary classification of CRISPR–Cas systems : a burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Kant, R.; Behera, S.P.; Dwivedi, G.R.; Singh, R. Next-Generation Diagnostic with CRISPR/Cas : Beyond Nucleic Acid Detection. Int. J. Mol. Sci. 2022, 23, 6052. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Silva, G.; Poirot, L.; Galetto, R.; Smith, J.; Montoya, G.; Duchateau, P.; Pâques, F. Meganucleases and Other Tools for Targeted Genome Engineering : Perspectives and Challenges for Gene Therapy. Current Gene Therapy 2011, 11, 11–27. [Google Scholar] [CrossRef]
- Carroll, D. Genome Engineering With Zinc-Finger Nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA Double-Strand Breaks with TAL Effector Nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Guha, T.K.; Wai, A.; Hausner, G. Programmable Genome Editing Tools and their Regulation for Efficient Genome Engineering. Computational and Structural Biotechnol. J. 2017, 15, 146–160. [Google Scholar] [CrossRef]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Uyhazi, K.E.; Bennett, J. A CRISPR view of the 2020 Nobel Prize in Chemistry. J. Clin. Invest. 2021, 131, e145214. [Google Scholar] [CrossRef]
- Kim, S.; Ji, S.; Koh, H.R. CRISPR as a diagnostic tool. In Biomolecules 2021, 11, 1162. [Google Scholar] [CrossRef]
- Liu, W.; Yu, H.; Zhou, X.; Xing, D. In vitro Evaluation of CRISPR/Cas9 Function by an Electrochemiluminescent Assay. Anal. Chem. 2016, 88, 8369–8374. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome engineering with RNA-targeting Type VI-D CRISPR effectors. Cell 2018, 173, 665–676.e14. [Google Scholar] [CrossRef]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; Zhang, F.; Koonin, E.V. Discovery and functional characterization of diverse Class 2 CRISPR-Cas systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; Koonin, E.V.; Zhang, F. Cpf1 is a single RNA-guided endonuclease of a Class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Ma, J.; Li, Z.; You, L.; Wang, J.; Wang, M.; Zhang, X.; Wang, Y. The molecular architecture for RNA-guided RNA cleavage by Cas13a. Cell 2017, 170, 714–726.e10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, S.; Wang, J.; Liu, G. CRISPR/Cas systems towards next-generation biosensing. Trends Biotechnol. 2019, 37, 730–743. [Google Scholar] [CrossRef]
- Bhattacharyya, R.P.; Thakku, S.; Hung, D.T. Harnessing CRISPR Effectors for Infectious Disease Diagnostics. A.C.S. Infect. Dis. 2018, 4, 1278–1282. [Google Scholar] [CrossRef]
- Angelo, C. Batista et Luis G. C. Pacheco. Detecting pathogens with zinc-Finger, TALE and CRISPR- based programmable nucleic acid binding proteins ». J. Microbiol. Methods 2018, 152, 98–104. [Google Scholar] [CrossRef]
- Kellner, M.J.; Koob, J.G.; Gootenberg, J.S.; Abudayyeh, O.O.; Zhang, F. Sherlock: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019, 14, 2986–3012. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, C.M.; Myhrvold, C.; Thakku, S.G.; Freije, C.A.; Metsky, H.C.; Yang, D.K.; Ye, S.H.; Boehm, C.K.; Kosoko-Thoroddsen, T.F.; Kehe, J.; Nguyen, T.G.; Carter, A.; Kulesa, A.; Barnes, J.R.; Dugan, V.G.; Hung, D.T.; Blainey, P.C.; Sabeti, P.C. Massively multiplexed nucleic acid detection with Cas13. Nature 2020, 582, 277–282. [Google Scholar] [CrossRef]
- Rauch, J.N.; Valois, E.; Ponce-Rojas, J.C.; Aralis, Z.; Lach, R.S.; Zappa, F.; Audouard, M.; Solley, S.C.; Vaidya, C.; Costello, M.; Smith, H.; Javanbakht, A.; Malear, B.; Polito, L.; Comer, S.; Arn, K.; Kosik, K.S.; Acosta-Alvear, D.; Wilson, M.Z.; Fitzgibbons, L.; Arias, C. Rapid CRISPR-based surveillance of SARS-CoV-2 in asymptomatic college students captures the leading edge of a community-wide outbreak. JAMA Network open 2021, 4, e2037129. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Valois, E.; Solley, S.C.; Braig, F.; Lach, R.S.; Audouard, M.; Ponce-Rojas, J.C.; Costello, M.S.; Baxter, N.J.; Kosik, K.S.; Arias, C.; Acosta-Alvear, D.; Wilson, M.Z. A scalable, easy-to-deploy protocol for Cas13-based detection of SARSCoV-2 genetic material. J. Clin. Microbiol. 2021, 59, e02402–20. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; Severinov, K.; Regev, A.; Lander, E.S.; Koonin, E.V.; Zhang, F. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.D.; Tjian, R.; Doudna, J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.G.; Lachenauer, A.E.; Nitido, A.; Siddiqui, S.; Gross, R.; Beitzel, B.; Siddle, K.J.; Freije, C.A.; Dighero-Kemp, B.; Mehta, S.B.; Carter, A.; Uwanibe, J.; Ajogbasile, F.; Olumade, T.; Odia, I.; Sandi, J.D.; Momoh, M.; Metsky, H.C.; Boehm, C.K.; Lin, A.E.; Kemball, M.; Park, D.J.; Branco, L.; Boisen, M.; Sullivan, B.; Amare, M.F.; Tiamiyu, A.B.; Parker, Z.F.; Iroezindu, M.; Grant, D.S.; Modjarrad, K.; Myhrvold, C.; Garry, R.F.; Palacios, G.; Hensley, L.E.; Schaffner, S.F.; Happi, C.T.; Colubri, A.; Sabeti, P.C. Deployable CRISPR-Cas13a diagnostic tools to detect and report Ebola and Lassa virus cases in real-time. Nat. Commun. 2020, 11, 4131. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.A.; Puig, H.D.; Nguyen, P.Q.; Angenent-Mari, N.M.; Donghia, N.M.; McGee, J.P.; Dvorin, J.D.; Klapperich, C.M.; Pollock, N.R.; Collins, J.J. Ultrasensitive CRISPR-based diagnostic for field-applicable detection of Plasmodium species in symptomatic and asymptomatic malaria. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 25722–25731. [Google Scholar] [CrossRef] [PubMed]
- Ganbaatar, U.; Liu, C. CRISPR-Based COVID-19 Testing: Toward Next-Generation Point-of-Care Diagnostics. Front. Cell. Infect. Microbiol. 2021, 11, 663949. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.L.; Paul, L.M.; Parham, L.A.; Garcia, K.F.; Barnes, K.G.; Chak, B.; Mondini, A.; Nogueira, M.L.; Isern, S.; Michael, S.F.; Lorenzana, I.; Yozwiak, N.L.; MacInnis, B.L.; Bosch, I.; Gehrke, L.; Zhang, F.; Sabeti, P.C. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef]
- Freije, C.A.; Myhrvold, C.; Boehm, C.K.; Lin, A.E.; Welch, N.L.; Carter, A.; Metsky, H.C.; Luo, C.Y.; Abudayyeh, O.O.; Gootenberg, J.S.; Yozwiak, N.L.; Zhang, F.; Sabeti, P.C. Programmable inhibition and detection of RNA viruses using Cas13. Mol. Cell 2019, 76, 826–837. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Wang, J.M.; Li, X.Y.; Zhang, Z.L.; Gao, S.; Cao, R.B.; Zhao, G.P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Lin, H.; Li, H.; Zhou, Y.; Liu, J.; Zhong, G.; Wu, L.; Jiang, W.; Du, H.; Yang, J.; Xie, Q.; Huang, L. Cas12a-based On-Site and rapid nucleic acid detection of African swine fever. Front. Microbiol. 2019, 10, 2830. [Google Scholar] [CrossRef]
- Brandsma, E.; Verhagen, H.J.M.P.; van de Laar, T.J.W.; Claas, E.C.J.; Cornelissen, M.; van den Akker, E. Rapid, sensitive, and specific severe acute respiratory syndrome coronavirus 2 detection: A multicenter comparison between standard quantitative reverse-transcriptase polymerase chain reaction and CRISPR-based DETECTR. J. Infect. Dis. 2021, 223, 206–213. [Google Scholar] [CrossRef]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; Zorn, K.; Gopez, A.; Hsu, E.; Gu, W.; Miller, S.; Pan, C.Y.; Guevara, H.; Wadford, D.A.; Chen, J.S.; Chiu, C.Y. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhou, X.; Wang, H.; Xing, D. Clustered regularly interspaced short palindromic repeats/Cas9 triggered isothermal amplification for site-specific nucleic acid detection. Anal. Chem. 2018, 90, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, R.; Sohail, M.; Kong, X.; Zhang, X.; Fu, N.; Li, B. CRISPR/Cas14 provides a promising platform in facile and versatile aptasensing with improved sensitivity. Talanta 2023, 254, 124120. [Google Scholar] [CrossRef]
- Yang, J.; Song, Y.; Deng, X.; Vanegas, J.A.; You, Z.; Zhang, Y.; Weng, Z.; Avery, L.; Dieckhaus, K.D.; Peddi, A.; Gao, Y.; Zhang, Y.; Gao, X. Engineered LwaCas13a with enhanced collateral activity for nucleic acid detection. Nat. Chem. Biol. 2023, 19, 45–54. [Google Scholar] [CrossRef]
- Kim, M.; Shin, H.H. Next-generation Molecular Diagnostic Technology for Infectious Disease: CRISPR-Cas System. KSBB J. 2023, 38, 77–89. [Google Scholar] [CrossRef]
- Alexandra, East-Seletsky; et al. RNA targeting by functionally orthogonal Type VI-A CRISPR-Cas enzymes. Mol. Cell 2017, 66, 373–383. [Google Scholar]
- Qin, P.; Park, M.; Alfson, K.J.; Tamhankar, M.; Carrion, R.; Patterson, J.L.; Griffiths, A.; He, Q.; Yildiz, A.; Mathies, R.; Du, K. Rapid and fully microfluidic Ebola virus detection with CRISPR-Cas13a. A.C.S. Sens. 2019, 4, 1048–1054. [Google Scholar] [CrossRef]
- Fozouni, P.; Son, S.; Díaz de León Derby, M.; Knott, G.J.; Gray, C.N.; D’Ambrosio, M.V.; Zhao, C.; Switz, N.A.; Kumar, G.R.; Stephens, S.I.; Boehm, D.; Tsou, C.L.; Shu, J.; Bhuiya, A.; Armstrong, M.; Harris, A.R.; Chen, P.Y.; Osterloh, J.M.; Meyer-Franke, A.; Joehnk, B.; Walcott, K.; Sil, A.; Langelier, C.; Pollard, K.S.; Crawford, E.D.; Puschnik, A.S.; Phelps, M.; Kistler, A.; DeRisi, J.L.; Doudna, J.A.; Fletcher, D.A.; Ott, M. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 2021, 184, 323–333.e9. [Google Scholar] [CrossRef]
- Katzmeier, F.; Aufinger, L.; Dupin, A.; Quintero, J.; Lenz, M.; Bauer, L.; Klumpe, S.; Sherpa, D.; Dürr, B.; Honemann, M.; Styazhkin, I.; Simmel, F.C.; Heymann, M. A low-cost fluorescence reader for in vitro transcription and nucleic acid detection with Cas13a. PLOS ONE 2019, 14, e0220091. [Google Scholar] [CrossRef]
- Johnston, M.; Ceren Ates, H.; Glatz, R.T.; Mohsenin, H.; Schmachtenberg, R.; Göppert, N.; Huzly, D.; Urban, G.A.; Weber, W.; Dincer, C. Multiplexed biosensor for point-of-care COVID-19 monitoring: CRISPRpowered unamplified RNA diagnostics and protein-based therapeutic drug management. Mater. Today 2022, 61, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.E.; Berendsen, J.T.W.; Steenbergen, R.D.M.; Wolthuis, R.M.F.; Eijkel, J.C.T.; Segerink, L.I. Point-of-care CRISPR/Cas nucleic acid detection: recent advances, challenges and opportunities. Biosens. Bioelectron. 2020, 166, 112445. [Google Scholar] [CrossRef]
- Torricelli, F.; Adrahtas, D.Z.; Bao, Z.; Berggren, M.; Biscarini, F.; Bonfiglio, A.; Bortolotti, C.A.; Frisbie, C.D.; Macchia, E.; Malliaras, G.G.; McCulloch, I.; Moser, M.; Nguyen, T.Q.; Owens, R.M.; Salleo, A.; Spanu, A.; Torsi, L. Electrolyte-gated transistors for enhanced performance bioelectronics. Nat. Rev. Methods Primers 2021, 1, 66. [Google Scholar] [CrossRef] [PubMed]
- Chennit, K.; Delavari, N.; Mekhmoukhen, S.; Boukraa, R.; Fillaud, L.; Zrig, S.; Battaglini, N.; Piro, B.; Noël, V.; Zozoulenko, I.; Mattana, G. Inkjet-printed, coplanar electrolyte-gated organic field-effect transistors on flexible substrates: fabrication, modeling, and applications in biodetection. Adv. Mater. Technol. 2023, 8, 2200300. [Google Scholar] [CrossRef]
- Takeya, J.; Yamada, K.; Hara, K.; Shigeto, K.; Tsukagoshi, K.; Ikehata, S.; Aoyagi, Y. High-density electrostatic carrier doping in organic single-crystal transistors with polymer gel electrolyte. Appl. Phys. Lett. 2006, 88, 112102. [Google Scholar] [CrossRef]
- Panzer, M.J.; Newman, C.R.; Frisbie, C.D. Low-voltage operation of a pentacene field-effect transistor with a polymer electrolyte gate dielectric. Appl. Phys. Lett. 2005, 86, 103503. [Google Scholar] [CrossRef]
- Rivnay, J.; Inal, S.; Salleo, A.; Owens, R.M.; Berggren, M.; Malliaras, G.G. Organic electrochemical transistors. Nat. Rev. Mater. 2018, 3, 17086. [Google Scholar] [CrossRef]
- Kim, D.J.; Lee, N.E.; Park, J.S.; Park, I.J.; Kim, J.G.; Cho, H.J. Organic electrochemical transistor based immunosensor for prostate specific antigen (PSA) detection using gold nanoparticles for signal amplification. Biosens. Bioelectron. 2010, 25, 2477–2482. [Google Scholar] [CrossRef]
- Liu, J.; Agarwal, M.; Varahramyan, K. Glucose sensor based on organic thin film transistor using glucose oxidase and conducting polymer. Sens. Actuators B 2008, 135, 195–199. [Google Scholar] [CrossRef]
- He, R.X.; Zhang, M.; Tan, F.; Leung, P.H.M.; Zhao, X.Z.; Chan, H.L.W.; Yang, M.; Yan, F. Detection of bacteria with organic electrochemical transistors. J. Mater. Chem. 2012, 22, 22072. [Google Scholar] [CrossRef]
- Hai, W.; Goda, T.; Takeuchi, H.; Yamaoka, S.; Horiguchi, Y.; Matsumoto, A.; Miyahara, Y. Human influenza virus detection using sialyllactose-functionalized organic electrochemical transistors. Sens. Actuators B 2018, 260, 635–641. [Google Scholar] [CrossRef]
- Tao, W.; Lin, P.; Hu, J.; Ke, S.; Song, J.; Zeng, X. A sensitive DNA sensor based on an organic electrochemical transistor using a peptide nucleic acid-modified nanoporous gold gate electrode. R.S.C. Adv. 2017, 7, 52118–52124. [Google Scholar] [CrossRef]
- Peng, J.; He, T.; Sun, Y.; Liu, Y.; Cao, Q.; Wang, Q.; Tang, H. An organic electrochemical transistor for determination of microRNA21 using gold nanoparticles and a capture DNA probe. Mikrochim. Acta 2018, 185, 408. [Google Scholar] [CrossRef]
- Sensi, M.; Migatti, G.; Beni, V.; Marchesi D'Alvise, T.; Weil, T.; Berto, M.; Greco, P.; Imbriano, C.; Biscarini, F.; Bortolotti, C.A. Monitoring DNA Hybridization with Organic Electrochemical Transistors Functionalized with Polydopamine. Macromol. Mater. Eng. 2022, 307, 2100880. [Google Scholar] [CrossRef]
- Lin, P.; Yan, F.; Yu, J.; Chan, H. L. W.; Yang, M. Organic Electrochemical Transistors Integrated in Flexible Microfluidic Systems and Used for Label-Free DNA Sensing. Adv. Mater. 2011, 23, 4035–4040. [Google Scholar] [CrossRef]
- Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Zhang, Y.; Dubonos, S. V.; Grigorieva, I. V.; Firsov, A. A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Song, Z.; Li, T.; Li, X.; Ogbazghi, A. Y.; Feng, R.; Dai, Z.; Marchenkov, A. N.; Conrad, E. H.; First, P. N.; de Heer, W. A. Ultrathin Epitaxial Graphite: 2D Electron Gas Properties and a Route toward Graphene-based Nanoelectronics. J. Phys. Chem. B 2004, 108, 19912–19916. [Google Scholar] [CrossRef]
- Schwierz, F. Graphene Transistors. Nat. Nanotechnol. 2010, 5, 487–496. [Google Scholar] [CrossRef]
- Wallace, P. R. The Band Theory of Graphite. Phys. Rev. 1947, 71, 622–634. [Google Scholar] [CrossRef]
- Novoselov, K. S.; Geim, A. K.; Morozov, S. V.; Jiang, D.; Katsnelson, M. I.; Grigorieva, I. V.; Dubonos, S. V.; Firsov, A. A. Two-Dimensional Gas of Massless Dirac Fermions in Graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef]
- Castro Neto, A. H.; Guinea, F.; Peres, N. M. R.; Novoselov, K. S.; Geim, A. K. The Electronic Properties of Graphene. Rev. Mod. Phys. 2009, 81, 109–162. [Google Scholar] [CrossRef]
- Lotya, M.; King, P. J.; Khan, U.; De, S.; Coleman, J. N. High-Concentration, Surfactant-Stabilized Graphene Dispersions. ACS Nano 2010, 4, 3155–3162. [Google Scholar] [CrossRef]
- Geim, A. K.; Novoselov, K. S.; Novoselov, K. S. The Rise of Graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Ławkowska, K.; Jabłońska-Trypuć, A.; Antonowicz, B.; Wołosiak, S.; Kocira, S.; Skórka-Majewicz, M. Application of Graphene in Tissue Engineering of the Nervous System. Int. J. Mol. Sci. 2021, 23, 33. [Google Scholar] [CrossRef] [PubMed]
- Dröscher, S.; Roulleau, P.; Molitor, F.; Güttinger, J.; Stampfer, C.; Ihn, T.; Ensslin, K. Quantum Capacitance and Density of States of Graphene. Appl. Phys. Lett. 2010, 96, 152104. [Google Scholar] [CrossRef]
- Vasilijević, S. Printed Graphene Field-Effect Transistors: Chemical and Electrochemical Control of Graphene’s Electronic Properties and Applications in Biosensing. Ph.D. Thesis, 2021.
- Vasilijević, S.; Wang, W.; Tatsi, E.; Arenal, R.; Happy, H.; Noël, V.; Noël, V. Electrochemical Tuning of Reduced Graphene Oxide in Printed Electrolyte-Gated Transistors: Impact on Charge Transport Properties. Electrochim. Acta 2021, 371, 137819. [Google Scholar] [CrossRef]
- Salvo, P.; Calisi, N.; Melai, B.; Cortigiani, B.; Mannini, M.; Caneschi, A.; Chiellini, F.; Di Francesco, F. Graphene-Based Devices for Measuring pH. Sens. Actuators B Chem. 2018, 256, 976–991. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, L.; Tang, H.; Zhang, Z.; Wang, M.; Zhang, Y.; Liu, X.; Wang, Q. Ultrasensitive Label-Free miRNA Sensing Based on a Flexible Graphene Field-Effect Transistor without Functionalization. ACS Appl. Electron. Mater. 2020, 2, 1090–1098. [Google Scholar] [CrossRef]
- Seo, G.; Lee, G.; Kim, M. J.; Baek, S.-H.; Choi, M.; Ku, K. B.; Lee, C.-S.; Jun, S.; Park, D.; Kim, H. G.; Kim, S. J.; Lee, J.-O.; Kim, B. T.; Park, E. C.; Kim, S. I. Rapid Detection of COVID-19 Causative Virus (SARS-CoV-2) in Human Nasopharyngeal Swab Specimens Using Field-Effect Transistor-Based Biosensor. ACS Nano 2020, 14, 5135–5142. [Google Scholar] [CrossRef]
- Tian, M.; Wang, Y.; Zhang, M.; Zhang, L.; Zhang, L. Highly-Sensitive Graphene Field-Effect Transistor Biosensor Using PNA and DNA Probes for RNA Detection. Appl. Surf. Sci. 2020, 527, 146839. [Google Scholar] [CrossRef]
- Chan, C.; Liu, Y.; Chia, W. S.; Tan, S. Y.; Yao, K.; Tan, L. P.; Verma, C.; Deng, J.; Wang, Y.; Liesbet, L.; Lim, C. T. A Microfluidic Flow-Through Chip Integrated with Reduced Graphene Oxide Transistor for Influenza Virus Gene Detection. Sensors and Actuators B: Chemical 2017, 251, 927–933. [Google Scholar] [CrossRef]
- Hensel, R. C.; Braun, M.; Klepper, C.; Langlois, A.; Adelhelm, P.; Hauke, F.; Hirsch, A. Graphene Acetic Acid-Based Hybrid Supercapacitor and Liquid-Gated Transistor. Adv. Electron. Mater. 2024, 10, 2300685. [Google Scholar] [CrossRef]
- Flauzino, J. M. R.; Chagas, R. C. R.; Vicentini, F. C.; Janegitz, B. C. Label-Free and Reagentless Electrochemical Genosensor Based on Graphene Acid for Meat Adulteration Detection. Biosens. Bioelectron. 2022, 195, 113628. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dong, X.; Chen, P.; Wang, J.; Li, L.-J. Work Function Engineering of Graphene Electrode via Chemical Doping. ACS Nano 2010, 4, 2689–2694. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Wang, S.; Huang, L.; Ning, Y.; Zhang, Z.; Zhang, G.-J. Gold Nanoparticles-Decorated Graphene Field-Effect Transistor Biosensor for Femtomolar MicroRNA Detection. Biosens. Bioelectron. 2015, 74, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cheng, R.; Lin, W.; Liu, Y.; Wei, L.; Gao, J.; Li, Y.; Fan, Q.; Li, H. Highly Sensitive Solution-Gated Graphene Transistors for Label-Free DNA Detection. Biosens. Bioelectron. 2019, 136, 91–96. [Google Scholar] [CrossRef]
- Hajian, R.; Balderston, S.; Tran, T.; DeBoer, T.; Etienne, J.; Sandhu, M.; Warden, A. R.; Langelier, C.; Peytavi, R.; Humphries, R. M.; Murthy, N.; Sahoo, M. K.; Hibbs, S.; Stevens, M. M.; Puschnik, A. S.; Fletcher, D. A.; Pellegrini, M.; Nair, V.; Hajian, B.; Aran, K. Detection of Unamplified Target Genes via CRISPR–Cas9 Immobilized on a Graphene Field-Effect Transistor. Nat. Biomed. Eng. 2019, 3, 427–437. [Google Scholar] [CrossRef]
- Ohshima, H.; Ohki, S. Donnan Potential and Surface Potential of a Charged Membrane. Biophys. J. 1985, 47, 673–678. [Google Scholar] [CrossRef]
- Kaisti, M. Detection Principles of Biological and Chemical FET Sensors. Biosens. Bioelectron. 2017, 98, 437–448. [Google Scholar] [CrossRef]
- Balderston, S.; Taulbee, J. J.; Celaya, E.; Lukianitskiy, A.; McMahan, S.; Liu, G.; Johnson, E.; Torrente-Rodríguez, R. M.; Tran, T.; Aran, K. Discrimination of Single-Point Mutations in Unamplified Genomic DNA via Cas9 Immobilized on a Graphene Field-Effect Transistor. Nat. Biomed. Eng. 2021, 5, 713–725. [Google Scholar] [CrossRef]
- Yu, H.; He, X.; Wang, L.; Ma, J.; Zhang, Y.; He, L.; Liu, Y.; Wang, P.; Liu, G. Rapid and Unamplified Detection of SARS-CoV-2 RNA via CRISPR-Cas13a-Modified Solution-Gated Graphene Transistors. ACS Sens. 2022, 7, 3923–3932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, X.; Wang, C.; Zhang, Y.; Wang, P.; Liu, G. Unamplified and Label-Free Detection of HPV16 DNA Using CRISPR-Cas12a-Functionalized Solution-Gated Graphene Transistors. Adv. Healthc. Mater. 2023, 12, 2300563. [Google Scholar] [CrossRef]
- Dai, Y.; Somoza, R. A.; Wang, L.; Welter, J. F.; Li, Y.; Caplan, A. I.; Liu, C. C. Exploring the Trans-Cleavage Activity of CRISPR-Cas12a (Cpf1) for the Development of a Universal Electrochemical Biosensor. Angew. Chem. Int. Ed. 2019, 58, 17399–17405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yan, Y.; Que, H.; Yang, Y.; Yang, Q.; Chen, J. CRISPR/Cas12a-Mediated Interfacial Cleaving of Hairpin DNA Reporter for Electrochemical Nucleic Acid Sensing. ACS Sens. 2020, 5, 557–562. [Google Scholar] [CrossRef]
- Kashefi-Kheyrabadi, L.; Mehrgardi, M. A.; Sheikhnejad, Y.; Baradaran, B.; Mobaraki, N.; Wen, Y. Ultrasensitive and Amplification-Free Detection of SARS-CoV-2 RNA Using an Electrochemical Biosensor Powered by CRISPR/Cas13a. Bioelectrochemistry 2023, 150, 108364. [Google Scholar] [CrossRef] [PubMed]
- Heo, W.; Shin, S.; Park, J. Y.; Kim, M.-I.; Kim, Y.-R. Electrochemical Biosensor for Nucleic Acid Amplification-Free and Sensitive Detection of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) RNA via CRISPR/Cas13a Transcleavage Reaction. Biosens. Bioelectron. 2022, 201, 113960. [Google Scholar] [CrossRef] [PubMed]
- Kashefi-Kheyrabadi, L.; Mehrgardi, M. A.; Sheikhnejad, Y.; Wen, Y. Rapid, Multiplexed, and Nucleic Acid Amplification-Free Detection of SARS-CoV-2 RNA Using an Electrochemical Biosensor. Biosens. Bioelectron. 2022, 195, 113649. [Google Scholar] [CrossRef]
- Li, H.; Zhang, H.; Li, Y.; Zhang, C.; Wang, P.; Liu, G. Amplification-Free Detection of SARS-CoV-2 and Respiratory Syncytial Virus Using CRISPR Cas13a and Graphene Field-Effect Transistors. Angew. Chem. Int. Ed. 2022, 61, e202203826. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Zhang, C.; Yu, H.; Li, Y.; Liu, G. Rapid and Ultrasensitive Detection of Mpox Virus Using CRISPR/Cas12b-Empowered Graphene Field-Effect Transistors. Appl. Phys. Rev. 2023, 10, 031409. [Google Scholar] [CrossRef]
- Weng, Z.; Li, Z.; Zhang, M.; Peng, Y.; Sun, L.; Li, Y.; Liu, G. CRISPR-Cas12a Biosensor Array for Ultrasensitive Detection of Unamplified DNA with Single-Nucleotide Polymorphic Discrimination. ACS Sens. 2023, 8, 1489–1499. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, C.; Liu, Y.; Wang, L.; Wang, P.; Liu, G. Unamplified System for Sensitive and Typing Detection of ASFV by the Cascade Platform That CRISPR-Cas12a Combined with Graphene Field-Effect Transistor. Biosens. Bioelectron. 2023, 240, 115637. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, C.; Wang, C.; Yang, Q.; Liu, G.; Zhang, Y.; Liu, Y.; Fan, X.; Li, H.; Sun, L. Tandem Cas13a/crRNA-Mediated CRISPR-FET Biosensor: A One-for-All Check Station for Virus without Amplification. ACS Sens. 2022, 7, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Sun, L.; Liu, Y.; Liu, G. Amplification-Free CRISPR/Cas Detection Technology: Challenges, Strategies, and Perspectives. Chem. Soc. Rev. 2023, 52, 361–382. [Google Scholar] [CrossRef] [PubMed]
- Ban, D. K.; Abdel-Rahman, M.; Sandwell, D.; Skrypka, A. V.; Hill, E. H.; Kudr, J.; Kostesha, N.; Englebienne, P.; Smith, A. A Single Multiomics Transistor for Electronic Detection of SARS-CoV-2 Variants Antigen and Viral RNA Without Amplification. Adv. Mater. Technol. 2023, 8, 2201945. [Google Scholar] [CrossRef]
- Sun, Y.; Li, H.; Zhang, C.; Liu, Y.; Wang, P.; Liu, G. High-Intensity Vector Signals for Detecting SARS-CoV-2 RNA Using CRISPR/Cas13a Coupled with Stabilized Graphene Field-Effect Transistor. Biosens. Bioelectron. 2022, 222, 114979. [Google Scholar] [CrossRef]
- Chen, D.; Han, Y.; Zhang, Y.; Zhang, F.; Zhang, H.; Liu, Y.; Wang, P.; Liu, G. CRISPR-Mediated Profiling of Viral RNA at Single-Nucleotide Resolution. Angew. Chem. Int. Ed. 2023, 62, e202304298. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the PCR process for nucleic acid amplification (dNTPs: desoxyNucleotides TriPhosphate, the various A, T, C and G nucleotides). Note that the first mandatory step is denaturation of the dsDNA (double-strand DNA, the only one naturally occurring) into ssDNA (single-strand DNA), which needs stringent conditions hardly attainable in POC devices. Reproduced from https://commons.wikimedia.org/wiki/File:Polymerase_chain_reaction-en.svg. Enzoklop, CC BY-SA 4.0, via Wikimedia Commons.
Figure 1.
Schematic representation of the PCR process for nucleic acid amplification (dNTPs: desoxyNucleotides TriPhosphate, the various A, T, C and G nucleotides). Note that the first mandatory step is denaturation of the dsDNA (double-strand DNA, the only one naturally occurring) into ssDNA (single-strand DNA), which needs stringent conditions hardly attainable in POC devices. Reproduced from https://commons.wikimedia.org/wiki/File:Polymerase_chain_reaction-en.svg. Enzoklop, CC BY-SA 4.0, via Wikimedia Commons.
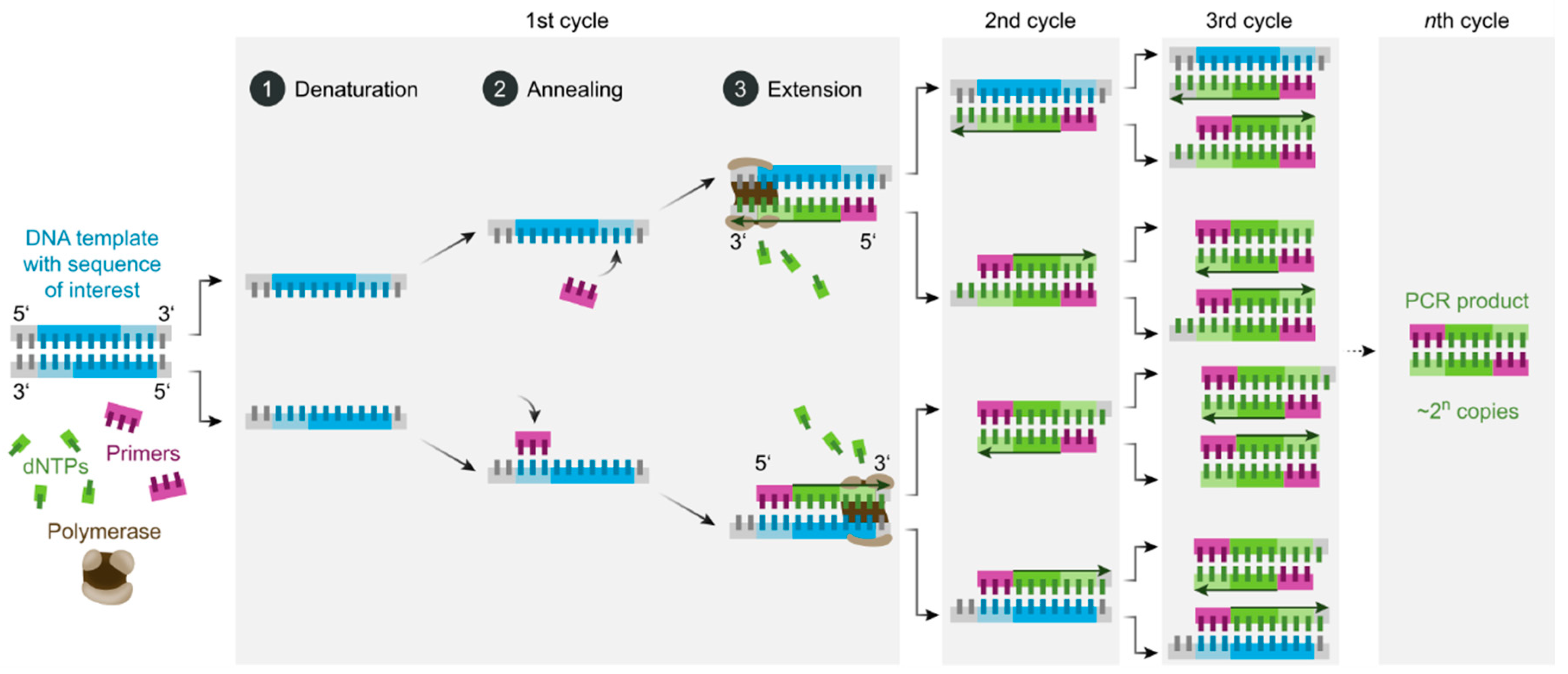
Figure 2.
Immune Response of CRISPR-Cas Systems, adapted from [24] with permission from Springer Nature.
Figure 2.
Immune Response of CRISPR-Cas Systems, adapted from [24] with permission from Springer Nature.
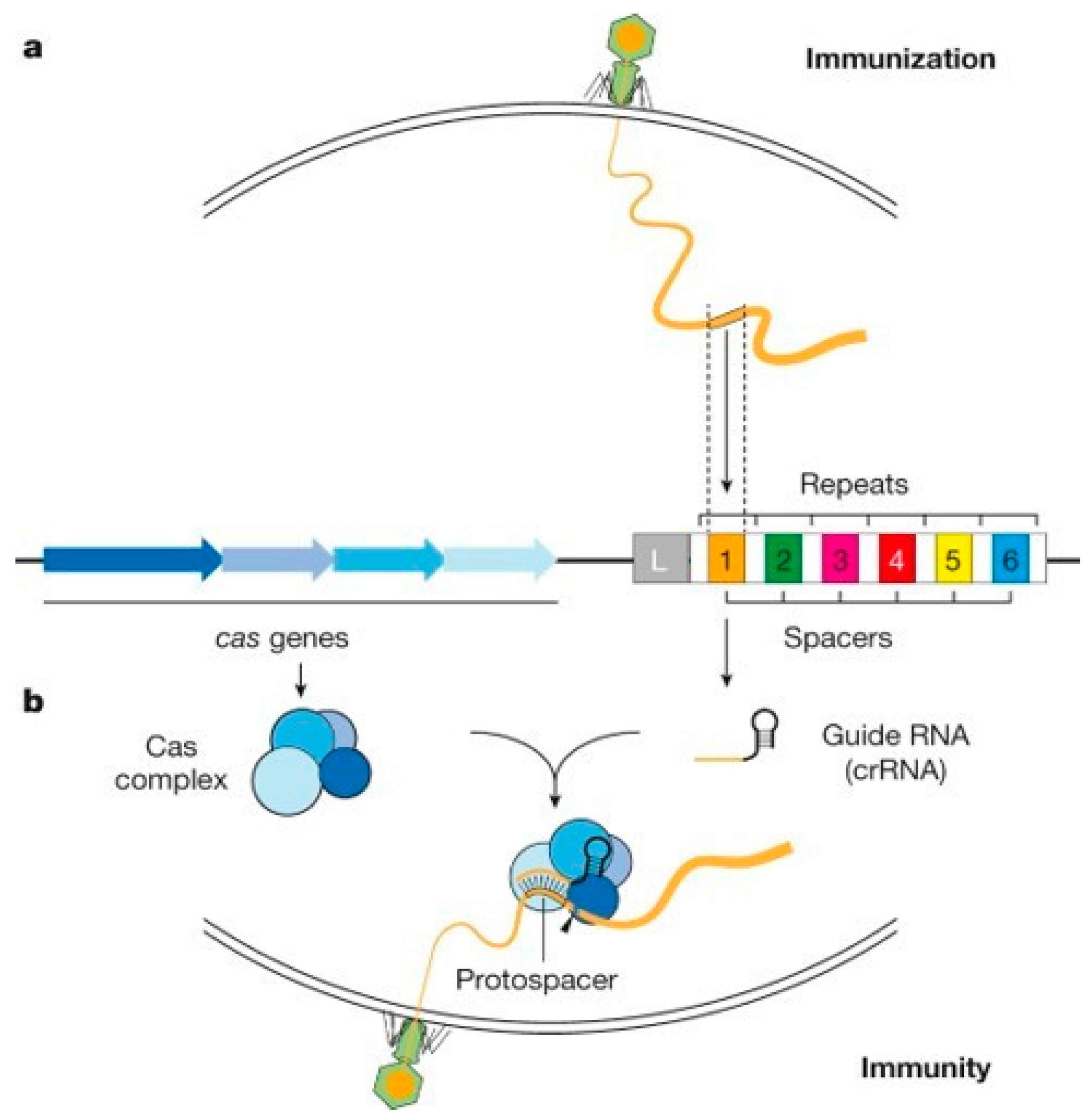
Figure 3.
Simplified classification of current CRISPR-Cas systems. Adapted from [28] under CC BY licence.
Figure 3.
Simplified classification of current CRISPR-Cas systems. Adapted from [28] under CC BY licence.
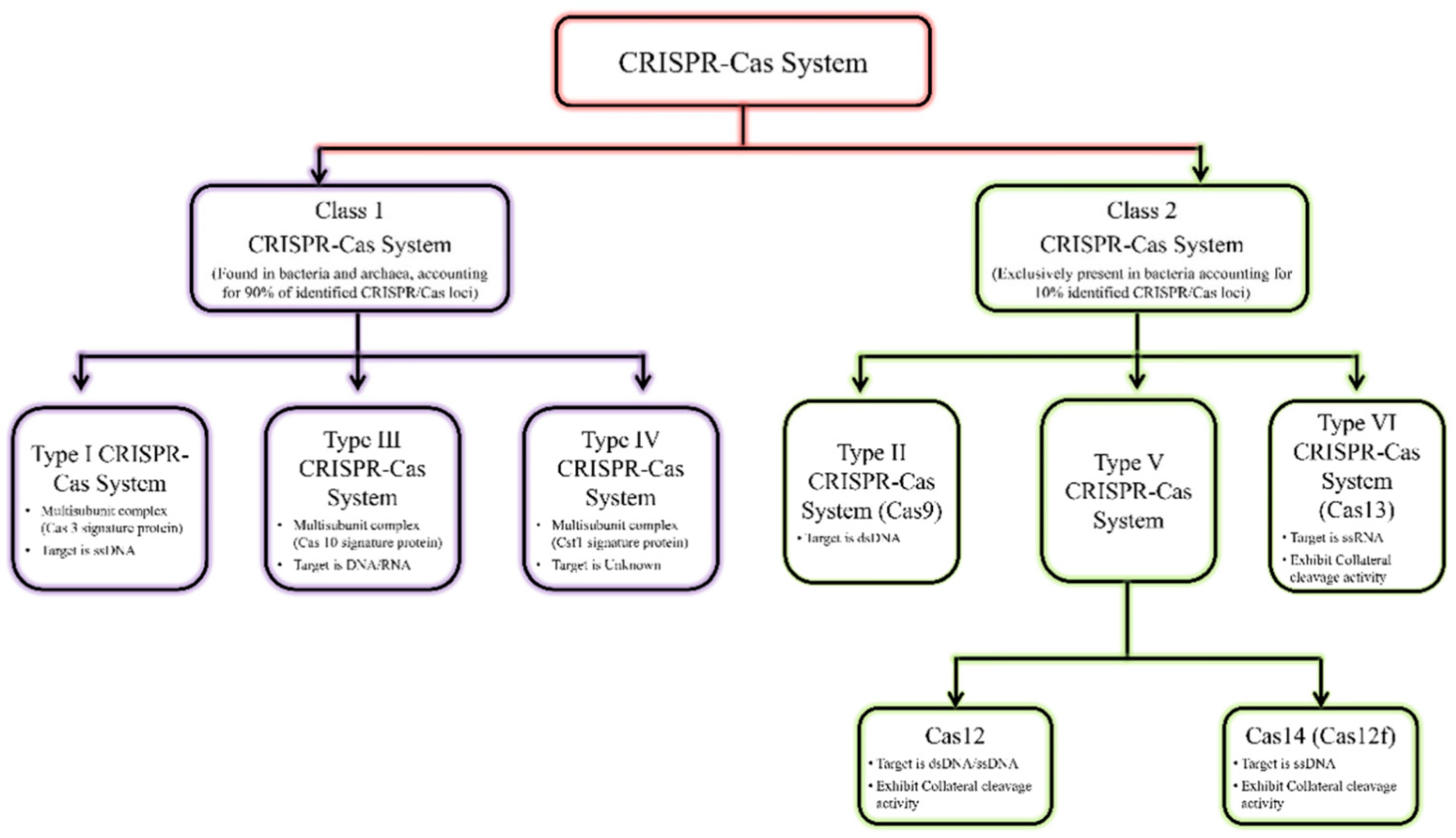
Figure 4.
(A) Schematic diagram showing the various steps involved in transcription, guide RNA recognition and binding, and activation of Cas13 RNAse activity (rU: ribose Uracile). (B) Structure of an RNA chain around an Uracile nucleotide. Adapted from [42] with permission from Elsevier.
Figure 4.
(A) Schematic diagram showing the various steps involved in transcription, guide RNA recognition and binding, and activation of Cas13 RNAse activity (rU: ribose Uracile). (B) Structure of an RNA chain around an Uracile nucleotide. Adapted from [42] with permission from Elsevier.
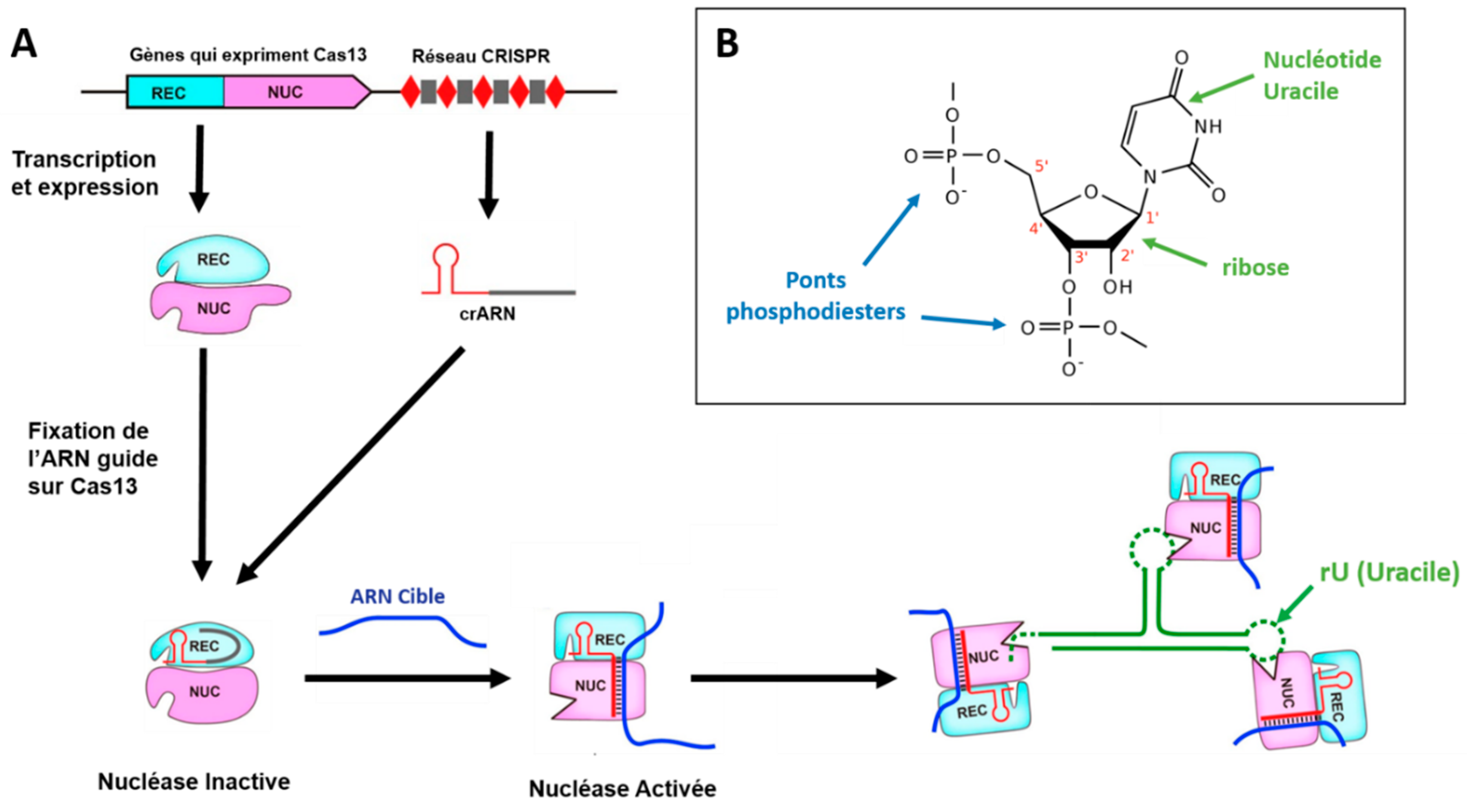
Figure 5.
Schematic diagram of the SHERLOCK method. Adapted from [3] under CC BY-NC-SA 4.0 license.
Figure 5.
Schematic diagram of the SHERLOCK method. Adapted from [3] under CC BY-NC-SA 4.0 license.
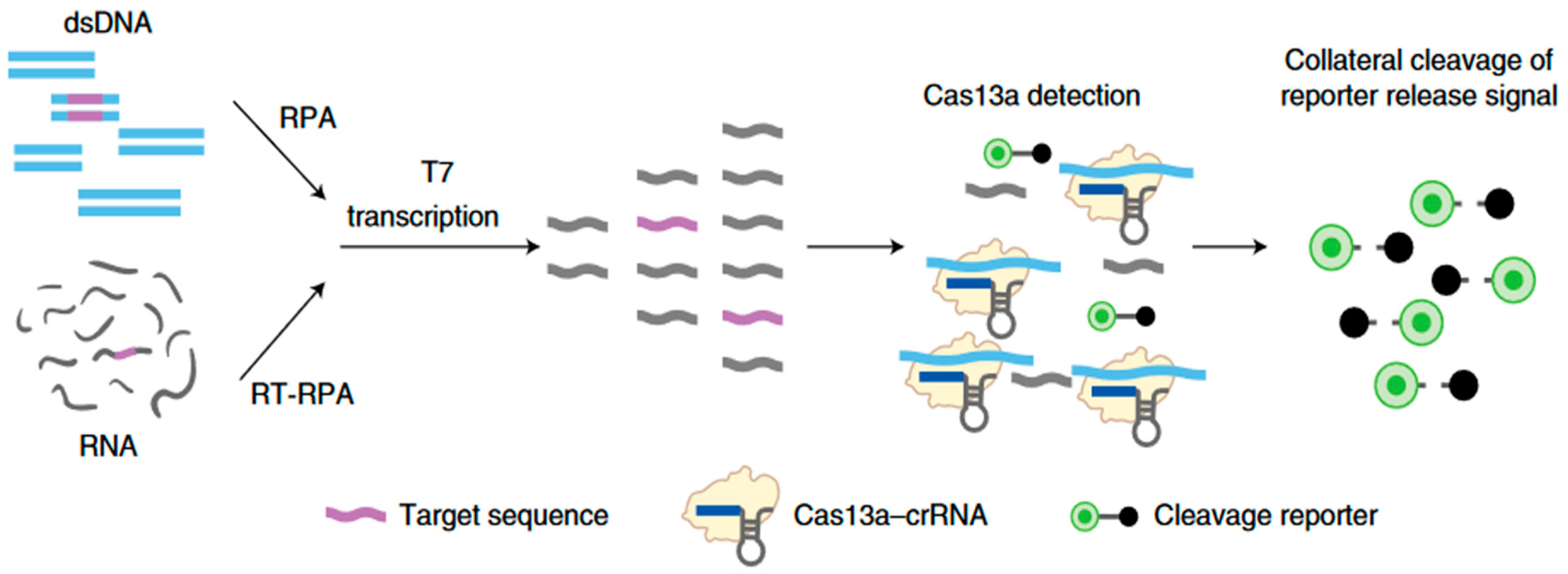
Figure 6.
A) Schematic diagram of the SHERLOCKv2 method (FAM: carboxyfluorescein, fluorophore). B) Detection of synthetic Zika virus (ZIKV) RNA using the SHERLOCKv2 method by immunochromatographic strip. C) Quantification of the fluorescence of the detection bands read on B) linked to the presence of the FAM fluorophore, captured by its specific antibody. The RPA pre-amplification step is not shown. Adapted from [55] with permission from The American Association for the Advancement of Science, © 2018.
Figure 6.
A) Schematic diagram of the SHERLOCKv2 method (FAM: carboxyfluorescein, fluorophore). B) Detection of synthetic Zika virus (ZIKV) RNA using the SHERLOCKv2 method by immunochromatographic strip. C) Quantification of the fluorescence of the detection bands read on B) linked to the presence of the FAM fluorophore, captured by its specific antibody. The RPA pre-amplification step is not shown. Adapted from [55] with permission from The American Association for the Advancement of Science, © 2018.
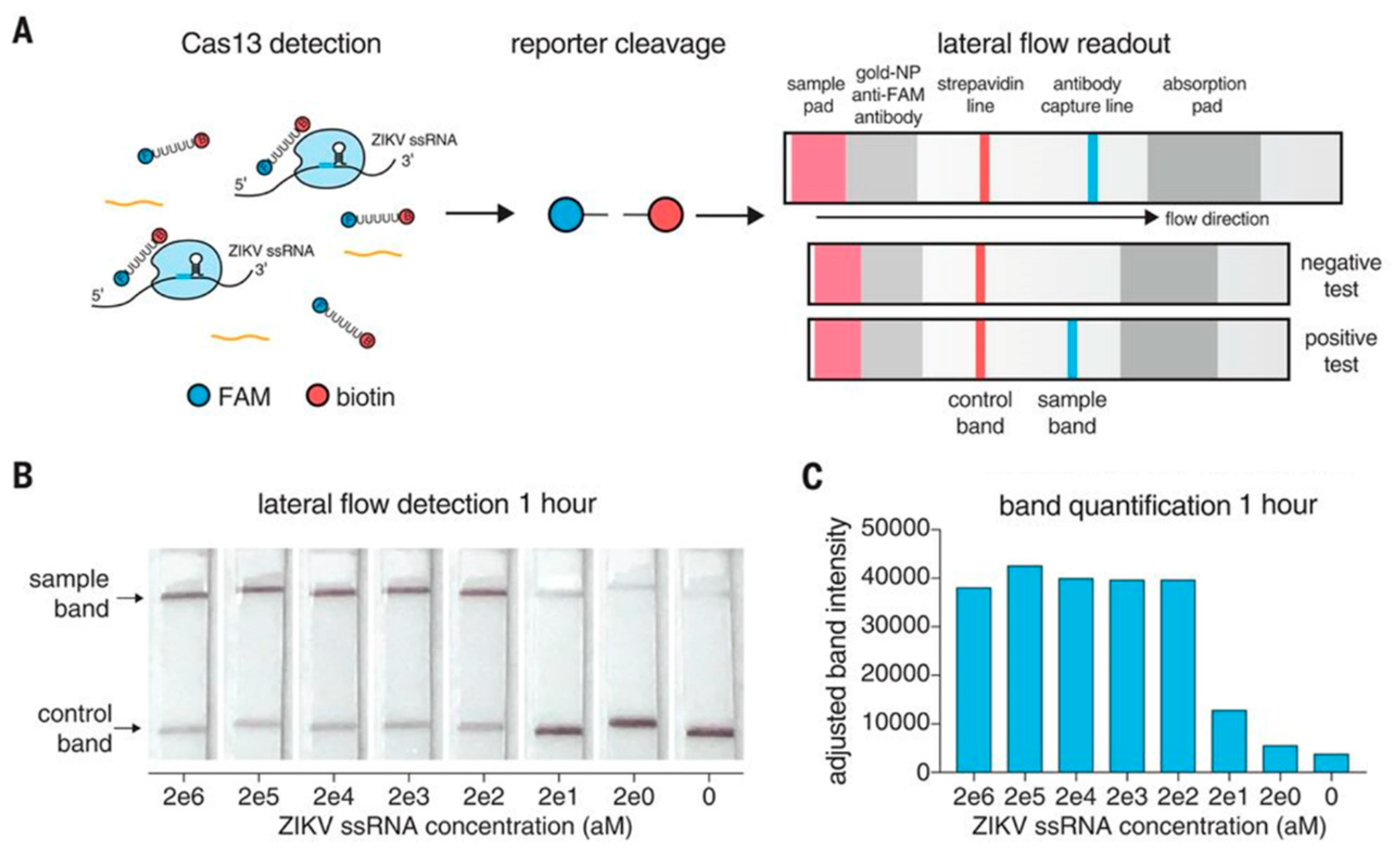
Figure 8.
Basic architectures for electrolytic gate field effect transistors (EGFETs). The various components such as gate, electrolyte, source, drain and semiconductor channel are illustrated. VG and VD are the respective potentials of the gate and drain, with the source connected to ground. a) Top-gate configuration. b) Top-gate configuration with a recognition layer on the semiconductor channel. c) Top-gate configuration with a recognition layer on the gate. d) Bottom-gate configuration. e) Side-gate configuration. f) Extended-gate (or floating-gate) configuration. Adapted from [73] form Springer Nature with permission © 2021.
Figure 8.
Basic architectures for electrolytic gate field effect transistors (EGFETs). The various components such as gate, electrolyte, source, drain and semiconductor channel are illustrated. VG and VD are the respective potentials of the gate and drain, with the source connected to ground. a) Top-gate configuration. b) Top-gate configuration with a recognition layer on the semiconductor channel. c) Top-gate configuration with a recognition layer on the gate. d) Bottom-gate configuration. e) Side-gate configuration. f) Extended-gate (or floating-gate) configuration. Adapted from [73] form Springer Nature with permission © 2021.
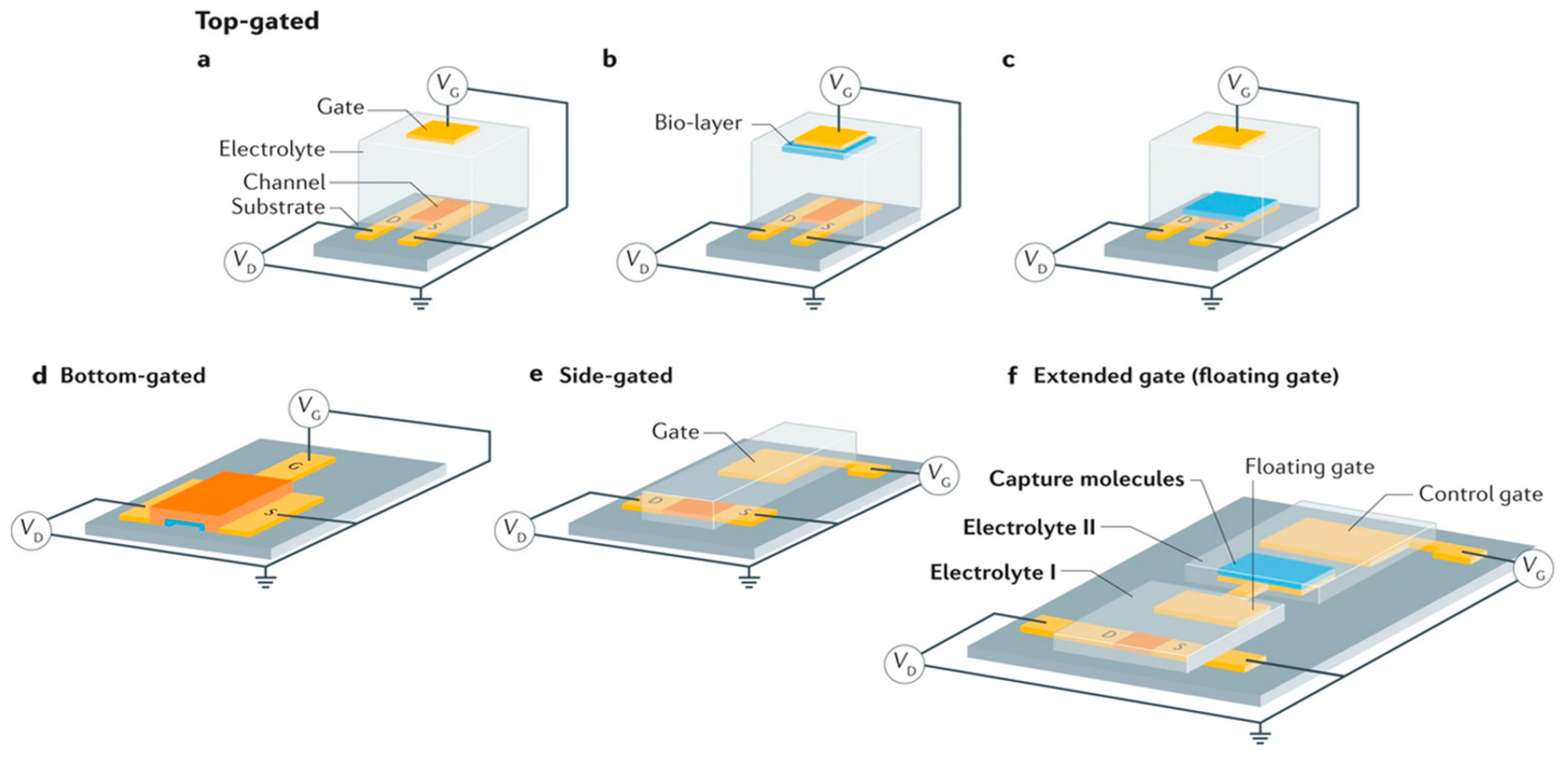
Figure 9.
a) Cross-section of an EGFET. Operation of an EGFET in charge carrier accumulation mode for an ion-impermeable (b) and ion-permeable (c) semiconductor.
Figure 9.
a) Cross-section of an EGFET. Operation of an EGFET in charge carrier accumulation mode for an ion-impermeable (b) and ion-permeable (c) semiconductor.
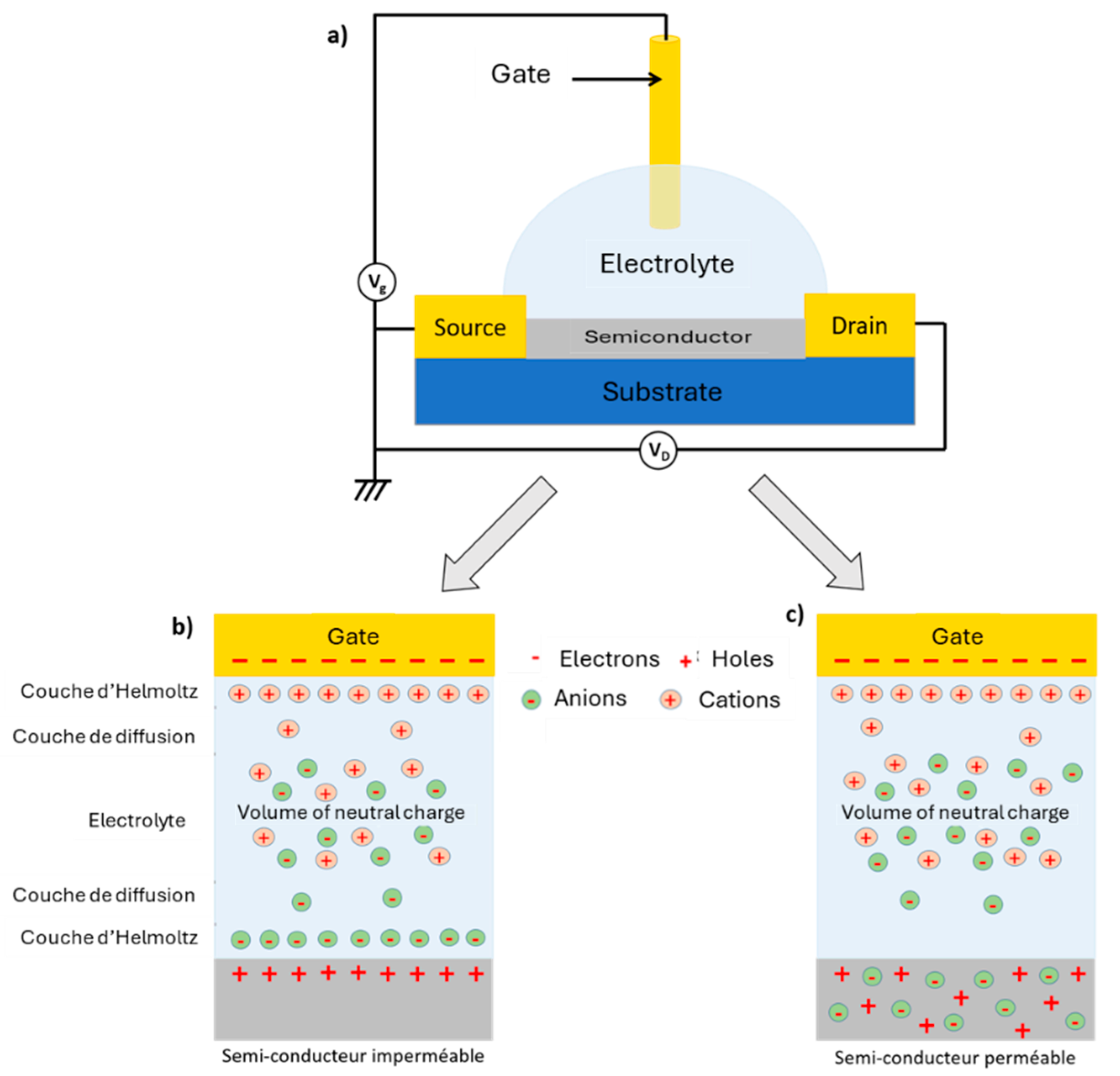
Figure 10.
a) Representation of sp2 hybridization in graphene. b) Construction of reciprocal space from the primitive graphene lattice. c) Structure of energy bands in the reciprocal lattice. Adapted with permission from [91], Copyright 2009 by the American Physical Society.
Figure 10.
a) Representation of sp2 hybridization in graphene. b) Construction of reciprocal space from the primitive graphene lattice. c) Structure of energy bands in the reciprocal lattice. Adapted with permission from [91], Copyright 2009 by the American Physical Society.
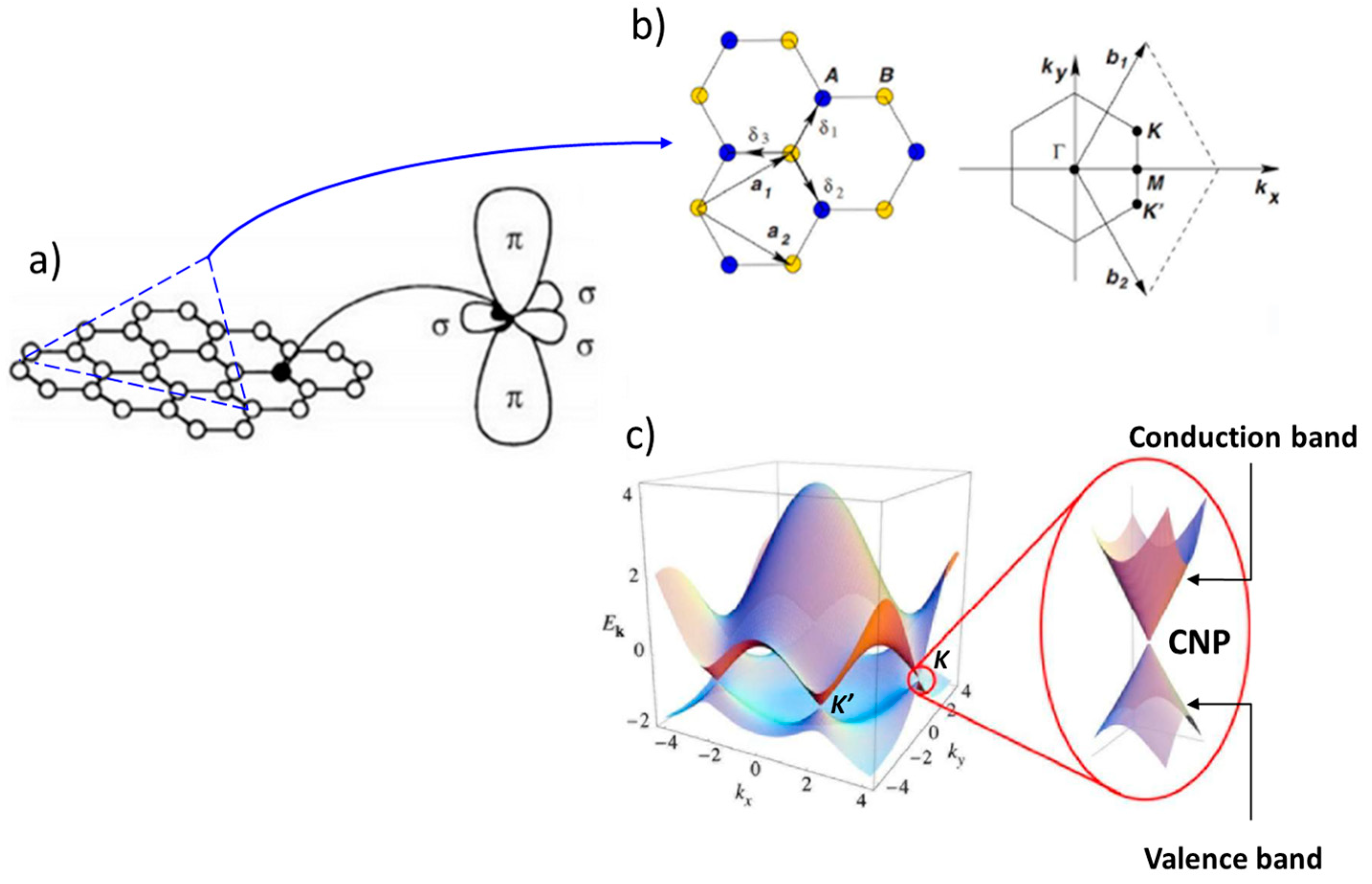
Figure 11.
Dirac fermions. a) Representation of the Dirac cone. b) Evolution of charge carrier mass (normalized to m0, the mass of a free electron) as a function of concentration n in the conduction band (holes, red circles) and valence band (electrons, blue circles). The green curve is an extrapolated trend towards the charge neutrality point (CNP), where n = 0. c). Evolution of the resistivity ρ of graphene as a function of the voltage VG applied to the gate of the transistor. The rapid decrease in resistivity when charge carriers are added indicates their high mobility. Dirac cones show the evolution of the Fermi level as a function of VG. Adapted from [90,93] with permission from Springer Nature, ©2005,2007.
Figure 11.
Dirac fermions. a) Representation of the Dirac cone. b) Evolution of charge carrier mass (normalized to m0, the mass of a free electron) as a function of concentration n in the conduction band (holes, red circles) and valence band (electrons, blue circles). The green curve is an extrapolated trend towards the charge neutrality point (CNP), where n = 0. c). Evolution of the resistivity ρ of graphene as a function of the voltage VG applied to the gate of the transistor. The rapid decrease in resistivity when charge carriers are added indicates their high mobility. Dirac cones show the evolution of the Fermi level as a function of VG. Adapted from [90,93] with permission from Springer Nature, ©2005,2007.
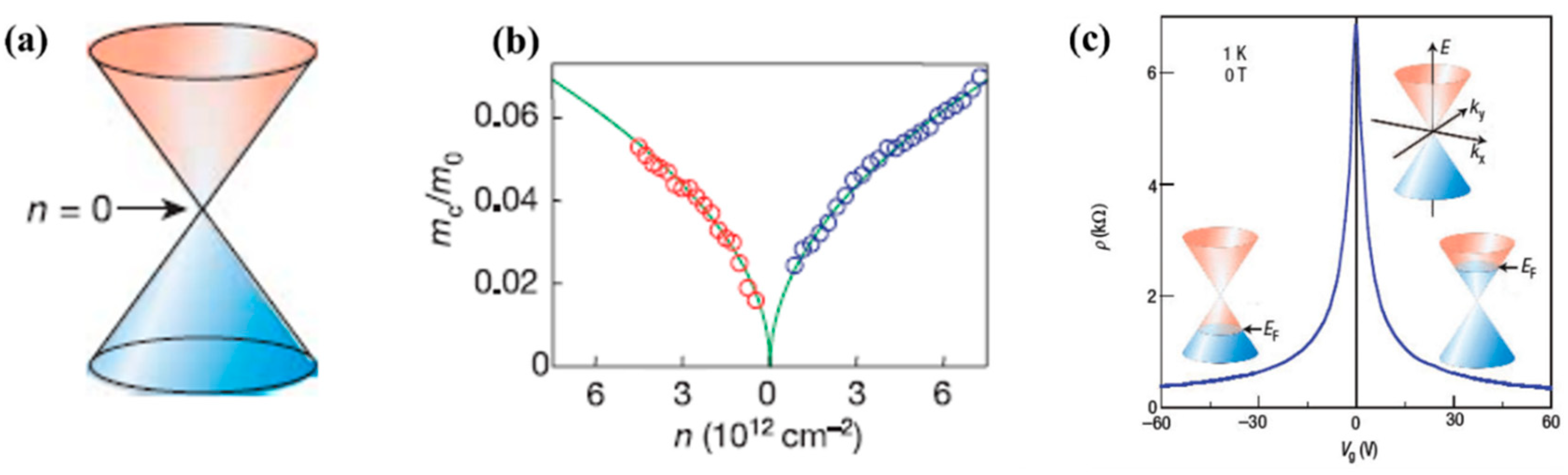
Figure 13.
Graphene structure, GO and rGO. Reproduced from [94] under CC BY license.
Figure 13.
Graphene structure, GO and rGO. Reproduced from [94] under CC BY license.
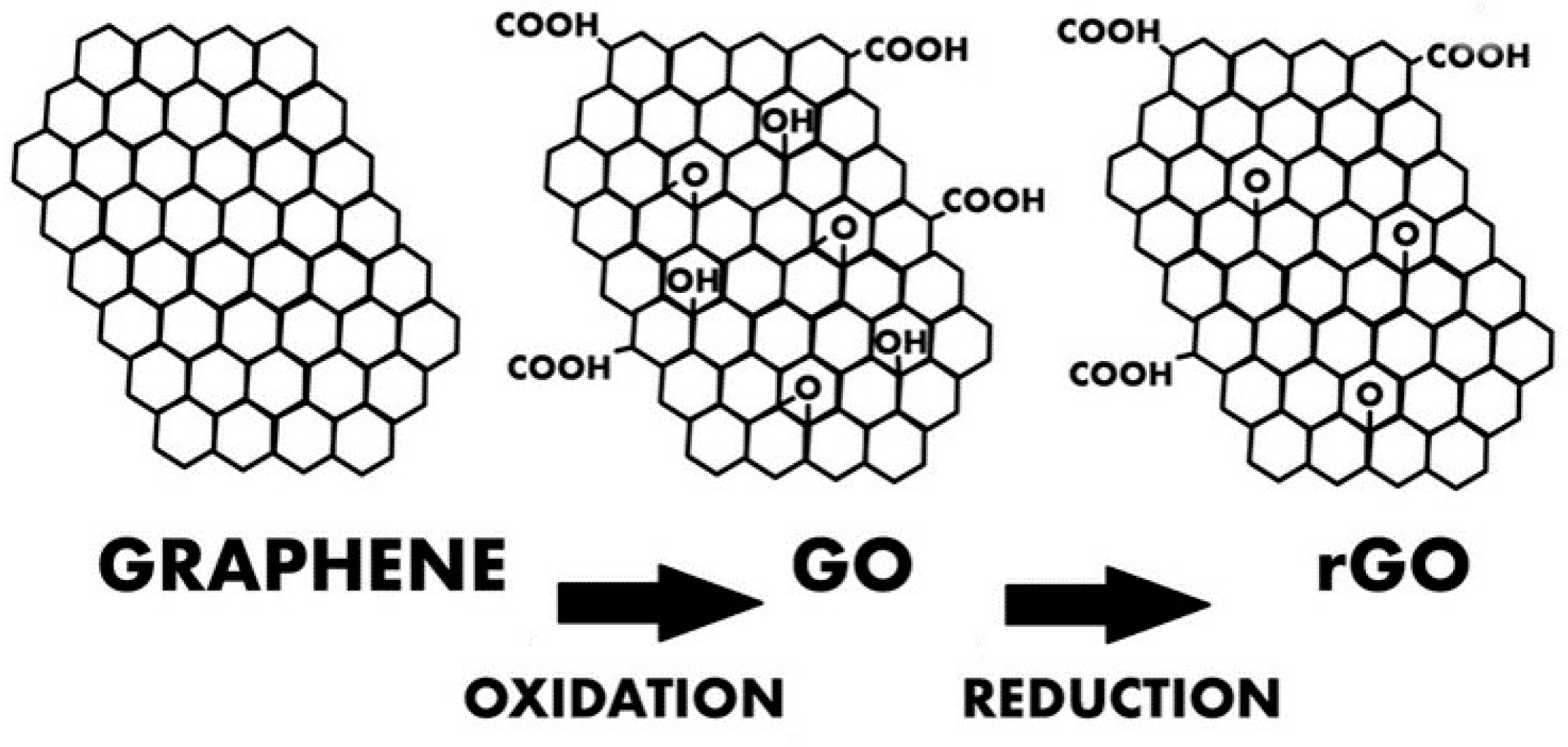
Figure 14.
a) Structure and operating principle of an EGGFET b) Capacitive model of the transistor. The total capacitance of the transistor is a function of the capacitances of the 3 capacitors in series.
Figure 14.
a) Structure and operating principle of an EGGFET b) Capacitive model of the transistor. The total capacitance of the transistor is a function of the capacitances of the 3 capacitors in series.
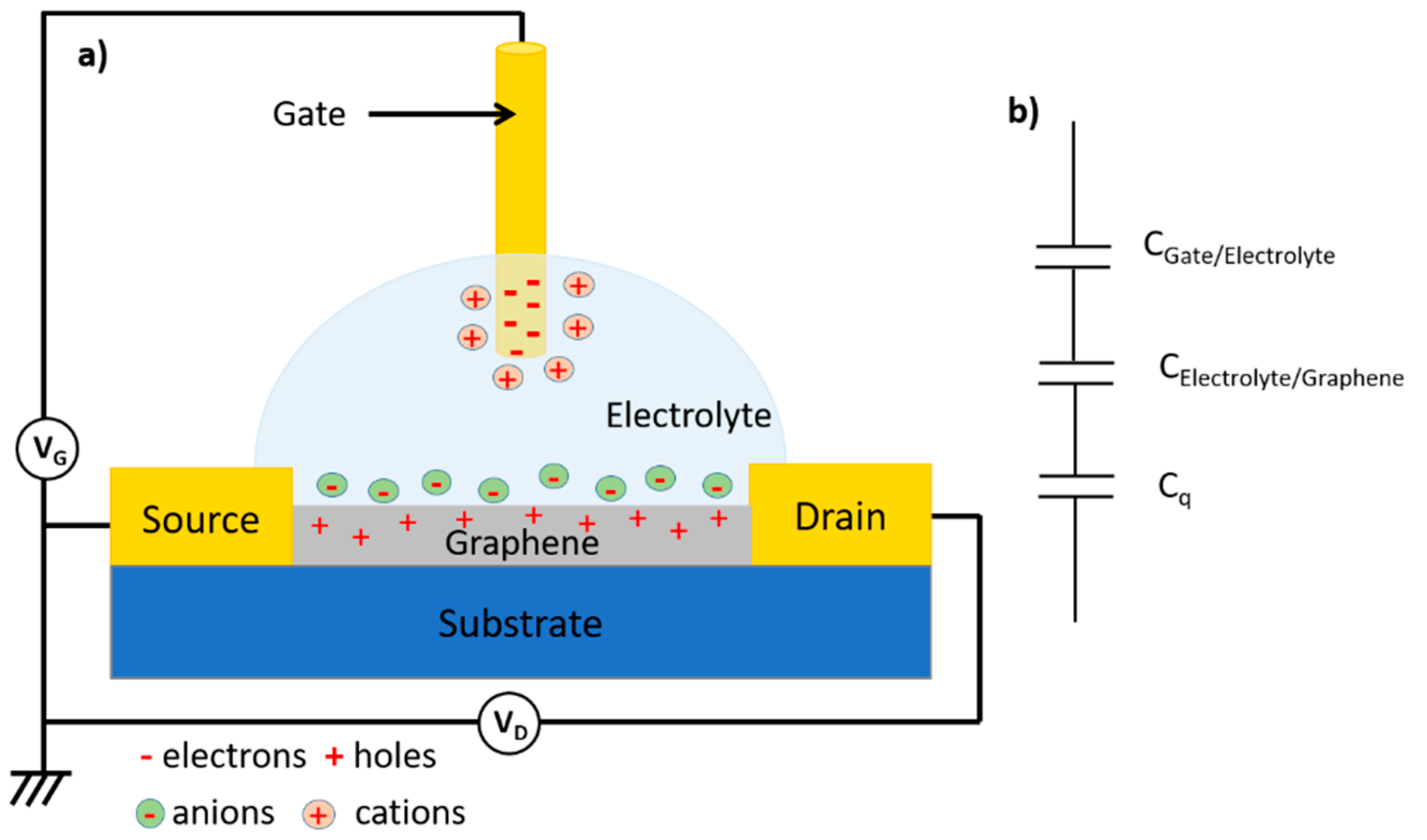
Figure 15.
Typical a) output b) and transfer curves of an EGGFET with rGO as active material. Adapted from [97] with permission from Elsevier © 2021.
Figure 15.
Typical a) output b) and transfer curves of an EGGFET with rGO as active material. Adapted from [97] with permission from Elsevier © 2021.
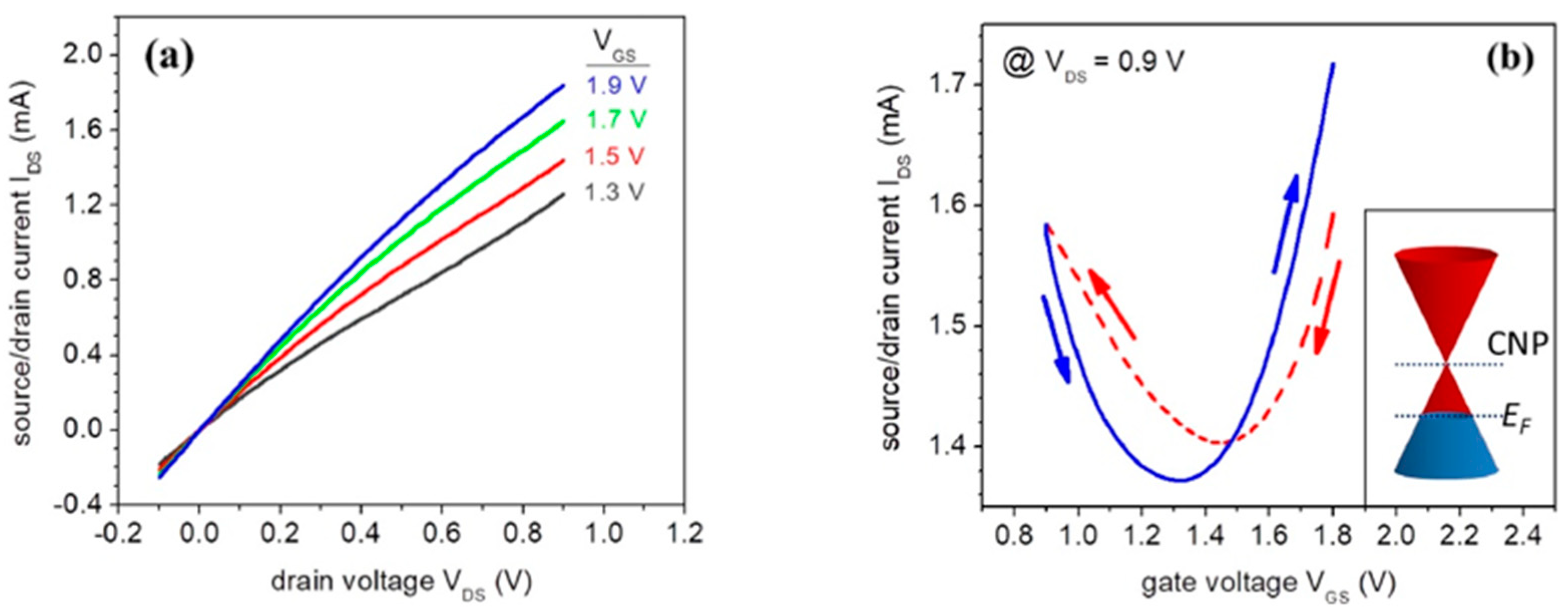
Figure 16.
Detection mechanism of EGGFETs in the case of charge transfer (left) or electrostatic interaction (right).
Figure 16.
Detection mechanism of EGGFETs in the case of charge transfer (left) or electrostatic interaction (right).
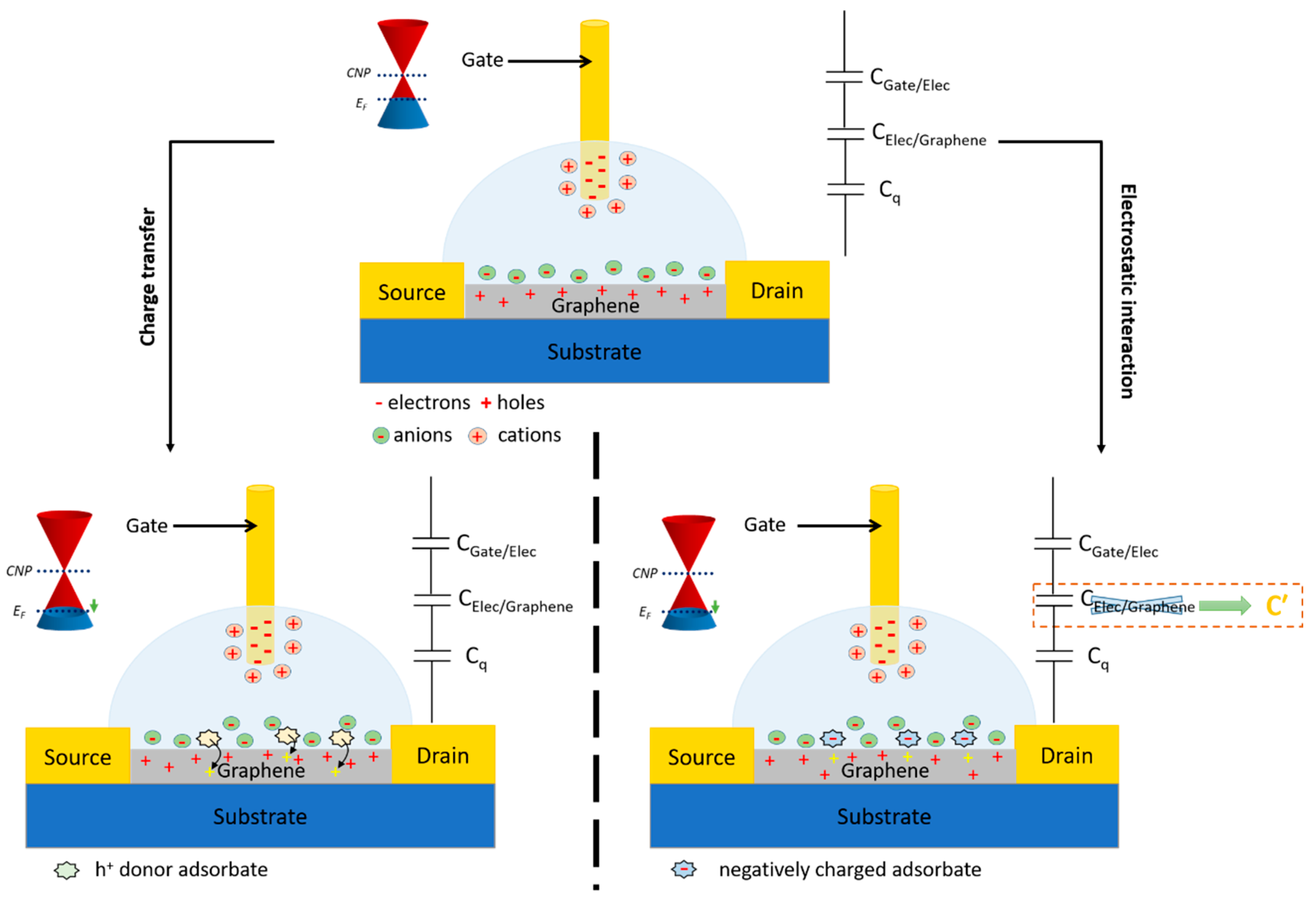
Figure 17.
pH sensor based on an EGGFET. a) Example of Dirac point VDirac (or V*) shift with increasing pH. b) Ion and charge carrier distribution at the graphene/electrolyte interface at alkaline and acid pH. Adapted from [98] Elsevier with permission, © 2018.
Figure 17.
pH sensor based on an EGGFET. a) Example of Dirac point VDirac (or V*) shift with increasing pH. b) Ion and charge carrier distribution at the graphene/electrolyte interface at alkaline and acid pH. Adapted from [98] Elsevier with permission, © 2018.
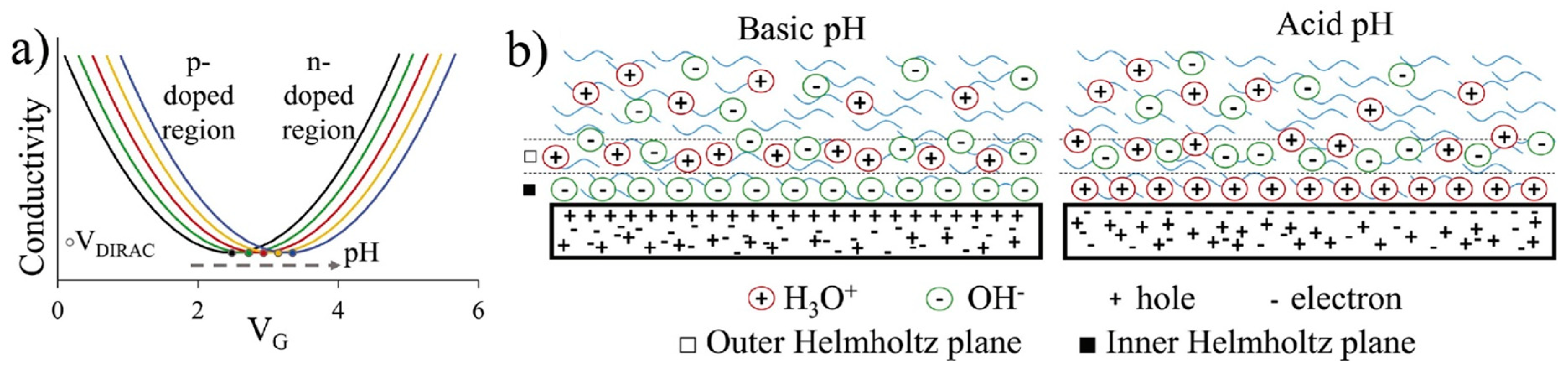
Figure 18.
(a) PBASE structure and (b) strategy for functionalizing an EGGFET with PBASE and a DNA probe linked to PBASE by an amide function. Adapted from [101] with permission for Elsevier © 2020.
Figure 18.
(a) PBASE structure and (b) strategy for functionalizing an EGGFET with PBASE and a DNA probe linked to PBASE by an amide function. Adapted from [101] with permission for Elsevier © 2020.
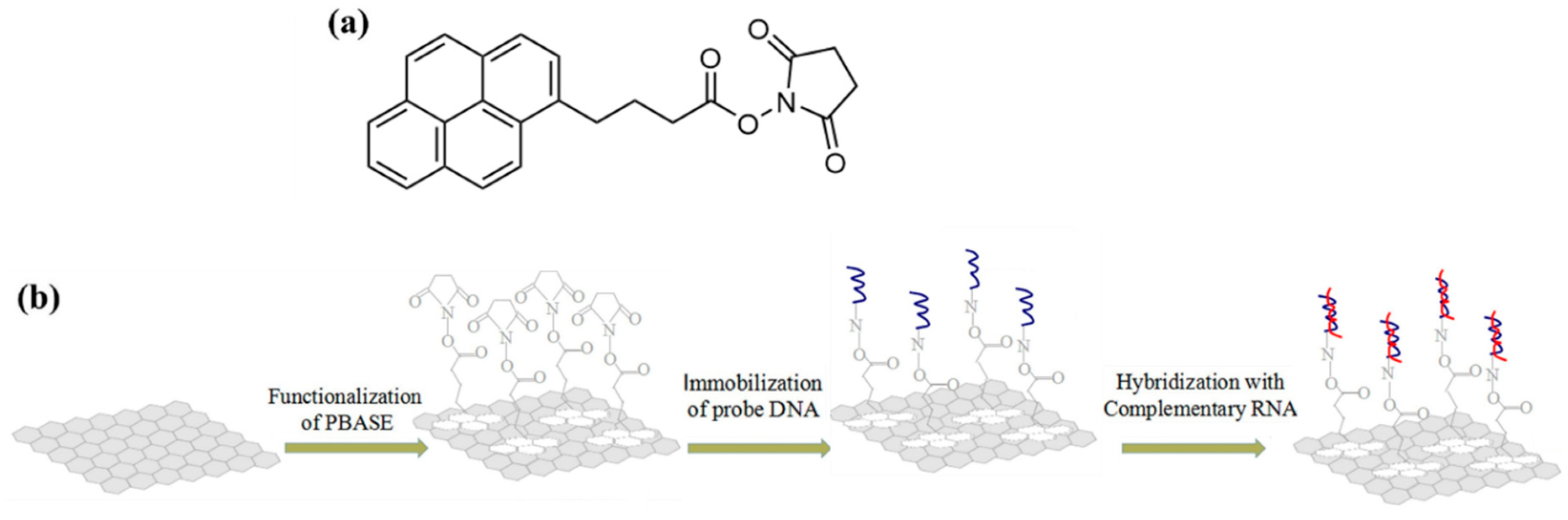
Figure 19.
DNA probe immobilization strategy described in Chan et al. (a) The use of a DNA probe immobilization section maintains π-stacking interaction with the rGO after hybridization. (b) In the presence of a flux, short capture probes whose sequence fully matches that of the target DNA are removed from the rGO surface after hybridization. Long capture probes remain on the rGO surface. Adapted from [102] with permission from Elsevier © 2017.
Figure 19.
DNA probe immobilization strategy described in Chan et al. (a) The use of a DNA probe immobilization section maintains π-stacking interaction with the rGO after hybridization. (b) In the presence of a flux, short capture probes whose sequence fully matches that of the target DNA are removed from the rGO surface after hybridization. Long capture probes remain on the rGO surface. Adapted from [102] with permission from Elsevier © 2017.
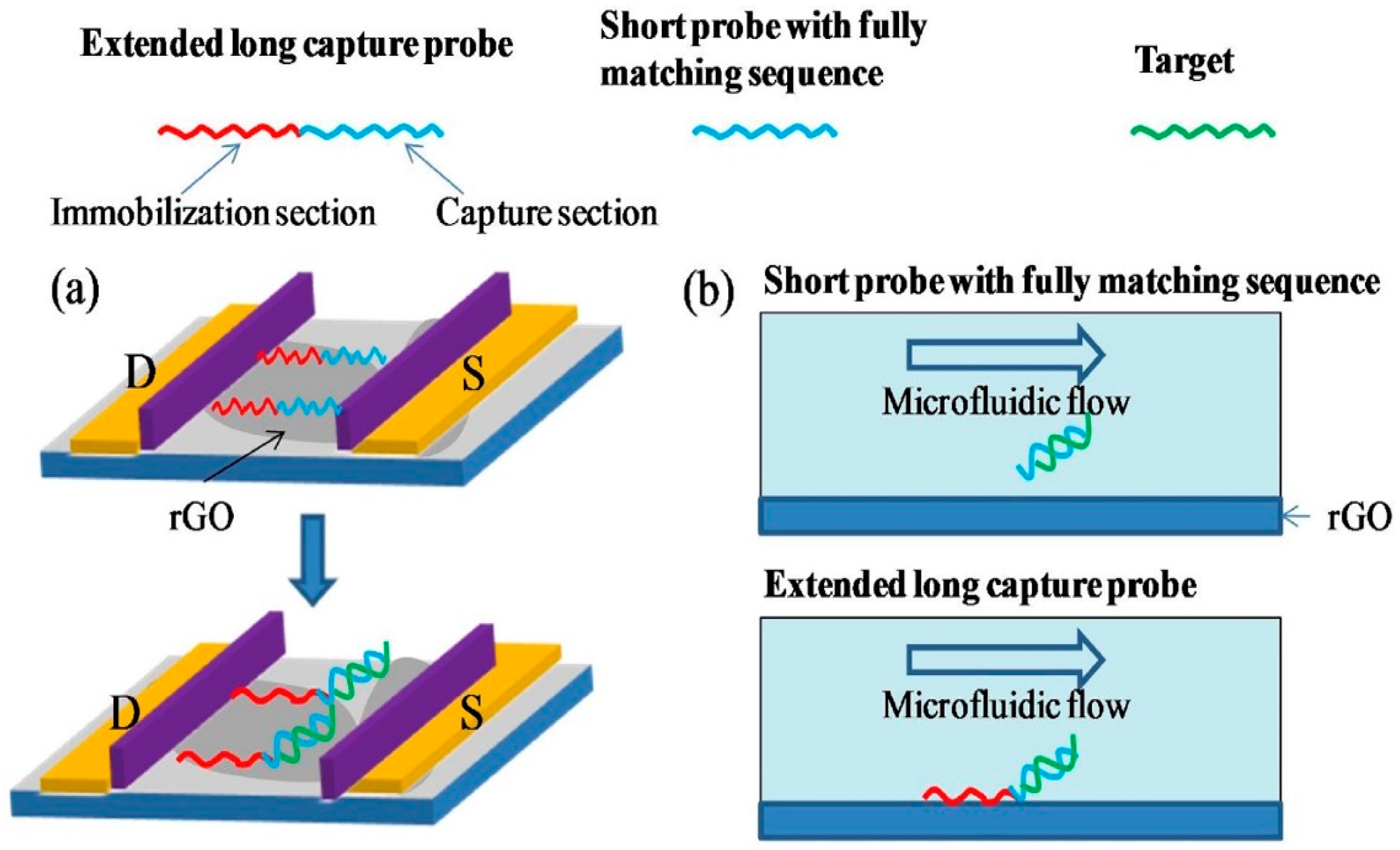
Figure 20.
Structure of "formic acid" and "acetic acid" graphene compounds.
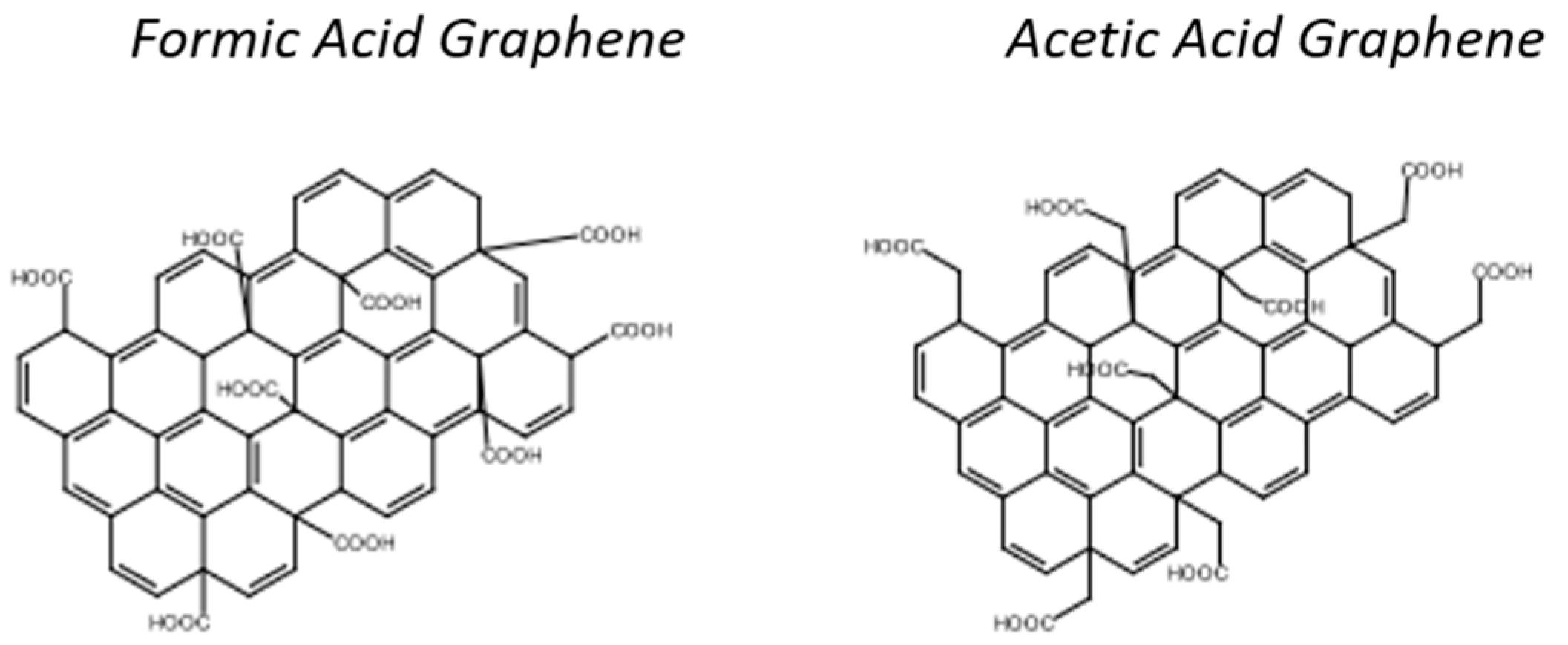
Figure 21.
(a) Simplified process for functionalizing the channel of an EGGFET for DNA detection described by Cai et al. (b) Transfer curves (the PNA curve corresponds to the functionalized transistor) and (c) shifts in the associated ΔVCNP charge neutrality point as a function of the different species brought into contact with the transistor: PBS buffer without target strand (red), PBS + non-complementary DNA (navy blue), PBS + DNA carrying a mismatch (light blue) and PBS + complementary DNA (purple). Adapted from [106] with permission from Elsevier © 2015.
Figure 21.
(a) Simplified process for functionalizing the channel of an EGGFET for DNA detection described by Cai et al. (b) Transfer curves (the PNA curve corresponds to the functionalized transistor) and (c) shifts in the associated ΔVCNP charge neutrality point as a function of the different species brought into contact with the transistor: PBS buffer without target strand (red), PBS + non-complementary DNA (navy blue), PBS + DNA carrying a mismatch (light blue) and PBS + complementary DNA (purple). Adapted from [106] with permission from Elsevier © 2015.
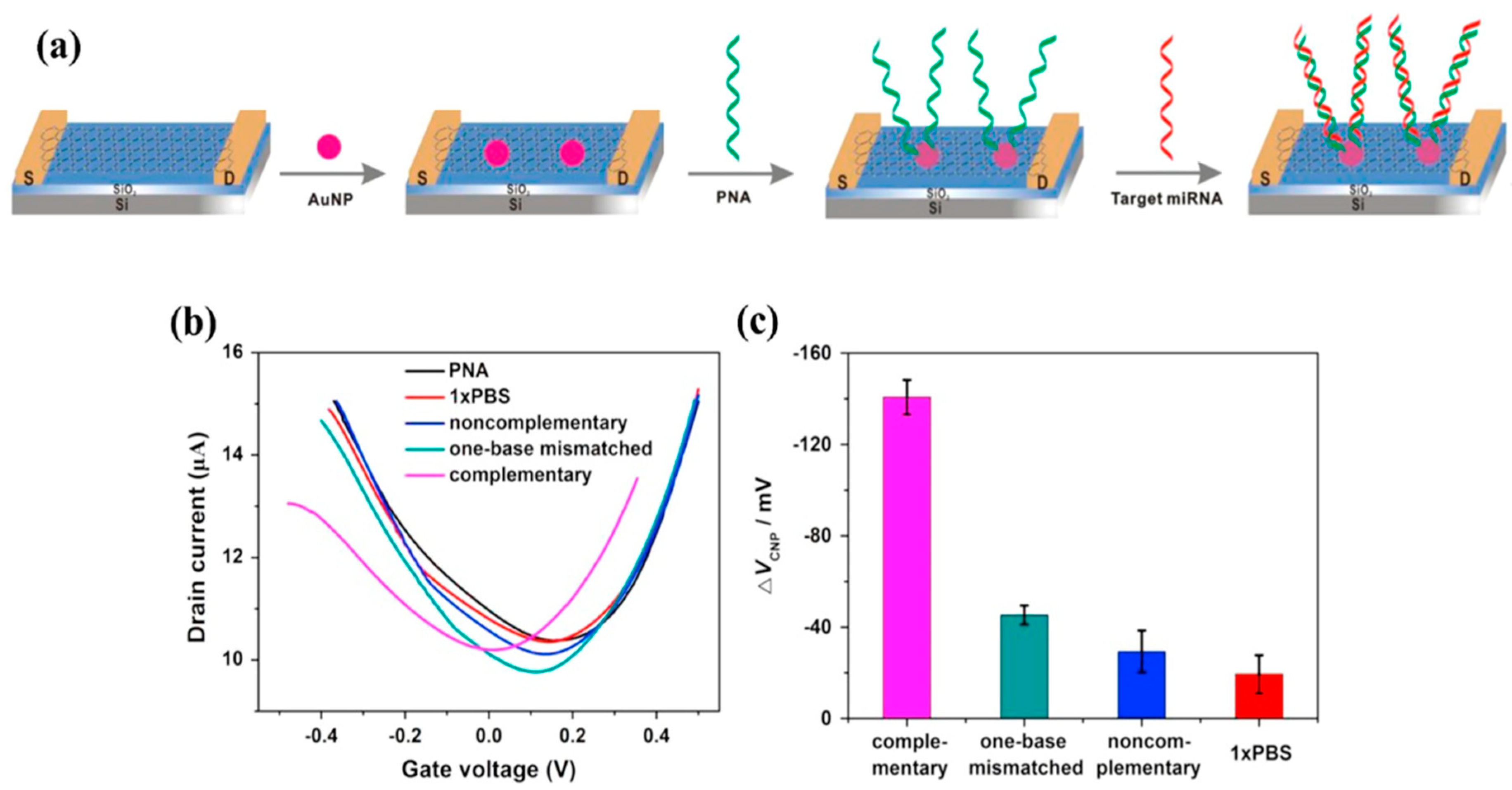
Figure 22.
Operating principle of the DNA-sensing EGGFET described by Li et al. (a) Structure and functionalization of the EGGFET. (b) Transistor transfer curves measured with a bare gate (control), after functionalization with single-stranded DNA (ssDNA) and after hybridization with its complement (dsDNA). (c) Evolution of the current response of the functionalized EGGFET during hybridization with the complementary DNA chains. Gate and drain potentials are fixed at VG = 0.8 V and VD = 0.1 V. Adapted from [107] with permission from © 2019 Elsevier B.V. All rights reserved.
Figure 22.
Operating principle of the DNA-sensing EGGFET described by Li et al. (a) Structure and functionalization of the EGGFET. (b) Transistor transfer curves measured with a bare gate (control), after functionalization with single-stranded DNA (ssDNA) and after hybridization with its complement (dsDNA). (c) Evolution of the current response of the functionalized EGGFET during hybridization with the complementary DNA chains. Gate and drain potentials are fixed at VG = 0.8 V and VD = 0.1 V. Adapted from [107] with permission from © 2019 Elsevier B.V. All rights reserved.
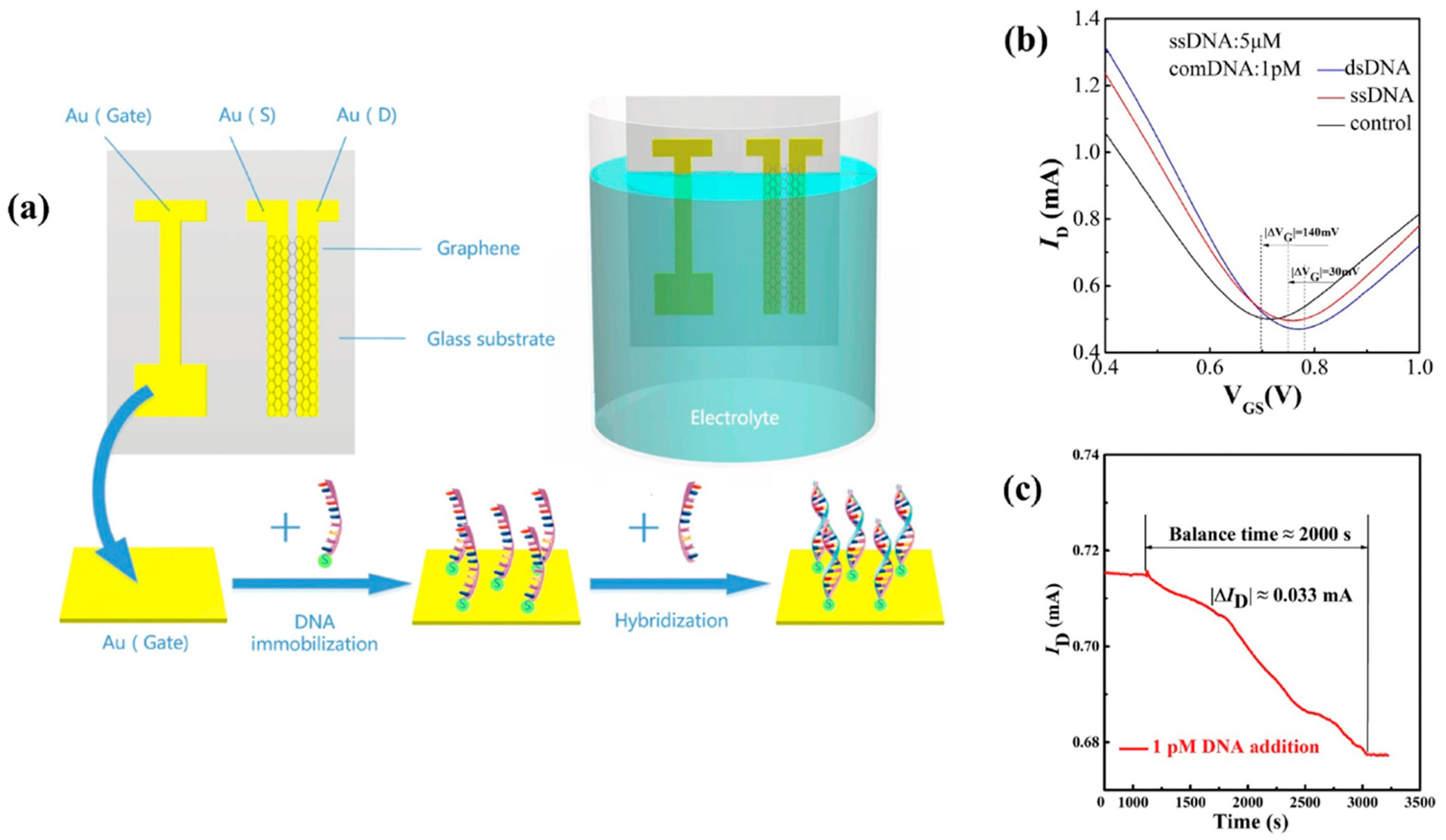
Figure 23.
(a) CRISPR-Chip diagnostic platform proposed by Hajian et al. enables DNA detection in less than 15 minutes. The dCas9 enzyme complexed with a DNA target-specific guide RNA is immobilized on the graphene surface of an EGGFET. The immobilized CRISPR-dCas9 complex scans the entire genomic DNA until it identifies its target sequence. Hybridization between the target DNA and CRISPR-dCas9 modulates the electrical characteristics of the EGGFET, including the ID output current. Target DNA detection is possible with a LOD of 1.7 fM. Reproduced from [108] with permission from Springer Nature © 2019.
Figure 23.
(a) CRISPR-Chip diagnostic platform proposed by Hajian et al. enables DNA detection in less than 15 minutes. The dCas9 enzyme complexed with a DNA target-specific guide RNA is immobilized on the graphene surface of an EGGFET. The immobilized CRISPR-dCas9 complex scans the entire genomic DNA until it identifies its target sequence. Hybridization between the target DNA and CRISPR-dCas9 modulates the electrical characteristics of the EGGFET, including the ID output current. Target DNA detection is possible with a LOD of 1.7 fM. Reproduced from [108] with permission from Springer Nature © 2019.
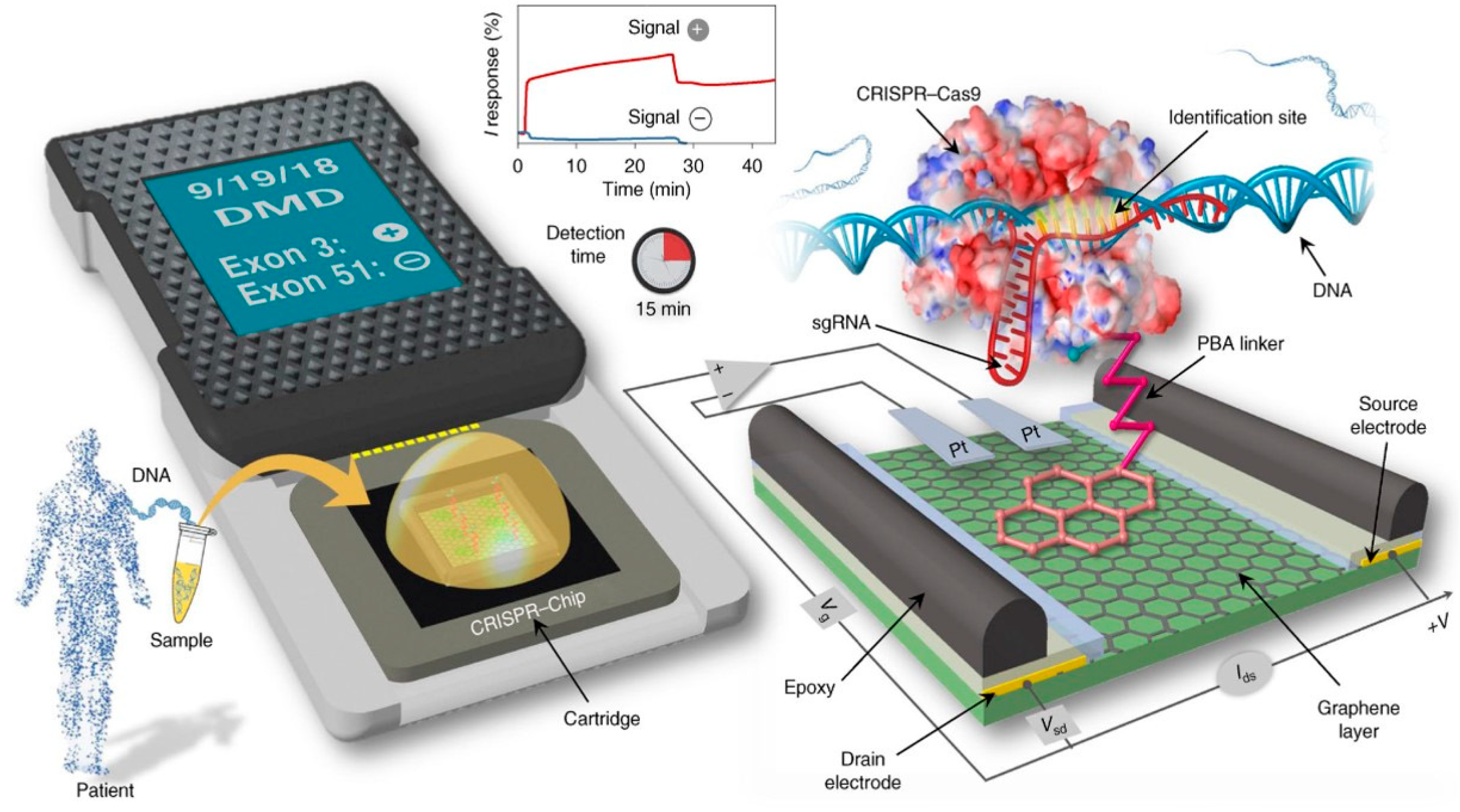
Figure 24.
Schematic diagram of the CRISPR-SNP-Chip transistor functionalized with dCas9, proposed by Balderston et al. gRNA-HTYa and gRNA-SCDa are the guide RNAs. In the presence of the single polymorphism associated with the gRNA-SCDa guide RNA, dCas9-HTYa does not hybridize completely with its target DNA, which then dissociates from the dCas9-gRNA complex. Adapted from [111] with permission from Springer Nature © 2021.
Figure 24.
Schematic diagram of the CRISPR-SNP-Chip transistor functionalized with dCas9, proposed by Balderston et al. gRNA-HTYa and gRNA-SCDa are the guide RNAs. In the presence of the single polymorphism associated with the gRNA-SCDa guide RNA, dCas9-HTYa does not hybridize completely with its target DNA, which then dissociates from the dCas9-gRNA complex. Adapted from [111] with permission from Springer Nature © 2021.
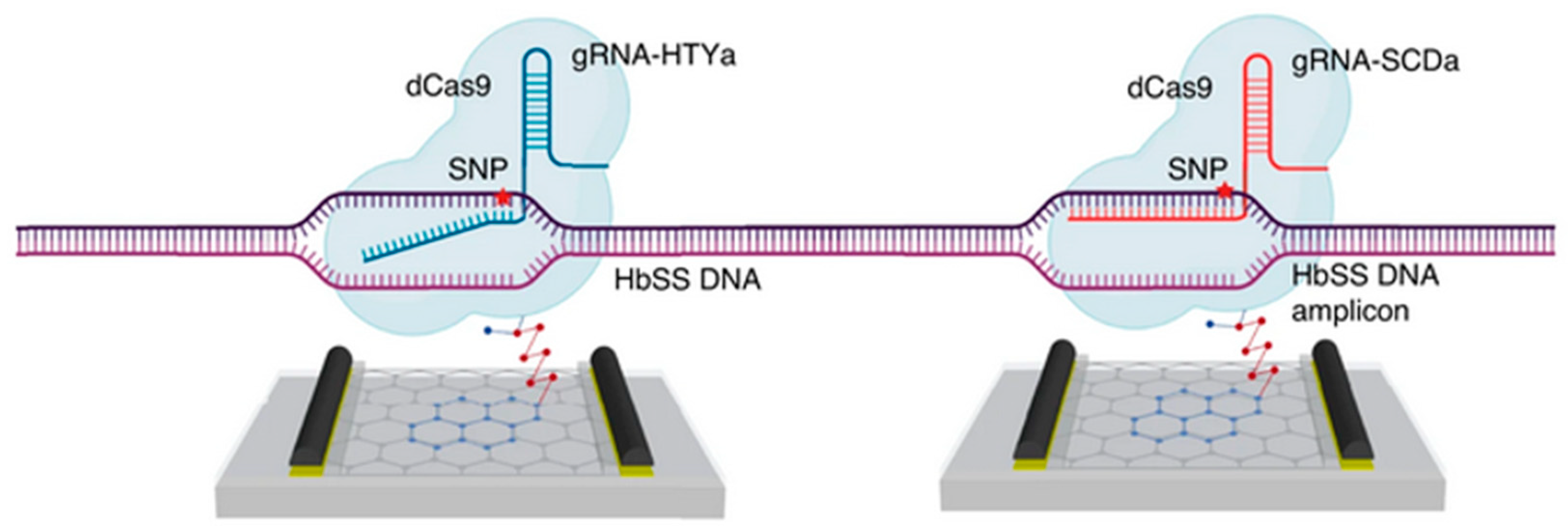
Figure 25.
Design of the CRISPR-Cas13a-based sensor proposed by Yu et al. a) Functionalization of the transistor gate by CRISPR-Cas13a. b) Detection principle between RNA target and CRISPR-Cas13a. c) Sensor structure with interchangeable gate. Characterization of the biological signal into an electrical signal by the EGGFET transistor. Reproduced with permission from [112]. Copyright © 2022, American Chemical Society.
Figure 25.
Design of the CRISPR-Cas13a-based sensor proposed by Yu et al. a) Functionalization of the transistor gate by CRISPR-Cas13a. b) Detection principle between RNA target and CRISPR-Cas13a. c) Sensor structure with interchangeable gate. Characterization of the biological signal into an electrical signal by the EGGFET transistor. Reproduced with permission from [112]. Copyright © 2022, American Chemical Society.
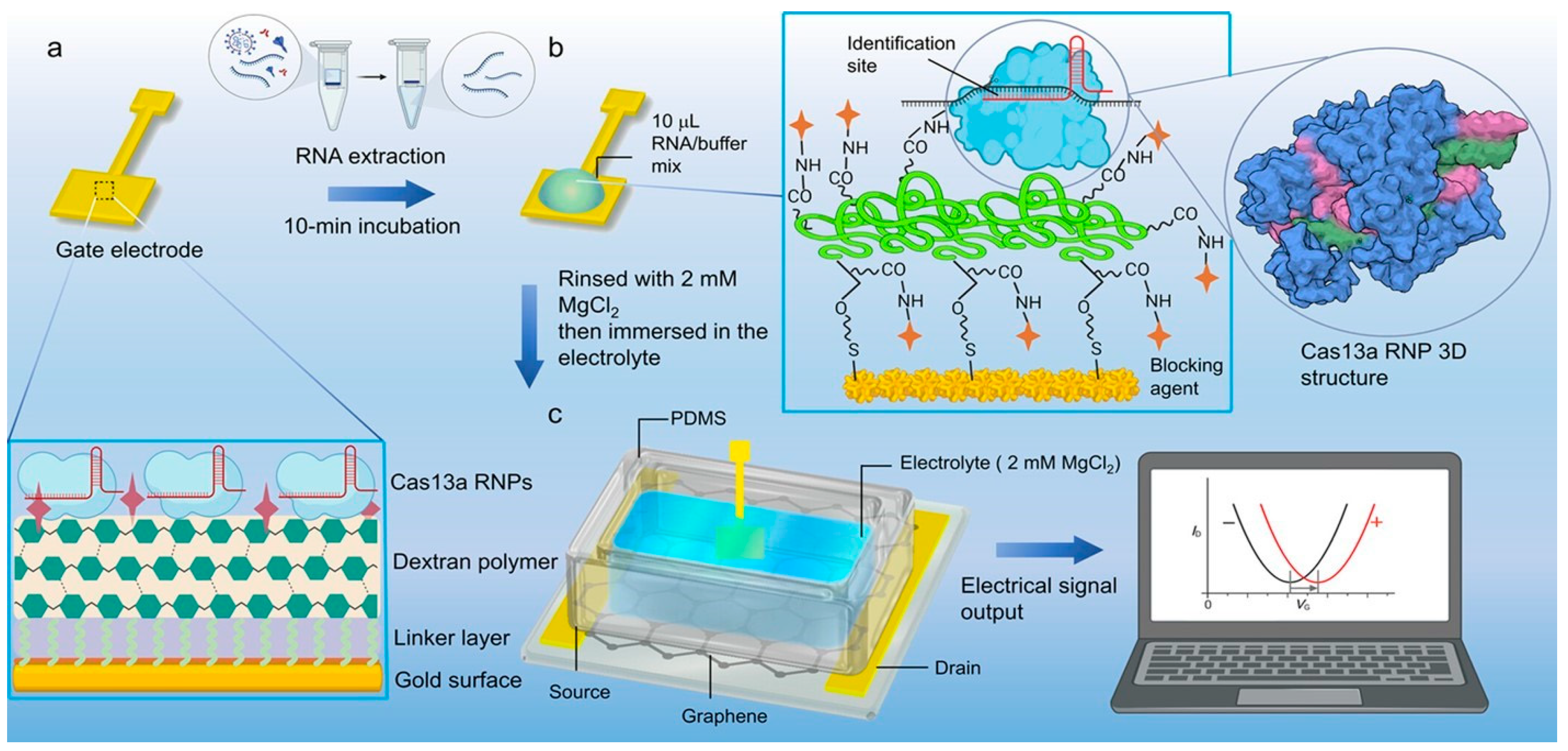
Figure 26.
Design of the CRISPR-Cas13a-based sensor proposed by Yu et al. a) Functionalization of the transistor gate by CRISPR-Cas13a. b) Detection principle between RNA target and CRISPR-Cas13a. c) Sensor structure with interchangeable gate. Characterization of the biological signal into an electrical signal by the EGGFET transistor. Reproduced with permission from [122], © 2023 Elsevier.
Figure 26.
Design of the CRISPR-Cas13a-based sensor proposed by Yu et al. a) Functionalization of the transistor gate by CRISPR-Cas13a. b) Detection principle between RNA target and CRISPR-Cas13a. c) Sensor structure with interchangeable gate. Characterization of the biological signal into an electrical signal by the EGGFET transistor. Reproduced with permission from [122], © 2023 Elsevier.
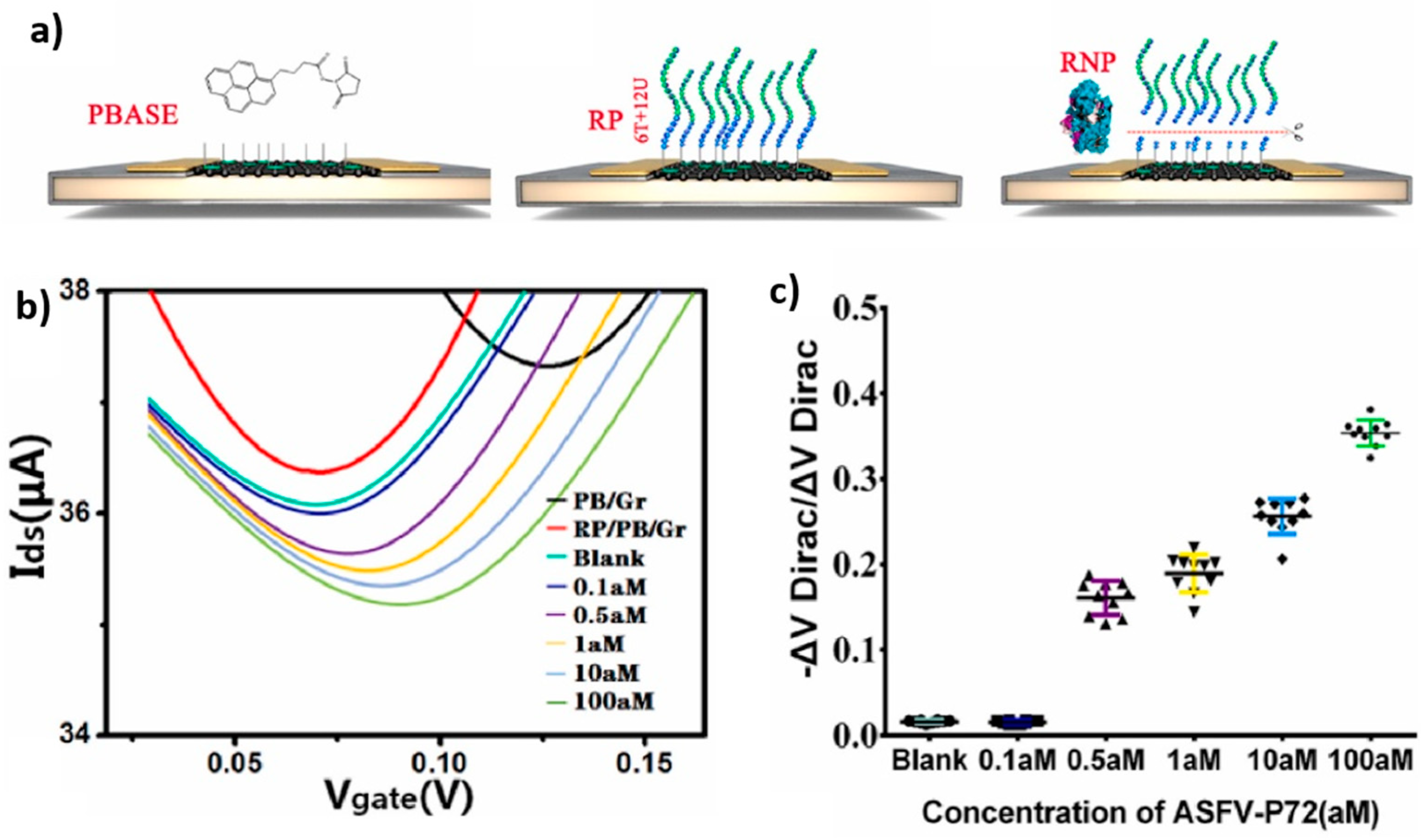
Figure 27.
Schematics of the biosensor proposed by Li et al. which uses two different crRNAs for accelerated and more sensitive detection of SARS-CoV-2. The RNA probes present a hairpin structure. Reproduced with permission from [123]. Copyright © 2022, American Chemical Society.
Figure 27.
Schematics of the biosensor proposed by Li et al. which uses two different crRNAs for accelerated and more sensitive detection of SARS-CoV-2. The RNA probes present a hairpin structure. Reproduced with permission from [123]. Copyright © 2022, American Chemical Society.
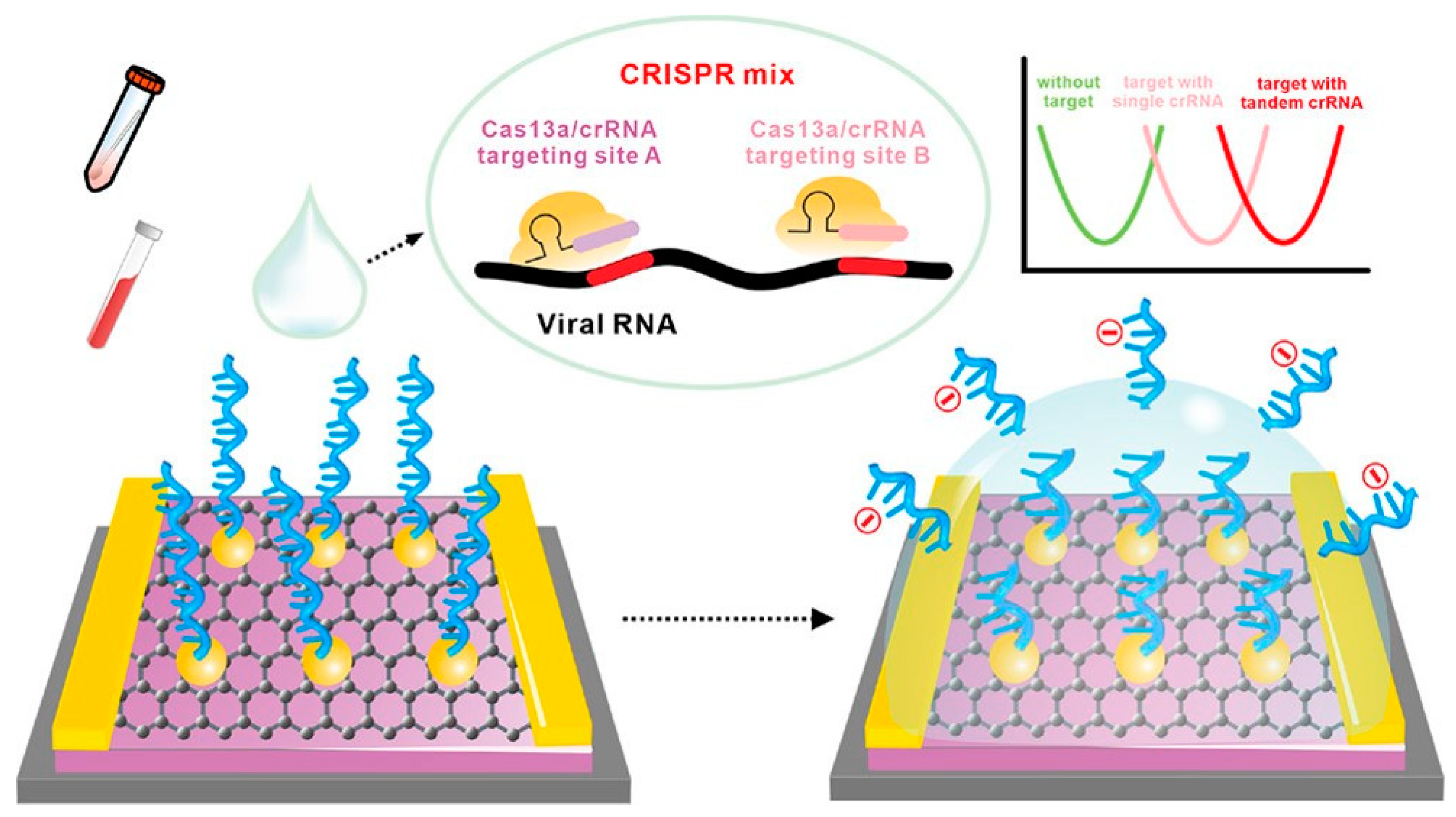
Figure 28.
Schematics of the EGGFET proposed by Ban et al. for the detection of SARS-CoV-2 RNA and virus particles. Reproduced with permission from [125], © 2023 John Wiley and Sons.
Figure 28.
Schematics of the EGGFET proposed by Ban et al. for the detection of SARS-CoV-2 RNA and virus particles. Reproduced with permission from [125], © 2023 John Wiley and Sons.

Table 1.
Summary table of the main nucleic acid amplification methods. Reverse transcriptase enzymes are also used in each process when the target to be amplified is RNA (except for NASBA, which amplifies RNA directly). Adapted from [19].
Table 1.
Summary table of the main nucleic acid amplification methods. Reverse transcriptase enzymes are also used in each process when the target to be amplified is RNA (except for NASBA, which amplifies RNA directly). Adapted from [19].
| Method | PCR | RPA | LAMP | NASBA |
| Temperature (°C) | 40-95 | 37-42 | 60-65 | 40-55 |
| Reaction time (minutes) |
60-180 | 20-60 | 20-60 | 60-120 |
| Proteins implied | TAQpolymerase | Recombinase, Single-stranded binding protein, Strand-displacement DNA polymerase |
Strand-displacement DNA polymerase | Inverse-transcriptase inverse, RNAse H, T7 RNA polymerase |
| References | [20] | [13] | [14] | [3,15] |
Table 2.
Overview of different sensors using CRISPR-Cas13 for RNA detection without gene amplification.
Table 2.
Overview of different sensors using CRISPR-Cas13 for RNA detection without gene amplification.
| Enzyme | Name of the method | Pathogen | Preamplification | Measurement | LOD | Refs |
| CRISPRCas13 | SHERLOCK | ZIKV, DENV | RT-RPA | Fluorescence | 10 aM | [3] |
| SHERLOCKv2 | ZIKV, DENV | RT-RPA | Fluorescence | 8 zM | [55] | |
| Colorimetric strip | 2 aM | [55] | ||||
| - | Bacteriophage RNA, Human RNA |
None | Fluorescence | 1 pM | [50,51] | |
| - | ZIKV, DENV | None | Fluorescence | 50 fM | [3] | |
| - | Ebola | None | Fluorescence | 10 fM | [68] | |
| - | Not specified | None | Fluorescence | 10 fM | [62] | |
| - | SARS-CoV-2 | Naone | Fluorescence on smartphone | 1 fM | [69] | |
| - | SARS-Cov-2 (Gene E) | None | Electrochemical 4 fM | [71] | ||
Table 3.
Summary of the various sensors proposed for nucleic acid detection, which exploit the cis-cleavage property of Cas enzymes. All the transistors used in these devices are EGGFETs.
Table 3.
Summary of the various sensors proposed for nucleic acid detection, which exploit the cis-cleavage property of Cas enzymes. All the transistors used in these devices are EGGFETs.
| Enzyme | Target | Functionnalisation area | Grafting scheme | LOD | Reaction time | Refs |
| dCas9 | ADN | Channel | Graphene/PBA/Cas | 1.7 fM | 15 min | [108,111] |
| Cas12a | ADN | Gate | Au/SAM/Cas | 8.3 aM | 20 min | [113] |
| Cas13a | ARN | Gate | Au/SAM/Dextran/Cas | 13 aM | 10 min | [112] |
Table 4.
An overview of the various EGGFETs proposed for nucleic acid detection using the trans-cleavage property of Cas enzymes.
Table 4.
An overview of the various EGGFETs proposed for nucleic acid detection using the trans-cleavage property of Cas enzymes.
| Enzyme | Target | Transistor | Temperature | LOD | Reaction time | Refs |
| Cas12b | MPXV | Graphene-EGFET | 52°C | 1 aM | 20 min | [120] |
| Cas12a | AFSV | Graphene-EGFET | TA | 0.5 aM | 30 min | [122] |
| Cas12a | HPV-16 | Graphene-EGFET | TA | 1 aM | 30 min | [121] |
| Cas13a | SARS-CoV-2 HCV | rGO-EGFET | 37°C | 1.56 aM | 30 min | [123] |
| Cas13a | SARS-CoV-2 | Graphene-EGFET | 37°C | 0.25 aM | 120 min | [126] |
| Cas13a | SARS-CoV-2 RSV | Graphene-EGFET | 37°C | 1 aM | 30 min | [119] |
| Cas13a | SARS-CoV-2 | Graphene-EGFET | TA | 65 aM | 30 min | [125] |
| Cas13a | SARS-CoV-2 | IGZO-EGFET | TA | 1.7 aM | 20 min | [127] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated