
Submitted:
29 September 2024
Posted:
30 September 2024
You are already at the latest version
Abstract
Primary hyperparathyroidism represents the third more prevalent endocrine disease in general population consisting of an excessive secretion of parathyroid hormone from one, more frequently, or more of the parathyroid glands, leading to a dysregulation of calcium homeostasis. Schematically, its development occurs primarily by pathophysiological events with genetic mutation, at germline and/or somatic level, that favor the neoplastic transformation of parathyroid cells, and promoting their aberrant proliferation, and mutations determining the shift of PTH "set-point”, thus interfering with the normal pathways of PTH secretion, and leading to a “resetting” of Ca2+-dependent PTH secretion or to a secretion of PTH insensitive to changes in extracellular Ca2+ levels. Familial syndromic and non-syndromic forms of primary hyperparathyroidism are responsible for approximately 2-5%, of primary hyperparathyroidism cases and most of them are inherited forms. The history of the genetic/molecular studies of parathyroid tumorigenesis associated with Multiple Endocrine Neoplasia type 1 Syndrome (MEN1) represents a fascinating topic and model to understand genetic-epigenetic-molecular aspects underlying the pathophysiology of primary hyperparathyroidism, both in relation to the syndromic and non-syndromic forms. This minireview aims to take a quick and simplified look at the MEN1-associated parathyroid tumorigenesis, focusing on the molecular underlying mechanisms. Clinical, epidemiological, and observational studies, as well as specific guidelines, molecular-genetics studies, and reviews, have been considered. Only studies submitted to PubMed in English language were included, without time constraints.
Keywords:
parathyroid tumorigenesis
; primary hyperparathyrodism
; genetics of primary hyperparathyrodism
; MEN1 parathyroid tumors
; loss of heterozygosity
; clonality or parathyroid tumors
; menin
; epigenetics of parathyroid tumorigenesis
; microRNAs
; famillial parathyroid tumors
1. Introduction
Primary Hyperparathyroidism (PHPT) is distinguished by an excessive secretion of parathyroid hormone (PTH) from one or more of the parathyroid glands, leading to a dysregulation of calcium homeostasis. PHPT is one of the most prevalent endocrine disorders in the general population, with an estimated frequency of 3 cases per 1000 people in Europe. This prevalence increases to 21 cases per 1000 in post-menopausal women[1]. Regardless of its genetic or molecular characteristics, the development of PHPT is primarily the result of two factors: a) a genetic mutation, whether germline or somatic, that enables the neoplastic transformation of parathyroid tissue, resulting in an aberrant proliferation of parathyroid cells and an increase in parathyroid cell mass; b) a shift of PTH "set-point” caused by mutations that interfere with the normal pathways of PTH secretion, leading to a “resetting” of Ca2+-dependent PTH secretion or to a secretion of PTH that is insensitive to changes in extracellular Ca2+ levels [2,3] (Figure 1).
The development of PHPT is influenced by a complex interplay of genetic, molecular, and environmental factors, representing a captivating puzzle for both researchers and clinicians. When the delicate equilibrium of parathyroid function is disrupted, the stage is set for the emergence of parathyroid tumors, most notably adenomas and only rarely carcinomas, precipitating the cascade of events leading to PHPT. While most cases of PHPT arise sporadically, a subset exhibits familial clustering. Familial PHPT (FPHPT) can be classified in two groups: syndromic forms, where PHPT is part of a complex clinical picture involving several other organs, and non-syndromic forms, where PHPT occurs as an isolated disorder. Familial syndromic and non-syndromic forms of PHPT are responsible for approximately 2-5%, of PHPT cases [2,3] (Table 1). A further distinction within FPHPT should be made between inherited and heritable forms. This difference is subtle and can be summarized as follows: a) the inherited form is when the known causal germinal genetic alteration, generally heterozygous, has been transmitted to offspring; b) a heritable form indicates the existence of an identified de novo germline mutation in a patient with PHPT, without suspicion/evidence of familial cases, but potentially transmissible to offspring, triggering a future FPHPT.
The investigation of FPHPT yielded numerous insights into the genetic pathways that contribute to parathyroid tumorigenesis, including modifications to critical genes such as CDC73, MEN1, and CASR. Each of these genes orchestrates a unique role in the dysregulation of cell cycle control, apoptosis, and calcium sensing [2,3]. Unfortunately, no clear genotype-phenotype correlation is observed in PHPT. Indeed, a single genetic mutation can result in a variety of parathyroid pathological conditions, including adenomas, diffuse hyperplasia, and, in rare cases, carcinomas. Additionally, the clinical presentation of these conditions is frequently overlapping, making it challenging to differentiate malignant and benign disease at a pre-operative level. Even after surgery is performed, no specific histological characteristics allow to define the parathyroid carcinoma[4].
Therefore, it is vital to increase our insight on genetic, epigenetic, and molecular signatures driving parathyroid tumorigenesis, to identify novel diagnostic biomarkers that can distinguish the different kinds of parathyroid neoplasms, as well as to develop innovative therapeutic strategies. The study of parathyroid tumorigenesis associated with Multiple Endocrine Neoplasia type 1 Syndrome (MEN1) represents a fascinating model to understand genetic-epigenetic-molecular aspects underlying the pathophysiology of PHPT, both in relation to the syndromic and non-syndromic forms.
This minireview aims to take a quick look at the history of parathyroid tumorigenesis associated with MEN1, focusing on the molecular underlying mechanisms. Clinical, epidemiological, and observational studies, as well as specific guidelines, molecular-genetics studies, and reviews, have been considered. Only studies submitted to PubMed in English language were included, without time constraints.
The following keywords have been used: parathyroid tumorigenesis; parathyroid tumors; Multiple Endocrine Neoplasia type 1 Syndrome (MEN1); MEN1 gene; allelic loss; loss of heterozygosity; MEN1 parathyroid tumors; tumor suppressor genes; oncogenes; epigenetics; oncomir; microRNAs/miRNAs.
2. Multiple Endocrine Neoplasia Type 1 (MEN1) Syndrome and MEN1 Gene
MEN1 syndrome (OMIM#131100) is a highly penetrating rare autosomal dominant inherited tumor syndrome, typically characterized by the presence of parathyroid, pituitary, and gastro-entero-pancreatic (GEP) neuroendocrine tumors (NETs) [5,6]. A combination of other endocrine and non-endocrine tumors may also be present, such as thymic, bronchial carcinoids, adrenocortical tumors, facial angiofibromas, lipomas, and collagenomas [7,8,9], resulting in a broad spectrum of phenotypic presentations. The majority of patients with the inherited form of MEN1 hosts germline mutations of the MEN1 gene [10]. Several germline or somatic mutations of MEN1 gene have been described to date, both in inherited and de novo MEN1 cases[11,12]. These mutations are mainly heterozygous inactivating mutations located in the coding region of the MEN1 gene [13,14,15]. Specifically, around 10-15% are nonsense mutations, 40% are frameshift insertions and deletions, 25% are missense mutations, while approximately 11% are splice site defects [3].
3. MEN1-PHPT
The parathyroid tumors linked to MEN1 syndrome typically exhibit benign growth and affect the whole parathyroid tissue. In particular, an asynchronous and asymmetric hyperplasia of all parathyroid glands is observed, explaining why the multiglandular parathyroid involvement may be missed at the time of initial surgery, and be confused with a single adenoma[16]. (Figure 2). Less than 20 cases of parathyroid carcinoma have been published to date in MEN1 patients[17]. Although extremely rare in general, the presence of PC is always possible and should be considered in patients with symptomatic FPHPT and PHPT.
4. Epidemiological Aspects
In line with an autosomal dominant transmission, an equal distribution between the sexes is observed. Frequently, PHPT is the first biochemical-clinical presentation of the syndrome, since the onset of PHPT in MEN1 typically occurs during the second decade of life[2,3]. Penetrance reaches approximately 50% within 20 years of age, and 100% within 50 years, occurring approximately 3 decades in advance with respect to sporadic PHPT in the general population[5]. In an observational study on 160 MEN1 patients, PHPT was present in 58% of subjects aged 15-20 years, in 32% between 10-15 years, and in 10% during the first decade of life, including also reports on PHPT in a 4-year-old child[18].
5. Clinical Presentation
Primary hyperparathyroidism in MEN1 (MEN1-PHPT) is distinguished by early onset and clinical aggressiveness. Rapid loss of bone mass is observed, particularly at the cortical level as in classic PHPT, but with a more severe bone impairment and increased risk for fragility fractures than sporadic forms[19,20]. Up to 50% of patients with MEN1 present reduced bone mineral density (BMD) before 50 years of age[19,21]. Moreover, recurrent nephrolithiasis is one of the most frequent clinical manifestations of MEN1, frequently occurring before 30 years of age. Up to two-third of patients with MEN1 present complications of urolithiasis (renal/ureteric colic, urinary tract infection) as the first clinical manifestation of the syndrome, and in half of cases urolithiasis is reported as the only clinical manifestation[22]. Notably, PTH and calcium elevation in MEN1-PHPT is often milder than in sporadic PHPT, especially in younger patient. Indeed, up to one third of patients <50 years with MEN1-PHPT actually exhibit PTH levels within the normal range[19]. On this regard, it has been suggested that a MEN-1 patient aged <50 years with normal PTH levels is 13.5 times more likely to develop PHPT than a non-MEN-1 patient aged >50 years with elevated PTH levels[2,4,24].
A comprehensive evaluation of complications associated to PHPT should be therefore performed in all MEN-1 patients, regardless of PTH levels. Furthermore, genetic screening for MEN1 should be offered to patients with familial cases of PHPT, with early-onset hyperparathyroidism and those with multi-gland hyperparathyroidism, regardless of age[24].
6. MEN1 Gene
The MEN1 gene (NM_130799.2) is a tumor suppressor gene located on chromosome 11q13 approximatively 9,000 bp of genomic DNA and organized in 10 exons transcribed into a 2.8 kb mRNA with the translational start codon located in exon-2 and the stop-codon in exon-10 spanning [10,16]. Its encoded product is the menin protein, of which different isoforms have been reported: a long isoform consisting of 1,615 amino acids, the canonical sequence, a short isoform of 2, 610 amino acids length, and another isoform consisting of 575 amino acids[11]. Menin has three nuclear localization signals (NLSs), located in its C-terminal region, respectively at codons 479–497 (NLS1), 546–572 (NLSa) and 588–608 (NLS2) and five putative guanosine triphosphatase (GTPase) sites (G1–G5) . Such NLSs directly interact, in a sequence-independent manner, with DNA as a scaffold protein controlling gene expression and cell signaling. Different menin regions have been implicated in the binding to different interacting proteins, such as JunD, GFAP, NFB, Smad1/5, Pam, NM23H1, RPA2, Runx2, MLL, NMMHC II-A, FANC2, mSin3A, HDAC1, Ask, Vimentin, CHES1, and estrogen receptor-alpha (ER-alpha). Their functions, within the menin’s partnership, may vary from the genomic stability control (RPA2, FANC2), to cell division (Vimentin, NMMHC II-A, GFAP), cell cycle control (NM23, ASK), gene transcription regulation (JunD, NFB, Smad proteins, Runx2, MLL, ER-alpha, CHES1), and epigenetic regulation (MLL, HDAC). Furthermore, menin has an important role within the selective mediation of chromatin remodeling and, consequently, in both gene expression and cell proliferation regulation [10]. The disruption and/or alteration of one or more of these molecular partnerships may account for tumor susceptibility, including the parathyroid tissue.
7. MEN1 Gene’s Role in Parathyroid Tumorigenesis
Parathyroid tumorigenesis in MEN1 results from the combination of a germline MEN1 mutation together with a somatic mutation occurring in a parathyroid tumor precursor cell. Typically, loss of heterozygosity (LOH) for polymorphic DNA markers is found at chromosome 11q13 region, where MEN1 gene has been mapped (Figure 3). This genetic feature aligns with the expected behavior of a tumor suppressor gene (TSG)[11,26]. The time interval, probably variable and multifactorial, between the occurrence of the second mutational event in the parathyroid tissue and the onset of clinical or biochemical manifestation of PHPT is unknown[10].
Interestingly, whole-exome studies identified MEN1 mutation/inactivation as a common molecular event also in sporadic parathyroid adenomas, unravelling the occurrence of somatic MEN1 mutations in about 35-40% of investigated sporadic tumors[27,28,29]. Truncating mutation of the MEN1 gene are the most frequent, together with LOH at the 11q13 locus, which has been reported in about 40% of sporadic parathyroid adenomas[30,31].
8. Molecular Genetic Studies of MEN1-PHPT Tissues
They can be "scholastically" divided into before and after the cloning of the MEN1 gene.
8.1. Before the MEN1 Gene Cloning
Before the identification of MEN1 gene in late 1990s, parathyroid tumors in MEN1 were considered as the result of a polyclonal expansion[32,33,34]. A contribution to the current genetic approach to parathyroid tumorigenesis in MEN1 was provided by a study involving in patients affected by MEN1-PHPT, in whom DNA extracted from parathyroid tissues was compared with the constitutive DNA extracted from whole blood [35]. The results of this study suggested that MEN1-hyperplastic parathyroid tumors could be regarded to as monoclonal lesions, progressing, or beginning by germline inactivation of MEN1 gene, followed by somatic loss of the wild copy of the gene. Lately, clonal composition analyses of micro-dissected sections from abnormal MEN1 parathyroid tissue on DNA obtained from single nodules and non-nodular areas, using by both chromosome 11q13 microsatellite-PCR and patterns of X chromosome-inactivation, highlighted a genetic heterogeneity in MEN 1 parathyroid microareas, exhibiting a "clonal" pattern for allelic losses[36,37]. Specifically, the cytohistological structure did not result to be useful predictors of clonality but these findings strongly suggested that in familial MEN1 parathyroid tumors the nodular pattern growth, the large size of chief cells and the nuclear atypia associated with monoclonality. Interestingly, the LOH at 11q13 region is observed in the majority of MEN 1 parathyroid tumors, while it less frequent in the sporadic counterpart, confirming the role of an inactivation of a 11q13-located tumor suppressor gene (TSG) in parathyroid tumorigenesis, but also highlighting an apparent heterogeneity within sporadic parathyroid tumors[38]. These findings may be considered prodromal to the more recent redefinition of clonality of parathyroid hyperplasia reported in the 2022 WHO guidelines, endorsing PHPT-related multiglandular parathyroid disease as a germline susceptibility-driven neoplasm[39].
8.2. After the MEN1 Gene Cloning
At the Catalogue of Somatic Mutations in Cancer (COSMIC), only a subset of all the MEN1 germline mutations has been reported as somatic mutations in tumors, suggesting that the mutations likely contribute to clinical pathologies rather than to neoplasia[40]. Interestingly, only few germline mutations of MEN1 have been specifically associated to parathyroid tumors, including P325L (P320L in isoform 2), which is predicted to significantly decrease the protein stability of menin by targeting the protein for degradation [41], and D423N (D418N in isoform 2) whose molecular consequence is still unknown[40]. Nevertheless, more recent research has demonstrated that tumorigenesis in MEN1, including development of parathyroid tumors, may be influenced by both genetic and epigenetic factors. Indeed, menin engages in a multifaceted epigenetic regulation that encompasses a variety of processes, summarized in Table 2.
9. Menin and Epigenetics
Menin acts either as an activator or repressor of DNA transcription by the interacting with several factors, including the mixed lineage leukemia (MLL) complex, the histone deacetylase (HDAC), or the histone lysine methyltransferase SUV39H1. Specifically, menin interacts with MLL promoting the methylation of the histone H3 (H3K4), which in turn regulates suppression of the cyclin D kinase inhibitors (CDKi) p18 and p27, and Hox genes, negatively affecting the suppression of cell proliferation[10]. More recently, the loss of menin in MEN1-related tumors was also associated with an increased activity of the DNA (cytosine-5)-methyltransferase 1 (DNMT1), accounting for methylation of cytosine residues of the CpG islands of DNA, thus resulting in silencing gene expression. The hypermethylation of promoters of TSGs following the loss of menin, can be regarded to as a common pro-oncogenic epigenetic change in MEN1 pancreatic neuroendocrine tumors[42], and, similarly, it could also occur in MEN1 loss-driven parathyroid tumorigenesis. Furthermore, menin was found to interact with HDAC, inducing repression of the transcription factor JunD, involved in negative control of cell proliferation. Moreover, menin is able to silence SUV39H1, a histone 3 lysine 9 (H3K9) methyltransferase regulating both chromatin remodeling and the expression transcription factors, such as interleukin 6 and Gastrulation brain homeobox 2 (Gbx2), involved in cell differentiation[43].
10. Haploinsufficiency of Menin and the Role of Micro-RNAs
Menin could have a role also in tissue-specific haploinsufficiency that occurs after the mutation/loss of one copy of the MEN1 gene, either at germline or somatic level. In primary cultures of skin fibroblasts obtained from MEN1 patients, mRNAs encoded by the wild type retained MEN1 allele were expressed, while mutant alleles did not, being partially degraded in a nonsense-mediated mRNA decay pathway[44]. This phenomenon results in a reduced expression of MEN1 mRNA and extremely low levels of menin in MEN1-PHPT with respect to the sporadic PHPT forms and normal parathyroid gland. Furthermore, menin enables the formation of a ribonucleoprotein structure in which multiple mRNAs are coordinately regulated by RNA binding proteins and miRNAs[44]. Such findings suggest that the expression of the MEN1 gene may be regulated through a mechanism of feedback from its product menin, possibly compensating the haploinsufficiency derived from the allelic loss though an up-regulation of wild-type menin expression at post-transcriptional level [45].
Many studies have pointed out that also micro-RNAs (miRNAs) expression may be involved in parathyroid tumorigenesis, unravelling a wide spectrum of differentially expressed miRNAs in parathyroid tumors [46,47,48,49,50]. A study on both healthy and MEN1-parathyroid tissues, with and without LOH at 11q13, has revealed that miR-24-1 may act as an oncomir by blocking menin expression and, consequently, it may contribute to parathyroid tumorigenesis via an epigenetic mechanism mimicking the “Knudson’s second hit”. In synthesis, after the first inherited germinal “hit”, the onset and progression of MEN1-associated neoplasms, including those involving parathyroids, could depend on an impairment of the ‘‘negative feedback loop’’ between menin and miR-24-1[51]. Specifically, MEN1 parathyroid tumors still having the wild type allele of the MEN1 gene may exhibit miR-24-1 increased levels in association with the menin complete silencing, thus suggesting a miR-24-1-dependent inhibition direct on the MEN1 mRNA translation, suggesting that such epigenetic events may drive to an intermediate molecular step before the genetic LOH of the wild type MEN1 allele occur, consequently inhibiting the menin expression and triggering MEN1 parathyroid tumorigenesis by mimicking the “Knudson’s second inactivating hit” on the MEN1 gene expression [52]. Therefore, the existence of an autoregulatory network between miR-24-1, MEN1-mRNA, and menin has been proposed. Menin promotes the transcription of the primary transcript of miR-24-1 (pri-miR-24-1) by binding the upstream region of the miR-24-1-encoding gene cluster on chromosome 9. Menin directly interacts with pri-miR-24-1, facilitating the DROSHA-mediated processing to pre-miR-24-1, in a positive feedforward loop in which menin promotes the maturation of its own repressor. The mature miR-24-1, together with the RISC complex, binds the 3’ UTR of MEN1 mRNA blocking the translation of menin (Figure 4). In this context, miR-24 may act as a possible effector of tumor development. Thus, the expression of this miRNA only in MEN1 parathyroid adenomas could account for selective parathyroid tumorigenesis, mimicking and substituting the second somatic ‘‘hit’’ of the TSG inactivation, even before the occurrence of LOH[51].
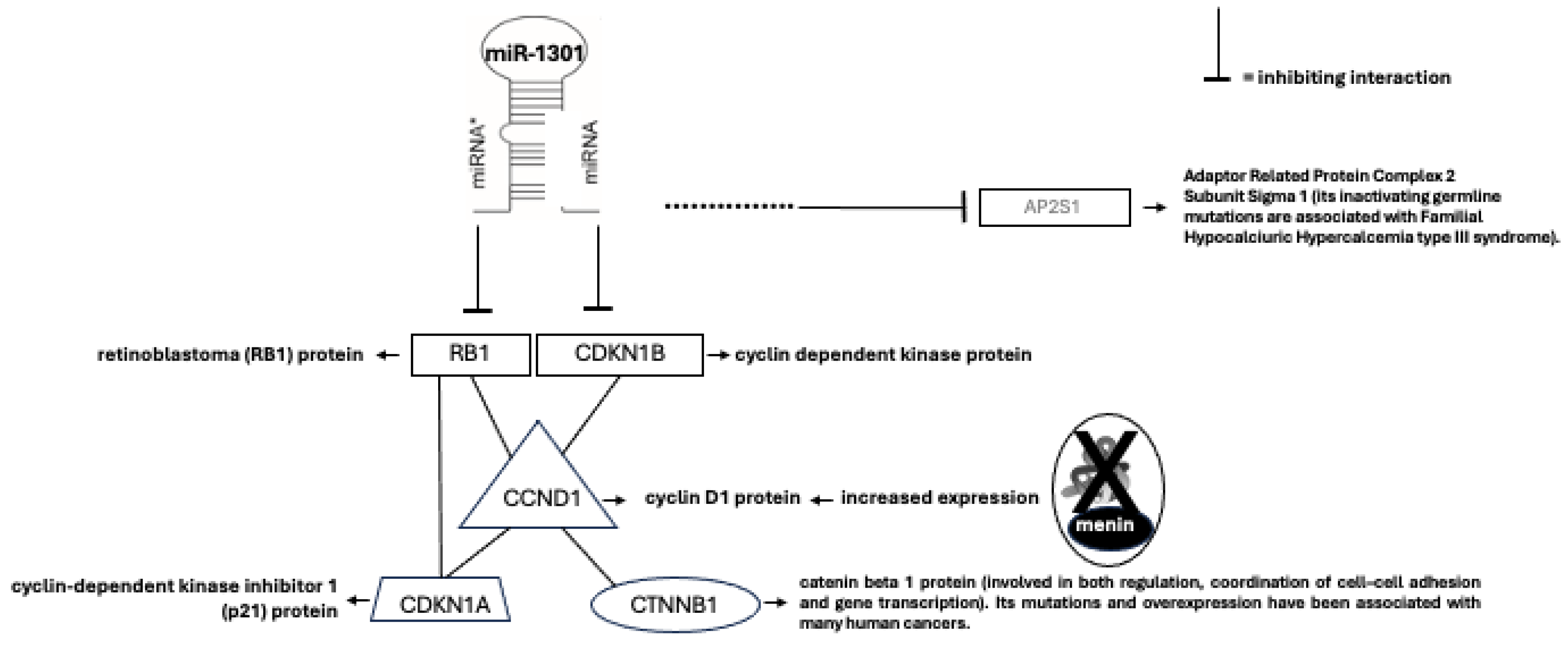
A global miRNA expression profiling in MEN1 parathyroid samples, with and without LOH, identified specific deregulated miRNAs in LOH positive tumors (LOH+) and in LOH negative tumors (LOH-), as compared with control samples. The most differentially expressed were miR-1301, miR-664 and miR-4258[28,53,54]. In-silico microarray analyses suggested that miR-1301, as well as miR-664, may potentially target several genes involved in parathyroid tumorigenesis (Figure 5)[55]. In addition, miR-4258 was reported to be downregulated in MEN1 LOH+ parathyroid adenomas, and it is predicted to target and inhibit the CCDN1 gene encoding the cyclin D1, which is positive regulator of cell cycle and cell growth [28]. Lacking the wild type menin expression, the parathyroid cell no longer shows the miR-4258-driven negative control of CCND1 expression, and, consequently, reveals an increased expression of cyclin D1 and an uncontrolled cell growth.
11. Long Non-Coding RNAs (lncRNAs)
A profile analysis of lncRNAs expression in parathyroid tumors, including some with MEN1 mutations, showed that the tumors exhibiting MEN1 gene mutations had an increased expression of six lncRNAs, including BC200, HAR1B, HOXA3as, NEAT1, SNHG6, and ZFAS1. These findings suggest that MEN1 gene may also act as a potential modulator of the lncRNAs expression. In particular, LncRNA profiling has been suggested as a potential biomarker to distinguish different kinds of parathyroid neoplasms, such as carcinomas, atypical adenomas, and typical adenomas, since these conditions have shown distinct lncRNA profiles. In particular, the expression of BC200 resulted increased in parathyroid carcinomas compared to adenomas, suggesting a potential role of BC200 as a novel circulating biomarker for parathyroid carcinoma[54].
12. Conclusions
Beyond the realm of genetics, it is essential to considered that also environmental influences contribute significantly to the nuanced manifestation of parathyroid tumors, either at sporadic or MEN1 level, contributing to the overall variable clinical expressivity of the MEN1 syndrome and, of course, of the MEN1-PHPT. Such variability is observed both at an intrafamilial and interfamilial level, regardless of the type of germline mutation of the MEN1 gene. As we delve into the labyrinthine world of parathyroid tumorigenesis, it becomes evident that unraveling its mysteries holds promise for not only comprehending the origins of PHPTH, but also tailoring precision therapies. The deep exploration of the underlying molecular mechanisms is essential not only for refining diagnostic strategies or to precociously identify subjects at “hereditary” risk to develop PHPT, but also for advancing targeted interventions that may mitigate the impact of parathyroid tumors on bone health, renal function, and overall well-being.
This journey into the heart of parathyroid tumorigenesis associated with MEN1 syndrome invites us to navigate the intricate molecular landscapes, decode the genetic signatures, and appreciate the dynamic interplay between inherent susceptibilities and external triggers. In doing so, we embark on a path towards a deeper understanding of this fascinating endocrine disorder, laying the groundwork for innovative approaches to diagnosis, treatment, and perhaps one day, prevention.
References
- S. Adami, C. Marcocci, e D. Gatti, «Epidemiology of primary hyperparathyroidism in Europe», J Bone Miner Res, vol. 17 Suppl 2, pp. N18-23, nov. 2002.
- J. P. Bilezikian, N. E. Cusano, A. A. Khan, J.-M. Liu, C. Marcocci, e F. Bandeira, «Primary hyperparathyroidism», Nat Rev Dis Primers, vol. 2, p. 16033, mag. 2016. [CrossRef]
- Falchetti, «Genetics of parathyroids disorders: Overview», Best Pract Res Clin Endocrinol Metab, vol. 32, fasc. 6, pp. 781–790, dic. 2018. [CrossRef]
- Falchetti, «New Perspective on the Genetic Dissection Underlying the Development of Parathyroid Cancer», J Clin Endocrinol Metab, vol. 108, fasc. 12, pp. e1751–e1752, nov. 2023. [CrossRef]
- M. L. Brandi et al., «Guidelines for diagnosis and therapy of MEN type 1 and type 2», J Clin Endocrinol Metab, vol. 86, fasc. 12, pp. 5658–5671, dic. 2001. [CrossRef]
- Falchetti, F. Marini, F. Tonelli, e M. L. Brandi, «Lessons from genes mutated in multiple endocrine neoplasia (MEN) syndromes», Ann Endocrinol (Paris), vol. 66, fasc. 3, pp. 195–205, giu. 2005. [CrossRef]
- S. K. Agarwal, «Multiple endocrine neoplasia type 1», Front Horm Res, vol. 41, pp. 1–15, 2013. [CrossRef]
- S. Marx, A. M. Spiegel, M. C. Skarulis, J. L. Doppman, F. S. Collins, e L. A. Liotta, «Multiple endocrine neoplasia type 1: clinical and genetic topics», Ann Intern Med, vol. 129, fasc. 6, pp. 484–494, set. 1998. [CrossRef]
- P. Langer et al., «Adrenal involvement in multiple endocrine neoplasia type 1», World J Surg, vol. 26, fasc. 8, pp. 891–896, ago. 2002. [CrossRef]
- R. V. Thakker, «Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4)», Mol Cell Endocrinol, vol. 386, fasc. 1–2, pp. 2–15, apr. 2014. [CrossRef]
- S. C. Chandrasekharappa et al., «Positional cloning of the gene for multiple endocrine neoplasia-type 1», Science, vol. 276, fasc. 5311, pp. 404–407, apr. 1997. [CrossRef]
- Lemmens et al., «Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1», Hum Mol Genet, vol. 6, fasc. 7, pp. 1177–1183, lug. 1997. [CrossRef]
- M. C. Lemos e R. V. Thakker, «Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene», Hum Mutat, vol. 29, fasc. 1, pp. 22–32, gen. 2008. [CrossRef]
- P. Concolino, A. Costella, e E. Capoluongo, «Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years», Cancer Genet, vol. 209, fasc. 1–2, pp. 36–41, 2016. [CrossRef]
- F. Marini et al., «Multiple endocrine neoplasia type 1», Orphanet J Rare Dis, vol. 1, p. 38, ott. 2006. [CrossRef]
- Arnold AM, Levine MA, The Parathyroids: Basic and Clinical Concepts (ed. Bilezikian JP). Academic Press, 2015.
- Christakis et al., «Parathyroid carcinoma and atypical parathyroid neoplasms in MEN1 patients; A clinico-pathologic challenge. The MD Anderson case series and review of the literature», Int J Surg, vol. 31, pp. 10–16, lug. 2016. [CrossRef]
- P. Goudet et al., «MEN1 disease occurring before 21 years old: a 160-patient cohort study from the Groupe d’étude des Tumeurs Endocrines», J Clin Endocrinol Metab, vol. 100, fasc. 4, pp. 1568–1577, apr. 2015. [CrossRef]
- Eller-Vainicher et al., «Sporadic and MEN1-related primary hyperparathyroidism: differences in clinical expression and severity», J Bone Miner Res, vol. 24, fasc. 8, pp. 1404–1410, ago. 2009. [CrossRef]
- M. Lourenço, F. L. Coutinho, R. A. Toledo, F. L. M. Montenegro, J. E. M. Correia-Deur, e S. P. A. Toledo, «Early-onset, progressive, frequent, extensive, and severe bone mineral and renal complications in multiple endocrine neoplasia type 1-associated primary hyperparathyroidism», J Bone Miner Res, vol. 25, fasc. 11, pp. 2382–2391, nov. 2010. [CrossRef]
- F. Marini, F. Giusti, T. Iantomasi, F. Cioppi, e M. L. Brandi, «Bone phenotypes in multiple endocrine neoplasia type 1: survey on the MEN1 Florentine database», Endocr Connect, vol. 11, fasc. 5, p. e210456, mag. 2022. [CrossRef]
- Christopoulos, N. Antoniou, A. Thempeyioti, A. Calender, e P. Economopoulos, «Familial multiple endocrine neoplasia type I: the urologist is first on the scene», BJU Int, vol. 96, fasc. 6, pp. 884–887, ott. 2005. [CrossRef]
- E.-V. Cristina e F. Alberto, «Management of familial hyperparathyroidism syndromes: MEN1, MEN2, MEN4, HPT-Jaw tumour, Familial isolated hyperparathyroidism, FHH, and neonatal severe hyperparathyroidism», Best Pract Res Clin Endocrinol Metab, vol. 32, fasc. 6, pp. 861–875, dic. 2018. [CrossRef]
- T. Lassen, L. Friis-Hansen, A. K. Rasmussen, U. Knigge, e U. Feldt-Rasmussen, «Primary hyperparathyroidism in young people. When should we perform genetic testing for multiple endocrine neoplasia 1 (MEN-1)?», J Clin Endocrinol Metab, vol. 99, fasc. 11, pp. 3983–3987, nov. 2014. [CrossRef]
- C. Larsson, B. Skogseid, K. Oberg, Y. Nakamura, e M. Nordenskjöld, «Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma», Nature, vol. 332, fasc. 6159, pp. 85–87, mar. 1988. [CrossRef]
- G. Knudson, «Antioncogenes and human cancer», Proc Natl Acad Sci U S A, vol. 90, fasc. 23, pp. 10914–10921, dic. 1993. [CrossRef]
- Scarpelli et al., «Novel somatic MEN1 gene alterations in sporadic primary hyperparathyroidism and correlation with clinical characteristics», J Endocrinol Invest, vol. 27, fasc. 11, pp. 1015–1021, dic. 2004. [CrossRef]
- M. I. Alvelos et al., «MEN1 intragenic deletions may represent the most prevalent somatic event in sporadic primary hyperparathyroidism», Eur J Endocrinol, vol. 168, fasc. 2, pp. 119–128, feb. 2013. [CrossRef]
- M. K. Cromer et al., «Identification of somatic mutations in parathyroid tumors using whole-exome sequencing», J Clin Endocrinol Metab, vol. 97, fasc. 9, pp. E1774-1781, set. 2012. [CrossRef]
- N. Hendy e D. E. C. Cole, «Genetic defects associated with familial and sporadic hyperparathyroidism», Front Horm Res, vol. 41, pp. 149–165, 2013. [CrossRef]
- P. J. Newey et al., «Whole-exome sequencing studies of nonhereditary (sporadic) parathyroid adenomas», J Clin Endocrinol Metab, vol. 97, fasc. 10, pp. E1995-2005, ott. 2012. [CrossRef]
- Rouleau et al., «Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22», Nature, vol. 329, fasc. 6136, pp. 246–248, set. 1987. [CrossRef]
- R. Rizzoli, J. Green, e S. J. Marx, «Primary hyperparathyroidism in familial multiple endocrine neoplasia type I. Long-term follow-up of serum calcium levels after parathyroidectomy», Am J Med, vol. 78, fasc. 3, pp. 467–474, mar. 1985. [CrossRef]
- M. L. Brandi et al., «Parathyroid mitogenic activity in plasma from patients with familial multiple endocrine neoplasia type 1», N Engl J Med, vol. 314, fasc. 20, pp. 1287–1293, mag. 1986. [CrossRef]
- E. Friedman et al., «Clonality of parathyroid tumors in familial multiple endocrine neoplasia type 1», N Engl J Med, vol. 321, fasc. 4, pp. 213–218, lug. 1989. [CrossRef]
- Morelli et al., «Clonal analysis by chromosome 11 microsatellite-PCR of microdissected parathyroid tumors from MEN 1 patients», Biochem Biophys Res Commun, vol. 227, fasc. 3, pp. 736–742, ott. 1996. [CrossRef]
- Lubensky et al., «Allelic deletions on chromosome 11q13 in multiple tumors from individual MEN1 patients», Cancer Res, vol. 56, fasc. 22, pp. 5272–5278, nov. 1996.
- M. L. Brandi, A. Falchetti, F. Tonelli, e C. Bordi, «Are allelic losses at 11q13 universal in MEN 1 tumors?», J Clin Endocrinol Metab, vol. 81, fasc. 9, pp. 3162–3163, set. 1996. [CrossRef]
- L. A. Erickson, O. Mete, C. C. Juhlin, A. Perren, e A. J. Gill, «Overview of the 2022 WHO Classification of Parathyroid Tumors», Endocr Pathol, vol. 33, fasc. 1, pp. 64–89, mar. 2022. [CrossRef]
- D. D. Nelakurti, A. L. Pappula, S. Rajasekaran, W. O. Miles, e R. C. Petreaca, «Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer», Cancers (Basel), vol. 12, fasc. 9, p. 2616, set. 2020. [CrossRef]
- Yaguchi, N. Ohkura, M. Takahashi, Y. Nagamura, I. Kitabayashi, e T. Tsukada, «Menin Missense Mutants Associated with Multiple Endocrine Neoplasia Type 1 Are Rapidly Degraded via the Ubiquitin-Proteasome Pathway», Mol Cell Biol, vol. 24, fasc. 15, pp. 6569–6580, ago. 2004. [CrossRef]
- E. B. Conemans et al., «DNA methylation profiling in MEN1-related pancreatic neuroendocrine tumors reveals a potential epigenetic target for treatment», Eur J Endocrinol, vol. 179, fasc. 3, pp. 153–160, set. 2018. [CrossRef]
- Z. Feng, J. Ma, e X. Hua, «Epigenetic regulation by the menin pathway», Endocr Relat Cancer, vol. 24, fasc. 10, pp. T147–T159, ott. 2017. [CrossRef]
- E. Luzi et al., «Ribozyme-mediated compensatory induction of menin-oncosuppressor function in primary fibroblasts from MEN1 patients», Cancer Gene Ther, vol. 17, fasc. 11, pp. 814–825, nov. 2010. [CrossRef]
- R. Rahbari, A. K. Holloway, M. He, E. Khanafshar, O. H. Clark, e E. Kebebew, «Identification of differentially expressed microRNA in parathyroid tumors», Ann Surg Oncol, vol. 18, fasc. 4, pp. 1158–1165, apr. 2011. [CrossRef]
- S. M. Sadowski et al., «Identification of Differential Transcriptional Patterns in Primary and Secondary Hyperparathyroidism», J Clin Endocrinol Metab, vol. 103, fasc. 6, pp. 2189–2198, giu. 2018. [CrossRef]
- S. Corbetta et al., «Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue», Endocr Relat Cancer, vol. 17, fasc. 1, pp. 135–146, mar. 2010. [CrossRef]
- V. Vaira et al., «The microRNA cluster C19MC is deregulated in parathyroid tumours», J Mol Endocrinol, vol. 49, fasc. 2, pp. 115–124, ott. 2012. [CrossRef]
- C. Verdelli et al., «The aberrantly expressed miR-372 partly impairs sensitivity to apoptosis in parathyroid tumor cells», Endocr Relat Cancer, vol. 25, fasc. 7, pp. 761–771, lug. 2018. [CrossRef]
- Wang et al., «Expression profile of serum-related exosomal miRNAs from parathyroid tumor», Endocrine, vol. 72, fasc. 1, pp. 239–248, apr. 2021. [CrossRef]
- Costa-Guda, I. Marinoni, S. Molatore, N. S. Pellegata, e A. Arnold, «Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas», J Clin Endocrinol Metab, vol. 96, fasc. 4, pp. E701-706, apr. 2011. [CrossRef]
- E. Luzi, F. Marini, F. Giusti, G. Galli, L. Cavalli, M.L. Brandi. The negative feedback-loop between the oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by mimicking the “Knudson’s second hit”. PLoS ONE 2012, 7, e39767.
- Costa-Guda, C.-P. Soong, V. I. Parekh, S. K. Agarwal, e A. Arnold, «Germline and somatic mutations in cyclin-dependent kinase inhibitor genes CDKN1A, CDKN2B, and CDKN2C in sporadic parathyroid adenomas», Horm Cancer, vol. 4, fasc. 5, pp. 301–307, ott. 2013. [CrossRef]
- E. Luzi, S. Ciuffi, F. Marini, C. Mavilia, G. Galli, e M. L. Brandi, «Analysis of differentially expressed microRNAs in MEN1 parathyroid adenomas», Am J Transl Res, vol. 9, fasc. 4, pp. 1743–1753, 2017.
- Morotti et al., «The Long Non-Coding BC200 Is a Novel Circulating Biomarker of Parathyroid Carcinoma», Front Endocrinol (Lausanne), vol. 13, p. 869006, 2022. [CrossRef]
Figure 1.
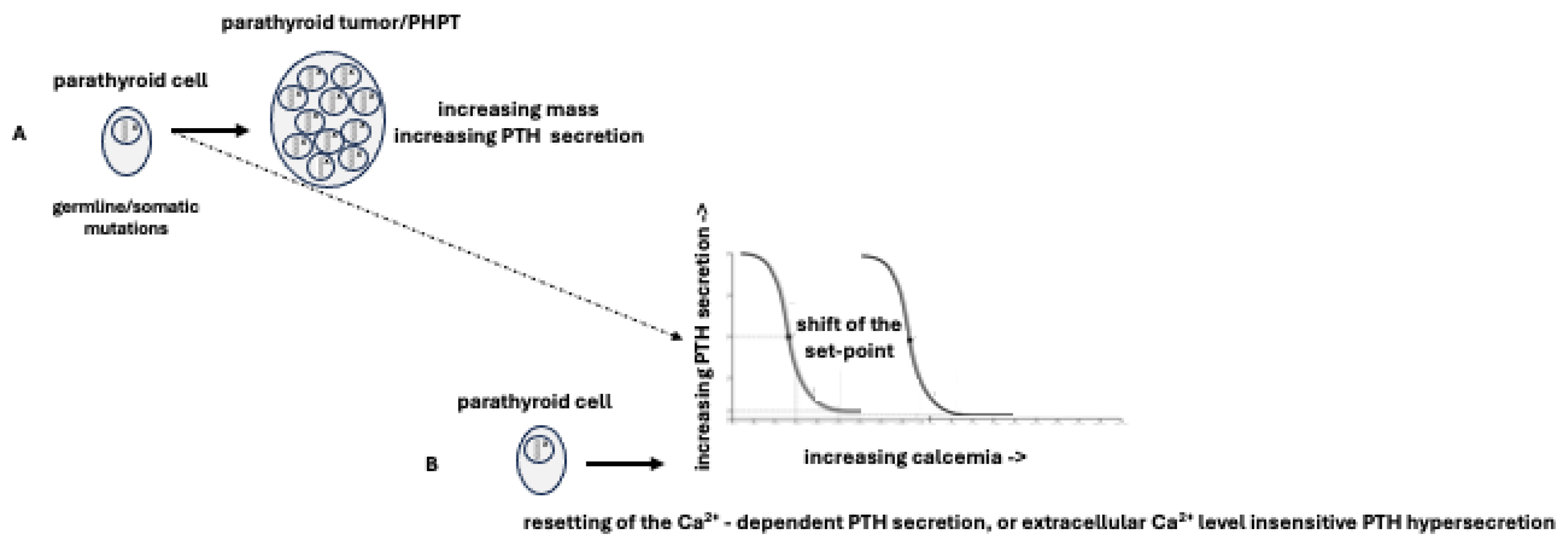
Figure 2.
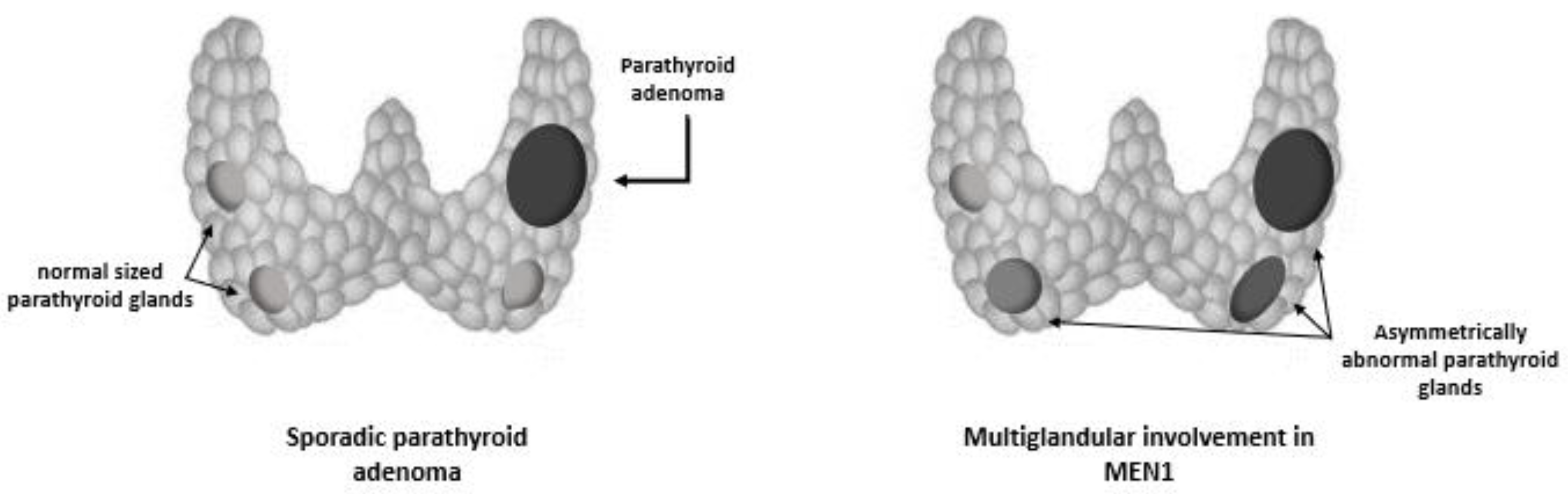
Figure 3.
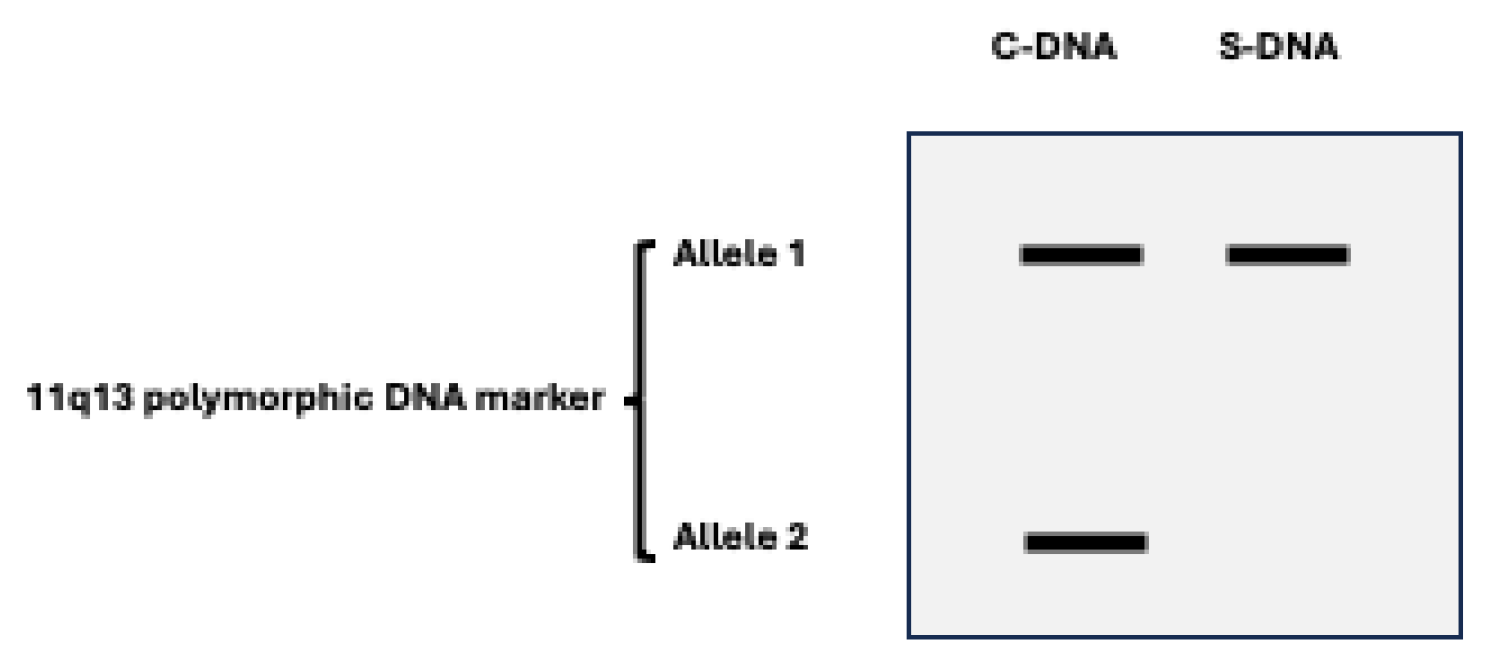
Figure 4.
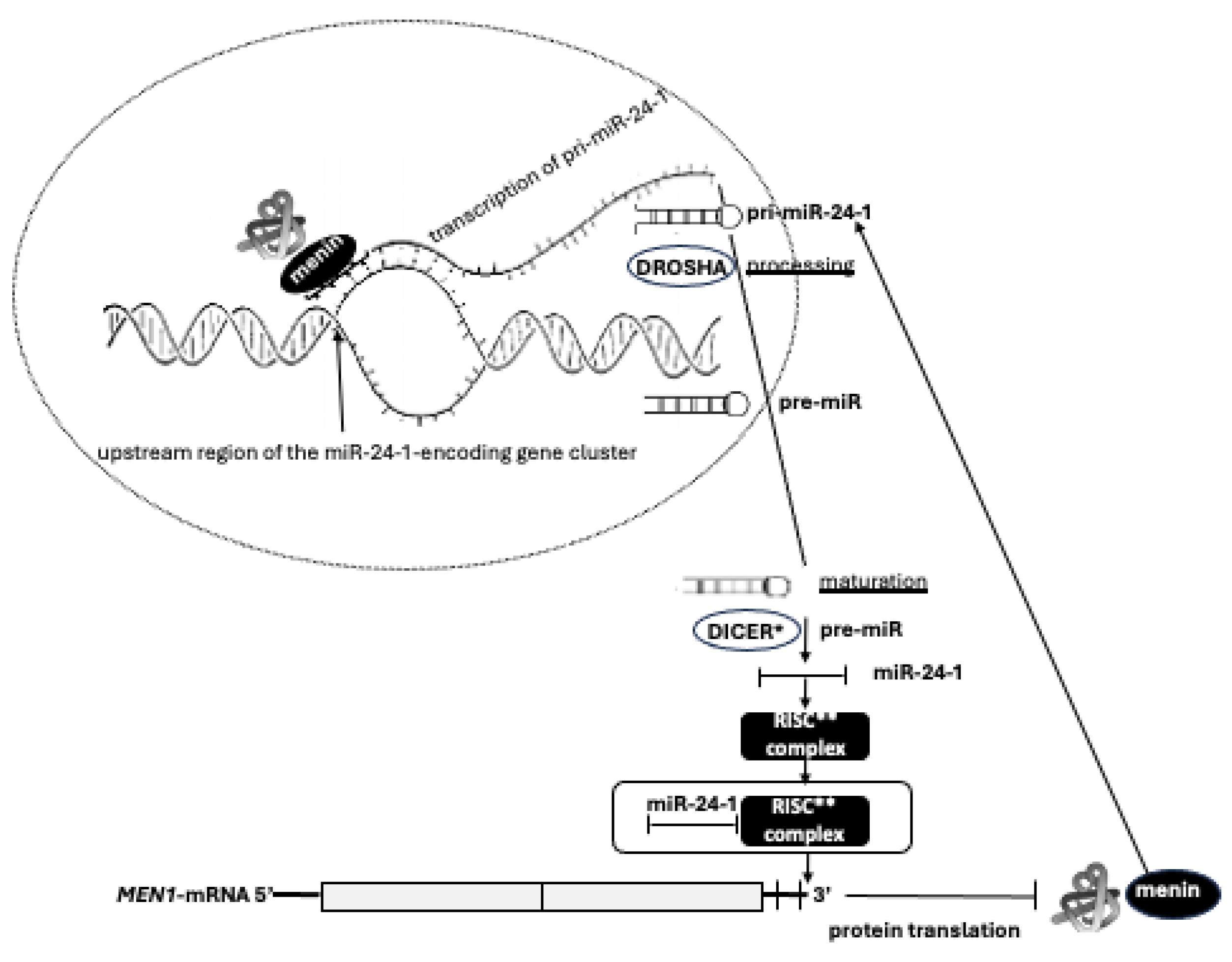
Figure 5.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



