Submitted:
28 September 2024
Posted:
30 September 2024
You are already at the latest version
Abstract
Amyotrophic lateral sclerosis (ALS) is a refractory neurodegenerative disease characterized by the de-generation and loss of motor neurons, leading to death within five years of onset. There have been few effective treatments, making the development of robust therapies an urgent challenge. Genetic mutations have been identified as contributors to ALS, with mutations in superoxide dismutase 1 (SOD1), which neutralizes the harmful reactive oxygen species superoxide, accounting for approximately 2% of all ALS cases. To counteract the toxic gain of function caused by SOD1 mutations, therapeutic strategies aimed at suppressing SOD1 gene expression have shown promise. Antisense oligonucleotide (ASO) is an arti-ficially synthesized short single-stranded DNA/RNA molecule that binds to target RNA to alter gene expression, representing a next-generation therapeutic approach. In 2023, tofersen became the first ASO drug approved by the FDA for ALS. Administered intrathecally, tofersen specifically binds to SOD1 mRNA, inhibiting the production of toxic SOD1 protein, thereby improving biomarkers of ALS. The long-term efficacy and safety of tofersen require further validation, and the development of more opti-mized treatment protocols is essential. A series of studies and therapeutic developments related to SOD1 mutations have advanced the understanding of ALS pathophysiology and significantly contributed to treatment strategies for central nervous system disorders. This review focuses on the overview of SOD1 mutations and the development process of tofersen, aiming to deepen the understanding of advancements in ALS research and discuss future challenges and directions for ASO therapy.
Keywords:
amyotrophic lateral sclerosis (ALS)
; superoxide dismutase 1 (SOD1)
; antisense oligonucleotide (ASO)
; tofersen
; central nervous system
1. Introduction
Amyotrophic lateral sclerosis (ALS) is one of the neurodegenerative diseases first described by Jean-Martin Charcot in 1869 [1]. ALS is relatively rare, with an incidence rate of about 2 per 100,000 person-years worldwide [2,3]. It is characterized by the gradual degeneration and loss of both upper and lower motor neurons, leading to atrophy of voluntary muscles, paralysis, and ultimately death within 2 to 5 years due to swallowing and respiratory muscle paralysis [4,5]. Advances in the development and improvement of respiratory assist devices, as well as the emergence of enteral nutrition and gastrostomy, have enabled longer-term care than in the past. However, the underlying cause of motor neuron degeneration remains completely unknown even 150 years after the initial description of the disease, and no fundamental treatment based on its etiology has been established.
Since the identification of superoxide dismutase 1 (SOD1) as a cause of familial ALS in 1993 [6], the significance of genetic mutations in ALS has become increasingly clear. Consequently, there has been a surge in the exploration of therapeutic strategies that target specific DNA and RNA as novel treatment approaches for ALS. The development of antisense oligonucleotide (ASO) therapies for SOD1 mutation-related ALS has progressed through various preclinical and clinical trials, culminating in the Food and Drug Administration (FDA) approval of tofersen in April 2023 [7].
This review aims to deepen the understanding of the role of genetic mutations in ALS and the development of next-generation therapies, focusing specifically on SOD1 mutations and the journey of tofersen. We also discuss the future directions of ASO in ALS.
2. Genetic Background of ALS
2.1. Overview
Approximately 10% of ALS patients have a family history of the disease, and more than 30 ALS-related genes have been identified in familial ALS cases to date [2,3,8]. Notably, many of the mutated gene products are involved in RNA metabolism, intracellular transport, or protein degradation mechanisms [9]. The remaining 90% of cases are sporadic, but pathogenic mutations identified in familial ALS have also been observed in many sporadic cases [10]. Genetic studies, including family aggregation studies, twin studies, and genome-wide association studies (GWAS), have confirmed that ALS has a substantial genetic component (61% in twin studies, 21% in GWAS) [11,12,13]. Four representative ALS-related genes are superoxide dismutase 1 (SOD1), TAR DNA-binding protein (TARDBP), fused in sarcoma (FUS), and chromosome 9 open reading frame 72 (C9orf72). These pathogenic mutations account for approximately 60% of familial cases and about 10% of sporadic ALS cases [2]. Additionally, methods such as whole exome sequencing have led to the discovery of less frequent genetic mutations, identifying genetic mutations in 76% of familial cases and 25% of sporadic cases [2,9,14]. Moreover, environmental and lifestyle factors also contribute to ALS, in addition to genetic mutations [15]. For instance, alcohol consumption, educational attainment, physical exercise, dyslipidemia, and smoking have been identified as risk factors for ALS and are thought to interact with the genetic background to influence the onset of the disease [15,16,17,18].
2.1. ALS with SOD1 Mutations
The SOD1 gene was first identified as a causative gene for ALS in 1993 [6]. This finding was significant as it was the first to show that genetic abnormalities play a role in the development of ALS. SOD1 encodes a protein of 153 amino acids that acts as an enzyme, catalyzing the breakdown of superoxide radicals into hydrogen peroxide and oxygen [19]. To date, over 200 mutations in the SOD1 gene have been reported. In Europe, SOD1 mutations are the second most common cause of ALS after C9orf72 mutations, accounting for 12-15% of familial ALS and 1-2% of sporadic ALS cases [20,21,22]. In Japan, SOD1 mutations are the most prevalent, representing about 30% of familial ALS and 1-2% of sporadic ALS cases [22,23]. Most pathogenic SOD1 mutations are missense mutations [20]. These mutations typically follow an autosomal dominant pattern with high penetrance, though some mutations can have asymptomatic carriers [23]. The most common clinical presentation in patients with SOD1 mutations is leg weakness with prominent lower motor neuron signs [24]. Certain SOD1 mutations are associated with distinct clinical phenotypes of ALS, which is important for developing personalized treatment strategies [25,26,27]. For instance, the p.A4V mutation, prevalent in ALS patients in North America, leads to a rapidly progressive dominant form of the disease [28,29]. Conversely, the p.H46R mutation, more common in Japan, results in a slowly progressive form [30].
The precise mechanism by which SOD1 mutations cause toxicity is not yet fully understood, but it is widely accepted that these mutations involve a toxic gain of function [19,31,32]. Just one year after the identification of SOD1 as an ALS causative gene, transgenic mice carrying the mutated SOD1 gene were developed [33]. These transgenic mice have significantly advanced our understanding of the mechanisms underlying ALS pathogenesis due to SOD1 mutations. They exhibit disease features similar to those in ALS patients, such as muscle atrophy, glial cell activation, and motor neuron loss in the spinal cord, which has made them valuable model organisms for ALS research [33,34,35]. SOD1 mutations are thought to cause neuronal cell death through several mechanisms, including glutamate excitotoxicity, oxidative stress, non-cell-autonomous toxicity of glial cells, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, axonal transport defects, and extracellular toxicity of mutant SOD1 proteins [36,37,38,39]. Interestingly, while the loss of SOD1 causes motor neuron dysfunction, it does not result in motor neuron death [40,41].
3. Treatment of ALS
There is currently no cure for ALS. Prior to the approval of tofersen, the available treatments were limited to riluzole, a glutamate antagonist, and edaravone, a free radical scavenger (Table 1). Riluzole inhibits the release of glutamic acid from neurons, partly by inactivating voltage-dependent sodium channels on glutamatergic nerve terminals, and also blocks some of the post-synaptic effects of glutamate through noncompetitive blockade of N-methyl-D-aspartate (NMDA) receptors [42]. While riluzole has been shown to modestly extend survival in Phase 3 trials and subsequent population studies, its effect remains limited [43,44,45]. Edaravone, administered intravenously or orally, functions as an antioxidant. The initial randomized, double-blind, placebo-controlled Phase 3 trial did not show significant efficacy for edaravone [46]. However, post-hoc analyses suggested potential benefits in a specific ALS subgroup with a shorter disease duration [47]. Consequently, additional Phase 3 trials focused on a more specific subgroup of patients with a shorter disease duration. These trials demonstrated a modest but statistically significant positive effect of edaravone on the rate of functional decline over 6 months, although there was no significant impact on respiratory function [48]. As a result, edaravone received FDA approval for intravenous treatment in 2017 and for oral administration in 2022.
In 2022, the FDA approved a combination of sodium phenylbutyrate and taurursodiol (PB/TURSO) [49]. PB/TURSO is an oral drug with potential neuroprotective effects, believed to inhibit motor neuron apoptosis by reducing oxygen free radical production, decreasing ER stress, and inhibiting caspase activation [50]. However, this medication was voluntarily removed from the market based on topline results from the Phase 3 PHOENIX trial (NCT05021536). Research into more effective ALS drugs is ongoing, with current focuses on a variety of approaches including antioxidants, cell therapy, mitochondrial dysfunction, neuroinflammation, and proteostasis [51].
In recent years, the development of molecular therapies targeting specific genes has advanced. It is known that the majority of monogenic causes of ALS act through a toxic gain of function of the mutated gene. In this context, ASO therapies, which aim to directly modify disease-causing genes and neutralize toxic gene products, hold significant promise [52]. Currently, there are antisense therapies at various stages of development targeting the transcripts of SOD1, C9orf72, and FUS [51].

Table 1.
Summary of FDA-approved drug for ALS.
| Drug | Year | Administration | Mechanism | Note |
|---|---|---|---|---|
| riluzole | 1995 | Oral | Glutamate antagonist | Thickened riluzole was approved in 2018. Oral film was approved in 2019. |
| edaravone | 2017 | Intravenous, Oral | Free radical scavenger | Oral formation was approved in 2022. |
| PB/TURSO | 2022 | Oral | Inhibition of motor neuron apoptosis | Voluntarily removed from the market in 2024 base on the results from Phase 3 PHOENIX trial. |
| tofersen | 2023 | Intrathecal | ASO (2′-MOE gapmer) | For ALS patients with SOD1 mutation |
FDA, food and drug administration; ALS, amyotrophic lateral sclerosis; PB/TURSO, sodium phenylbutyrate and taurursodiol; ASO, antisense oligonucleotide; 2′-MOE, 2′-O-Methoxyethyl; SOD1, superoxide dismutase 1;
4. ASO in CNS Disorders
4.1. ASO Overview
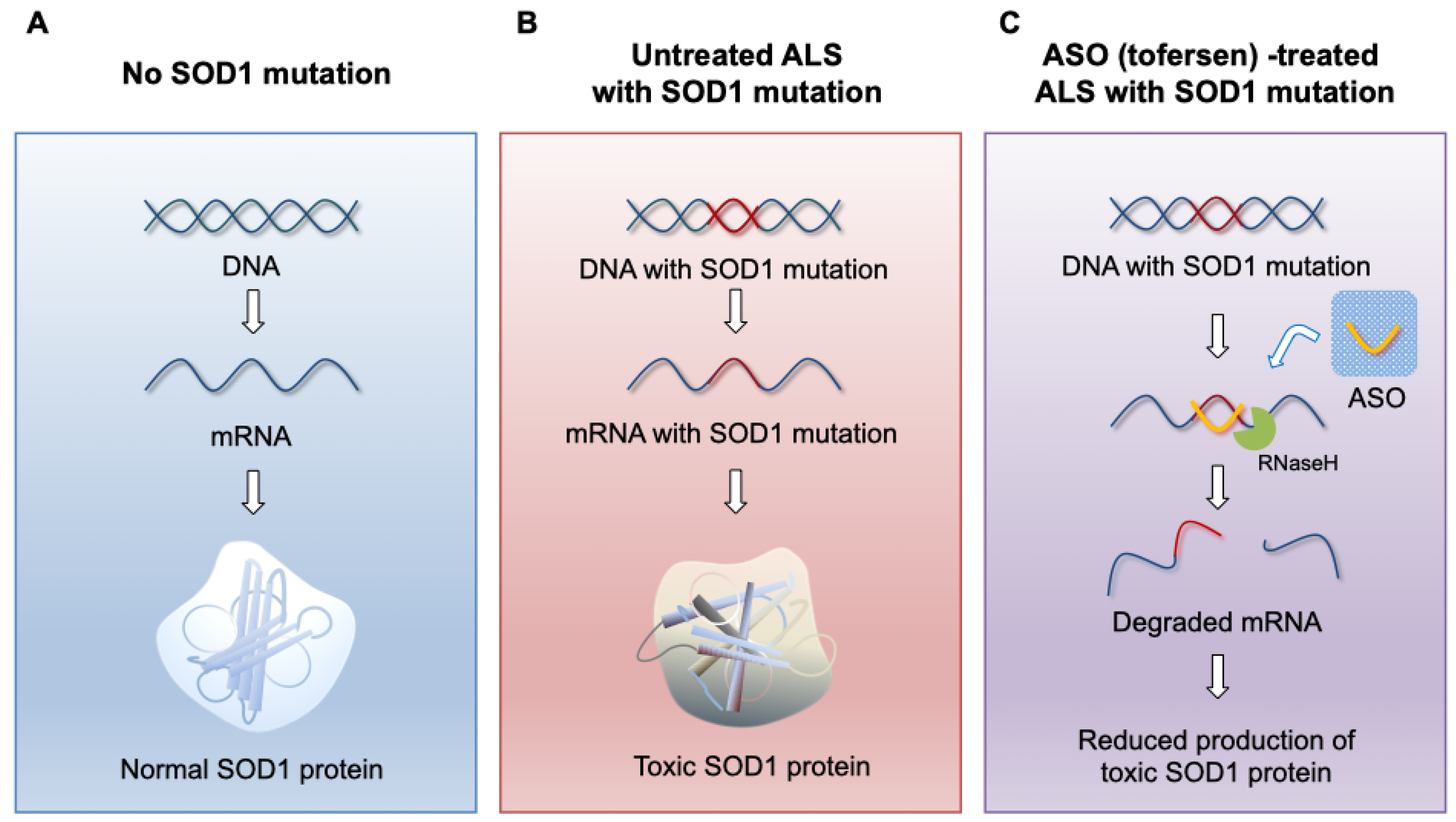
As previously mentioned, considering that the pathogenesis of SOD1 ALS involves a dominant gain of function, therapies aimed at reducing the expression of the SOD1 gene are promising options. Among the various methods for gene silencing, ASO therapy is currently the most promising approach (Figure 1). ASOs are single-stranded oligonucleotides, typically composed of 14 to 25 synthetic nucleotides. They bind specifically to target mRNA through Watson-Crick hybridization, with minimal off-target pairing [53,54]. Once ASOs bind to the target RNA, they alter gene expression primarily through two mechanisms: RNase H-mediated degradation of the target RNA or steric blockage of specific sites on pre-mRNA or mRNA to modulate splicing or prevent translation [55]. RNase H is an enzyme found in all eukaryotic cells, responsible for hydrolyzing RNA bound to DNA during DNA replication [56]. By having a complementary sequence to the target RNA, ASOs utilize the RNA degradation capability of RNase H. When an ASO-RNA heteroduplex is formed, RNase H can bind to the RNA and hydrolyze it, preventing the translation of the disease-causing protein [57]. However, if all DNA bases within the ASO are chemically modified, RNase H cannot recognize the DNA-RNA heteroduplex and thus cannot induce enzymatic activity [58,59]. In the absence of RNase H activity, ASOs bound to target RNA can block mechanisms involved in RNA transcript translation or splicing (steric block). The most widely used application of steric block ASOs is the modulation of selective splicing, allowing for the selective exclusion or inclusion of specific exons [55]. The first ASO drug approved by the FDA for motor neuron disease is nusinersen, used for spinal muscular atrophy (SMA). Nusinersen binds to the intron 7 of the survival motor neuron 2 (SMN2) pre-mRNA, suppressing the skipping of exon 7, thereby producing SMN2 mRNA that includes exon 7 and facilitating the expression of full-length SMN protein [60].
Figure 1.
ASO (tofersen) mechanism of action. (A) In the absence of the SOD1 gene mutation, functional SOD1 protein is produced by normal transcription and translation. (B) In the presence of SOD1 gene mutation, toxic SOD1 protein is produced by its transcription and translation. (C) When tofersen, an ASO gapmer against the SOD1 gene, specifically binds to SOD1 mRNA, SOD1 mRNA is degraded by the activity of RNase H, inhibiting the production of toxic SOD1 protein.
Figure 1.
ASO (tofersen) mechanism of action. (A) In the absence of the SOD1 gene mutation, functional SOD1 protein is produced by normal transcription and translation. (B) In the presence of SOD1 gene mutation, toxic SOD1 protein is produced by its transcription and translation. (C) When tofersen, an ASO gapmer against the SOD1 gene, specifically binds to SOD1 mRNA, SOD1 mRNA is degraded by the activity of RNase H, inhibiting the production of toxic SOD1 protein.
4.2. Challenges of ASO Therapy for CNS Disorders
A significant issue, not limited to ASOs, when delivering drugs to the central nervous system (CNS) is the presence of the blood-brain barrier (BBB). The BBB poses a significant delivery barrier to the CNS due to non-fenestrated capillaries, dense junctional proteins, and continuous basement membranes, making large molecule extravasation into the brain extremely challenging [61]. Molecules that can pass through the BBB need to be hydrophobic low molecular weight compounds (below 500 Da) [62]. Thus, how to deliver hydrophilic high molecular weight ASOs to the CNS is an area of ongoing research. Intrathecal administration is a method that does not require consideration of the BBB, allowing for high efficacy in delivering ASOs to the CNS. Studies in rodents and primates have shown that intrathecal delivery of ASOs results in wide distribution and tissue penetration throughout the CNS [63,64,65]. It is common to administer ASOs alone without the aid of delivery molecules. However, a major drawback of this method is its invasiveness. In clinical trials, side effects such as headaches and back pain from lumbar punctures have been frequently reported, which may hinder treatment continuation or necessitate schedule changes [66,67].
Unfortunately, there is currently no established method for delivering ASOs to the CNS via alternative routes that can cross the BBB, but advancements in drug delivery technologies are underway [68]. For example, cell-penetrating peptides that can bind to ASOs may provide a promising approach by facilitating the delivery of ASOs across BBB, thereby bypassing the need for invasive intrathecal injections and the associated side effects, which could potentially improve treatment efficacy for CNS disorders [54].
5. ASO Therapy for SOD1 ALS
5.1. Background Leading to the Development of Tofersen
ASO therapy targeting SOD1 has been explored for nearly 20 years. ASO333611 is a 2′-O-Methoxyethyl (2′-MOE) gapmer against SOD1 and was developed as the first ASO therapy for the CNS [63]. Initial studies demonstrated that when ASOs are administered intrathecally, they distribute widely throughout the CNS and penetrate tissues effectively. Using transgenic rats expressing human mutant SOD1G93A gene, ASO333611 was continuously infused at a dose of 100 µg/day into the lateral ventricle via an osmotic pump starting at postnatal day 65, approximately 30 days before the expected onset of disease [69]. This led to a significant reduction in SOD1G93A mRNA levels in the brainstem and across all levels of the spinal cord, as well as a significant decrease in SOD1G93A protein levels in the cervical cord. Although there was no observed slowing of early disease progression, a 37% increase in survival was noted.
These promising preclinical data prompted a Phase 1 double-blind, placebo-controlled clinical trial (NCT01041222) to evaluate the safety and tolerability of intrathecally administered ASO333611 in SOD1 ALS patients [70]. Thirty-two ALS patients with SOD1 mutations were enrolled, including eight in the placebo group. Low doses of ASO333611 (0.15-3 mg) were administered via intrathecal infusion over 11.5 hours. Adverse events were reported in 88% patients in the placebo group and 83% in the ASO333611 group. The most common events included post-lumbar puncture syndrome (38% vs. 33%), back pain (50% vs. 17%), and nausea (0% vs. 13%). No dose-limiting toxic effects or any safety or tolerability concerns related to ASO333611 were observed. Due to the low dose administered, a reduction in SOD1 protein levels was not demonstrated. This trial was the first-in-man study of intrathecal ASO administration, confirming the safety and tolerability of ASO333611.
The series of preclinical data and Phase 1 trials for SOD1 ASO have collectively demonstrated the safety of intrathecal administration and its efficacy in rodents, established protocols and evaluation methods for intrathecal administration studies, and indicated the feasibility of ASO therapy for CNS disorders [71].
5.2. Preclinical Study of Tofersen
Tofersen (BIIB067) is an ASO developed by Ionis Pharmaceuticals and licensed to Biogen. As mentioned, the feasibility of SOD1 ASO therapy has been suggested; however, the effects of ASO333611 were modest. With advancements in ASO development technologies over time, there was a demand for more effective SOD1 ASOs, leading to the progression of next-generation therapeutics.
In the research, more than 2,000 ASOs targeting the human SOD1 gene were screened in vitro, followed by optimization, resulting in the selection of a potent 2′-MOE mixed backbone ASO targeting the 3' UTR [72]. In a human neuroblastoma cell line (SH-SY5Y), BIIB067 demonstrated a dose-dependent reduction in SOD1 mRNA levels, showing significant effects compared to ASO333611. In vivo experiments involved bolus injections of BIIB067 into the ventricles of SOD1G93A transgenic mice to deliver it into the cerebrospinal fluid (CSF). BIIB067 reduced human SOD1 mRNA levels in a dose-dependent manner, achieving up to approximately 75% reduction after a single injection. BIIB067 was significantly more active and effective than ASO333611. Similarly, in experiments where BIIB067 was bolus injected into the intrathecal space of SOD1G93A rats, a dose-dependent decrease in SOD1 mRNA was observed, demonstrating higher activity than ASO333611. Sustained reductions in SOD1 mRNA were observed for about eight weeks after a single bolus administration. Furthermore, administration of BIIB067 before disease onset in SOD1G93A mice and rats maintained motor function and significantly prolonged the time until weight loss occurred. The SOD1 ASO extended the lifespan of SOD1G93A rats by more than 50 days (32%) and that of SOD1G93A mice by approximately 40 days (22%). The initial loss of compound muscle action potential in SOD1G93A mice was also reversed after a single dose of tofersen. In addition, increases in serum phosphorylated neurofilament heavy (pNFH), a cytoskeletal protein known to be released into the CSF and serum during axonal injury and correlated with disease severity, were ameliorated by the ASO treatment. [73,74]. Intrathecal injection of BIIB067 in non-human primates (cynomolgus monkeys) resulted in a dose-dependent reduction of SOD1 mRNA and protein levels in CNS tissues and CSF.
5.3. Clinical Study of Tofersen
5.3.1. Phase 1/2 VALOR
Based on the groundbreaking effects observed in rodents, a randomized, double-blind, placebo-controlled Phase 1/2 trial (VALOR; NCT02623699) was initiated to evaluate the tolerability and pharmacokinetics of intrathecal administration of tofersen in patients with familial ALS carrying SOD1 gene mutations [75]. Fifty participants were randomly assigned in a 3:1 ratio to receive either tofersen or placebo, with five doses administered over 12 weeks. Participants in the tofersen group received doses of 0, 20, 40, 60, or 100 mg. The primary outcomes were safety and pharmacokinetics, while the secondary outcome was the change from baseline in SOD1 protein concentration in CSF at day 85. Forty-eight participants completed all five scheduled doses. There were three reported deaths: one in the placebo group due to respiratory failure during the trial, one in the 20 mg group due to pulmonary embolism during the follow-up period, and another in the 60 mg group due to respiratory failure during the follow-up. Most patients experienced adverse events related to lumbar puncture, and increased CSF protein and white cell counts were also observed; however, tolerability was generally good and deemed safe. The difference at day 85 in the change from baseline in CSF SOD1 concentration between the tofersen groups and the placebo group was -33 percentage points (95% confidence interval [CI], -47 to -16) for the 100 mg dose; a decrease in CSF SOD1 concentration was observed in the higher concentration group than in the lower concentration group.
5.3.2. Phase 3 VALOR
Following the promising results from the Phase 1/2 trial, tofersen advanced to a randomized, double-blind, placebo-controlled Phase 3 trial (VALOR; NCT02623699) utilizing the maximum dose from the previous study [25]. In this Phase 3 trial, 108 patients with ALS carrying SOD1 mutations were randomly assigned in a 2:1 ratio to receive either 100 mg of tofersen or placebo, administered intrathecally for a total of eight doses over 24 weeks. To reduce the impact of ALS heterogeneity, participants were divided into fast- and slow-progressing subgroups. The primary endpoint was the change from baseline to week 28 in the total score on the ALS Functional Rating Scale–Revised (ALSFRS-R; range, 0 to 48, with higher scores indicating better function) [76] among fast-progressing group. Secondary endpoints included changes in the total concentration of SOD1 protein in CSF, concentration of neurofilament light chains in plasma (pNFL), slow vital capacity, and handheld dynamometry in 16 muscles. pNFL is a marker known to be strongly associated with death and progression of ALS [77]. Among the 60 participants in the faster-progression primary analysis subgroup, the change in the ALSFRS-R total score from baseline to week 28 was –6.98 points in the tofersen group and –8.14 points in the placebo group (difference, 1.2 points; 95% CI, –3.2 to 5.5; P=0.97), indicating that the primary endpoint was not achieved. However, regarding some secondary endpoints, tofersen resulted in a greater reduction in CSF SOD1 concentration and pNFL levels compared to placebo. Specifically, in the faster-progressing group, CSF SOD1 protein levels decreased by 29% in the tofersen-treated participants, while they increased by 16% in the placebo group (P value not reported due to the primary endpoint not being achieved). Additionally, pNFL levels decreased by 60% in the tofersen group, whereas they increased by 20% in the placebo group. Results for secondary clinical endpoints did not show significant differences between the two groups. Neurologic serious adverse events were reported in 7% of patients receiving tofersen, including myelitis, chemical or aseptic meningitis, lumbar radiculopathy, increased intracranial pressure, and papilledema. Thus, while the Phase III VALOR study did not achieve the primary clinical endpoint, tofersen was approved by the FDA in April 2023 and subsequently by the Committee for Medicinal Products for Human Use (CHMP) in February 2024, based on biomarker responses [7].
5.3.3. Phase 3 Extension Study
An open-label Phase 3 extension study (NCT03070119) is currently ongoing to assess the long-term safety and tolerability of tofersen. The results from this study up to 52 weeks are being integrated with the VALOR trial data, comparing participants who started tofersen at the beginning of the trial (early-start cohort) with those who switched from placebo to treatment at week 28 (delayed-start cohort) [25]. At 52 weeks, earlier initiation of tofersen was associated with less decline in functional measures, including ALSFRS-R, slow vital capacity, and handheld dynamometry compared to the delayed start open-label extension cohort. However, the open-label extension design did not allow for direct comparison with the placebo group, limiting interpretation. The potential effects of earlier compared to delayed initiation of tofersen are being further evaluated in the extension phase.
5.4. Trends after Tofersen Approval
In a multicenter cohort study conducted in Germany involving 24 patients who received tofersen in a real-world setting, clinical parameters, laboratory findings, and biomarkers were tracked. Similar to the VALOR trial and its open-label extension, a decrease in pNFL levels under treatment with tofersen was observed, along with a reduction of pNFH in CSF [78]. The therapy was deemed safe, as no persistent symptoms were noted. However, pleocytosis and immunoglobulin synthesis in CSF with clinical symptoms related to myeloradiculitis in two patients warranted careful monitoring.
In a multicenter observational study involving more than 18 months of tofersen treatment, the ALS progression rate (ALS-PR), as measured by the monthly change in ALSFRS-R [79], showed a mean change of 0.2 (range 0 to 1.1) and a relative reduction of 25%. Among the 16 patients, seven demonstrated an increase in ALSFRS-R [80]. Furthermore, patient-reported outcomes suggested a favorable perception of tofersen treatment in clinical practice.
5.4.1. Phase 3 ATLAS
Currently, a clinical trial is underway for asymptomatic SOD1 mutation carriers (ATLAS; NCT04856982) [81]. The Phase 3 ATLAS study is designed to evaluate the impact of initiating tofersen in presymptomatic carriers of SOD1 variants associated with high or complete penetrance and rapid disease progression who also have biomarker evidence of disease activity (elevated pNFL levels). Specifically, ATLAS will test the hypothesis that presymptomatic initiation of tofersen will delay the emergence of clinically manifest ALS and/or reduce the loss of function over time compared to the initiation of treatment after the emergence of clinically manifest ALS. ATLAS is the first interventional trial in presymptomatic ALS. This randomized controlled trial plans to enroll approximately 150 presymptomatic carriers by August 2027. pNFL levels will be frequently measured, and upon detecting an increase, participants will receive either 100 mg of tofersen or a placebo monthly for two years. The primary outcomes include the proportion of participants who develop clinically manifest ALS within 12 and 24 months after randomization and the time from randomization to the onset of clinically manifest ALS. During the open-label extension period, participants who develop clinically manifest ALS will be able to receive tofersen. ATLAS will expand our understanding of ALS biomarkers and the natural history of the disease, particularly in its earlier stages, making it a valuable study, and its results are eagerly awaited.
Table 2 summarizes a series of clinical trials related to SOD1 ALS.
6. Future Directions
Thirty years after the discovery of SOD1 gene mutation, tofersen, an ASO for SOD1 ALS, has finally been approved. The series of research and development efforts for treatments has not been a smooth journey, but it has significantly contributed to the understanding of the pathophysiology of ALS, providing great hope for a disease that has had almost no effective treatments to date. SOD1 ALS, which is the target for tofersen, accounts for about 2% of all ALS cases, and it is hoped that the approval of ASOs for other mutations will be accelerated. The establishment of the utility of intrathecal administration and the clinical trial protocol that led to FDA approval is expected to greatly benefit future research not only in ALS but also in CNS diseases and hereditary rare diseases as a whole.
The approval of tofersen is by no means the end goal; rather, it is merely a milestone. Verifying the long-term safety and efficacy of tofersen remains an important challenge ahead. The VALOR study showed improvements in biomarkers with tofersen treatment, but clinical endpoints did not demonstrate improvement, highlighting a discrepancy. Some findings suggesting clinical efficacy of tofersen are beginning to emerge in real-world settings, which is noteworthy [80,82]. pNFL levels increase 6–12 months before phenoconversion, and there may be a delay between pNFL reduction and clinical benefit [83]. This suggests that the timing of the primary endpoint evaluation in VALOR study might have been too early. In any case, it is crucial to accumulate and scrutinize data from clinical trials and real-world settings to establish evidence for the optimal use of this costly medication. The ongoing ATLAS study, which targets pre-symptomatic carriers, holds significant potential. If the efficacy of pre-symptomatic administration of tofersen is confirmed, the focus of ALS treatment and development may significantly shift towards the prevention stage.
Over 90% of rare diseases lack standard treatment options [84]. Many rare diseases are caused by pathogenic mutations in a single gene, making ASO-based therapies a promising treatment approach moving forward. Research and development of ASO therapies with greater efficacy and safety remain important themes, especially for CNS diseases, where alternative approaches to the burdensome intrathecal injections are highly desirable, and future outcomes are eagerly anticipated.
Author Contributions
Conceptualization, H.M. and T.Y.; writing—original draft preparation, H.M.; writing—review and editing, H.N. and T.Y.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. T.Y. is supported by the Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, Canadian Institute of Health Research (CIHR), Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, the Women and Children’s Health Research Institute (WCHRI), the Heart and Stroke Foundation Canada, and the US Department of Defense. H.M. is supported by scholarships from the University of Alberta Faculty of Graduate and Postdoctoral Studies.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank the University of Alberta Faculty of Medicine and Dentistry, the Canadian Institute of Health Research, the Friends of Garrett Cumming Research Funds, HM Toupin Neurological Science Research Funds, Muscular Dystrophy Canada, and the Women and Children’s Health Research Institute for their support.
Conflicts of Interest
T.Y. is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Kumar, D. R.; Aslinia, F.; Yale, S. H.; Mazza, J. J., Jean-Martin Charcot: the father of neurology. Clin Med Res 2011, 9, (1), 46-9. [CrossRef]
- Akçimen, F.; Lopez, E. R.; Landers, J. E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B. J., Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat Rev Genet 2023, 24, (9), 642-658. [CrossRef]
- Rowland, L. P.; Shneider, N. A., Amyotrophic lateral sclerosis. N Engl J Med 2001, 344, (22), 1688-700.
- Chiò, A.; Logroscino, G.; Traynor, B. J.; Collins, J.; Simeone, J. C.; Goldstein, L. A.; White, L. A., Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2013, 41, (2), 118-30. [CrossRef]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S., Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol 2020, 267, (4), 944-953.
- Rosen, D. R., Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, (6435), 362.
- Blair, H. A., Tofersen: First Approval. Drugs 2023, 83, (11), 1039-1043. [CrossRef]
- Goutman, S. A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M. G.; Kiernan, M. C.; Feldman, E. L., Emerging insights into the complex genetics and pathophysiology of amyotrophic lateral sclerosis. Lancet Neurol 2022, 21, (5), 465-479. [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B. J., Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol 2018, 17, (1), 94-102. [CrossRef]
- Renton, A. E.; Chiò, A.; Traynor, B. J., State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014, 17, (1), 17-23. [CrossRef]
- Keller, M. F.; Ferrucci, L.; Singleton, A. B.; Tienari, P. J.; Laaksovirta, H.; Restagno, G.; Chiò, A.; Traynor, B. J.; Nalls, M. A., Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol 2014, 71, (9), 1123-34. [CrossRef]
- Al-Chalabi, A.; Fang, F.; Hanby, M. F.; Leigh, P. N.; Shaw, C. E.; Ye, W.; Rijsdijk, F., An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry 2010, 81, (12), 1324-6. [CrossRef]
- Fang, F.; Kamel, F.; Lichtenstein, P.; Bellocco, R.; Sparén, P.; Sandler, D. P.; Ye, W., Familial aggregation of amyotrophic lateral sclerosis. Ann Neurol 2009, 66, (1), 94-9. [CrossRef]
- Grassano, M.; Calvo, A.; Moglia, C.; Sbaiz, L.; Brunetti, M.; Barberis, M.; Casale, F.; Manera, U.; Vasta, R.; Canosa, A.; D'Alfonso, S.; Corrado, L.; Mazzini, L.; Dalgard, C.; Karra, R.; Chia, R.; Traynor, B.; Chiò, A., Systematic evaluation of genetic mutations in ALS: a population-based study. J Neurol Neurosurg Psychiatry 2022, 93, (11), 1190-3. [CrossRef]
- Vasta, R.; Chia, R.; Traynor, B. J.; Chiò, A., Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine 2022, 75, 103795. [CrossRef]
- van Rheenen, W.; van der Spek, R. A. A.; Bakker, M. K.; van Vugt, J.; Hop, P. J.; Zwamborn, R. A. J.; de Klein, N.; Westra, H. J.; Bakker, O. B.; Deelen, P.; Shireby, G.; Hannon, E.; Moisse, M.; Baird, D.; Restuadi, R.; Dolzhenko, E.; Dekker, A. M.; Gawor, K.; Westeneng, H. J.; Tazelaar, G. H. P.; van Eijk, K. R.; Kooyman, M.; Byrne, R. P.; Doherty, M.; Heverin, M.; Al Khleifat, A.; Iacoangeli, A.; Shatunov, A.; Ticozzi, N.; Cooper-Knock, J.; Smith, B. N.; Gromicho, M.; Chandran, S.; Pal, S.; Morrison, K. E.; Shaw, P. J.; Hardy, J.; Orrell, R. W.; Sendtner, M.; Meyer, T.; Başak, N.; van der Kooi, A. J.; Ratti, A.; Fogh, I.; Gellera, C.; Lauria, G.; Corti, S.; Cereda, C.; Sproviero, D.; D'Alfonso, S.; Sorarù, G.; Siciliano, G.; Filosto, M.; Padovani, A.; Chiò, A.; Calvo, A.; Moglia, C.; Brunetti, M.; Canosa, A.; Grassano, M.; Beghi, E.; Pupillo, E.; Logroscino, G.; Nefussy, B.; Osmanovic, A.; Nordin, A.; Lerner, Y.; Zabari, M.; Gotkine, M.; Baloh, R. H.; Bell, S.; Vourc'h, P.; Corcia, P.; Couratier, P.; Millecamps, S.; Meininger, V.; Salachas, F.; Mora Pardina, J. S.; Assialioui, A.; Rojas-García, R.; Dion, P. A.; Ross, J. P.; Ludolph, A. C.; Weishaupt, J. H.; Brenner, D.; Freischmidt, A.; Bensimon, G.; Brice, A.; Durr, A.; Payan, C. A. M.; Saker-Delye, S.; Wood, N. W.; Topp, S.; Rademakers, R.; Tittmann, L.; Lieb, W.; Franke, A.; Ripke, S.; Braun, A.; Kraft, J.; Whiteman, D. C.; Olsen, C. M.; Uitterlinden, A. G.; Hofman, A.; Rietschel, M.; Cichon, S.; Nöthen, M. M.; Amouyel, P.; Traynor, B. J.; Singleton, A. B.; Mitne Neto, M.; Cauchi, R. J.; Ophoff, R. A.; Wiedau-Pazos, M.; Lomen-Hoerth, C.; van Deerlin, V. M.; Grosskreutz, J.; Roediger, A.; Gaur, N.; Jörk, A.; Barthel, T.; Theele, E.; Ilse, B.; Stubendorff, B.; Witte, O. W.; Steinbach, R.; Hübner, C. A.; Graff, C.; Brylev, L.; Fominykh, V.; Demeshonok, V.; Ataulina, A.; Rogelj, B.; Koritnik, B.; Zidar, J.; Ravnik-Glavač, M.; Glavač, D.; Stević, Z.; Drory, V.; Povedano, M.; Blair, I. P.; Kiernan, M. C.; Benyamin, B.; Henderson, R. D.; Furlong, S.; Mathers, S.; McCombe, P. A.; Needham, M.; Ngo, S. T.; Nicholson, G. A.; Pamphlett, R.; Rowe, D. B.; Steyn, F. J.; Williams, K. L.; Mather, K. A.; Sachdev, P. S.; Henders, A. K.; Wallace, L.; de Carvalho, M.; Pinto, S.; Petri, S.; Weber, M.; Rouleau, G. A.; Silani, V.; Curtis, C. J.; Breen, G.; Glass, J. D.; Brown, R. H., Jr.; Landers, J. E.; Shaw, C. E.; Andersen, P. M.; Groen, E. J. N.; van Es, M. A.; Pasterkamp, R. J.; Fan, D.; Garton, F. C.; McRae, A. F.; Davey Smith, G.; Gaunt, T. R.; Eberle, M. A.; Mill, J.; McLaughlin, R. L.; Hardiman, O.; Kenna, K. P.; Wray, N. R.; Tsai, E.; Runz, H.; Franke, L.; Al-Chalabi, A.; Van Damme, P.; van den Berg, L. H.; Veldink, J. H., Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 2021, 53, (12), 1636-1648.
- Bandres-Ciga, S.; Noyce, A. J.; Hemani, G.; Nicolas, A.; Calvo, A.; Mora, G.; Tienari, P. J.; Stone, D. J.; Nalls, M. A.; Singleton, A. B.; Chiò, A.; Traynor, B. J., Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann Neurol 2019, 85, (4), 470-481. [CrossRef]
- Pingault, J. B.; O'Reilly, P. F.; Schoeler, T.; Ploubidis, G. B.; Rijsdijk, F.; Dudbridge, F., Using genetic data to strengthen causal inference in observational research. Nat Rev Genet 2018, 19, (9), 566-580. [CrossRef]
- Bunton-Stasyshyn, R. K.; Saccon, R. A.; Fratta, P.; Fisher, E. M., SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist 2015, 21, (5), 519-29.
- Abel, O.; Powell, J. F.; Andersen, P. M.; Al-Chalabi, A., ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum Mutat 2012, 33, (9), 1345-51. [CrossRef]
- McCann, E. P.; Henden, L.; Fifita, J. A.; Zhang, K. Y.; Grima, N.; Bauer, D. C.; Chan Moi Fat, S.; Twine, N. A.; Pamphlett, R.; Kiernan, M. C.; Rowe, D. B.; Williams, K. L.; Blair, I. P., Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J Med Genet 2020. [CrossRef]
- Suzuki, N.; Nishiyama, A.; Warita, H.; Aoki, M., Genetics of amyotrophic lateral sclerosis: seeking therapeutic targets in the era of gene therapy. J Hum Genet 2023, 68, (3), 131-152. [CrossRef]
- Nishiyama, A.; Niihori, T.; Warita, H.; Izumi, R.; Akiyama, T.; Kato, M.; Suzuki, N.; Aoki, Y.; Aoki, M., Comprehensive targeted next-generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol Aging 2017, 53, 194.e1-194.e8. [CrossRef]
- Bernard, E.; Pegat, A.; Svahn, J.; Bouhour, F.; Leblanc, P.; Millecamps, S.; Thobois, S.; Guissart, C.; Lumbroso, S.; Mouzat, K., Clinical and Molecular Landscape of ALS Patients with SOD1 Mutations: Novel Pathogenic Variants and Novel Phenotypes. A Single ALS Center Study. Int J Mol Sci 2020, 21, (18).
- Miller, T. M.; Cudkowicz, M. E.; Genge, A.; Shaw, P. J.; Sobue, G.; Bucelli, R. C.; Chiò, A.; Van Damme, P.; Ludolph, A. C.; Glass, J. D.; Andrews, J. A.; Babu, S.; Benatar, M.; McDermott, C. J.; Cochrane, T.; Chary, S.; Chew, S.; Zhu, H.; Wu, F.; Nestorov, I.; Graham, D.; Sun, P.; McNeill, M.; Fanning, L.; Ferguson, T. A.; Fradette, S., Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 2022, 387, (12), 1099-1110. [CrossRef]
- Radunovíc, A.; Leigh, P. N., Cu/Zn superoxide dismutase gene mutations in amyotrophic lateral sclerosis: correlation between genotype and clinical features. J Neurol Neurosurg Psychiatry 1996, 61, (6), 565-72.
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P. M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M., SOD1 mutations associated with amyotrophic lateral sclerosis analysis of variant severity. Sci Rep 2022, 12, (1), 103. [CrossRef]
- Cudkowicz, M. E.; McKenna-Yasek, D.; Sapp, P. E.; Chin, W.; Geller, B.; Hayden, D. L.; Schoenfeld, D. A.; Hosler, B. A.; Horvitz, H. R.; Brown, R. H., Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol 1997, 41, (2), 210-21.
- Saeed, M.; Yang, Y.; Deng, H. X.; Hung, W. Y.; Siddique, N.; Dellefave, L.; Gellera, C.; Andersen, P. M.; Siddique, T., Age and founder effect of SOD1 A4V mutation causing ALS. Neurology 2009, 72, (19), 1634-9. [CrossRef]
- Zou, Z. Y.; Liu, M. S.; Li, X. G.; Cui, L. Y., H46R SOD1 mutation is consistently associated with a relatively benign form of amyotrophic lateral sclerosis with slow progression. Amyotroph Lateral Scler Frontotemporal Degener 2016, 17, (7-8), 610-613. [CrossRef]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; Rusmini, P.; Bolzoni, E.; Bendotti, C.; Poletti, A., Mutation of SOD1 in ALS: a gain of a loss of function. Hum Mol Genet 2007, 16, (13), 1604-18. [CrossRef]
- Ekhtiari Bidhendi, E.; Bergh, J.; Zetterström, P.; Forsberg, K.; Pakkenberg, B.; Andersen, P. M.; Marklund, S. L.; Brännström, T., Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol 2018, 136, (6), 939-953. [CrossRef]
- Gurney, M. E.; Pu, H.; Chiu, A. Y.; Dal Canto, M. C.; Polchow, C. Y.; Alexander, D. D.; Caliendo, J.; Hentati, A.; Kwon, Y. W.; Deng, H. X.; et al., Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, (5166), 1772-5. [CrossRef]
- Bruijn, L. I.; Cleveland, D. W., Mechanisms of selective motor neuron death in ALS: insights from transgenic mouse models of motor neuron disease. Neuropathol Appl Neurobiol 1996, 22, (5), 373-87. [CrossRef]
- Matsumoto, A.; Okada, Y.; Nakamichi, M.; Nakamura, M.; Toyama, Y.; Sobue, G.; Nagai, M.; Aoki, M.; Itoyama, Y.; Okano, H., Disease progression of human SOD1 (G93A) transgenic ALS model rats. J Neurosci Res 2006, 83, (1), 119-33. [CrossRef]
- Hayashi, Y.; Homma, K.; Ichijo, H., SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul 2016, 60, 95-104. [CrossRef]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guégan, C.; Nagai, M.; Xu, Z.; Sosunov, A. A.; McKhann, G. M., 2nd; Przedborski, S., Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A 2006, 103, (15), 6025-30. [CrossRef]
- Ilieva, H.; Polymenidou, M.; Cleveland, D. W., Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 2009, 187, (6), 761-72. [CrossRef]
- Masrori, P.; Van Damme, P., Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol 2020, 27, (10), 1918-1929. [CrossRef]
- Fischer, L. R.; Li, Y.; Asress, S. A.; Jones, D. P.; Glass, J. D., Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp Neurol 2012, 233, (1), 163-71. [CrossRef]
- Reaume, A. G.; Elliott, J. L.; Hoffman, E. K.; Kowall, N. W.; Ferrante, R. J.; Siwek, D. F.; Wilcox, H. M.; Flood, D. G.; Beal, M. F.; Brown, R. H., Jr.; Scott, R. W.; Snider, W. D., Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 1996, 13, (1), 43-7. [CrossRef]
- Doble, A., The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, (6 Suppl 4), S233-41. [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V., A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994, 330, (9), 585-91. [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P. N.; Guillet, P.; Meininger, V., Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996, 347, (9013), 1425-31. [CrossRef]
- Hinchcliffe, M.; Smith, A., Riluzole: real-world evidence supports significant extension of median survival times in patients with amyotrophic lateral sclerosis. Degener Neurol Neuromuscul Dis 2017, 7, 61-70. [CrossRef]
- Exploratory double-blind, parallel-group, placebo-controlled study of edaravone (MCI-186) in amyotrophic lateral sclerosis (Japan ALS severity classification: Grade 3, requiring assistance for eating, excretion or ambulation). Amyotroph Lateral Scler Frontotemporal Degener 2017, 18, (sup1), 40-48.
- Takahashi, F.; Takei, K.; Tsuda, K.; Palumbo, J., Post-hoc analysis of MCI186-17, the extension study to MCI186-16, the confirmatory double-blind, parallel-group, placebo-controlled study of edaravone in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2017, 18, (sup1), 32-39. [CrossRef]
- Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2017, 16, (7), 505-512.
- Paganoni, S.; Hendrix, S.; Dickson, S. P.; Knowlton, N.; Macklin, E. A.; Berry, J. D.; Elliott, M. A.; Maiser, S.; Karam, C.; Caress, J. B.; Owegi, M. A.; Quick, A.; Wymer, J.; Goutman, S. A.; Heitzman, D.; Heiman-Patterson, T. D.; Jackson, C. E.; Quinn, C.; Rothstein, J. D.; Kasarskis, E. J.; Katz, J.; Jenkins, L.; Ladha, S.; Miller, T. M.; Scelsa, S. N.; Vu, T. H.; Fournier, C. N.; Glass, J. D.; Johnson, K. M.; Swenson, A.; Goyal, N. A.; Pattee, G. L.; Andres, P. L.; Babu, S.; Chase, M.; Dagostino, D.; Hall, M.; Kittle, G.; Eydinov, M.; McGovern, M.; Ostrow, J.; Pothier, L.; Randall, R.; Shefner, J. M.; Sherman, A. V.; St Pierre, M. E.; Tustison, E.; Vigneswaran, P.; Walker, J.; Yu, H.; Chan, J.; Wittes, J.; Yu, Z. F.; Cohen, J.; Klee, J.; Leslie, K.; Tanzi, R. E.; Gilbert, W.; Yeramian, P. D.; Schoenfeld, D.; Cudkowicz, M. E., Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve 2021, 63, (1), 31-39. [CrossRef]
- Ketabforoush, A.; Faghihi, F.; Azedi, F.; Ariaei, A.; Habibi, M. A.; Khalili, M.; Ashtiani, B. H.; Joghataei, M. T.; Arnold, W. D., Sodium Phenylbutyrate and Tauroursodeoxycholic Acid: A Story of Hope Turned to Disappointment in Amyotrophic Lateral Sclerosis Treatment. Clin Drug Investig 2024, 44, (7), 495-512. [CrossRef]
- Mead, R. J.; Shan, N.; Reiser, H. J.; Marshall, F.; Shaw, P. J., Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov 2023, 22, (3), 185-212. [CrossRef]
- Lauffer, M. C.; van Roon-Mom, W.; Aartsma-Rus, A., Possibilities and limitations of antisense oligonucleotide therapies for the treatment of monogenic disorders. Commun Med (Lond) 2024, 4, (1), 6. [CrossRef]
- Shadid, M.; Badawi, M.; Abulrob, A., Antisense oligonucleotides: absorption, distribution, metabolism, and excretion. Expert Opin Drug Metab Toxicol 2021, 17, (11), 1281-1292. [CrossRef]
- Leckie, J.; Yokota, T., Potential of Cell-Penetrating Peptide-Conjugated Antisense Oligonucleotides for the Treatment of SMA. Molecules 2024, 29, (11). [CrossRef]
- Roberts, T. C.; Langer, R.; Wood, M. J. A., Advances in oligonucleotide drug delivery. Nat Rev Drug Discov 2020, 19, (10), 673-694. [CrossRef]
- Stein, H.; Hausen, P., Enzyme from calf thymus degrading the RNA moiety of DNA-RNA Hybrids: effect on DNA-dependent RNA polymerase. Science 1969, 166, (3903), 393-5. [CrossRef]
- Wu, H.; Lima, W. F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S. T., Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem 2004, 279, (17), 17181-9. [CrossRef]
- Rigo, F.; Hua, Y.; Chun, S. J.; Prakash, T. P.; Krainer, A. R.; Bennett, C. F., Synthetic oligonucleotides recruit ILF2/3 to RNA transcripts to modulate splicing. Nat Chem Biol 2012, 8, (6), 555-61. [CrossRef]
- Stephenson, M. L.; Zamecnik, P. C., Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci U S A 1978, 75, (1), 285-8. [CrossRef]
- Haque, U. S.; Yokota, T., Recent Progress in Gene-Targeting Therapies for Spinal Muscular Atrophy: Promises and Challenges. Genes (Basel) 2024, 15, (8). [CrossRef]
- Sweeney, M. D.; Zhao, Z.; Montagne, A.; Nelson, A. R.; Zlokovic, B. V., Blood-Brain Barrier: From Physiology to Disease and Back. Physiol Rev 2019, 99, (1), 21-78. [CrossRef]
- Barchet, T. M.; Amiji, M. M., Challenges and opportunities in CNS delivery of therapeutics for neurodegenerative diseases. Expert Opin Drug Deliv 2009, 6, (3), 211-25. [CrossRef]
- Smith, R. A.; Miller, T. M.; Yamanaka, K.; Monia, B. P.; Condon, T. P.; Hung, G.; Lobsiger, C. S.; Ward, C. M.; McAlonis-Downes, M.; Wei, H.; Wancewicz, E. V.; Bennett, C. F.; Cleveland, D. W., Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest 2006, 116, (8), 2290-6. [CrossRef]
- Kordasiewicz, H. B.; Stanek, L. M.; Wancewicz, E. V.; Mazur, C.; McAlonis, M. M.; Pytel, K. A.; Artates, J. W.; Weiss, A.; Cheng, S. H.; Shihabuddin, L. S.; Hung, G.; Bennett, C. F.; Cleveland, D. W., Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron 2012, 74, (6), 1031-44.
- Passini, M. A.; Bu, J.; Richards, A. M.; Kinnecom, C.; Sardi, S. P.; Stanek, L. M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; Kaye, E. M.; Shihabuddin, L. S.; Krainer, A. R.; Bennett, C. F.; Cheng, S. H., Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med 2011, 3, (72), 72ra18. [CrossRef]
- Chiriboga, C. A.; Swoboda, K. J.; Darras, B. T.; Iannaccone, S. T.; Montes, J.; De Vivo, D. C.; Norris, D. A.; Bennett, C. F.; Bishop, K. M., Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 2016, 86, (10), 890-7. [CrossRef]
- Fox, D.; To, T. M.; Seetasith, A.; Patel, A. M.; Iannaccone, S. T., Adherence and Persistence to Nusinersen for Spinal Muscular Atrophy: A US Claims-Based Analysis. Adv Ther 2023, 40, (3), 903-919. [CrossRef]
- Cantara, S.; Simoncelli, G.; Ricci, C., Antisense Oligonucleotides (ASOs) in Motor Neuron Diseases: A Road to Cure in Light and Shade. Int J Mol Sci 2024, 25, (9). [CrossRef]
- Liu, J.; Shinobu, L. A.; Ward, C. M.; Young, D.; Cleveland, D. W., Elevation of the Hsp70 chaperone does not effect toxicity in mouse models of familial amyotrophic lateral sclerosis. J Neurochem 2005, 93, (4), 875-82. [CrossRef]
- Miller, T. M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S. H.; Andres, P. L.; Mahoney, K.; Allred, P.; Alexander, K.; Ostrow, L. W.; Schoenfeld, D.; Macklin, E. A.; Norris, D. A.; Manousakis, G.; Crisp, M.; Smith, R.; Bennett, C. F.; Bishop, K. M.; Cudkowicz, M. E., An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 2013, 12, (5), 435-42. [CrossRef]
- Rinaldi, C.; Wood, M. J. A., Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol 2018, 14, (1), 9-21. [CrossRef]
- McCampbell, A.; Cole, T.; Wegener, A. J.; Tomassy, G. S.; Setnicka, A.; Farley, B. J.; Schoch, K. M.; Hoye, M. L.; Shabsovich, M.; Sun, L.; Luo, Y.; Zhang, M.; Comfort, N.; Wang, B.; Amacker, J.; Thankamony, S.; Salzman, D. W.; Cudkowicz, M.; Graham, D. L.; Bennett, C. F.; Kordasiewicz, H. B.; Swayze, E. E.; Miller, T. M., Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J Clin Invest 2018, 128, (8), 3558-3567. [CrossRef]
- Lu, C. H.; Petzold, A.; Kalmar, B.; Dick, J.; Malaspina, A.; Greensmith, L., Plasma neurofilament heavy chain levels correlate to markers of late stage disease progression and treatment response in SOD1(G93A) mice that model ALS. PLoS One 2012, 7, (7), e40998. [CrossRef]
- McCombe, P. A.; Pfluger, C.; Singh, P.; Lim, C. Y.; Airey, C.; Henderson, R. D., Serial measurements of phosphorylated neurofilament-heavy in the serum of subjects with amyotrophic lateral sclerosis. J Neurol Sci 2015, 353, (1-2), 122-9. [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P. J.; Andersen, P. M.; Atassi, N.; Bucelli, R. C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A. L.; Maragakis, N. J.; McDermott, C. J.; Pestronk, A.; Ravits, J.; Salachas, F.; Trudell, R.; Van Damme, P.; Zinman, L.; Bennett, C. F.; Lane, R.; Sandrock, A.; Runz, H.; Graham, D.; Houshyar, H.; McCampbell, A.; Nestorov, I.; Chang, I.; McNeill, M.; Fanning, L.; Fradette, S.; Ferguson, T. A., Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 2020, 383, (2), 109-119. [CrossRef]
- Cedarbaum, J. M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A., The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci 1999, 169, (1-2), 13-21. [CrossRef]
- Thouvenot, E.; Demattei, C.; Lehmann, S.; Maceski-Maleska, A.; Hirtz, C.; Juntas-Morales, R.; Pageot, N.; Esselin, F.; Alphandéry, S.; Vincent, T.; Camu, W., Serum neurofilament light chain at time of diagnosis is an independent prognostic factor of survival in amyotrophic lateral sclerosis. Eur J Neurol 2020, 27, (2), 251-257. [CrossRef]
- Wiesenfarth, M.; Dorst, J.; Brenner, D.; Elmas, Z.; Parlak, Ö.; Uzelac, Z.; Kandler, K.; Mayer, K.; Weiland, U.; Herrmann, C.; Schuster, J.; Freischmidt, A.; Müller, K.; Siebert, R.; Bachhuber, F.; Simak, T.; Günther, K.; Fröhlich, E.; Knehr, A.; Regensburger, M.; German, A.; Petri, S.; Grosskreutz, J.; Klopstock, T.; Reilich, P.; Schöberl, F.; Hagenacker, T.; Weyen, U.; Günther, R.; Vidovic, M.; Jentsch, M.; Haarmeier, T.; Weydt, P.; Valkadinov, I.; Hesebeck-Brinckmann, J.; Conrad, J.; Weishaupt, J. H.; Schumann, P.; Körtvélyessy, P.; Meyer, T.; Ruf, W. P.; Witzel, S.; Senel, M.; Tumani, H.; Ludolph, A. C., Effects of tofersen treatment in patients with SOD1-ALS in a "real-world" setting - a 12-month multicenter cohort study from the German early access program. EClinicalMedicine 2024, 69, 102495. [CrossRef]
- Kimura, F.; Fujimura, C.; Ishida, S.; Nakajima, H.; Furutama, D.; Uehara, H.; Shinoda, K.; Sugino, M.; Hanafusa, T., Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006, 66, (2), 265-7. [CrossRef]
- Meyer, T.; Schumann, P.; Weydt, P.; Petri, S.; Weishaupt, J. H.; Weyen, U.; Koch, J. C.; Günther, R.; Regensburger, M.; Boentert, M.; Wiesenfarth, M.; Koc, Y.; Kolzarek, F.; Kettemann, D.; Norden, J.; Bernsen, S.; Elmas, Z.; Conrad, J.; Valkadinov, I.; Vidovic, M.; Dorst, J.; Ludolph, A. C.; Hesebeck-Brinckmann, J.; Spittel, S.; Münch, C.; Maier, A.; Körtvélyessy, P., Clinical and patient-reported outcomes and neurofilament response during tofersen treatment in SOD1-related ALS-A multicenter observational study over 18 months. Muscle Nerve 2024, 70, (3), 333-345. [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P. M.; Bucelli, R. C.; Andrews, J. A.; Otto, M.; Farahany, N. A.; Harrington, E. A.; Chen, W.; Mitchell, A. A.; Ferguson, T.; Chew, S.; Gedney, L.; Oakley, S.; Heo, J.; Chary, S.; Fanning, L.; Graham, D.; Sun, P.; Liu, Y.; Wong, J.; Fradette, S., Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: the ATLAS Study. Neurotherapeutics 2022, 19, (4), 1248-1258. [CrossRef]
- Meyer, T.; Schumann, P.; Weydt, P.; Petri, S.; Koc, Y.; Spittel, S.; Bernsen, S.; Günther, R.; Weishaupt, J. H.; Dreger, M.; Kolzarek, F.; Kettemann, D.; Norden, J.; Boentert, M.; Vidovic, M.; Meisel, C.; Münch, C.; Maier, A.; Körtvélyessy, P., Neurofilament light-chain response during therapy with antisense oligonucleotide tofersen in SOD1-related ALS: Treatment experience in clinical practice. Muscle Nerve 2023, 67, (6), 515-521. [CrossRef]
- Oliveira Santos, M.; de Carvalho, M., Profiling tofersen as a treatment of superoxide dismutase 1 amyotrophic lateral sclerosis. Expert Rev Neurother 2024, 24, (6), 549-553. [CrossRef]
- Aartsma-Rus, A.; Dooms, M.; Le Cam, Y., Orphan Medicine Incentives: How to Address the Unmet Needs of Rare Disease Patients by Optimizing the European Orphan Medicinal Product Landscape Guiding Principles and Policy Proposals by the European Expert Group for Orphan Drug Incentives (OD Expert Group). Front Pharmacol 2021, 12, 744532. [CrossRef]
Table 2.
Summary of ASO clinical trials for SOD1 ALS.
| Drug | Phase Study type Study objective |
Number of patients Administration, dose |
Outcome |
|---|---|---|---|
| ASO 333611 | Phase 1 (NCT01041222) Randomised, double-blind, placebo-controlled Safety and tolerability study of ASO 333611 in SOD1 ALS |
33 patients 12hr intrathecal infusion of 0.15, 0.5, 1.5 and 3 mg |
No drug-related safety issues. No reduction in SOD1 protein levels in CSF. |
| BIIB067 tofersen |
Phase 1/2 VALOR (NCT02623699) Randomised, double-blind, placebo-controlled Safety, tolerability and pharmacokinetics study of tofersen in SOD1 ALS |
50 patients Intrathecal injection of 20, 40, 60, 100 mg |
Tofersen was generally well tolerated and safe. The highest concentration of tofersen decreased CSF SOD1 concentrations the most. |
| BIIB067 tofersen |
Phase 3 VALOR (NCT02623699) Randomised, double-blind, placebo-controlled Efficacy study of tofersen in SOD1 ALS |
108 patients Intrathecal injection of 100 mg |
Tofersen did not improve the ALSFRS-R total score from baseline to week 28 in faster progression group. Tofersen resulted in a greater reduction in CSF SOD1 and pNFL concentrations. |
| BIIB067 tofersen |
Phase 3 (NCT03070119) Open-label extension Long-term Evaluation of tofersen in SOD1 ALS |
138 patients Intrathecal injection of 100 mg |
Ongoing |
| BIIB067 tofersen |
Phase 3 ATLAS (NCT04856982) Randomised, double-blind, placebo-controlled and subsequent open-label extension Long-term, efficacy study of tofersen in presymptomatic carriers of SOD1 |
150 patients (estimated) Intrathecal injection of 100 mg |
Ongoing |
ASO, antisense oligonucleotide; SOD1, superoxide dismutase 1; ALS, amyotrophic lateral sclerosis; CSF, cerebrospinal fluid; ALSFRS-R, ALS Functional Rating Scale–Revised.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



