
Submitted:
30 September 2024
Posted:
01 October 2024
You are already at the latest version
Abstract
Diabetic foot ulcers (DFUs) are a multifactorial pathophysiologic condition that occurs in patients with diabetes mellitus (DM). Approximately 9.1 to 26.1 million people are affected by DFUs annually. This condition can lead to progressive foot infections and, ultimately, foot amputation. Various microbes contribute to DFUs, including methicillin-resistant Staphylococcus. aureus, Pseudomonas aeruginosa, and Escherichia coli can form biofilms composed of complex matrices that create a protective shield around the microorganisms, enhancing their survival and resistance to treatment. Biofilm formation is a critical virulence factor directly associated with the onset and persistence of DFUs. It not only complicates the clinical management of these ulcers but also facilitates antibiotic resistance, necessitating a comprehensive approach to treatment. Effective management strategies must therefore target biofilm formation and the virulence factors associated with these pathogens. By addressing these elements, conventional antibiotics can be rendered more effective in treating diabetic foot ulcers. This approach aims not only to promote wound healing but also to address underlying causes and prevent further complications. This article seeks to highlight the significance of biofilm formation in DFUs, explore methodologies for studying this condition, as well as discuss demographic considerations and treatment options.
Keywords:
Diabetic Foot Ulcers
; DFU
; Diabetic Foot Infections
; DFI
; Biofilm
; Recalcitrant Wounds
; Multidrug resistance
; Antimicrobial Resistance
Introduction
Diabetic Foot Ulcer (DFU) is a degenerative disease of the foot affecting patients with diabetes mellitus. DFUs are a severe complication of diabetes mellitus, characterized by painful and persistent wounds that can occur anywhere on the foot. Diabetic foot ulcer disease manifests when the blood glucose level in a patient is high, providing nutrition and upregulating the essential genes for production of Extracellular Polymeric substances (EPS) in the DFU pathogens, leading to the formation of biofilms, and therefore formation of chronically infected wounds due to resulting reduction in response to antimicrobial treatment. In this disease an ulcer can form at the bottom or top of the foot due to excessive infection which is mediated by formation of biofilms. From here the tissue necrosis starts, can progress to gangrene if not treated properly and ultimately leading to amputation. The aim of this review is to highlight the effect of biofilm formation leading to wound formation in the Diabetic Foot Ulcers (DFUs), focusing on the factors responsible for biofilm formation and difficulty of treatment of this disease due to antimicrobial resistance, recent and conventional detection methods, the precise mechanism of biofilm formation in the major organisms involved in pathogenesis, conventional and newer treatment methods.
- The disease
Patients with diabetes are often in a chronic hyperglycemic state, which creates an ideal environment for the growth of microbes, specifically Pseudomonas aeruginosa and Staphylococcus aureus. These microbes thrive and produce toxins that form biofilms [1,2]. Biofilms are complex structures formed by a variety of pathogens, notably S. aureus, P. aeruginosa, and E. coli. The biofilms formed by S. aureus and P. aeruginosa are particularly problematic due to their thicker and more resilient nature compared to those formed by other organisms [8]. Biofilms generally consist of extracellular polymeric substances (EPS), including DNA, glycoproteins, proteins, and polysaccharides, produced by sessile microorganisms [3]. Elevated glucose levels stimulate EPS production, causing microorganisms to become sessile and embed into the biofilm matrix [4,5]. This matrix hinders antibiotic penetration and reduces treatment effectiveness due to both the thickness of the biofilm and the presence of efflux pumps. Additionally, EPS reduces the phagocytic activity of leukocytes [6], impeding their ability to penetrate the biofilm [7].
P. aeruginosa produces alginate, which not only protects its own biofilm but also enhances the protection of S. aureus biofilms, creating a synergistic and heterogeneous biofilm environment [8,9,10]. Moreover, this biofilm environment favors horizontal gene transfer [10], a key mechanism for the spread of antibiotic resistance among microorganisms. The mutation rate of sessile organisms within biofilms is higher compared to planktonic cells, leading to the acquisition of resistance genes by microbes that were initially non-resistant to antibiotics [11]. Since the most common pathogens are S. aureus, P. aeruginosa, E. coli which are potent biofilm formers [12], the consequent increase in the ability of the horizontal gene transfer among the biofilm forming organisms leads to initially susceptible microbes becoming resistant to antimicrobial compounds. This complicates treatment and accelerates foot deformity and ulceration, potentially progressing to gangrene if not effectively managed.
DFUs typically begin with callus formation, which progresses into a subcutaneous hemorrhage as the skin becomes dry, followed by erosion of the callus, finally becoming an ulcer, often leading to foot deformities and sensory neuropathy [13]. Ulcers have about a 50%-60% chance of developing into an infection which could be polymicrobial, and form biofilms consisting of S. aureus and P. aeruginosa, which play a critical role in the pathogenesis of DFUs. Gangrene can also be caused by Panton-Valentine leukocidin produced by S. aureus, as well as hydrogen cyanide (HCN), pyocyanin, and pyoverdin produced by P. aeruginosa. These toxins induce cell death, leading to tissue necrosis and ultimately gangrene formation [14]. In addition, biofilms accelerate the progression of foot deformities, causing lower-grade ulcers to intensify into more severe ulcers, potentially reaching grade 4 or 5 and leading to gangrene [15]. Gangrenous tissue is severe and often necessitates amputation to prevent its spread to the rest of the body [16].
Current treatment for DFUs involves addressing both the infection and the underlying biofilm-related resistance issues. Conservative treatments may fail, requiring amputation for advanced ulcers (grades 4 and 5) to prevent the spread of gangrene throughout the body [16]. Recalcitrant wounds that become gangrenous are often difficult to treat due to factors such as increased biofilm formation and simultaneous antimicrobial resistance [17]. The need for amputation depends on the condition of the foot and the extent of gangrene, with amputation being necessary for grade 5 ulcers and potentially for grades 4 and 3 depending on the situation [18].
Emerging therapies, such as placental-derived materials [19] and human umbilical allografts [20], promise to reduce wound size and promote healing. Therefore, to treat diabetic foot ulcers, special care must be implemented to eradicate polymicrobial biofilms, reducing the chance of infection of the wound and aiding in wound healing. Effective management requires a deep understanding of biofilm dynamics and antimicrobial resistance. Ongoing research and comprehensive reviews of treatment options are crucial for improving disease burden and patient outcomes for those with persistent antibiotic-resistant diabetic foot ulcers.
- Burden of the disease
As of 2021, there are approximately 537 million cases of diabetes mellitus reported globally [21]. Annually, between 9.1 and 26.1 million new cases of diabetic foot ulcers (DFUs) are reported, with an occurrence rate of 19%-34% [13], and an annual incidence rate of 1.9%-4.0% [22,23]. The cost of treating DFUs varies significantly by country. In the UK, it averages $7,539 per patient, in India about $1,960 per patient, and in the US, it totals between $3.93 and $10.9 billion per year [24]. This highlights the global threat that DFUs pose to patient safety and the economic burden they impose.
Infection is a major complication, affecting 50-60% of DFU cases [13], which often progresses to amputation due to necrosis. The likelihood of developing a DFU in a hyperglycemic patient range from 15-25%, with an amputation incidence between 25%-50% [25].
Diabetic foot ulcers (DFUs) and diabetes mellitus most commonly affect individuals around 45 years of age, particularly males. Patients over 50 are at a greater risk of developing DFUs [26]. Additionally, male patients generally show higher susceptibility and those with lower educational levels are also at an increased risk [26]. Understanding the pathophysiology and progression of the disease is crucial for managing and preventing DFUs. On searching the published reports on the incidence of biofilms in DFUs globally in the last 10 years, we found that a significant majority of currently available literature mentioned the presence of monomicrobial or polymicrobial biofilms in DFU patients.
A breakdown of 88 articles found on PubMed using specific parameters, (last 10 years, biofilms in DFUs) are summarized showing the number of articles that mentioned whether biofilms were present or not. The 66 articles that did report the occurrence of biofilms in were further separated by whether the biofilm composition was discussed. The 45 articles that provided information on biofilm makeup are further separated based on whether they provided information on the genetic makeup or specific species within the biofilms.
As shown in Table 1, within those 88 articles, 75% talked about biofilms in general. Of those 66 articles, 68% talked about the genetic makeup of the biofilms that were studied. Of those 45 articles, 93% of the articles mentioned Staphylococcus, Pseudomonas, and/or Pseudomonas aeruginosa as the bulk of the biofilm formation that have antibiotic resistance. Biofilms are present on every wound that appears in the human body. Understanding the genetic make-up of wounds and ulcers in addition to the role biofilms play on DFUs can dramatically lessen recovery time, prevent DFUs leading to amputation, and improve prognosis. By seeing a common pattern of microorganisms, we can better generalize treatment in cases where microbiology reports are not yet concluded. In addition, knowing that most biofilms in DFUs are made up by specific microorganisms, we can better target biofilms using a narrower spectrum of antibiotics.
- Pathogenesis of DFU
The development of a diabetic foot ulcer generally occurs in three stages:
- Callus Formation: The initial stage involves the formation of a callus, a thickening of the skin under the foot that appears as a yellowish tinge. This condition is often caused by peripheral neuropathy, resulting from nerve damage in the foot, which can occasionally affect leg nerves as well. Symptoms of peripheral neuropathy include numbness, cramps, and muscle fatigue.
- Motor Neuropathy: In this stage, motor neurons in the foot are damaged, leading to weakness and deformation of the feet.
- Sensory Neuropathy: The final stage involves damage to sensory neurons, resulting in loss of sensation, which can lead to trauma, skin drying, and autonomic neuropathy. Frequent damage to the callus can result in subcutaneous hemorrhage, ultimately forming an ulcer [13].
The progression of DFUs is heavily influenced by bacterial infections. Poor hygiene and foot damage in diabetic patients create ideal conditions for DFUs. Bacterial associations and biofilm formation play a significant role in the rapid progression of DFUs. Bacteria involved in DFUs can form biofilms, which contribute to the disease's severity. DFUs are classified into five grades based on their severity:
- Grade 0: No visible ulcer
- Grade 1: Superficial ulcer
- Grade 2: Deep tissue ulceration
- Grade 3: Abscess formation involving bone
- Grade 4: Gangrene formation at the toe
- Grade 5: Extensive gangrene and necrosis throughout the foot [27]
About 32.4% of patients have Grade 1 ulcers, 29.2% have Grade 2 ulcers, and only 8.2% have ulcers graded higher than Grade 2 [28]. Developing effective therapies requires creating disease models based on both in-vivo and in-vitro studies to address the various grades of DFUs effectively. Figure 1 illustrates the mechanisms of diabetic foot ulcers (DFUs), including their causes and effects.
- Detection of biofilms in DFU
MolecuLight™, an autofluorescence device, is the latest technology used to assess bacterial load in diabetic foot ulcers by employing the principle of autofluorescence. This non-invasive method enables the detection of biofilm formation, as bacterial load is proportional to biofilm presence. The device has a specificity and sensitivity of approximately 78% when guided by autofluorescence imaging. MolecuLight™ features a slide-based mechanism to switch between white light and autofluorescence modes, utilizing a 405 nm LED light along with dual-band fluorescence emission filters (500 nm – 545 nm and 600 nm – 665 nm) during autofluorescence mode. This method functionality aids in accurately identifying the wound area and sampling bacterial load [29].
In contrast, traditional biopsy methods are less frequently used due to the risk of contamination, although they may be warranted in cases of moderate to severe wound infections. In such cases, tissue from the debrided wound base can be collected and analyzed using techniques such as Fluorescent In-Situ Hybridization (FISH), Confocal Laser Scanning Microscopy, and Scanning Electron Microscopy [30]. Additionally, a microtiter plate assay combined with FISH can provide insights into biofilm thickness and composition [31]. The simplest method for assessing biofilm samples involves staining with Crystal Violet and measuring optical density to estimate bacterial load [32]. Furthermore, combining microtiter assays with ELISA, Scanning Electron Microscopy, and XTT Formazan assays allows for the detection of non-Candida albicans species in biofilms [33].
- Methods to Study Biofilms in Diabetic Foot Ulcers
Diabetic foot ulcers can be studied by in vitro and in vivo analyses. Both methods involve studies that culture patient cells under hyperglycemic conditions, along with various pathogens, (both individually and in mixed populations). Additionally, pre-existing data, as well as statistical analysis from patients with diabetic foot ulcers, can provide insights into the demographics of the disease and patterns of disease progression
In vitro methods include 2-Dimensional cell culture model and 3-D DFU model. In vivo methods include The Ischemic and neuropathic animal ulcer models as well as the infected diabetic foot ulcer model. A synopsis of these methods is presented in Table 1. In vivo methods include studying the progression of DFU in animal models which have been induced to develop Type 2 Diabetes through spontaneous and targeted mutations by utilizing various approaches as listed in Table 1 and then studied for different types of DFUs.
- In Vitro Methods
- 2-Dimensional cell culture model
Fibroblast and keratin-producing cells are obtained from the patient and cultured in a hyperglycemic environment to mimic the high glucose concentration characteristic of hyperglycemic patients. This method studies various markers of diabetic foot ulcers (DFU), such as the glycosylation of collagen fibers in the skin [34]. It is a straightforward approach to evaluate the mechanisms of wound formation and healing, as well as to test different therapies for DFU.
- 3-D DFU model
The physiological parameters of cells grown in a 2D cell culture model can be studied using a 3D diabetic foot ulcer (DFU) model. This three-dimensional model allows for the examination of various phenotypes related to angiogenesis, extracellular matrix deposition, and wound healing through re-epithelialization, among other processes [35].
- In Vivo methods
Rats such as the Zucker diabetic fatty (ZDF) and Goto-Kakizaki (GK) develop Type 2 diabetes, which can be induced using streptozotocin. In contrast, both ob/ob and db/db mice carry mutations related to the leptin pathway. The mutation in ob/ob mice prevents them from producing leptin, while db/db mice have a mutation in the leptin receptor that renders them insensitive to leptin. Both mutations lead to obesity and the development of Type 2 diabetes [36]. Db/db mice are often preferred for research because their lack of functional leptin receptors results in a Type 2 diabetic phenotype that closely resembles that of humans [37,38]. Once the rats or mice are induced into a Type 2 diabetes model using either method, they can be studied for various types of diabetic foot ulcers.
- The Ischemic animal ulcer model
This model is created by resecting or ligating the femoral artery in mice, resulting in acute severe necrosis of the artery. This method utilizes Laser Doppler measurements and is primarily used to differentiate the ischemic model, serving as a reference in ischemic model trials for therapy [39,40].
- The Neuropathic animal ulcer model
In this method, the sciatic nerves of the mice are clamped with hemostatic forceps approximately 0.5 cm above the site of nerve bifurcation. The clamping is maintained for about one minute, after which the area is sutured. Ulcers are then observed in the right foot after seven days. The detection of diabetic neuropathy is performed using the single filament test and paw retraction test, following hot and cold stimulation [41,42].
- Diabetic Ulcer Model
Infection in diabetic foot ulcers is primarily caused by MRSA (methicillin resistant staph aureus) (is there a source for this) . The method for generating an infected diabetic foot ulcer (DFU) mouse model involves anesthetizing the mouse followed by applying approximately 10 µL of a microbial suspension to the hind limb of diabetic mice. Similarly, models of Pseudomonas aeruginosa induced DFU can be created by inoculating the mice with bacterial cultures obtained from diabetic foot tissue [44,45,46]. Cultures of Staphylococcus aureus and P. aeruginosa have proven to slow wound healing and produce extensive biofilms in db/db mice [47,48].
- Data Analysis Methodology
Statistical data was collected from patients admitted to the hospital for Type 2 diabetes-related complications associated with diabetic foot ulcers (DFUs). To meet the inclusion criteria, patients must have diagnostic codes corresponding to the pathophysiology of DFUs, as well as discharge diagnoses and soft tissue lesion classifications, in accordance with the International Classification of Diseases, 10th edition [49].
Key risk factors include age, gender, residential area, history of diabetes, and the presence of conditions such as retinopathy, neuropathy, nephropathy, peripheral artery disease, and hypertension. Biological markers such as glycated hemoglobin, white blood cell count, and fibrinogen levels are also recorded. DFU disease progression and severity is diagnosed and categorized following systems such as the Wagner-Meggitt classification [50] or the SINBAD classification (Site, Ischemia, Neuropathy, Bacterial infection, Area, Depth) [51]. Grading systems, including the Society for Vascular Surgery Lower Extremity Threatened Limb Classification [52] and the Saint Elian Wound Score System, may also be utilized [53].
Statistical analysis can be performed using the Statistical Package for Social Sciences (SPSS) by IBM or the Statistical Analysis System (SAS) developed by SAS Institute in North Carolina, USA. This analysis reveals the incidence of biofilm formation in wounds and its impact on wound healing, underscoring the need to understand the biofilm-forming mechanisms of the most common pathogens.
- Biofilm formation
The primary criterion for biofilm formation in the foot of a patient is the presence of exogenous glucose, which is commonly observed in individuals with diabetes mellitus. Elevated glucose levels in the blood upregulate genes responsible for producing extracellular polymeric substances, which are the building blocks found in biofilms [1]. The most common microorganisms found in diabetic foot ulcers are Methicillin-resistant Staphylococcus aureus (MRSA) and Pseudomonas aeruginosa [54]; other organisms include E. coli and K. pneumoniae.
P. aeruginosa produces excessive alginate, which protects S. aureus biofilms from the effects of antibiotics [9]. This overproduction is primarily due to mutations in the mucA gene, which upregulate the alginate-synthesizing operon [10]. Additionally, there is a cooperative interaction between S. aureus and P. aeruginosa, as the production of virulence factors by P. aeruginosa, such as hydrogen cyanide and pyocyanin, along with factors from S. aureus, including alpha-hemolysin and Panton-Valentine leukocidin, enhances the antibiotic resistance of biofilms formed by both organisms [14]. The antibiotic resistance associated with these biofilms are summarized in Table 2.
E. coli and K. pneumoniae biofilms share many genes necessary for biofilm formation. The incidence of glycosuria in diabetes mellitus further facilitates the biofilm formation of E. coli [55]. Moreover, it has been observed that diabetes mellitus often lead to urinary tract infections caused by E. coli [56]. Additionally, the prevailing conditions in diabetes mellitus can reduce the antimicrobial activity of certain molecules like that of the antimicrobial peptide psoriasin [57]. Therefore, diabetes mellitus significantly contributes to biofilm formation which progressively leads to various complications, including diabetic foot ulcers.
- Biofilm formation of gram-positive organisms in Diabetic Foot Ulcer
- Mechanism of Biofilm Formation in Staphylococcus aureus
There are two types of biofilm-forming mechanisms demonstrated by S. aureus. These include the ica gene dependent mechanism of biofilm formation as well as another mechanism that is independent of the involvement of the ica gene.
- Ica mediated biofilm formation
The ica-dependent method of biofilm formation is prevalent in both Staphylococcus epidermidis and Staphylococcus aureus, relying on the overproduction of extracellular polysaccharide adhesins known as PIA (polysaccharide intercellular adhesin) and PNAG (polymeric N-acetylglucosamine) [62]. These polysaccharides are produced by the ica operon, which helps the bacteria evade the host immune system, making it beneficial during the early stages of infection.
The ica operon consists of several components, including the icaAD complex, as well as the icaB and icaC genes. The icaA and icaD genes function synergistically as activators. IcaA, IcaD, and IcaC are transmembrane proteins, while IcaB is a surface-attached protein involved in the deacetylation and transport of PIA or PNAG [63].
Ica-dependent biofilm formation is primarily triggered by high osmotic stress resulting from increased NaCl concentrations, which upregulates the sigma factor B. This, in turn, leads to the upregulation of the SarA protein and subsequently the icaA gene. The IcaA protein acts as an N-acetylglucosaminyl transferase and functions optimally in the presence of IcaD. The IcaAD complex produces a 20-subunit oligomeric peptide chain, while IcaC elongates the peptide chain and translocates it outside the cell [63]. IcaB, a surface-bound protein, deacetylates the peptide chain, generating short peptide chains of poly-β-(1,6)-acetylglucosamine [64].
Several downregulators of the icaADBC operon exist, including TcaR (teicoplanin-associated regulator) and the IcaR repressor protein, which is produced following the upregulation of TcaR by the Spx protein [65]. Another downregulator is the luxS gene, which inhibits biofilm formation by suppressing the icaADBC operon [66]. RsbU is another regulator that upregulates the global stress response regulator sigma B in methicillin-sensitive Staphylococcus epidermidis and represses the icaR gene [67]. Mutations in the Spx protein can lead to upregulation of the icaR gene, resulting in reduced expression of the ica operon; however, RsbU does not exert the same effect in S. aureus [68].
Spx is cleaved proteolytically by the ClpXP enzyme in methicillin-sensitive Staphylococcus aureus (MSSA), and any mutations in this enzyme can impair biofilm formation [69]. Accessory components, such as the ArlRS protein, also play a role by upregulating icaADBC and suppressing icaR, aiding in the autolysis of older cells and promoting biofilm formation. Ica mediated biofilm formation is depicted in Figure 2.
- Ica independent Biofilm formation in MRSA
Alternatively, biofilms can be synthesized without the use of the ica operon. This type of biofilm formation is the typical mechanism for MRSA. However, the SarA gene remains essential, as it activates the agr gene set in MRSA [70]. Deletion of the ica genes does not affect the ability of S. aureus to form biofilms, as first demonstrated by the University of Arkansas for Medical Sciences [71]. MRSA possesses a gene encoding penicillin-binding proteins, specifically the mecA gene [72]. The main operon responsible for biofilm formation in MRSA is the agrBDCA operon, which is transcribed by RNA III [73].
Interestingly, both MRSA and MSSA strains can switch from PIA-dependent to PIA-independent biofilm formation in environments with high glucose concentrations [74,75]. The LPXTG proteins, such as Sortase A [76], along with fnbA and fnbB, are necessary for inducing biofilm formation in glucose-rich environments [2].
The peptide precursor autoinducer protein, AgrD, is cleaved by AgrB, a membrane-bound peptidase. This cleavage produces an eight-residue peptide chain, with a thiolactone ring formed by the last five residues. This peptide then phosphorylates AgrC, a membrane-bound histidine kinase, which undergoes autophosphorylation in the presence of the autoinducer molecule. This autophosphorylation relays a signal to AgrA, which binds to the promoters P2 and P3, initiating the upregulation of agrBDCA and subsequently increasing RNA III production, which produces the delta toxin [73].
A key component of MRSA biofilm generation is the release of extracellular DNA upon cellular lysis by CidA hydrolases, which occurs independently of PIA [77]. MRSA also produces the autolysin Atl [78], which aids in biofilm formation and promotes intercellular aggregation with a specific phenotype associated with fnbA and fnbB [79].
Therefore, it can be inferred that even if a diabetic foot ulcer patient reduces their sugar intake, a high salt concentration in the blood may prompt MRSA to switch from ica-independent to ica-dependent biofilm formation. Conversely, high sugar concentrations and low salt concentrations may lead to the opposite effect [80]. Consequently, a comprehensive approach, including proper diet and treatment, is crucial for the complete recovery of the patient. Non-ica-mediated biofilm formation is demonstrated in Figure 3.
- Biofilm formation of Gram-negative organisms in Diabetic Foot Ulcer
- Mechanism of Biofilm Formation in Pseudomonas aeruginosa
Recent studies indicate that the presence of glucose significantly enhances the biofilm formation of Pseudomonas aeruginosa. This upregulation is primarily dependent on the extracellular polymeric substance PslA, which is produced during glucose metabolism. PslA is a branched pentasaccharide composed of D-glucose, D-mannose, and D-rhamnose in a ratio of 3:1:1 [81]. Notably, glucose selectively increases the concentration of PslA without affecting the synthesis of the pel and alg genes. Furthermore, exogenous glucose has been shown to stimulate Ofloxacin resistance in Pseudomonas aeruginosa [1].
Biofilm formation occurs through three primary pathways: las, rhl, and pqs systems. The las system requires 3-oxo-C12 homoserine lactone, while the rhl system depends on C4 homoserine lactone. The pqs system involves the interaction of acyl homoserine lactones and quinolone molecules [82]. C12 and C4 HSL are synthesized via a lactonization process, which is directly linked to the increase in cell population. As the cell count rises, the threshold for C12 HSL is reached, allowing the LasR receptor protein to bind to C12 HSL and form the LasR-C12 HSL complex. This complex initiates the transcription of las genes and expresses pslA-L genes, leading to the production of more extracellular polymeric substances in the form of PslA [83].
The LasR-C12 HSL complex also stimulates the rhlR gene, resulting in the production of the RhlR receptor protein. When the C4 HSL threshold is met, the RhlR-C4 HSL complex forms and further transcribes the rhl genes. This complex additionally transcribes the pelA-G genes, which produce extracellular polymeric substances. The rhl genes encode rhamnolipids, pyocyanin, hydrogen cyanide, Lectins A and B, exoenzymes A and B, and proteins involved in twitching and swarming motility [84]. Moreover, the LasR-C12 HSL complex promotes the pqs system and releases extracellular DNA through the autolysis of older cells [85].
Biofilm regulation is mediated by the GacS/A system. GacS functions as a sensor kinase that, upon autophosphorylation, transfers a phosphate group to GacA, which upregulates small regulatory RNAs such as RsmZ and RsmY [86]. These RNAs bind to unbound RsmA, thereby reducing its repression of autoinducer formation. The RetS/LadS system also plays a role in determining the phosphorylating state of GacS, influencing AHL production, biofilm formation, and the production of virulence factors [88]. This regulatory system can lead to both chronic infections and acute infections in patients.
In patients with diabetic foot ulcers, elevated blood glucose levels contribute to the production of PslA through increased transcription of the pslA gene [83]. High glucose concentrations promote PslA formation, which subsequently regulates the production of cyclic di-GMP by combining two molecules of guanosine triphosphate via diguanylate cyclase. Therefore, PslA acts as a signaling molecule for cyclic di-GMP production [89]. Cyclic di-GMP further stimulates the production of alg and pel genes, which generate Alg44 and PelD, both of which are extracellular polymeric substances. Conversely, when cyclic di-GMP levels are low, bacterial dispersion occurs, disrupting the biofilm, a scenario that may arise with low glucose concentrations (Figure 4).
- Mechanism of Biofilm Formation in E.coli_
Initial adhesion is mediated by genes such as fli, flg, and flh. At this stage, the bacteria are in a planktonic state. The flhDC gene encodes proteins for the structural assembly of flagella, representing the class 1 gene set [90,91,92]. The class 2 gene set includes the fli gene set, which is related to the formation of the basal body and hook, comprising flgAMN, fliFGHIJK, fliMNOPQR, fliE, flgBCDEFGHIJ, fliAZY, and flhBAE [90]. The class 3 gene set includes motAB-cheAW, fliDST, flgKL, fliC, tar-tap-cheRBYZ, and flgMN. The expression of these gene sets is regulated by the class 2 gene sets and is important to produce chemotactic signals and flagellar filaments [90].
Irreversible attachment is facilitated by fimbriae-encoding genes, specifically the fim genes, which include the fimAICDFGH gene sets. Here, FimA serves as the major subunit forming the rod of type 1 fimbriae, FimC acts as a chaperone that binds to the SecYEG channel, FimD is the translocon subunit, and FimF, FimG, and FimH are located at the tips of the fimbriae [93]. FimH binds to the lectin domain of eukaryotic cells, allowing E. coli to adhere to eukaryotic cell surfaces and other abiotic components [94]. FimI acts as the terminator component of this type of fimbriae [95]. Additionally, biofilm formation is mediated through the production of curli fimbriae by the csgBAC operon, which generates fibrous components of the fimbriae, and csgDEFG, where CsgD is the regulatory protein and produces cellulose, while CsgEG functions as the transport protein [96,97]. Antigen 43, encoded by the flu genes, also contributes to cellular aggregation alongside AidA and TibA proteins, promoting biofilm formation [98].
Maturation occurs with the production of poly-β-1,6-N-acetyl-D-glucosamine (PGA) polymer and cellulose. PGA is synthesized by the PgaC glycosyltransferase encoded by the pgaABCD operon [100,101]. Cellulose is synthesized by the bcsABZC operon, which encodes BcsA cellulose synthase, contributing to the rigidity of the biofilm [102]. Cyclic di-GMP inhibits flagellar movement by increasing the production of the YgcR protein. Conversely, PdeH proteins inactivate YgcR, reducing the concentration of cyclic di-GMP and increasing flagellar movement [103]. The two-component system also facilitates the production of poly-β-1,6-N-acetyl-D-glucosamine through the CpxAR complex, which downregulates curli fimbriae production by activating OmpC, inhibiting flagellar motion and thereby promoting biofilm formation [104,105]. The EnvZ/OmpR system primarily functions to inhibit flagellar motion [106]. The RcsCDB proteins regulate the synthesis of colanic acid and inhibit the flhDC operon [107].
Biofilm formation is also significantly influenced by the production of autoinducer 2 (AI-2), a furanosyl borate diester, as E. coli cannot produce autoinducer 1. E. coli encodes an AI-1 sensor through the sdiA gene, which is a luxR homolog. AI-2 is produced by the luxS gene, pumped out of the cell, and then taken up by the LsrABCD proteins and ABC transporters. LsrK kinase subsequently phosphorylates AI-2, with LsrK being repressed by LsrR, which enhances the uptake of the AI-2 molecule [108].
- Mechanism of Biofilm Formation in Klebsiella pneumoniae
Klebsiella pneumoniae shares similarities with Escherichia coli in that it cannot produce autoinducer 1 (AI-1) molecules. Instead, it possesses the sdiA gene, which produces the SdiA protein, a homolog of the LuxR protein that senses AI-1 molecules from other bacteria [109]. AI-2 is produced through a LuxS synthase-dependent mechanism, as observed in E. coli [110]. This system is crucial for biofilm formation and lipopolysaccharide (LPS) generation in K. pneumoniae. The SdiA protein also represses fimbriae production, thereby reducing biofilm formation. Specifically, the genes sdiA, fimK, and kpfR promote type 1 fimbriae formation; fimK reduces cyclic di-GMP levels, and lower cyclic di-GMP concentrations decrease type 3 fimbriae formation, ultimately reducing biofilm formation [111,112]. The mrkH and mrkI genes are essential for inducing biofilm formation, as they encode type 3 fimbriae [113]. The wbbM gene is responsible for LPS formation, the wzm gene is involved in transport, and the wcaG gene contributes to the cell surface properties.
MrkH and MrkI can actively function in the presence of cyclic di-GMP due to their cyclic di-GMP binding domains. In this context, MrkH activates MrkA, which induces type 3 fimbriae formation [116]. The IcsR protein acts as a repressor within this system [113,117].
- Pathogenesis of Biofilm-Infected Diabetic Foot Ulcers (DFUs)
As biofilm-forming cells continue to produce extracellular polysaccharides, the efficacy of antibiotics is significantly limited [118]. Biofilms are inherently impermeable to antibiotics, and the presence of efflux pumps in these cells further counters antibiotic entry [119]. The formation of biofilms enhances the capacity for horizontal gene transfer, thereby increasing the likelihood of acquiring virulence genes [11]. This process contributes to antimicrobial resistance, complicating treatment efforts and allowing infections to progress from lower-grade ulcers to higher-grade ones. Higher-grade ulcers correlate with increased gangrene spread and a greater likelihood of amputation [16].
In severe cases where treatment fails to eradicate the infection, amputation becomes a last resort to prevent the disease from spreading to other body regions. Patients with Wagner Grade 5 ulcers are typically advised to undergo amputation of the necrotic foot region, which may involve below-knee, above-knee, or hip amputations [18]. The rate of amputation in diabetic foot infections ranges from 14% to 24% [120]. For Wagner Grade 4 ulcers, the need for amputation depends on the site and condition of the gangrene. In cases of Grade 3 ulcers and below, efforts should focus on treating the infection and preventing amputation [121].
- Treatment of Biofilm in DFU
There are various treatment options for addressing wounds affected by biofilms, including conventional methods, alternative therapies, and anti-biofilm agents. These different options target different aspects of the biofilm are illustrated in Table 4 and Figure 5.
- Conventional Treatment
The conventional treatment for methicillin-resistant Staphylococcus aureus (MRSA) is vancomycin. However, due to the widespread resistance observed in S. aureus [122,123], linezolid is often prescribed for MRSA infections. Linezolid is an oxazolidinone effective against both methicillin-resistant and vancomycin-resistant S. aureus, as well as against streptococci and enterococci. Other treatment options include combinations of piperacillin and tazobactam, ticarcillin and clavulanic acid, or ampicillin and sulbactam, which are broad-spectrum antibiotics used for moderate to severe infections. Carbapenem antibiotics are commonly used for treating multidrug-resistant Gram-negative organisms. However, due to extensive resistance to these drugs, treatment guidelines now recommend clindamycin combined with piperacillin-tazobactam or cefoperazone-sulbactam [125]. Other options include cefepime with amikacin, imipenem, gentamicin, and tazobactam [126].
Debridement is another effective method for removing bacterial biofilms [127]. Active debridement involves removing necrotic tissue around the wound using either a scalpel or hydro-surgical debridement with a jet spray of water [128]. Topical antimicrobial solutions, such as 10% povidone-iodine, are effective in eradicating bacterial biofilms [129]. Cadexomer iodine, encapsulated in small polysaccharide beads, can also effectively reduce biofilm formation [130,131].
- Alternative Treatment Methods
A novel treatment for diabetic foot ulcers is the use of sono-catalytically activated C₃N₄ sheets powered by ultrasonic waves. This method utilizes NADH present in cells to generate H₂, which oxidizes the cells and perforates their membranes. The resulting NADH deficiency impairs the electron transport chain, reducing cellular respiration and ATP production, ultimately leading to cellular lysis and biofilm degradation. These sheets can be applied to the wound area for up to 15 days [132].
Photodynamic therapy using toluidine blue-chitosan coated gold-silver nanoparticles has been shown to effectively eradicate polymicrobial biofilms of P. aeruginosa and S. aureus [133]. This treatment works primarily through the generation of reactive oxygen species [134], making it effective against multidrug-resistant organisms that form biofilms.
Surfactin-associated antibiotic treatment is a new approach that has shown promise. It has been observed that traditional antibiotics often fail due to antimicrobial resistance, such as that seen in MRSA. However, when surfactin is used, oxacillin can effectively combat MRSA. It is hypothesized that surfactin downregulates the expression of the icaADBC operon [135]. Surfactin also inhibits the expression of SortaseA, preventing the production of adhesion-promoting proteins like fibronectin proteins FnbA and FnbB, thereby disrupting both ica-dependent and ica-independent biofilm formation. Additionally, surfactin makes MRSA sensitive to beta-lactam drugs by inhibiting the expression of mecA gene sets and tetracycline destructase enzymes, allowing for effective eradication of MRSA biofilms when used with oxacillin [136].
Nisin, a 34-amino-acid cationic antibiotic peptide, disrupts bacterial cell walls by interacting with the cell wall precursor lipid II. This interaction alters the electrostatic potential of the transmembrane domain, increasing cell wall permeability. Consequently, EDTA and other antibiotics can enter the cytoplasm and induce cellular lysis. Nisin Z, a variant with an asparagine residue at position 27, has shown inhibitory effects against dual cultures of S. aureus and P. aeruginosa when combined with EDTA (0.4% or 4000 µg/ml), exhibiting both bactericidal and antibiofilm effects [137]. When Nisin Z is embedded in a polyelectrolyte membrane, complete eradication of MRSA biofilms is observed [138].
Silver sulfadiazine is another treatment option that inhibits the electron transport chain in bacteria, thereby obstructing respiration. This makes it suitable for patients with extensive bacterial biofilm-mediated infections [139]. Silver also disrupts bacterial replication and transcription by binding to DNA [140]. It has been shown to be effective against planktonic forms of S. aureus and P. aeruginosa, with P. aeruginosa being particularly sensitive. Consequently, silver sulfadiazine can be administered to patients with diabetic foot ulcers to eradicate biofilms present in the wound [141].
- Other Anti-Biofilm Agents
Ginkgo biloba extracts, along with ginkgolic acid, are effective anti-adhesion agents that prevent the binding of bacteria such as E. coli O157. They also suppress the expression of curli genes and disrupt fimbriae-producing genes [142,143]. Eugenol independently inhibits csgABDFG and fimCDH, thereby preventing adhesion by E. coli. Phloretin is another anti-adhesion molecule that inhibits the expression of lsrACDBF in E. coli, which is essential for the uptake of autoinducer 2 molecules. It also suppresses curli genes csgA and csgB and prevents the expression of toxin genes hlyE and stx2 [142,144].
AMP 108 is a synthetic peptide that represses alarmone signals in bacteria such as Acinetobacter baumannii, P. aeruginosa, K. pneumoniae, and S. aureus. This peptide is effective in eradicating mature biofilms, as the absence of the ppGpp alarmone signal reduces antibiotic resistance, virulence, and disrupts the biofilm-forming capacity of the bacteria [145,146,147].

Table 4.
Treatment strategies for targeting biofilms in DFI.
| Treatment | Description | References |
|---|---|---|
|
Vancomycin Powder Bolus |
Large initial antibiotic concentration Concentration of Antibiotics decreases steadily and rapidly dropping below detectable levels (Rapid washout) No Zone of inhibition Potential side effect:
|
[219,221] |
| Calcium sulphate beads with PMMA loaded space (Vancomycin) |
Greater area under the concentration-time curve (AUC) compared to antibiotics-loaded PMMA space alone. Excessive wound drainage Potentially cytotoxic |
[221] |
| Tobramycin powder bolus | Large initial antibiotic concentration | [221] |
| Calcium sulphate beads with PMMA loaded space (Tobramycin) |
Greater area under the concentration-time curve (AUC) compared to antibiotics loaded PMMA space alone Largest concentration of antibiotics Potentially cytotoxic |
[221] |
| 26% (26 percent degree of substitution of the quaternary ammonium) HACC- loaded PMMA | Cytotoxic and interferes with proliferation and osteogenic differentiation of human bone marrow-derived mesenchymal stem cells with increasing degree of substitution of the quaternary ammonium. Potential obstacle:
|
[222] |
| Gentamicin-loaded PMMA |
|
[222] |
| Limonene |
|
[223] |
| Silver nanoparticle functionalized silicone elastomer |
|
[224] |
| Cold atmospheric plasma | Break peptidoglycan bond of gram-positive bacteria in biofilm | [225] |
| Phage lysins |
|
[226] |
| Mannosidase and glucanasa | Hydrolyses mannan-glucan in C.auris biofilm | [226] |
| Alginate lyase | Removes exopolysaccharide from the surface promoting biofilm eradication | [226] |
| Magnetic iron oxide nanoparticles with magnet fields | Causes mechanical damage to the matrix of the biofilm leading to eradication. | [226] |
| Magneto-responsive gallium-based liquid metal droplet | Disrupts matrix, results in bacterial lysis | [226] |
| DNase |
|
[226] |
| Carolacton |
|
[226] |
| Rhamnolipid | Disrupts and eradicate S. aureus biofilms | [226] |
| D-amino acids incorporation |
Potential side effect:
|
[226,227] |
| Rhamnolipid coated silver and iron oxide nanoparticles |
|
[226,228] |
| Linezolid | Targets Methicillin resistant S.aureus, Streptococci, Enterococci | [122,123] |
| Piperacillin /Tazobactam, Ticarcillin/ Clavulanic acid, Ampicillin/ Sulbactam |
Targets both gram-negative and gram-positive bacteria | [124,125] |
| Active Debridement | Removes necrotic tissue containing bacterial biofilms | [127,128] |
| Iodine solutions (Povidone Iodine 10%, Cadexomer Iodine) | Targets bacterial biofilms | [129,130,131] |
| Sono-catalytically activated C3N4 | Eradication of all types of bacterial biofilms and planktonic cells |
[132] |
| Photodynamic Therapy by Toluidine blue-chitosan coated Gold-Silver nano particles | Eradication of polymicrobial biofilms of P.aeruginosa and S.aureus |
[133,134] |
| Surfactin mediated Oxacillin treatment | Specifically targets S.aureus biofilms and cells (both multi-drug resistant and sensitive) | [135,136] |
| Nisin-EDTA mediated Treatment | Targets polymicrobial biofilms of S.aureus and P.aeruginosa | [137,138] |
| Silver Sulfadiazine | Effective for both S.aureus and P.aeruginosa (P.aeruginosa is more sensitive) | [139,140,141] |
| AMP 108 | Eradicates biofilms of A.baumanii , P.aeruginosa , K.pneumoniae ,S.aureus | [145,146,147] |
| Gingkgo biloba extract | Inhibits adhesion and curli genes of E.coli | [142,143] |
| Eugenol | Inhibits adhesion of E.coli | [142] |
| Phloretin | Inhibits adhesion of E.coli | [142,144] |
The inhibition and eradication of biofilm in foot ulcers is effective with a combination therapy rather than a single drug therapy to properly and effectively treat the chronicity of diabetic foot ulcers caused by bacterial/fungal/mixed biofilm formation.
Conclusions
Diabetic foot ulcers (DFUs) necessitate careful management due to the complexities associated with their treatment. Microorganisms such as Staphylococcus aureus and Pseudomonas aeruginosa are significant biofilm formers, creating protective biofilms that obstruct the penetration of antibiotics and leukocytes into the wound. The extracellular polymeric substance (EPS) produced by these biofilms diminishes the phagocytic activity of leukocytes. Additionally, these organisms engage in horizontal gene transfer, which increases antimicrobial resistance by facilitating the transfer of resistance genes among them. This biofilm formation represents a major challenge in the treatment of DFUs.
Adherence to prescribed antibiotic regimens is crucial, as any deviation must be avoided. This review has detailed the significance of biofilms in DFUs, methods for detecting biofilms, and various treatment options. Recent advancements, such as the use of placental-derived materials and human umbilical cord allografts, offer promising treatments with little to no side effects.
It is critical to note that if a low-grade ulcer progresses to a high-grade ulcer, amputation may be necessary [18]. While lower-grade ulcers can often be treated conservatively, there remains a 25-50% chance of amputation for patients with DFUs [25], alongside a 50-60% infection rate [13]. Therefore, optimal care is essential, including routine hospital visits and debridement of necrotic tissue [128].
Proper wound care, including the use of iodine solutions, can effectively reduce polymicrobial biofilm formation [130,131]. Maintaining an oxygenated environment is vital to prevent the growth of anaerobic bacteria. A clean, hygienic setting, combined with appropriate treatments, will facilitate faster recovery. The primary goal of DFU treatment is to eradicate polymicrobial biofilms, as their presence significantly delays wound healing [58]. Consequently, direct approaches to eliminate biofilms in diabetic foot ulcers are essential for effective treatment and improved patient outcomes.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Conflicts of Interest
The authors declare no conflict of interest exists.
References
- P. She et al., ‘Effects of exogenous glucose on Pseudomonas aeruginosa biofilm formation and antibiotic resistance’, MicrobiologyOpen, vol. 8, no. 12, p. e933, Dec. 2019. [CrossRef]
- E. O’Neill et al., ‘A Novel Staphylococcus aureus Biofilm Phenotype Mediated by the Fibronectin-Binding Proteins, FnBPA and FnBPB’, J. Bacteriol., vol. 190, no. 11, pp. 3835–3850, Jun. 2008. [CrossRef]
- T. Bjarnsholt, ‘The role of bacterial biofilms in chronic infections’, APMIS, vol. 121, no. s136, pp. 1–58, May 2013. [CrossRef]
- L. Hall-Stoodley, J. W. Costerton, and P. Stoodley, ‘Bacterial biofilms: from the Natural environment to infectious diseases’, Nat. Rev. Microbiol., vol. 2, no. 2, pp. 95–108, Feb. 2004. [CrossRef]
- T.-F. C. Mah and G. A. O’Toole, ‘Mechanisms of biofilm resistance to antimicrobial agents’, Trends Microbiol., vol. 9, no. 1, pp. 34–39, Jan. 2001. [CrossRef]
- J. Hirschfeld, ‘Dynamic interactions of neutrophils and biofilms’, J. Oral Microbiol., vol. 6, no. 1, p. 26102, Jan. 2014. [CrossRef]
- K. K. Jefferson, ‘What drives bacteria to produce a biofilm?’, FEMS Microbiol. Lett., vol. 236, no. 2, pp. 163–173, Jul. 2004. [CrossRef]
- C. Mottola, J. J. Mendes, J. M. Cristino, P. Cavaco-Silva, L. Tavares, and M. Oliveira, ‘Polymicrobial biofilms by diabetic foot clinical isolates’, Folia Microbiol. (Praha), vol. 61, no. 1, pp. 35–43, Jan. 2016. [CrossRef]
- C. E. Price, D. G. Brown, D. H. Limoli, V. V. Phelan, and G. A. O’Toole, ‘Exogenous Alginate Protects Staphylococcus aureus from Killing by Pseudomonas aeruginosa’, J. Bacteriol., vol. 202, no. 8, Mar. 2020. [CrossRef]
- D. H. Limoli et al., ‘Pseudomonas aeruginosa Alginate Overproduction Promotes Coexistence with Staphylococcus aureus in a Model of Cystic Fibrosis Respiratory Infection’, mBio, vol. 8, no. 2, pp. e00186-17, May 2017. [CrossRef]
- M. Berlanga and R. Guerrero, ‘Living together in biofilms: the microbial cell factory and its biotechnological implications’, Microb. Cell Factories, vol. 15, no. 1, p. 165, Dec. 2016. [CrossRef]
- A. Banu, M. M. N. Hassan, J. Rajkumar, and S. Srinivasa, ‘Spectrum of bacteria associated with diabetic foot ulcer and biofilm formation: A prospective study’, Australas. Med. J., pp. 280–285, Sep. 2015. [CrossRef]
- D. G. Armstrong, A. J. M. Boulton, and S. A. Bus, ‘Diabetic Foot Ulcers and Their Recurrence’, N. Engl. J. Med., vol. 376, no. 24, pp. 2367–2375, Jun. 2017. [CrossRef]
- A. Hotterbeekx, S. Kumar-Singh, H. Goossens, and S. Malhotra-Kumar, ‘In vivo and In vitro Interactions between Pseudomonas aeruginosa and Staphylococcus spp.’, Front. Cell. Infect. Microbiol., vol. 7, Apr. 2017. [CrossRef]
- A. Atlaw, H. B. Kebede, A. A. Abdela, and Y. Woldeamanuel, ‘Bacterial isolates from diabetic foot ulcers and their antimicrobial resistance profile from selected hospitals in Addis Ababa, Ethiopia’, Front. Endocrinol., vol. 13, p. 987487, Aug. 2022. [CrossRef]
- F. W. Wagner, ‘The Dysvascular Foot: A System for Diagnosis and Treatment’, Foot Ankle, vol. 2, no. 2, pp. 64–122, Sep. 1981. [CrossRef]
- S. Vazeille, L. Hawker, R. Chandrasekar, and U. Srinivas-Shankar, ‘Recalcitrant Foot Ulceration in a Patient With Type 1 Diabetes Mellitus’, Cureus, Jun. 2020. [CrossRef]
- X. Liao, S.-H. Li, M. M. El Akkawi, X. Fu, H. Liu, and Y. Huang, ‘Surgical amputation for patients with diabetic foot ulcers: A Chinese expert panel consensus treatment guide’, Front. Surg., vol. 9, p. 1003339, Nov. 2022. [CrossRef]
- N. M. Protzman et al., ‘Placental-Derived Biomaterials and Their Application to Wound Healing: A Review’, Bioengineering, vol. 10, no. 7, p. 829, Jul. 2023. [CrossRef]
- W. Tettelbach et al., ‘A multicentre prospective randomised controlled comparative parallel study of dehydrated human umbilical cord (EpiCord) allograft for the treatment of diabetic foot ulcers’, Int. Wound J., vol. 16, no. 1, pp. 122–130, Feb. 2019. [CrossRef]
- D. Magliano and E. J. Boyko, IDF diabetes atlas, 10th edition. Brussels: International Diabetes Federation, 2021.
- F. Crawford et al., ‘The risk of foot ulceration in people with diabetes screened in community settings: findings from a cohort study’, QJM Int. J. Med., vol. 104, no. 5, pp. 403–410, May 2011. [CrossRef]
- C. A. Abbott et al., ‘The North-West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community-based patient cohort’, Diabet. Med., vol. 19, no. 5, pp. 377–384, May 2002. [CrossRef]
- A. Raghav, Z. A. Khan, R. K. Labala, J. Ahmad, S. Noor, and B. K. Mishra, ‘Financial burden of diabetic foot ulcers to world: a progressive topic to discuss always’, Ther. Adv. Endocrinol. Metab., vol. 9, no. 1, pp. 29–31, Jan. 2018. [CrossRef]
- A. Ibrahim, ‘IDF Clinical Practice Recommendation on the Diabetic Foot: A guide for healthcare professionals’, Diabetes Res. Clin. Pract., vol. 127, pp. 285–287, May 2017. [CrossRef]
- Z. Yekta, Pourali, and M. Ghasemi-rad, ‘Comparison of demographic and clinical characteristics influencing health-related quality of life in patients with diabetic foot ulcers and those without foot ulcers’, Diabetes Metab. Syndr. Obes. Targets Ther., p. 393, Dec. 2011. [CrossRef]
- P. Shah et al., ‘Wagner’s Classification as a Tool for Treating Diabetic Foot Ulcers: Our Observations at a Suburban Teaching Hospital’, Cureus, Jan. 2022. [CrossRef]
- O. Niță et al., ‘Evaluating Classification Systems of Diabetic Foot Ulcer Severity: A 12-Year Retrospective Study on Factors Impacting Survival’, Healthcare, vol. 11, no. 14, p. 2077, Jul. 2023. [CrossRef]
- K. Ottolino-Perry et al., ‘Improved detection of clinically relevant wound bacteria using autofluorescence image-guided sampling in diabetic foot ulcers’, Int. Wound J., vol. 14, no. 5, pp. 833–841, Oct. 2017. [CrossRef]
- N. Høiby et al., ‘ESCMID∗ guideline for the diagnosis and treatment of biofilm infections 2014’, Clin. Microbiol. Infect., vol. 21, pp. S1–S25, May 2015. [CrossRef]
- S. Sugimoto et al., ‘Staphylococcus epidermidis Esp Degrades Specific Proteins Associated with Staphylococcus aureus Biofilm Formation and Host-Pathogen Interaction’, J. Bacteriol., vol. 195, no. 8, pp. 1645–1655, Apr. 2013. [CrossRef]
- T. S. Murali et al., ‘Characteristics of microbial drug resistance and its correlates in chronic diabetic foot ulcer infections’, J. Med. Microbiol., vol. 63, no. 10, pp. 1377–1385, Oct. 2014. [CrossRef]
- D. Kumar, T. Banerjee, J. Chakravarty, S. Singh, A. Dwivedi, and R. Tilak, ‘Identification, antifungal resistance profile, in vitro biofilm formation and ultrastructural characteristics of Candida species isolated from diabetic foot patients in Northern India’, Indian J. Med. Microbiol., vol. 34, no. 3, pp. 308–314, Jul. 2016. [CrossRef]
- C.-C. E. Lan, C.-S. Wu, H.-Y. Kuo, S.-M. Huang, and G.-S. Chen, ‘Hyperglycaemic conditions hamper keratinocyte locomotion via sequential inhibition of distinct pathways: new insights on poor wound closure in patients with diabetes’, Br. J. Dermatol., vol. 160, no. 6, pp. 1206–1214, Jun. 2009. [CrossRef]
- A. G. Maione et al., ‘Altered ECM deposition by diabetic foot ulcer-derived fibroblasts implicates fibronectin in chronic wound repair’, Wound Repair Regen., vol. 24, no. 4, pp. 630–643, Jul. 2016. [CrossRef]
- B. Wang, P. Chandrasekera, and J. Pippin, ‘Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes’, Curr. Diabetes Rev., vol. 10, no. 2, pp. 131–145, May 2014. [CrossRef]
- P. M. Pitale and M. S. Gorbatyuk, ‘Diabetic Retinopathy: From Animal Models to Cellular Signaling’, Int. J. Mol. Sci., vol. 23, no. 3, p. 1487, Jan. 2022. [CrossRef]
- J. Michaels et al., ‘db/db mice exhibit severe wound-healing impairments compared with other murine diabetic strains in a silicone-splinted excisional wound model’, Wound Repair Regen., vol. 15, no. 5, pp. 665–670, Sep. 2007. [CrossRef]
- C.-H. Jung, Y. Y. Cho, D. Choi, B.-Y. Kim, C.-H. Kim, and J.-O. Mok, ‘Relationship of Sarcopenia with Microcirculation Measured by Skin Perfusion Pressure in Patients with Type 2 Diabetes’, Endocrinol. Metab., vol. 35, no. 3, pp. 578–586, Sep. 2020. [CrossRef]
- L. M. Rodrigues, C. Rocha, H. Ferreira, and H. Silva, ‘Different lasers reveal different skin microcirculatory flowmotion - data from the wavelet transform analysis of human hindlimb perfusion’, Sci. Rep., vol. 9, no. 1, p. 16951, Nov. 2019. [CrossRef]
- S. A. Cudlip, F. A. Howe, J. R. Griffiths, and B. A. Bell, ‘Magnetic resonance neurography of peripheral nerve following experimental crush injury, and correlation with functional deficit’, J. Neurosurg., vol. 96, no. 4, pp. 755–759, Apr. 2002. [CrossRef]
- S. Jin, M. Zhang, Y. Gao, X. Zhang, G. Cui, and Y. Zhang, ‘The Efficacy of Jing Wan Hong Ointment for Nerve Injury Diabetic Foot Ulcer and Its Mechanisms’, J. Diabetes Res., vol. 2014, pp. 1–9, 2014. [CrossRef]
- J. Rich and J. C. Lee, ‘The Pathogenesis of Staphylococcus aureus Infection in the Diabetic NOD Mouse’, Diabetes, vol. 54, no. 10, pp. 2904–2910, Oct. 2005. [CrossRef]
- M. Y. El-Naggar, Y. M. Gohar, M. A. Sorour, and M. G. Waheeb, ‘Hydrogel Dressing with a Nano-Formula against Methicillin-Resistant Staphylococcus aureus and Pseudomonas aeruginosa Diabetic Foot Bacteria’, J. Microbiol. Biotechnol., vol. 26, no. 2, pp. 408–420, Feb. 2016. [CrossRef]
- J.-P. Rasigade et al., ‘A Prophage in Diabetic Foot Ulcer–Colonizing Staphylococcus aureus Impairs Invasiveness by Limiting Intracellular Growth’, J. Infect. Dis., vol. 214, no. 10, pp. 1605–1608, Nov. 2016. [CrossRef]
- J.-F. Huon et al., ‘Phages versus Antibiotics To Treat Infected Diabetic Wounds in a Mouse Model: a Microbiological and Microbiotic Evaluation’, mSystems, vol. 5, no. 6, pp. e00542-20, Dec. 2020. [CrossRef]
- D. E. Snow et al., ‘The presence of biofilm structures in atherosclerotic plaques of arteries from legs amputated as a complication of diabetic foot ulcers’, J. Wound Care, vol. 25, no. Sup2, pp. S16–S22, Feb. 2016. [CrossRef]
- F. A. Al-Joufi et al., ‘Microbial spectrum, antibiotic susceptibility profile, and biofilm formation of diabetic foot infections (2014–18): a retrospective multicenter analysis’, 3 Biotech, vol. 10, no. 7, p. 325, Jul. 2020. [CrossRef]
- S. J. Steindel, ‘International classification of diseases, 10th edition, clinical modification and procedure coding system: descriptive overview of the next generation HIPAA code sets’, J. Am. Med. Inform. Assoc., vol. 17, no. 3, pp. 274–282, May 2010. [CrossRef]
- Dr. M. Mehraj, ‘A review of Wagner classification and current concepts in management of diabetic foot’, Int. J. Orthop. Sci., vol. 4, no. 1n, pp. 933–935, Jan. 2018. [CrossRef]
- P. Ince et al., ‘Use of the SINBAD Classification System and Score in Comparing Outcome of Foot Ulcer Management on Three Continents’, Diabetes Care, vol. 31, no. 5, pp. 964–967, May 2008. [CrossRef]
- J. L. Mills et al., ‘The Society for Vascular Surgery Lower Extremity Threatened Limb Classification System: Risk stratification based on Wound, Ischemia, and foot Infection (WIfI)’, J. Vasc. Surg., vol. 59, no. 1, pp. 220-234.e2, Jan. 2014. [CrossRef]
- F. R. Martínez-De Jesús, ‘A Checklist System to Score Healing Progress of Diabetic Foot Ulcers’, Int. J. Low. Extrem. Wounds, vol. 9, no. 2, pp. 74–83, Jun. 2010. [CrossRef]
- K. E. Macdonald, S. Boeckh, H. J. Stacey, and J. D. Jones, ‘The microbiology of diabetic foot infections: a meta-analysis’, BMC Infect. Dis., vol. 21, no. 1, p. 770, Dec. 2021. [CrossRef]
- M. J. Islam, K. Bagale, P. P. John, Z. Kurtz, and R. Kulkarni, ‘Glycosuria Alters Uropathogenic Escherichia coli Global Gene Expression and Virulence’, mSphere, vol. 7, no. 3, pp. e00004-22, Jun. 2022. [CrossRef]
- L. Korbel and J. D. Spencer, ‘Diabetes mellitus and infection: an evaluation of hospital utilization and management costs in the United States’, J. Diabetes Complications, vol. 29, no. 2, pp. 192–195, Mar. 2015. [CrossRef]
- S. Mohanty et al., ‘Diabetes downregulates the antimicrobial peptide psoriasin and increases E. coli burden in the urinary bladder’, Nat. Commun., vol. 13, no. 1, p. 4983, Sep. 2022. [CrossRef]
- C. Pouget, C. Dunyach-Remy, A. Pantel, S. Schuldiner, A. Sotto, and J.-P. Lavigne, ‘Biofilms in Diabetic Foot Ulcers: Significance and Clinical Relevance’, Microorganisms, vol. 8, no. 10, p. 1580, Oct. 2020. [CrossRef]
- C. Khadka et al., ‘Extended-spectrum β-lactamases producing Enterobacteriaceae (ESBL-PE) prevalence in Nepal: A systematic review and meta-analysis’, Sci. Total Environ., vol. 901, p. 166164, Nov. 2023. [CrossRef]
- R. Dziri et al., ‘Characterization of extended-spectrum β-lactamase (ESBL)-producing Klebsiella, Enterobacter , and Citrobacter obtained in environmental samples of a Tunisian hospital’, Diagn. Microbiol. Infect. Dis., vol. 86, no. 2, pp. 190–193, Oct. 2016. [CrossRef]
- I. Kyriakidis, E. Vasileiou, Z. D. Pana, and A. Tragiannidis, ‘Acinetobacter baumannii Antibiotic Resistance Mechanisms’, Pathogens, vol. 10, no. 3, p. 373, Mar. 2021. [CrossRef]
- D. Mack et al., ‘The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis’, J. Bacteriol., vol. 178, no. 1, pp. 175–183, Jan. 1996. [CrossRef]
- C. Gerke, A. Kraft, R. Süßmuth, O. Schweitzer, and F. Götz, ‘Characterization of theN-Acetylglucosaminyltransferase Activity Involved in the Biosynthesis of the Staphylococcus epidermidisPolysaccharide Intercellular Adhesin’, J. Biol. Chem., vol. 273, no. 29, pp. 18586–18593, Jul. 1998. [CrossRef]
- C. Vuong et al., ‘A Crucial Role for Exopolysaccharide Modification in Bacterial Biofilm Formation, Immune Evasion, and Virulence’, J. Biol. Chem., vol. 279, no. 52, pp. 54881–54886, Dec. 2004. [CrossRef]
- K. K. Jefferson, D. B. Pier, D. A. Goldmann, and G. B. Pier, ‘The Teicoplanin-Associated Locus Regulator (TcaR) and the Intercellular Adhesin Locus Regulator (IcaR) Are Transcriptional Inhibitors of the ica Locus in Staphylococcus aureus’, J. Bacteriol., vol. 186, no. 8, pp. 2449–2456, Apr. 2004. [CrossRef]
- L. Xu et al., ‘Role of the luxS Quorum-Sensing System in Biofilm Formation and Virulence of Staphylococcus epidermidis’, Infect. Immun., vol. 74, no. 1, pp. 488–496, Jan. 2006. [CrossRef]
- J. K.-M. Knobloch, K. Bartscht, A. Sabottke, H. Rohde, H.-H. Feucht, and D. Mack, ‘Biofilm Formation by Staphylococcus epidermidis Depends on Functional RsbU, an Activator of the sigB Operon: Differential Activation Mechanisms Due to Ethanol and Salt Stress’, J. Bacteriol., vol. 183, no. 8, pp. 2624–2633, Apr. 2001. [CrossRef]
- J. Valle et al., ‘SarA and not σ B is essential for biofilm development by Staphylococcus aureus’, Mol. Microbiol., vol. 48, no. 4, pp. 1075–1087, May 2003. [CrossRef]
- D. Frees et al., ‘Clp ATPases are required for stress tolerance, intracellular replication and biofilm formation in Staphylococcus aureus’, Mol. Microbiol., vol. 54, no. 5, pp. 1445–1462, Dec. 2004. [CrossRef]
- A. L. Cheung and S. J. Projan, ‘Cloning and sequencing of sarA of Staphylococcus aureus, a gene required for the expression of agr’, J. Bacteriol., vol. 176, no. 13, pp. 4168–4172, Jul. 1994. [CrossRef]
- K. E. Beenken et al., ‘Global Gene Expression in Staphylococcus aureus Biofilms’, J. Bacteriol., vol. 186, no. 14, pp. 4665–4684, Jul. 2004. [CrossRef]
- R. H. Deurenberg and E. E. Stobberingh, ‘The evolution of Staphylococcus aureus’, Infect. Genet. Evol., vol. 8, no. 6, pp. 747–763, Dec. 2008. [CrossRef]
- C. Vuong, H. L. Saenz, F. Götz, and M. Otto, ‘Impact of the agr Quorum-Sensing System on Adherence to Polystyrene in Staphylococcus aureus’, J. Infect. Dis., vol. 182, no. 6, pp. 1688–1693, Dec. 2000. [CrossRef]
- E. O’Neill et al., ‘Association between Methicillin Susceptibility and Biofilm Regulation in Staphylococcus aureus Isolates from Device-Related Infections’, J. Clin. Microbiol., vol. 45, no. 5, pp. 1379–1388, May 2007. [CrossRef]
- F. Fitzpatrick, H. Humphreys, and J. P. O’Gara, ‘Environmental regulation of biofilm development in methicillin-resistant and methicillin-susceptible Staphylococcus aureus clinical isolates’, J. Hosp. Infect., vol. 62, no. 1, pp. 120–122, Jan. 2006. [CrossRef]
- N. Merino et al., ‘Protein A-Mediated Multicellular Behavior in Staphylococcus aureus’, J. Bacteriol., vol. 191, no. 3, pp. 832–843, Feb. 2009. [CrossRef]
- K. C. Rice et al., ‘The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus’, Proc. Natl. Acad. Sci., vol. 104, no. 19, pp. 8113–8118, May 2007. [CrossRef]
- J. L. Bose, M. K. Lehman, P. D. Fey, and K. W. Bayles, ‘Contribution of the Staphylococcus aureus Atl AM and GL Murein Hydrolase Activities in Cell Division, Autolysis, and Biofilm Formation’, PLoS ONE, vol. 7, no. 7, p. e42244, Jul. 2012. [CrossRef]
- P. Houston, S. E. Rowe, C. Pozzi, E. M. Waters, and J. P. O’Gara, ‘Essential Role for the Major Autolysin in the Fibronectin-Binding Protein-Mediated Staphylococcus aureus Biofilm Phenotype’, Infect. Immun., vol. 79, no. 3, pp. 1153–1165, Mar. 2011. [CrossRef]
- M. Vergara-Irigaray et al., ‘Relevant Role of Fibronectin-Binding Proteins in Staphylococcus aureus Biofilm-Associated Foreign-Body Infections’, Infect. Immun., vol. 77, no. 9, pp. 3978–3991, Sep. 2009. [CrossRef]
- D. Passos Da Silva et al., ‘The Pseudomonas aeruginosa lectin LecB binds to the exopolysaccharide Psl and stabilizes the biofilm matrix’, Nat. Commun., vol. 10, no. 1, p. 2183, May 2019. [CrossRef]
- P. Nadal Jimenez, G. Koch, J. A. Thompson, K. B. Xavier, R. H. Cool, and W. J. Quax, ‘The Multiple Signaling Systems Regulating Virulence in Pseudomonas aeruginosa’, Microbiol. Mol. Biol. Rev., vol. 76, no. 1, pp. 46–65, Mar. 2012. [CrossRef]
- K. B. Gilbert, T. H. Kim, R. Gupta, E. P. Greenberg, and M. Schuster, ‘Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR’, Mol. Microbiol., vol. 73, no. 6, pp. 1072–1085, Sep. 2009. [CrossRef]
- D. H. Dusane, S. S. Zinjarde, V. P. Venugopalan, R. J. Mclean, M. M. Weber, and P. K. S. M. Rahman, ‘Quorum sensing: implications on Rhamnolipid biosurfactant production’, Biotechnol. Genet. Eng. Rev., vol. 27, no. 1, pp. 159–184, Jan. 2010. [CrossRef]
- M. Allesen-Holm et al., ‘A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms’, Mol. Microbiol., vol. 59, no. 4, pp. 1114–1128, Feb. 2006. [CrossRef]
- A. Brencic et al., ‘The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs’, Mol. Microbiol., vol. 73, no. 3, pp. 434–445, Aug. 2009. [CrossRef]
- Y. Irie, M. Starkey, A. N. Edwards, D. J. Wozniak, T. Romeo, and M. R. Parsek, ‘Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA’, Mol. Microbiol., vol. 78, no. 1, pp. 158–172, Oct. 2010. [CrossRef]
- A. L. Goodman, B. Kulasekara, A. Rietsch, D. Boyd, R. S. Smith, and S. Lory, ‘A Signaling Network Reciprocally Regulates Genes Associated with Acute Infection and Chronic Persistence in Pseudomonas aeruginosa’, Dev. Cell, vol. 7, no. 5, pp. 745–754, Nov. 2004. [CrossRef]
- D.-G. Ha and G. A. O’Toole, ‘c-di-GMP and its Effects on Biofilm Formation and Dispersion: a Pseudomonas Aeruginosa Review’, Microbiol. Spectr., vol. 3, no. 2, p. 3.2.27, Apr. 2015. [CrossRef]
- D. Apel and M. G. Surette, ‘Bringing order to a complex molecular machine: The assembly of the bacterial flagella’, Biochim. Biophys. Acta BBA - Biomembr., vol. 1778, no. 9, pp. 1851–1858, Sep. 2008. [CrossRef]
- R. M. Macnab, ‘GENETICS AND BIOGENESIS OF BACTERIAL FLAGELLA’, Annu. Rev. Genet., vol. 26, no. 1, pp. 131–158, Dec. 1992. [CrossRef]
- D. M. Fitzgerald, R. P. Bonocora, and J. T. Wade, ‘Comprehensive Mapping of the Escherichia coli Flagellar Regulatory Network’, PLoS Genet., vol. 10, no. 10, p. e1004649, Oct. 2014. [CrossRef]
- G. Capitani, O. Eidam, R. Glockshuber, and M. G. Grütter, ‘Structural and functional insights into the assembly of type 1 pili from Escherichia coli’, Microbes Infect., vol. 8, no. 8, pp. 2284–2290, Jul. 2006. [CrossRef]
- C. Beloin, A. Roux, and J.-M. Ghigo, ‘Escherichia coli Biofilms’, in Bacterial Biofilms, vol. 322, T. Romeo, Ed., in Current Topics in Microbiology and Immunology, vol. 322. , Berlin, Heidelberg: Springer Berlin Heidelberg, 2008, pp. 249–289. [CrossRef]
- M. L. Valenski, S. L. Harris, P. A. Spears, J. R. Horton, and P. E. Orndorff, ‘The Product of the fimI Gene Is Necessary for Escherichia coli Type 1 Pilus Biosynthesis’, J. Bacteriol., vol. 185, no. 16, pp. 5007–5011, Aug. 2003. [CrossRef]
- M. M. Barnhart and M. R. Chapman, ‘Curli Biogenesis and Function’, Annu. Rev. Microbiol., vol. 60, no. 1, pp. 131–147, Oct. 2006. [CrossRef]
- L. Gualdi, L. Tagliabue, S. Bertagnoli, T. Ieranò, C. De Castro, and P. Landini, ‘Cellulose modulates biofilm formation by counteracting curli-mediated colonization of solid surfaces in Escherichia coli’, Microbiology, vol. 154, no. 7, pp. 2017–2024, Jul. 2008. [CrossRef]
- P. Klemm and M. Schembri, ‘Type 1 Fimbriae, Curli, and Antigen 43: Adhesion, Colonization, and Biofilm Formation’, EcoSal Plus, vol. 1, no. 1, p. 10.1128/ecosalplus.8.3.2.6, Dec. 2004. [CrossRef]
- L. Laganenka, R. Colin, and V. Sourjik, ‘Chemotaxis towards autoinducer 2 mediates autoaggregation in Escherichia coli’, Nat. Commun., vol. 7, no. 1, p. 12984, Sep. 2016. [CrossRef]
- Y. Itoh et al., ‘Roles of pgaABCD Genes in Synthesis, Modification, and Export of the Escherichia coli Biofilm Adhesin Poly-β-1,6- N -Acetyl- d -Glucosamine’, J. Bacteriol., vol. 190, no. 10, pp. 3670–3680, May 2008. [CrossRef]
- X. Wang, J. F. Preston, and T. Romeo, ‘The pgaABCD Locus of Escherichia coli Promotes the Synthesis of a Polysaccharide Adhesin Required for Biofilm Formation’, J. Bacteriol., vol. 186, no. 9, pp. 2724–2734, May 2004. [CrossRef]
- D. O. Serra, A. M. Richter, and R. Hengge, ‘Cellulose as an Architectural Element in Spatially Structured Escherichia coli Biofilms’, J. Bacteriol., vol. 195, no. 24, pp. 5540–5554, Dec. 2013. [CrossRef]
- U. Römling, M. Y. Galperin, and M. Gomelsky, ‘Cyclic di-GMP: the First 25 Years of a Universal Bacterial Second Messenger’, Microbiol. Mol. Biol. Rev., vol. 77, no. 1, pp. 1–52, Mar. 2013. [CrossRef]
- J. A. Markova, E. V. Anganova, A. L. Turskaya, V. A. Bybin, and E. D. Savilov, ‘Regulation of Escherichia coli Biofilm Formation (Review)’, Appl. Biochem. Microbiol., vol. 54, no. 1, pp. 1–11, Jan. 2018. [CrossRef]
- C. Prigent-Combaret et al., ‘Complex Regulatory Network Controls Initial Adhesion and Biofilm Formation in Escherichia coli via Regulation of the csgD Gene’, J. Bacteriol., vol. 183, no. 24, pp. 7213–7223, Dec. 2001. [CrossRef]
- B. M. Prüß, C. Besemann, A. Denton, and A. J. Wolfe, ‘A Complex Transcription Network Controls the Early Stages of Biofilm Development by Escherichia coli’, J. Bacteriol., vol. 188, no. 11, pp. 3731–3739, Jun. 2006. [CrossRef]
- N. Majdalani and S. Gottesman, ‘THE RCS PHOSPHORELAY: A Complex Signal Transduction System’, Annu. Rev. Microbiol., vol. 59, no. 1, pp. 379–405, Oct. 2005. [CrossRef]
- C. S. Pereira, J. A. Thompson, and K. B. Xavier, ‘AI-2-mediated signalling in bacteria’, FEMS Microbiol. Rev., vol. 37, no. 2, pp. 156–181, Mar. 2013. [CrossRef]
- T. Pacheco et al., ‘SdiA, a Quorum-Sensing Regulator, Suppresses Fimbriae Expression, Biofilm Formation, and Quorum-Sensing Signaling Molecules Production in Klebsiella pneumoniae’, Front. Microbiol., vol. 12, p. 597735, Jun. 2021. [CrossRef]
- C. De Araujo, D. Balestrino, L. Roth, N. Charbonnel, and C. Forestier, ‘Quorum sensing affects biofilm formation through lipopolysaccharide synthesis in Klebsiella pneumoniae’, Res. Microbiol., vol. 161, no. 7, pp. 595–603, Sep. 2010. [CrossRef]
- J. G. Johnson and S. Clegg, ‘Role of MrkJ, a Phosphodiesterase, in Type 3 Fimbrial Expression and Biofilm Formation in Klebsiella pneumoniae’, J. Bacteriol., vol. 192, no. 15, pp. 3944–3950, Aug. 2010. [CrossRef]
- D. A. Rosen, J. S. Pinkner, J. M. Jones, J. N. Walker, S. Clegg, and S. J. Hultgren, ‘Utilization of an Intracellular Bacterial Community Pathway in Klebsiella pneumoniae Urinary Tract Infection and the Effects of FimK on Type 1 Pilus Expression’, Infect. Immun., vol. 76, no. 7, pp. 3337–3345, Jul. 2008. [CrossRef]
- J. G. Johnson, C. N. Murphy, J. Sippy, T. J. Johnson, and S. Clegg, ‘Type 3 Fimbriae and Biofilm Formation Are Regulated by the Transcriptional Regulators MrkHI in Klebsiella pneumoniae’, J. Bacteriol., vol. 193, no. 14, pp. 3453–3460, Jul. 2011. [CrossRef]
- J. Zheng et al., ‘Biofilm Formation in Klebsiella pneumoniae Bacteremia Strains Was Found to be Associated with CC23 and the Presence of wcaG’, Front. Cell. Infect. Microbiol., vol. 8, p. 21, Feb. 2018. [CrossRef]
- C. Vuotto et al., ‘Biofilm formation and antibiotic resistance in Klebsiella pneumoniae urinary strains’, J. Appl. Microbiol., vol. 123, no. 4, pp. 1003–1018, Oct. 2017. [CrossRef]
- J. J. Wilksch et al., ‘MrkH, a Novel c-di-GMP-Dependent Transcriptional Activator, Controls Klebsiella pneumoniae Biofilm Formation by Regulating Type 3 Fimbriae Expression’, PLoS Pathog., vol. 7, no. 8, p. e1002204, Aug. 2011. [CrossRef]
- T.-H. Lin, C.-Y. Tseng, Y.-C. Lai, C.-C. Wu, C.-F. Huang, and C.-T. Lin, ‘IscR Regulation of Type 3 Fimbriae Expression in Klebsiella pneumoniae CG43’, Front. Microbiol., vol. 8, p. 1984, Oct. 2017. [CrossRef]
- J. F. González, M. M. Hahn, and J. S. Gunn, ‘Chronic biofilm-based infections: skewing of the immune response’, Pathog. Dis., vol. 76, no. 3, Apr. 2018. [CrossRef]
- I. Alav, J. M. Sutton, and K. M. Rahman, ‘Role of bacterial efflux pumps in biofilm formation’, J. Antimicrob. Chemother., vol. 73, no. 8, pp. 2003–2020, Aug. 2018. [CrossRef]
- Z. Yekta, Pourali, M. Ghasemi-rad, Ravanyar, and Nezhadrahim, ‘Clinical and behavioral factors associated with management outcome in hospitalized patients with diabetic foot ulcer’, Diabetes Metab. Syndr. Obes. Targets Ther., p. 371, Oct. 2011. [CrossRef]
- J. V. Eggert, E. R. Worth, and C. C. Van Gils, ‘Cost and mortality data of a regional limb salvage and hyperbaric medicine program for Wagner Grade 3 or 4 diabetic foot ulcers’, Undersea Hyperb. Med. J. Undersea Hyperb. Med. Soc. Inc, vol. 43, no. 1, pp. 1–8, 2016.
- A. Shariati, M. Dadashi, M. T. Moghadam, A. Van Belkum, S. Yaslianifard, and D. Darban-Sarokhalil, ‘Global prevalence and distribution of vancomycin resistant, vancomycin intermediate and heterogeneously vancomycin intermediate Staphylococcus aureus clinical isolates: a systematic review and meta-analysis’, Sci. Rep., vol. 10, no. 1, p. 12689, Jul. 2020. [CrossRef]
- B. P. Howden, J. K. Davies, P. D. R. Johnson, T. P. Stinear, and M. L. Grayson, ‘Reduced Vancomycin Susceptibility in Staphylococcus aureus , Including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: Resistance Mechanisms, Laboratory Detection, and Clinical Implications’, Clin. Microbiol. Rev., vol. 23, no. 1, pp. 99–139, Jan. 2010. [CrossRef]
- K. Bush and P. A. Bradford, ‘β-Lactams and β-Lactamase Inhibitors: An Overview’, Cold Spring Harb. Perspect. Med., vol. 6, no. 8, p. a025247, Aug. 2016. [CrossRef]
- M. Karmaker, S. K. Sanyal, M. Sultana, and M. A. Hossain, ‘Association of bacteria in diabetic and non-diabetic foot infection – An investigation in patients from Bangladesh’, J. Infect. Public Health, vol. 9, no. 3, pp. 267–277, May 2016. [CrossRef]
- W. D. Aumiller and H. A. Dollahite, ‘Pathogenesis and management of diabetic foot ulcers’, J. Am. Acad. Physician Assist., vol. 28, no. 5, pp. 28–34, May 2015. [CrossRef]
- D. G. Armstrong, L. A. Lavery, B. P. Nixon, and A. J. M. Boulton, ‘It’s Not What You Put On, but What You Take Off: Techniques for Debriding and Off-Loading the Diabetic Foot Wound’, Clin. Infect. Dis., vol. 39, no. Supplement_2, pp. S92–S99, Aug. 2004. [CrossRef]
- K. A. Tayeb, S. D. Bateman, S. Hampton, M. Malone, M. Malone, and J. Fletcher, ‘Managing infection: a holistic approach’, J. Wound Care, vol. 24, no. Sup5b, pp. 20–30, May 2015. [CrossRef]
- L. Gryson, S. Meaume, I. Feldkaemper, and F. Favalli, ‘Anti-biofilm Activity of Povidone-Iodine and Polyhexamethylene Biguanide: Evidence from In Vitro Tests’, Curr. Microbiol., vol. 80, no. 5, p. 161, May 2023. [CrossRef]
- J. A. Schwartz, J. C. Lantis, C. Gendics, A. M. Fuller, W. Payne, and D. Ochs, ‘A prospective, non comparative, multicenter study to investigate the effect of cadexomer iodine on bioburden load and other wound characteristics in diabetic foot ulcers’, Int. Wound J., vol. 10, no. 2, pp. 193–199, Apr. 2013. [CrossRef]
- M. Malone et al., ‘Effect of cadexomer iodine on the microbial load and diversity of chronic non-healing diabetic foot ulcers complicated by biofilm in vivo’, J. Antimicrob. Chemother., vol. 72, no. 7, pp. 2093–2101, Jul. 2017. [CrossRef]
- Q. Xu et al., ‘Sonocatalytic hydrogen/hole-combined therapy for anti-biofilm and infected diabetic wound healing’, Natl. Sci. Rev., vol. 10, no. 5, p. nwad063, Apr. 2023. [CrossRef]
- F. Akhtar et al., ‘A nano phototheranostic approach of toluidine blue conjugated gold silver core shells mediated photodynamic therapy to treat diabetic foot ulcer’, Sci. Rep., vol. 11, no. 1, p. 24464, Dec. 2021. [CrossRef]
- S. Khan, S. N. Khan, R. Meena, A. M. Dar, R. Pal, and A. U. Khan, ‘Photoinactivation of multidrug resistant bacteria by monomeric methylene blue conjugated gold nanoparticles’, J. Photochem. Photobiol. B, vol. 174, pp. 150–161, Sep. 2017. [CrossRef]
- J. Liu et al., ‘Surfactin effectively inhibits Staphylococcus aureus adhesion and biofilm formation on surfaces’, Appl. Microbiol. Biotechnol., vol. 103, no. 11, pp. 4565–4574, Jun. 2019. [CrossRef]
- Z. Li et al., ‘Antibacterial Activity of Surfactin and Synergistic Effect with Conventional Antibiotics Against Methicillin-Resistant Staphylococcus aureus Isolated from Patients with Diabetic Foot Ulcers’, Diabetes Metab. Syndr. Obes., vol. Volume 16, pp. 3727–3737, Nov. 2023. [CrossRef]
- I. Serrano, B. Alhinho, E. Cunha, L. Tavares, A. Trindade, and M. Oliveira, ‘Bacteriostatic and Antibiofilm Efficacy of a Nisin Z Solution against Co-Cultures of Staphylococcus aureus and Pseudomonas aeruginosa from Diabetic Foot Infections’, Life, vol. 13, no. 2, p. 504, Feb. 2023. [CrossRef]
- J. L. Webber et al., ‘Incorporation and antimicrobial activity of nisin Z within carrageenan/chitosan multilayers’, Sci. Rep., vol. 11, no. 1, p. 1690, Jan. 2021. [CrossRef]
- J. B. Wright, K. Lam, and R. E. Burrell, ‘Wound management in an era of increasing bacterial antibiotic resistance: A role for topical silver treatment’, Am. J. Infect. Control, vol. 26, no. 6, pp. 572–577, Dec. 1998. [CrossRef]
- K. Chaloupka, Y. Malam, and A. M. Seifalian, ‘Nanosilver as a new generation of nanoproduct in biomedical applications’, Trends Biotechnol., vol. 28, no. 11, pp. 580–588, Nov. 2010. [CrossRef]
- E. G. Di Domenico et al., ‘Silver Sulfadiazine Eradicates Antibiotic-Tolerant Staphylococcus aureus and Pseudomonas aeruginosa Biofilms in Patients with Infected Diabetic Foot Ulcers’, J. Clin. Med., vol. 9, no. 12, p. 3807, Nov. 2020. [CrossRef]
- L. Lu et al., ‘Developing natural products as potential anti-biofilm agents’, Chin. Med., vol. 14, no. 1, p. 11, Dec. 2019. [CrossRef]
- J.-H. Lee, Y.-G. Kim, S. Y. Ryu, M. H. Cho, and J. Lee, ‘Ginkgolic acids and Ginkgo biloba extract inhibit Escherichia coli O157:H7 and Staphylococcus aureus biofilm formation’, Int. J. Food Microbiol., vol. 174, pp. 47–55, Mar. 2014. [CrossRef]
- J.-H. Lee et al., ‘Apple Flavonoid Phloretin Inhibits Escherichia coli O157:H7 Biofilm Formation and Ameliorates Colon Inflammation in Rats’, Infect. Immun., vol. 79, no. 12, pp. 4819–4827, Dec. 2011. [CrossRef]
- R. Srinivasan, S. Santhakumari, P. Poonguzhali, M. Geetha, M. Dyavaiah, and L. Xiangmin, ‘Bacterial Biofilm Inhibition: A Focused Review on Recent Therapeutic Strategies for Combating the Biofilm Mediated Infections’, Front. Microbiol., vol. 12, p. 676458, May 2021. [CrossRef]
- D. Fleming and K. Rumbaugh, ‘Approaches to Dispersing Medical Biofilms’, Microorganisms, vol. 5, no. 2, p. 15, Apr. 2017. [CrossRef]
- R. Roy, M. Tiwari, G. Donelli, and V. Tiwari, ‘Strategies for combating bacterial biofilms: A focus on anti-biofilm agents and their mechanisms of action’, Virulence, vol. 9, no. 1, pp. 522–554, Dec. 2018. [CrossRef]
- T. J. Koob et al., ‘Angiogenic properties of dehydrated human amnion/chorion allografts: therapeutic potential for soft tissue repair and regeneration’, Vasc. Cell, vol. 6, no. 1, p. 10, 2014. [CrossRef]
- Y. Mao et al., ‘An in vitro comparison of human corneal epithelial cell activity and inflammatory response on differently designed ocular amniotic membranes and a clinical case study’, J. Biomed. Mater. Res. B Appl. Biomater., vol. 111, no. 3, pp. 684–700, Mar. 2023. [CrossRef]
- T. Ž. Ramuta, T. Šket, M. Starčič Erjavec, and M. E. Kreft, ‘Antimicrobial Activity of Human Fetal Membranes: From Biological Function to Clinical Use’, Front. Bioeng. Biotechnol., vol. 9, p. 691522, May 2021. [CrossRef]
- K. M. Mamede and L. B. Sant’Anna, ‘Antifibrotic effects of total or partial application of amniotic membrane in hepatic fibrosis’, An. Acad. Bras. Ciênc., vol. 91, no. 3, p. e20190220, 2019. [CrossRef]
- C.-H. Wassmer and E. Berishvili, ‘Immunomodulatory Properties of Amniotic Membrane Derivatives and Their Potential in Regenerative Medicine’, Curr. Diab. Rep., vol. 20, no. 8, p. 31, Aug. 2020. [CrossRef]
- J. C. Warning, S. A. McCracken, and J. M. Morris, ‘A balancing act: mechanisms by which the fetus avoids rejection by the maternal immune system’, REPRODUCTION, vol. 141, no. 6, pp. 715–724, Jun. 2011. [CrossRef]
- A. Clinton and T. Carter, ‘Chronic Wound Biofilms: Pathogenesis and Potential Therapies’, Lab. Med., vol. 46, no. 4, pp. 277–284, Nov. 2015. [CrossRef]
- A. Roy, M. Mantay, C. Brannan, and S. Griffiths, ‘Placental Tissues as Biomaterials in Regenerative Medicine’, BioMed Res. Int., vol. 2022, pp. 1–26, Apr. 2022. [CrossRef]
- Z. Chen, Y. Wang, and C. Shi, ‘Therapeutic Implications of Newly Identified Stem Cell Populations from the Skin Dermis’, Cell Transplant., vol. 24, no. 8, pp. 1405–1422, Aug. 2015. [CrossRef]
- L. E. Kokai, K. Marra, and J. P. Rubin, ‘Adipose stem cells: biology and clinical applications for tissue repair and regeneration’, Transl. Res., vol. 163, no. 4, pp. 399–408, Apr. 2014. [CrossRef]
- J. D. Bullard, J. Lei, J. J. Lim, M. Massee, A. M. Fallon, and T. J. Koob, ‘Evaluation of dehydrated human umbilical cord biological properties for wound care and soft tissue healing’, J. Biomed. Mater. Res. B Appl. Biomater., vol. 107, no. 4, pp. 1035–1046, May 2019. [CrossRef]
Figure 1.
Mechanism of Diabetic Foot Ulcer.
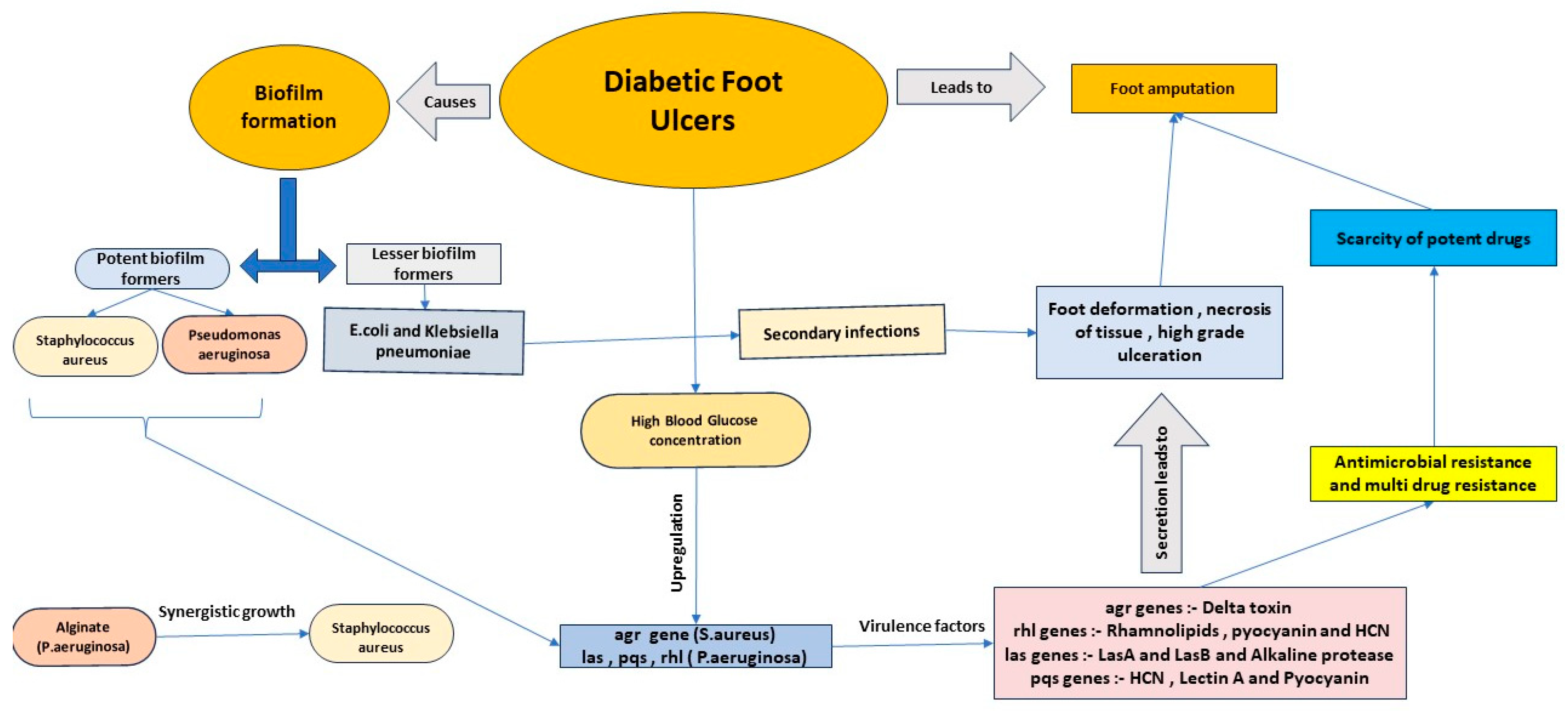
Figure 2.
ica mediated biofilm formation.
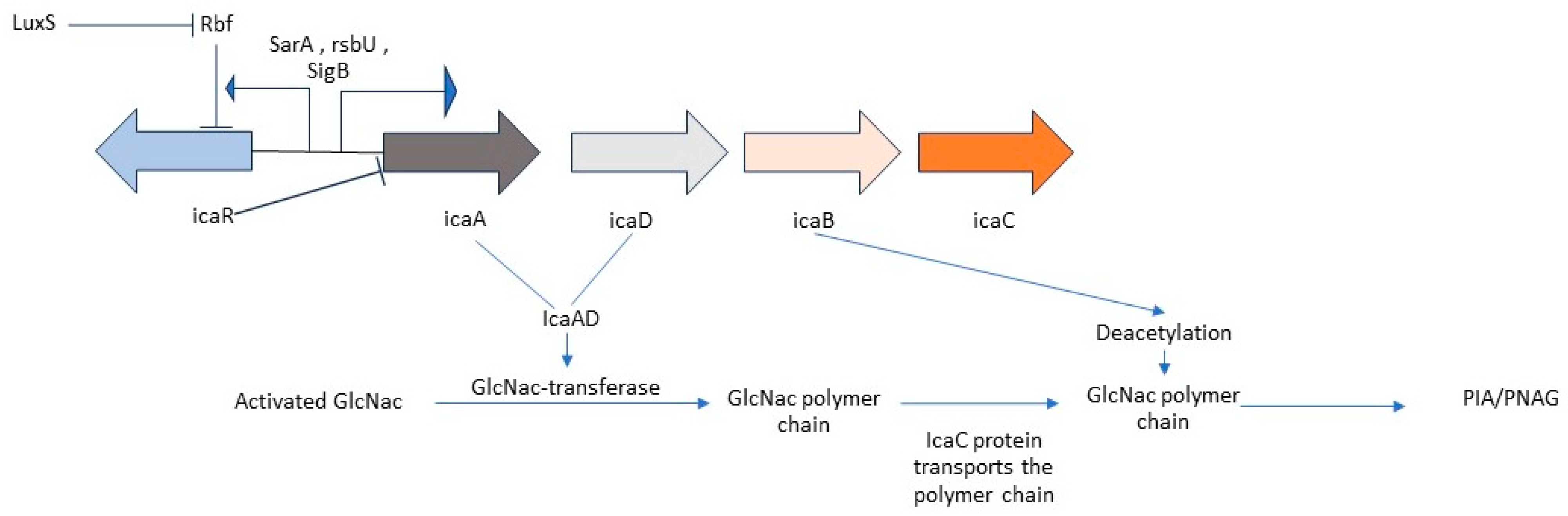
Figure 3.
ica independent biofilm formation.
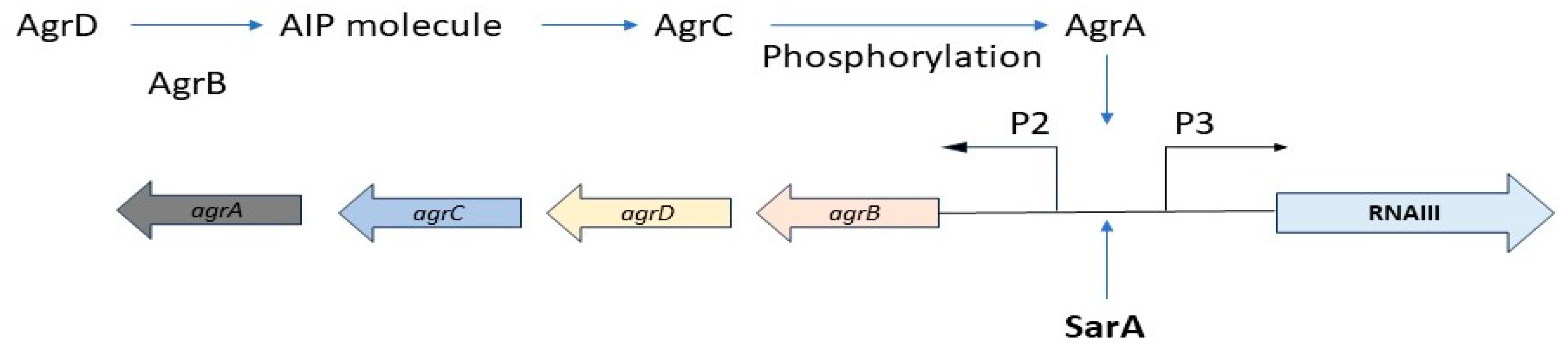
Figure 4.
Biofilm formation in Pseudomonas aeruginosa.
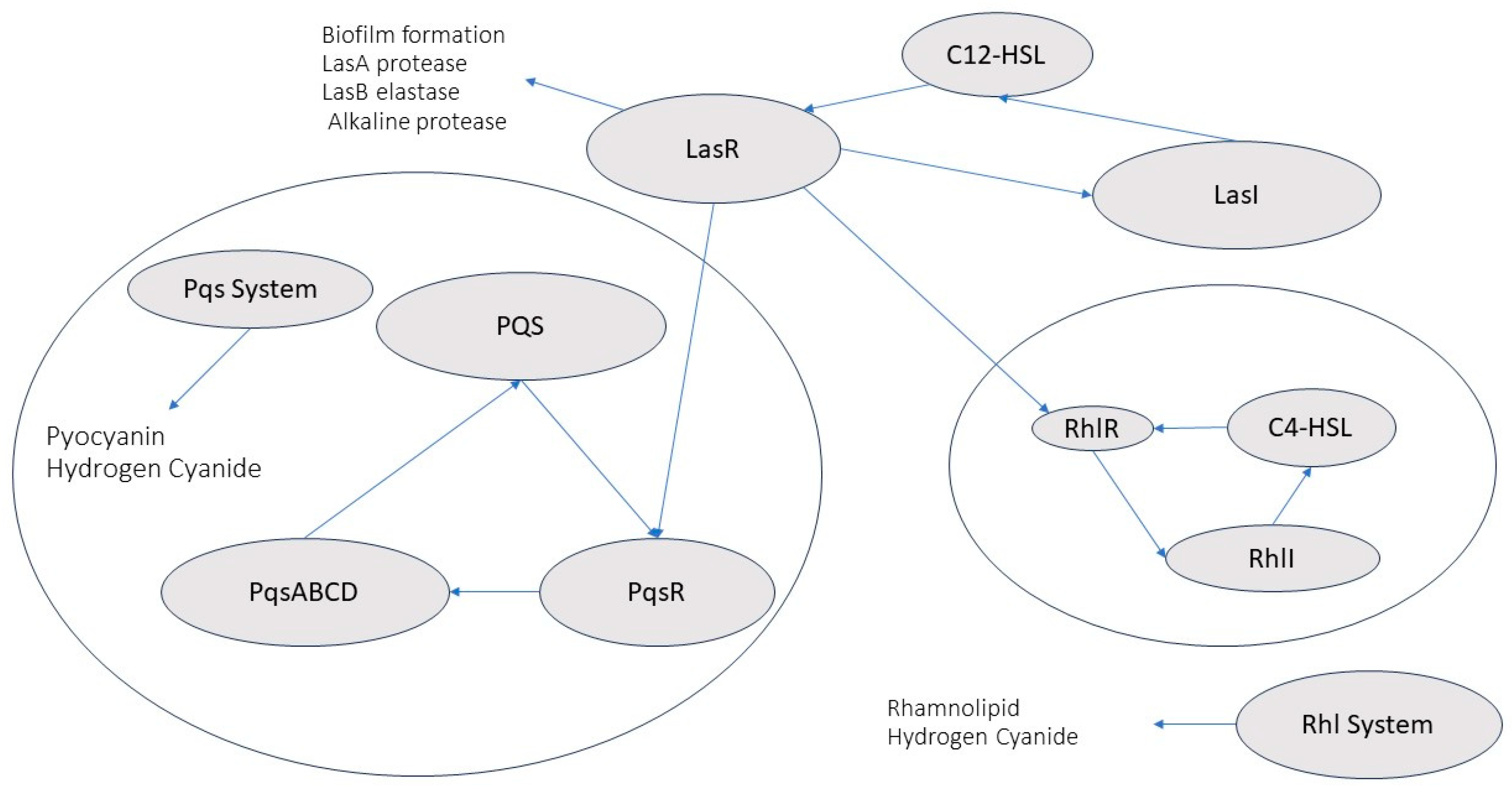
Figure 5.
The treatment methods for Diabetic foot ulcers.
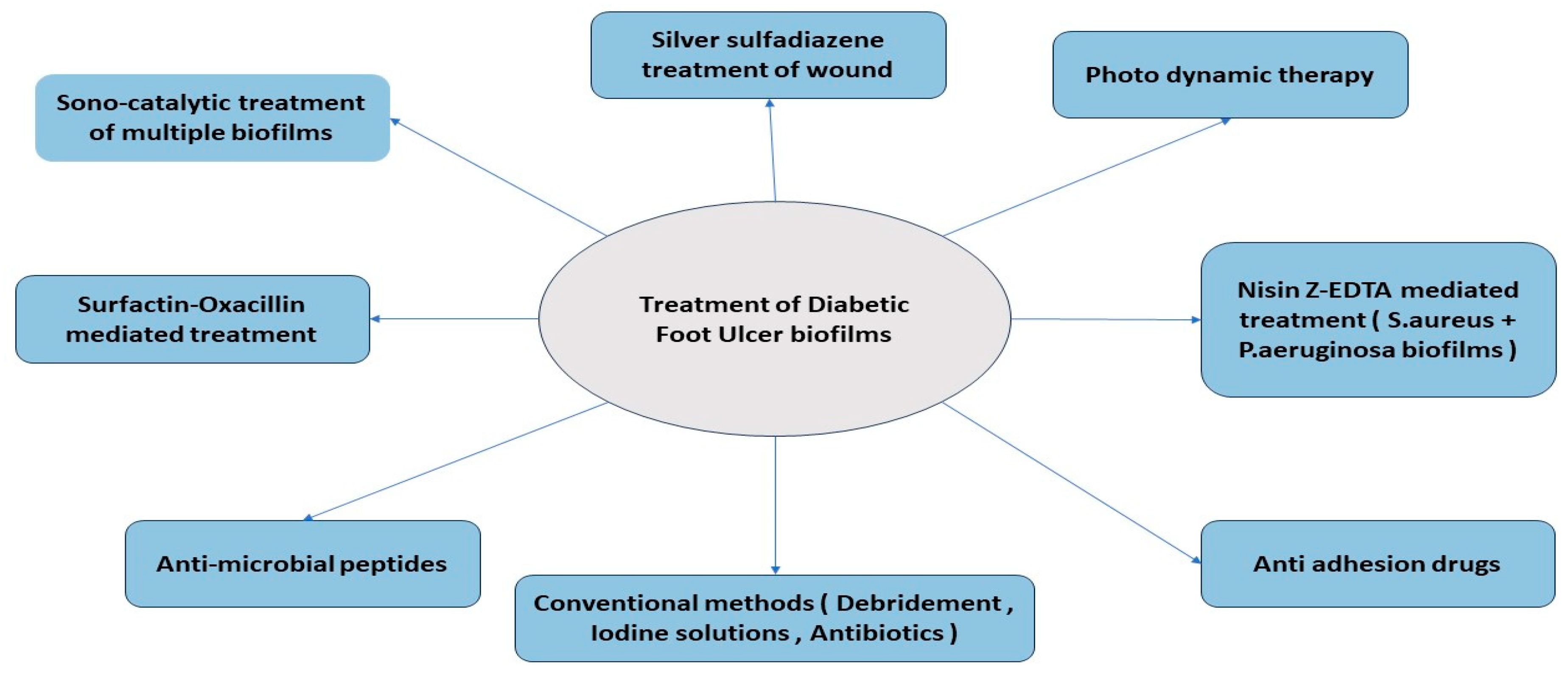
Table 1.
Global survey of articles on the incidence of biofilms in DFUs (2014-24).
| Occurrence of Biofilms |
No of articles | No of articles: Biofilm makeup |
No of articles: Specific species |
||
|---|---|---|---|---|---|
|
Present |
66/88 |
Provided | 45/66 |
Provided | 42/45 |
| Not provided | 21/66 |
Not provided | 3/45 | ||
|
Absent |
22/88 |
Not applicable |
Not applicable |
||
Table 2.
Methods Of Studying Diabetic Foot Ulcers.
| Method of Study | Aim of Study | References |
|---|---|---|
| In Vitro method | ||
| 2-Dimensional Cell Culture | Wound healing and wound formation | [34] |
| 3-Dimensional Cell Culture | Mechanism of wound healing (angiogenesis , re-epithelialization) | [35] |
| In Vivo method | [37],[38] |
|
| Ischemic animal ulcer model | primary ischemic therapy | [39],[40] |
| Neuropathic animal Ulcer model | mechanism of neuropathic infections | [41],[42] |
| Infected Diabetic Ulcer Model | wound formation, progression and healing in diabetic mice | [43], [44], [45], [46], [47], [48] |
| Statistical Data Analysis | Statistical significance of Diabetic foot ulcer data | [49],[50],[51],[52],[53] |
Table 3.
Presence Of Antibiotic resistance and Biofilm forming ability.
| Bacteria | Multi Drug resistance status | Biofilm formation | References |
|---|---|---|---|
| Escherichia coli | Yes | ++ | [12], [8], [58], [59], [60] |
| Pseudomonas aeruginosa | Yes | ++++ | [12], [8], [58] |
| Proteus sp. | Yes | + | [12], [8], [58] |
| Klebsiella pneumoniae | Yes | + | [12], [8], [60], [58] |
| Staphylococcus aureus | Yes | ++++ | [12], [8], [58] |
| Citrobacter sp. | Yes | ++ | [12], [8], [60], [58] |
| Acinetobacter baumanii | Yes | +++ | [12], [8], [61], [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



