
Submitted:
01 October 2024
Posted:
01 October 2024
You are already at the latest version
Abstract
Accurate determination of an individual’s unique human leukocyte antigen (HLA) allele holds important significance in evaluating the risk associated with autoimmune and infectious diseases, such as human immunodeficiency virus (HIV) infection. Several allelic variants within the HLA system have been linked to either increased protection or susceptibility in the context of infectious and autoimmune diseases. This study aimed to determine the frequency and association of HLA alleles between people living with HIV (PLHIV) as the case group and HIV-uninfected Peruvian individuals with high-risk behavior of sexual transmission diseases as the control group. Whole exome sequencing (WES) was used to determine high-resolution HLA allelotypes using OptiType and arcasHLA tools. The HLA alleles present in HLA classes I (A, B, and C loci) and II (DPA1, DPB1, DQA1, DQB1 and DRB1 loci) were determined in a cohort of 59 PLHIV (cases) and 44 HIV-uninfected individuals (controls). The most frequent HLA alleles were A*02:01, DPB1*04:02 and DQB1*03:419 at 36%, 30% and 28% prevalence in general population.
We found that C*07:01 (p = 0.0101; OR = 10.222, 95% IC: 1.40–74.55), DQA1*03:02 (p = 0.0051; OR = 5.297, 95% IC: 1.48–19.02) and DRB1*09:01 (p = 0.0119; OR = 4.788, 95% IC: 1.39–16.44) showed an association with susceptibility to HIV infection, while DQB1*03:419 (p = 0.0478; OR = 0.327, 95% IC: 0.11–0.96) was associated with protection from HIV infection. Our findings contribute to the knowledge of HLA allele diversity in the Peruvian population (around 70% South American indigenous ancestry), lay the groundwork for further valuable large-scale use of HLA typing and offer a novel association of HIV infection that is relevant to vaccine studies.
Keywords:
HIV
; HLA genes
; WES
; susceptibility
; Peruvian population
1. Introduction
According to the estimation of the Joint United Nations Programme on HIV/AIDS (UNAIDS), there are 39 million people living with HIV (PLHIV), and 29.58 million of them have access to antiretroviral therapy (ART) around the world. Of these, 2.2 million are from Latin America [1].
The human leukocyte antigen (HLA) is situated in a region spanning approximately 4 million base pairs on the short arm of human chromosome 6 (6p21.3). This region encompasses over 200 protein-coding genes [2], with more than 35,000 alleles present in the HLA locus [3,4,5]. HLA class I and class II genes play a central role in host adaptive immune responses to infectious pathogens. HLA class I genes are also involved in the host innate immune response. Class I genes encode proteins that facilitate the presentation of antigenic peptides from pathogens to CD8+ T cells, leading to the destruction of infected cells and pathogens, while HLA class II antigens contribute to the initiation of adaptive immune responses by presenting antigenic epitopes to CD4+ T cells. The remarkable polymorphism of HLA class I and class II genes contributes to the diverse host immune responses to many infectious pathogens [6].
In recent years, advancements in next-generation sequencing (NGS) have brought significant improvements, allowing for rapid, high-throughput sequencing of genomes, exomes, and targeted gene panels, with the ability to process multiple samples simultaneously. Within this framework, whole exome sequencing (WES) can be adapted for various configurations, tailored to clinical or research needs, particularly for HLA alleles determination [7]. The incorporation of NGS, a sophisticated, high-resolution assay, has been a significant advancement in HLA typing and has markedly improved the performance and data collection of many HLA laboratories [3,8,9].
Considering the potential impact of various HLA alleles on both HIV acquisition and pathogenesis, these findings will provide deeper insights into host genetics implicated in differential immunological responses to infection and disease progression. Such insights are crucial to optimizing treatment and vaccine effectiveness, as well as contextualizing the human host determinants that influence the epidemiology of HIV in distinct populations. Our aim is to identify HLA alleles that are associated with susceptibility or resistance to HIV in the Peruvian population with a major South American autochthonous ancestry, part of the 80% of the world population neglected under population genetics studies. This will contribute to the knowledge of alleles that would serve as markers in this population and those of the neighboring countries, as well as to their response to HIV.
2. Materials AND Methods
2.1. Study Population and Design
We conducted a cross-sectional study involving 59 PLHIV from the Santa Rosa Hospital (Lima, Perú) and 46 HIV-uninfected individuals with high-risk behaviors from the NGO “MCC Voluntades Lima Norte” (Lima, Perú). The study participants were enrolled, from December 2019 to October 2021.
Eligibility criteria included being of Peruvian nationality, aged 18 years or older, male or female, and gave written informed consent. HIV seropositive individuals were referred to medical care and assigned to the PLHIV group. The inclusion criteria for the HIV-uninfected group were a negative rapid HIV test, exhibiting high-risk sexual behaviors, and perception of HIV risk. Transgender women who met the eligibility criteria were included in this group.
Data from patients (socio-demographic, clinical, immunological and laboratory results) were obtained from the clinical history of each patient and the CD4 T cell count results were provided by the National Reference Laboratory for STD/HIV-AIDS at the Instituto Nacional de Salud (INS) in Lima.
For the HIV-uninfected group, all individuals completed a brief questionnaire about their high-risk sexual behaviors regarding HIV transmission, such as their history of sexual contact with sex workers, diagnosis or treatment of a sexually-transmitted infection (STI), condom use, and having multiple sexual partners.
2.2. Biological Samples and DNA Extraction
Blood samples were collected in 3 ml ethylenediaminetetraacetic acid (EDTA) tubes from all the enrolled participants in both groups, and DNA was extracted using the salting-out procedure with minor modifications [10]. Samples were then quantified and checked for purity using a NanoDrop spectrophotometer (Thermo Scientific).
2.3. Whole-Exome Sequencing (WES)
Extracted DNA was sent to two different service providers, who carried out library construction and WES: Macrogen (Korea) and Novogene (Cambridge, UK). DNA libraries were prepared using SureSelect XT Human All Exon V6 - bait library: S07604514 (Agilent Technologies, Santa Clara, CA, USA), and WES was performed using 150 bp paired-end reads and sequenced on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA) according to the manufacturer’s instructions.
2.4. Bioinformatics Pipeline to HLA Typing
We received the exome data and established an in-house bioinformatics pipeline for HLA typing. First, the WES FastQ files were preprocessed by fastp (version 0.23.2) [11] and then mapped to the human reference genome GRCh37/hg19 using BWA software (version 0.7.17-r1188, bwa mem) [12]. The filtered and mapped reads were sorted by coordinates using SAMtools (version 1.7) [12], and possible PCR duplicate reads were marked by Picard (version 2.27.4, Broad Institute). The reads that matched the targeted chromosome 6 sequences were extracted from the bam file and mapped on a comprehensive reference panel from the IMGT/HLA database (v3.52.0) [4]. In addition, we evaluated the germline variants (single-nucleotide polymorphisms [SNPs] and small insertions/deletions [indels]) in the HLA region.
We performed the HLA typing analysis with the reads from chromosome 6 that were generated as an input. We imputed HLA class I alleles for three loci (HLA-A, HLA-B and HLA-C) using OptiType software (version 1.3.3) [13] and HLA class II alleles for four loci (HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1) were imputed with arcasHLA [14]. The HLA class I and class II alleles were assigned at the 2-field resolution. We set a threshold for the depth (≥20X) of HLA alleles and samples with a depth lower than 20 were excluded. After these considerations, we included 56 cases and 44 controls.
2.5. Statistical Analysis
Socio-demographic and clinical data were expressed as mean ± standard deviation. Comparisons between PLHIV and HIV-uninfected groups were performed using the chi-squared test for categorical/qualitative variables and the Mann–Whitney U test (or Student’s t-test) for quantitative variables. Likewise, HLA allele frequencies and HLA association analysis for performing case–control studies were determined with PyHLA software [15] using additive logistic regression models. Age, sex, and CD4 values were included as covariates in the association test.
In all analyses, a p-value less than 0.05 and a confidence interval of 95% were considered statistically significant. R software (version: 4.3.1) was used for statistical analysis and plot creation.
2.6. Hardware and Software Environment
All software was run according to instructions on the Linux server (Ubuntu 18.04) with the following hardware configuration: Intel(R) Xeon(R) CPU E5-2697 v2 @ 3.50 GHz each with 24 physical CPU cores, and 94 GiB RAM installed. All computational resources were provided by the Research Center of Genetic and Molecular Biology – Universidad de San Martin de Porres.
2.7. Ethics Statement
The study received ethical approval from the “Comité de Ética en Investigación de la Universidad de San Martín de Porres” (IRB IORB00003251 OHRP/FDA). Likewise, this study was also approved by the Ethics Committees of the following institutes that participated in the study: the National Institute of Health of Peru and Santa Rosa Hospital. Written informed consent from patients was obtained locally at Santa Rosa Hospital and the NGO “MCC Voluntades Lima Norte”. An explanation session for each participant was held to clarify doubts about the procedure and to explain the results obtained.
3. Results
The study sample included 59 PLHIV (cases) and 46 HIV-uninfected (control) subjects. The relevant socio-demographic, clinical and ART treatment characteristics are provided in Table 1; the distribution of these characteristics varied according to gender, age, and CD4 cell count between the case and control groups. The majority of participants were male (73.0%) and born in Lima (71.43%). The median age was 41 years in the case group and 36 years in the control groups. At the time of enrollment, the median CD4 count was 634 cells/mm3 and 952.67 cells/mm3 in the PLHIV and HIV-uninfected groups, respectively.
Based on the inclusion criteria concerning HIV risk behaviors presented in the HIV-uninfected group, all participants reported having sex with sex workers. Of these participants, 26 (56.52%) self-identified as heterosexual, 14 (30.43%) as homosexual, and 6 (13.04%) as bisexual. Regarding sexual behavior, 76% of participants reported having had multiple sexual partnerships (5–15 sexual partners in the last 12 months).
High-resolution HLA typing was performed in all 105 enrolled subjects to yield at least 2-field resolution HLA alleles for HLA-A, HLA-B and HLA-C in class I, and HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1 in class II. The number of HLA alleles present across the six HLA loci is provided in Table 2. A total of 1394 2-field HLA alleles were observed across the seven loci in the population study. In HLA alleles class I, 200, 200, and 198 alleles were counted in HLA-A, HLA-B and HLA-C genes, respectively (Table 2). In class II alleles, 200, 196, 200, and 200 alleles were counted in HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1 genes, respectively (Table 2).
The three most frequent alleles for each HLA locus were A*02:01 (35.5%), A*24:02 (11.0%), A*02:11 (10.0%); B*35:01 (11.5%), B*48:01 (6.50%), B*15:04 (6.0%); C*04:01 (22.73%), C*07:02 (18.69%), C*01:02 (12.12%); DPB1*04:02 (29.50%), DPB1*04:01 (16.00%), DPB1*14:01 (13.50%), DQA1*03:01 (22.45%), DQA1*05:03 (11.73%), DQA1*04:01 (11.22%), DQB1*03:419 (28.00%), DQB1*03:02 (21.00%), DQB1*03:96 (10.00%), DRB1*09:01 (14.50%), DRB1*04:07 (13.00%) and DRB1*08:02 (11.0%) prevalence in general population (complete HLA allele frequencies are detailed in Supplementary Table S1).
We identified three novel SNP-type variants in two patients and one control, respectively: NM_002116.8(HLA-A):c.619+8G>T in splice region variant (Qual:16.5), NM_001243961.2(HLA-DBQ1):c.109+71G>C in region variant (Qual:11.7) and NM_002117.6(HLA-C):c.620-177G>T in region variant (Qual:10). Additionally, we detected one novel deletion variant in a patient: NM_002117.6(HLA-C):c.1097-21del in the intron variant. However, the novel variants had a variant quality score below 20. No novel alleles were identified in the evaluated HLA genes; all detected alleles had already been reported in the IPD-IMGT/HLA database.
The allele frequencies of class I (Figure 1) and class II (Figure 2) in the PLHIV sample (pink bar) were compared to those in the HIV-uninfected sample (sky-blue bar). Alleles with frequencies less than 1% were omitted.
We found four HLA alleles that were associated with HIV infection; these are shown in Table 3 (results for other HLA alleles are detailed in Supplementary Table 2). The HLA-C, HLA-DQA1 and HLA-DRB1 alleles associated with the risk of HIV infection were C*07:01 (p = 0.0101, OR = 10.222), DQA1*03:02 (p = 0.0051, OR = 5.297) and DRB1*09:01 (p = 0. 0119, OR = 4.788), respectively. Only the HLA-DQB1*03:419 (p = 0.0412, OR = 0.3273) allele conferred protection (Table 3).
4. Discussion
In this study, we performed high-resolution HLA allele-calling using a WES approach in a sample of a Peruvian population. HLA typing through WES proves to be a powerful tool that offers a comprehensive view of an individual’s genetic composition. This approach allows for the identification of variants in HLA genes and other genes associated with susceptibility to HIV infection. Additionally, it is considered a cost-effective strategy for genomic sequencing projects [16].
Our results show diverse HLA profiles and frequencies between PLHIV and HIV-uninfected individuals. We analyzed the association of HLA class I and II alleles to determine the relationship of HLA alleles that confer HIV protection or susceptibility in our population. We detected HLA alleles for three class I loci (HLA-A, HLA-B, HLA-C) and four loci in class II (HLA-DPB1, HLA-DQA1, HLA-DQB1, and HLA-DRB1) because these genes have crucial roles in antigen presentation and high degrees of polymorphism [17]. HLA allele composition is key for association studies and helps us understand the genetic risk of autoimmune and infectious diseases, such as HIV infection [6].
The relevant socio-demographic, clinical and ART treatment characteristics varied in distribution according to gender, age, and CD4 cell count between the case and control groups. The majority of participants were male (73.0%) and born in Lima (71.43%). The median age was 41 years in the case group and 36 years in the control group. At the time of enrolment, the median CD4 count was 634 cells/mm3 and 952.67 cells/mm3 in the PLHIV and HIV-uninfected groups, respectively. This finding is consistent with a study carried out in Peru, where 162 patients with HIV infection were recruited, the median age was 42 years, 61% patients were male, 71% were heterosexual, and 58% were born in Lima [18].
On the other hand, the imputed HLA alleles for PLHIV and HIV-uninfected in this Peruvian sample, were in the range of 196 to 200 alleles (Table 2, Figure 1 and Figure 2). The most frequent alleles for each HLA locus were A*02:01 (35.5%), B*35:01 (11.5%), C*04:01 (22.73%), DPB1*04:02 (29.50%), DQA1*03:01 (22.45%), DQB1*03:419 (28.00%) and DRB1*09:01 (14.50%) prevalence in general population. The Allele Frequency Net Database (http://www.allelefrequencies.net/) [19] reports the frequencies of HLA alleles detected in different populations. In a study carried out in the Uros population localized in Puno City (Peru), the five most frequent alleles were found to be: B*35:05 (51.5%), A*02:01 (50%), DQB1*03:02 (38.6%), DQB1*04:02 (33.4%) and DRB1*08:02 (31.9%) [20]. Similar allele frequencies of HLA class I alleles were reported by another study in Lima, with 468 individuals recruited. Of these, 222 were seronegative for HIV-1 (HIV-negative) and 246 were infected with HIV-1. The most common alleles for the different loci were HLA-A*02:01 (46.8%), HLA-B*35:01 (12.0%) and HLA-C*04:01 (37.6%) [21].
We were able to identify significant associations of common HLA variants in our cohort. We identified significant associations of HLA-C*07:01 (p = 0.0101, OR = 10.222), HLA-DQA1*03:02 (p = 0.0051, OR = 5.297) and HLA-DRB1*09:01 (p = 0.0119, OR = 4.788) with susceptibility to HIV infection while HLA-DQB1*03:419 (p = 0.0478, OR = 0.327) was associated with protection from HIV exposure (Table 3). A similar study from Peru concluded that HLA-B*35:43 showed the strongest association with HIV acquisition (p = 0.012), while HLA-A*02:01 and HLA-C*04:01 were both associated with high viral loads (p = 0.0313 and 0.0001, respectively) [21].
HLA-C belongs to class I and encodes a protein composed of a membrane-bound mature heavy chain and a light chain, β2-microglobulin (β2M). HLA-C plays a role in presenting peptides to virus-specific T-cells, although much less is known about the CD8+ T-cell recognition of peptides restricted by HLA-C. This function is crucial for the initiation and maintenance of adaptive immunity [22]. In addition, our analysis has shown that the HLA-C*07:01 allele might be associated with HIV susceptibility, which is consistent with the results of a genome-wide association study (GWAS) on HIV susceptibility in European individuals [23], where this allelic variant was classified as a risk allele. Another study in a European population showed that the HLA-C*07:01 allele generates susceptibility to autoimmune hepatitis (AIH) [34]. Our study confirmed the influence of HLA-C*07:01 on susceptibility to HIV-1 infection.
HLA-DQ molecules form a transmembrane protein composed of an α chain that is encoded by HLA-DQA and consists of α1 and α2 domains and HLA-DQB encoded a β chain consists of β1 and β2 domains. Both chains are anchored in the membrane and form an antigen-presenting groove. No association was found between the DQA1*03:02 allele and infectious diseases. However, this allele has been reported to be associated with systemic lupus erythematosus (SLE) in Chinese Han patients [25].
The primary function of HLA class II molecules (HLA-DRB1, HLA-DQA1, and HLA-DQB1) is to process exogenous peptides for presentation to CD4+ T cells, which are crucial in antiviral cellular and humoral immunity [26]. The HLA-DRB1 gene encodes the DRβ1 chain. The association of HLA-DRB1 alleles may affect the specific structure of the HLA-DR molecule and its binding affinity to epitopes [27]. Previous studies have reported associations of the HLA-DRB1*09:01 allele with infectious diseases. For example, Anzurez et al. conducted a study that found that this allelic variant was associated with the risk of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in a Japanese population [26]. In contrast, Nguyen et al. reported that HLA-DRB1*09:01 showed a protective effect against the development of Dengue Shock Syndrome (DSS), particularly in patients with DEN-2 infection in a Vietnamese population [28]. Although there is no association between DRB1*09:01 and HIV infection, new associations between the DQA1*03:02 and DRB1*09:01 allelic variants and HIV infection have been identified, which could be attributed to an increased affinity and specificity of the peptide-binding region (PBR), influencing the recognition of pathogen-derived antigens [29].
In contrast to susceptibility alleles, in the HLA-DQB1 locus, the HLA-DQB1*03:419 allele was identified as a novel protective allele against HIV infection in this study. The HLA-DQB1*03032 and HLA-DQB1*0602 alleles have shown protective associations against HIV infection in Caucasian and African American ethnic groups, respectively [30]. Rallón et al. showed that the HLA-DQB1*03:02 allele was implicated in protection from HIV infection in a Spanish population [31], and Hardie et al. showed that the HLA-DQB1*0603 allele conferred protection from HIV-1 infection in a Pumwani cohort [32]. Therefore, the combination of HLA-DQA1 (encodes alpha chain) and HLA-DQB1 (encodes beta chain) forms a binding pocket that determines the DQ molecule’s specificity and diversity for antigen presentation. These allelic variants can affect peptide binding, leading to differential antigen presentation by the DQ molecule, which may be associated with resistance and susceptibility to HIV-1 infection.
Our results add information about a population with scarce, namely the Peruvian population, whose genetic composition is represented by that of Lima, the capital city of Peru, with about one third of the country’s population mostly coming from all over the country in the last 70 years. Lima has a mixed population, characterized by a high Amerindian component, around 70%; the rest are admixed with European, African and Asian ancestry [33]. This is the first report of the predisposing associations of C*07:01, DQA1*03:02, and DRB1*09:01 and the protective association of DQB1*03:419 with HIV infection in Peruvian patients.
The present study has limitations worth noting. First, our HIV-uninfected group was selected based on self-reported risk behavior, which may underestimate actual risk behavior; therefore, we could have introduced biased information. Second, the recruitment methodology did not consider the determination of the patients’ HIV viral load. Thus, we were unable to compare the viral load with other variables. Additionally, the small sample size is also a limitation of this study, although these two parameters did not influence the test results according to the statistical analysis. This study only determined the four-digit resolution prediction for HLA typing. However, a high-throughput HLA typing resolution such as six or eight-digit resolution is crucial to increasing variant accuracy. We employed high sequencing depth and coverage (≥20X) to generate high-resolution HLA alleles.
5. Conclusions
In conclusion, we identified three HLA alleles (HLA C*07:01, DQA1*03:02, and DRB1*09:01) that were associated with susceptibility to HIV infection, and one HLA allele (HLA DQB1*03:419) that showed a significant protective effect against HIV infection. Our findings contribute to the knowledge of HLA allele diversity in the Peruvian population with 70% of autochthonous South American ancestry, lay the groundwork for further valuable large-scale use of HLA typing, and offer novels associations of the HIV infection that is relevant to treatment and vaccine studies.
Conflict of Interests
The authors report no conflict of interests.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceived and designed the experiments: DOA, OAC, CYV, and RFA. Performed the experiments: DOA, MLGG, and OAC. Analyzed and interpreted the data: DOA, MLGG, OAC, and RFA. Data collection: SEC, CYV, and SEA. Wrote the paper: DOA, OAC, MLGG, and RFA. Manuscript review and revisions: DOA, OAC, MLGG, SEC, CYV, SEA, and RFA.
Funding
This research was funded by Peruvian Fondecyt (Fondo Nacional de Desarrollo Científico, Tecnológico y de Innovación) [contract N°012-2019-FONDECYT].
Acknowledgments
We are grateful to the study staff, Santa Rosa Hospital, and the MCC “Voluntades” Lima Norte community, who partnered with us to conduct the recruitment process of the study.
References
- UNAIDS (2023). UNAIDS 2023 Data. Available online at: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed October 7, 2023).
- Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, Khodiyar VK, Lush MJ, Povey S, Talbot CC Jr, Wright MW, Wain HM, Trowsdale J, Ziegler A, Beck S. Gene map of the extended human MHC. Nat Rev Genet 2004; 5(12):889-99. [CrossRef]
- Kim, J. Y., Lee, S. Y., Kim, G. G., Song, H. I., Jang, M. M., Lee, C. S., Hong, J. Y., Shin, M. G., & Choi, H. J. Validation and application of new NGS-based HLA genotyping to clinical diagnostic practice. HLA 2023, 101(5), 496–506. [CrossRef]
- Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P,Marsh SGE. IPD-IMGT/HLA Database. Nucleic Acids Res 2020; 48(D1):D948-D955. [CrossRef]
- Marsh SG, Albert ED, Bodmer WF, et al. Nomenclature for factors of the HLA system, 2010.Tissue Antigens. 2010; 75(4):291-455.
- Luo, M. Natural Immunity against HIV-1: Progression of Understanding after Association Studies. Viruses. 2022 Jun 8;14(6):1243. Erratum in: Viruses. 2023 ;15(6). [CrossRef]
- Dashti M, Malik MZ, Nizam R, Jacob S, Al-Mulla F and Thanaraj TA (2024), Evaluation of HLA typing content of next-generation sequencing datasets from family trios and individuals of arab ethnicity. Front. Genet. 15:1407285. [CrossRef]
- Bentley G, Higuchi R, Hoglund B, Goodridge D, Sayer D, Trachtenberg EA, et al. High-resolution, high-throughput HLA genotyping by next-generation sequencing. Tissue Antigens 2009; 74:393-403.
- Erlich H. HLA DNA typing: past, present, and future. Tissue Antigens 2012; 80:1-11.
- Miller S, Dykes D, Polesky H. A simple salting out procedure for extracting DNA from human nucleated cells. Nuc Acids Res 1988; 16:1215.
- Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:884–90.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [CrossRef]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [CrossRef]
- Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: Precision HLA typing from next-generation sequencing data. Bioinformatics. 2014; 30(23):3310–6.
- Orenbuch R, Filip I, Comito D, Shaman J, Pe’er I, Rabadan R. arcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics. 2020; 36(1):33-40. [CrossRef]
- Fan Y, Song YQ. PyHLA: tests for the association between HLA alleles and diseases. BMC Bioinformatics. 2017; 18:90. [CrossRef]
- Liu, P., Yao, M., Gong, Y., Song, Y., Chen, Y., Ye, Y., Liu, X., Li, F., Dong, H., Meng, R., Chen, H., & Zheng, A. (2021). Benchmarking the Human Leukocyte Antigen Typing Performance of Three Assays and Seven Next-Generation Sequencing-Based Algorithms. Frontiers in immunology, 12, 652258. [CrossRef]
- Sanchez-Mazas, A. A review of HLA allele and SNP associations with highly prevalent infectious diseases in human populations. Swiss Med Wkly 2020; 150:w20214. [CrossRef]
- Amanzo-Vargas MP, Arellano-Veintemilla T, González-Lagos E, Echevarría J, Mejía F, Graña A, Gotuzzo E. Socio-Demographic, Clinical, and Mortality Differences between HIV-Infected and HIV/HTLV-1 Co-Infected Patients in Peru. Pathogens 2023; 12(7):869. [CrossRef]
- Gonzalez-Galarza FF, McCabe A, Santos EJ, Jones J, Takeshita LY, Ortega-Rivera ND, Del Cid-Pavon GM, Ramsbottom K, Ghattaoraya GS, Alfirevic A, Middleton D and Jones AR. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acid Res 2020, 48:D783-8.
- Arnaiz-Villena A, Gonzalez-Alcos V, Serrano-Vela JI, Reguera R, Barbolla L, Parga-Lozano C, Gómez-Prieto P, Abd-El-Fatah-Khalil S, Moscoso J. HLA genes in Uros from Titikaka Lake, Peru: origin and relationship with other Amerindians and worldwide populations. Int J Immunogenet. 2009; 36(3):159-67. [CrossRef]
- Olvera A, Pérez-Álvarez S, Ibarrondo J, Ganoza C, Lama JR, Lucchetti A, Cate S, Hildebrand W, Bernard N, Gomez L, et al. The HLA-C*04: 01/KIR2DS4 gene combination and human leukocyte antigen alleles with high population frequency drive rate of HIV disease progression. AIDS 2015, 29:507–517. [CrossRef]
- Blais, M. E., Dong, T., & Rowland-Jones, S. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player?. Immunology 2011, 133(1), 1–7. https://doi.org/10.1111/j.1365-2567.2011.03422.x. [CrossRef]
- International HIV Controllers Study, Pereyra, F., Jia, X., McLaren, P. J., Telenti, A., de Bakker, P. I., Walker, B. D., Ripke, S., Brumme, C. J., Pulit, S. L., Carrington, M., Kadie, C. M., Carlson, J. M., Heckerman, D., Graham, R. R., Plenge, R. M., Deeks, S. G., Gianniny, L., Crawford, G., Sullivan, J., … Zhao, M. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 2010, 330(6010): 1551–1557. [CrossRef]
- M.D. Strettell, L.J. Thomson, P.T. Donaldson, M. Bunce, C.M. O’Neill, R. Williams. HLA-C genes and susceptibility to type 1 autoimmune hepatitis. Hepatology 1997, 26: 1023–1026.
- Qian, J., Chen, Y., Yang, X., Wang, Q., Zhao, J., Deng, X., Ding, Y., Li, S., Liu, Y., Tian, Z., Shen, J., Liao, Q., Wang, Y., Zuo, X., Zhang, X., Li, M., Cui, Y., Yu, X., & Zeng, X. Association Study Identified HLA-DQA1 as a Novel Genetic Risk of Systemic Lupus Erythematosus-Associated Pulmonary Arterial Hypertension. Arthritis Rheumatol 2023, 75(12), 2207–2215. [CrossRef]
- Anzurez, A., Naka, I., Miki, S., Nakayama-Hosoya, K., Isshiki, M., Watanabe, Y., Nakamura-Hoshi, M., Seki, S., Matsumura, T., Takano, T., Onodera, T., Adachi, Y., Moriyama, S., Terahara, K., Tachikawa, N., Yoshimura, Y., Sasaki, H., Horiuchi, H., Miyata, N., Miyazaki, K., … Kawana-Tachikawa, A. Association of HLA-DRB1*09:01 with severe COVID-19. HLA 2021, 98(1): 37–42. [CrossRef]
- Choo S., Y. The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med J 2007, 48(1), 11–23. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T. P., Kikuchi, M., Vu, T. Q., Do, Q. H., Tran, T. T., Vo, D. T., Ha, M. T., Vo, V. T., Cao, T. P., Tran, V. D., Oyama, T., Morita, K., Yasunami, M., & Hirayama, K. Protective and enhancing HLA alleles, HLA-DRB1*0901 and HLA-A*24, for severe forms of dengue virus infection, dengue hemorrhagic fever and dengue shock syndrome. PLoS Negl Trop Dis 2008, 2(10): e304. [CrossRef]
- Sarri CA, Giannoulis T, Moutou KA, Mamuris Z. HLA class II peptide-binding-region analysis reveals funneling of polymorphism in action. Immunol Lett. 2021; 238: 75–95.
- Roe, D. L., Lewis, R. E., & Cruse, J. M. Association of HLA-DQ and -DR alleles with protection from or infection with HIV-1. Exp Mol Pathol 2000, 68(1): 21–28. [CrossRef]
- Rallón N, Restrepo C, Vicario J, Romero J, Rodríguez C, García-Samaniego J, et al. Human leucocyte antigen (HLA)-DQB1*03:02 and HLA-A*02:01 have opposite patterns in their effects on susceptibility to HIV infection. HIV Med. 2017; 18(8):587-594. [CrossRef]
- Hardie R, Luo M, Bruneau B, Knight E, Nagelkerke N, Kimani J, Wachihi C, Ngugi E, Plummer F. Human leukocyte antigen-DQ alleles and haplotypes and their associations with resistance and susceptibility to HIV-1 infection. AIDS 2008, 22: 807–816. [CrossRef]
- Sandoval J, Salazar-Granara A, Acosta O, Castillo-Herrera W, Fujita R, Pena SDJ, et al. Tracing the genomic ancestry of Peruvians reveals a major legacy of pre-Columbian ancestors. J Hum Genet 2013; 58(9):627-34. [CrossRef]
Figure 1.
Frequency distribution of HLA class I alleles in PLHIV and HIV-uninfected based on 2-field resolution HLA imputation.
Figure 1.
Frequency distribution of HLA class I alleles in PLHIV and HIV-uninfected based on 2-field resolution HLA imputation.
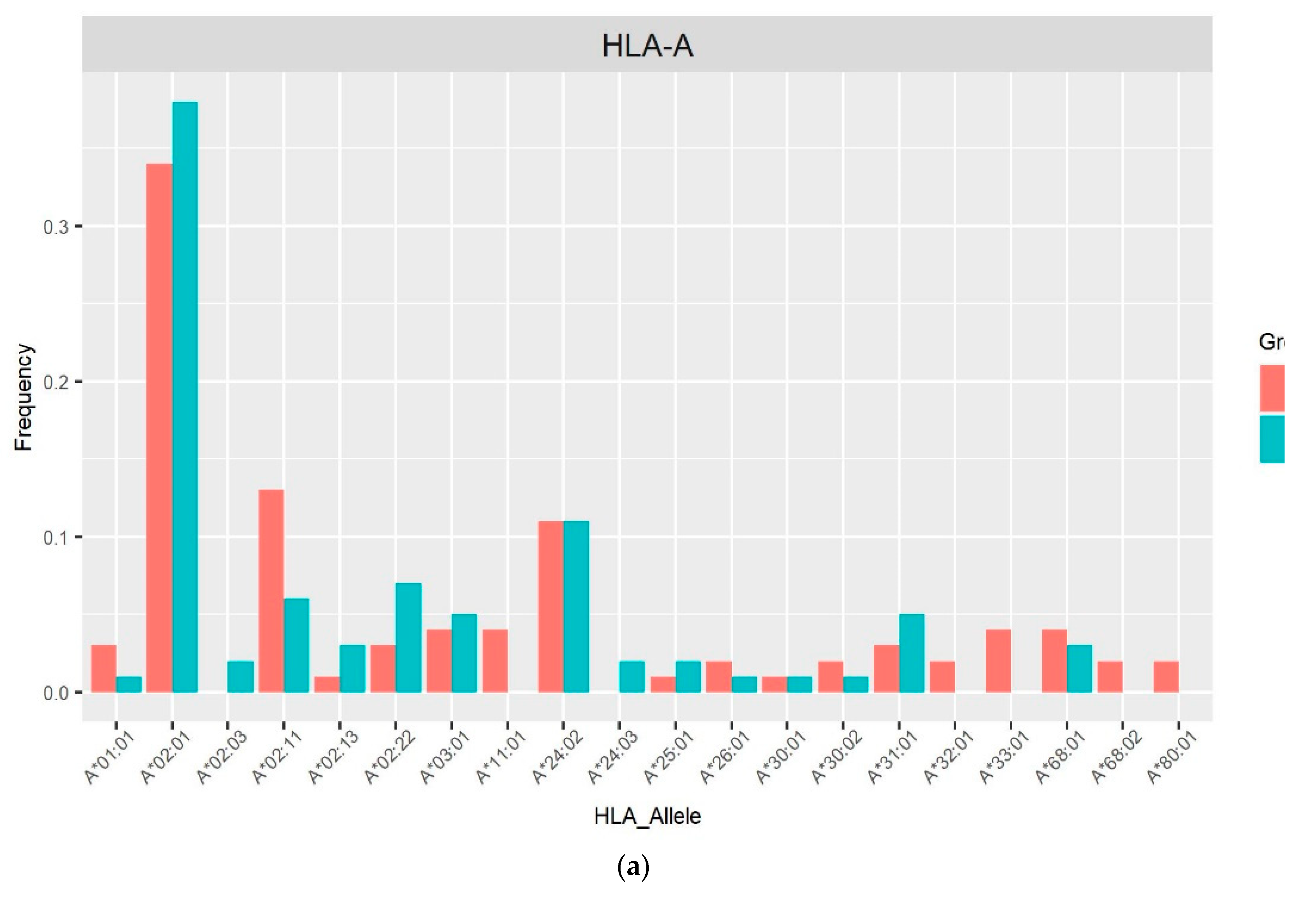
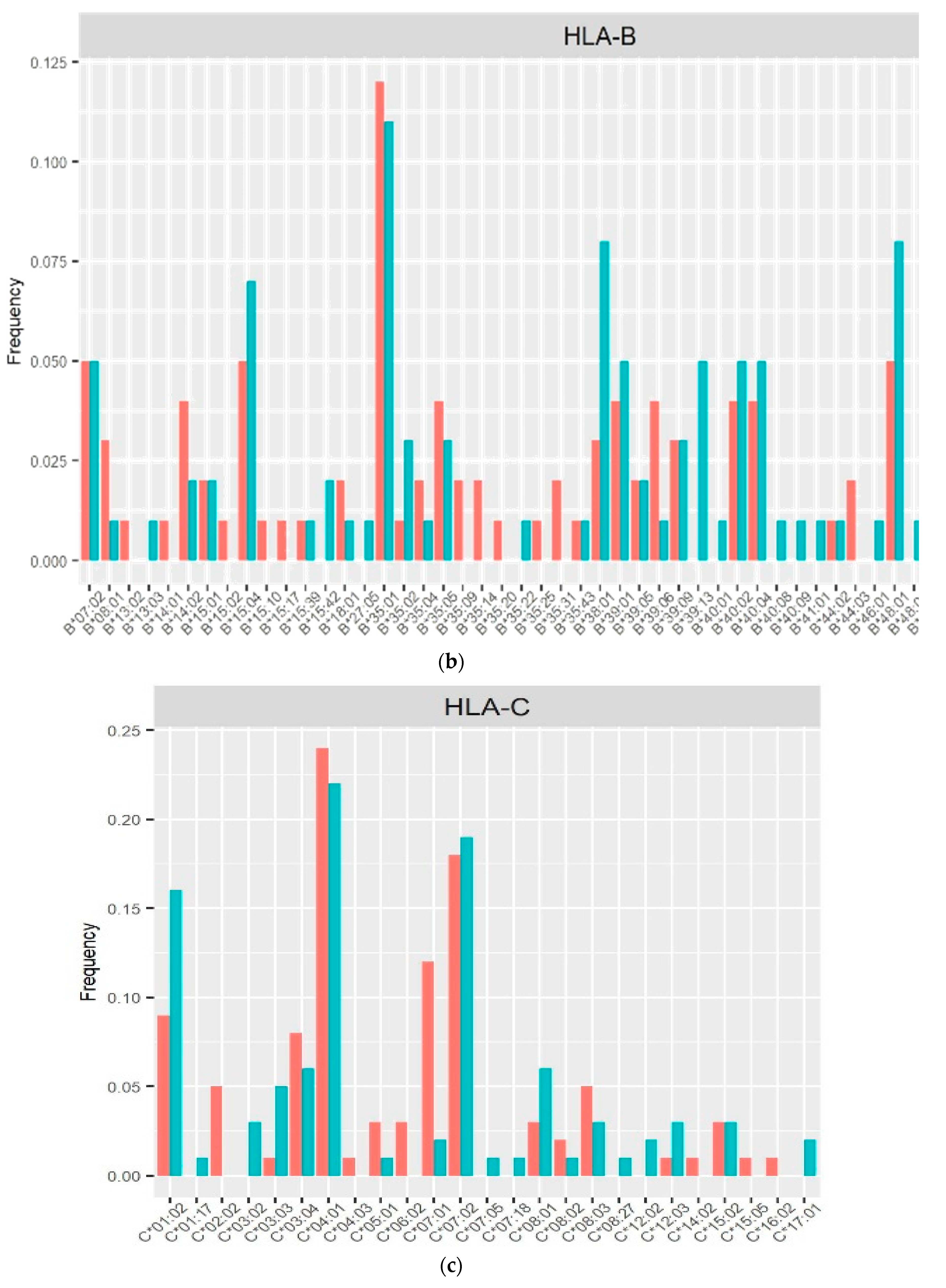
Figure 2.
Frequency distribution of HLA alleles class II in PLHIV and HIV-uninfected based on 2-field resolution HLA imputation.
Figure 2.
Frequency distribution of HLA alleles class II in PLHIV and HIV-uninfected based on 2-field resolution HLA imputation.
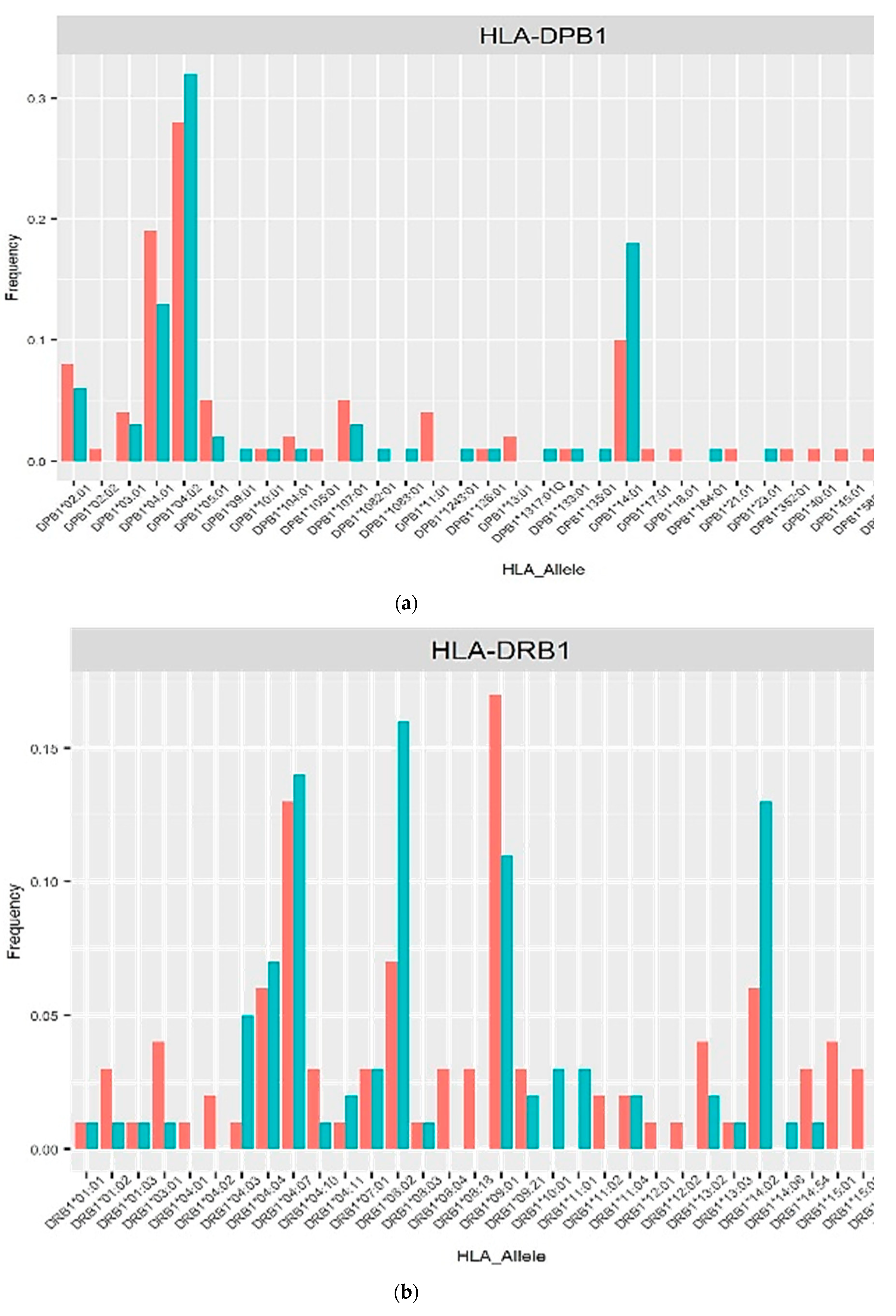
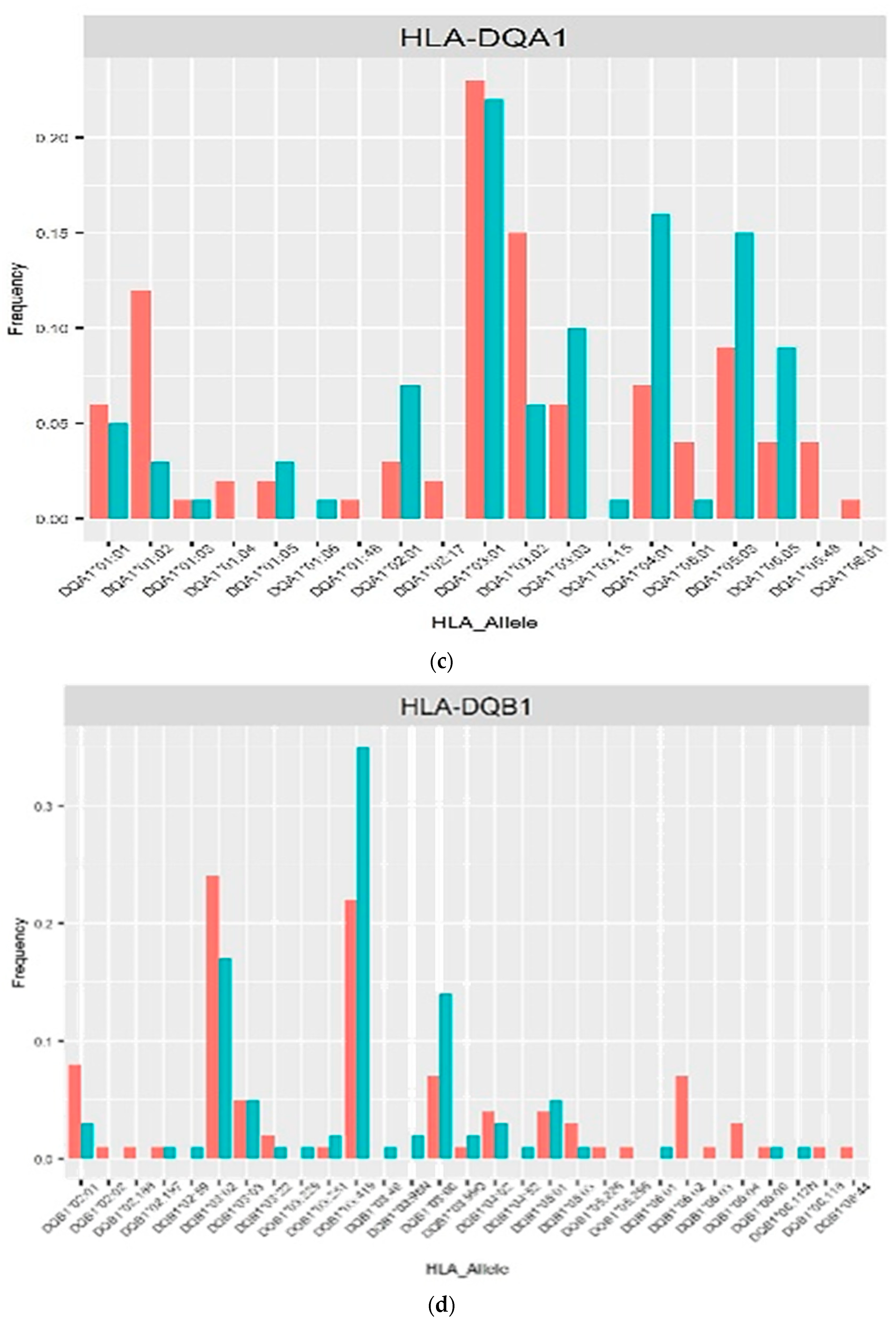

Table 1.
Sociodemographic, clinical and ART treatment characteristics of PLHIV and HIV-uninfected in Peru.
Table 1.
Sociodemographic, clinical and ART treatment characteristics of PLHIV and HIV-uninfected in Peru.
| Characteristics | PLHIV (n=59) |
HIV-uninfected (n=46) |
p |
|---|---|---|---|
| Sex, n (%) | |||
| Female | 36 (61.02) | 1 (2.17) | 0.0000 *a |
| Male | 23 (38.98) | 45 (97.83) | |
| Age at entry (Mean ± SD) | 41.05 ± 11.08 | 35.78 ± 10.23 | 0.0132 *a |
| Initial CD4 cell count (cells/mm3), n (%) | 634.00 ± 291.50 | 952.67 ± 316.36 | 0.0000 *b |
| < 200 | 4 (6.78) | 0 | |
| 200–499 | 13 (22.03) | 2 (4.35) | |
| ≥ 500 | 42 (71.19) | 44 (95.65) | |
| Birthplace, n (%) | |||
| Central Coast region: Lima | 37 (62.71) | 38 (82.61) | |
| North Coast region | 15 (25.42) | 5 (10.87) | |
| Andean region | 7 (11.86) | 1 (2.17) | |
| Amazon region | 0 | 2 (4.35) | |
| Use of ART, n (%) | |||
| Yes | 59 (100.00) ** | - | |
| No | - | - | |
| ART regimen, n (%) | |||
| 2 PIs + 1 IIs | 1 (1.69) | - | |
| 2 PIs + 2 NRTIs | 5 (8.47) | - | |
| 2 NRTIs + 1 IIs | 24 (40.68) | - | |
| 2 NRTIs + 1 NNRTIs | 26 (44.07) | - | |
| Drugs, n (%) | |||
| NRTIs | |||
| Lamivudine (3TC) | 50 (84.75) | - | |
| Tenofovir (TDF) | 51 (86.44) | - | |
| Emtricitabine (FTC) | 5 (8.47) | - | |
| Abacavir (ABC) | 3 (5.08) | - | |
| Zidovudine (AZT) | 1 (1.69) | - | |
| NNRTIs | |||
| Efavirenz (EFV) | 26 (44.07) | - | |
| PIs | |||
| Ritonavir (RTV) | 6 (10.17) | - | |
| Lopinavir (LPV) | 4 (6.78) | - | |
| Atazanavir (ATV) | 1 (1.69) | - | |
| Darunavir (DRV) | 1 (1.69) | - | |
| IIs | |||
| Dolutegravir (DTG) | 21 (35.59) | - | |
| Raltegravir (RAL) | 4 (6.78) | - | |
| Sexual Orientation, n (%) | |||
| Homosexual | - | 14 (30.43) | |
| Heterosexual | - | 26 (56.52) | |
| Bisexual | - | 6 (13.04) | |
| Sex encounters with sex workers, n (%) | |||
| Yes | - | 46 (100.00) | |
| No | - | - | |
| No. of sex partners in last 12 month | |||
| Less than 5 sexual partners | - | 3 (6.52) | |
| 5 – 15 sexual partners | - | 35 (76.09) | |
| More than 15 sexual partners | - | 8 (17.39) |
a Pearson’s chi-squared test; b Mann–Whitney U test; * p-value < 0.05. ART: antiretroviral therapy; PLHIV: people living with HIV; NNRTIs: non-nucleoside reverse transcriptase inhibitors; NRTIs: nucleoside reverse transcriptase inhibitors; PIs: protease inhibitors; IIs: integrase inhibitors. **Two patients did not specify their medications.
Table 2.
Summary of imputed HLA locus for PLHIV and HIV-uninfected in Peru.
| HLA locus | Number of alleles | Alleles by locus, total | |
|---|---|---|---|
| PLHIV (n=56) |
HIV-uninfected (n=44) |
||
| HLA-A | 112 | 88 | 200 |
| HLA-B | 112 | 88 | 200 |
| HLA-C | 110 | 88 | 198 |
| HLA-DPB1 | 112 | 88 | 200 |
| HLA-DQA1 | 108 | 88 | 196 |
| HLA-DQB1 | 112 | 88 | 200 |
| HLA-DRB1 | 112 | 88 | 200 |
Table 3.
Haplotype frequencies of HLA class I and II systems and the association of PLHIV and HIV-uninfected in Peru.
Table 3.
Haplotype frequencies of HLA class I and II systems and the association of PLHIV and HIV-uninfected in Peru.
| HLA alleles | Allele frequency | Allele effect | |||
|---|---|---|---|---|---|
| PLHIV | HIV-uninfected | OR (95% CI) | p | p adj | |
| Susceptible | |||||
| C*07:01 | 0.1182 | 0.0227 | 10.222 (1.40-74.55) | 0.0219 | 0.0101 |
| DQA1*03:02 | 0.1481 | 0.0568 | 5.2972 (1.48-19.02) | 0.0106 | 0.0051 |
| DRB1*09:01 | 0.1696 | 0.1136 | 4.7880 (1.39-16.44) | 0.0129 | 0.0119 |
| Protective | |||||
| DQB1*03:419 | 0.2232 | 0.3523 | 0.3273 (0.11-0.96) | 0.0412 | 0.0478 |
OR: odds ratio; CI: confidence interval. P-values were calculated using a logistic regression model. The adjusted p-values (P adj) were calculated using 10,000 permutation results and by imposing a false discovery rate (FDR) of 0.05. A p-value < 0.05 was considered statistically significant (highlighted in bold).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



