
Submitted:
30 September 2024
Posted:
02 October 2024
You are already at the latest version
Abstract
Atherosclerosis and cardiovascular disease are associated with high morbidity and mortality in industrialized nations. The Tyro3, Axl, and Mer (TAM) family of receptor tyrosine kinases is involved in amplification or resolution of atherosclerosis pathology and other cardiovascular pathology. The ligands of these receptors, Protein S (PS) and growth arrest specific protein 6 (Gas6), are essential for TAM receptor functions in amplification and resolution of atherosclerosis. The Axl-Gas6 interaction has various effects on cardiovascular disease. Mer and PS dampen inflammation, thereby protecting against atherosclerosis progression. Tyro3, the least studied TAM receptor in cardiovascular disease, appears to protect against fibrosis in post-myocardial infarction injury. Ultimately, PS, Gas6, and TAM receptors present an exciting avenue of potential therapeutic targets against inflammation associated with atherosclerosis and cardiovascular disease.
Keywords:
Protein S
; Gas6
; atherosclerosis
; TAM receptors
; cardiovascular disease
; myocardial infarction
; apoptosis
Abstract Atherosclerosis and cardiovascular disease are associated with high morbidity and mortality in industrialized nations. The Tyro3, Axl, and Mer (TAM) family of receptor tyrosine kinases is involved in amplification or resolution of atherosclerosis pathology and other cardiovascular pathology. The ligands of these receptors, Protein S (PS) and growth arrest specific protein 6 (Gas6), are essential for TAM receptor functions in amplification and resolution of atherosclerosis. The Axl-Gas6 interaction has various effects on cardiovascular disease. Mer and PS dampen inflammation, thereby protecting against atherosclerosis progression. Tyro3, the least studied TAM receptor in cardiovascular disease, appears to protect against fibrosis in post-myocardial infarction injury. Ultimately, PS, Gas6, and TAM receptors present an exciting avenue of potential therapeutic targets against inflammation associated with atherosclerosis and cardiovascular disease.
1. Introduction
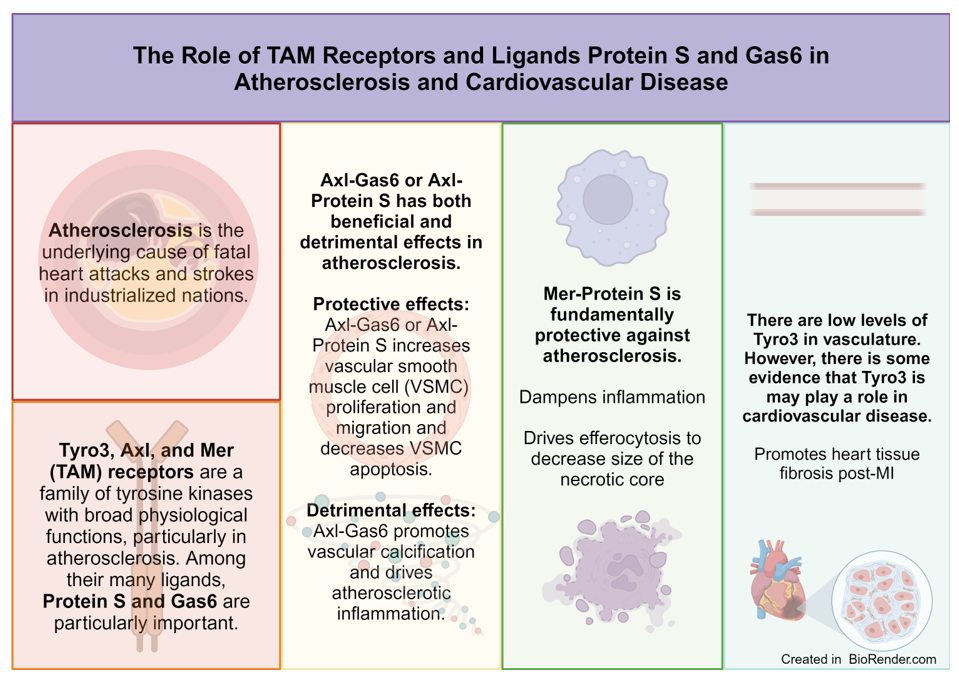
Advanced atherosclerosis is the cause of fatal heart attacks and strokes, which are responsible for most deaths in industrialized nations [1,2]. From cell and mouse models to clinical studies of humans, there is extensive evidence suggesting that the immune system has a decisive function in the development of atherosclerotic plaques. Atherosclerosis is characterized by non-resolving chronic inflammation of the arterial intima, both the innate and adaptive immune systems are essential in promoting and mitigating cardiovascular pathology [3].
Cardiovascular pathology begins at the level of the blood vessel. Each artery wall has three layers: 1) the inner layer or intima, 2) the media layer, and 3) the outer layer or adventitia. The intima is composed of a single layer of endothelial cells that are firmly attached to a thin basal membrane and accompanied by a subendothelial layer of collagen fibers [4]. The media layer consists of smooth muscle cells (SMCs) and a fibrous network of collagen and elastin [5]. Adventitia is loose connective tissue that forms a sleeve around the vessel [4]. Normally, immune cells patrol the vessel wall and return to the blood circulation [6].
Retention of apolipoprotein B in the subendothelial layer of arteries leads to oxidation of accumulated fatty acids and thus initiates an inflammatory response in the blood vessel [7]. As the atherosclerotic plaque grows, leukocytes, primarily monocyte-derived macrophages, enter the wall and phagocytose oxidized and unoxidized cholesterol-rich lipoproteins [8,9]. Excess ingestion of lipid transforms macrophages into foam cells that secrete extracellular matrix to trap lipoprotein and sequester proinflammatory cytokines [2,3,10]. The increase in inflammatory mediators and endothelial cell expression of adhesion molecules promotes recruitment of additional monocytes, T cells, and neutrophils, thereby sustaining inflammatory stimuli [11]. Extracellular neutrophil traps composed of DNA and proteins appear to promote inflammation by increasing interleukin-1β production [12]. Chronic inflammation of the vessel wall is promoted by adaptive immune cells such as T and B lymphocytes. In atherosclerotic plaques, there are abnormal T helper (Th) types and two varieties of B cells, B1 and B2 cells [10,13].The B1 cells generate antibodies to intra-lesional lipids, whereas B2 cells promote the progression of disease-causing lesions [13].
Because of the release of sustained cytotoxic factors, lesional cells are unable to leave the plaque, and they become apoptotic [14]. In early stages of atherosclerotic lesions, apoptotic cells are successfully cleared by neighboring macrophages to limit lesion cellularity. However, with time, efferocytosis fails and apoptotic cells accumulate to form a large, highly necrotic “core”, the hallmark of advanced atherosclerotic disease [14,15,16]. The necrotic nucleus of the plaque causes an imbalance in the tight plaque structure and eventually leads to dilaceration or rupture. Blood clotting then causes partial or complete vessel blockage and leads to acute thrombotic cardiovascular events, such as myocardial infarction, unstable angina, cardiac death, or stroke [14]. Thus, clearance of dead cells is essential to prevent clinically significant atherosclerotic plaque development [14,16].
Global changes to the blood vessel occur because of attempts by the immune system to resolve the insult. Proliferation of smooth muscle cells (SMCs) surrounding the plaque thickens the vessel and the adventitia, and the vessel wall also accumulates activated immune cells. Various immune cells normally are present in the arterial wall, and progressive atherosclerosis elevates their number significantly [17]. Clinically, progressive atherosclerosis is indicated by high plasma levels of low-density lipoproteins (LDL) [18]. Advanced atherosclerosis is often present in conjunction with high blood pressure, which also activates endothelial cells because of unresolved inflammation [19]. Hypertensive conditions in blood vessels promote foam cell formation and subsequent cytokine production, enhancing atherosclerosis [20]. Figure 1 illustrates these concepts.
The TAM receptor family was originally described as a group of orphan receptors. In the 1990s, the TAM ligands were identified, namely growth arrest protein 6 (Gas6) and Protein S (PS). The ligands have different affinities for the individual TAM receptors [21,22]. Gas6 associates with all receptors; its affinity is greatest for Axl, then Tyro3, and lastly Mer. Conversely, PS does not bind to Axl and has a greater affinity for Tyro3 compared with Mer [23]. Different tissues and immune cells exhibit various levels of expression in each of these receptors. Mer is highly expressed in macrophages, whereas Axl and Tyro3 are prominently expressed in dendritic cells [24,25].
TAM receptors have essential functions in homeostasis, particularly in driving immunosuppression by inhibiting T cells and promoting efferocytosis to elicit immunosuppressive cytokines [26]. Thus, pan-inhibition of TAM receptors is expected to remove their immunosuppressive properties and has been suggested as an alternative cancer therapy by improving antitumor immunity [27].
Both Protein S and Gas6 proteins undergo vitamin K-mediated γ-carboxylation, and they share structural homology of approximately 42% with similar domain composition. Despite structural similarity, each protein has distinct functions. Protein S circulates in the plasma at ~350 nM and is involved in hemostasis, apoptosis, inflammation, and atherosclerosis [28] [29]. In its hemostasis function, PS, encoded by the PROS1 gene in humans is predominantly expressed in the liver and secreted to mediate anticoagulation. Protein S is expressed as a signaling molecule in several other tissues [30]. In human plasma, 40% of PS exists in a free (physiologically active) form to mediate anticoagulation, and 60% of PS is bound to C4 binding protein (C4-BP), an acute phase protein, to mediate other physiological roles of PS [31]. Protein S contains an amino terminal γ carboxyglutamic acid (GLA) domain, followed by a thrombin-sensitive loop region and 4 epidermal growth factor-like domains [29]. The C-terminal region consists of 2 laminin G domains that comprise the sex hormone-binding globulin domain and is sufficient for TAM receptor binding and receptor autophosphorylation [32].
Gas6 also has a thrombin-sensitive region (a disulfide-bridged thumb loop) but, unlike PS, the corresponding Gas6 region cannot be cleaved by serine proteases [33]. Gas6 circulates in plasma at a much lower concentration of 0.25 nM and is elevated in patients with severe sepsis [34]. Unlike Protein S, Gas6 is not produced in the liver but instead it is expressed in the lungs, kidneys, and heart. Gas6 has several functions in endothelial cells (ECs), vascular smooth muscle cells (VSMCs) and bone marrow [35]. Human platelets aggregate via TAM activation by Gas6 [36].
Defects in TAM-induced efferocytosis and the resulting inflammation promote atherosclerosis progression [37]. Recent results from the CANTOS trial (Canakinumab Anti-Inflammatory Thrombosis and Outcomes Study) have shown that interleukin-1β inhibition could reduce cardiovascular events. Although the CANTOS trial showed a significant increase in lethal infections in study participants, the CANTOS trial provided the first evidence that targeting inflammation to lower the frequency of cardiovascular events is effective regardless of a reduction in lipid levels [38]. Future therapeutic advances targeting inflammation in atherosclerosis treatment may indicate treatments that modulate TAM receptors. Herein, we discuss recent research regarding the functions of TAM receptors and their PS and Gas6 ligands in atherosclerosis.
2. Discussion
1. TAM Receptors
a. Structure
The structure of TAM receptors includes an extracellular N-terminal region with two immunoglobulin (Ig)-like domains. Following the Ig-like domains, there are two fibronectin type III domains, a hydrophobic domain that crosses the cell membrane, and an intracellular tyrosine kinase C-terminal domain [39].
Although Gas6 and PS differ in their functions, both proteins contain a 50-residue Gla-domain. The Gla-domain has high affinity for calcium and promotes binding of phosphatidylserine (PtdSer) molecules to the surface of platelets as well as to the outer leaf of cell membranes on apoptotic cells [40]. Structurally opposed to the Gla domain is the C-terminus of Gas6 and PS that possess laminin G (globular) domains that facilitate interactions with the TAM receptor Ig-like domains. Like other tyrosine kinase receptors, ligand binding facilitates dimerization, autophosphorylation, and coupling with proteins from different signaling pathways [41].
b. Function
Unlike most receptor tyrosine kinases, TAM receptors are not required for embryonic development. However, adult expression of TAM receptors is essential. TAM receptor triple knockout (KO) mice develop common phenotypes like male infertility and blindness [42,43]. Infertility results from inability to clear apoptotic gamete cells during spermatogenesis in the testis [44]. Similarly, retinal epithelial cells are unable to engulf the outer segment of photoreceptors and toxic byproducts of phototransduction remain. Thus, retinal cells undergo apoptosis leading to blindness [43].
Particularly, in atherosclerosis, TAM receptors are critically important to help clear apoptotic cells in atherosclerotic lesions. However, in advanced atherosclerosis, efferocytosis is less functional, which contributes to post-apoptotic necrosis of lesional cells [45]. Large areas of plaque necrosis develop, making plaques susceptible to rupture or erosion [22].
Surprisingly, TAM receptors also have an important function in preventing autoimmunity [46]. Failure to clear apoptotic bodies from damaged or dying cells leads to necrosis and development of self-antigen [47]. For example, TAM triple KO mice develop lymphoproliferative disorder from broad spectrum autoimmunity due to chronic hyperactivation of dendritic cells (DCs), monocytes, and macrophages [48].
Likewise, deregulation of TAM-mediated suppression strongly influences cardiovascular immunity. Under normal conditions, TAM receptors are upregulated upon innate immune cell activation to prime the system for negative feedback. After the adaptive response is initiated, increased TAM receptor ligand availability promotes taming of the immune response [49].
After activation of innate immune cells, ligand-mediated autophosphorylation of TAM receptors blocks activation of the type 1 interferon receptor (IFNAR)-STAT1 complex. Normally, IFNAR-STAT1 drives inflammation by association with the R1 subunit and IFNAR, and anti-inflammatory molecular signaling begins. Transcription of suppressor of cytokine signaling (SOCS) 1 and SOCS3 proteins begins and ultimately suppresses both cytokine receptor and Toll-Like receptor (TLR) 3, TLR4, and TLR9 pro-inflammatory pathways [50]. Downstream signaling of IFNAR-STAT1 is essential for T cells, which upon activation in T cells produces PS which acts on dendritic cell (DC) TAM receptors [51]. Deregulation of the interaction between T cells and DC in atherosclerotic lesions enhances inflammation surrounding the plaque; thus, interruption of the interaction and presents a potential therapeutic approach [52].
c. Regulation
Regulation of TAM receptors occurs at the transcriptional, post-transcriptional, and protein levels [53]. Various cytokines such as transforming growth factor beta (TGF-β) and granulocyte-macrophage colony-stimulating factors (G-CSF) upregulate expression of Axl. Cytokines interleukin-17A and IL-10 and dexamethasone drive Mer expression [54]. Wang et al. suggested that downregulation of Tyro3 occurs in inflammation associated with autoimmune disorders. However, little is known about cytokines that affect Tyro3 expression [55]. Other studies have reported microRNA regulation of Axl and Tyro3 in different disease pathologies. Endisha et al. reported that microRNA-34a suppresses Axl protein expression, which contributes to rheumatoid arthritis and osteoarthritis [56]. Similarly, microRNA7 inhibits Tyro3 expression and is being examined as an RNA-based therapeutic for treating aberrant Tyro3 overexpression in human colorectal carcinoma [57].
Once TAM receptors are fully synthesized and activated, their extracellular domain can be cleaved by the metalloproteinase A Disintegrin and Metalloproteinase-17 (ADAM-17). The cleavage liberates a soluble extracellular domain bound to soluble Axl or active Axl and destroys TAM receptor activity [58]. Conversely, secretory leukocyte protease inhibitor increases expression of Mer on the cell surface by inhibiting cleavage [59].
1. Gas6-Axl in Cardiovascular Disease
a. Gas6-Axl and Regulation of Cardiovascular Remodeling and Vascular Calcification
Vascular remodeling involves structural changes to the vascular wall in response to injury or inflammatory mediators; chronic conditions such as hypertension or atherosclerosis drive vascular remodeling [60]. Endothelial cells (ECs), vascular smooth muscle cells (VSMCs), fibroblasts, and myofibroblasts contribute to vascular remodeling by four main processes: 1) cellular migration, 2) proliferation, 3) survival, and 4) modification of extracellular matrix. In each of these processes, Axl and its ligands have key functions [61].
Protein S and Gas6 are secreted by, and drive proliferation of, vascular smooth muscle cells. Melaragno et al. showed that, after balloon injury in a rat carotid artery, Axl expression was increased in VSMCs and upregulation of Gas6 and Axl was temporally correlated with neointima formation [62]. In Axl double KO mice, Chen et al. found that vessel injury resulted in reduced intimal thickening compared with control mice, and Axl deficiency led to reduced systolic blood pressure and reduced modeling of the mesenteric artery in a mouse model of hypertension [63]. We need further understanding of the function of Gas6-Axl signaling in VSMC proliferation and migration.
In hypertension, Angiotensin II (Ang II) is produced as a byproduct of the renin-angiotensin system. Angiotensin II has an important function in several cardiovascular pathologies, including vascular remodeling and neointima formation [64]. Interestingly, Ang II also activates the Gas6-Axl pathway and is needed for the effects of Axl on VSMC proliferation [65]. Likewise, reactive oxygen species (ROS) elicit pathological effects on vasculature partially through Axl expression in VSMCs [66]. By inducing interactions between Axl glutathiolated non-muscle myosin heavy chain (MHC)-IIB, ROS increases VSMC migration in vascular injury. In targeting Axl in vitro, oxidative stress significantly decreases and reduces VSMC migration [67]. Lee et al. found that Axl deficiency showed an increase in VSMC apoptosis and reduces intima-media thickening following arterial ligation, suggesting that Axl/Gas6 may prolong VSMC survival [68].
After vascular injury, VSMCs proliferate and migrate, aggravating vascular disease. Although the molecular mechanisms of neointima formation are not fully understood, Lee et al. reported that extracellular vesicles (EVs) are mediators of intercellular communication that aids neointima formation [69]. McShane et al. showed that small extracellular vesicles increase following ligation of the right carotid artery in rats, and subsequently neointima formation is aggravated via interaction with membrane PtdSer [23]. Furthermore, incubation of damaged vessels with EVs enhanced Axl and Mer phosphorylation in VSMCs and increased downstream signaling pathways associated with Akt, extracellular signal-regulated kinase (ERK), and focal adhesion kinase (FAK) [69]. Okamoto et al. reported that transcription factor YAP increases expression of Axl, constituting an Axl-positive feedback loop [70]. Small molecule inhibitors of both Axl and Mer hindered VSMC proliferation and migration [71]. This evidence shows the potential of Axl inhibitors to prevent aggravation of vascular injury associated with atherosclerosis.
Similarly, Liu et al. showed that Axl-Gas6 signaling promoted mitogenic effects on fibroblasts, protecting cells from apoptosis [72]. Signaling also affects survival and migration of endothelial cells by upregulation of vascular endothelial growth factor A [73]. Axl also directly regulates cytokine/chemokine expression and ECM remodeling in the vessel wall. Ultimately, Axl mediates these functions by activation of phosphatidylinositol-3-OH kinase (PI3K)/protein kinase B (Akt), sarcoma (SRC) signaling pathways and extracellular signal-regulated kinases (ERKs), like other receptor tyrosine kinase-mediated processes [74].
Excessive stretching of endothelial cells releases Gas6 which activates Axl on monocytes and promotes inflammation [75,76]. With a mouse model of hypertension, Chen et al. demonstrated that brief hypertensive episodes induced chronic aortic remodeling consistent with persistent low-grade inflammation of the aorta and kidneys. Furthermore, blockade of Axl and subsequent blockade of Gas/Axl signaling lessened the severity of hypertension [63].
Interestingly, glucose concentration affects Axl behavior in vitro. Zdzalik-Bielecka et al. exposed VSMCs to low glucose concentration (5.5 mmol/L) and high glucose concentration (27.5 mmol/L). They found that Axl preferentially interacted with proteins in PI3K signaling pathway in the low glucose condition [77]. In low-glucose conditions, Axl stimulates anti-apoptotic signaling and enhances survival of VSMCs. In contrast, Axl is associated with signaling proteins of the ERK1/2 pathway in high glucose conditions, driving VSMC migration. This finding may have important implications in our understanding of metabolic syndrome [78].
Vascular calcification is another process that enhances inflammation associated with advanced atherosclerosis. Calcium build-up in the vasculature correlates with worse clinical outcomes [79]. However, in the absence of calcification in coronary arteries, persons have a low risk of cardiovascular disease even if they have high levels of traditional risk factors [80]. Vascular pericytes undergo osteogenic differentiation during calcification and in vitro Axl is downregulated during this osteogenic differentiation [81]. Levels of Axl also decrease when cultured VSMCs calcify their matrix, and Axl overexpression prevents calcification [82]. Notably, Badi et al. showed that miR-34a promotes VSMC calcification, and calcification correlated with decreased Axl expression in cultured VSMCs [83]. Interestingly, Son et al. Found that hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins) prevent phosphate-induced calcification by VSMCs in vitro via restoration of the Gas6-Axl mediated survival pathway [84]. It is unknown whether Axl affects vascular calcification in vivo.
b. Gas6-Axl and Inflammation in Cardiovascular Disease
High levels of vasculature inflammation promote cardiovascular pathology. Although there are no studies of the impact of Axl on atherosclerosis pathology, Gerloff et al. found that Axl expression was downregulated in atherosclerotic plaques compared with normal carotid arteries [85]. Interestingly, Axl expression is higher in vessels such as the left internal mammary artery, which is less prone to develop atherosclerosis compared with vessels more prone to developing atherosclerosis, such as the aorta [86]. Conversely, Liu et al. detected elevated levels of soluble Axl in acute coronary syndrome, and Gas6 expression increases in ECs, VSMCs, and macrophages associated with atherosclerosis development [87]. Some groups suggest that genetic knockout of Gas6 increases plaque stability although in clinical studies, Sunbul et al. found that levels of Gas6 correlated with increased risk of coronary artery disease in patients with psoriasis [88]. However, recently You et al. showed that Axl signaling dampened dendritic cell maturation when exposed to cholesterol and Axl may be a target to prevent stimulation of dendritic cells [89]. Axl is also necessary for the survival of T lymphocytes, and Axl influences vascular remodeling and inflammation in a mouse model of hypertension. Axl drives pro-inflammatory activation of VSMCs in vein graft remodeling. Depletion of Axl in the vein graft donor and recipient mouse leads to decreased levels of Axl, less immune activation, and subsequent downregulation of various pro-inflammatory cytokines [90]. Axl deficiency increases the expression of SOCS1 in VSMC, contrary to what is observed in immune cells [23,90].
Axl also has a function in hypertension [91]. Axl-expressing immune cells drive pro-inflammatory gene expression and increase immune cell infiltration in the kidneys at early stages of hypertension and was detrimental in late stages of hypertensive disease [92]. Li et al. showed in a recent clinical study that decreased serum concentrations of Gas6, Axl, and soluble Axl were associated with high HbA1c values and could predict the severity of diabetic nephropathy, which is often associated with hypertension [93].
In patients with heart failure and post ST-segment elevation myocardial infarction, Axl and soluble Axl levels are elevated in cardiac tissue [94]. Axl level is predictive of adverse pathology, such as the extent of left ventricle remodeling and future cardiovascular events [95,96]. Decreased hypertrophy, fibrosis, and contractile dysfunction from chronic stress induced by aortic banding was demonstrated in Gas6 deficient-mice, and cardiac-specific Gas6 overexpression enhanced these pathologies. Gas6 acts through the ERK pathway to drive cardiac hypertrophy, and reversal of hypertrophy was shown with ERK inhibitor treatment [65]. Hence, the Gas6-Axl interaction might be a therapeutic target in heart failure.
Axl and Mer are necessary for macrophage activation following sterile wound healing associated with myocardial infarction [97]. In myocardial ischemia/reperfusion infarction, cross signaling between Axl and TLR4 in cardiac macrophages directed a switch from glycolysis to lipid metabolism and secretion of proinflammatory IL-1β [97,98]. This maladaptive condition promoted increased intramyocardial inflammation, impaired contractile function, and adverse ventricular remodeling [97]. Although Mer is cardioprotective, Axl functions independently to reduce the efficacy of cardiac repair. However, small molecule inhibitors of Axl and Mer cleavage significantly improved cardiac healing [23]. Figure 2 summarizes key roles of Axl-Gas6 in cardiovascular disease.
2. PS and Mer in Cardiovascular Disease
a. Mer-Mediated Resolution of Inflammation in Atherosclerosis
Resolution of inflammation is a highly coordinated process that counterbalances excess inflammation without compromising host defense to restore tissue homeostasis after injury [99]. When inflammation fails to resolve, it exacerbates numerous chronic inflammatory diseases including atherosclerosis [100].
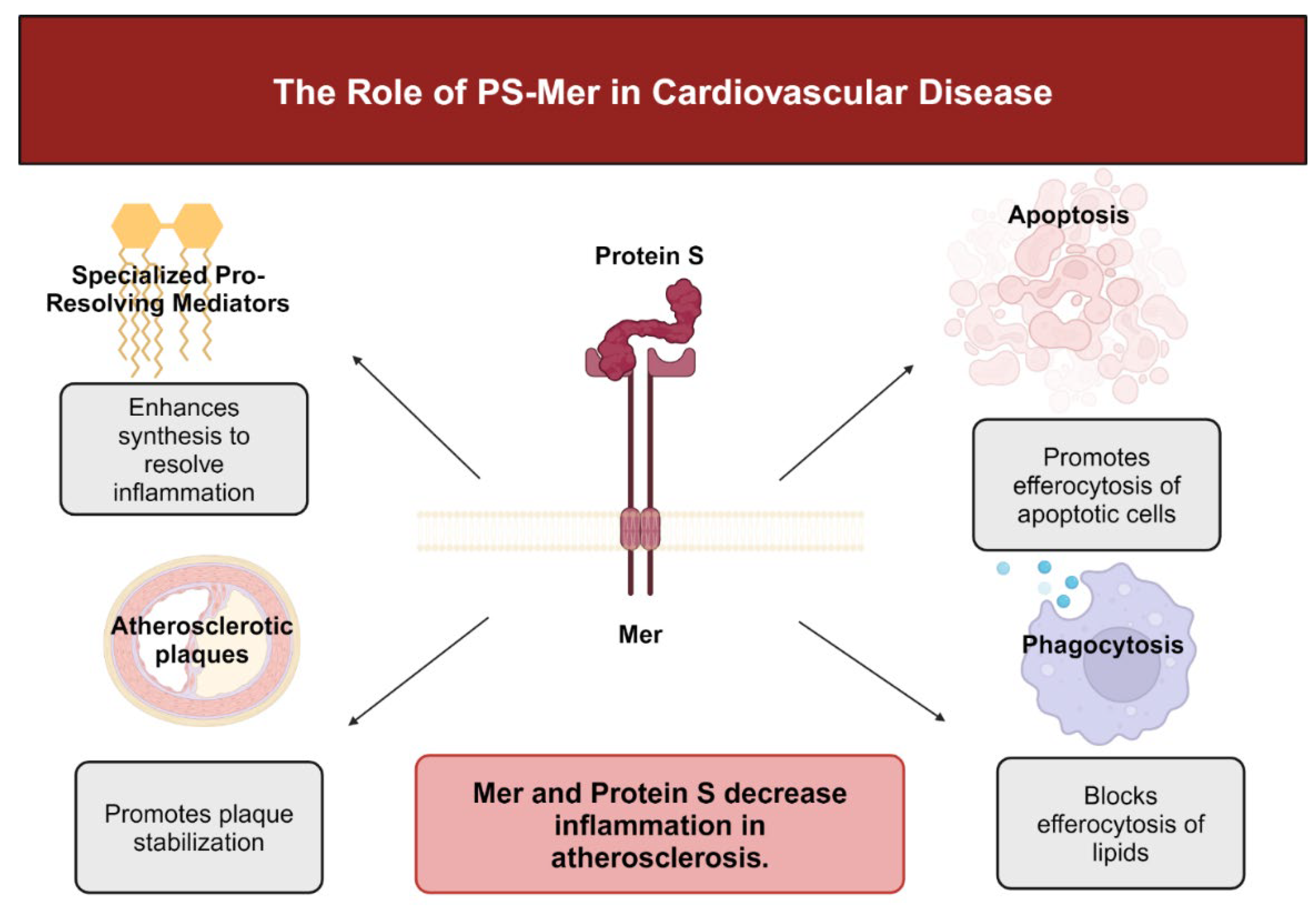
Senescent cells release factors that promote Mer cleavage, thus promoting impaired efferocytosis. However, specialized pro-resolving mediator (SPM) resolvin 1 (RvD1) prevents Mer cleavage [101]. By downstream signaling, Mer promotes SPM production to resolve inflammation [102]. Specialized pro-resolving mediators (SPMs) include lipoxins, protectins, maresins like MaR1, and resolvins such as resolvin 1 and are crucial anti-inflammatory lipid mediators. Lipoxygenase enzymes such as 5-lipoxygenase and 12/15-lipoxygenase convert arachidonic acid to lipoxins such as LX4A and docosahexanoic acid to form resolvins such as RvD1 [103]. Upon release, SPMs block inflammatory cell influx and promote the emigration of inflammatory cells to limit tissue damage and begin the process of healing [104]. In advanced atherosclerosis, pro-inflammatory mediators such as leukotrienes predominate over pro-resolving mediators. Consequently, the ratio of leukotrienes to SPMs is significantly decreased in advanced atherosclerotic plaques in humans and mice [105].
Without effective resolution, plaques become clinically dangerous. Lack of effective efferocytosis in association with damage associated molecular pattern (DAMP)-mediated inflammation promotes formation of a necrotic core and thinning of the protective collagen cap that overlies the core [106]. Evidence suggests that SPMs decrease plaque necrosis and inflammation [107]. For example, administration of RvD1 decreased the size of necrotic cores, improved intralesional efferocytosis and thickened collagenous fibrous caps in a mouse model of atherosclerosis. Consequently, lesion size decreased, and platelet and leukocyte influx lessened [108]. When treated simultaneously with RdV1 and maresin MaR1, plaque stability prevailed by decreasing lesional macrophage numbers, thickening collagen caps, and halting necrotic core growth [109]. Maresin-1 prevents atherosclerotic progression by suppressing necrosis and limiting macrophage numbers while simultaneously increasing the fibrotic strength of the smooth muscle coat [110].
Unlike the contradictory functions of Axl in promoting and inhibiting cardiovascular pathology, Mer-mediated efferocytosis and inflammation resolution has a protective function in atherosclerosis by limiting formation of an inflammatory necrotic core [111]. Liao et al showed that Mer KO mice had decreased clearance of apoptotic bodies. Consequently, plaque necrosis and inflammation were accelerated leading to advanced atherosclerotic lesions [37]. Because of downregulation of Mer in atherosclerotic plaques, atherosclerosis becomes more severe. Mer-mediated efferocytosis in atherosclerosis is prevented by macrophages that express calcium/calmodulin-dependent protein kinase II gamma (CaMKII-gamma). CaMKII-gamma prevents Mer expression by inhibiting transcription factors ATF6 and LXR-α [112]. Similarly, the immunoproteasome subunit β-5i decreases Mer expression in macrophages and enhances necrotic core area in atherosclerotic lesions [113]. Patel et al. suggested that C-C chemokine receptor type 2 expressed by infiltrating circulatory monocytes negatively affects the ability of Mer to drive phagocytic repair following ischemia-reperfusion [114].
When Mer-mediated efferocytosis functions in an atherosclerotic plaque, Mer binds apoptotic cells by way of bridging molecules Gas6 or PS and mediates uptake of apoptotic debris via actin signaling pathways [115]. Unlike Axl, Mer is the dominant efferocytosis receptor in lesional clearance of apoptotic debris, whereas Axl expressed on dendritic cells does not affect efferocytosis [116]. Mer deficiency enhances pathology in mouse models of atherosclerosis, showing a marked decrease in apoptotic body clearance and subsequent development of large necrotic plaque lesions [117].
Efferocytosis via the Mer receptor stimulates formation of specialized pro-resolving mediators (SPM) [118]. Activation by apoptotic cells or Mer ligands such as Gas6 and Protein S promotes the cytoplasmic localization of non-phosphorylated 5-lipoxygenase in macrophages. Upon activation of ERK in macrophages, expression of sarcoplasmic/endoplasmic reticulum calcium ATPase 2 increases consequently decreasing the cytosolic calcium concentration and suppressing CaMKII activity [118,119]. In turn, decreased cytosolic calcium reduces p38 MAP kinase and MAPKAP kinases MK2 activity. Thus, the unphosphorylated cytoplasmic form of 5-lipoxygenase predominates and enhances SPM biosynthesis [118]. Release of SPMs resolves inflammation in the environment surrounding apoptotic and necrotic cells [120]. Mer deficiency in sterile peritonitis and ischemia-reperfusion injury delays healing by suppressing synthesis of SPMs such as lipoxins and resolvins [121,122].
In the development of advanced atherosclerotic lesions, macrophage Mer deficiency occurs near the necrotic cores of human plaques because of ADAM17-mediated Mer cleavage. Mer is cleaved at proline-485, and cells that express Mer lacking residues 483–488 are resistant to ADAM17 cleavage [123]. ADAM17 is activated by the products of polyunsaturated fatty acid oxidation and by inflammatory mediators trapped in the necrotic core. The presence of inflammatory stimuli promotes ADAM17 cleavage of the Mer ectodomain [124]. Levels of the resulting soluble Mer are elevated in murine models of atherosclerosis and in patients experiencing symptomatic atherosclerotic plaques in the carotid artery. Cai et al. showed that soluble Mer, which is the inactive version of the Mer receptor, is marker of defective Mer-mediated efferocytosis and is associated with advanced plaque progression [112].
Kawai et al. showed that macrophages near the necrotic core of an atheroma have lower expression of Mer and higher expression of ADAM17 compared with macrophages on the periphery of atherosclerotic lesions [125]. Furthermore, lower levels of soluble Mer in individual plaques were correlated with worse necrosis, and mice resistant to Mer cleavage showed improvement in lesional efferocytosis, stable plaque formation and inflammation resolution [126]. Thus, in advanced atherosclerosis Mer cleavage may contribute to defective efferocytosis and failed inflammation resolution.
Remarkably, cleavage-resistant Mer in the presence of elevated ADAM17 is protective in different mouse models [127]. In an ischemia-reperfusion-induced lung injury model and the sterile peritonitis model, cleavage-resistant Mer promoted efferocytosis, inflammation resolution, and high circulating levels of SPMs [121]. An atherosclerosis model demonstrated lesions with improved efferocytosis, thicker fibrous caps, increased pro-resolving lipid mediators, and smaller necrotic cores. After myocardial ischemia-reperfusion injury, these mice also had improved efferocytosis, reduced overall infarct size, and improved cardiac function [112,118]. Sufit et al. presented evidence that cleavage-resistant Mer promoted pro-resolving mediators and restored senescence-reduced efferocytosis in models of aged mice, potentially impacting aging [128]. Conversely, Mer KO mice had inherent increased production of pro-inflammatory cytokines such as IFN-γ, IL-12, and tumor necrosis factor α [129]. Consequently, anti-atherogenic IL-10 production is downregulated, demonstrating the function of Mer in dampening inflammation [127].
Regulation of Mer-mediated efferocytosis may also arise from soluble Axl. Weinger et al. showed that, as levels of soluble Axl rise in association with Gas6 in mouse serum, soluble Axl may neutralize Gas6 in serum. By blocking the interaction of Gas6 with TAM receptors, particularly to Mer, soluble Axl indirectly decreases the interaction between Mer and apoptotic cells and limits efferocytosis. Thus, soluble Axl may influence atherosclerosis pathology by a soluble Axl/Gas6/Mer axis [130].
Recently, Wu et al. demonstrated that, in vessels with disturbed flow characteristic of atherosclerosis, absence of Mer resulted in endothelial and mitochondrial dysfunction in the human aorta. Absence of Mer also induced abnormal endothelial thickening associated with decreased endothelial efferocytosis, and Mer absence promoted development of atherosclerosis [131]. Thus, a strategy to regulate Mer in EC efferocytosis may be of therapeutic promise in atherosclerosis.
The relation between Mer and PS also has an important function in modulation of atheroma development [28]. Because PS from plasma accumulates in the necrotic core of a developed plaque, PS interacts with negatively charged phospholipids on the membranes of dying cells. By interaction with Mer, PS inhibits acetylated LDL uptake by macrophages and downregulates scavenger receptor A, which is mainly expressed by macrophages [132]. Thus, PS could have a dual function in advanced lesions to prevent inflammation. Although PS blocks LDL phagocytosis, PS promotes Mer-mediated efferocytosis of apoptotic cells [28].
Protein S-Mer may participate in atherosclerosis by preventing LDL phagocytosis and inhibiting inflammation. Khatana et al. reported that, in the presence of excess lipoprotein, LXR expression is activated, upregulating Mer and mitigating pro-inflammatory cytokine release upon engagement of cholesterol loaded macrophages [133]. By way of Mer, PS also blocks expression of macrophage scavenger receptor A and reduces uptake of modified lipoproteins [119]. Thus, Mer and PS are crucial for attenuation of inflammation in atherosclerosis [28,134]. Figure 3 summarizes these concepts.
b. Mer-Mediated Efferocytosis in Myocardial Infarction
Following myocardial infarction (MI), an essential first step in the healing process is clearance of dead cardiomyocytes by several types of immune cells. DeBerge et al. showed that Mer-expressing monocytes/macrophages are needed to clear injured cardiomyocytes and improve cardiac tissue remodeling after MI in mice [135]. After an experimental MI, Mer KO mice experienced accumulation of apoptotic cardiac cells, resulting in larger infarct sizes and worse cardiac functional outcomes [136]. During homeostasis, cardiomyocytes release nonfunctional mitochondria, known as exophers, in subcellular particles [137]. Exophers are engulfed and cleared by macrophages in cardiac tissue, preventing inflammation and phagocytosis by surrounding cardiomyocytes and maintaining cardiac homeostasis [138]. When Mer is ablated on cardiac macrophages, metabolic function is impaired due to residual exophers in cardiac tissue [139].
Neutrophils also are critical for influencing Mer expression on cardiac macrophages. Secretion of neutrophil gelatinase-associated lipocalin (NGAL) activates Mer on cardiac macrophages to facilitate efferocytosis. Absence of NGAL reduces expression of Mer on the cardiac macrophage membrane thereby impairing efferocytosis [140].
Dying cardiomyocytes induce expression of Mer on their surface, which prevents their engulfment. Subsequently, macrophages upregulate CD47, a “don’t eat me” marker for dying cells that prevents efferocytosis [141]. Cell marker CD47 is often found on lingering dead and necroptotic cells [142]. Treatment with anti-CD47 antibody leads to increase in levels of SPMs such as RvD1 in plaques and suggests another feedforward circuit to inhibit inflammation [126]. Although residual necrotic cardiomyocytes in MI is pathological and may lead to worsening clinical outcomes, preventing efferocytosis may preserve cardiomyocyte numbers in lieu of low regenerative capacity [22]. Treatment of mice with anti-CD47 enhanced cardiomyocyte phagocytosis and decreased infarct size, leading to better post-MI outcomes [126]. Importantly, anti-CD47 treatment may be a therapeutic approach focused on pro-resolving mechanisms instead of direct inhibition of inflammation.
3. Tyro3 in Cardiovascular Disease
Because of low levels of expression of Tyro3 in the vasculature compared with the central nervous system and the reproductive system, the function of Tyro3 in cardiovascular disease has not been studied in detail [143]. Hurtado et al. showed association of Tyro3 and Mer variants with carotid atherosclerosis [144].
However, Tyro3 is thought to suppress a subset of CD11c+ DC that express programmed cell death protein 2 and decrease Th2-associated molecule production [145]. Part of a negative feedback loop, IL-4 increases expression of Protein S in T cells and then activated Tyro3-mediated suppression of Th2 activation. Although IL-4 and IL-5 protect against atherosclerosis, IL-9 may promote atherosclerosis [146]. Furthermore, fibrosis of heart tissue in the process of aging and reperfusion injury is promoted by IL-13 secreted by regulatory T cells which may also promote efferocytosis in atherosclerotic lesions [23,147]. Further studies are needed to elucidate the function of Tyro3 in cardiovascular disease.
3. Conclusions
The TAM receptors, Gas6, and PS have many functions that suppress or drive cardiovascular pathology. There is no consensus to suggest that a detrimental function or protective effect of Axl-Gas6 stems from differences in activation of inflammation to signaling to suppress the immune response. Although Axl may protect against vascular calcification, the function of Axl-Gas6 in taming inflammation in vascular cells is unclear. However, Mer-PS is fundamentally protective in its function against cardiovascular disease. From immune response suppression to immune response resolution, Mer is instrumental in preventing the inflammation characteristic of atherosclerotic lesions. The protective property of Mer in cardiac tissue presents a new therapeutic consideration for heart disease. Of the TAM receptors, Tyro3 remains the least studied in cardiovascular disease, and the few studies that exist suggest that Tyro3 could be a therapeutic target after myocardial injury. In sum, the functions of TAM receptors, Gas6, and PS present an exciting opportunity of therapeutic interventions in cardiovascular setting.
Future Directions
The high mortality and morbidity associated with advanced atherosclerosis necessitates broader understanding of novel therapeutic targets to treat disease that can develop in the absence of major clinical symptoms. Although representing a broad range of physiological activities, the functions of Tyro3, Axl, and Mer receptors may be critically important to treat the clinical complications of cardiovascular disease.
Author Contributions
Conceptualization, TP; Investigation, T.P.; Writing – Original Draft Preparation, T.P.; Writing – Review & Editing, R.M. and S.M.; Supervision, R.M. and S.M.
Funding
This research received no external funding.
Acknowledgments
We thank Dr. Howard Fried for editing the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38(2), 208–211. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and unknown. Pathol. Int. 2022, 72(3), 151–160. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592(7855), 524–533. [Google Scholar] [CrossRef] [PubMed]
- Pugsley, M.K.; Tabrizchi, R. The vascular system. An overview of structure and function. J. Pharmacol. Toxicol. Methods 2000, 44(2), 333–440. [Google Scholar] [CrossRef]
- Pries, A.R.; Secomb, T.W. Control of blood vessel structure: Insights from theoretical models. Am. J. Physiol. Heart Circ. Physiol. 2005, 288(3), H1010–H1015. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100(2), 158–173. [Google Scholar] [CrossRef]
- Mehta, A.; Shapiro, M.D. Apolipoproteins in vascular biology and atherosclerotic disease. Nat. Rev. Cardiol. 2022, 19(3), 168–179. [Google Scholar] [CrossRef]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, (8 Suppl), C7–12. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19(11), 42. [Google Scholar] [CrossRef]
- Hinkley, H.; Counts, D.A.; VonCanon, E.; Lacy, M. T Cells in Atherosclerosis: Key Players in the Pathogenesis of Vascular Disease. Cells 2023, 12(17), 2152. [Google Scholar] [CrossRef]
- Galindo, C.L.; Khan, S.; Zhang, X.; Yeh, Y.S.; Liu, Z.; Razani, B. Lipid-laden foam cells in the pathology of atherosclerosis: Shedding light on new therapeutic targets. Expert. Opin. Ther. Targets 2023, 27(12), 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349(6245), 316–320. [Google Scholar] [PubMed]
- Sage, A.P.; Mallat, Z. Multiple potential roles for B cells in atherosclerosis. Ann. Med. 2014, 46(5), 297–303. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276(6), 618–632. [Google Scholar] [CrossRef]
- Vergallo, R.; Crea, F. Atherosclerotic Plaque Healing. Reply. N. Engl. J. Med. 2021, 384(3), 294. [Google Scholar] [CrossRef]
- Puylaert, P.; Zurek, M.; Rayner, K.J.; De Meyer, G.R.Y.; Martinet, W. Regulated Necrosis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42(11), 1283–1306. [Google Scholar] [CrossRef] [PubMed]
- Sterpetti, A.V. Inflammatory Cytokines and Atherosclerotic Plaque Progression. Therapeutic Implications. Curr. Atheroscler. Rep. 2020, 22(12), 75. [Google Scholar] [CrossRef]
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; Nordestgaard, B.G.; Watts, G.F.; Bruckert, E.; Fazio, S.; Ference, B.A.; Graham, I.; Horton, J.D.; Landmesser, U.; Laufs, U.; Masana, L.; Pasterkamp, G.; Raal, F.J.; Ray, K.K.; Schunkert, H.; Taskinen, M.R.; van de Sluis, B.; Wiklund, O.; Tokgozoglu, L.; Catapano, A.L.; Ginsberg, H.N. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41(24), 2313–2330. [Google Scholar] [CrossRef]
- Leong, X.F.; Ng, C.Y.; Jaarin, K. Animal Models in Cardiovascular Research: Hypertension and Atherosclerosis. Biomed. Res. Int. 2015, 2015, 528757. [Google Scholar] [CrossRef]
- Hurtubise, J.; McLellan, K.; Durr, K.; Onasanya, O.; Nwabuko, D.; Ndisang, J.F. The Different Facets of Dyslipidemia and Hypertension in Atherosclerosis. Curr. Atheroscler. Rep. 2016, 18(12), 82. [Google Scholar] [CrossRef]
- Tutusaus, A.; Mari, M.; Ortiz-Perez, J.T.; Nicolaes, G.A.F.; Morales, A.; Garcia de Frutos, P. Role of Vitamin K-Dependent Factors Protein S and GAS6 and TAM Receptors in SARS-CoV-2 Infection and COVID-19-Associated Immunothrombosis. Cells 2020, 9(10), 2186. [Google Scholar] [CrossRef]
- Cai, B.S.; Kasikara, C. TAM receptors and their ligand-mediated activation: Role in atherosclerosis. Tam. Receptors in Health and Disease 2020, 357, 21–33. [Google Scholar]
- McShane, L.; Tabas, I.; Lemke, G.; Kurowska-Stolarska, M.; Maffia, P. TAM receptors in cardiovascular disease. Cardiovasc. Res. 2019, 115(8), 1286–1295. [Google Scholar] [CrossRef]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19(9), 539–549. [Google Scholar] [CrossRef]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 2007, 178(9), 5635–5642. [Google Scholar] [CrossRef]
- Pu, Y.; Ji, Q. Tumor-Associated Macrophages Regulate PD-1/PD-L1 Immunosuppression. Front. Immunol. 2022, 13, 874589. [Google Scholar] [CrossRef]
- Deryckere, D.; Huelse, J.M.; Earp, H.S.; Graham, D.K. TAM family kinases as therapeutic targets at the interface of cancer and immunity. Nature Reviews Clinical Oncology 2023, 20(11), 755–779. [Google Scholar] [CrossRef]
- Suleiman, L.; Négrier, C.; Boukerche, H. Protein S: A multifunctional anticoagulant vitamin K-dependent protein at the crossroads of coagulation, inflammation, angiogenesis, and cancer. Critical Reviews in Oncology Hematology 2013, 88(3), 637–654. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, A.; Pilli, V.S.; Fried, H.; Majumder, R. Protein S: A Multifunctional Anticoagulant. Biomed. Res. Clin. Pract. 2017, 2(5). [Google Scholar] [CrossRef] [PubMed]
- van der Meer, J.H.; van der Poll, T.; van’t Veer, C. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123(16), 2460–2469. [Google Scholar] [CrossRef]
- Petri, A.; Sasikumar, P.; Folgado, P.B.; Jones, D.; Xu, Y.X.; Ahnström, J.; Salles-Crawley, I.I.; Crawley, J.T.B. TFPIα anticoagulant function is highly dependent on protein S in vivo. Sci. Adv. 2024, 10(5). [Google Scholar]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18(1), 94. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Taheri, M.; Mokhtari, M. A review on the role of GAS6 and GAS6-AS1 in the carcinogenesis. Pathol. Res. Pract. 2021, 226, 153596. [Google Scholar] [CrossRef]
- Ni, J.; Lin, M.; Jin, Y.; Li, J.; Guo, Y.; Zhou, J.; Hong, G.; Zhao, G.; Lu, Z. Gas6 Attenuates Sepsis-Induced Tight Junction Injury and Vascular Endothelial Hyperpermeability via the Axl/NF-kappaB Signaling Pathway. Front. Pharmacol. 2019, 10, 662. [Google Scholar] [CrossRef]
- Dihingia, A.; Kalita, J.; Manna, P. Implication of a novel Gla-containing protein, Gas6 in the pathogenesis of insulin resistance, impaired glucose homeostasis, and inflammation: A review. Diabetes Res. Clin. Pract. 2017, 128, 74–82. [Google Scholar] [CrossRef]
- Zhai, X.; Pu, D.; Wang, R.; Zhang, J.; Lin, Y.; Wang, Y.; Zhai, N.; Peng, X.; Zhou, Q.; Li, L. Gas6/AXL pathway: Immunological landscape and therapeutic potential. Front. Oncol. 2023, 13, 1121130. [Google Scholar] [CrossRef]
- Liao, J.W.; Xie, Y.P.; Lin, Q.Y.; Yang, X.L.; An, X.B.; Xia, Y.L.; Du, J.; Wang, F.; Li, H.H. Immunoproteasome subunit β5i regulates diet-induced atherosclerosis through altering MERTK-mediated efferocytosis in knockout mice. J. Pathol. 2020, 250(3), 275–287. [Google Scholar] [CrossRef]
- Weber, C.; von Hundelshausen, P. CANTOS Trial Validates the Inflammatory Pathogenesis of Atherosclerosis: Setting the Stage for a New Chapter in Therapeutic Targeting. Circ. Res. 2017, 121(10), 1119–1121. [Google Scholar] [CrossRef]
- Burstyn-Cohen, T.; Fresia, R. TAM receptors in phagocytosis: Beyond the mere internalization of particles. Immunol. Rev. 2023, 319(1), 7–26. [Google Scholar] [CrossRef]
- Geng, K. Post-translational modifications of the ligands: Requirement for TAM receptor activation. Int. Rev. Cell Mol. Biol. 2020, 357, 35–55. [Google Scholar]
- Burstyn-Cohen, T.; Maimon, A. TAM receptors, Phosphatidylserine, inflammation, and Cancer. Cell Commun. Signal 2019, 17(1), 156. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, N.; Chen, Q.; Yan, K.; Liu, Z.; Zhang, X.; Liu, P.; Chen, Y.; Han, D. Breakdown of immune homeostasis in the testis of mice lacking Tyro3, Axl and Mer receptor tyrosine kinases. Immunol. Cell Biol. 2013, 91(6), 416–426. [Google Scholar] [CrossRef] [PubMed]
- Yefimova, M.G.; Ravel, C.; Rolland, A.D.; Bourmeyster, N.; Jegou, B. MERTK-Mediated LC3-Associated Phagocytosis (LAP) of Apoptotic Substrates in Blood-Separated Tissues: Retina, Testis, Ovarian Follicles. Cells 2021, 10(6), 1443. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Chen, Q.; Han, D. The roles of TAM receptor tyrosine kinases in the mammalian testis and immunoprivileged sites. Front Biosci (Landmark Ed), 2016; 21, 2, 316–327. [Google Scholar]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Interplay between efferocytosis and atherosclerosis. Arch. Cardiovasc. Dis. 2023, 116(10), 474–484. [Google Scholar] [CrossRef]
- Ranta, A.; Kumar, S. Recent advancements in role of TAM receptors on efferocytosis, viral infection, autoimmunity, and tissue repair. Int. Rev. Cell Mol. Biol. 2020, 357, 1–19. [Google Scholar] [PubMed]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Malawista, A.; Wang, X.; Trentalange, M.; Allore, H.G.; Montgomery, R.R. Coordinated expression of tyro3, axl, and mer receptors in macrophage ontogeny. Macrophage (Houst) 2016, 3. [Google Scholar]
- Peeters, M.J.W.; Rahbech, A.; Thor Straten, P. TAM-ing T cells in the tumor microenvironment: Implications for TAM receptor targeting. Cancer Immunol. Immunother. 2020, 69(2), 237–244. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131(6), 1124–1136. [Google Scholar] [CrossRef]
- Peeters, M.J.W.; Dulkeviciute, D.; Draghi, A.; Ritter, C.; Rahbech, A.; Skadborg, S.K.; Seremet, T.; Simoes, A.M.C.; Martinenaite, E.; Halldórsdóttir, H.R.; Andersen, M.H.; Olofsson, G.H.; Svane, I.M.; Rasmussen, L.J.; Met, Ö.; Becker, J.C.; Donia, M.; Desler, C.; Straten, P.T. MERTK Acts as a Costimulatory Receptor on Human CD8 T Cells. Cancer Immunol. Res. 2019, 7(9), 1472–1484. [Google Scholar] [CrossRef]
- Carrera Silva, E.A.; Chan, P.Y.; Joannas, L.; Errasti, A.E.; Gagliani, N.; Bosurgi, L.; Jabbour, M.; Perry, A.; Smith-Chakmakova, F.; Mucida, D.; Cheroutre, H.; Burstyn-Cohen, T.; Leighton, J.A.; Lemke, G.; Ghosh, S.; Rothlin, C.V. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity 2013, 39(1), 160–170. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.R.; Rankin, E.B.; Giaccia, A.J. Therapeutic targeting of the functionally elusive TAM receptor family. Nat. Rev. Drug Discov. 2024, 23(3), 201–217. [Google Scholar] [CrossRef] [PubMed]
- Gadiyar, V.; Patel, G.; Davra, V. Immunological role of TAM receptors in the cancer microenvironment. Int. Rev. Cell Mol. Biol. 2020, 357, 57–79. [Google Scholar] [PubMed]
- Wang, Z.; Zhao, Z.; Li, Z.; Xu, L.; Li, H.; Zhu, H.; Cheng, G.; Yao, R.; Pei, W.; Liang, R.; Liang, R.; Ye, H.; Jiang, S.; Niu, H.; Sun, X.; Su, Y. Tyro3 receptor tyrosine kinase contributes to pathogenic phenotypes of fibroblast-like synoviocytes in rheumatoid arthritis and disturbs immune cell balance in experimental arthritis. Clin. Immunol. 2023, 255, 109753. [Google Scholar] [CrossRef] [PubMed]
- Endisha, H.; Datta, P.; Sharma, A.; Nakamura, S.; Rossomacha, E.; Younan, C.; Ali, S.A.; Tavallaee, G.; Lively, S.; Potla, P.; Shestopaloff, K.; Rockel, J.S.; Krawetz, R.; Mahomed, N.N.; Jurisica, I.; Gandhi, R.; Kapoor, M. MicroRNA-34a-5p Promotes Joint Destruction During Osteoarthritis. Arthritis Rheumatol. 2021, 73(3), 426–439. [Google Scholar] [CrossRef] [PubMed]
- Qin, A.; Qian, W. MicroRNA-7 inhibits colorectal cancer cell proliferation, migration and invasion via TYRO3 and phosphoinositide 3-kinase/protein B kinase/mammalian target of rapamycin pathway suppression. Int. J. Mol. Med. 2018, 42(5), 2503–2514. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Cheng, X.; Li, F.; Guan, Z.; Xu, J.; Wu, D.; Gao, Y.; Zhan, X.; Wang, P.; Zhou, H.; Rao, Z.; Cheng, F. Defective efferocytosis by aged macrophages promotes STING signaling mediated inflammatory liver injury. Cell Death Discov. 2023, 9(1), 236. [Google Scholar] [CrossRef]
- Lahey, K.C.; Varsanyi, C.; Wang, Z.; Aquib, A.; Gadiyar, V.; Rodrigues, A.A.; Pulica, R.; Desind, S.; Davra, V.; Calianese, D.C.; Liu, D.; Cho, J.H.; Kotenko, S.V.; De Lorenzo, M.S.; Birge, R.B. Regulation of Mertk Surface Expression via ADAM17 and gamma-Secretase Proteolytic Processing. Int. J. Mol. Sci. 2024, 25(8), 4404. [Google Scholar] [CrossRef]
- Ye, C.; Zheng, F.; Wu, N.; Zhu, G.Q.; Li, X.Z. Extracellular vesicles in vascular remodeling. Acta Pharmacol. Sin. 2022, 43(9), 2191–2201. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar]
- Melaragno, M.G.; Wuthrich, D.A.; Poppa, V.; Gill, D.; Lindner, V.; Berk, B.C.; Corson, M.A. Increased expression of Axl tyrosine kinase after vascular injury and regulation by G protein-coupled receptor agonists in rats. Circ. Res. 1998, 83(7), 697–704. [Google Scholar] [CrossRef]
- Chen, W.; Van Beusecum, J.P.; Xiao, L.; Patrick, D.M.; Ao, M.F.; Zhao, S.L.; Lopez, M.G.; Billings, F.T.; Cavinato, C.; Caulk, A.W.; Humphrey, J.D.; Harrison, D.G. Role of Axl in target organ inflammation and damage due to hypertensive aortic remodeling. Am. J. Physiol. -Heart Circ. Physiol. 2022, 323(5), H917–H933. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.F.; Xu, D.C.; Zhu, G.F.; Zhu, M.Y.; Tang, K.; Li, W.M.; Xu, Y.W. Growth Arrest-Specific 6 Exacerbates Pressure Overload-Induced Cardiac Hypertrophy. Hypertension 2016, 67(1), 118–129. [Google Scholar] [CrossRef] [PubMed]
- Smolock, E.M.; Korshunov, V.A. Pharmacological inhibition of Axl affects smooth muscle cell functions under oxidative stress. Vascul Pharmacol. 2010, 53(3-4), 185–192. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Cho, C.Y.; Hong, C.C.; Yan, M.D.; Hsieh, M.C.; Lay, J.D.; Lai, G.M.; Cheng, A.L.; Chuang, S.E. Oxidative stress enhances Axl-mediated cell migration through an Akt1/Rac1-dependent mechanism. Free Radical Biology and Medicine 2013, 65, 1246–1256. [Google Scholar] [CrossRef]
- Lee, C.H.; Hung, Y.J.; Shieh, Y.S.; Chien, C.Y.; Hsu, Y.J.; Lin, C.Y.; Chiang, C.F.; Huang, C.L.; Hsieh, C.H. Cilostazol inhibits uremic toxin-induced vascular smooth muscle cell dysfunction: Role of Axl signaling. Am. J. Physiol. Renal Physiol. 2017, 312(3), F398–F406. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, M.; Kim, H.Y.; Kim, J.K.; Kim, W.K.; Lim, S.C.; Kang, K.W. Circulating small extracellular vesicles promote proliferation and migration of vascular smooth muscle cells via AXL and MerTK activation. Acta Pharmacol. Sin. 2023, 44(5), 984–998. [Google Scholar] [CrossRef]
- Okamoto, K.; Ando, T.; Izumi, H.; Kobayashi, S.S.; Shintani, T.; Gutkind, J.S.; Yanamoto, S.; Miyauchi, M.; Kajiya, M. AXL activates YAP through the EGFR-LATS1/2 axis and confers resistance to EGFR-targeted drugs in head and neck squamous cell carcinoma. Oncogene 2023, 42(39), 2869–2877. [Google Scholar] [CrossRef] [PubMed]
- Saab, S.; Chang, O.S.; Nagaoka, K.; Hung, M.C.; Yamaguchi, H. The potential role of YAP in Axl-mediated resistance to EGFR tyrosine kinase inhibitors. Am. J. Cancer Res. 2019, 9(12), 2719–2729. [Google Scholar]
- Liu, Q.; Niu, Y.; Pei, Z.; Yang, Y.; Xie, Y.; Wang, M.; Wang, J.; Wu, M.; Zheng, J.; Yang, P.; Hao, H.; Pang, Y.; Bao, L.; Dai, Y.; Niu, Y.; Zhang, R. Gas6-Axl signal promotes indoor VOCs exposure-induced pulmonary fibrosis via pulmonary microvascular endothelial cells-fibroblasts cross-talk. J Hazard Mater 2024, 474, 134786. [Google Scholar] [CrossRef]
- Perez-Gutierrez, L.; Ferrara, N. Biology and therapeutic targeting of vascular endothelial growth factor A. Nat. Rev. Mol. Cell Biol. 2023, 24(11), 816–834. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.B.; Peng, Y.Q.; Zhou, X.M.; Yang, B.; Zheng, Z.; Liu, L.M.; Song, F.L.; Li, J.M.; Zhou, K.; Meng, J.C.; Yuan, L.Q.; Xie, H. Taurine restores Axl/Gas6 expression in vascular smooth muscle cell calcification model. Amino Acids 2010, 39(2), 375–383. [Google Scholar] [CrossRef] [PubMed]
- Happonen, K.E.; Tran, S.; Morgelin, M.; Prince, R.; Calzavarini, S.; Angelillo-Scherrer, A.; Dahlback, B. The Gas6-Axl Protein Interaction Mediates Endothelial Uptake of Platelet Microparticles. J. Biol. Chem. 2016, 291(20), 10586–10601. [Google Scholar] [CrossRef] [PubMed]
- Alciato, F.; Sainaghi, P.P.; Sola, D.; Castello, L.; Avanzi, G.C. TNF-α, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J Leukocyte Biol 2010, 87(5), 869–875. [Google Scholar] [CrossRef] [PubMed]
- Zdzalik-Bielecka, D.; Poswiata, A.; Kozik, K.; Jastrzebski, K.; Schink, K.O.; Brewinska-Olchowik, M.; Piwocka, K.; Stenmark, H.; Miaczynska, M. The GAS6-AXL signaling pathway triggers actin remodeling that drives membrane ruffling, macropinocytosis, and cancer-cell invasion. P. Natl. Acad. Sci. USA 2021, 118(28). [Google Scholar] [CrossRef]
- Pei-Yuan, Z.; Yu-Wei, L.; Xiang-Nan, Z.; Song, T.; Rong, Z.; Xiao-Xiao, H.; Sheng-Shuai, S.; Kun, W.; Cheng-Yun, L. Overexpression of Axl reverses endothelial cells dysfunction in high glucose and hypoxia. J. Cell Biochem. 2019, 120(7), 11831–11841. [Google Scholar] [CrossRef]
- Leopold, J.A. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc. Med. 2015, 25(4), 267–274. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.K.; Jeon, J.H. Vascular Calcification-New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21(8), 2685. [Google Scholar] [CrossRef]
- Collett, G.; Wood, A.; Alexander, M.Y.; Varnum, B.C.; Boot-Handford, R.P.; Ohanian, V.; Ohanian, J.; Fridell, Y.W.; Canfield, A.E. Receptor tyrosine kinase Axl modulates the osteogenic differentiation of pericytes. Circ. Res. 2003, 92(10), 1123–1129. [Google Scholar] [CrossRef]
- Qiu, C.; Zheng, H.; Tao, H.; Yu, W.; Jiang, X.; Li, A.; Jin, H.; Lv, A.; Li, H. Vitamin K2 inhibits rat vascular smooth muscle cell calcification by restoring the Gas6/Axl/Akt anti-apoptotic pathway. Mol. Cell Biochem. 2017, 433(1-2), 149–159. [Google Scholar] [CrossRef]
- Badi, I.; Mancinelli, L.; Polizzotto, A.; Ferri, D.; Zeni, F.; Burba, I.; Milano, G.; Brambilla, F.; Saccu, C.; Bianchi, M.E.; Pompilio, G.; Capogrossi, M.C.; Raucci, A. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler. Thromb. Vasc. Biol. 2018, 38(9), 2079–2090. [Google Scholar] [CrossRef]
- Son, B.K.; Kozaki, K.; Iijima, K.; Eto, M.; Nakano, T.; Akishita, M.; Ouchi, Y. Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur. J. Pharmacol. 2007, 556(1-3), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gerloff, J.; Korshunov, V.A. Immune modulation of vascular resident cells by Axl orchestrates carotid intima-media thickening. Am. J. Pathol. 2012, 180(5), 2134–2143. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Shieh, Y.S.; Tsai, C.S.; Hung, Y.J.; Tsai, Y.T.; Lin, C.Y. Expression of growth arrest-specific protein 6 and Axl molecules in the left internal mammary artery of patients undergoing coronary artery bypass grafting. J. Clin. Pathol. 2014, 67(6), 506–511. [Google Scholar] [CrossRef]
- Liu, Y.W.; Yang, Q.F.; Zuo, P.Y.; Xiao, C.L.; Chen, X.L.; Liu, C.Y. Elevated serum levels of soluble Axl in acute coronary syndrome. Am. J. Med. Sci. 2015, 349(2), 124–129. [Google Scholar] [CrossRef] [PubMed]
- Sunbul, M.; Cagman, Z.; Gerin, F.; Ozgen, Z.; Durmus, E.; Seckin, D.; Ahmad, S.; Uras, F.; Agirbasli, M. Growth arrest-specific 6 and cardiometabolic risk factors in patients with psoriasis. Cardiovasc. Ther. 2015, 33(2), 56–61. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Chi, H. Lipid metabolism in dendritic cell biology. Immunol. Rev. 2023, 317(1), 137–151. [Google Scholar] [CrossRef] [PubMed]
- Batchu, S.N.; Xia, J.; Ko, K.A.; Doyley, M.M.; Abe, J.; Morrell, C.N.; Korshunov, V.A. Axl modulates immune activation of smooth muscle cells in vein graft remodeling. Am. J. Physiol. Heart Circ. Physiol. 2015, 309(6), H1048–H1058. [Google Scholar] [CrossRef]
- Van Beusecum, J.P.; Barbaro, N.R.; Smart, C.D.; Patrick, D.M.; Loperena, R.; Zhao, S.; de la Visitacion, N.; Ao, M.F.; Xiao, L.; Shibao, C.A.; Harrison, D.G. Growth Arrest Specific-6 and Axl Coordinate Inflammation and Hypertension. Circulation Research 2021, 129(11), 975–991. [Google Scholar] [CrossRef]
- Batchu, S.N.; Dugbartey, G.J.; Wadosky, K.M.; Mickelsen, D.M.; Ko, K.A.; Wood, R.W.; Zhao, Y.; Yang, X.; Fowell, D.J.; Korshunov, V.A. Innate Immune Cells Are Regulated by Axl in Hypertensive Kidney. Am. J. Pathol. 2018, 188(8), 1794–1806. [Google Scholar] [CrossRef]
- Li, W.N.; Wang, J.L.; Ge, L.N.; Shan, J.H.; Zhang, C.B.; Liu, J.P. Growth arrest-specific protein 6 (Gas6) as a noninvasive biomarker for early detection of diabetic nephropathy. Clin. Exp. Hypertens. 2017, 39(4), 382–387. [Google Scholar] [CrossRef]
- Batlle, M.; Recarte-Pelz, P.; Roig, E.; Castel, M.A.; Cardona, M.; Farrero, M.; Ortiz, J.T.; Campos, B.; Pulgarín, M.J.; Ramírez, J.; Pérez-Villa, F.; de Frutos, P.G. AXL receptor tyrosine kinase is increased in patients with heart failure. Int. J. Cardiol. 2014, 173(3), 402–409. [Google Scholar] [CrossRef]
- Cristobal, H.; Enjuanes, C.; Batlle, M.; Tajes, M.; Campos, B.; Francesch, J.; Moliner, P.; Farrero, M.; Andrea, R.; Ortiz-Perez, J.T.; Morales, A.; Sabate, M.; Comin-Colet, J.; Garcia de Frutos, P. Prognostic Value of Soluble AXL in Serum from Heart Failure Patients with Preserved and Reduced Left Ventricular Ejection Fraction. J. Pers. Med. 2023, 13(3), 446. [Google Scholar] [CrossRef]
- Caldentey, G.; Garcia De Frutos, P.; Cristobal, H.; Garabito, M.; Berruezo, A.; Bosch, X.; San Antonio, R.; Flores-Umanzor, E.; Perea, R.J.; De Caralt, T.M.; Rodriguez, J.; Ortiz-Perez, J.T. Serum levels of Growth Arrest-Specific 6 protein and soluble AXL in patients with ST-segment elevation myocardial infarction. Eur. Heart J. Acute Cardiovasc. Care 2019, 8(8), 708–716. [Google Scholar] [CrossRef]
- DeBerge, M.; Glinton, K.; Subramanian, M.; Wilsbacher, L.D.; Rothlin, C.V.; Tabas, I.; Thorp, E.B. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J. Clin. Investig. 2021, 131(6). [Google Scholar] [CrossRef]
- Vago, J.P.; Valdrighi, N.; Blaney-Davidson, E.N.; Hornikx, D.; Neefjes, M.; Barba-Sarasua, M.E.; Thielen, N.G.M.; van den Bosch, M.H.J.; van der Kraan, P.M.; Koenders, M.I.; Amaral, F.A.; van de Loo, F.A.J. Gas6/Axl Axis Activation Dampens the Inflammatory Response in Osteoarthritic Fibroblast-like Synoviocytes and Synovial Explants. Pharmaceuticals (Basel) 2023, 16(5), 703. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16(7), 389–406. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454(7203), 428–435. [Google Scholar] [CrossRef]
- Rymut, N.; Heinz, J.; Sadhu, S.; Hosseini, Z.; Riley, C.O.; Marinello, M.; Maloney, J.; MacNamara, K.C.; Spite, M.; Fredman, G. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J. 2020, 34(1), 597–609. [Google Scholar] [CrossRef]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Invest. 2018, 128(7), 2713–2723. [Google Scholar] [CrossRef]
- Castilla-Madrigal, R.; Gil-Iturbe, E.; Lopez de Calle, M.; Moreno-Aliaga, M.J.; Lostao, M.P. DHA and its derived lipid mediators MaR1, RvD1 and RvD2 block TNF-alpha inhibition of intestinal sugar and glutamine uptake in Caco-2 cells. J. Nutr. Biochem. 2020, 76, 108264. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Libreros, S.; Nshimiyimana, R. E-series resolvin metabolome, biosynthesis and critical role of stereochemistry of specialized pro-resolving mediators (SPMs) in inflammation-resolution: Preparing SPMs for long COVID-19, human clinical trials, and targeted precision nutrition. Semin. Immunol. 2022, 59. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Serhan, C.N. Specialized pro-resolving mediators in vascular inflammation and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2024. [CrossRef] [PubMed]
- Adkar, S.S.; Leeper, N.J. Efferocytosis in atherosclerosis. Nat. Rev. Cardiol. 2024. [CrossRef] [PubMed]
- Kotlyarov, S.; Kotlyarova, A. Molecular Pharmacology of Inflammation Resolution in Atherosclerosis. Int. J. Mol. Sci. 2022, 23(9). [Google Scholar]
- Gerlach, B.D.; Marinello, M.; Heinz, J.; Rymut, N.; Sansbury, B.E.; Riley, C.O.; Sadhu, S.; Hosseini, Z.; Kojima, Y.; Tang, D.D.; Leeper, N.J.; Spite, M.; Barroso, M.; Rayner, K.J.; Fredman, G. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. 2020, 27(2), 525–539. [Google Scholar] [CrossRef]
- Bouhadoun, A.; Manikpurage, H.D.; Deschildre, C.; Zalghout, S.; Dubourdeau, M.; Urbach, V.; Ho-Tin-Noe, B.; Deschamps, L.; Michel, J.B.; Longrois, D.; Norel, X. DHA, RvD1, RvD5, and MaR1 reduce human coronary arteries contractions induced by PGE(2). Prostaglandins Other Lipid Mediat 2023, 165, 106700. [Google Scholar] [CrossRef]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Doring, Y.; Drechsler, M.; Weber, C.; Zimmer, R.; Cenac, N.; Soehnlein, O. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ. Res. 2016, 119(9), 1030–1038. [Google Scholar] [CrossRef]
- Tajbakhsh, A.; Rezaee, M.; Kovanen, P.T.; Sahebkar, A. Efferocytosis in atherosclerotic lesions: Malfunctioning regulatory pathways and control mechanisms. Pharmacol. Ther. 2018, 188, 12–25. [Google Scholar] [CrossRef]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Sansbury, B.E.; Daemen, M.J.; Dorweiler, B.; Spite, M.; Fredman, G.; Tabas, I. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Invest. 2017, 127(2), 564–568. [Google Scholar] [CrossRef]
- Wu, H.; Zheng, J.; Xu, S.; Fang, Y.; Wu, Y.; Zeng, J.; Shao, A.; Shi, L.; Lu, J.; Mei, S.; Wang, X.; Guo, X.; Wang, Y.; Zhao, Z.; Zhang, J. Mer regulates microglial/macrophage M1/M2 polarization and alleviates neuroinflammation following traumatic brain injury. J. Neuroinflammation 2021, 18(1), 2. [Google Scholar] [CrossRef]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic. Transl. Sci. 2018, 3(2), 230–244. [Google Scholar] [CrossRef]
- Lundquist, M.R.; Jaffrey, S.R. Gas6-Axl Signaling Induces SRF/MRTF-A Gene Transcription via MICAL2. Genes-Basel 2023, 14(12), 2231. [Google Scholar] [CrossRef]
- Justynski, O.; Bridges, K.; Krause, W.; Forni, M.F.; Phan, Q.M.; Sandoval-Schaefer, T.; Carter, K.; King, D.E.; Hsia, H.C.; Gazes, M.; Vyce, S.D.; Driskell, R.R.; Miller-Jensen, K.; Horsley, V. Apoptosis recognition receptors regulate skin tissue repair in mice. Elife 2023, 12. [Google Scholar] [CrossRef]
- Qiu, S.; Liu, J.; Chen, J.; Li, Y.; Bu, T.; Li, Z.; Zhang, L.; Sun, W.; Zhou, T.; Hu, W.; Yang, G.; Yuan, L.; Duan, Y.; Xing, C. Targeted delivery of MerTK protein via cell membrane engineered nanoparticle enhances efferocytosis and attenuates atherosclerosis in diabetic ApoE(-/-) Mice. J. Nanobiotechnology 2024, 22(1), 178. [Google Scholar] [CrossRef]
- Cai, B.; Kasikara, C.; Doran, A.C.; Ramakrishnan, R.; Birge, R.B.; Tabas, I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci. Signal 2018, 11(549). [Google Scholar] [CrossRef]
- Penberthy, K.K.; Ravichandran, K.S. Apoptotic cell recognition receptors and scavenger receptors. Immunol. Rev. 2016, 269(1), 44–59. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 2018, 128(7), 2657–2669. [Google Scholar] [CrossRef]
- Choi, J.Y.; Seo, J.Y.; Yoon, Y.S.; Lee, Y.J.; Kim, H.S.; Kang, J.L. Mer signaling increases the abundance of the transcription factor LXR to promote the resolution of acute sterile inflammation. Sci. Signal 2015, 8(365), ra21. [Google Scholar] [CrossRef]
- Zhabyeyev, P.; McLean, B.; Bassiouni, W.; Valencia, R.; Paul, M.; Darwesh, A.M.; Seubert, J.M.; Hazra, S.; Oudit, G.Y. Loss of PI3Kalpha Mediates Protection From Myocardial Ischemia-Reperfusion Injury Linked to Preserved Mitochondrial Function. J. Am. Heart Assoc. 2023, 12(12), e022352. [Google Scholar] [CrossRef]
- !!! INVALID CITATION !!! [124–126].
- Zhang, Y.; Wang, Y.; Zhou, D.; Zhang, L.S.; Deng, F.X.; Shu, S.; Wang, L.J.; Wu, Y.; Guo, N.; Zhou, J.; Yuan, Z.Y. Angiotensin II deteriorates advanced atherosclerosis by promoting MerTK cleavage and impairing efferocytosis through the ATR/ROS/p38 MAPK/ADAM17 pathway. Am. J. Physiol-Cell Ph 2019, 317(4), C776–C787. [Google Scholar] [CrossRef]
- Kawai, T.; Elliott, K.J.; Scalia, R.; Eguchi, S. Contribution of ADAM17 and related ADAMs in cardiovascular diseases. Cell Mol. Life Sci. 2021, 78(9), 4161–4187. [Google Scholar] [PubMed]
- Tan, H.; Li, W.; Pang, Z.; Weng, X.; Gao, J.; Chen, J.; Wang, Q.; Li, Q.; Yang, H.; Dong, Z.; Wang, Z.; Zhu, G.; Tan, Y.; Fu, Y.; Han, C.; Cai, S.; Qian, J.; Huang, Z.; Song, Y.; Ge, J. Genetically Engineered Macrophages Co-Loaded with CD47 Inhibitors Synergistically Reconstruct Efferocytosis and Improve Cardiac Remodeling Post Myocardial Ischemia Reperfusion Injury. Adv. Healthc. Mater. 2024, 13(16), e2303267. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.S.; Spite, M.; Fredman, G.; Tabas, I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. U S A 2016, 113(23), 6526–6531. [Google Scholar] [CrossRef]
- Sufit, A.; Lee-Sherick, A.B.; DeRyckere, D.; Rupji, M.; Dwivedi, B.; Varella-Garcia, M.; Pierce, A.M.; Kowalski, J.; Wang, X.; Frye, S.V.; Earp, H.S.; Keating, A.K.; Graham, D.K. MERTK Inhibition Induces Polyploidy and Promotes Cell Death and Cellular Senescence in Glioblastoma Multiforme. PLoS One 2016, 11(10), e0165107. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.H.; Kuan, A.P.; Wang, C.; Abraham, V.; Waldman, M.A.; Vogelgesang, A.; Wittenburg, G.; Choudhury, A.; Tsao, P.Y.; Miwa, T.; Eisenberg, R.A.; Cohen, P.L. Disrupted Mer receptor tyrosine kinase expression leads to enhanced MZ B-cell responses. J. Autoimmun. 2010, 35(4), 368–374. [Google Scholar] [CrossRef] [PubMed]
- Weinger, J.G.; Omari, K.M.; Marsden, K.; Raine, C.S.; Shafit-Zagardo, B. Up-regulation of soluble Axl and Mer receptor tyrosine kinases negatively correlates with Gas6 in established multiple sclerosis lesions. Am. J. Pathol. 2009, 175(1), 283–293. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, S.; Banerjee, O.; Shi, H.; Xue, B.; Ding, Z. Disturbed flow impairs MerTK-mediated efferocytosis in aortic endothelial cells during atherosclerosis. Theranostics 2024, 14(6), 2427–2441. [Google Scholar] [CrossRef]
- Liao, D.; Wang, X.W.; Li, M.; Lin, P.H.; Yao, Q.Z.; Chen, C.Y. Human protein S inhibits the uptake of AcLDL and expression of SR-A through Mer receptor tyrosine kinase in human macrophages. Blood 2009, 113(1), 165–174. [Google Scholar] [CrossRef]
- Khatana, C.; Saini, N.K.; Chakrabarti, S.; Saini, V.; Sharma, A.; Saini, R.V.; Saini, A.K. Mechanistic Insights into the Oxidized Low-Density Lipoprotein-Induced Atherosclerosis. Oxid. Med. Cell Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Liao, C.C.; Xu, J.W.; Huang, W.C.; Chang, H.C.; Tung, Y.T. Plasma Proteomic Changes of Atherosclerosis after Exercise in ApoE Knockout Mice. Biology (Basel) 2022, 11(2), 253. [Google Scholar] [CrossRef]
- DeBerge, M.; Yeap, X.Y.; Dehn, S.; Zhang, S.; Grigoryeva, L.; Misener, S.; Procissi, D.; Zhou, X.; Lee, D.C.; Muller, W.A.; Luo, X.; Rothlin, C.; Tabas, I.; Thorp, E.B. MerTK Cleavage on Resident Cardiac Macrophages Compromises Repair After Myocardial Ischemia Reperfusion Injury. Circ. Res. 2017, 121(8), 930–940. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. A MERry response after myocardial infarction. Circ. Res. 2013, 113(8), 949–951. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Avila, J.A.; Sanchez-Diaz, M.; Hidalgo, A. Isolation of exophers from cardiomyocyte-reporter mouse strains by fluorescence-activated cell sorting. STAR Protoc. 2021, 2(1), 100286. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; Martinez-de-Mena, R.; Castejon-Vega, B.; Pun-Garcia, A.; Traves, P.G.; Bonzon-Kulichenko, E.; Garcia-Marques, F.; Cusso, L.; N, A.G.; Gonzalez-Guerra, A.; Roche-Molina, M.; Martin-Salamanca, S.; Crainiciuc, G.; Guzman, G.; Larrazabal, J.; Herrero-Galan, E.; Alegre-Cebollada, J.; Lemke, G.; Rothlin, C.V.; Jimenez-Borreguero, L.J.; Reyes, G.; Castrillo, A.; Desco, M.; Munoz-Canoves, P.; Ibanez, B.; Torres, M.; Ng, L.G.; Priori, S.G.; Bueno, H.; Vazquez, J.; Cordero, M.D.; Bernal, J.A.; Enriquez, J.A.; Hidalgo, A. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183(1), 94–109 e23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yeap, X.Y.; Grigoryeva, L.; Dehn, S.; DeBerge, M.; Tye, M.; Rostlund, E.; Schrijvers, D.; Zhang, Z.J.; Sumagin, R.; Tourtellotte, W.G.; Lee, D.; Lomasney, J.; Morrow, J.; Thorp, E.B. Cardiomyocytes induce macrophage receptor shedding to suppress phagocytosis. J. Mol. Cell Cardiol. 2015, 87, 171–179. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38(3), 187–197. [Google Scholar] [CrossRef]
- Wei, Z.; Chen, Z.; Zhao, Y.; Fan, F.; Xiong, W.; Song, S.; Yin, Y.; Hu, J.; Yang, K.; Yang, L.; Xu, B.; Ge, J. Mononuclear phagocyte system blockade using extracellular vesicles modified with CD47 on membrane surface for myocardial infarction reperfusion injury treatment. Biomaterials 2021, 275, 121000. [Google Scholar] [CrossRef]
- Kojima, Y.; Volkmer, J.P.; McKenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J.; Schadt, E.E.; Quertermous, T.; Betancur, P.; Maegdefessel, L.; Matic, L.P.; Hedin, U.; Weissman, I.L.; Leeper, N.J. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016, 536(7614), 86–90. [Google Scholar] [CrossRef]
- Smart, S.K.; Vasileiadi, E.; Wang, X.D.; DeRyckere, D.; Graham, D.K. The Emerging Role of TYRO3 as a Therapeutic Target in Cancer. Cancers 2018, 10(12), 474. [Google Scholar] [CrossRef]
- Hurtado, B.; Abasolo, N.; Muñoz, X.; García, N.; Benavente, Y.; Rubio, F.; de Frutos, P.G.; Krupinski, J.; Sala, N. Association study between polymorphims in GAS6-TAM genes and carotid atherosclerosis. Thromb. Haemost. 2010, 104(3), 592–598. [Google Scholar] [CrossRef]
- Chan, P.Y.; Silva, E.A.C.; De Kouchkovsky, D.; Joannas, L.D.; Hao, L.M.; Hu, D.L.; Huntsman, S.; Eng, C.; Licona-Limón, P.; Weinstein, J.S.; Herbert, D.R.; Craft, J.E.; Flavell, R.A.; Repetto, S.; Correale, J.; Burchard, E.G.; Torgerson, D.G.; Ghosh, S.; Rothlin, C.V. The TAM family receptor tyrosine kinase TYRO3 is a negative regulator of type 2 immunity. Science 2016, 352(6281), 99–103. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122(12), 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Matsushima, G.K. Tyro3, Axl, Mertk receptor-mediated efferocytosis and immune regulation in the tumor environment. Int. Rev. Cell Mol. Biol. 2021, 361, 165–210. [Google Scholar] [PubMed]
Figure 1.
Pathogenesis of atherosclerosis. Created with BioRender.com.
Figure 1.
Pathogenesis of atherosclerosis. Created with BioRender.com.
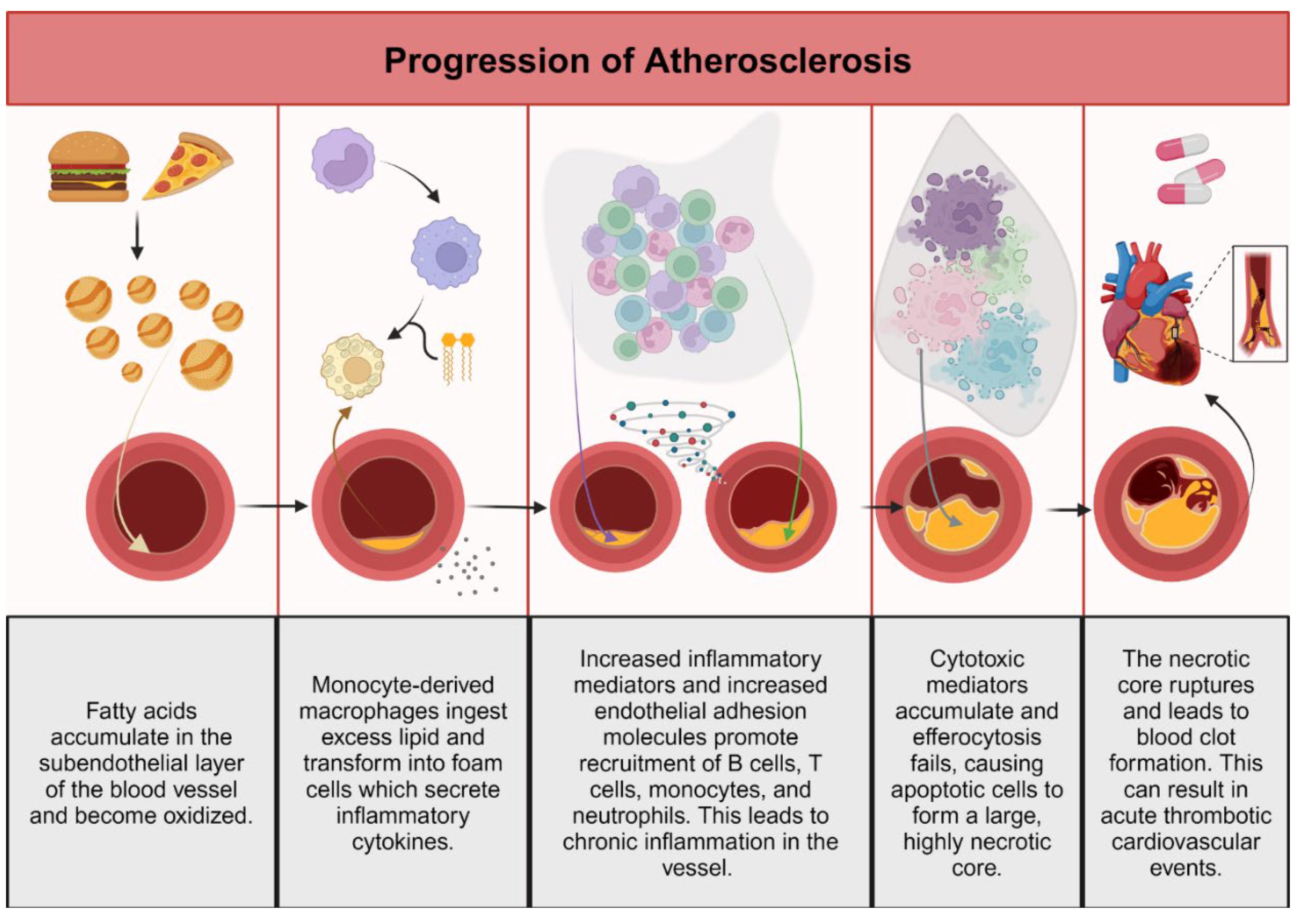
Figure 2.
Summary of the functions of Gas6-Axl in cardiovascular disease. Created with BioRender.com.
Figure 2.
Summary of the functions of Gas6-Axl in cardiovascular disease. Created with BioRender.com.
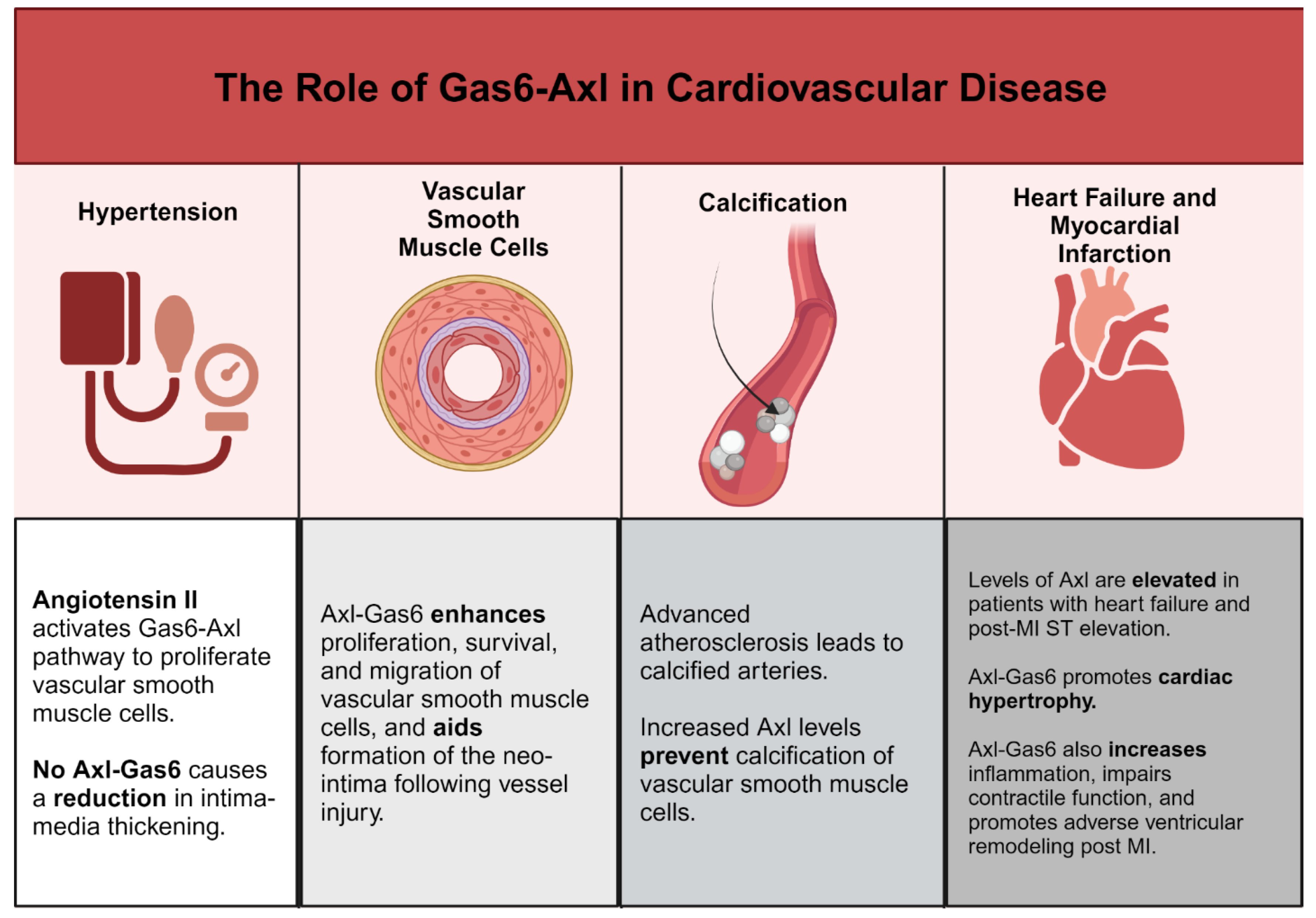
Figure 3.
Summary of the functions of PS-Mer in cardiovascular disease. Created with BioRender.com.
Figure 3.
Summary of the functions of PS-Mer in cardiovascular disease. Created with BioRender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



