
Submitted:
01 October 2024
Posted:
02 October 2024
You are already at the latest version
Abstract
Partial or transient cellular reprogramming is defined by the limited induction of pluripotency factors without fully de-differentiating cells into a pluripotent state. Comparing in vitro and in vivo mouse studies, and in vitro studies in humans, supported by visualizations of the interconnections among the data, we show consistent patterns in how such reprogramming modulates key biological processes. Generally, it leads to enhanced chromatin accessibility, upregulation of chromatin modifiers, and improved mitochondrial activity. These changes are accompanied by shifts in stress response programs, such as inflammation, autophagy, and cellular senescence, as well as dysregulation of extracellular matrix pathways. We also underscore the challenges in evaluating complex processes like aging and cellular senescence, given the variability in biomarkers used across studies. Overall, we highlight biological processes consistently influenced by reprogramming while noting that some effects are context-dependent, varying according to cell type, species, sex, and the reprogramming method employed. These insights inform future research and therapeutic applications in aging and regenerative medicine.
Keywords:
Partial Reprogramming
; Aging
; Rejuvenation
; Epigenetic clocks
1. Introduction
Recent studies have shown that limited use of Yamanaka factors or chemicals that mimic their effects can partially reverse cellular or organismal aging. This has been observed in both in vitro human and mouse models, as well as in vivo mouse studies, without fully de-differentiating cells into a pluripotent state. These studies encompass various models, including healthy aged and diseased mice, such as progeroid mice, and involve pulsed, short-term, and medium-term reprogramming regimes; these we call “partial reprogramming” as long as no induction of pluripotency (iPS) is observed (which we call “full reprogramming”; “partial reprogramming” is also called “transient reprogramming” in the literature). Here, we have collected and reviewed various biological processes and stress responses that are reported to change following partial reprogramming based on the literature (Supplementary Table 1).
1.1. Partial Reprogramming Overview
Partial reprogramming influences a plethora of biological processes (see Figure 1 for an overview). Generally, studies indicate that partial reprogramming leads to an upregulation of histone modifications, increased activity of transcription factors “pioneering” the opening of chromatin, enhanced chromatin accessibility, and, more generally, higher expression of chromatin modifiers and methyltransferase genes (Francesconi et al., 2019; Gill et al., 2022; Hishida et al., 2022; Kovatcheva et al., 2023; Paine et al., 2023; Wang et al., 2023). As full reprogramming results in the dedifferentiation of treated cells, these epigenetic changes can be viewed as a prerequisite. At the transcriptomic level, these changes manifest as the differential expression of developmental and differentiation pathways, including downregulation of identity markers of differentiated cells and upregulation of markers of stem or progenitor cells (for an overview of studies see ‘Development/Differentiation’ in Figure 1). For example, mouse cardiomyocytes partially reprogrammed in vivo for 6 or 12 days exhibit upregulation of embryonic genes and downregulation of muscle system process genes, indicating a shift from differentiated cardiac cells towards a more embryonic- and endodermal-like identity (Chen et al., 2021). Other in vivo partial reprogramming results linked to shifts in cell identity gene expression include downregulation of hepatocyte markers following reprogramming of mouse liver hepatocytes to earlier more progenitor-like cell states, downregulation of epidermal differentiation pathways in skin cells following whole body reprogramming in mice, and downregulation of muscle differentiation pathways in reprogrammed young mouse myofibers, accompanied by upregulation of genes linked to cytoskeletal organization (Browder et al., 2022; Hishida et al., 2022; Wang et al., 2021). In vitro studies recapitulate these results, with B-cells downregulating B-cell-specific genes in early reprogramming and upregulating proliferation- and histone-modifier-associated genes at later stages, adipose stem cells upregulating genes associated with development and fibroblasts downregulating fibroblast-specific genes while upregulating pluripotency markers throughout the reprogramming time course (Francesconi et al., 2019; Olova et al., 2019; Wang et al., 2023).
1.2. Temporal Dynamics of Reprogramming
Cellular reprogramming is likely a spectrum, akin to gradual movements in the Waddington landscape (Waddington, 2014/1957), rather than a transition between explicit well-defined stages. Unfortunately, we can only take snapshots of such gradual changes. Nonetheless, Gill et al. characterized various biomarkers at early, mid, and late ‘stages’ of reprogramming, and other studies have similarly reported specific phenotypes along the reprogramming process (Roux et al., 2022; Wang et al., 2023). The three stages defined by Gill et al. are as follows: i) the initiation phase (IP), characterized by the repression of somatic processes alongside the upregulation of mesenchymal-epithelial-transition (MET) pathways; ii) the maturation phase (MP), which involves expression of a subset of pluripotency genes; and iii) the stabilization phase (SP), characterized as full activation of the pluripotency program (Gill et al., 2022). Some studies suggest there can be an extra-embryonic endoderm-like stage between MP and SP, although this stage can be bypassed with more efficient reprogramming protocols (Guan et al., 2022; Liuyang et al., 2023). Before maturation phase transient reprogramming (MPTR) was investigated, various studies focused on reprogramming only to the IP (Ocampo et al., 2016; Sarkar et al., 2020). We find that various transcriptomic pathways associated with each ‘stage’ are consistently shared across studies, as follows.
Paine et al. identified downregulation of epithelial-mesenchymal transition (EMT) — a process that is going in the opposite direction compared to MET — during the first two days of reprogramming mouse tail tip fibroblasts with Yamanaka factors (Paine et al., 2023). Various studies (‘EMT’ in Figure 1) have similarly reported EMT downregulation or MET upregulation in early reprogramming stages, including in fibroblasts and adipose stem cells (Mitchell et al., 2024; Paine et al., 2023; Roux et al., 2022; Yang et al., 2023). Furthermore, Liuyang et al. performed time-course chemical reprogramming on mouse mesenchymal stem cells and found that knockdown of LIN28A impairs the emergence of epithelial-like characteristics and subsequent reprogramming stages, suggesting that development of epithelial characteristics is a key event in reprogramming in this context (Liuyang et al., 2023). Notably, prior research had already shown that LIN28A improves reprogramming efficiency (Wang et al., 2019).
Some studies report a decrease in MET pathways and an increase in EMT pathways at later stages of reprogramming (Francesconi et al., 2019; Paine et al., 2023). However, it is unclear if this is a universal feature indicating the termination of the IP, a marker of subsequent reprogramming stages, or a cell-type- or context-specific marker of reprogramming. Indeed, MET activation occurs at earlier time points in reprogrammed mouse cells compared to human cells (Teshigawara et al., 2017). While early-stage MET upregulation appears to be a common theme across cell types and tissues in early stages of reprogramming, further research is necessary to determine how these markers are expressed in cell-type or stage-dependent contexts.
1.3. Epigenetic Memory
How do cells retain their identity during reprogramming? Current evidence suggests that cells retain some epigenetic memory before induced pluripotent stem cell (iPSC) induction. Gill et al. found that fibroblasts reprogrammed to the MP preserved clusters of genes associated with fibroblast identity (Gill et al., 2022). They proposed that this residual identity allows the cells to re-differentiate into fibroblasts once the Yamanaka factors are removed. Similarly, Chen et al. showed that cardiomyocytes reprogrammed in vivo for six days re-differentiated into cardiomyocytes after six days of recovery. This process involved downregulating endodermal genes and upregulating muscle system genes (Chen et al., 2021).
This lingering epigenetic memory — especially at enhancer regions — potentially enables re-differentiation before any later reprogramming stages, where epigenetic memory is fully erased. The persistent expression of some cell identity genes until the SP may also contribute to this phenomenon (Gill et al., 2022; Jadhav et al., 2019; Olova et al., 2019). It is also possible that signals from surrounding cells and tissues support the re-differentiation process.
1.4. Impact of Partial Reprogramming on Surrounding Tissue
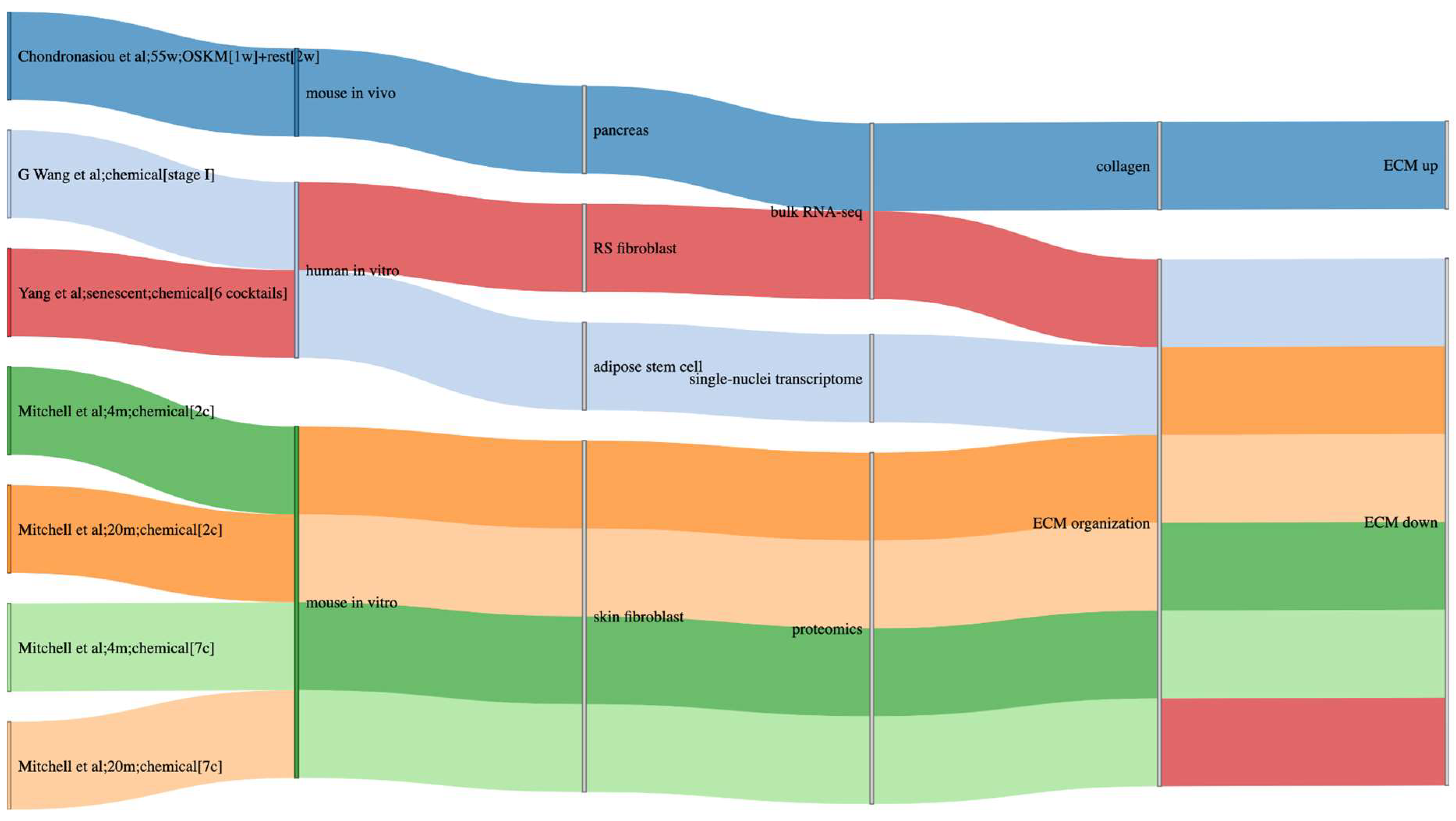
Reprogramming specific cells in vivo affects surrounding tissue. For example, C Wang et al. found that in vivo activation of OSKM in myofibers led to proliferation of satellite cells in the stem cell niche of the myofibers, without inducing myofiber dedifferentiation (Wang et al., 2021). It is likely that these changes are at least partially modulated via changes to the extracellular matrix (ECM). Chondronasiou et al. showed that as mice age, collagen-associated transcript levels decrease, and this is at least partially reversed following OSKM treatment (Chondronasiou et al., 2022a). Some ECM-associated processes are upregulated or remain continually expressed throughout partial reprogramming in fibroblasts and adipose mesenchymal cells, including pathways associated with collagen, alongside a decrease in fibrosis-associated gene expression (Gill et al., 2022; Roux et al., 2022; Wang et al., 2023) although various studies also show a downregulation in the ECM organization pathway across omics modalities (Mitchell et al., 2024; Wang et al., 2023; Yang et al., 2023). These discrepancies suggest that different components and regulatory mechanisms of the ECM may be dysregulated in distinct ways during reprogramming. Further research is necessary to elucidate the precise temporal dynamics and specific roles of ECM-related genes to better understand their regulation and function in the context of cellular reprogramming.
Figure 2.
Sankey plot of ECM-associated biological processes regulated during partial reprogramming in in vitro and in vivo experiments in mice, alongside in vitro human experiments.
Figure 2.
Sankey plot of ECM-associated biological processes regulated during partial reprogramming in in vitro and in vivo experiments in mice, alongside in vitro human experiments.
1.5. Cancer and Safety Concerns
Because the recent interest in partial in vivo reprogramming is triggered by the hope to reverse aging or induce regeneration, some studies aim to reprogram past the IP due to the greater rejuvenative effects observed (Gill et al., 2022), while avoiding the SP because overly dedifferentiated cells can negatively impact tissue function (Abad et al., 2013; Ohnishi et al., 2014). For example extended reprogramming of cardiomyocytes in vivo in mice for 12 days results in excessive dedifferentiation and cell death (Chen et al., 2021). Similarly, Hishida et al. demonstrated that excessive in vivo reprogramming of liver cells leads to loss of liver function and subsequent liver failure (Hishida et al., 2022). As such, the MP may be optimal for balancing rejuvenation benefits and minimizing risks. Nonetheless, there are still some cancer-associated risks with MP reprogramming; partial reprogramming has been associated with teratoma formation and cancer in vivo, just like full reprogramming (Abad et al., 2013; Chen et al., 2021; Ohnishi et al., 2014; Shibata et al., 2018).
By inducing low or short-term expression of Yamanaka factors it is possible to reprogram cells in vivo and avoid teratomas (Chondronasiou et al., 2022a). Nonetheless, partial reprogramming can also increase cancer risk for various reasons. The first reason is the use of MYC – an oncoprotein and chromatin remodeler (Dhanasekaran et al., 2022) – as part of the Yamanaka factor reprogramming regime. Cells expressing endogenous or exogenous MYC at the induction of reprogramming protocols reprogram more efficiently, and MYC targets are upregulated by partial reprogramming (Francesconi et al., 2019; Heffernan et al., 2012; Roux et al., 2022; Yang et al., 2023; Zviran et al., 2019). However, some reprogramming regimens avoid MYC and only reprogram with OSK (Li et al., 2011; Lu et al., 2020; Wernig et al., 2008). Moreover, it is also possible that reprogramming may facilitate pre-cancer cells to develop into cancer (Shibata et al., 2018). For example, single cell RNA-seq of pancreatic cells undergoing reprogramming in vivo has shown a subset of ‘intermediate reprogrammed’ cells that upregulate some transcription factors reminiscent of Kras-driven acinar metaplastic cells (Chondronasiou et al., 2022b). Reprogramming also allows senescent cells to re-enter the cell cycle, which we elaborate on in section 2.3.2 (Yang et al., 2023). Indeed, in vitro and in vivo partially/transiently reprogrammed cells exhibit an increase in gene expression linked to proliferation in general (see ‘Proliferation’ in Figure 1) (Chen et al., 2021; Hishida et al., 2022; Wang et al., 2023). Various studies have further shown an increase in cell number or the amount of Ki67 – a cellular marker of proliferation – following reprogramming (Browder et al., 2022; Chen et al., 2021; Liuyang et al., 2023; Xu et al., 2024). It is worth highlighting that partial reprogramming of cells with the 7c chemical cocktail treatment is not associated with increases in proliferation, implying that the cell cycle may be uncoupled from partial reprogramming (Mitchell et al., 2024). Finally, increases in cell proliferation could also be stage-dependent (Francesconi et al., 2019; Liuyang et al., 2023).
1.6. Differences in “Reprogrammability”
Various studies have shown that some cell types are more susceptible to reprogramming than others (Browder et al., 2022; Chondronasiou et al., 2022b; Wang et al., 2021; Xu et al., 2024). For example, human adult adipose-derived stromal cells are more susceptible to chemical reprogramming than human adult skin fibroblasts, and specific combinations of small molecules are needed to increase efficiency (Liuyang et al., 2023). In genetic manipulation experiments, this may be partially explained by the heterogeneous expression of the OSKM cassette across different cell types (Chondronasiou et al., 2022b); in chemical reprogramming, we may speculate that differences in pharmacokinetics play a role. Nonetheless, one study also found that young female mice are less susceptible to reprogramming, although these sex-specific differences disappear in aged mice (Mosteiro et al., 2018). As such, understanding the susceptibility and consequences of reprogramming different cell types to different phases is fundamental for translating partial reprogramming treatments to clinical trials in the future.
2. Health Benefits and Age Reversal Effects
Studies show that multiple molecular biomarkers of aging – including transcriptomics, epigenomics, functional protein expression and cell migration speed – can be reversed following reprogramming (Gill et al., 2022). This also includes various hallmarks of aging (Miliotou and de Lazaro, 2024). Here we review the impact of partial reprogramming on health and aging-associated pathways.
2.1. Aging Clocks
Partial and full reprogramming can partially reverse age-related transcriptomic and epigenetic changes (Chondronasiou et al., 2022b). Indeed, various transcriptomic and epigenetic clocks have been developed to track biological age (Horvath, 2013; Mitchell et al., 2024), and in vivo reprogramming via the Yamanaka factors has been shown to rewind clocks in mouse brain, kidney, skin, spleen, liver, and pancreas (see ‘Aging’ in Figure 1) (Browder et al., 2022; Chondronasiou et al., 2022b; Xu et al., 2024). These results are recapitulated at the in vitro level; Yamanaka-treated or chemically reprogrammed fibroblasts and adipose cells showcase reversal of epigenetic and transcriptomic clocks (Gill et al., 2022; Mitchell et al., 2024; Olova et al., 2019; Paine et al., 2023; Roux et al., 2022). Reprogramming also rewinds clocks in progeric cells (Paine et al., 2023).
It is unclear to what extent aging clocks are measuring true biological age. For example, aging clocks may partially be measuring inflammation, which increases with age (Cribb et al., 2022; Xu et al., 2024). Xu et al. found that transcriptomic aging clocks did not show clear reductions in age following partial reprogramming of microglia, which they hypothesize is due to increases in inflammation (Xu et al., 2024). Another issue in the field is to distinguish rejuvenation from dedifferentiation (Yucel and Gladyshev, 2024); some studies suggest that partial reprogramming may activate regenerative cellular programs while failing to reverse all aging-related omics changes (Niimi et al., 2024; Wang et al., 2023). Indeed, there is only partial reversion to a dedifferentiated state of transcriptomics and promoter methylation following reprogramming. For example, Roux et al found that young and aged but reprogrammed adipogenic and mesenchymal stem cells do not cluster together when analyzed using dimensional reduction embeddings, suggesting that some transcriptomic aging signals persist after reprogramming (Roux et al., 2022). Furthermore, Browder et al. found changes in skin and kidney epigenetic age of mice following long-term OSKM treatments, but other tissues did not show changes (Browder et al., 2022). Moreover, short-term OSKM treatment did not significantly impact epigenetic age in their study. It is worth highlighting that reductions in epigenetic age also depend on the clock used to measure ‘age’ (Browder et al., 2022; Kriukov et al., 2024; Mitchell et al., 2024). Moreover, clock behavior depends on the reprogramming stage (Gill et al., 2022), indicating that different clocks are measuring different biological processes. For example, Gill et al. found that the skin and blood clock, alongside the multi-tissue epigenetic clock, both showed decreases in epigenetic age at both the IP and MP. On the other hand, various clocks like the GrimAge, Hannum, and PhenoAge clocks did not show substantial declines in epigenetic age until the SP. This issue is further exacerbated when we consider that different studies use different clocks to measure ‘biological age’, making cross-study comparisons more challenging. Furthermore, there is a caveat with whole-body reprogramming regimens because it is unclear whether shifts towards young omics expression in any given tissue are due to reprogramming of the specified tissue, or whether they are a byproduct of other tissues rejuvenating (Chondronasiou et al., 2022a). Finally, aging clocks in general and their use in measuring rejuvenation effects in particular have been criticized for having high epistemic uncertainty, and lacking estimates for these uncertainties, amongst other criticisms (Kriukov et al., 2024).
2.2. Rejuvenation
Regardless of what epigenetic aging clocks measure exactly, there are other biomarkers of rejuvenation that can be measured in partial reprogramming experiments. Studies suggest that specifically if cycles of short-time reprogramming factor expression are followed by a recovery phase, rejuvenation effects can be observed. By default, rejuvenation markers must be evaluated on a tissue-by-tissue basis (Chondronasiou et al., 2022a; Ocampo et al., 2016; Sarkar et al., 2020). An intriguing example is the brain, where cycling OSKM with recovery restores the proportion of neuroblasts, blunts age-related transcriptomic changes, and improves the production of neurons in vivo (Xu et al., 2024). Moreover, in vivo studies performed on mouse neurons and rat dental gyrus cells suggest that OSKM can reverse age-associated neurological decline and enhance memory (Anton-Fernandez et al., 2024; Horvath et al., 2024). Other mouse in vivo studies have shown that reprogramming enhances liver regeneration, promotes the repair of crushed optic nerves and ameliorates aging-associated loss of visual acuity, allows for muscle fiber regeneration, improves skin wound healing in aged mice, and promotes heart rejuvenation following myocardial infarction (Browder et al., 2022; Chen et al., 2021; Hishida et al., 2022; Lu et al., 2020; Wang et al., 2021). The mechanism of rejuvenation appears to partially depend on how cells are reprogrammed. Indeed, G Wang et al. found that the mechanism of somatic cell reprogramming via small molecule regimens is distinct from transcription factor-mediated reprogramming (Wang et al., 2023). By constructing chromatin landscapes, they identified hierarchal histone modifications and sequential enhancer recommissioning which underlies regeneration programs following chemical reprogramming; this regeneration program appears to reverse the loss of regenerative potential in organismal aging but apparently it is not activated in OSKM reprogramming.
2.3. Stress Responses
Aging is also associated with a dysregulation of stress responses. For example, inflammation increases with age (Ferrucci and Fabbri, 2018), while senescent cell burden appears to increase and autophagy appears to decrease (Martinez-Lopez et al., 2015; Yousefzadeh et al., 2020). If reprogramming is reversing aging-related phenotypes, there should be a reversal of these phenotypes as well, which we evaluate in the next sections.
2.3.1. Inflammation
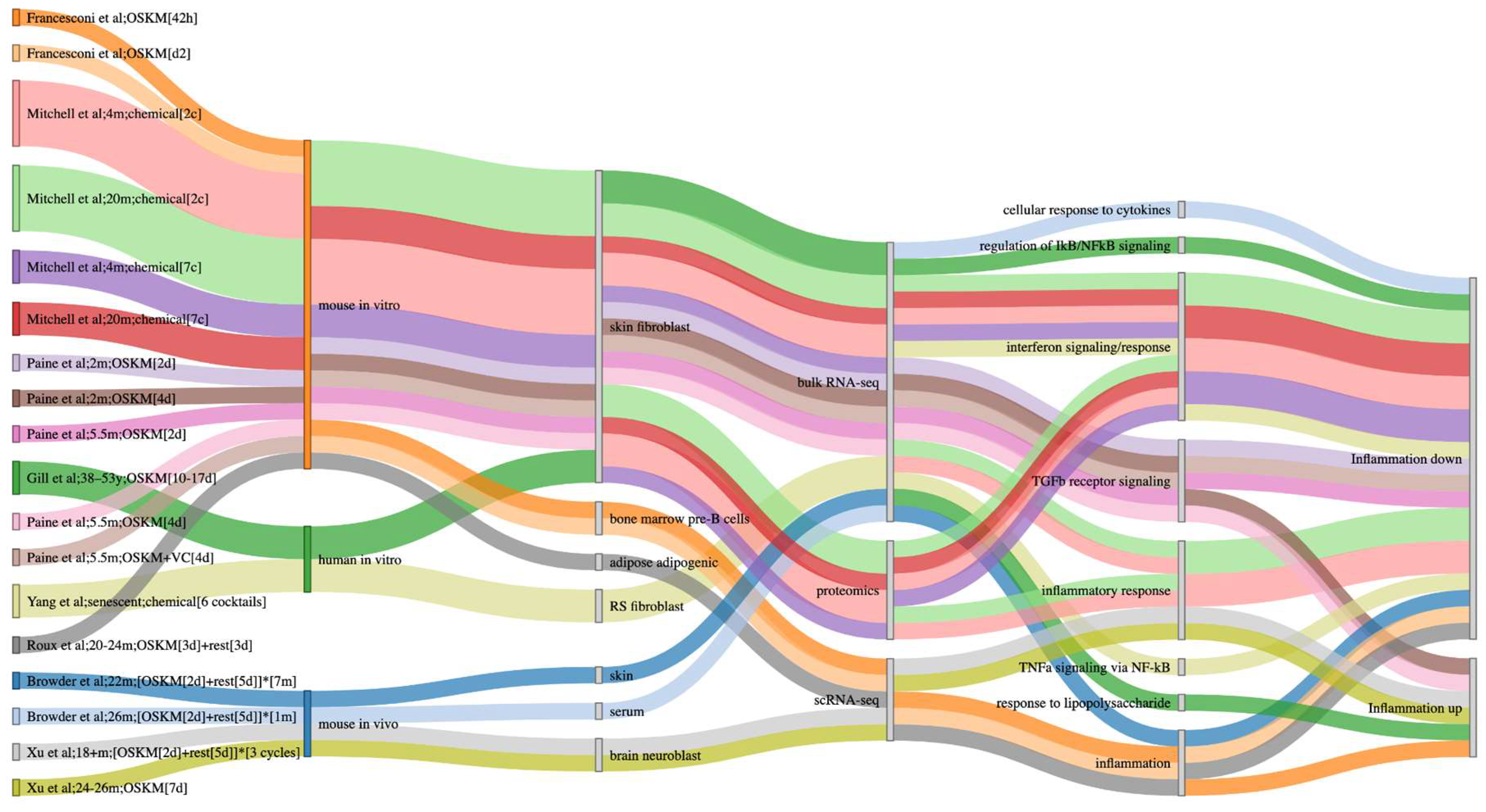
Various partial reprogramming studies have shown decreases in inflammation at the transcriptomic level (Figure 3). Paine et al. identified ‘TGFb receptor signaling’ as an overexpressed process in both aged progeroid and healthy skin fibroblasts, which was reversed following IP reprogramming (Paine et al., 2023). OSKM also reversed inflammation of old adipogenic and mesenchymal stem cells in mouse muscle following treatment (Roux et al., 2022). These results have been recapitulated in vitro in fibroblast and B-cells (Francesconi et al., 2019; Mitchell et al., 2024). Mitchell et al. further showed that the reversal of age-associated inflammation of mouse skin cells is detected at the proteomic level as well (Mitchell et al., 2024).
On the other hand, some studies report an increase in inflammation following reprogramming. For example, Gill et al. reports that genes associated with the ‘response to polysaccharides’ gene ontology (GO) term are temporarily upregulated in transient reprogramming of dermal fibroblasts, which they speculate is a response to reprogramming factors (Gill et al., 2022). Xu et al. found that neuroblasts of mouse brain subventricular zones showcase decreased inflammation with age, which increases during reprogramming (Xu et al., 2024). Intriguingly, Francesconi et al. identified increases in B cell inflammation during early reprogramming, followed by a drop in inflammation at later stages (Francesconi et al., 2019). There are at least two points to consider when assessing inflammation in reprogramming: 1) to what extent is the reprogramming protocol itself acting as a stressor on the cell, thereby increasing inflammation; and 2) whether an observed increase in inflammation in reprogrammed cells is due to inherently pro-inflammatory cells becoming more 'youthful.’ Finally, proinflammatory factors like IL6 and NF-κb have been shown to facilitate reprogramming in vivo (Chiche et al., 2017; Mosteiro et al., 2016), in a paracrine manner via downstream effectors. As such, while the general trend shows that inflammation decreases in reprogramming, more research is required to understand mechanisms in reducing inflammation, alongside contexts in which inflammation increases. Similar considerations apply to cellular senescence, which overlaps with inflammatory cellular phenotypes.
2.3.2. Cellular Senescence
Cellular senescence (CS) is a stress response modulated by various stimuli, including telomere attrition, DNA damage, and oncogene activation, characterized as a stable cell cycle arrest (Huang et al., 2022). Senescent cells are implicated in aging and virtually all aging-related chronic diseases (Huang et al., 2022; Mylonas and O'Loghlen, 2022). Senescent phenotypes are heterogeneous, dependent on cell type and insult, and CS lacks a universal biomarker (Gonzalez-Gualda et al., 2021).
Traditionally, senescent cells are seen as major barriers to partial and full cellular reprogramming (Banito et al., 2009; Haridhasapavalan et al., 2020). Multiple CS pathways — including the p53/p21 protein pathway, encoded by TP53 and CDKN1A respectively, alongside the p16 pathway encoded by CDKN2A — act as barriers towards reprogramming. Acute genetic ablation of p53 in mice in cell subpopulations that typically fail to reprogram allows for iPSC formation (Utikal et al., 2009). The CDKN2A gene, which encodes two transcripts that drive CS, is a key barrier to reprogramming of senescent cells as well. Importantly, the mouse p19ARF transcript of Cdkn2a — a driver of CS via its ability to stabilize p53 (Qian and Chen, 2013) — is a key inhibitor of reprogramming in mice (Utikal et al., 2009), whereas the p16INK4A transcript of CDKN2A — which activates CS via the retinoblastoma pathway and heterochromatic rearrangements (Kosar et al., 2011; Takahashi et al., 2006) — appears to be a more human-specific inhibitor of reprogramming (Haridhasapavalan et al., 2020; Li et al., 2009). On the other hand, Doeser et al. found that expression of senescence-associated genes showed high variability with no significant difference between controls and OSKM-reprogrammed senescent cells in a mouse wound healing model (Doeser et al., 2018). Then again, Chondronasiou et al. found that a single cycle of OSKM did not decrease various biomarkers of CS in aged mouse liver, including p16 expression (Chondronasiou et al., 2022a). These results indicate that overall, senescent cells are resistant to reprogramming.
On the other hand, various groups have successfully reduced biomarkers indicative of CS using the Yamanaka factors, alongside chemical reprogramming (Lapasset et al., 2011; Yang et al., 2023). Browder et al. identified a decrease in inflammation in skin cells following both short- and long-term partial reprogramming of mice in-vivo (Browder et al., 2022). Some pro-inflammatory genes that were downregulated in this context have been previously linked to senescence-associated secretory phenotypes (SASPs), like Il6 and Il1a (Coppe et al., 2010), alongside the in vivo downregulation of other senescence-associated genes including heat shock proteins and cyclin-dependent kinase inhibitors like Cdkn1a (Browder et al., 2022). Olova et al. also reported a decrease in CDKN1A and CDKN2A, alongside a decrease in pro-inflammatory SASP genes like IL6, IL1B, and IL8 in OSKM-treated human dermal fibroblasts (Olova et al., 2019). Intriguingly, the expression of specific aspects of senescence-associated gene expression changed throughout the particular data they analyzed; in the early stages of reprogramming (~day 11-15) senescence-associated gene expression increased, before decreasing at later stages of reprogramming. Yang et al. found that human fibroblasts induced into replicative senescence could be reprogrammed (Yang et al., 2023). While senescent fibroblasts downregulate cell cycle genes, these genes were upregulated following four days of chemical- and OSK-induced reprogramming. It is unclear to what extent these cells were cycling, or whether cells induced into senescence via other stimuli like oncogene overexpression showcase similar patterns following reprogramming treatments. It is worth highlighting that senescent cells are drivers of so-called inflammaging (Balistreri et al., 2013), and one potential reason as to why inflammation decreases following reprogramming is because senescent cells themselves may be reprogrammed.
Intriguingly, the secretory phenotype of senescent cells may facilitate the reprogramming process, such as in an in vivo model of skeletal muscle injury (Chiche et al., 2017; Mosteiro et al., 2016). Indeed, tissues lacking p16 expression are less susceptible to reprogramming, indicating that senescent cells may be necessary for efficient reprogramming in some in vivo contexts (Mosteiro et al., 2016). These results suggest that while senescence serves as a barrier to reprogramming at the level of individual cells, the paracrine signaling from senescent cells, particularly through the SASP, paradoxically facilitates reprogramming. This occurs despite the fact that SASP factors themselves are also capable of inducing senescence (Acosta et al., 2008). Given that SASP factors IL6 and IL8 induce EMT transitions via paracrine signaling (Coppe et al., 2008), this may be a potential mechanism by which senescent cells facilitate reprogramming (Mosteiro et al., 2018).
Overall, there does not appear to be a uniform effect on senescence biomarkers in partial reprogramming. Sarkar et al. reprogrammed aged human endothelial cells and fibroblasts using a cocktail of mRNAs including the Yamanaka factors, LIN28A, and NANOG (Sarkar et al., 2020). The authors found that transient expression of this cocktail led to a rapid and sustained reduction and reversal of cellular age in human somatic cells, as indicated by transcriptomic, epigenetic, and cellular biomarkers. However, while reprogrammed endothelial cells showcased a decrease in pro-inflammatory SASP markers – coupled with decreased senescence-associated β-galactosidase (SA β-gal) staining – aged fibroblasts did not. It is worth highlighting that the aged reprogrammed epithelial cells were not explicitly senescent, but rather showcased a decrease in biomarkers typically associated with senescence. Intriguingly, some studies suggest that early reprogramming stages are reminiscent of senescence induction (based on p21 and p16 expression, alongside SA β-gal staining and cell morphology) which nevertheless may act as a temporal barrier for reprogramming (Banito et al., 2009). Overall, the effects of reprogramming on senescent cells is context dependent; it is unclear to what extent reprogramming can reverse senescence phenotypes, what is the temporal dynamics of this reversal, and to what extent this reversal is cell-type, treatment, or senescence pathway-dependent. Finally, senescence itself is a heterogeneous process lacking a universal marker, and different studies measure senescence induction and reversal using different combinations of biomarkers (Avelar et al., 2020; Hernandez-Segura et al., 2017). As such, comparative analyses on the effect of reprogramming on cellular senescence are themselves hindered by the heterogeneous nature of CS.
2.3.3. Autophagy and mTOR Pathways
mTOR is a known repressor of autophagy and is implicated in aging (Carosi et al., 2022). Across various reprogramming studies, autophagy appears to increase, and some mTOR pathways are downregulated (Mitchell et al., 2024). For example, Sarkar et al. report an increase in autophagosomes following chemical reprogramming in human fibroblasts and endothelial cells, which they speculate is because reprogramming promotes clearance of degraded biomolecules (Sarkar et al., 2020). Chondronasiou et al. also reported a decrease in mTOR signaling following OSKM treatment in in vivo pancreas (Chondronasiou et al., 2022a).
2.3.4. DNA Repair
DNA repair is downregulated in aging and this downregulation can be counteracted by reprogramming in vivo (Chondronasiou et al., 2022a). A recent study showed that in vivo partial reprogramming of progeroid mice with an Ercc1 mutation – involved in DNA repair – resulted in upregulation of DNA repair processes and reversion of DNA damage (Paine et al., 2023). In this study, immunofluorescence staining of γH2AX also decreased, while nuclear area increased – both indicative of DNA damage – and these phenotypes were reversed following reprogramming. Other studies have also found significant increases in DNA repair pathways following reprogramming (Chondronasiou et al., 2022a). It is worth highlighting that this is not a universal effect though. Indeed, Mitchell et al. found via proteomics that in vitro 7c chemical reprogramming treatment in aged mouse skin fibroblasts downregulated the DNA repair and base excision repair REACTOME pathways, while reprogramming with 2c chemical reprogramming did not significantly increase the expression of genes linked with these pathways (Mitchell et al., 2024). It makes sense that the upregulation of DNA repair depends on the amount of DNA damage already featured by the cells undergoing reprogramming, and on the amount of DNA damage induced by the reprogramming factors, specifically due to the forced induction of the cell-cycle.
2.4. Metabolic Changes
Metabolism changes with age, and mitochondrial dysfunction is a hallmark of aging (Lopez-Otin et al., 2013). Various studies have reported changes to mitochondrial function and energetics pathways (see ‘Energetics’ in Figure 1), alongside other metabolic changes following various partial reprogramming protocols.
2.4.1. Mitochondrial Function and Energetic Pathways
Various energetic and mitochondrial pathways including fatty acid oxidation, tricarboxylic acid cycle, and oxidative phosphorylation (OXPHOS) have been shown to be downregulated with age and upregulated with reprogramming (Chondronasiou et al., 2022a; Mitchell et al., 2024; Roux et al., 2022). In particular, upregulation of OXPHOS pathways is commonly reported in reprogramming, including in vitro in skin fibroblasts and adipose stem cells (Liuyang et al., 2023; Mitchell et al., 2024; Roux et al., 2022) and in vivo in skin (Browder et al., 2022), by chemical and by TF-based reprogramming. Furthermore, Liuyang et al. showed that inhibition of OXPHOS inhibits proliferation in reprogramming, alongside subsequent formation of iPSCs, suggesting OXPHOS is necessary for reprogramming in this context (Liuyang et al., 2023). Various mitochondrial characteristics are rejuvenated by reprogramming, including increases in the activity and/or abundance of OXPHOS complexes, mitochondrial size, spare respiratory capacity, mitochondrial membrane potential, O2 consumption, and decreases in mitochondrial ROS and proton leakage (Liuyang et al., 2023; Mitchell et al., 2024; Sarkar et al., 2020).
Reprogramming is an energetically consumptive process, and rejuvenated cells may also require more energy to function. For instance, Cheng et al. discovered that short-term partial reprogramming activates an energy switch essential for cytoskeletal reorganization in nucleus pulposus cells (Cheng et al., 2022). Inhibition of glycolysis with 3-Bromopyruvic acid – an inhibitor of the rate-limiting glycolysis enzyme HK2 – led to disorganized cytoskeletal structures, increased SA β-gal staining, and higher expression of age-related stress response genes following reprogramming. Indeed, energetic pathways, including the TCA cycle, glycolysis, and lipid metabolism are consistently upregulated in reprogramming studies (Browder et al., 2022; Chen et al., 2021; Francesconi et al., 2019; Mitchell et al., 2024; Roux et al., 2022). On the other hand, metabolic processes associated with fat and lipid metabolism have also been shown to downregulate in vivo following reprogramming in some tissues, including in liver, muscle, and heart (Chen et al., 2021; Hishida et al., 2022; Wang et al., 2021). Furthermore, inhibition of glycolysis still allows for formation of iPSCs, suggesting that this pathway is dispensable for reprogramming in some contexts (Liuyang et al., 2023).
2.4.2. Metabolic Changes
Various other metabolic changes are reported in reprogrammed cells. From a metabolic panel of mouse serum from old mice before and one week after reprogramming, Chondronasiou et al. found that 4-hydroxyproline and trimethyl-lysine were downregulated with age and rescued following reprogramming, while indole-3-propionic acid was upregulated in aging and reversed following reprogramming (Chondronasiou et al., 2022a). Indeed, studies show a general change in amino acid metabolism although there is no clear trend. For example, Hishida et al. identified downregulation of amino acid metabolism in liver – this was coupled with upregulation of alanine aminotransferase and aspartate aminotransferase, two enzymes involved in amino acid catabolism, alongside a significant decrease in total protein levels (Hishida et al., 2022). On the other hand, Yang et al found upregulation of amino acid metabolism in both aged and reprogrammed senescent fibroblasts (Yang et al., 2023). Ultimately, a more systematic approach is necessary to understand whether uniform metabolic changes exist in reprogramming, or to what extent reprogramming-associated metabolic changes are cell-type, treatment-, or species-specific.
2.5. Similarities between Partial Reprogramming and Aging
Some studies have found similarities in aging and reprogramming, such as the aforementioned increase in inflammation in mouse brain following whole-body reprogramming (Xu et al., 2024). Furthermore, Roux et al. reprogrammed murine adipogenic stem cells using OSKM and found that ‘oxidation phosphorylation’ and ‘mTOR signaling’ gene sets were upregulated in both aging and reprogramming (Roux et al., 2022). Francesconi et al. and Mitchell et al. also found that mTORc1 signaling was upregulated following reprogramming, although in the latter study, reprogrammed fibroblasts also showcased downregulation of the ‘PI3K AKT mTOR signaling’ pathway, suggesting either negative feedback or that only some aspects of the mTOR signaling pathway are upregulated in reprogramming (Francesconi et al., 2019; Mitchell et al., 2024). Finally, Kriukov et al made similar observations, showing that aging clocks predicted that old liver samples reprogrammed in vivo were either of the same age or even significantly older than aged controls, although this may be an issue with aging clocks themselves (Kriukov et al., 2024).
Telomere length – a hallmark of aging and driver of cellular senescence (Lopez-Otin et al., 2023; Meena et al., 2015) – is not rejuvenated until the SP stage of reprogramming, and multiple studies find that partially reprogrammed cells either do not show increases in telomere length or showcase slight but statistically significant decreases in length, potentially due to reprogramming-associated stress or increases in cellular proliferation (Gill et al., 2022; Sarkar et al., 2020; Shimamoto et al., 2015).
Concluding Remarks
Ultimately, until we achieve a more robust understanding of aging at the molecular level – and identify much more reliable biomarkers of biological age – the extent to which reprogramming can reverse aging will remain unclear. The effects and potential side effects of reprogramming are context-dependent, varying with the specifics of the reprogramming protocol (such as duration) and the characteristics of the target, including species and tissue or cell types involved. Nonetheless, reprogramming holds significant promise in reversing various biomarkers of aging. Therefore, further research is crucial to unravel the mechanisms that can enhance the safety and efficacy of partial reprogramming protocols.
Supplementary Materials
Biological_pathway_reprogramming_changes.csv
Author Contributions
RAA wrote the manuscript. RAA, GF, DP, and AK compiled reprogramming-associated biological processes from the literature. RAA produced figures. GF, AK, and DP edited the manuscript. GF supervised the project.
Acknowledgments
GF: DP, and RAA are funded by the DFG, FU 583/7-1 and Exist-Gründer, FKZ 03EGTMV007.
Conflict of Interest
This work was funded in part by entrepreneurship-related scholarships, but no entrepreneurial activity has been scheduled yet.
References
- Abad, M.; Mosteiro, L.; Pantoja, C.; Canamero, M.; Rayon, T.; Ors, I.; Grana, O.; Megias, D.; Dominguez, O.; Martinez, D. , et al . ( 2013). Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 502, 340–345. [CrossRef]
- Acosta, J.C.; O'Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N. , et al. ( 2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. [CrossRef]
- Anton-Fernandez, A.; Roldan-Lazaro, M.; Valles-Saiz, L.; Avila, J.; Hernandez, F. (2024). In vivo cyclic overexpression of Yamanaka factors restricted to neurons reverses age-associated phenotypes and enhances memory performance. Commun Biol 7, 631. [CrossRef]
- Avelar, R.A.; Ortega, J.G.; Tacutu, R.; Tyler, E.J.; Bennett, D.; Binetti, P.; Budovsky, A.; Chatsirisupachai, K.; Johnson, E.; Murray, A. , et al. ( 2020). A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol 21, 91. [CrossRef]
- Balistreri, C.R.; Candore, G.; Accardi, G.; Colonna-Romano, G.; Lio, D. (2013). NF-kappaB pathway activators as potential ageing biomarkers: targets for new therapeutic strategies. Immun Ageing 10, 24.
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M. , et al. ( 2009). Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev 23, 2134–2139. [CrossRef]
- Browder, K.C.; Reddy, P.; Yamamoto, M.; Haghani, A.; Guillen, I.G.; Sahu, S.; Wang, C.; Luque, Y.; Prieto, J.; Shi, L. , et al. (2022). In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat Aging 2, 243-253. [CrossRef]
- Carosi, J.M.; Fourrier, C.; Bensalem, J.; Sargeant, T.J. (2022). The mTOR-lysosome axis at the centre of ageing. FEBS Open Bio 12, 739-757.
- Chen, Y.; Luttmann, F.F.; Schoger, E.; Scholer, H.R.; Zelarayan, L.C.; Kim, K.P.; Haigh, J.J.; Kim, J.; Braun, T. (2021). Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 373, 1537-1540. [CrossRef]
- Cheng, F.; Wang, C.; Ji, Y.; Yang, B.; Shu, J.; Shi, K.; Wang, L.; Wang, S.; Zhang, Y.; Huang, X. , et al. ( 2022). Partial reprogramming strategy for intervertebral disc rejuvenation by activating energy switch. Aging Cell 21, e13577. [CrossRef]
- Chiche, A.; Le Roux, I.; von Joest, M.; Sakai, H.; Aguin, S.B.; Cazin, C.; Salam, R.; Fiette, L.; Alegria, O.; Flamant, P. , et al. (2017). Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 20, 407-414 e404. [CrossRef]
- Chondronasiou, D.; Gill, D.; Mosteiro, L.; Urdinguio, R.G.; Berenguer-Llergo, A.; Aguilera, M.; Durand, S.; Aprahamian, F.; Nirmalathasan, N.; Abad, M. , et al. (2022a). Multi-omic rejuvenation of naturally aged tissues by a single cycle of transient reprogramming. Aging Cell 21, e13578. [CrossRef]
- Chondronasiou, D.; Martinez de Villarreal, J.; Melendez, E.; Lynch, C.J.; Pozo, N.D.; Kovatcheva, M.; Aguilera, M.; Prats, N.; Real, F.X.; Serrano, M. (2022b). Deciphering the roadmap of in vivo reprogramming toward pluripotency. Stem Cell Reports 17, 2501-2517. [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99-118. [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853-2868. [CrossRef]
- Cribb, L.; Hodge, A.M.; Yu, C.; Li, S.X.; English, D.R.; Makalic, E.; Southey, M.C.; Milne, R.L.; Giles, G.G.; Dugue, P.A. (2022). Inflammation and Epigenetic Aging Are Largely Independent Markers of Biological Aging and Mortality. J Gerontol A Biol Sci Med Sci 77, 2378-2386. [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. (2022). The MYC oncogene - the grand orchestrator of cancer growth and immune evasion. Nat Rev Clin Oncol 19, 23-36. [CrossRef]
- Doeser, M.C.; Scholer, H.R.; Wu, G. (2018). Reduction of Fibrosis and Scar Formation by Partial Reprogramming In Vivo. Stem Cells 36, 1216-1225. [CrossRef]
- Ferrucci, L.; Fabbri, E. (2018). Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol 15, 505-522. [CrossRef]
- Francesconi, M.; Di Stefano, B.; Berenguer, C.; de Andres-Aguayo, L.; Plana-Carmona, M.; Mendez-Lago, M.; Guillaumet-Adkins, A.; Rodriguez-Esteban, G.; Gut, M.; Gut, I.G. , et al. (2019). Single cell RNA-seq identifies the origins of heterogeneity in efficient cell transdifferentiation and reprogramming. Elife 8. [CrossRef]
- Gill, D.; Parry, A.; Santos, F.; Okkenhaug, H.; Todd, C.D.; Hernando-Herraez, I.; Stubbs, T.M.; Milagre, I.; Reik, W. (2022). Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. Elife 11. [CrossRef]
- Gonzalez-Gualda, E.; Baker, A.G.; Fruk, L.; Munoz-Espin, D. (2021). A guide to assessing cellular senescence in vitro and in vivo. FEBS J 288, 56-80.
- Guan, J.; Wang, G.; Wang, J.; Zhang, Z.; Fu, Y.; Cheng, L.; Meng, G.; Lyu, Y.; Zhu, J.; Li, Y. , et al. ( 2022). Chemical reprogramming of human somatic cells to pluripotent stem cells. Nature 605, 325–331. [CrossRef]
- Haridhasapavalan, K.K.; Raina, K.; Dey, C.; Adhikari, P.; Thummer, R.P. (2020). An Insight into Reprogramming Barriers to iPSC Generation. Stem Cell Rev Rep 16, 56-81. [CrossRef]
- Heffernan, C.; Sumer, H.; Malaver-Ortega, L.F.; Verma, P.J. (2012). Temporal Requirements of cMyc Protein for Reprogramming Mouse Fibroblasts. Stem Cells Int 2012, 541014. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. (2017). Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr Biol 27, 2652-2660 e2654. [CrossRef]
- Hishida, T.; Yamamoto, M.; Hishida-Nozaki, Y.; Shao, C.; Huang, L.; Wang, C.; Shojima, K.; Xue, Y.; Hang, Y.; Shokhirev, M. , et al. ( 2022). In vivo partial cellular reprogramming enhances liver plasticity and regeneration. Cell Rep 39, 110730. [CrossRef]
- Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biol 14, R115. [CrossRef]
- Horvath, S.; Lacunza, E.; Mallat, M.C.; Portiansky, E.L.; Gallardo, M.D.; Brooke, R.T.; Chiavellini, P.; Pasquini, D.C.; Girard, M.; Lehmann, M. , et al. (2024). Cognitive rejuvenation in old rats by hippocampal OSKM gene therapy. Geroscience. [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. (2022). Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 18, 611-627. [CrossRef]
- Jadhav, U.; Cavazza, A.; Banerjee, K.K.; Xie, H.; O'Neill, N.K.; Saenz-Vash, V.; Herbert, Z.; Madha, S.; Orkin, S.H.; Zhai, H. , et al. (2019). Extensive Recovery of Embryonic Enhancer and Gene Memory Stored in Hypomethylated Enhancer DNA. Mol Cell 74, 542-554 e545. [CrossRef]
- Kosar, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. (2011). Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 10, 457-468.
- Kovatcheva, M.; Melendez, E.; Chondronasiou, D.; Pietrocola, F.; Bernad, R.; Caballe, A.; Junza, A.; Capellades, J.; Holguin-Horcajo, A.; Prats, N. , et al. ( 2023). Vitamin B(12) is a limiting factor for induced cellular plasticity and tissue repair. Nat Metab 5, 1911–1930. [CrossRef]
- Kriukov, D.; Kuzmina, E.; Efimov, E.; Dylov, D.V.; Khrameeva, E.E. (2024). Epistemic uncertainty challenges aging clock reliability in predicting rejuvenation effects. Aging Cell, e14283. [CrossRef]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ait-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. (2011). Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev 25, 2248-2253. [CrossRef]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Canamero, M.; Blasco, M.A.; Serrano, M. (2009). The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460, 1136-1139.
- Li, H.Y.; Chien, Y.; Chen, Y.J.; Chen, S.F.; Chang, Y.L.; Chiang, C.H.; Jeng, S.Y.; Chang, C.M.; Wang, M.L.; Chen, L.K. , et al. (2011). Reprogramming induced pluripotent stem cells in the absence of c-Myc for differentiation into hepatocyte-like cells. Biomaterials 32, 5994-6005. [CrossRef]
- Liuyang, S.; Wang, G.; Wang, Y.; He, H.; Lyu, Y.; Cheng, L.; Yang, Z.; Guan, J.; Fu, Y.; Zhu, J. , et al. (2023). Highly efficient and rapid generation of human pluripotent stem cells by chemical reprogramming. Cell Stem Cell 30, 450-459 e459. [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194-1217.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. (2023). Hallmarks of aging: An expanding universe. Cell 186, 243-278. [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S. , et al. ( 2020). Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129. [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. (2015). Autophagy and aging. Adv Exp Med Biol 847, 73-87.
- Meena, J.K.; Cerutti, A.; Beichler, C.; Morita, Y.; Bruhn, C.; Kumar, M.; Kraus, J.M.; Speicher, M.R.; Wang, Z.Q.; Kestler, H.A. , et al. (2015). Telomerase abrogates aneuploidy-induced telomere replication stress, senescence and cell depletion. EMBO J 34, 1371-1384.
- Miliotou, E.; de Lazaro, I. (2024). A Youthful Touch: Reversal of Aging Hallmarks by Cell Reprogramming. Cells Tissues Organs, 1-13.
- Mitchell, W.; Goeminne, L.J.E.; Tyshkovskiy, A.; Zhang, S.; Chen, J.Y.; Paulo, J.A.; Pierce, K.A.; Choy, A.H.; Clish, C.B.; Gygi, S.P. , et al. (2024). Multi-omics characterization of partial chemical reprogramming reveals evidence of cell rejuvenation. Elife 12.
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marion, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Munoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J. , et al. (2016). Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 354. [CrossRef]
- Mosteiro, L.; Pantoja, C.; de Martino, A.; Serrano, M. (2018). Senescence promotes in vivo reprogramming through p16(INK)(4a) and IL-6. Aging Cell 17.
- Mylonas, A.; O'Loghlen, A. (2022). Cellular Senescence and Ageing: Mechanisms and Interventions. Front Aging 3, 866718. [CrossRef]
- Niimi, P.; Gould, V.; Thrush-Evensen, K.; Levine, M.E. (2024). The Latent Aging of Cells. bioRxiv.
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E. , et al. (2016). In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 167, 1719-1733 e1712. [CrossRef]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T. , et al. ( 2014). Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156, 663–677. [CrossRef]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. (2019). Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 18, e12877. [CrossRef]
- Paine, P.T.; Rechsteiner, C.; Morandini, F.; Desdin-Mico, G.; Mrabti, C.; Parras, A.; Haghani, A.; Brooke, R.; Horvath, S.; Seluanov, A. , et al. ( 2023). Initiation phase cellular reprogramming ameliorates DNA damage in the ERCC1 mouse model of premature aging. Front Aging 4, 1323194. [CrossRef]
- Qian, Y.; Chen, X. (2013). Senescence regulation by the p53 protein family. Methods Mol Biol 965, 37-61.
- Roux, A.E.; Zhang, C.; Paw, J.; Zavala-Solorio, J.; Malahias, E.; Vijay, T.; Kolumam, G.; Kenyon, C.; Kimmel, J.C. (2022). Diverse partial reprogramming strategies restore youthful gene expression and transiently suppress cell identity. Cell Syst 13, 574-587 e511. [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S. , et al. (2020). Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat Commun 11, 1545. [CrossRef]
- Shibata, H.; Komura, S.; Yamada, Y.; Sankoda, N.; Tanaka, A.; Ukai, T.; Kabata, M.; Sakurai, S.; Kuze, B.; Woltjen, K. , et al. (2018). In vivo reprogramming drives Kras-induced cancer development. Nat Commun 9, 2081. [CrossRef]
- Shimamoto, A.; Yokote, K.; Tahara, H. (2015). Werner Syndrome-specific induced pluripotent stem cells: recovery of telomere function by reprogramming. Front Genet 6, 10. [CrossRef]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. (2006). Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol 8, 1291-1297. [CrossRef]
- Teshigawara, R.; Cho, J.; Kameda, M.; Tada, T. (2017). Mechanism of human somatic reprogramming to iPS cell. Lab Invest 97, 1152-1157. [CrossRef]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. (2009). Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460, 1145-1148. [CrossRef]
- Waddington, C.H. (2014). The strategy of the genes (Routledge).
- Wang, C.; Rabadan Ros, R.; Martinez-Redondo, P.; Ma, Z.; Shi, L.; Xue, Y.; Guillen-Guillen, I.; Huang, L.; Hishida, T.; Liao, H.K. , et al. ( 2021). In vivo partial reprogramming of myofibers promotes muscle regeneration by remodeling the stem cell niche. Nat Commun 12, 3094. [CrossRef]
- Wang, G.; Wang, Y.; Lyu, Y.; He, H.; Liuyang, S.; Wang, J.; Sun, S.; Cheng, L.; Fu, Y.; Zhu, J. , et al. (2023). Chemical-induced epigenome resetting for regeneration program activation in human cells. Cell Rep 42, 112547. [CrossRef]
- Wang, L.; Su, Y.; Huang, C.; Yin, Y.; Chu, A.; Knupp, A.; Tang, Y. (2019). NANOG and LIN28 dramatically improve human cell reprogramming by modulating LIN41 and canonical WNT activities. Biol Open 8. [CrossRef]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. (2008). c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2, 10-12. [CrossRef]
- Xu, L.; Ramirez-Matias, J.; Hauptschein, M.; Sun, E.D.; Lunger, J.C.; Buckley, M.T.; Brunet, A. (2024). Restoration of neuronal progenitors by partial reprogramming in the aged neurogenic niche. Nat Aging 4, 546-567. [CrossRef]
- Yang, J.H.; Petty, C.A.; Dixon-McDougall, T.; Lopez, M.V.; Tyshkovskiy, A.; Maybury-Lewis, S.; Tian, X.; Ibrahim, N.; Chen, Z.; Griffin, P.T. , et al. ( 2023). Chemically induced reprogramming to reverse cellular aging. Aging (Albany NY) 15, 5966–5989. [CrossRef]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F. , et al. ( 2020). Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19, e13094. [CrossRef]
- Yucel, A.D.; Gladyshev, V.N. (2024). The long and winding road of reprogramming-induced rejuvenation. Nat Commun 15, 1941. [CrossRef]
- Zviran, A.; Mor, N.; Rais, Y.; Gingold, H.; Peles, S.; Chomsky, E.; Viukov, S.; Buenrostro, J.D.; Scognamiglio, R.; Weinberger, L. , et al. (2019). Deterministic Somatic Cell Reprogramming Involves Continuous Transcriptional Changes Governed by Myc and Epigenetic-Driven Modules. Cell Stem Cell 24, 328-341 e329. [CrossRef]
Figure 1.
Sankey plot of various biological processes that are affected across reprogramming studies in in vitro and in vivo mouse studies, alongside in vitro human studies. The first column indicates the study and the age of cells or mice reprogrammed, when available, in weeks (w), months (m) or years (y). The fourth column indicates the tissue analyzed.
Figure 1.
Sankey plot of various biological processes that are affected across reprogramming studies in in vitro and in vivo mouse studies, alongside in vitro human studies. The first column indicates the study and the age of cells or mice reprogrammed, when available, in weeks (w), months (m) or years (y). The fourth column indicates the tissue analyzed.
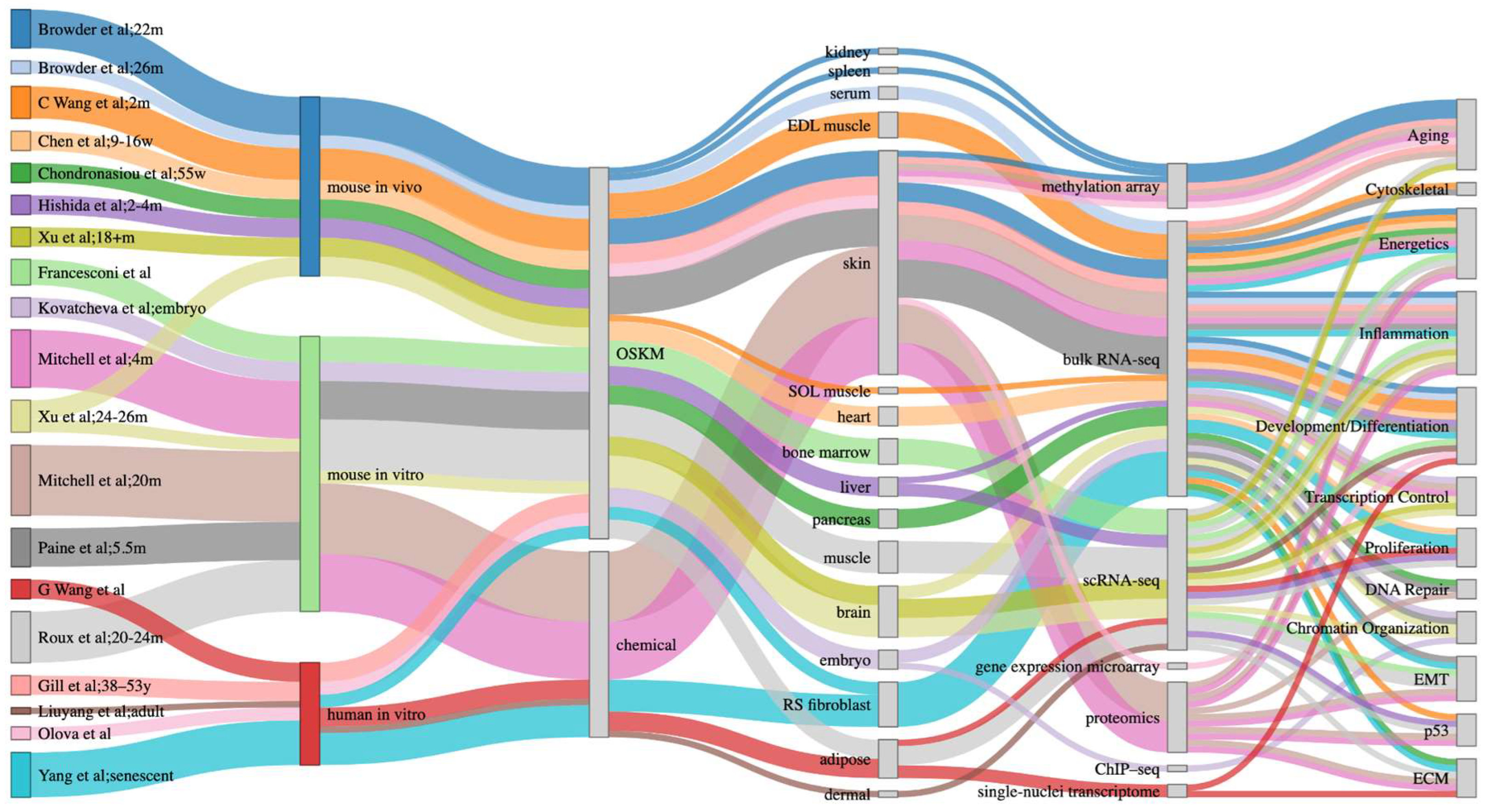
Figure 3.
Sankey plot of inflammation-associated biological processes regulated during partial reprogramming in in vitro and in vivo experiments in mice, alongside in vitro human experiments.
Figure 3.
Sankey plot of inflammation-associated biological processes regulated during partial reprogramming in in vitro and in vivo experiments in mice, alongside in vitro human experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



