
Submitted:
01 October 2024
Posted:
02 October 2024
You are already at the latest version
Abstract
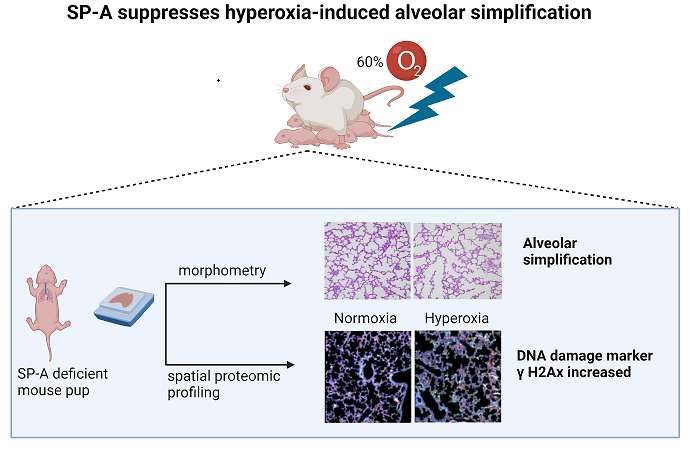
Background-Bronchopulmonary dysplasia (BPD) is a condition of poor alveolar formation that causes chronic breathing impairment in infants born prematurely. Preterm lungs lack surfactant and are vulnerable to oxidative injuries driving the development of BPD. Our recent studies reported that surfactant protein A (SP-A) genetic variants influence susceptibility to neonatal lung disease. SP-A modulates activation of alveolar macrophages and parturition onset in late gestation. We asked whether a lack of SP-A alters alveolarization in a mouse model of hyperoxia-induced BPD. Methods- SP-A-deficient and control newborn mice were exposed to either clinically relevant 60% O2 hyperoxia or normoxia for 5-7 days. Alveolar formation was then assessed by mean linear intercept (MLI) and radial alveolar count (RAC) measurements in lung tissue sections. Results- We report that the combination of SP-A deficiency and hyperoxia reduces alveolar growth compared to WT mice. The morphometric analysis of normoxic SP-A-deficient lungs showed lower RAC compared to controls, indicating reduced alveolar number. In the presence of hyperoxia, MLI was higher in SP-A-deficient lungs compared to controls. Differences were statistically significant for female pups. Spatial proteomic profiling of lung tissue sections showed that hyperoxia caused a 4-fold increase in the DNA damage marker gammaH2Ax in macrophages of SP-A-deficient lungs compared to normoxia. Conclusions- Our short report suggests an important role for SP-A in perinatal lung development and in protecting lung macrophages from oxidant injury. These studies warrant future investigation to discern the temporal interaction of SP-A, gender, oxidant injury, and lung macrophages in perinatal alveolar formation and development of BPD.
Keywords:
bronchopulmonary dysplasia
; hyperoxia
; surfactant protein A (SP-A)
; prematurity
; neonatal
1. Introduction
Bronchopulmonary dysplasia (BPD) is the most common chronic respiratory morbidity of prematurity. BPD is characterized by an arrest in lung growth and disruption of alveolar development [1]. BPD is associated with high mortality, increased healthcare costs, and poor neurodevelopmental outcomes [2]. Despite advances in critical care, there is increasing BPD incidence with challenges in developing newer diagnostics and therapeutics. The innate immune system likely plays an important role in BPD pathogenesis in the setting of inflammation and oxidative stress [3]. Premature neonates are born with surfactant deficiency and lack immune protection by surfactant protein A (SP-A). SP-A plays a dual role in surfactant ultrastructure and modulation of inflammation and innate host defense by alveolar macrophages (AMs) [4]. SP-A gene polymorphisms have been associated with an increased risk for the development of BPD in preterm infants [5,6]. SP-A is not incorporated in commercially available surfactant replacement therapies that are used to treat preterm infants at risk of BPD.
SP-A is expressed and secreted by alveolar type II epithelial cells of the fetal lung in late gestation. SP-A modulates the activation of AMs and the onset of parturition in late gestation [7,8,9]. SP-A mediates bacterial phagocytosis [10] and secretion of inflammatory mediators by macrophages [11]. These SP-A functions are relevant to BPD pathogenesis as infection and inflammation play an important role in causing lung injury. Additionally, SP-A affects the NAD(H) redox status of AMs in response to ozone exposure [12]. Oxidative stress due to hyperoxia is an important factor in the pathology of BPD [3]. Here, we report that lack of SP-A affects neonatal lung growth in response to hyperoxia in SP-A-deficient mice.
2. Materials and Methods
2.1. Mice
All animal work was approved by the Institutional Animal Care and Use Committee (Protocol No 101786). and performed within an American Association for the Accreditation of Laboratory Animal Care-certified barrier facility at Pennsylvania State University College of Medicine. Mice were housed under specific pathogen-free conditions in micro-isolator cages, provided food and sterile water ad-libitum, and maintained on a 12-hour-light/12-hour-dark cycle. The generation of SP-A-deficient mice has been described previously [13]. SP-A-deficient mice were backcrossed to the C57BL/6J background [14]. WT male and female C57BL/6J controls were purchased from JAX labs at 4-6 weeks of age and bred in-house. Male and female mice were used for mating at the age of 6-8 weeks at a ratio of 1:1 male to female.
2.2. Exposure to Hyperoxia or Normoxia
Dams and pups were exposed to either hyperoxia (60% O2) or maintained at normoxia in room air (21% O2) from post-natal day 1 (P1) until post-natal day 5-7 (P5-P7). The litter size was maintained at 6-10 pups per exposure. Nursing dams received food ad libitum. If nursing dams were unable to nurse due to oxygen toxicity, they were transferred between room air and hyperoxia. Dams did not exhibit overt signs of illness at 60% O2 during the 7-day exposure period.
2.3. Histology and Morphometry
Mice were anesthetized by intraperitoneal injection of Ketamine/Xylazine (90/10 mg/kg/dose) followed by laparotomy and exsanguination via inferior vena cava incision. The lungs were then exposed via thoracotomy, intubated using a tracheal cannula, and fixed with 10% (v/v) formalin or 4% (w/v) paraformaldehyde at 25 cm H2O hydrostatic pressure. The fixed lungs were then processed for paraffin embedding, sectioned at 6 μm, and stained with the periodic acid Schiff (PAS) stain. Sections were evaluated blinded to the genotype. The quantitative measures included mean linear intercept (MLI) and radial alveolar count (RAC). MLI was calculated by determining the average distance between the intersections of alveolar septal tissue. To achieve this, we used a stereological method that involves randomly placing a counting grid over the lung tissue. The number of times the grid intersects the alveolar septal tissue was counted, and the average distance between these intersections was computed to obtain the MLI. RAC was determined by counting the number of alveoli between the pleural surface of the lung and the nearest terminal bronchiole. We employed a systematic sampling method to determine RAC, which involves counting the number of alveoli in a series of parallel lines intersecting the lung parenchyma. RAC was expressed as the number of alveoli per unit length of the line, and it provides a measure of the density of alveoli in the lung tissue.
2.4. Spatial Proteomic Profiling by GeoMx® Nanostring
The changes in CD11b+ and ABAC3+ populations in the lungs exposed to hyperoxia and normoxia were assessed using Nanostring spatial proteomics. Two representative slides of mouse lungs from SP-A knockout male pups were analyzed. A total of 24 AOI (area of interest) with 8 CD11b+, 8ABCA3+, and 8DNA+ populations were assessed. The mouse immune-oncogene core protein panel plus immune activation status, immune cell typing, cell death, MAPK signaling, PI3k/AKT signaling, and pan-tumor panels that included 72 protein targets and 6 housekeeping targets were analyzed. The morphology markers that were used were αSMA, CD11b, ABCA3, and DAPI to visualize airway smooth muscle and blood vessels, macrophages, alveolar type II epithelial cells, and DNA, respectively. The gene expression patterns were assessed by the DSP data analysis tool (GeoMx® Nanostring). Data normalization for the DSP protein assay was done by normalizing to housekeeping protein controls – GAPDH, Histone H3, S6 and scaling to isotype negative controls- Rat IgG2a, Rat IgG2b, Rabbit IgG. Housekeeping and isotype control normalization were used to control for within-slide differences in the region of interest (ROI) cellularity and between-slide variability (differences in signal volume due to tissue quality, fixation, processing, etc.). Further normalization was done by scaling to the area to normalize the geomean of the masked area to correct for variable surface area, and scaling to nuclei was done to normalize the geomean of nuclei count of masked ROI.
2.5. Immunohistochemistry
Immunofluorescent staining of lung tissues was performed on formalin-fixed paraf-fin-embedded sections. The sections were washed in xylene and gradually rehydrated in ethanol (100%, 95%, 70%, 50%) and processed per the modified Chapman protocol. Samples were washed with Phosphate Buffered Saline (PBS), blocked with 1 % BSA/5 % horse serum (45 min, room temperature), and then incubated overnight at 4°C with primary antibody (1:500 dilutions in 0·5 % BSA), washed 3 times with PBS, incubated with appropriate fluorescent-labeled secondary antibodies (1:1000 dilution in 0·5 % BSA, Life Technologies Inc) and the nuclear marker, 4`,6-diamidino-2-phenylindole, dihydrochloride (DAPI, Invitrogen cat# P36962). The primary antibody used was γH2AX (Cell signaling #9718). Slides were imaged using fluorescent microscopy (Nikon Eclipse TE2000-U). Fluorescence intensity was quantified and analyzed via ImageJ software.[15]
2.6. Statistical Analysis
Descriptive characteristics were computed as means +/- standard deviation (SD). Two-way ANOVA was used to compare groups using GraphPad Prism software. All tests were 2-tailed, and significance was assigned for p <0.05. The p-value was adjusted using Tukey’s multiple comparisons test. There were 3-10 mouse pups per sex per group.
3. Results
3.1. Lack of SP-A Enhances Hyperoxia-Induced Arrest in Alveolar Formation
To assess the impact of SP-A deficiency in alveolar simplification, we exposed mice to 60% O2 for 5-7 days after birth when the saccular to alveolar transition of alveolar development is known to occur [16]. This period corresponds to 24-36 weeks gestation of human neonates when premature infants are at high risk of developing BPD. The 60% O2 dose is comparable to clinical settings and sufficient to induce alveolar arrest in WT mouse lungs [17]. We used MLI and RAC as hallmark morphometric measures of alveolar development and growth, respectively [18,19]. Data were pooled together or analyzed separately for male and female pups (Figure 1). Hyperoxia resulted in a significant increase in MLI to 47.4±12.4 µm (Mean±SD) in SP-A-deficient lungs compared to 34.7±2.3 µm (adjusted p=0.0005) and 34.5±4.1 µm (adjusted p=0.0006) in hyperoxic and normoxic WT lungs, respectively (Figure 2A). The SP-A-deficient female lungs were the most sensitive to hyperoxia with an MLI at 50.1±13.8 µm compared to 33.8±4.7 µm (adjusted p= 0.018) in normoxic WT male lungs, 35.3±3.7 µm (adjusted p= 0.043) in normoxic WT female lungs, 34.4±3.6 µm (adjusted p= 0.014) in hyperoxic WT male lungs, and 35.14±2.9 µm (adjusted p= 0.039) in hyperoxic WT female lungs (Figure 2B).
Measurements of RAC, however, showed that lack of SP-A impacts alveolar growth independent of supplemental oxygen exposure (Figure 2C) and gender (Figure 2D). The combined RAC indices of male and female mice of 3.9±1.2 (Mean±SD) and 4.0±1.5 (Mean±SD) in normoxic and hyperoxic SP-A-deficient lungs were significantly lower than 8.7±2.0 (adjusted p<0.0001) and 7.9±2.0 (adjusted p<0.0001) in WT normoxic and hyperoxic pups, respectively (Figure 2C). Alveolar growth under conditions of hyperoxia appeared to occur faster in female lungs, although differences were not statistically different (Figure 2D). This, however, reflects a significant difference in the RAC index between WT and SP-A-deficient female pups (Figure 2D).
Taken together, these results indicate that the underdeveloped SP-A-deficient lungs are vulnerable to hyperoxia-induced injury with more severe impact in the female premature lung.
3.2. Lack of SP-A Enhances the DNA Damage Response to Hyperoxia in Lung Macrophages
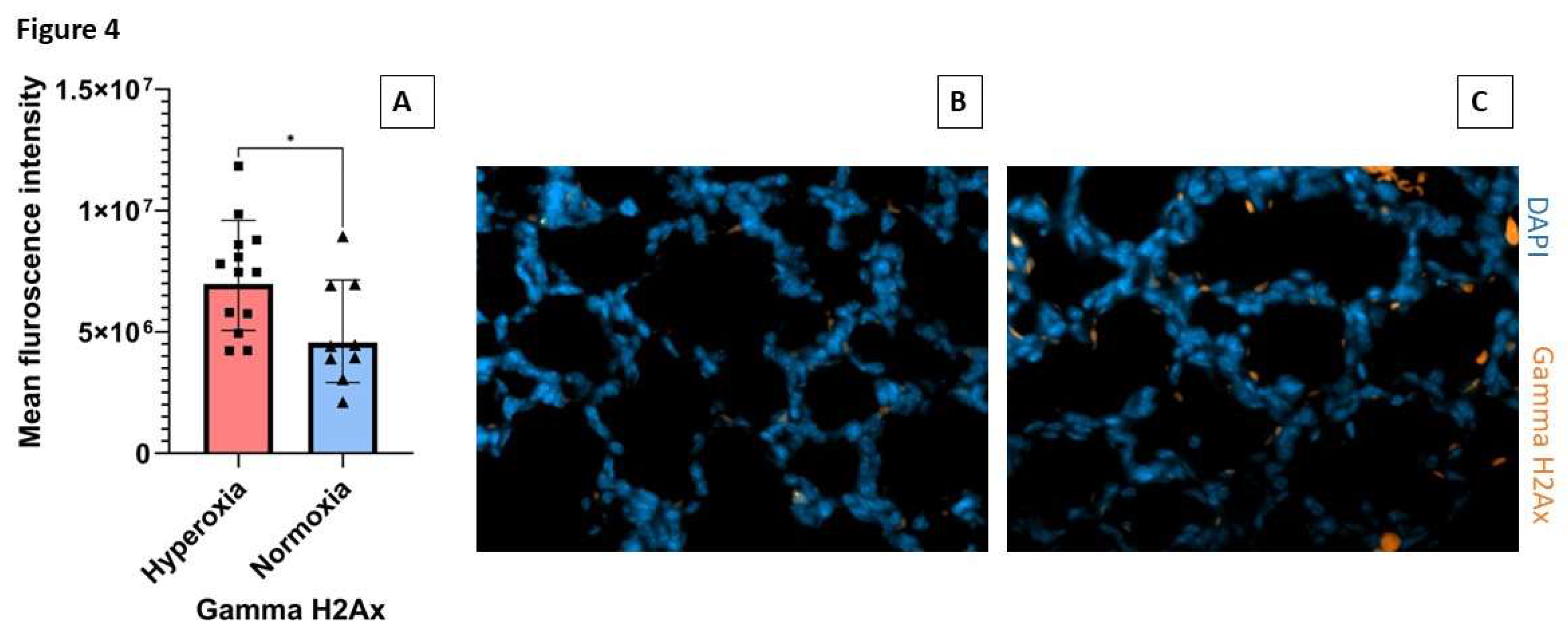
SP-A is the principal oxidant-sensitive component and modulator of reactive oxygen species by AMs in pulmonary surfactant [11,20,21]. Here, we utilized spatial proteomics to assess whether lack of SP-A impacts the hyperoxia response in neonatal lung immune microenvironment (Figure 3). To address this question, we applied spatial proteomic profiling using GeoMx® Nanostring to assess protein expression in ABCA3+ type II cells, DNA, CD11b+ macrophages in hyperoxic and normoxic lungs of SP-A-deficient mice (Figure 3A-B and Supplemental Figure S1). The cluster analysis shows the differential protein expression profile among the 72 targeted proteins after exposure to hyperoxia vs normoxia in CD11b+ macrophages and ABAC3+ alveolar type II epithelial cells (Figure 3A). The volcano plot in Figure 3B shows a 2- and 4-fold increase in phosphorylated S6 and the formation of gamma-H2Ax in CD11b+ macrophages in SP-A-deficient lungs exposed to hyperoxia(p<0.05) (Supplemental Table). These results indicate that lung macrophages in SP-A-deficient mice experience increased levels of hyperoxic stress compared to normoxia. Oxidative stress via hyperoxia in SP-A KO mouse pups resulted in increased expres-sion of γH2AX as 7.3x106 mean intensity when compared to normoxia SP-A KO mouse pups as 4.9x106 mean intensity (p=0.026) (Figure 4.) These results indicate that lung macrophages in SP-A-deficient mice experience increased levels of hyperoxic stress compared to normoxia.
4. Discussion
To our knowledge, we present the first known evaluation of the effects of hyperoxia on neonatal mouse pups in the absence of SP-A. We demonstrate that SP-A-deficient neonatal mice exhibit greater arrest in alveolar development at 60% O2 compared to WT mice. The O2 dosage was shown to cause morphological changes in WT neonatal lungs following prolonged exposure from P1 to P14 days after birth [17]. Our study shows that a lack of SP-A accelerates the effect of hyperoxia on alveolar arrest within a shorter time frame of oxygen exposure. Our findings reflect those of preterm infants at risk of BPD, who may develop lung injury even with lower oxygen exposure. These results warrant further investigation into the mechanisms by which SP-A attenuates the development of BPD in early infancy.
Lack of SP-A reduced RAC similarly in both male and female hyperoxic lungs compared to normoxia. The RAC index of normoxic SP-A-deficient lungs, however, was lower than that of WT lungs, suggesting a novel role of SP-A in perinatal lung development. This hypomorphism may contribute to increased susceptibility to oxidant injury in the immature preterm lung when SP-A is not yet induced. Recent studies showed that SP-A supports the integrity of the developing airspace from inflammation through the SP-A receptor SP-R210 [22].
Data analysis according to gender indicates a greater impact of gender in hyperoxic female lungs of SP-A-deficient mice. Previous studies reported sexual dimorphism at higher 95% O2 [23]. Biochemical assays and secretion of inflammatory mediators on day 7 in this study showed increased inflammation in females, although alveolar simplification was not quantitated on this day. Sex-dependent differences in alveolar simplification were inferred by immunohistochemical differences in endothelial cell remodeling with lesser impact in females [23]. The present results show that WT female lungs had the highest increase in RAC index under hyperoxia, consistent with a more resilient endothelium in female lungs. Lack of SP-A, however, also resulted in the highest MLI in female lungs. Adult female SP-A-deficient mice are also noted to be affected more by oxidative stress compared to their male counterparts[24]. However, clinically, males are affected more with severe BPD, and WT male mice exposed to hyperoxia were noted to have increased alveolar simplification compared to females [23]. The present results, however, indicate that SP-A deficient males exposed to hyperoxia exhibit increased interstitial infiltration comparable to WT counterparts, indicating that sex-specific response to hyperoxia needs further exploration beyond lung morphometry. Additional studies are therefore needed to decipher the intersection of SP-A and gender in alveolar expansion, hyperoxic saccular injury, and inflammation.
Alveolar macrophages (AMs) play central roles in post-natal lung function and development [25,26,27,28,29,30,31,32,33] and are critically involved in the pathogenesis of BPD [25,34,35,36]. Preterm infant lung macrophages primed with 65% O2 display a persistent inflammatory response to LPS [35]. Prior studies have reported γH2Ax as a response to DNA damage resulting from oxidative stress in lung tissues associated with chronic pulmonary obstructive disease [37,38] and pulmonary arterial hypertension[39]. Our preliminary spatial proteomic profiling indicating an increase in γH2Ax in macrophages in hyperoxic SP-A-deficient lungs warrants additional studies to determine whether SP-A modulates the DNA damage response in lung macrophages that would indicate SP-A administration as a treatment to promote lung development in premature neonates.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1:
Author Contributions
Conceptualization, SA. and ZC.; methodology, SA, ML, TMU.; software, SA, ML.; validation, SA, ML; formal analysis, SA.; writing—original draft preparation, SA writing—review and editing, SA, ZC, TMU, ML; visualization, SA.; supervision, ZC.; funding acquisition, SA. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the Children’s Miracle Network Grant Funds, the Department of Pediatrics, and the College of Medicine.
Institutional Review Board Statement
All animal work was approved by the Institutional Animal Care and Use Committee (Protocol No 101786). and performed within an American Association for the Accreditation of Laboratory Animal Care-certified barrier facility at Pennsylvania State University College of Medicine.
Data Availability Statement
The data will be made available by Shaili Amatya and can be reached out at samatya@pennstatehealth.psu.edu
Acknowledgments
We would like to acknowledge GeoMx Nanostring (Seattle, WA) for the digital spatial proteomic profiling and analysis of the mouse lung FFPE sections.
Conflicts of Interest
Zissis C. Chroneos is co-founder of Respana Therapeutic, Inc. (http://respana-therapeutics.com/) an early-stage company developing immune-therapeutics. Dr. Chroneos and The Pennsylvania State University own equity in Respana Therapeutics. These financial interests have been reviewed by the University’s Institutional and Individual Conflict of Interest Committees and are currently being managed by the University. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Jobe, A.H., The new BPD. NeoReviews, 2006. 7(10): p. e531-e545.
- Schmidt, B., et al., Prediction of late death or disability at age 5 years using a count of 3 neonatal morbidities in very low birth weight infants. The Journal of pediatrics, 2015. 167(5): p. 982-986. e2.
- Higgins, R.D., et al., Bronchopulmonary dysplasia: executive summary of a workshop. The Journal of pediatrics, 2018. 197: p. 300-308. [CrossRef]
- Wright, J.R., Immunoregulatory functions of surfactant proteins. Nature Reviews Immunology, 2005. 5(1): p. 58-68. [CrossRef]
- Silveyra, P. and J. Floros, Genetic variant associations of human SP-A and SP-D with acute and chronic lung injury. Frontiers in bioscience: a journal and virtual library, 2012. 17: p. 407. [CrossRef]
- Liu, D.-Y., et al., Expression of IL-8, SP-A and TGF-beta1 in bronchoalveolar lavage fluid of neonates with bronchopulmonary dysplasia. Zhongguo Dang dai er ke za zhi= Chinese Journal of Contemporary Pediatrics, 2010. 12(6): p. 444-446.
- Montalbano, A.P. Hawgood, and C.R. Mendelson, Mice deficient in surfactant protein A (SP-A) and SP-D or in TLR2 manifest delayed parturition and decreased expression of inflammatory and contractile genes. Endocrinology, 2013. 154(1): p. 483-498. [CrossRef]
- Yadav, A.K. Madan, and A.L. Bernal, Surfactant proteins A and D in pregnancy and parturition. Front Biosci (Elite Ed), 2011. 3: p. 291-300. [CrossRef]
- Condon, J.C., et al., Surfactant protein secreted by the maturing mouse fetal lung acts as a hormone that signals the initiation of parturition. Proceedings of the National Academy of Sciences, 2004. 101(14): p. 4978-4983. [CrossRef]
- Mikerov, A.N., et al., SP-A1 and SP-A2 variants differentially enhance association of Pseudomonas aeruginosa with rat alveolar macrophages. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2005. 288(1): p. L150-L158.
- Wang, G., et al., The effect of ozone exposure on the ability of human surfactant protein a variants to stimulate cytokine production. Environmental health perspectives, 2002. 110(1): p. 79-84. [CrossRef]
- Xu, H.N., et al., Sex and SP-A2 dependent NAD (H) redox alterations in mouse alveolar macrophages in response to ozone exposure: Potential implications for COVID-19. Antioxidants, 2020. 9(10): p. 915. [CrossRef]
- Korfhagen, T.R., et al., Altered surfactant function and structure in SP-A gene targeted mice. Proceedings of the National Academy of Sciences, 1996. 93(18): p. 9594-9599. [CrossRef]
- Goto, H., et al., The role of surfactant protein A in bleomycin-induced acute lung injury. American journal of respiratory and critical care medicine, 2010. 181(12): p. 1336-1344. [CrossRef]
- Chhinder, P. Sodhi1, Andres J. Gonzalez Salazar1,2, Mark L. Kovler1,2, William B. Fulton1,2, Yukihiro Yamaguchi1,2, Asuka Ishiyama1,2, Sanxia Wang1,2, Thomas Prindle Jr.1,2, Mustafa Vurma3, Tapas Das3, Hongpeng Jia1,2, Peng Lu1,2 and David J. Hackam1,2*, The administration of a pre-digested fat-enriched formula prevents necrotising enterocolitis-induced lung injury in mice. British Journal of Nutrition, 2022. 128: p. 1050-1063.
- Berger, J. and V. Bhandari, Animal models of bronchopulmonary dysplasia. The term mouse models. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2014. 307(12): p. L936-L947.
- Nardiello, C., et al., Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia. Disease models & mechanisms, 2017. 10(2): p. 185-196.
- Cooney, T.P. and W.M. Thurlbeck, The radial alveolar count method of Emery and Mithal: a reappraisal 1--postnatal lung growth. Thorax, 1982. 37(8): p. 572-579. [CrossRef]
- Hsia, C.C., et al., An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. American journal of respiratory and critical care medicine, 2010. 181(4): p. 394-418. [CrossRef]
- Kuzmenko, A.I., et al., Surfactant Protein A Is a Principal and Oxidation-sensitive MicrobialPermeabilizing Factor in the Alveolar LiningFluid. Journal of Biological Chemistry, 2005. 280(27): p. 25913-25919. [CrossRef]
- Crowther, J.E., et al., Pulmonary surfactant protein a inhibits macrophage reactive oxygen intermediate production in response to stimuli by reducing NADPH oxidase activity. The Journal of Immunology, 2004. 172(11): p. 6866-6874. [CrossRef]
- Guan, Z., et al., Disruption of the SP-A/SP-R210L (MYO18Aα) pathway prolongs gestation and reduces fetal survival during lipopolysaccharide-induced parturition in late gestation. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2024. 326(4): p. L508-L513.
- Lingappan, K., et al., Sex-specific differences in neonatal hyperoxic lung injury. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2016. 311(2): p. L481-L493.
- Mikerov, A.N., et al., Ablation of SP-A has a negative impact on the susceptibility of mice to Klebsiella pneumoniae infection after ozone exposure: sex differences. Respiratory Research, 2008. 9: p. 1-16.
- Kalymbetova, T.V., et al., Resident alveolar macrophages are master regulators of arrested alveolarization in experimental bronchopulmonary dysplasia. The Journal of pathology, 2018. 245(2): p. 153-159. [CrossRef]
- Bonfield, T.L., et al., PU. 1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2003. 285(5): p. L1132-L1136. [CrossRef]
- Guilliams, M., et al., Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. Journal of Experimental Medicine, 2013. 210(10): p. 1977-1992. [CrossRef]
- Schneider, C., et al., Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS pathogens, 2014. 10(4): p. e1004053. [CrossRef]
- Tan, S.Y. and M.A. Krasnow, Developmental origin of lung macrophage diversity. Development, 2016. 143(8): p. 1318-1327. [CrossRef]
- Gschwend, J., et al., Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. Journal of Experimental Medicine, 2021. 218(10): p. e20210745. [CrossRef]
- Philipp, H., et al., Perinatal development of innate immune topology. eLife, 2021. 10.
- Martin, F.P., et al., Alveolar macrophages: adaptation to their anatomic niche during and after inflammation. Cells, 2021. 10(10): p. 2720. [CrossRef]
- Yang, Y. and Y. Wang, Autocrine, Paracrine, and Endocrine Signals That Can Alter Alveolar Macrophages Function. Reviews of Physiology, Biochemistry and Pharmacology, 2022: p. 177-198.
- Hallman, M., The surfactant system protects both fetus and newborn. Neonatology, 2013. 103(4): p. 320-326. [CrossRef]
- Twisselmann, N., et al., Hyperoxia/Hypoxia exposure primes a sustained pro-inflammatory profile of preterm infant macrophages upon LPS stimulation. Frontiers in Immunology, 2021. 12: p. 762789.
- Willis, G.R., et al., Extracellular vesicles protect the neonatal lung from hyperoxic injury through the epigenetic and transcriptomic reprogramming of myeloid cells. American Journal of Respiratory and Critical Care Medicine, 2021. 204(12): p. 1418-1432. [CrossRef]
- Caramori, G., et al., Unbalanced oxidant-induced DNA damage and repair in COPD: a link towards lung cancer. Thorax, 2011. 66(6): p. 521-527. [CrossRef]
- Pastukh, V.M., et al., Oxidative DNA damage in lung tissue from patients with COPD is clustered in functionally significant sequences. International journal of chronic obstructive pulmonary disease, 2011: p. 209-217. [CrossRef]
- Van Houten, B., Pulmonary arterial hypertension is associated with oxidative stress–induced genome instability. 2015, American Thoracic Society. p. 129-130.
Figure 1.
Prominent hyperoxia-induced alveolar simplification in SP-A-deficient lungs. Representative images of PAS-stained formalin-fixed lungs from WT (A-H) and SP-A-deficient (I-P) pups exposed to normoxia (A-D, I-L) or hyperoxia (E-H, M-P). Images were captured at 200x (left columns) or 400x (right columns) magnification from female and male pups, as indicated. All images were captured on postnatal day 7 except images I and J, which were captured on day 6.
Figure 1.
Prominent hyperoxia-induced alveolar simplification in SP-A-deficient lungs. Representative images of PAS-stained formalin-fixed lungs from WT (A-H) and SP-A-deficient (I-P) pups exposed to normoxia (A-D, I-L) or hyperoxia (E-H, M-P). Images were captured at 200x (left columns) or 400x (right columns) magnification from female and male pups, as indicated. All images were captured on postnatal day 7 except images I and J, which were captured on day 6.
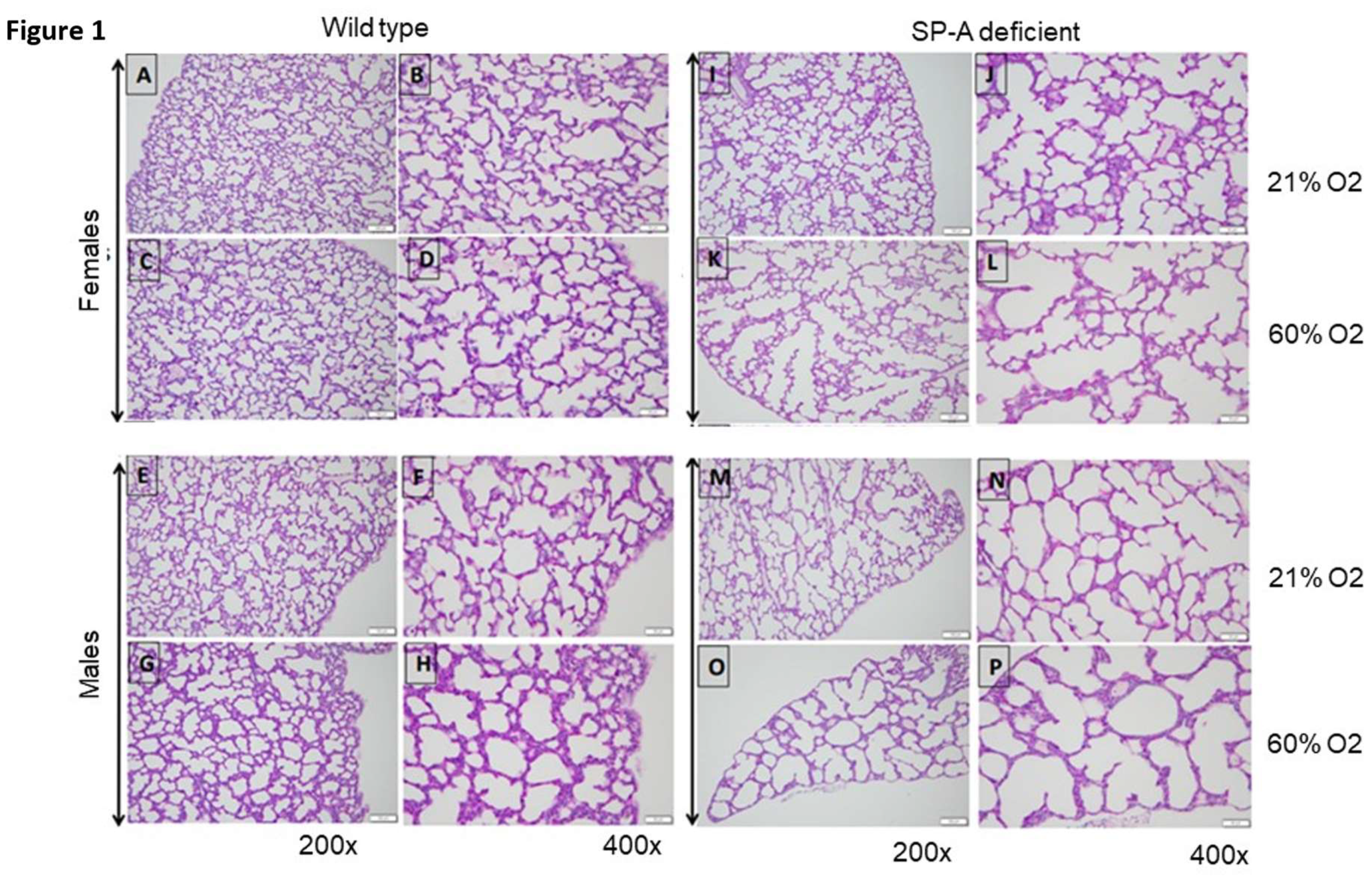
Figure 2.
Lack of surfactant protein A (SP-A) modulates hyperoxia induced alveolar simplification. Neonatal pups were exposed to either normoxia or hyperoxia with oxygen exposure at 60% from birth till post-natal day 5-7. Lack of SP-A causes worsening of alveolar simplification secondary to hyperoxia as noted by increased mean linear intercept (A) and reduced radial alveolar count (C) compared to wild type. Lack of SP-A noted to contribute to sex differences in hyperoxia induced neonatal lung injury, as female SP-A knockout (KO) mice are more prone to alveolar simplification (B) and (D) when exposed to hyperoxia when compared to wild type (WT) genotype. Data shown as Means ± SD. N= 3-10 mouse pups per sex per group. *adjusted p<0.05, ***adjusted p<0.001, ****adjusted p<0.0001.
Figure 2.
Lack of surfactant protein A (SP-A) modulates hyperoxia induced alveolar simplification. Neonatal pups were exposed to either normoxia or hyperoxia with oxygen exposure at 60% from birth till post-natal day 5-7. Lack of SP-A causes worsening of alveolar simplification secondary to hyperoxia as noted by increased mean linear intercept (A) and reduced radial alveolar count (C) compared to wild type. Lack of SP-A noted to contribute to sex differences in hyperoxia induced neonatal lung injury, as female SP-A knockout (KO) mice are more prone to alveolar simplification (B) and (D) when exposed to hyperoxia when compared to wild type (WT) genotype. Data shown as Means ± SD. N= 3-10 mouse pups per sex per group. *adjusted p<0.05, ***adjusted p<0.001, ****adjusted p<0.0001.
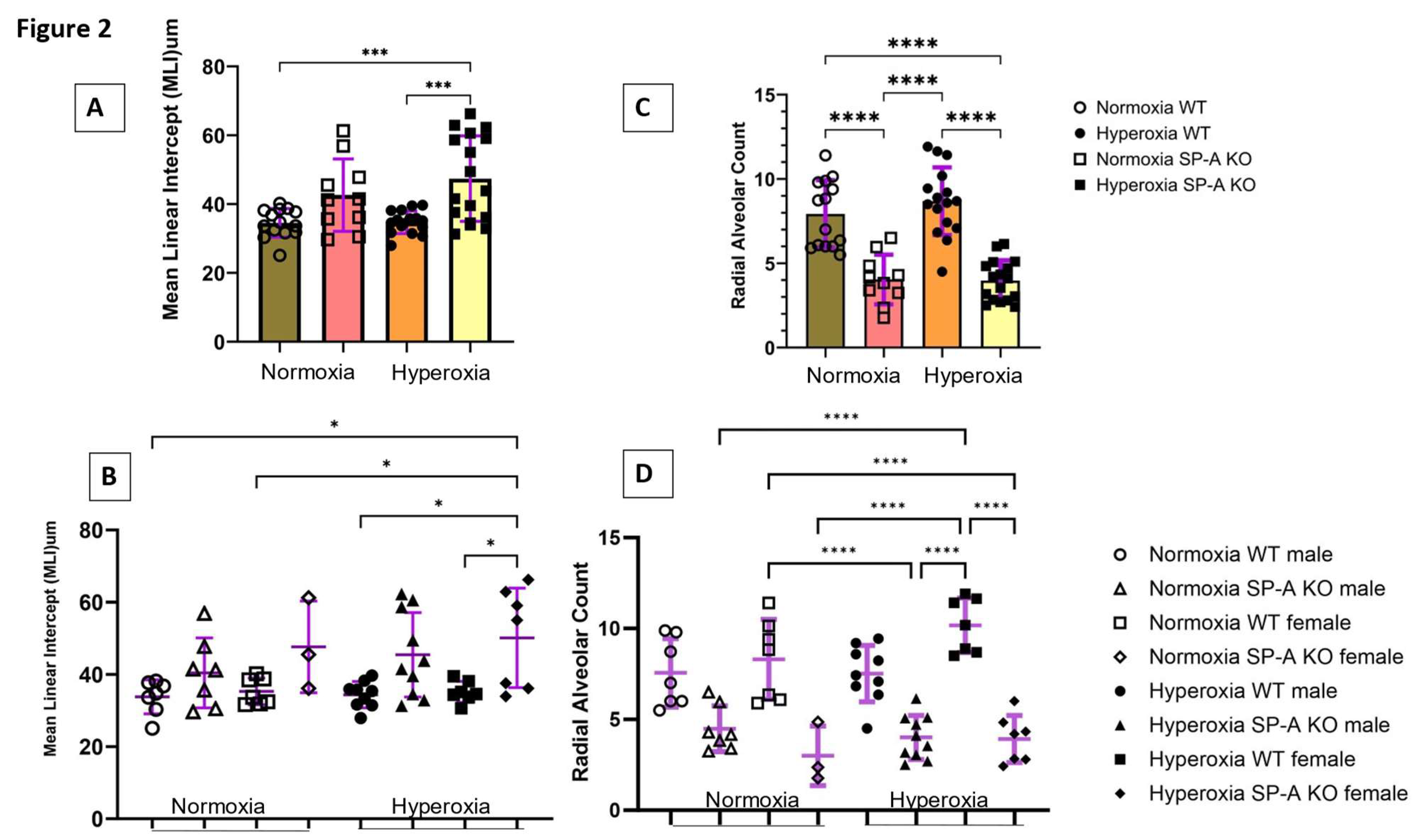
Figure 3.
Spatial proteomic profiling of neonatal mouse lungs shows that lack of SP-A alters the response to hyperoxia and contributes to lung injury. SP-A deficient neonatal pups were exposed to either normoxia or hyperoxia with oxygen exposure at 60% from birth till post-natal day 7. (A) The heatmap analysis demonstrates the differential protein expression profile of hyperoxia versus normoxia among the 72 targeted proteins that were compared in different lung populations such as macrophage (CD11b+), Type II cells (ABCA3+) and DNA (B) The volcano plot depicts the proteins that have been upregulated and downregulated in the SP-A deficient mouse alveolar macrophage upon exposure to hyperoxia in comparison to normoxia. The blue color depicts the proteins with a –log 10 p-value less than 2, and the red dot shows a –log 10 p-value of more than 2.
Figure 3.
Spatial proteomic profiling of neonatal mouse lungs shows that lack of SP-A alters the response to hyperoxia and contributes to lung injury. SP-A deficient neonatal pups were exposed to either normoxia or hyperoxia with oxygen exposure at 60% from birth till post-natal day 7. (A) The heatmap analysis demonstrates the differential protein expression profile of hyperoxia versus normoxia among the 72 targeted proteins that were compared in different lung populations such as macrophage (CD11b+), Type II cells (ABCA3+) and DNA (B) The volcano plot depicts the proteins that have been upregulated and downregulated in the SP-A deficient mouse alveolar macrophage upon exposure to hyperoxia in comparison to normoxia. The blue color depicts the proteins with a –log 10 p-value less than 2, and the red dot shows a –log 10 p-value of more than 2.
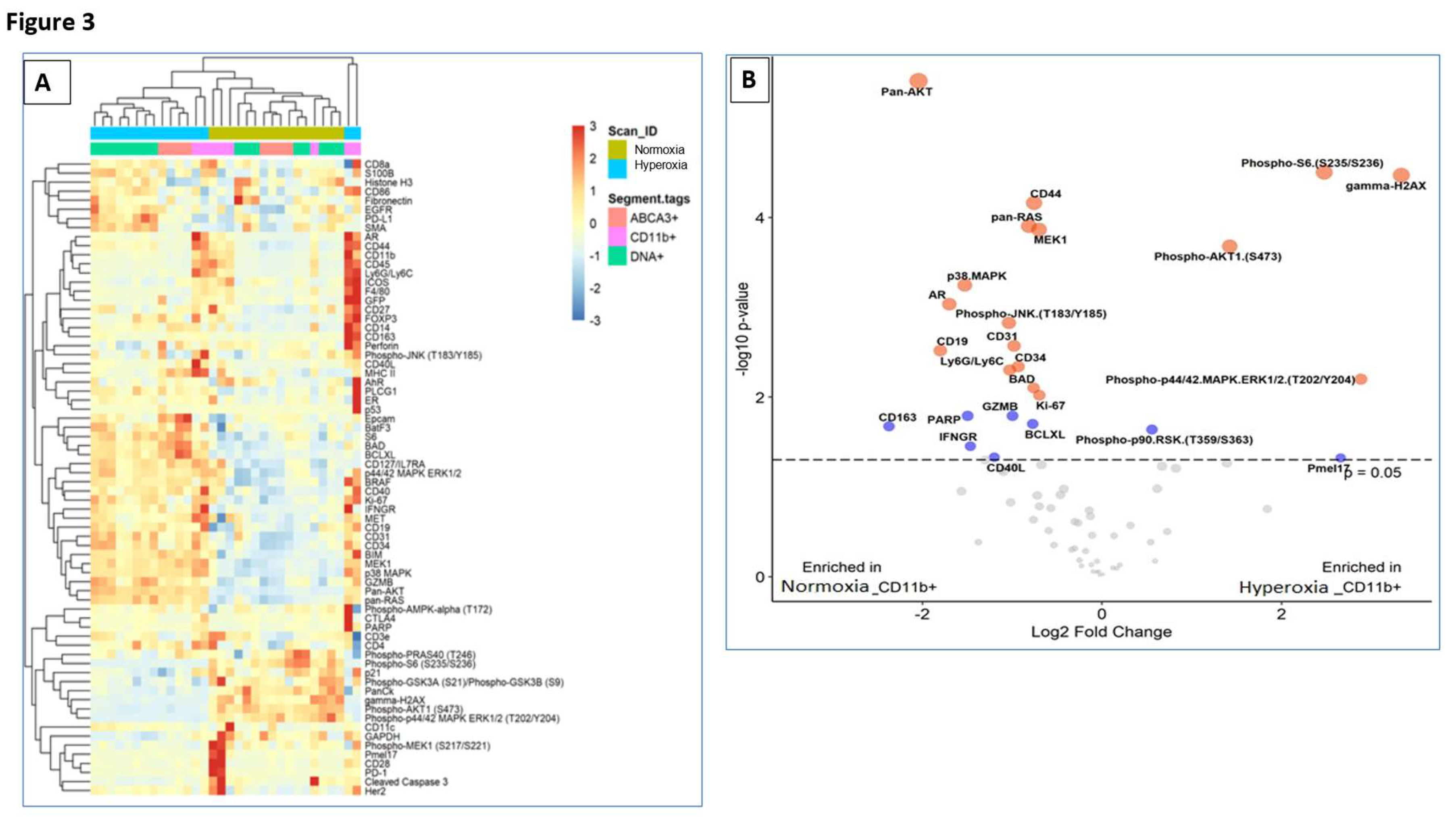
Figure 4.
SP-A deficiency in hyperoxia-exposed mouse pups simulates alveolar injury in a mouse model of BPD. Neonatal pups were exposed to normoxia or hyperoxia (O2 at 60%) from birth till PND7. (A) Quantification of mean fluorescence intensity of γH2Ax in SP-A deficient mouse pups exposed to normoxia and hyperoxia (one or more separate areas from each mouse formalin fixed paraffin embedded lung section was imaged and quantified, total mice n>2 mice per group). (B)-(C) immunofluorescence images of γH2Ax staining at 20x magnification (γH2Ax orange, DAPI nuclei blue) in normoxia (B) and hyperoxia (C).
Figure 4.
SP-A deficiency in hyperoxia-exposed mouse pups simulates alveolar injury in a mouse model of BPD. Neonatal pups were exposed to normoxia or hyperoxia (O2 at 60%) from birth till PND7. (A) Quantification of mean fluorescence intensity of γH2Ax in SP-A deficient mouse pups exposed to normoxia and hyperoxia (one or more separate areas from each mouse formalin fixed paraffin embedded lung section was imaged and quantified, total mice n>2 mice per group). (B)-(C) immunofluorescence images of γH2Ax staining at 20x magnification (γH2Ax orange, DAPI nuclei blue) in normoxia (B) and hyperoxia (C).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



