
Submitted:
03 October 2024
Posted:
04 October 2024
You are already at the latest version
Abstract
Jararhagin-C (JarC) is a protein from the venom of Bothrops jararaca consisting of disintegrin-like and cysteine-rich domains. JarC shows a modulating effect on angiogenesis and remodeling of extracellular matrix constituents improving wound healing in mice experimental model. JarC is purified from crude venom and the yield is less than 1 %. The aim of this work was to obtain the recombinant form of JarC and to test its biological activity. For this purpose, the bicistronic vector pSUMOUlp1 was used. This vector allowed the expression of the recombinant toxin JarC (rJarC) in fusion with the small ubiquitin-related modifier (SUMO) as well as the SUMO protease Ulp1. After expression, this protease was able to efficiently remove SUMO from rJarC inside the bacteria. rJarC free from SUMO was purified at the expected molecular mass, and recognized by polyclonal anti-Jararhagin antibodies. In terms of biological activity, both the native and recombinant forms showed no toxicity to the HUVEC cell line CRL1730 and were effective in modulating cell migration activity in experimental in vitro model. These results demonstrate the successful production of rJarC and the preservation of its biological activity, which may facilitate further investigations into the therapeutic potential of this snake venom-derived protein.
Keywords:
snake venom disintegrins
; recombinant protein
; Jararhagin-C
; endothelial cells
1. Introduction
Disintegrins are a family of highly homologous soluble polypeptides found in various snake venoms [1,2]. Most of the known disintegrins seem to be compatible with the hypothesis that their free forms are released from larger precursors containing a metalloproteinase domain, while others are directly synthesized from mRNA without the metalloproteinase region [3]. Jararhagin C (JarC) is a 28 kD protein containing disintegrin-like and cysteine-rich domains, derived from the SVMP PIII Jararhagin [4], which has an ECD integrin-targeting tripeptide [5] and a domain that recognizes selectively the α2β1 integrin present in endothelial cells [6]. Integrins are transmembrane non-enzymatic receptors that are essential to form cell-cell and cell-matrix adhesions [7]. Among the main actions triggered by integrins activation, considering physiological and pathological conditions, are migration, adhesion, proliferation and cell invasion playing key roles in development, immune responses, leucocyte traffic, hemostasis and cancer metastasis [8]. The disruption of integrin function by soluble integrin ligand can act as a potent agonist or antagonists of several integrin receptors. Previous studies have shown that JarC is a versatile molecule that can be explored as a therapeutic strategy for wound healing, including the treatment of chronic wounds where processes such as inflammation, angiogenesis and the deposition/remodeling of matrix components are unregulated [9,10,11,12,13].
To obtain JarC in its native form, numerous purification procedures are carried out by hydrophobic interaction and ion exchange chromatography, and at the end of the process, the fraction corresponding to this protein represents about 1% of the crude venom. Cloning and heterologous expression of this toxin in a bacterial system could circumvent this problem. However, it is very difficult to express a snake venom toxin in recombinant form in such a way that this recombinant toxin retains the structure, conformation and biological activity compared to the native toxin. This is due to the fact that when a recombinant protein is synthesized in E. coli, the microenvironment differs from the native environment in terms of pH, osmolarity, redox potential, cofactors and folding mechanisms. Furthermore, at a high expression rate, hydrophobic regions of the produced protein are present in high concentration and are available for interactions with similar regions. All these factors can lead to the instability and aggregation of some recombinant proteins, known as inclusion bodies [14,15]. The use of fusion proteins can increase the solubility and stability of recombinant proteins such as glutathione S-transferase (GST) [16]), Nus A [17], thioredoxin (Trx) [18], maltose binding protein (MBP) [19] and ubiquitin-related modifying protein (SUMO) [20]. SUMO is a Saccharomyces cerevisiae-derived protein that belongs to the family of ubiquitin-like proteins, which are not found in prokaryotes but are highly conserved in eukaryotes and are responsible for several cellular processes [21]. One of the advantages of SUMO is its easy removal by SUMO protease 1 (Ulp1), as this enzyme specifically recognizes and cleaves the tertiary structure of SUMO [22], avoiding the possibility of non-specific cleavages. Thus, the SUMO protease Ulp1 perfectly removes SUMO in vitro without leaving additional amino acids in parts of the cleavage sites, facilitating the production of target proteins with a native N-terminus. Therefore, SUMO can serve as an important tool for the expression of improved functional proteins in both eukaryotes and prokaryotes [23].
To obtain sufficient amounts of JarC for further pharmacological studies, in this work, we expressed it in a bacterial expression system using the bicistronic vector pSUMOUlp1 developed by our group [24], which allowed us to obtain this toxin in recombinant active form.
2. Results
Expression and Purification of rJar-C
The pSUMOUlp1 vector containing the sequence of JarC (pSUMOUlp1-JarC) successfully expressed the recombinant JarC (rJarC) in E. coli BL 21 Star (DE3) cells. During the expression, due to the concomitant expression of SUMO protease Ulp1, the rJarC was obtained free from SUMO and ready for purification due to the presence of a histidine tag at its C-terminal. The purified rJarC was analyzed by SDS-PAGE, which showed a band at the expected molecular size of approximately 28 kDa (Figure 1A), which was also detected by anti-His antibodies in Western blot analysis (Figure 1B). The final yield of the purified rJarC was approximately 5 mg per liter of bacterial culture.
The proteomic analysis confirmed that the recombinant protein is JarC, as shown in Figure 2. The identified peptides mapped across the entire sequence show a high degree of coverage, further confirming the successful expression and accurate identification of the recombinant protein.
Tertiary Structure of rJarC Is Important for Antibody Recognition
The polyclonal anti-jararhagin antibody detected the rJarC, native JarC, and jararhagin only under non-reduced condition, revealing that the conformational structure is important and was preserved on the recombinant protein (Figure 3). These results confirm the maintenance of the native tertiary structure of the toxins for recognition by the anti-jararhagin antibody.
Native and Recombinant JarC Exhibit Non-Toxic Effects on HUVEC Cells
The results shown in Figure 4 demonstrate that both native and recombinant JarC are non-toxic to HUVEC cells, as shown by the MTT cell viability assay. Here, the recombinant green fluorescent protein (GFP) was used as an unrelated recombinant protein control.
Native and Recombinant JarC Promote HUVEC Cell Migration on Collagen Substrates
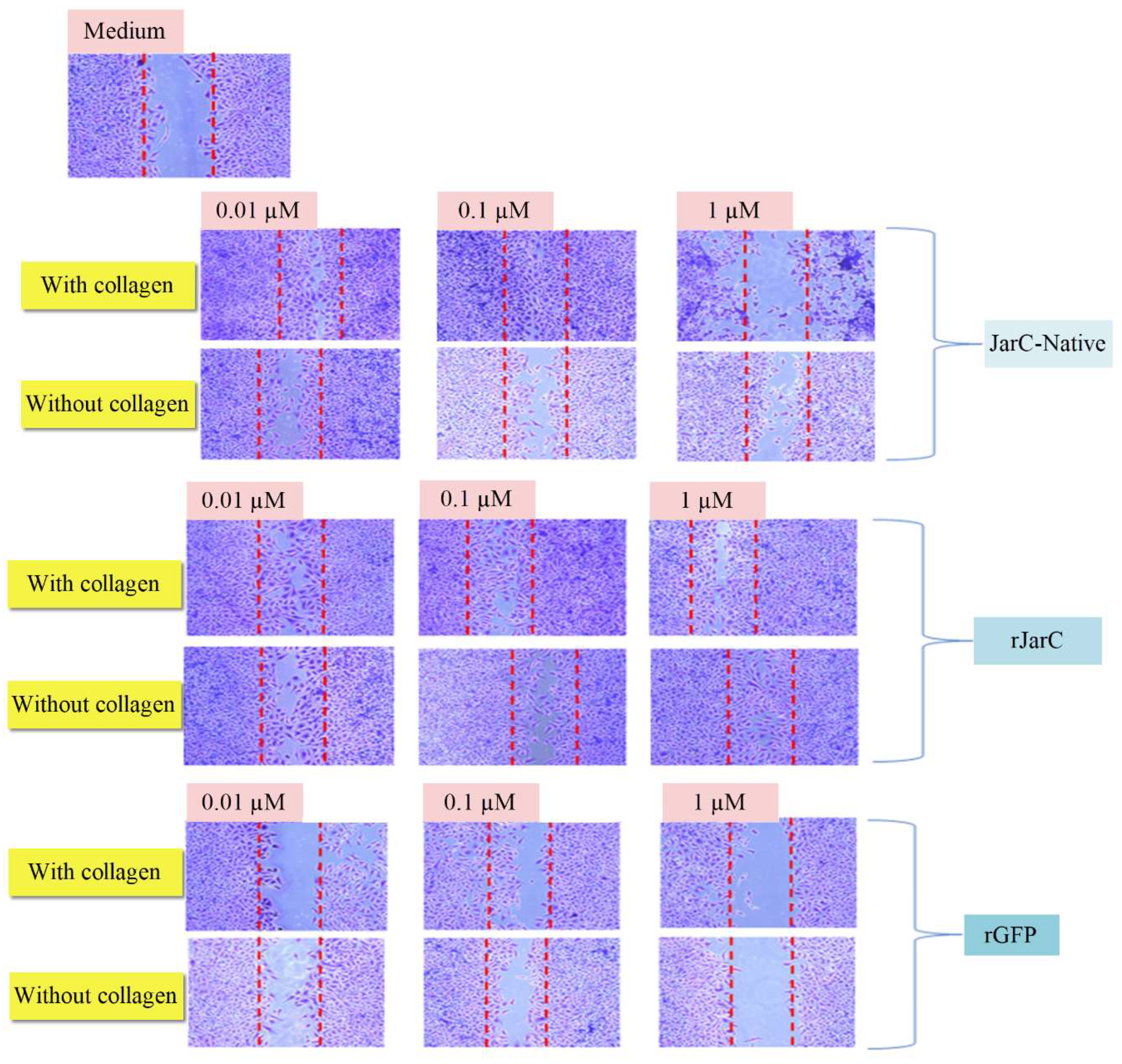
Both native JarC and rJarC were effective in stimulating the migration of HUVEC cells attached to a type I collagen substrate or directly on the culture plate surface (Figure 5). Lower doses of native JarC (0.1 µM) were more efficient in promoting cell migration on the collagen-coated surface compared to the untreated surface, while higher doses (1 µM) appeared to induce cell detachment. In contrast, rJarC stimulated cell migration at all tested doses on both substrates, with no significant differences observed in the migration patterns. The rGFP protein, on the other hand, did not stimulate HUVEC cell migration. These results collectively demonstrate the ability of rJarC to induce in vitro cell migration, which is highly similar to the effects observed with the native JarC protein, suggesting that the recombinant protein retains the pro-migratory properties of the native toxin.
3. Discussion
Recombinant disintegrins have been addressed for the comprehension of biological activities dependent on integrin receptors, such as hemostasis, thrombosis, cell adhesion, angiogenesis, proliferation and tumor [29].
In a previous study conducted in our laboratory, JarC (jarhagin ECD-containing disintegrin domain) was successfully cloned and biologically actively expressed in the pET32 vector fused to thioredoxin (Trx-JD9) and expressed in E. coli [30]. Although obtaining the recombinant protein was successful, it was necessary to proceed with time-consuming steps to obtain rJarC in free form, such as the in vitro cleavage with enterokinase as well as a further step to purify it from the fusion protein thioredoxin. At the end of the process, after cleavage and purification of the TRX-JD9 protein, the yield was 850 µg/L of culture, approximately 5 times less than the yield obtained in the present study (5mg/L of culture), which was a limitation to obtain the recombinant protein on a large scale. Therefore, the successful expression and purification of recombinant Jar-C (rJarC) using the bicistronic pSUMOUlp1 vector system is an important advancement. The concomitant Ulp1 protease expression of this vector allowed the removal of SUMO from JarC, and after a single purification step, a good yield of this disintegrin-like toxin was obtained for further biological characterization. The fact that JarC has no predicted glycosylation makes it suitable for expression in bacterial system. However, the fact that it contains 28 cysteines, which correspond to 13.2% of its amino acid composition, makes obtaining this molecule in a soluble and active form is quite a challenge, since mis-pairing of cysteines can cause misfolding, aggregation and ultimately result in low yields. Nevertheless, the use of pSUMOUlp1 vector was very effective to produce this molecule in soluble free form with a yield of 5mg/L of culture. The maintenance of the native tertiary structure, as evidenced by the recognition of rJarC by anti-jararhagin polyclonal antibodies under non-reducing conditions, indicates that the tertiary structure is apparently preserved, which is crucial for the functional properties of the toxin.
In our study, both the native and recombinant forms of JarC exhibited non-toxic effects on HUVEC endothelial cells, consistent with previous reports on the effect of disintegrin-like/cysteine-rich protein on endothelial cells such as Alternagin-C, isolated from Bothrops alternatus venom [31]. The ability of rJarC to stimulate cell migration on collagen substrates (wound healing assay), similar to the pro-migratory properties observed with the native toxin, suggests that the recombinant protein retains the key functional activities associated with JarC. This is in line with studies demonstrating the role of Alternagin-C in promoting cell migration [32].
Key limitations of this study include the lack of in vivo validation to assess the translation of the in vitro results. Therefore, addressing this limitation in future research would improve the understanding of the recombinant toxin and its similarities to the native form, which is critical for evaluating its potential therapeutic applications.
4. Conclusion
In conclusion, the successful expression and purification of rJarC using the bicistronic pSUMOUlp1 vector system has enabled the production of sufficient quantities of this disintegrin-like toxin for further investigation of its biological activities. The recombinant protein maintains key functional properties, such as the ability to stimulate one-dimensional cell migration, similar to the native JarC. These findings provide a foundation for more in-depth studies on the potential therapeutic applications of this recombinant toxin, particularly in the context of wound healing, angiogenesis, and extracellular matrix remodeling.
5. Material and Methods
Cloning of JarC into the pSUMOUlp1 Vector
The JarC sequence (1077 to 1713 pb) from the database (GenBank: X68251.1) was optimized and synthesized by GeneArt (Thermo Fisher Scientific) for expression in E. coli. The gene was inserted into the commercial vector pUC57. To clone the JarC sequence into the pSUMOUlp1 vector, it was amplified from pUC57 by polymerase chain reaction (PCR) using the primers JarC_Forward: 5’ggatccATTATTTCACCTCC3´ and JarC_Reverse: 5´ctcgagAAGCTTTGTAGCC3´, containing the restriction site BamHI and XhoI (underlined), respectively. After amplification, the JarC-PCR product and the pSUMOUlp1 vector were digested with the restriction enzymes BamHI and XhoI. The digested products were subjected to a 1% agarose gel, and the bands corresponding to JarC and pSUMOUlp1 were cut and purified using the Wizard® plus Miniprep DNA purification system kit (Promega®) according to the manufacturer’s instructions. Subsequently, 25 μg of vector and 75 μg of insert (1:3 ratio), 1 μL ligase, 2 μL ligase buffer (50 mM Tris-HCl pH 7.5, 10 mM MgCl2, 10 mM dithiothreitol, 1 mM ATP, 25 µg/mL BSA) were used for a 15 µL final volume reaction and incubated overnight at 4 °C. Chemically competent top 10 bacteria were transformed with the JarC-pSUMOUlp1 construct, and colonies were analyzed by PCR to identify those with the correct insert. Subsequently, the plasmids of the colonies that showed amplification were purified and subjected to sequencing by the Center for Human Genome Research of the University of São Paulo - Institute of Life Sciences.
rJarC Expression in pSUMOUlp1 Vector
The pSUMOUlp1 vector was transformed into the competent bacterium Escherichia coli BL 21 Star (DE3) using the thermal shock transformation method, whereby 1 ml of LB medium (Luria Bertani) was added to the transformed bacteria and shaken in a dry bath at 37 °C for 1 hour at 400 rpm. Subsequently, 1 ml of this bacterial volume was added to 30 ml of LB medium containing the antibiotic chloramphenicol (34 mg/ml) as well as calcium chloride and magnesium chloride (both at a concentration of 1 mM) and kept overnight at 30 °C with shaking at 180 rpm. The following day, 4 ml of the preinoculum was added to a volume of 200 ml of LB medium and maintained at 30 °C with shaking at 180 rpm until bacterial growth reached an optical density of 0.5 to 0.6 at an absorbance of 600 nm, when rJAR-C expression was induced with 1 mM IPTG (isopropyl-β-D-thiogalactopyranoside) for 4 hours. After this time, the entire bacterial volume was centrifuged at 12,000 g and the precipitate was stored at -20°C until processing. The precipitate was resuspended in wash and equilibration buffer (20 mM sodium phosphate, 500 mM NaCl, with 20 mM imidazole pH 7.40). Lysozyme at final concentration of 0.2 mg/mL was added and homogenized for 30 minutes at room temperature. The bacterial pellet was then sonicated in an intermittent procedure (20% amplitude with a 3-second pulse and a 4-second interval between each pulse) in an ice bath with a 4-minute cooling break. This procedure was repeated five times and the lysate material was centrifuged at 12000 g for 15 minutes at 4 °C. The supernatant was used for the purification of the recombinant protein.
Purification of rJarC
Purification was performed by Immobilized Metal Affinity Chromatography (IMAC) using Ni Sepharose® 6 Fast Flow Resin (GE® Healthcare) according to the manufacturer’s recommendations. The total volume of supernatant was applied to 1.5 ml of resin, which was previously washed and equilibrated in binding buffer (20 mM sodium phosphate, 500 mM NaCl, with 20 mM imidazole pH 7.40). The protein binding reaction to the resin was carried out for 30 minutes in a carousel homogenizer. It was washed twice with 70 mM imidazole and then eluted with 1M imidazole. The eluate was dialyzed overnight at 4 °C with phosphate-buffered saline (PBS) containing NaCl and MgCl (both 1 mM, pH 7.40). After this step, the protein was analyzed on a 12% polyacrylamide/acrylamide gel with SDS (sodium dodecyl sulfate) under reducing conditions. The Page Ruler Unstained Protein Ladder (Thermo Fisher Scientific) was used as a molecular weight standard. The gel was stained with Coomassie blue (40 % methanol, 7 % glacial acetic acid, and blue dye R-250), and the protein concentration was estimated using the Bicinchoninic Acid (BCA) method (Thermo Fisher Scientific).
In-Solution Digestion and Proteomic Analysis
The sample was subjected to in-solution digestion under the following conditions: (1) 5 μL of DTT (100 mM dithiothreitol) was added and maintained for 30 minutes at 60 ºC; (2) 2.5 μL of iodoacetamide (200 mM) was added and the samples were kept for 30 minutes at room temperature and protected from light; (3) the sample was incubated for at least 12 hours at room temperature with 10 μL of trypsin (40 ng/μL in 100 mM ammonium bicarbonate). The reaction was stopped by adding 50% ACN/5% TFA. Samples were analyzed by a liquid chromatography-mass spectrometry system on an ESI-IT-TOF instrument coupled to a UFLC (20A Prominence, Shimadzu). Each sample was injected into a C18 column (Kinetex C18, 5 μm; 50 × 2.1 mm) in a binary solvent system: (A) water: DMSO: acid (949:50:1) and (B) ACN: DMSO: water: acid (850: 50: 99: 1). A gradient elution of 0-40% of solvent B was used for 35 minutes at a constant flow rate of 0.2 ml.min-1. The samples were monitored by a Shimadzu SPD-M20A PDA detector before being injected into the mass spectrometer.
The interface voltage was 4.5 kV; capillary voltage was 1.95 kV, at 200 ºC; and collision-induced fragmentation by argon was set to 55% “energy”. MS spectra were acquired in positive mode, in the mass range of 350-1400 / charge (m/z). MS/MS spectra were collected in the range of 50 to 1950 m/z. The raw Shimadzu LCD data (LCMS Protein Postrun, Shimadzu) was converted to .mzXML files before analysis. Peaks Studio V7.0 software (BSI, Toronto, ON, Canada) was used for processing (de novo peptide sequencing and proteomic identification). Proteomic identification was carried out according to the following parameters: mass error (MS and MS/MS) adjusted to 0.1 Da; oxidation of methionine and carbamidomethylation as variable and fixed modifications, respectively; trypsin method; maximum missed cleavages (3), maximum variable post-translational modifications (PTMs) per peptide (3), and non-specific cleavage (1). The samples were analyzed against the “Animal Toxins” database compiled from the UniProt database.
Western Blot Analysis
The Western Blot analysis using anti-His antibody was performed according to the description by Calabria et al., 2019 [25]. Samples of recombinant JarC were analyzed on a 12% SDS-PAGE under reducing conditions. Subsequently, the samples were transferred to nitrocellulose membranes using the Trans-Blot® SD Semi-Dry Transfer Cell (Bio-Rad® Laboratories, Hercules, CA, USA) following the manufacturer’s recommendations. After transfer, the nitrocellulose membranes were stained with Ponceau S® (Merck Millipore Corporation, Darmstadt, Germany) 1:20 to verify the transfer of the proteins. To remove the dye, the membranes were washed with TBS-Tween (20 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.5) until complete removal. Subsequently, the membranes were blocked with incubation buffer (Tris/NaCl, pH 7.5 with 5% powder skimmed milk) for 2 h at room temperature and then washed 3 times with TBS-Tween. Afterward, the membranes were incubated for 2 h with mouse monoclonal anti-polyhistidine antibody (Sigma Life Science, Merck Corporation, Darmstadt, Germany) at a 1:1000 dilution in an incubation buffer. After, the membranes were washed with TBS-Tween and incubated for 2 h with the peroxidase-labeled anti-mouse IgG (Sigma Life Science, Merck Corporation, Darmstadt, Germany) at a 1:5000 dilution in incubation buffer. Then a new wash cycle was performed and the antigenic components were revealed with 0.05% (w/v) 4-chloro-1α-naphthol in 15% (v/v) methanol in the presence of 0.03% H2O2 (v/v).
rJarC Detection by Polyclonal Anti-Jararhagin Antibodies (Dot Blot)
The similarity of the conformational structure between rJarC and native JarC was investigated using an anti-Jararhagin antibody in a dot-blot assay. The assay was performed with the proteins under reducing and non-reducing conditions to evaluate the conformational structure. For analysis under reducing conditions, 2 µg of each sample was added to 5 µl of sample buffer containing reducing agent (2.5 % DTT, 62.5 mM Tris pH 6.8, 10 % glycerol, 2 % SDS) and boiled at 100 °C for five minutes. For analysis under non-reducing conditions, 2 µg of each sample was added to 5 µl of PBS and applied directly to the nitrocellulose membrane. The following proteins were used: rJarC, JarC and Jararhagin. These proteins were diluted in PBS and the concentration was adjusted to 2 µg/5 µl.
The total volume of 5 µl of each sample was applied directly to the nitrocellulose membrane and incubated at 37°C until the samples had dried. The membranes were blocked with Tris-phosphate buffered saline (TBS) and 5% soluble skim milk (Molico®-Nestle) for 2 hours with agitation, then three washes of 10 minutes each were performed with 0.1% TBS/Tween 20. After washing, the membranes were incubated overnight at 4°C with two different primary antibodies: a homemade polyclonal anti-jararhagin antibody prepared in rabbits, diluted 1:1000, or an anti-histidine antibody prepared in mice (SIGMA-USA), diluted 1:3000 in TBS solution and 5% soluble skim milk overnight at 4°C. The next day, the membranes were washed three times for 10 minutes each with 0.1% TBS/Tween 20. The membranes were incubated with the secondary antibody: Peroxidase-labeled anti-rabbit IgG or anti-mouse IgG diluted 1:1000 in TBS solution and 5% soluble skim milk and shaken for 2 hours. It was washed three times for 10 minutes, each time with TBS-Tween. Peroxidase detection was performed by adding a solution containing the chromogenic substrate 4-chloro-1-naphthol (Sigma-USA) and H2O2, prepared according to the manufacturer’s recommendations. The reaction was interrupted with distilled water immediately after the stains developed on the nitrocellulose membrane.
Purification of Native JarC (JarC)
Native JarC was purified from freeze-dried Bothrops jararaca venom provided by the Herpetology Laboratory of the Butantan Institute. Purification was performed as previously described [12]. The protein concentration of native and rJarC was measured using the Bradford assay (Bio-Rad) according to the manufacturer’s instructions. The purity of native and rJarC was analyzed by SDS-PAGE electrophoresis under denaturing conditions (SDS) according to the technique described previously [26]. Toxins were analyzed with a 15% SDS-PAGE gel at 110 V, 55 mA, for 3 hours and 10 minutes using the Page Ruler Unstained Protein Ladder Molecular Mass Marker (Thermo Fisher Scientific). A solution of 0.2% (m/v) Coomassie Blue R-250 in water and methanol in a 1:1 (v/v) ratio was used to stain the gel bands, and the gel was bleached with methanol diluted in distilled water with 30%/10% acetic acid.
HUVEC Cell Culture
Human vascular endothelial cells (HUVEC) of the ATCC - CRL1730 line were grown in RPMI 1640 culture medium (Gibco®) supplemented with 10 % complement-inactivated fetal bovine serum (FBS), 2 mM L-glutamine (Gibco®), 100 U/ml penicillin/100 µg/ml streptomycin (Gibco®) in an incubator at 37 ºC and 5 % CO2. The culture medium supplemented with 10 % FBS was used only for the maintenance and proliferation of the cells and was replaced during the experiments by a culture medium supplemented only with antibiotics and L-glutamine. After reaching 80-100 % confluence, the cells were harvested with trypsin/EDTA (Gibco®) according to the manufacturer’s instructions.
Cell Viability Assay
A total of 2 x 104 CRL1730 HUVEC cells per well, diluted in RPMI supplemented with 10 % FBS, were seeded in a 96-well plate. These cells were kept at 37 °C for 2 hours in the presence of CO2 to adhere to the bottom of the plate. Then, the culture medium was removed, the cells were washed twice with PBS buffer and RPMI culture medium was added. Cells were incubated for 24 hours in the presence of 1µM JarC, rJarC, rGFP (recombinant green fluorescent protein used as an unrelated recombinant protein) or 0.1% Triton X-100 (a negative control toxic to the cells). RPMI medium supplemented with 10 % FBS was used as a positive control. For the MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) assay, the Chemicon International kit was used according to the manufacturer’s instructions. Finally, the plate was analyzed with a Spectramax spectrophotometer at 570 nm.
In Vitro Migration of Endothelial Cells: Wound Healing Assay
The assay to test cell movement in an empty field mimicking an in vitro model of wound healing was performed using CRL 1730 HUVEC cells as described in [27,28]. An amount of 1 x 106 CRL1730 HUVEC cells per well, diluted in RPMI supplemented with 10% FBS, was seeded into a 24-well culture plate that had been previously sensitized with collagen type I (50 µg/mL) or had not received any sensitization treatment. The cells were maintained in complete RPMI medium at 37 °C and 5 % CO2 for 24 hours to attach to the substrate. After this time, the cells were washed twice with PBS buffer and RPMI culture medium containing only the antibiotics was added, and a line was scraped into each well using a sterile tip (200 µl). Samples of JarC and rJarC were added to these wells at concentrations of 0.01 µM, 0.1 µM and 1 µM or RPMI medium. The recombinant EGFP protein was used as an unrelated recombinant protein control. After 24 hours, cells were fixed and stained using the HEMA 3 Stain Set Protocol (Thermo Fisher Scientific) according to the manufacturer’s recommendations and analyzed under an inverted microscope with phase contrast (LEICA). Images were captured in triplicate using a CCD camera attached to the microscope in 6 central fields for each sample. A representative field was selected to create the image. The presence of cells within the scratched area indicates whether the cell migration process has taken place.
References
- Juárez, P.; Comas, I.; González-Candelas, F.; Calvete, J.J. Evolution of snake venom disintegrins by positive Darwinian selection. Mol Biol Evol 2008, 25, 2391-2407. [CrossRef]
- Clissa, P.B.; Della-Casa, M.S.; Zychar, B.C.; Sanabani, S.S. The Role of Snake Venom Disintegrins in Angiogenesis. Toxins (Basel) 2024, 16. [CrossRef]
- Lima-Dos-Santos, I.; Della-Casa, M.S.; Portes-Junior, J.A.; Calabria, P.A.; Magalhães, G.S.; Moura-da-Silva, A.M. Characterization of Neuwiedin, a new disintegrin from Bothrops neuwiedi venom gland with distinct cysteine pattern. Toxicon 2015, 104, 57-64. [CrossRef]
- Usami, Y.; Fujimura, Y.; Miura, S.; Shima, H.; Yoshida, E.; Yoshioka, A.; Hirano, K.; Suzuki, M.; Titani, K. A 28 kDa-protein with disintegrin-like structure (jararhagin-C) purified from Bothrops jararaca venom inhibits collagen- and ADP-induced platelet aggregation. Biochem Biophys Res Commun 1994, 201, 331-339. [CrossRef]
- Paine, M.J.; Desmond, H.P.; Theakston, R.D.; Crampton, J.M. Purification, cloning, and molecular characterization of a high molecular weight hemorrhagic metalloprotease, jararhagin, from Bothrops jararaca venom. Insights into the disintegrin gene family. J Biol Chem 1992, 267, 22869-22876. [CrossRef]
- Moura-da-Silva, A.M.; Marcinkiewicz, C.; Marcinkiewicz, M.; Niewiarowski, S. Selective recognition of alpha2beta1 integrin by jararhagin, a Metalloproteinase/disintegrin from bBothrops jararaca venom. Thrombosis research 2001, 102, 153-159. [CrossRef]
- Munger, J.S.; Sheppard, D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol 2011, 3, a005017. [CrossRef]
- Hynes, R.O. Integrins: bidirectional, allosteric signaling machines. Cell 2002, 110, 673-687. [CrossRef]
- Clissa, P.B.; Lopes-Ferreira, M.; Della-Casa, M.S.; Farsky, S.H.; Moura-da-Silva, A.M. Importance of jararhagin disintegrin-like and cysteine-rich domains in the early events of local inflammatory response. Toxicon 2006, 47, 591-596. [CrossRef]
- Zychar, B.C.; Clissa, P.B.; Carvalho, E.; Baldo, C.; Gonçalves, L.R.C. Leukocyte recruitment induced by snake venom metalloproteinases: Role of the catalytic domain. Biochem Biophys Res Commun 2020, 521, 402-407. [CrossRef]
- Zychar, B.C.; Clissa, P.B.; Carvalho, E.; Alves, A.S.; Baldo, C.; Faquim-Mauro, E.L.; Gonçalves, L.R.C. Modulation of Adhesion Molecules Expression by Different Metalloproteases Isolated from. Toxins (Basel) 2021, 13. [CrossRef]
- Ferreira, B.A.; Deconte, S.R.; de Moura, F.B.R.; Tomiosso, T.C.; Clissa, P.B.; Andrade, S.P.; Araújo, F.A. Inflammation, angiogenesis and fibrogenesis are differentially modulated by distinct domains of the snake venom metalloproteinase jararhagin. Int J Biol Macromol 2018, 119, 1179-1187. [CrossRef]
- Ferreira, B.A.; De Moura, F.B.R.; Tomiosso, T.C.; Corrêa, N.C.R.; Goulart, L.R.; Barcelos, L.S.; Clissa, P.B.; Araújo, F.A. Jararhagin-C, a disintegrin-like protein, improves wound healing in mice through stimulation of M2-like macrophage, angiogenesis and collagen deposition. Int Immunopharmacol 2021, 101, 108224. [CrossRef]
- Hartley, D.L.; Kane, J.F. Properties of inclusion bodies from recombinant Escherichia coli. Biochem Soc Trans 1988, 16, 101-102. [CrossRef]
- Carrió, M.M.; Villaverde, A. Construction and deconstruction of bacterial inclusion bodies. J Biotechnol 2002, 96, 3-12. [CrossRef]
- Zhang, H.; Huang, P.F.; Meng, E.; Li, W.Y.; Zhou, L.; Zhu, L.Y.; Wu, L.; Li, M.J.; Liang, S.P.; Zhang, D.Y. An efficient strategy for heterologous expression and purification of active peptide hainantoxin-IV. PLoS One 2015, 10, e0117099. [CrossRef]
- Jiang, X.; Xu, J.; Yang, Q. Soluble expression, purification, and characterization of Gloydius shedaoensis venom gloshedobin in Escherichia coli by using fusion partners. Appl Microbiol Biotechnol 2010, 85, 635-642. [CrossRef]
- Yuan, S.; Duan, H.; Liu, C.; Liu, X.; Liu, T.; Tao, H.; Zhang, Z. The role of thioredoxin and disulfide isomerase in the expression of the snake venom thrombin-like enzyme calobin in Escherichia coli BL21 (DE3). Protein Expr Purif 2004, 38, 51-60. [CrossRef]
- Vu, T.T.; Jeong, B.; Yu, J.; Koo, B.K.; Jo, S.H.; Robinson, R.C.; Choe, H. Soluble prokaryotic expression and purification of crotamine using an N-terminal maltose-binding protein tag. Toxicon 2014, 92, 157-165. [CrossRef]
- Hernandez-Cuebas, L.M.; White, M.M. Expression of a biologically-active conotoxin PrIIIE in Escherichia coli. Protein Expr Purif 2012, 82, 6-10. [CrossRef]
- Müller, S.; Hoege, C.; Pyrowolakis, G.; Jentsch, S. SUMO, ubiquitin’s mysterious cousin. Nat Rev Mol Cell Biol 2001, 2, 202-210. [CrossRef]
- Marblestone, J.G.; Edavettal, S.C.; Lim, Y.; Lim, P.; Zuo, X.; Butt, T.R. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci 2006, 15, 182-189. [CrossRef]
- Peroutka Iii, R.J.; Orcutt, S.J.; Strickler, J.E.; Butt, T.R. SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes. Methods Mol Biol 2011, 705, 15-30. [CrossRef]
- Shimokawa-Falcão, L.H.; Caporrino, M.C.; Barbaro, K.C.; Della-Casa, M.S.; Magalhães, G.S. Toxin Fused with SUMO Tag: A New Expression Vector Strategy to Obtain Recombinant Venom Toxins with Easy Tag Removal inside the Bacteria. Toxins (Basel) 2017, 9. [CrossRef]
- Calabria, P.A.L.; Shimokava-Falcao, L.H.A.L.; Colombini, M.; Moura-da-Silva, A.M.; Barbaro, K.C.; Faquim-Mauro, E.L.; Magalhaes, G.S. Design and Production of a Recombinant Hybrid Toxin to Raise Protective Antibodies Against. Toxins (Basel) 2019, 11. [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680-685. [CrossRef]
- Sato, Y.; Rifkin, D.B. Autocrine activities of basic fibroblast growth factor: regulation of endothelial cell movement, plasminogen activator synthesis, and DNA synthesis. J Cell Biol 1988, 107, 1199-1205. [CrossRef]
- Zhong, Z.F.; Hoi, P.M.; Wu, G.S.; Xu, Z.T.; Tan, W.; Chen, X.P.; Cui, L.; Wu, T.; Wang, Y.T. Anti-angiogenic effect of furanodiene on HUVECs in vitro and on zebrafish in vivo. J Ethnopharmacol 2012, 141, 721-727. [CrossRef]
- David, V.; Succar, B.B.; de Moraes, J.A.; Saldanha-Gama, R.F.G.; Barja-Fidalgo, C.; Zingali, R.B. Recombinant and Chimeric Disintegrins in Preclinical Research. Toxins (Basel) 2018, 10. [CrossRef]
- Moura-da-Silva, A.M.; Línica, A.; Della-Casa, M.S.; Kamiguti, A.S.; Ho, P.L.; Crampton, J.M.; Theakston, R.D. Jararhagin ECD-containing disintegrin domain: expression in escherichia coli and inhibition of the platelet-collagen interaction. Arch Biochem Biophys 1999, 369, 295-301. [CrossRef]
- Cominetti, M.R.; Terruggi, C.H.; Ramos, O.H.; Fox, J.W.; Mariano-Oliveira, A.; De Freitas, M.S.; Figueiredo, C.C.; Morandi, V.; Selistre-de-Araujo, H.S. Alternagin-C, a disintegrin-like protein, induces vascular endothelial cell growth factor (VEGF) expression and endothelial cell proliferation in vitro. J Biol Chem 2004, 279, 18247-18255. [CrossRef]
- Selistre-de-Araujo, H.S.; Cominetti, M.R.; Terruggi, C.H.; Mariano-Oliveira, A.; De Freitas, M.S.; Crepin, M.; Figueiredo, C.C.; Morandi, V. Alternagin-C, a disintegrin-like protein from the venom of Bothrops alternatus, modulates alpha2beta1 integrin-mediated cell adhesion, migration and proliferation. Braz J Med Biol Res 2005, 38, 1505-1511. [CrossRef]
Figure 1.
A) 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel showing expression and purification of the recombinant JarC overexpressed in E. coli BL21 Star™ (DE3) at 37 °C. Protein was visualized on a 12.5% SDS/polyacrylamide gel under reducing conditions and stained with Coomassie blue. M: Molecular mass markers in kDa (Thermo Scientific); 1 and 2: Extract from BL21 Star™ (DE3) before and after isopropyl-β-D-thiogalactoside (IPTG) (1 mM) induction, respectively; 3: Recombinant JarC purified by IMAC. B) Analysis of rJarC expression by Western blot. Proteins were separated by 12% SDS-PAGE, transferred onto nitrocellulose membrane and incubated with anti-His antibody. Numbers on the left correspond to the position of molecular mass markers (M). 1 and 2: Extract from BL21 Star™ (DE3) before and after isopropyl-β-D-thiogalactoside (IPTG) (1 mM) induction, respectively; 3: Recombinant JarC purified by IMAC.
Figure 1.
A) 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel showing expression and purification of the recombinant JarC overexpressed in E. coli BL21 Star™ (DE3) at 37 °C. Protein was visualized on a 12.5% SDS/polyacrylamide gel under reducing conditions and stained with Coomassie blue. M: Molecular mass markers in kDa (Thermo Scientific); 1 and 2: Extract from BL21 Star™ (DE3) before and after isopropyl-β-D-thiogalactoside (IPTG) (1 mM) induction, respectively; 3: Recombinant JarC purified by IMAC. B) Analysis of rJarC expression by Western blot. Proteins were separated by 12% SDS-PAGE, transferred onto nitrocellulose membrane and incubated with anti-His antibody. Numbers on the left correspond to the position of molecular mass markers (M). 1 and 2: Extract from BL21 Star™ (DE3) before and after isopropyl-β-D-thiogalactoside (IPTG) (1 mM) induction, respectively; 3: Recombinant JarC purified by IMAC.
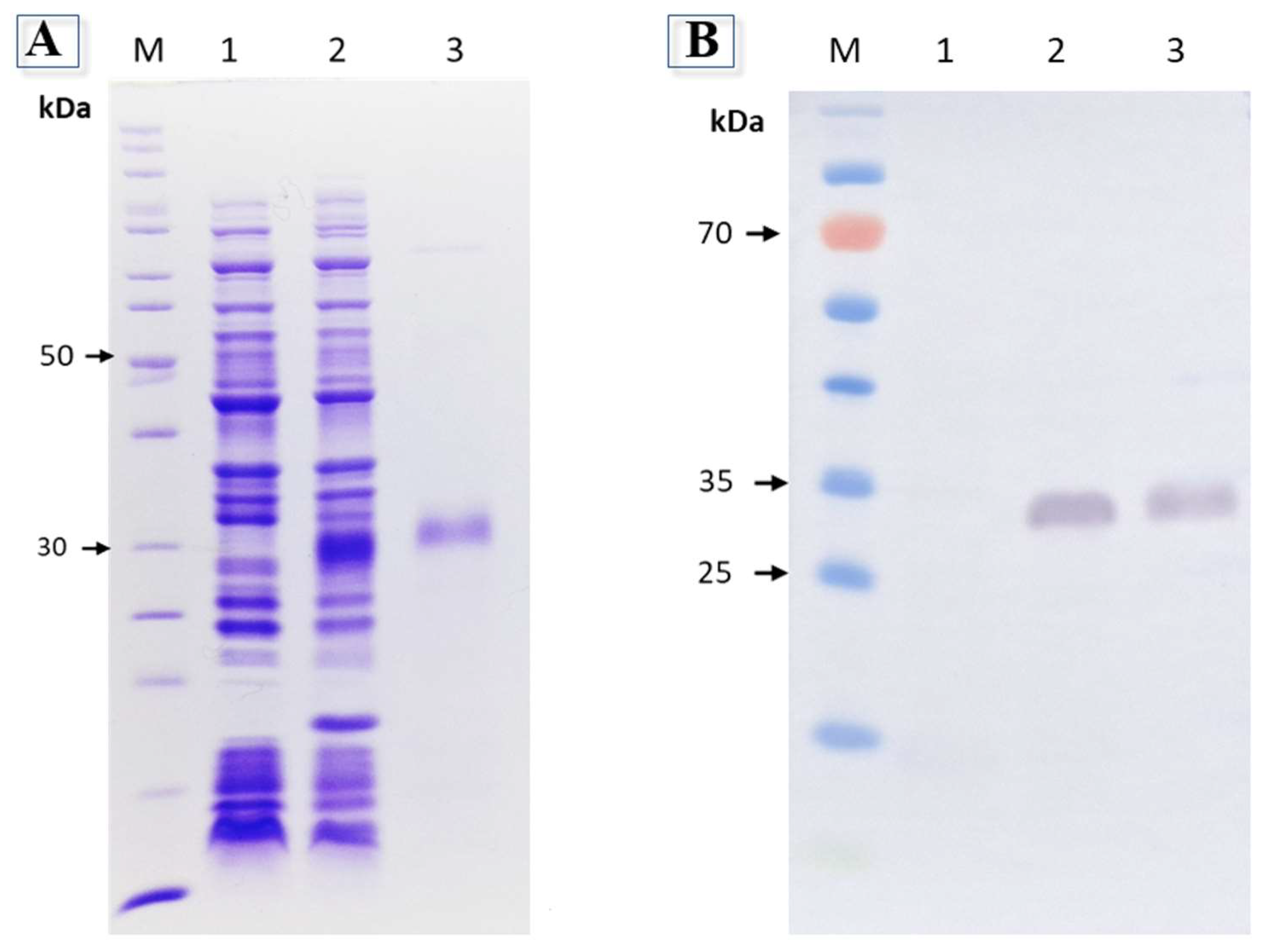
Figure 2.
Coverage map representing the proteomic identification of Jar-C (P30431), according to Peaks Studio analyses. Blue bars represent the proteomically matched peptides, over the deposited sequence from UniProt database.
Figure 2.
Coverage map representing the proteomic identification of Jar-C (P30431), according to Peaks Studio analyses. Blue bars represent the proteomically matched peptides, over the deposited sequence from UniProt database.
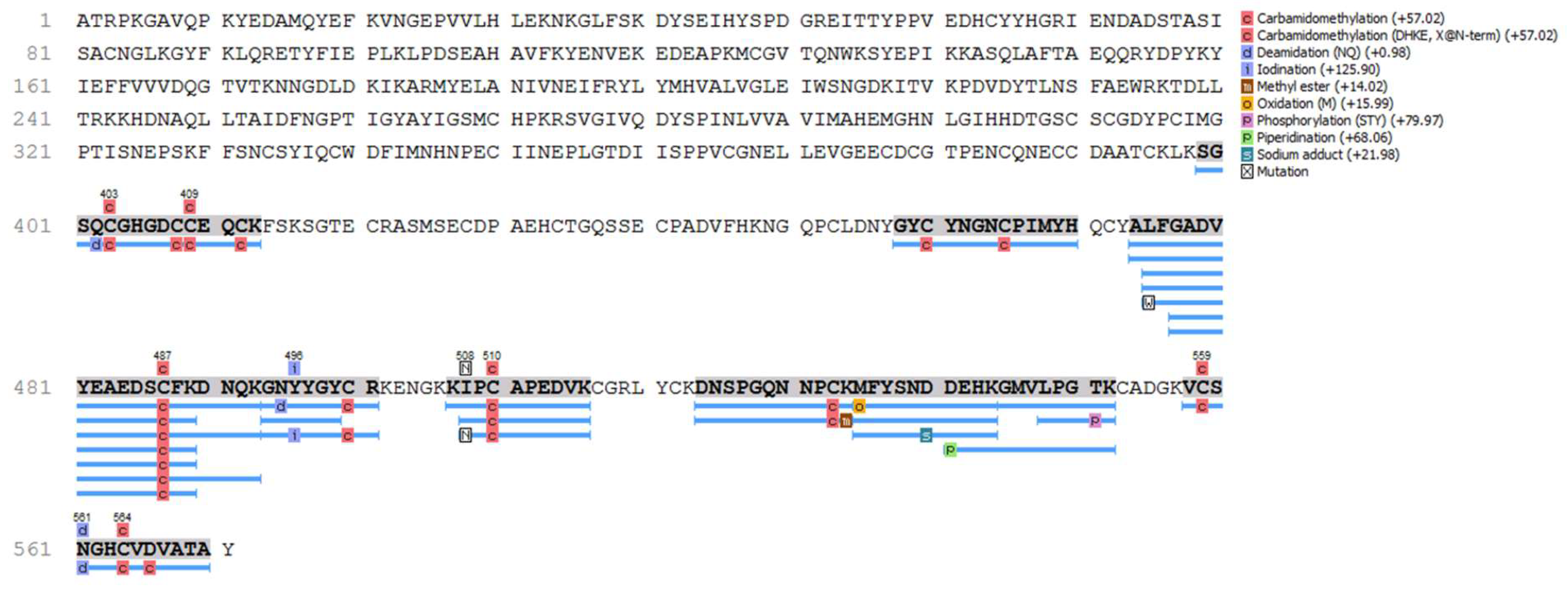
Figure 3.
Dot blotting of native (JarC and Jararhagin) and recombinant proteins (rJarC) recognized by a polyclonal anti-jararhagin or anti-histidine antibody under reduced (10µM Dithiothreitol - DTT) and non-reduced conditions.
Figure 3.
Dot blotting of native (JarC and Jararhagin) and recombinant proteins (rJarC) recognized by a polyclonal anti-jararhagin or anti-histidine antibody under reduced (10µM Dithiothreitol - DTT) and non-reduced conditions.

Figure 4.
Viability of HUVECs treated with Jar and rJarC. Cell viability was determined using the MTT assay. HUVECs were not treated (RPMI) or treated with JarC; rJarC; rGFP at concentrations of 1µM or Triton X 100 1% for 24h.
Figure 4.
Viability of HUVECs treated with Jar and rJarC. Cell viability was determined using the MTT assay. HUVECs were not treated (RPMI) or treated with JarC; rJarC; rGFP at concentrations of 1µM or Triton X 100 1% for 24h.
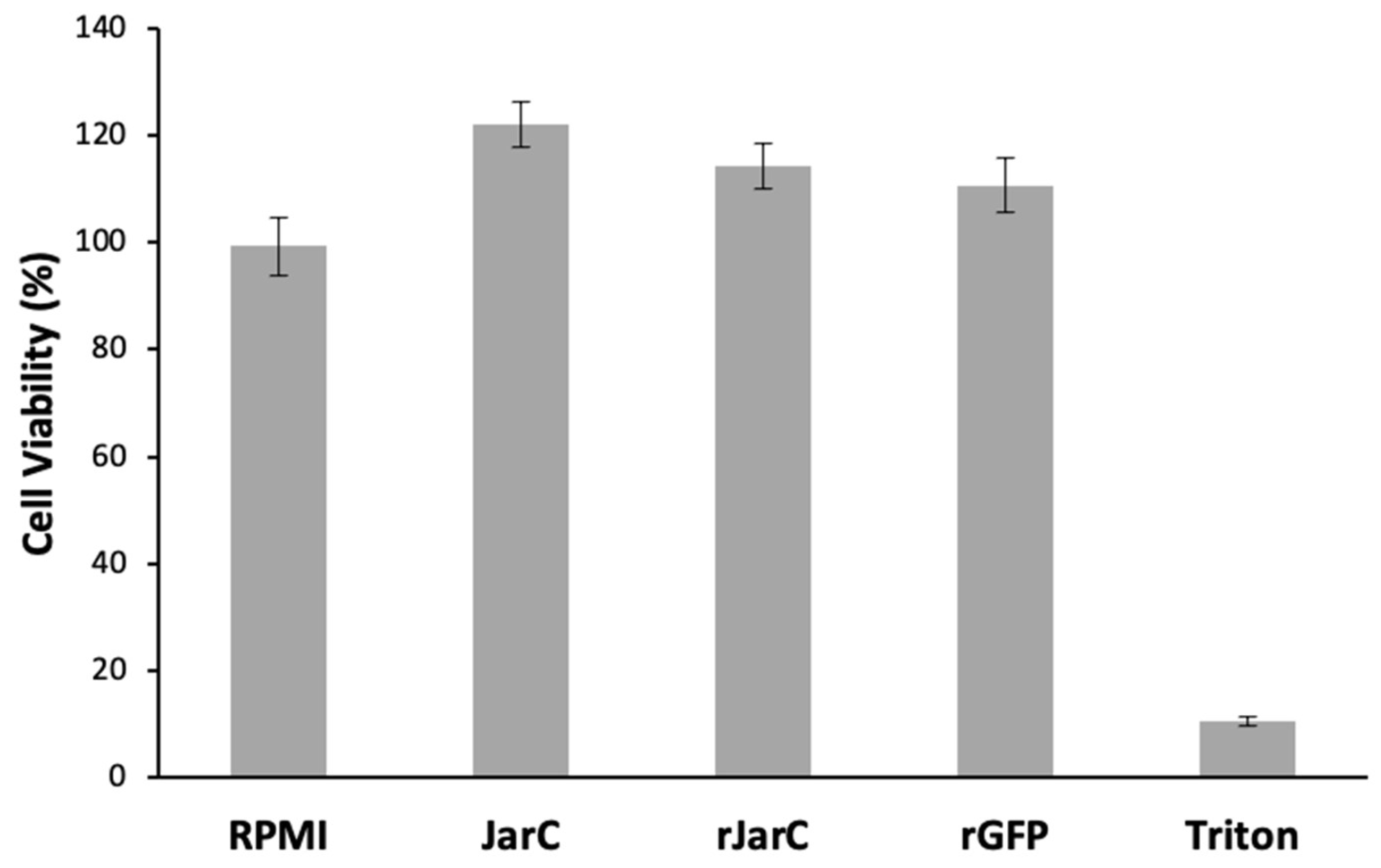
Figure 5.
Cell migration induced by JarC and rJarC in vitro (wound healing assay): HUVEC cells CRL1730 were cultured in a 24-well plate until they reached approximately 100% confluence, and then a line was drawn down the center of the wells with a tip. The cells in culture were treated with native JarC, rJarC, or rGFP at concentrations of 0.01 µM, 0.1 µM, and 1 µM or with culture medium as a negative control. After 24 hours, the culture medium was removed and the cells were fixed and stained with the Protocol Hema 3 staining kit. The plates were examined under the microscope and images were captured using a CCD camera coupled to the inverted microscope.
Figure 5.
Cell migration induced by JarC and rJarC in vitro (wound healing assay): HUVEC cells CRL1730 were cultured in a 24-well plate until they reached approximately 100% confluence, and then a line was drawn down the center of the wells with a tip. The cells in culture were treated with native JarC, rJarC, or rGFP at concentrations of 0.01 µM, 0.1 µM, and 1 µM or with culture medium as a negative control. After 24 hours, the culture medium was removed and the cells were fixed and stained with the Protocol Hema 3 staining kit. The plates were examined under the microscope and images were captured using a CCD camera coupled to the inverted microscope.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



