
Submitted:
03 October 2024
Posted:
04 October 2024
You are already at the latest version
Abstract
Fragile X Syndrome (FX) is the most common form of inherited cognitive impairment and falls under the broader category of Autism Spectrum Disorders (ASD). FX is caused by a CGG trinucleotide repeat expansion in the non-coding region of the X-linked Fragile X Messenger Ribonucleoprotein 1 (FMR1) gene, leading to its hypermethylation and epigenetic silencing. Animal models of FX rely on the deletion of the Fmr1 gene which fail to replicate the epigenetic silencing mechanism of the FMR1 gene seen in human patients. Human stem cells carrying FX repeat expansions have provided a better understanding of the basis of the epigenetic silencing of FMR1. Previous studies have found that 5-Azacytidine (5Azac) can reverse this methylation; however, 5Azac can be toxic and may limit its therapeutic potential. Here, we show that the dietary factor Ascorbic Acid (AsA) can reduce DNA methylation in the FMR1 locus and lead to an increase in FMR1 gene expression in FX iPSCs and cerebral organoids. In addition, AsA treatment rescued neuronal gene expression and the morphological defects observed in FX iPSC-derived cerebral organoids. Hence, we demonstrate that the dietary co-factor AsA can partially revert molecular and morphological defects seen in human FX models in vitro. Our findings have implications for the development of novel therapies for FX in the future.
Keywords:
FMR1
; Fragile X Syndrome
; Methylation
; Ascorbic Acid
; Gene Silencing
; Autism Spectrum Disorders (ASD)
; Induced Pluripotent Stem Cells
; Cerebral Organoids
; Neurodevelopmental Disorders
1. Introduction
Fragile X Syndrome (FX) is the most common form of genetically inherited cognitive impairment and mental retardation with a prevalence of 1 in 3600 in the population (varying by geographic region) and is one of the major forms of Autism Spectrum Disorders (ASD) [1]. FX patients display a constellation of symptoms and phenotypes including severe intellectual disability, attention deficits, problems with speech and social interaction, aggression, hyperactivity, and a high susceptibility to seizures [2,3]. FX has a massive impact on the lives of the patients and their families.
Unlike most forms of ASD, FX has a well established monogenic cause. A CGG trinucleotide repeat expansion in the 5’ untranslated region (UTR) of the Fragile X Messenger Ribonucleoprotein 1 (FMR1) gene leads to its epigenetic silencing and consequently results in FX [4,5]. However, FMR1 gene expression shows a CGG repeat expansion dosage effect. The normal FMR1 gene contains 5-50 CGG repeats. An expansion to 55-200 repeats is referred to as the pre-mutation state resulting in a reduction in Fragile X Mental Retardation Protein (FMRP) levels and mild cognitive defects but not FX [6,7,8]. Expansion beyond 200 CGG repeats induces DNA hypermethylation and epigenetic changes in the FMR1 promoter, silencing the gene and causing FX (Figure 1A) [4,5,9]. The DNA hypermethylation spreads to the entire locus causing chromatin condensation and making the FMR1 gene and the locus inaccessible to the transcriptional machinery [9]. This genetic causation in FX presents advantages for identifying genes involved in autism-like disorders and devising therapeutic approaches. Since FX is caused by epigenetic silencing of FMR1 and not a mutation in the coding region, a therapeutic strategy could be to reverse the hypermethylation of the gene locus and thereby reactivating the silenced gene. Mouse models of FX have partial or complete deletions of the Fmr1 gene to prevent the expression of a functional FMRP [10,11,12,13]. Hence, these models are not suitable for studying the mechanism controlling hypermethylation or epigenetic silencing of the FMR1 gene in humans. Human embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) that carry >200 CGG repeat expansions are powerful models to study FX [14,15]. This is supported by the finding that FX ESCs and iPSCs-derived neurons have epigenetic silencing of FMR1 [14,15]. Screens performed on FX iPSCs and their neuronal derivatives have identified several candidate compounds that can reactivate FMR1 gene expression [16,17,18]. The cytosine analog and methyltransferase inhibitor 5-AzaC increased FMR1 expression from 0% to 15-45% of control levels in FX cells [16]. Genetic approaches, such as CRISPR-editing to excise the expanded CGG repeat region in the FX FMR1 gene [19] and dCas9 to guide demethylases to the locus have also demonstrated success in reactivating FMR1 expression [20]. These studies highlight that demethylation of the hypermethylated FMR1 gene in FX cells can restore its activity [16,17,18,19,20].
In previous studies, Ascorbic Acid (AsA) has been shown to affect DNA methylation and chromatin conformation [21,22,23,24,25,26,27]. We hypothesized that AsA may be able to demethylate the FMR1 locus in FX cells. In this study, we treated FX iPSCs and cerebral organoids with AsA and addressed the effects on FMR1 methylation status, mRNA expression and organoid morphology. We found that AsA reduces FMR1 methylation and increases FMR1 expression in FX iPSCs and has similar a similar effect in FX cerebral organoids. Furthermore, AsA treatment upregulated genes previously identified to be downregulated in FX, and partially rescued the morphological development of FX cerebral organoids.
2. Results
2.1. Ascorbic Acid Restores FMR1 Expression by Reversing Hypermethylation in FX iPSCs
The FMR1 gene contains 5-50 CGG repeats in the 5’UTR of the gene and an upstream CpG island in the gene promoter (Figure 1A). Expansion of CGG triplicate repeats to >200 in the FMR1 gene leads to hypermethylation of the CGG repeats which spreads to the upstream CpG island and the entire locus resulting in transcriptional silencing and loss of FMRP protein [4,5,9] (Figure 1A). To investigate whether Ascorbic Acid (AsA) can reverse the hypermethylation and transcriptional silencing of the FMR1 gene in FX iPSCs, we treated FX iPSCs with different concentrations of AsA and quantified FMR1 mRNA levels by quantitative RT-PCR.
AsA was administered to FX iPSCs daily for 6 days. To prevent acidification of the medium by AsA, HEPES was added to the culture medium, serving as a vehicle-control. AsA induced FMR1 transcription in a dose-dependent manner with the highest levels of FMR1 mRNA expression observed at 500µM AsA (Figure 1B). We speculated that prolonged treatment of iPSCs with AsA could further increase the activation of the FMR1 gene in FX iPSCs. To test this, we plated FX and wild-type iPSCs in the presence or absence of 500μM AsA and cultured them for 6 days until confluency. We expanded the cells for 6 passages in the presence or absence of AsA and isolated genomic DNA for pyrosequencing to analyze methylation status and RNA to determine FMR1 gene expression by quantitative RT-PCR (Figure 1C). Long-term AsA treatment over 6 passages resulted in a statistically significant increase in FMR1 transcript levels compared to FX iPSCs treated with the vehicle-control (Figure 1D).
We next examined whether the long-term exposure of FX iPSCs to AsA could lead to demethylation of the FMR1 gene locus. Due to the repetitive nature and length of the CGG repeats in FMR1, the CpG island upstream of the CGG repeats was used in pyrosequencing as a proxy to assess the methylation status of the FMR1 gene (Figure 1E) [20]. The CpG island in wild-type iPSCs is unmethylated but hypermethylated in FX iPSCs (Figure 1F). AsA treatment of FX significantly reduced DNA methylation of the CpG island in FX iPSCs suggesting that AsA has the potential to restore FMR1 gene expression in FX iPSCs by reducing DNA methylation (Figure1F).
2.2. Ascorbic Acid Reactivates FMR1 and Reduces Methylation in FX Cerebral Organoids
Given that FX is a neurodevelopmental disorder, we moved to a relevant human model and assessed the effects of AsA treatment on FMR1 in cerebral organoids. Cerebral organoids have been shown to be an effective system to model human neurodevelopmental disorders [28,29,30]. We differentiated FX iPSCs that had been chronically treated with ±AsA for 6 passages into cerebral organoids. The cerebral organoids were exposed to ±500μM AsA throughout their differentiation and collected on day 50 for analysis (Figure 2A).
We assessed the formation and morphology of the cerebral organoids derived from wild-type, FX vehicle-treated and AsA-treated FX iPSCs at day 50 of differentiation (Figure 2A). We classified the organoids into 2 categories: (category 1) cerebral organoids with smooth contours and distinct visible neural structures; or (category 2) cerebral organoids that are disorganized, mostly cystic and lacking visible neural structures (Supplementary Figure 1A). These criteria were based on previously established phenotypic scoring using bright field microscopy [28,29,30,31,32,33,34].
The majority of cerebral organoids derived from wild-type iPSCs (83.6 ± 6.6%) showed organized structures with smooth, well-defined borders (Figure 2B), and were classified as category 1 (Figure 2C). In contrast, only a small fraction of FX vehicle-treated cerebral organoids (7.3 ± 0.6%) were classified as category 1, with most appearing highly disorganized and cystic (Figure 2B,C). Notably, AsA-treated FX iPSC-derived cerebral organoids showed a marked improvement in phenotype with defined morphology (38.3 ± 5.7%) (Figure 2B) had defined organization and smooth boundaries and were classified as category 1 (Figure 2C). However, the phenotypic rescue of FX cerebral organoids by AsA-treatment was highly variable.
In order to assess the effects of AsA treatment on the FMR1 gene expression, we quantified FMR1 mRNA levels in 50-day-old cerebral organoids. An initial comparison of FMR1 transcript levels between FX vehicle and AsA treated cerebral organoids indicated a trend towards increased FMR1 expression in response to AsA treatment (Figure 2D). However, the effects of AsA treatment were variable across individual organoids. Therefore, we used a threshold of the 95% confidence interval (CI) of the median of FMR1 expression in vehicle-treated FX cerebral organoids to define a threshold and identify AsA-treated cerebral organoids with increased FMR1 expression. 38% of AsA-treated FX cerebral organoids exceeded the threshold of the 95% CI (Figure 2D). These cerebral organoids were considered to be AsA responders. However, we identified two distinct populations of FX cerebral organoids: AsA responders and non-responders.
We next analyzed the methylation status of the CpG island upstream of the FMR1 gene in cerebral organoids derived from AsA-treated FX, vehicle-treated FX, and wild-type iPSC using pyrosequencing. In wild-type cerebral organoids, the CpG island showed a low average methylation level (3.5 ± 1.3%) (Figure 2E). In contrast, the CpG island in FX cerebral organoids is highly methylated (86 ± 1.7%) (Figure 2E, Supplementary Figure 1B).
Wild-type cerebral organoids displayed a homogeneous and low methylation range (1.8 - 4.4%) except for one organoid a higher average methylation of 17.1% (Figure 2E, Supplementary Figure 1B). FX vehicle-treated cerebral organoids exhibited a uniformly high level of methylation in the CpG island (73.2 - 94.1%, average 86 ± 1.7%, Figure 2E, Supplementary Figure 1B).
AsA-treated cerebral organoids displayed a broader range of FMR1 CpG island methylation (29.0 - 94.6%) with an average trend towards a decreased methylation (Figure 2E, Supplementary Figure 1C). As AsA treatment of FX cerebral organoids resulted in heterogeneity within the population, we again used the 95% CI of the vehicle-treated FX cerebral organoids to set a threshold and separate putative AsA responders from non-responders based on methylation status. 45% of AsA-treated FX cerebral organoids were classified as responders, with methylation levels similar to those of wild-type cerebral organoids (Figure 2E, Supplementary Figure 1C). Four AsA-treated cerebral organoids had CpG island methylation levels very similar to that of wild-type organoids (29.0, 31.5, 53.6 and 60.6% residue methylation over the entire island). In conclusion, combining the FMR1 expression and methylation data confirms that cerebral organoids derived from FX iPSCs respond differentially to AsA treatment resulting in two populations: responders and non-responders.
2.3. Ascorbic Acid Treatment Modulates Gene Expression in FX Cerebral Organoids Related to Neurodevelopment
In order to characterize the changes induced by AsA treatment in FX cerebral organoids, we performed RNASeq on individual AsA responders identified by their higher FMR1 expression levels. We compared the gene expression profiles of these AsA-treated FX cerebral organoids to that of vehicle-treated FX and wild-type cerebral organoids.
Principal component analysis (PCA) of the variation across the three groups revealed a clear genotype-based separation (Figure 3A). Wild-type cerebral organoids clustered distinctly from both the FX vehicle- and AsA-treated cerebral organoids in the 1st principal component (PC1) indicating that FMR1 silencing in FX cerebral organoids affected their gene expression profiles. AsA-treated FX cerebral organoids showed more variability, with looser clustering both PC1 and PC2, reflecting individual differences in their response to AsA treatment (Figure 3A). Notably, one AsA-treated cerebral organoid clustered closer to the wild-type organoids in PC1 suggesting that this organoid had responded strongest to AsA and thus had a more robust correction of its transcriptome.
An initial comparison of the cerebral organoid transcriptomes showed FMR1 to be the most significantly dysregulated gene between FX vehicle-treated and wild-type cerebral organoids thus supporting the approach (Figure 3B). Most downregulated genes in FX cerebral organoids were involved in neuronal lineage specification and differentiation including EMX1, NEUROD6, EPHA5, NEUROD2, and FOXG1 (Figure 3B). Enrichment analysis showed that these dysregulated gene were related to neuronal anatomy, function and development (Supplementary Figure 2A and 2B). Most pronounced was the downregulation of genes involved in synaptic signaling, in cognition and forebrain development (Figure 3C), with 30 of the downregulated genes in FX cerebral organoids are involved in synaptic signaling (Supplementary Figure 2A). Gene Set Enrichment Analysis (GSEA) further identified that the most dysregulated genes were related to neuronal development, anatomy and function (Supplementary Figure 2B).
AsA treatment significantly upregulated FMR1 by 12.6 ± 0.2-fold compared to vehicle-treated FX cerebral organoids (Figure 3D and 3E). When analyzing the downregulated genes in the FX cerebral organoids post-AsA treatment, we observed increased expression of genes involved in neuronal development and synaptic signaling (Figure 3F). Importantly, AsA treatment enhanced the expression of many of the gene involved in neuronal development and synaptic signaling including GABRA1, NNAT, GABRG2, CRCH2 and SLC24A2 (Figure 3F). STRING analysis of AsA-induced genes identified gene networks related to ionotropic glutamate receptor complex, GABA receptor activation, and positive regulators of synaptic plasticity (Figure 3G). This supports the notion that AsA can not only promote FMR1 re-expression, but also result in rescue of downstream neuronal aberrations associated with FX.
3. Discussion
FX is the most common inherited form of cognitive impairment and mental retardation [1]. Thus, there is a need to find novel therapeutic strategies to treat the epigenetic silencing of the FMR1 gene which is the root cause of the condition [4,5,9]. The majority of approaches used to model FX and devise therapeutic strategies have centered around animal models [2,35,36]. However, as animals fail to recapitulate the epigenetic silencing of the FMR1 gene observed in humans due to the CGG trinucleotide repeat expansion, therapeutic strategies based on gene knockout models have not been highly effective in the clinic [37]. Therefore, human models using FX iPSCs and their derivatives are promising as they recapitulate the epigenetic silencing of FMR1 in FX.
Compound screens in both FX iPSCs and early born neurons showed that DNMT inhibitor 5AzaC can block methylation of the FMR1 gene [16,17,18]. Furthermore, deletion of the expanded CGG repeats in FMR1 can lead to demethylation of the gene [19]. Furthermore, it was shown that de novo R-loops can form in human FX iPSCs and DNA replication and integrity check systems can excise the CGG repeats resulting in re-expression of FMR1 [38]. These studies exemplified the reversible nature of FMR1 silencing and the importance of the CGG repeats in the process. This is supported by the finding that targeting demethylases toward the FMR1 locus in FX iPSCs using a dCAS9 can selectively demethylate the locus and reinstate FMR1 expression [20].
These studies provide a foundation for possible approaches to treat FX in humans. However, 5AzaC can be cytotoxic, and may have consequences for developing FX children. In addition, genetic approach of editing- out the CGG repeats, for example using CRISPR technology, or capitalizing of R-loops for de novo editing and using guided demethylases to target the methylated FMR1 locus are technically challenging to target all cells of the brain. Hence, a pharmacological approach could help to circumvent these problems.
Our human FX patient-derived iPSCs and cerebral organoid models recapitulated the epigenetic silencing of FMR1. We show that both FX iPSCs and cerebral organoids responded to AsA treatment with reduced methylation of the FMR1 locus and increased levels of FMR1 transcripts. In addition, AsA treatment results in a degree of rescue of organoid morphology in the FX organoids and an increase in defective neuronal gene expression. Whereas the demethylation of the CpG island upstream of the FMR1 gene in FX iPSCs as a population was uniform and homogenous in response to AsA treatment, the responses of individual cerebral organoids were variable. We observed that 38-45% of organoids analyzed showed a positive response in organoid morphology, CpG island demethylation, and induction of FMR1 gene expression.
These findings show that AsA treatment has a pronounced effect on FX neural tissue, however, only a fraction of the cells seem to be responders in these assays. Why FX organoids show heterogeneity in response to AsA remains to be determined. One reason could be that cerebral organoids are not uniform and some are much larger than others. Technically, organoid size and the ensuing differences in efficacy of AsA penetration could play a role in the response of cells within the organoids. Additionally, cerebral organoids are heterogeneous in their differentiation stage and cellular composition which could also lead to differences in their biological response. In contrast, iPSCs cultures grow as homogeneous monolayers, and therefore likely have fewer problems with AsA penetration resulting in them responding in a more homogeneous fashion. In the future, single-cell analyses at the genomic, RNA, and protein levels may be able to address differences in cell-type specific responses to AsA.
Our findings demonstrate that AsA treatment can counteract to some degree the FX-induced neurodevelopment gene expression profiles and particularly neuronal differentiation and synapse formation. It is conceivable that a reduction in the expression of these genes in FX contribute to the developmental and cognitive abnormalities observed in FX patients. It is also plausible that AsA modification of FMR1 methylation and expression could have a positive effect on neurodevelopment in FX patients. However, we cannot exclude that the changes induced by AsA are not restricted to activation of the FMR1 gene and may have more global effects of gene expression. In the future, genome wide restricted bisulfite sequencing should be performed to asses changes in methylation status on other loci.
Two previous studies have used FX iPSCs and differentiated them into cerebral organoids to study and characterize the fundamental processes of human FX. One study generated FMR1 knockout iPSCs by truncating the FMR1 gene by CRISPR/Cas9 gene editing. Although useful to examine the role of FMRP in human neural cells in vitro, this model system does not utilize epigenetic silencing that causes FX [39]. Another study found that molecular defects in FX cerebral organoids could be rescued by inhibiting the phosphoinositide 3-kinase pathway [40]. How the phosphoinositide 3-kinase pathway is potentially involved in FX remain unclear, and whether the methylation status of FMR1 was altered was not addressed [40]. Our study, in summary, showed for the first time that AsA can target the root cause of FX, the epigenetic silencing of FMR1 in both FX iPSCs and cerebral organoids. To our knowledge, our study is the first time that a human cerebral organoid model has been used to demonstrate a reversal of the epigenetic silencing of the endogenous FMR1 gene without genetic intervention.
4. Materials and Methods
4.1. iPSC Cell Culture
FX52 (FX) and IPSO (wild-type) cell lines were used in the experiments [15]. FX52 and IPSO were cultured and maintained as described in previous studies [15]. Plating of cells for testing Ascorbic Acid concentration was done by plating 100,000 cells per well of 6-well plate. Cultures were maintained for 6-days until being collected for harvesting DNA and RNA. FX52 and IPSO iPSCs were cultured using mTeSR1 media (Stem Cell Technologies, mTeSR1 Basal Medium, Cat#05850 and mTeSR1 5x supplement, Cat#05851) on Matrigel (BD Bioscience, hESC qualified Matrigel, Cat#354277) at 37°C, 5% CO 2 with daily media change. Cells were passaged enzymatically every 6 days using StemPro Accutase (Gibco, Life Technology, Cat#A1110501). Cells were grown in mTeSR1 medium in presence 10μM Y- 27632 ROCK inhibitor (Miltenyi Biotec, Cat #130-104-169) after passage.
iPSCs vehicle-controls had mTeSR1 supplemented with an additional 15mM HEPES Buffer (Sigma, HEPES 1M, Cat#H0887). Ascorbic Acid (Sigma, L-Ascorbic Acid 100G, Cat#A92902-100G) was solubilized in ddH2O and supplemented at the appropriate concentration to mTeSR1 plus 15mM HEPES supplementation.
4.2. Cerebral Organoid Differentiation
Cerebral organoid differentiation was performed exactly according to Lancaster et al.l [33]. Only deviation was with every media change, organoid culture media was supplemented with 15mM HEPES (vehicle-control) (Sigma, HEPES 1M, Cat#H0887)and 500µM Ascorbic Acid (Sigma, L-Ascorbic Acid 100G, Cat#A92902-100G). Organoids were harvested at day 50 for analysis.
4.3. DNA and RNA Extraction
DNA extraction for cerebral organoids was performed using QIAamp DNA Mini Kit (50) (Qiagen, QIAamp DNA Mini Kit (50), Cat# 51304) following instructions exactly as described in the protocol. DNA was stored at -20°C long term.
RNA extraction for cerebral organoids was performed using Trizol extraction. Organoids were lysed directly in Trizol (Invitrogen, Trizol, Cat #15596018). RNA was isolated according to the manufacturer’s instructions. Cells were directly sorted into 1mL of Trizol. RNA was extracted in the aqueous phase after addition of 200µL chloroform, precipitated with an equal volume of isopropanol and washed once in fresh 80% ethanol. The RNA pellet was re-suspended in 20µL ddH2O at 50C for 10 minutes and then kept on ice. RNA was stored at -20°C long term.
Simultaneous DNA and RNA extraction was performed for iPSCs using the AllPrep DNA/RNA Micro Kit (50) (Qiagen, AllPrep DNA/RNA Micro Kit (50), Cat#80284) and performed exactly according to protocol instructions. DNA and RNA were stored at -20°C long term.
4.4. RT-PCR
RT-PCR to characterize gene expression levels of FMR1 were performed using TaqMan Assay primers (FMR1: Hs00924547_m1, ThermoFisher), and TaqMan Fast Advanced Master Mix for qPCR (ThermoFisher, REF#4444557 ). FMR1 expression levels were normalized to GAPDH (GAPDH: Hs02786624_g1, ThermoFisher).
4.4. Pyrosequencing
Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany),including RNase A treatment, according to the manufacturer’s protocol. Two micrograms of genomic DNA from each sample were bisulfite-treated using the EZ DNA Methylation Kit (Zymo Research, Orange, CA), following the manufacturer’s instructions, and stored at -80°C until further use.
Pyrosequencing was used to quantify DNA methylation levels in the CpG island at the FMR1 transcription start site. Primers were designed to amplify a 190 bp sequence containing 22 CpGs, located 150 bp upstream of the CGG repeat sequence. PCR was performed in two steps. The first PCR used a forward primer (5′-GTTATTGAGTGTATTTTTGTAGAAATG-3′) and a reverse primer with an 11-base tag sequence (5′-GCCCCCGCCCGCCCTCTCTCTTCAAATAACCTAAAAAC-3′). The second PCR used a nested forward primer (5′-GAGTGTATTTTTGTAGAAATGGG-3′) and a universal reverse primer with the same 11-base tag sequence (5′-(biotin)GCCCCCGCCCG-3′), which was biotin-labeled at the 5′ end and HPLC-purified (Microsynth AG, Switzerland). The first PCR was performed in a 25 μL reaction volume using FastStart Taq DNA polymerase (Roche Diagnostics, USA). After an initial denaturation at 95°C for 6 minutes, the cycling conditions consisted of 35 cycles of denaturation at 95°C for 30 seconds, annealing at 57°C for 30 seconds, and elongation at 72°C for 45 seconds. The amplified fragment was diluted 1000-fold and used as a template for the second PCR. The second PCR was carried out in a 50 μL reaction volume, with initial denaturation at 95°C for 5 minutes, followed by 25 cycles of denaturation at 95°C for 30 seconds, annealing at 52°C for 30 seconds, and elongation at 72°C for 45 seconds. A 10 μL aliquot of the PCR product was directly used for pyrosequencing with the PyroMark Q24 Pyrosequencing system, following the manufacturer’s instructions (Qiagen). For each sample, two sequencing runs were performed, using either the nested forward primer as the sequencing primer or an internal sequencing primer S1 (5′-GTTTTTTATTAAGTT-3′), to assess the methylation status of all 22 CpGs. The ratio of cytosine to thymine at each analyzed CpG site, quantified by assessing peak height, was exported using PyroMark Q24 Advanced Application Software Version 3.0.0. The percentage represents the average methylation level of all sequenced PCR products for the respective CpG site analyzed within the assay.
4.5. RNASeq and Bioinformatic Analysis
All the graphs for figures 3 and supplementary Figure 2 were made using RStudio 4.4.1. The data were prepared by collecting the RNAseq analysis, aligning it to human (GRCh38 human; https://www.gencodegenes.org/human/) and mouse (GRCm39 mouse; https://www.gencodegenes.org/ mouse) genomes (STAR) and then to eliminate the mouse aligned reads (XenofilteR), and finally we conducted Genome-wide expression analysis and normalization by TTM. Then the values table were taken and processed to build in the figures. SRA identification will be provided shortly.
4.6. Statistical Analysis
All statistical analysis for Figure 1 and Figure 2 were performed using the statistics functions of GraphPad Prism Version 9.2.0 (283). All tests were performed as one-tailed t-tests comparing FX versus FX Ascorbic Acid treated. Error bars are all plotted as ± Standard Error of the Mean (SEM). For statistical significance in figures, standard convention follows: * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, K. G., V. T. and N. B.; Methodology, K. G., V. T. and N. B.; Validation, K.G., F. N. and A. G.; Formal Analysis, K. G., F. N. and A. G., N. B. and V. T.; Investigation, K. G. F. N. and A. G.; Resources, P. S., N. B. and V. T.; Data Curation, K. G.; Writing—Original Draft Preparation, K. G.; Writing—Review & Editing, K. G., F. N., N. B. and V. T.; Visualization, K. G., F. N. and A. G.; Supervision, K. G., N. B. and V. T.; Project Administration: K. G. and V. T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Basel Research Centre for Child Health by Postdoctoral Excellence Programme Fellowship to K. G. This work was partially supported by the Azrieli Foundation, the Rosetrees Trust, the Israel Science Foundation (2054/22), and the ISF-Israel Precision Medicine Partnership (IPMP) Program (3605/21).
Data Availability Statement
SRA for all RNASeq samples will be shortly deposited and provided with access codes for the reviewers.
Acknowledgements
We thank members of the Taylor lab and Benvenisty lab for reading the manuscript and giving thoughtful feedback. We thank the DBM Microscopy Core Facility for providing excellent equipment and services. We thank specifically Dr. Aikaterini Lampada (Taylor lab) and Dr. Sara R. Riog (DB Microscopy Core Facility) for advice and assistance with imaging organoids. We thank Dr. Akanksha Jain for advice on organoid culturing. We would like dedicate this manuscript in memory of our friend and former lab manager of the Benvenisty lab the late Dr. Ofra Yanuka who helped K. G. to learn how to culture FX iPSCs and start this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Crawford, D. C., J. M. Acuna, and S. L. Sherman. “Fmr1 and the Fragile X Syndrome: Human Genome Epidemiology Review.”. Genet Med 2001, 3, 359–71. [Google Scholar] [CrossRef]
- Bhakar, A. L., G. Dolen, and M. F. Bear. “The Pathophysiology of Fragile X (and What It Teaches Us About Synapses).”. Annu Rev Neurosci 2012, 35, 417–43. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M., S. A. Sebastian, Y. Benitez, S. Desai, J. Quinonez, S. Ruxmohan, J. D. Stein, and W. Cueva. “Behavioral Problems in Fragile X Syndrome: A Review of Clinical Management.”. Cureus 2022, 14, e21840. [Google Scholar]
- Verkerk, A. J., M. Pieretti, J. S. Sutcliffe, Y. H. Fu, D. P. Kuhl, A. Pizzuti, O. Reiner, S. Richards, M. F. Victoria, F. P. Zhang, and et al. “Identification of a Gene (Fmr-1) Containing a Cgg Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome.”. Cell 1991, 65, 905–14. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, M., F. P. Zhang, Y. H. Fu, S. T. Warren, B. A. Oostra, C. T. Caskey, and D. L. Nelson. “Absence of Expression of the Fmr-1 Gene in Fragile X Syndrome.”. Cell 1991, 66, 817–22. [Google Scholar] [CrossRef]
- Hagerman, R. J., E. Berry-Kravis, H. C. Hazlett, D. B. Bailey, Jr., H. Moine, R. F. Kooy, F. Tassone, I. Gantois, N. Sonenberg, J. L. Mandel, and P. J. Hagerman. “Fragile X Syndrome.”. Nat Rev Dis Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Tassone, F., A. Beilina, C. Carosi, S. Albertosi, C. Bagni, L. Li, K. Glover, D. Bentley, and P. J. Hagerman. “Elevated Fmr1 Mrna in Premutation Carriers Is Due to Increased Transcription.”. RNA 2007, 13, 555–62. [Google Scholar] [CrossRef]
- Hagerman, R., and P. Hagerman. “Advances in Clinical and Molecular Understanding of the Fmr1 Premutation and Fragile X-Associated Tremor/Ataxia Syndrome.”. Lancet Neurol 2013, 12, 786–98. [Google Scholar] [CrossRef]
- Sutcliffe, J. S., D. L. Nelson, F. Zhang, M. Pieretti, C. T. Caskey, D. Saxe, and S. T. Warren. “DNA Methylation Represses Fmr-1 Transcription in Fragile X Syndrome.”. Hum Mol Genet 1992, 1, 397–400. [Google Scholar] [CrossRef]
- Willemsen, R. , and R. F. Kooy. “Mouse Models of Fragile X-Related Disorders.”. Dis Model Mech 2023, 16. [Google Scholar] [CrossRef]
- Colvin, S. Lea, Q. Zhang, M. Wienisch, T. Kaiser, T. Aida, and G. Feng. “341 Repeats Is Not Enough for Methylation in a New Fragile X Mouse Model.”. eNeuro 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Kooy, R. F., R. D’Hooge, E. Reyniers, C. E. Bakker, G. Nagels, K. De Boulle, K. Storm, G. Clincke, P. P. De Deyn, B. A. Oostra, and P. J. Willems. “Transgenic Mouse Model for the Fragile X Syndrome.”. Am J Med Genet 1996, 64, 241–5. [Google Scholar] [CrossRef]
- “Fmr1 Knockout Mice: A Model to Study Fragile X Mental Retardation The Dutch-Belgian Fragile X Consortium.”. Cell 1994, 78, 23–33.
- Eiges, R., A. Urbach, M. Malcov, T. Frumkin, T. Schwartz, A. Amit, Y. Yaron, A. Eden, O. Yanuka, N. Benvenisty, and D. Ben-Yosef. “Developmental Study of Fragile X Syndrome Using Human Embryonic Stem Cells Derived from Preimplantation Genetically Diagnosed Embryos.”. Cell Stem Cell 2007, 1, 568–77. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A., O. Bar-Nur, G. Q. Daley, and N. Benvenisty. “Differential Modeling of Fragile X Syndrome by Human Embryonic Stem Cells and Induced Pluripotent Stem Cells.”. Cell Stem Cell 2010, 6, 407–11. [Google Scholar] [CrossRef]
- Bar-Nur, O., I. Caspi, and N. Benvenisty. “Molecular Analysis of Fmr1 Reactivation in Fragile-X Induced Pluripotent Stem Cells and Their Neuronal Derivatives.”. J Mol Cell Biol 2012, 4, 180–3. [Google Scholar] [CrossRef]
- Vershkov, D., N. Fainstein, S. Suissa, T. Golan-Lev, T. Ben-Hur, and N. Benvenisty. “Fmr1 Reactivating Treatments in Fragile X Ipsc-Derived Neural Progenitors in Vitro and in Vivo.”. Cell Rep 2019, 26, 2531–39. [Google Scholar] [CrossRef]
- Vershkov, D., A. Yilmaz, O. Yanuka, A. L. Nielsen, and N. Benvenisty. “Genome-Wide Screening for Genes Involved in the Epigenetic Basis of Fragile X Syndrome.”. Stem Cell Reports 2022, 17, 1048–58. [Google Scholar] [CrossRef]
- Park, C. Y., T. Halevy, D. R. Lee, J. J. Sung, J. S. Lee, O. Yanuka, N. Benvenisty, and D. W. Kim. “Reversion of Fmr1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X Ipsc-Derived Neurons.”. Cell Rep 2015, 13, 234–41. [Google Scholar] [CrossRef]
- Liu, X. S., H. Wu, M. Krzisch, X. Wu, J. Graef, J. Muffat, D. Hnisz, C. H. Li, B. Yuan, C. Xu, Y. Li, D. Vershkov, A. Cacace, R. A. Young, and R. Jaenisch. “Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the Fmr1 Gene.”. Cell 2018, 172, 979–92. [Google Scholar] [CrossRef]
- Chong, T. L. L. Ahearn, and L. Cimmino. “Reprogramming the Epigenome with Vitamin C. Frontiers in Cell and Developmental Biology 2019, 7. [Google Scholar]
- Tsukada, Y., J. Fang, H. Erdjument-Bromage, M. E. Warren, C. H. Borchers, P. Tempst, and Y. Zhang. “Histone Demethylation by a Family of Jmjc Domain-Containing Proteins.”. Nature 2006, 439, 811–6. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M. A., T. Wang, B. Qin, J. Yang, D. Qin, J. Cai, W. Li, Z. Weng, J. Chen, S. Ni, K. Chen, Y. Li, X. Liu, J. Xu, S. Zhang, F. Li, W. He, K. Labuda, Y. Song, A. Peterbauer, S. Wolbank, H. Redl, M. Zhong, D. Cai, L. Zeng, and D. Pei. “Vitamin C Enhances the Generation of Mouse and Human Induced Pluripotent Stem Cells.”. Cell Stem Cell 2010, 6, 71–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, T., K. Chen, X. Zeng, J. Yang, Y. Wu, X. Shi, B. Qin, L. Zeng, M. A. Esteban, G. Pan, and D. Pei. “The Histone Demethylases Jhdm1a/1b Enhance Somatic Cell Reprogramming in a Vitamin-C-Dependent Manner.”. Cell Stem Cell 2011, 9, 575–87. [Google Scholar] [CrossRef]
- Stadtfeld, M., E. Apostolou, F. Ferrari, J. Choi, R. M. Walsh, T. Chen, S. S. Ooi, S. Y. Kim, T. H. Bestor, T. Shioda, P. J. Park, and K. Hochedlinger. “Ascorbic Acid Prevents Loss of Dlk1-Dio3 Imprinting and Facilitates Generation of All-Ips Cell Mice from Terminally Differentiated B Cells.”. Nat Genet 2012, 44, 398–405, S1. [Google Scholar] [CrossRef]
- Yue, X., and A. Rao. “Tet Family Dioxygenases and the Tet Activator Vitamin C in Immune Responses and Cancer.”. Blood 2020, 136, 1394–401. [Google Scholar] [CrossRef]
- Kaplanek, R., Z. Kejik, J. Hajduch, K. Vesela, K. Kucnirova, M. Skalickova, A. Venhauerova, B. Hosnedlova, R. Hromadka, P. Dytrych, P. Novotny, N. Abramenko, V. Antonyova, D. Hoskovec, P. Babula, M. Masarik, P. Martasek, and M. Jakubek. “Tet Protein Inhibitors: Potential and Limitations.”. Biomed Pharmacother 2023, 166, 115324. [Google Scholar]
- Lancaster, M. A., and J. A. Knoblich. “Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies.”. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Lancaster, M. A., M. Renner, C. A. Martin, D. Wenzel, L. S. Bicknell, M. E. Hurles, T. Homfray, J. M. Penninger, A. P. Jackson, and J. A. Knoblich. “Cerebral Organoids Model Human Brain Development and Microcephaly.”. Nature 2013, 501, 373–9. [Google Scholar] [CrossRef]
- Luo, C., M. A. Lancaster, R. Castanon, J. R. Nery, J. A. Knoblich, and J. R. Ecker. “Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain.”. Cell Rep 2016, 17, 3369–84. [Google Scholar] [CrossRef]
- Chiaradia, I., I. Imaz-Rosshandler, B. S. Nilges, J. Boulanger, L. Pellegrini, R. Das, N. D. Kashikar, and M. A. Lancaster. “Tissue Morphology Influences the Temporal Program of Human Brain Organoid Development.”. Cell Stem Cell 2023, 30, 1351–67. [Google Scholar] [CrossRef] [PubMed]
- Giandomenico, S. L., S. B. Mierau, G. M. Gibbons, L. M. D. Wenger, L. Masullo, T. Sit, M. Sutcliffe, J. Boulanger, M. Tripodi, E. Derivery, O. Paulsen, A. Lakatos, and M. A. Lancaster. “Cerebral Organoids at the Air-Liquid Interface Generate Diverse Nerve Tracts with Functional Output.”. Nat Neurosci 2019, 22, 669–79. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M. A., and J. A. Knoblich. “Generation of Cerebral Organoids from Human Pluripotent Stem Cells.”. Nat Protoc 2014, 9, 2329–40. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L. Bonfio, J. Chadwick, F. Begum, M. Skehel, and M. A. Lancaster. “Human Cns Barrier-Forming Organoids with Cerebrospinal Fluid Production.”. Science 2020, 369. [Google Scholar] [CrossRef] [PubMed]
- Drozd, M. Bardoni, and M. Capovilla. “Modeling Fragile X Syndrome in Drosophila.”. Front Mol Neurosci 2018, 11, 124. [Google Scholar] [CrossRef]
- Dahlhaus, R. “Of Men and Mice: Modeling the Fragile X Syndrome. ” Front Mol Neurosci 2018, 11, 41. [Google Scholar] [CrossRef]
- Berry-Kravis, E. M., L. Lindemann, A. E. Jonch, G. Apostol, M. F. Bear, R. L. Carpenter, J. N. Crawley, A. Curie, V. Des Portes, F. Hossain, F. Gasparini, B. Gomez-Mancilla, D. Hessl, E. Loth, S. H. Scharf, P. P. Wang, F. Von Raison, R. Hagerman, W. Spooren, and S. Jacquemont. “Drug Development for Neurodevelopmental Disorders: Lessons Learned from Fragile X Syndrome.”. Nat Rev Drug Discov 2018, 17, 280–99. [Google Scholar] [CrossRef]
- Lee, H. G., S. Imaichi, E. Kraeutler, R. Aguilar, Y. W. Lee, S. D. Sheridan, and J. T. Lee. “Site-Specific R-Loops Induce Cgg Repeat Contraction and Fragile X Gene Reactivation.”. Cell 2023, 186, 2593–609. [Google Scholar] [CrossRef]
- Brighi, C., F. Salaris, A. Soloperto, F. Cordella, S. Ghirga, V. de Turris, M. Rosito, P. F. Porceddu, C. D’Antoni, A. Reggiani, A. Rosa, and S. Di Angelantonio. “Novel Fragile X Syndrome 2d and 3d Brain Models Based on Human Isogenic Fmrp-Ko Ipscs.”. Cell Death Dis 2021, 12, 498. [Google Scholar] [CrossRef]
- Kang, Y., Zhou, Y. Li, Y. Han, J. Xu, W. Niu, Z. Li, S. Liu, H. Feng, W. Huang, R. Duan, T. Xu, N. Raj, F. Zhang, J. Dou, C. Xu, H. Wu, G. J. Bassell, S. T. Warren, E. G. Allen, P. Jin, and Z. Wen. “A Human Forebrain Organoid Model of Fragile X Syndrome Exhibits Altered Neurogenesis and Highlights New Treatment Strategies.”. Nat Neurosci 2021, 24, 1377–91. [Google Scholar] [CrossRef]
Figure 1.
Ascorbic Acid restores FMR1 expression by reversing hypermethylation in FX iPSCs. (A). Scheme representing the gene structure of human FMR1 in wild-type and FX. In wild-type FMR1 (top section of panel) there are less than 50 CGG repeats and no methylation of the CGG repeats and CpGs upstream and therefore the gene is transcriptional active, producing FMR1 mRNA and the FMRP protein product. In FX FMR1 (lower section of panel) there are >200 CGG repeats which are hypermethylated, hypermethylation spreads upstream to the CpGs upstream and therefore the gene is transcriptionally inactive. Unmethylated Cytosine is represented by transparent circular marks over the CpG and CGG sequence; methylated Cytosine is represented by red circular marks over the CpG and CGG sequence. (B). Dose response curve for FX iPSCs treated for 1 passage (6 days) with Ascorbic Acid (AsA). FX vehicle-control with 15mM HEPES (red), 10μM AsA (n=6, turquoise), 100μM AsA (n=6, navy blue), and 500μM AsA (n=6, blue). Values plotted are fold change of FMR1 expression normalized to FX vehicle-control. (C). Scheme representing the timeline and experimental setup for long term exposure to AsA. Low density of iPSCs are plated and kept in culture for 6 days and passaged, repeating until the sixth passage when cells are then collected for either DNA extraction (to perform pyrosequencing) or RNA extraction (to perform qRT-PCR). (D). FMR1 gene expression fold change (in log10) from FX vehicle-control (n=6, red) and FX +500μM AsA (n=6, blue) and wild-type (n=6, green). (E). Scheme of FMR1 gene structure with upstream CGG repeats and CpG island (in green) 150base pairs upstream of the CGG. CpG island in green is used as a proxy to quantify methylation of the FMR1 locus. Grey box highlights region of the CpG that was sequenced to quantify methylation levels by pyrosequencing. The arrows (black) show the location of the forward and reverse primer. (F). Left panel, average methylation levels per CpG site across all samples of iPSCs. FX vehicle-control (n=6, red); FX +500μM AsA (n=6, blue) and wild-type (n=6, green). Data are presented for 14 CpG sites, with error bars indicating the standard error of the mean (± SE). Grey dashed lines represent the median percentage methylation across all CpGs for each group. Right panel, boxplot showing the percentage methylation level for each individual sample; FX vehicle-control (n=6, red) and FX +500μM AsA (n=6, blue) and wild-type (n=6, green). All boxplots display the interquartile range (IQR) and median, with the mean indicated by white circles. P value calculated as Wilcox rank sum exact test.
Figure 1.
Ascorbic Acid restores FMR1 expression by reversing hypermethylation in FX iPSCs. (A). Scheme representing the gene structure of human FMR1 in wild-type and FX. In wild-type FMR1 (top section of panel) there are less than 50 CGG repeats and no methylation of the CGG repeats and CpGs upstream and therefore the gene is transcriptional active, producing FMR1 mRNA and the FMRP protein product. In FX FMR1 (lower section of panel) there are >200 CGG repeats which are hypermethylated, hypermethylation spreads upstream to the CpGs upstream and therefore the gene is transcriptionally inactive. Unmethylated Cytosine is represented by transparent circular marks over the CpG and CGG sequence; methylated Cytosine is represented by red circular marks over the CpG and CGG sequence. (B). Dose response curve for FX iPSCs treated for 1 passage (6 days) with Ascorbic Acid (AsA). FX vehicle-control with 15mM HEPES (red), 10μM AsA (n=6, turquoise), 100μM AsA (n=6, navy blue), and 500μM AsA (n=6, blue). Values plotted are fold change of FMR1 expression normalized to FX vehicle-control. (C). Scheme representing the timeline and experimental setup for long term exposure to AsA. Low density of iPSCs are plated and kept in culture for 6 days and passaged, repeating until the sixth passage when cells are then collected for either DNA extraction (to perform pyrosequencing) or RNA extraction (to perform qRT-PCR). (D). FMR1 gene expression fold change (in log10) from FX vehicle-control (n=6, red) and FX +500μM AsA (n=6, blue) and wild-type (n=6, green). (E). Scheme of FMR1 gene structure with upstream CGG repeats and CpG island (in green) 150base pairs upstream of the CGG. CpG island in green is used as a proxy to quantify methylation of the FMR1 locus. Grey box highlights region of the CpG that was sequenced to quantify methylation levels by pyrosequencing. The arrows (black) show the location of the forward and reverse primer. (F). Left panel, average methylation levels per CpG site across all samples of iPSCs. FX vehicle-control (n=6, red); FX +500μM AsA (n=6, blue) and wild-type (n=6, green). Data are presented for 14 CpG sites, with error bars indicating the standard error of the mean (± SE). Grey dashed lines represent the median percentage methylation across all CpGs for each group. Right panel, boxplot showing the percentage methylation level for each individual sample; FX vehicle-control (n=6, red) and FX +500μM AsA (n=6, blue) and wild-type (n=6, green). All boxplots display the interquartile range (IQR) and median, with the mean indicated by white circles. P value calculated as Wilcox rank sum exact test.
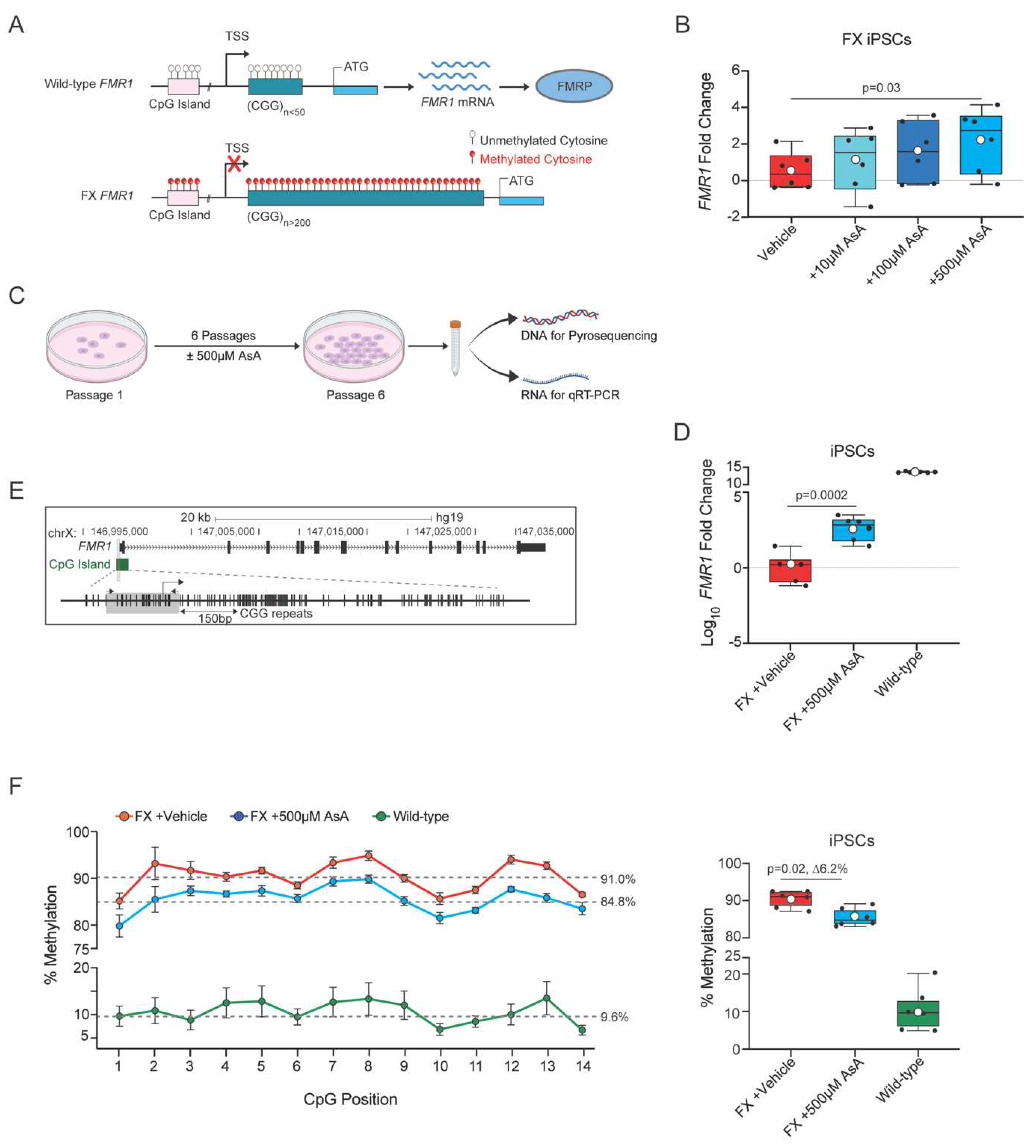
Figure 2.
Ascorbic Acid reactivates FMR1 and reduces methylation in FX cerebral organoids. (A). Scheme representing the timeline and experimental setup for long term exposure to AsA. Low density of iPSCs are plated and kept in culture for 6 days and passaged, and repeated until the sixth passage where cells are then used to start the cerebral organoid differentiation protocol. Cerebral organoids are cultured until day 50 where they are collected for either DNA extraction (to perform pyrosequencing) or RNA extraction (to perform qRT-PCR) or imaged for phenotype assessment. (B). Representative images of wild-type (left panel), FX vehicle-control (center panel), and FX +AsA (right panel) cerebral organoids. Scale bar for wild-type 2200μM, 3000μM for FX and 3000μM for FX +AsA. (C). Percent of cerebral organoids per plate that had morphologically organized neuronal tissue by visual scoring of phenotype; FX vehicle-control (n=6, red), FX +500μM AsA (n=6, blue) and wild-type (n=6, green). n = 6 plates per genotype and condition; each plate contained 10-20 cerebral organoids. (D). FMR1 gene expression fold change between FX vehicle-control cerebral organoids (n=13, red); all FX +500μM AsA treated cerebral organoids (n=26, navy blue); and FX +500μM AsA treated cerebral organoids stratified by the 95% CI of FX vehicle-controls into FX +AsA non responders (n=16, grey) and FX +AsA responders (n=10, blue). (E). Left panel, average percentage of methylation levels at each CpG site across all samples for FX (n = 14, red), non-responders (n = 11, grey), responders (n = 9, blue), and wild-type (n = 14, green). Data are presented for 14 CpG sites, with error bars indicating the standard error of the mean (± SE). Grey dashed lines represent the median percentage methylation across all CpGs for each group. Right panel, boxplot showing the percentage methylation level for each sample. Individual samples are represented by black circles. The boxplot displays the interquartile range (IQR) and median, with the mean indicated by white circles. P value is calculated as Wilcoxon rank sum exact test.
Figure 2.
Ascorbic Acid reactivates FMR1 and reduces methylation in FX cerebral organoids. (A). Scheme representing the timeline and experimental setup for long term exposure to AsA. Low density of iPSCs are plated and kept in culture for 6 days and passaged, and repeated until the sixth passage where cells are then used to start the cerebral organoid differentiation protocol. Cerebral organoids are cultured until day 50 where they are collected for either DNA extraction (to perform pyrosequencing) or RNA extraction (to perform qRT-PCR) or imaged for phenotype assessment. (B). Representative images of wild-type (left panel), FX vehicle-control (center panel), and FX +AsA (right panel) cerebral organoids. Scale bar for wild-type 2200μM, 3000μM for FX and 3000μM for FX +AsA. (C). Percent of cerebral organoids per plate that had morphologically organized neuronal tissue by visual scoring of phenotype; FX vehicle-control (n=6, red), FX +500μM AsA (n=6, blue) and wild-type (n=6, green). n = 6 plates per genotype and condition; each plate contained 10-20 cerebral organoids. (D). FMR1 gene expression fold change between FX vehicle-control cerebral organoids (n=13, red); all FX +500μM AsA treated cerebral organoids (n=26, navy blue); and FX +500μM AsA treated cerebral organoids stratified by the 95% CI of FX vehicle-controls into FX +AsA non responders (n=16, grey) and FX +AsA responders (n=10, blue). (E). Left panel, average percentage of methylation levels at each CpG site across all samples for FX (n = 14, red), non-responders (n = 11, grey), responders (n = 9, blue), and wild-type (n = 14, green). Data are presented for 14 CpG sites, with error bars indicating the standard error of the mean (± SE). Grey dashed lines represent the median percentage methylation across all CpGs for each group. Right panel, boxplot showing the percentage methylation level for each sample. Individual samples are represented by black circles. The boxplot displays the interquartile range (IQR) and median, with the mean indicated by white circles. P value is calculated as Wilcoxon rank sum exact test.
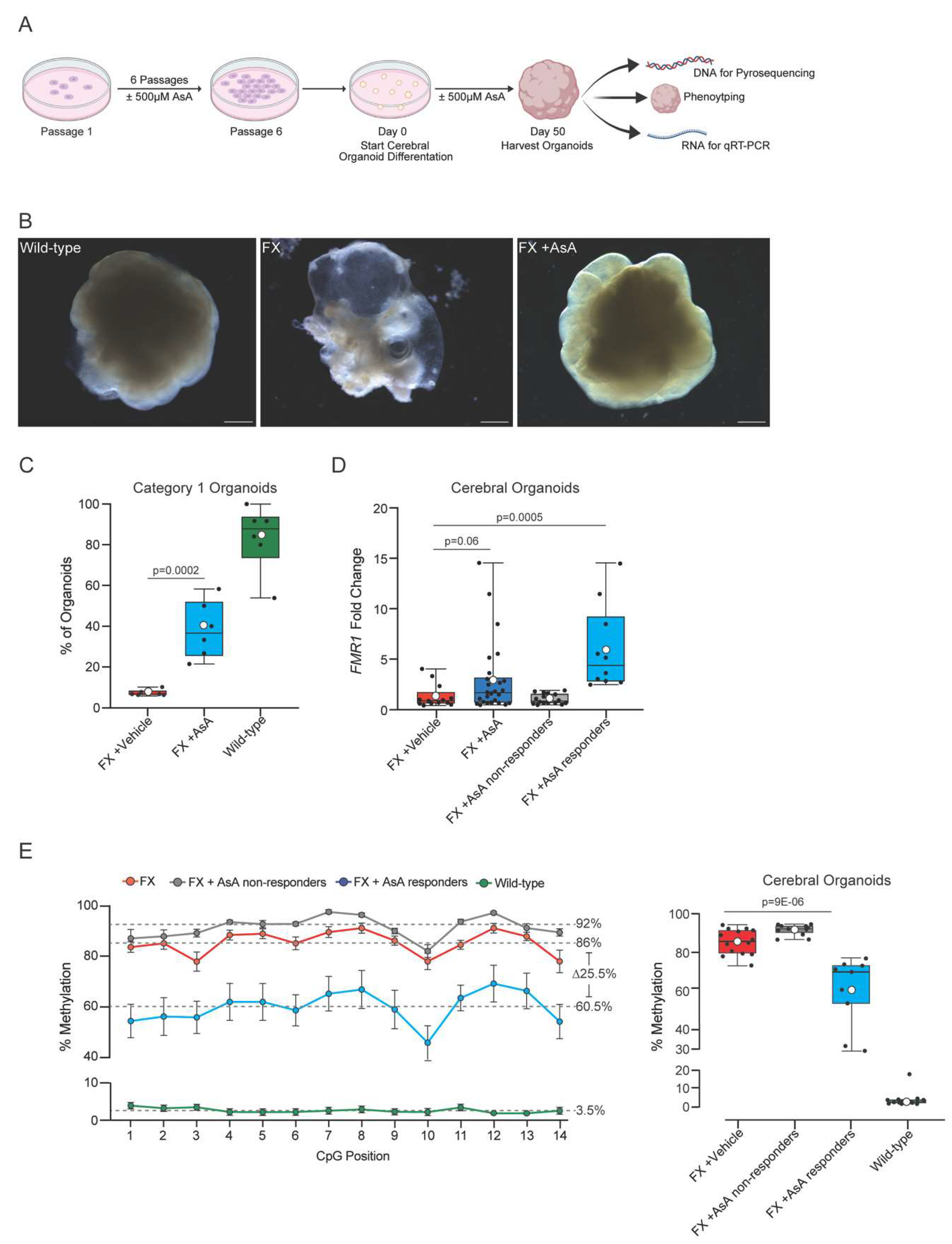
Figure 3.
Ascorbic Acid treatment causes changes in gene expression related to neurodevelopment. (A). Principal Component Analysis (PCA) of the 2 most significant principal components (PC1 and PC2) identified by comparing FX vehicle cerebral organoids (n=4, red), FX +500μM AsA cerebral organoids (n=4, blue) and wild-type cerebral organoids (n=4, green). (B). Volcano plot showing genes that are differentially expressed in FX vehicle cerebral organoids compared to wild-type cerebral organoids. Red indicates genes significantly downregulated in FX cerebral organoids compared with wild-type cerebral organoids. (C). Gene enrichment analysis showing families of genes downregulated in FX cerebral organoids compared to wild-type cerebral organoids. (D). Volcano plot showing genes that are differentially expressed in FX +500μM AsA cerebral organoids compared to FX vehicle cerebral organoids). Blue dots indicate genes with a significant upregulation due to AsA treatment. (E). Relative expression levels of FMR1 between FX (red) and FX +500μM AsA (blue) cerebral organoids. (F). Change in expression levels of genes found to be most downregulated in FX cerebral organoids compared to wild-type cerebral organoids. Comparison is made for FX cerebral organoids (red) and FX +500μM AsA cerebral organoids (blue). (G). String analysis of most upregulated genes in FX +500μM AsA cerebral organoids showing gene families involved in neuronal development and synapse formation and maturation. All statistics performed were unpaired one-tail t-test between FX vehicle-control (red) and 500μM AsA treatments (blue. Error bars plotted are ± standard error of the mean.
Figure 3.
Ascorbic Acid treatment causes changes in gene expression related to neurodevelopment. (A). Principal Component Analysis (PCA) of the 2 most significant principal components (PC1 and PC2) identified by comparing FX vehicle cerebral organoids (n=4, red), FX +500μM AsA cerebral organoids (n=4, blue) and wild-type cerebral organoids (n=4, green). (B). Volcano plot showing genes that are differentially expressed in FX vehicle cerebral organoids compared to wild-type cerebral organoids. Red indicates genes significantly downregulated in FX cerebral organoids compared with wild-type cerebral organoids. (C). Gene enrichment analysis showing families of genes downregulated in FX cerebral organoids compared to wild-type cerebral organoids. (D). Volcano plot showing genes that are differentially expressed in FX +500μM AsA cerebral organoids compared to FX vehicle cerebral organoids). Blue dots indicate genes with a significant upregulation due to AsA treatment. (E). Relative expression levels of FMR1 between FX (red) and FX +500μM AsA (blue) cerebral organoids. (F). Change in expression levels of genes found to be most downregulated in FX cerebral organoids compared to wild-type cerebral organoids. Comparison is made for FX cerebral organoids (red) and FX +500μM AsA cerebral organoids (blue). (G). String analysis of most upregulated genes in FX +500μM AsA cerebral organoids showing gene families involved in neuronal development and synapse formation and maturation. All statistics performed were unpaired one-tail t-test between FX vehicle-control (red) and 500μM AsA treatments (blue. Error bars plotted are ± standard error of the mean.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



