Submitted:
05 October 2024
Posted:
07 October 2024
You are already at the latest version
Abstract
In 2021, the World Health Organization classified isocitrate dehydrogenase (IDH) mutant gliomas as a distinct subgroup of tumors with genetic changes sufficient to enable a complete diagnosis. Patients with an IDH mutant glioma have improved survival which has been further enhanced by the advent of targeted therapies. IDH enzymes contribute to cellular metabolism, and mutations to specific catalytic residues result in the neomorphic production of D-2-hydroxyglutarate (D-2-HG). The accumulation of D-2-HG results in epigenetic alterations, oncogenesis and impacts the tumor microenvironment via immunological modulations. Here, we summarize the molecular, cellular, and clinical implications of IDH mutations in gliomas as well as current diagnostic techniques.
Keywords:
Isocitrate Dehydrogenase
; IDH1
; IDH2
; D-2-Hydroxyglutarate
; D-2-HG
; Epigenetics
; Glioma
Introduction
Gliomas are brain tumors associated with high mortality and life altering symptoms including seizures, cognitive/motor deficits, dysphagia, and aphasia [1]. Current treatment methods for glioma include surgical resection, radiation therapy, and chemotherapy with the use of temozolomide (TMZ). Despite these approaches, the long-term survival of patients remains poor. Approximately 30% of primary brain tumors are gliomas, which are believed to arise from neuroglial stem or progenitor cells [2]. Gliomas may be categorized as astrocytomas, oligodendrogliomas, oligo-astrocytoma, or ependymomas depending upon the cell type(s) from which they originate (Figure 1). When classifying these tumors, World Health Organization Central Nervous System 5 (WHO CNS5) grades ranging from I to IV are traditionally used, with grade I tumors being the least malignant [3]. To determine tumor grade, characteristics such as invasiveness, rate of growth, and degree of necrosis within the tumor are utilized. Gliomas may be diffuse in nature, which causes difficulty in both visualization via magnetic resonance imaging (MRI) and determination of margins during surgery, further complicating the identification and removal of malignant tissue.
Molecular analysis of the tumor allows for further classification of gliomas into subgroups. Classification of tumors provides valuable insight toward prognosis, and in some cases, the possibility of treatment with targeted therapies. Molecular classification may be accomplished with techniques including DNA/RNA sequencing, PCR, and DNA methylome profiling [4,5,6]. Various biomarkers have been established through the molecular classification of tumors, an example of which is isocitrate dehydrogenase (IDH). IDH enzymes play essential roles in metabolic processes such as the citric acid cycle, lipogenesis, glutamine metabolism, and redox regulation [7]. Oncogenic mutations to IDH1 were identified in 2008 following an integrated genomic analysis of glioblastoma multiforme (GBM) [8], and have since been targeted with pharmaceutical inhibitors [9,10]. The prognosis and treatment for IDH mutant gliomas differs from their IDH wildtype counterparts significantly, such that screening for them has become an important standard of care. Mutations to IDH have been documented in various types of cancer [11,12,13] but are more prevalent in glioma and acute myeloid leukemia (AML) [14,15]. In general, IDH mutations are associated with less aggressive cancer, as they render cells more vulnerable to death [16] and demonstrate conservative levels of migration, angiogenesis, and invasion [17]. In this review, we describe the molecular, cellular, and clinical consequences of IDH mutations in glioma as well as the current diagnostic methods and treatments.
Normal Function of IDH & the Cancer-Associated Accumulation of D-2-HG
IDH1/2 are the most frequently mutated cancer-associated metabolic genes. IDH exists in three isoforms: IDH1, IDH2, and IDH3. IDH1 and IDH2 enzymes function as homodimers and share approximately 70% sequence homology [18]. Conversely, IDH3 is a more distantly related heterotetrameric protein that is not known to be associated with the development of cancer [19]. IDH1 is expressed in the cytosol and peroxisomes and produces NADPH for fatty acid biosynthesis, with oncogenic mutations commonly associated with glioma. Alternatively, IDH2 localizes within the mitochondrial matrix, with mutations often driving AML, or less commonly, glioma [20]. IDH1 and IDH2 mutations are mutually exclusive, with rare exceptions [21], due to a common biochemical mechanism [22] that drives similar downstream effects. Ordinarily, IDH1/2 catalyze the conversion of isocitrate into α-ketoglutarate (αKG) while reducing NADP+ to NADPH as a byproduct. Missense mutations yield amino acid changes at positions R132 in IDH1 and R140 or R172 in IDH2. These mutations result in the replacement of conserved arginine residues in the catalytic site with an alternative amino acid. The consequence of these mutations is an altered protein with neomorphic activity- the conversion of αKG to D-2-Hydroxyglutarate (D-2-HG) [14,23] (Figure 2). D-2-HG is an oncometabolite that accumulates in primary IDH mutant tumors at concentrations ranging from 1 to 30 mM [14,23]. D-2-HG causes a plethora of downstream effects that mediate the development of cancer, as detailed below.
Epigenetic Alterations
Epigenetic alterations are a hallmark of IDH mutant-mediated oncogenesis and are largely driven by the production of D-2-HG. Due to its structural similarity to αKG and accumulation within IDH mutant cells, D-2-HG competitively inhibits various αKG-dependent dioxygenases. These dioxygenases include the Jumonji-C (JmjC) family of histone lysine demethylases (KDMs), ten-eleven translocation (TET) DNA cytosine-oxidizing enzymes, AlkB homologs (ABH), and prolyl hydroxylases (PHD) [24] (Figure 3). αKG-dependent dioxygenases play critical roles in epigenetics [24], biosynthesis [25], post-translational modifications [26], and hypoxic response [27]. The collective antagonization of demethylases by D-2-HG leads to instability of RNA transcripts and characteristic patterns of hypermethylation of DNA and histone proteins [28]. Due to these epigenetic modifications, mutant IDH is highly associated with the Glioma CpG island methylator phenotype (G-CIMP) [29], a locus-specific pattern of DNA hypermethylation at CpG-rich promoter regions.
JmjC-KDMs
JmjC-KDMs are enzymes that regulate gene expression through demethylation and chromatin scaffolding functions [30]. The JmjC protein family contains >30 members [31], several of which may actively contribute to the development of cancers including glioma [32], acute myeloid leukemia [33], and lung cancer [31] when dysregulated. Numerous silenced, mutated, or deleted JmjC-KDM genes have been identified in cancer [34] and appear to act as either a driver or suppressor of gene expression, depending upon cellular context. Since JmjC-KDMs aid in the control of genome-wide expression patterns, aberrant protein function may result in significant changes to the transcriptomic profile of the cell [35]. Potential targets of dysregulated JmjC-KDMs include pro-inflammatory pathways such as signal transducer and activator of transcription (STAT)-dependent response or proliferation associated pathways such as mitogen-activated protein kinase (MAPK) [31].
In some cases, D-2-HG mediated inhibition of JmjC-KDMs results in downstream effects which function in a manner similar to knockout models. This phenomenon has been demonstrated by several members of the JmjC-KDM5 family of H3K4 histone lysine demethylases (KDM5A, KDM5C, and KDM5D). The inhibition of these JmjC-KDM5 enzymes occurs at physiologically relevant concentrations of D-2-HG (less than 1 mM) and is thought to contribute to IDH mutant-mediated transformation [36]. Of all potential αKG analogs (for example, fumarate, L-2-HG, succinate), D-2-HG is the most potent inhibitor of KDM4A/B and KDM6A with IC50 values <200 µM. Collectively, KDMs inhibited by D-2-HG contribute to the regulation of diverse cellular processes to include differentiation [37], growth and development [37,38], adipogenesis [39], adhesion, and proliferation [33]. The competitive inhibition of several JmjC-KDMs by D-2-HG has been shown to increase dimethylation of H3K79, H3K27, and H3K9 as well as mono and trimethylation of H3K4 [40]. In combination with high levels of acetylation, H3K79 and H3K4 methylation are associated with active chromatin, with H3K79 found within the coding regions of genes and H3K4 often enriched at enhancers and promoters of actively transcribed genes. Conversely, H3K27 and H3K9 are associated with inactive chromatin [41]; Thus, the pleiotropic effects of D-2-HG accumulation impacts a variety of different biochemical pathways resulting in oncogenesis.
Although the epigenetic changes mediated by JmjC-KDMs in IDH mutant cancers are profound, they are also readily reversible by the addition of αKG. 300 µM αKG has been shown to negate the inhibition of KDM7A caused by up to 50 mM D-2-HG [40]. This reversibility demonstrates the relatively weak antagonism of JmjC-KDMs by D-2-HG despite occupying the same active site in the catalytic core. Regardless, IDH1-R132H mutations have shown a near 60% reduction of αKG and >20-fold increase in D-2-HG [40], suggesting that this competitive inhibition is due to metabolite quantity rather than changes to enzyme kinetics. Despite the inhibition of several JmjC-KDMs by D-2-HG, it is important to note that αKG-dependent dioxygenases vary greatly in their susceptibility to inhibition by D-2-HG, with some not significantly affected by relevant concentrations [34], such as KDM5B (IC50 3.6 mM) [42]. Recently, pharmaceutical inhibitors have been screened for their therapeutic potential toward inhibiting relevant JmjC-KDMs IDH mutant cancers, which we address later in this review.
Histone Deacetylases
Histone deacetylases (HDACs) possess chromatin remodeling activity and contribute to silencing genes in an epigenetic manner. HDACs hydrolyze acetate from lysine on histone tails [43], strengthening the interaction between histone proteins and DNA. HDACs are recruited to methylated DNA through their association with methyl-CpG binding proteins [44]. As hypermethylation is abundant in IDH mutant cancers, it is thought that HDACs play a role in transcriptomic alterations by compounding the effects of methylation to further condense chromatin near CpG islands. Expression profiles of IDH mutant glioma have demonstrated the upregulation of genes promoting HDAC function or related pathways in comparison to their IDH wildtype counterparts, including three of the six HDAC genes that are expressed in gliomas [45]. Additional studies have shown that overexpression of the IDH mutant enzyme results in histone hypoacetylation [46], further supporting the notion that HDACs play a role in IDH mutant oncogenesis. Several groups have presented HDAC inhibitors as a potential therapeutic for IDH mutant glioma, which we discuss later in this review.
TET Enzymes
TET enzymes are involved in various biological processes to include post-transcriptional regulation [47] and are essential for immune cell development [48] and embryogenesis [49]. TET enzymes generate oxidized derivatives of 5-methylcytosine (5mC) in a Fe(II)/αKG-dependent manner. In these reactions, 5mC is sequentially oxidized to 5-hydroxymethylcytosine, 5-formylcytosine, then 5-carboxylcytosine. This oxidation typically occurs at CpG dinucleotide sites, where thymine DNA glycosylase excises 5-formylcytosine and 5-carboxylcytosine then base excision repair is utilized to yield unmethylated cytosine [47,50]. Rather than being inert intermediates, the oxidation products of 5mC play unique and distinct biological roles and may also actively contribute to cancer in some cases [51]. For example, Johnson et al. observed the enrichment of 5-hydroxymethylcytosine in GBM at disease-specific enhancer elements to unexpectedly elicit open chromatin and increased gene expression [52]. To our knowledge, no data are currently available that explore the relationship between IDH mutant cancers and 5mC oxidation products.
The dysregulation of TET enzymes contributes to development of the G-CIMP through an increase in methylation [53] and subsequently alters the transcriptomic profile of IDH mutant cancers. Similar to JmjC-KDMs, TET enzymes are often mutated in various types of cancer such as AML and myelodysplastic/myeloproliferative neoplasms [54,55]. TET and IDH driver mutations are mutually exclusive [56], suggesting that the inhibition of TET enzymes contributes significantly to mutant IDH mediated oncogenesis [46]. While TET or IDH mutations routinely occur in leukemia [47,57], TET mediated oncogenesis is not typically thought to contribute to the development of glioma. TET enzymes are susceptible to competitive inhibition by the accumulation of D-2-HG, fumarate or succinate that result from oncogenic mutations to IDH, fumarase hydratase or succinate dehydrogenase, respectively [58]; however, L-2-HG, a naturally occurring enantiomer of D-2-HG that is upregulated under hypoxic conditions [59], does not appear to interfere with TET activity intracellularly. To this end, the use of cell-permeable L-2-HG to quantify TET enzymatic inhibition demonstrates that treatment with up to 3 mM of L-2-HG elicits little to no inhibition of TET2 or TET3 in situ [60].
Despite the oncogenic consequences of TET dysregulation, not all members of the TET family are prone to inhibition by clinically relevant concentrations of D-2-HG. For example, TET2 has demonstrated inhibition at low levels of D-2-HG in vitro (IC50= ~15 µM) while TET1 (IC50= ~800 µM) and TET3 (IC50= ~100 µM) are less susceptible. Although TET2 is the most prone to competitive inhibition by D-2-HG, TET1 and TET3 are highly expressed in specific areas of the brain including the cerebellum and cerebral cortex, respectively, while the expression of TET2 in the brain is somewhat modest [47]. Another potential variable regarding the inhibition of TET enzymes in IDH mutant cancers is the availability of ascorbic acid (vitamin C). Ascorbic acid modulates TET enzymes via a direct interaction with their catalytic domain [61] and reduces Fe(III) to Fe(II), an essential cofactor of TET. Indeed, co-administration of pharmaceutical mutant IDH inhibitors and ascorbic acid has been shown to restore TET activity in IDH1-R132H colorectal cancer cell lines [62]. Additional studies have demonstrated that supplementation with an oxidatively stable form of vitamin C (Ascorbate-2-phosphate) can restore 2-HG induced epigenetic remodeling [63]. Because IDH mutations alter NADP+/NADPH ratios and NADPH is a cofactor in recycling ascorbate, it is possible that vitamin C availability and D-2-HG accumulation have a combinatorial effect towards the dampened TET activity witnessed in IDH mutant cancers. To this end, an in vivo knock in model of IDH1-R132H has demonstrated altered NADP+/NADPH ratios with a coinciding decrease in ascorbate within the brain[64].
Prolyl Hydroxylases & Hypoxic Response
In addition to epigenetic modifications, αKG-dependent dioxygenases may also play key roles facilitating decisions regarding cell fate such as normal differentiation [65] and regulation of hypoxia inducible factor 1 (HIF-1α) [66]. Due to the rapid proliferation of tumors, hypoxia is frequently observed in the microenvironment as existing blood supplies are outgrown. HIF-1α is a transcription factor that is upregulated in response to low oxygen, including under inflammatory conditions [67], to induce the expression of various genes essential for cell survival and adaptation to a hypoxic environment [68]. PHDs utilize αKG as a substrate to selectively hydroxylate targets and are susceptible to dysregulation by the accumulation of D-2-HG [69]. Under ordinary conditions, HIF-1α is constitutively expressed and rapidly degraded by PHDs. Low levels of oxygen inhibit PHDs, resulting in elevated expression of HIF-1α and subsequently the upregulation of hypoxia associated genes.
EglN prolyl-4-hydroxylases ordinarily mark HIF-1α for polyubiquitylation and proteasomal degradation [70]; however, literature disagrees on whether D-2-HG stimulates or inhibits these PHDs. Some studies suggest that D-2-HG acts as a co-substrate to PHDs resulting in the destabilization of HIF-1α and reduced expression of associated genes [71]. The knockdown of PHDs in IDH mutant astrocytes was also shown to result in a marked reduction in proliferation, suggesting a potential role for HIF-1α as a tumor suppressor [69]. As ascorbic acid availability is lowered by the altered NADP+/NADPH ratios in IDH mutant cancers [64], it should also be noted that ascorbate encourages hydroxylase activity to suppress the transcriptional response of HIF-1α [72,73]. To this end, a decrease in ascorbate availability could also contribute to a decrease in the expression of hypoxia associated pathways; however, this relationship has never been explored.
Contrary to findings that HIF-1α is downregulated in IDH mutant cancers, many others have shown that HIF-1α is upregulated in IDH mutant cells, cells treated with exogenous 2-HG, IDH mutant tumors, and brains of mouse embryos expressing IDH1-R132H [40,64,74,75]. The notion of an increase in HIF-1α resulting from metabolically driven oncogenesis such as IDH is supported by cancers driven by a similar mechanism. Oncogenic mutations to fumarase hydratase or succinate dehydrogenase result in the accumulation of fumarate or succinate, respectively, which act in a similar manner to D-2-HG by competitively inhibiting αKG-dependent dioxygenases. In these instances, fumarate or succinate have been shown to inhibit αKG-dependent PHDs that target HIF-1α for degradation [76,77], resulting in its subsequent accumulation.
RNA Transcript Stability
The methylation of adenosine at the nitrogen-6 positions of RNA (N6-Methyladenosine, m6A) serves as an essential posttranslational regulatory mark of mRNAs and noncoding RNAs. In addition to playing a role in most RNA-related processes such as splicing and nuclear export, m6A is also associated with biological processes including transcriptional regulation, signal transduction, DNA damage response [78], and cancer [79]. m6A is a frequent internal modification of mRNA which recruits m6A binding proteins such as YTHDF1/2/3, YTHDC2, or IGF2BP1/2/3 to regulate stability and/or translation efficiency [79]. m6A modifications are ordinarily facilitated by methyltransferases and removed by demethylases such as fat mass and obesity-associated protein (FTO) or ALKBH5 which are Fe(II)/ αKG-dependent dioxygenases [80].
IDH wildtype cells treated with D-2-HG or IDH mutant cells producing D-2-HG exhibit an increase in m6A levels that can be reversed through treatment with IDH mutant pharmaceutical inhibitors such as Vorasidenib [81]. Since IDH mutant pharmaceutical inhibitors are known to deplete D-2-HG, these results support the role of the oncometabolite toward m6A accumulation. Moreover, D-2-HG has demonstrated a dose-dependent inhibition of FTO [81] that causes an increase in m6A levels [82,83,84]. The knockdown of endogenous FTO recapitulates the impacts of D-2-HG on cell viability, increases m6A levels [83], and promotes apoptosis and cell-cycle arrest at the G0/G1 phase [84]. Similarly, pharmacologic inhibition of FTO using FB23-2 or meclofenamic acid result in decreased proliferation and increased apoptosis. In addition to FTO, D-2-HG also inhibits ALKBH5; however, D-2-HG has a weaker binding affinity for ALKBH5 than FTO and the knockdown of ALKBH5 alone does not recapitulate the increase in m6A seen in FTO knockout lines or IDH mutant cells [81]. These results suggest that FTO inhibition is the primary mechanism of m6A accumulation in IDH mutant cells.
Increased levels of m6A result in lower stability of mRNA transcripts with a notable target of D-2-HG mediated transcript decay being the oncogene MYC [83]. Interestingly, ectopically expressing MYC rescues D-2-HG mediated growth inhibition [84]. Further studies have shown that m6A-mediated downregulation of activating transcription factor 5 (ATF5) mRNA may also play an important role in regulating proliferation and apoptosis in IDH mutant glioma [81]. In addition to impacting proliferation, m6A levels modify the metabolic profile of cells, with the largest impacts being the downregulation of phosphofructokinase platelet (PFKP) and lactate dehydrogenase B (LDHB) [84].
While TET enzymes are largely known for facilitating the active demethylation of DNA, recent studies have also demonstrated that TET2 contains an RNA-binding domain [85] and can oxidize 5-methylcytosine RNA into 5-hydroxymethylcytosine. Since 5-hydroxymethylcytosine is associated with active translation of mRNA [86], aberrant post-translational mRNA modifications by TET2 reduce transcript stability which may increase susceptibility to certain diseases [87]. With these recent findings, further studies are necessary to determine the specific impact of TET-mediated mRNA alterations in IDH mutant glioma.
Patterns of Transcription
The many epigenetic alterations within the IDH mutant genome have various downstream implications for the transcriptome. Tran et al., assessed RNA sequencing (RNAseq) and microarray data for 1032 gliomas from the cancer genome atlas and 395 gliomas from REMBRANDT and found four distinct transcriptomic profile groups. Interestingly, IDH mutant gliomas with codeletions were grouped with oligodendrogliomas with high tumor purity. Transcriptomic data for this group was enriched for neurotransmission, G-protein coupled receptor signaling, and insulin secretion pathways. Alternatively, IDH mutant gliomas without codeletions generally corresponded with astrocytoma and transcriptomic data enriched for immune, cell cycle, NOTCH signaling, transcription, and translation [88]. In a similar study, Cheng et al., utilized RNAseq data to establish a six-gene risk signature for IDH mutant low-grade glioma to assist in determining risk and prognosis. The six genes found to be highly significant for prognosis in IDH mutant patients included: cell division cycle (CDC) 20, Wiskott-Aldrich syndrome protein family (WASF)3, deleted in breast cancer (DBC)1, engrailed (EN)2, vimentin (VIM), and carboxypeptidase (CPE). Higher expression of CDC20, EN2, and VIM was found to be associated with risk while WASF3, DBC1, and CPE were considered protective genes with higher expression levels associated with better prognosis [89]. Another study focusing on transcriptomic profiles compared IDH mutant gliomas and IDH mutant AML, melanoma, and cholangiocarcinoma. Interestingly, these profiles showed pro-malignant genes unique to IDH mutant gliomas while genes associated with differentiation and immune response were suppressed in all IDH mutant cancers. Moreover, IDH mutant gliomas demonstrated a higher degree of genes that were both hypermethylated and differentially expressed in comparison to other types of IDH mutant cancers. Unruh et al. also noted variances in differential gene expression between IDH mutant oligodendrogliomas and astrocytoma, with oligodendrogliomas downregulating genes linked to angiogenesis, cell proliferation, and integrin binding [90]. Additionally, analysis of transcriptomic profiles of IDH mutant glioma patients revealed the decreased expression of HIF-1α targets as well as the inhibition of angiogenesis and vasculogenesis. Specifically, the expression of EGLN1 and EGLN3 PHDs, which serve to degrade HIF-1α was upregulated while pro-angiogenic targets such as vascular endothelial growth factor A, angiopoietin-2 and platelet-derived growth factor A was decreased [27].
Metabolic Reprogramming in IDH Mutant Glioma
Comparison of Wildtype and Mutant IDH Reactions
Wildtype IDH catalyzes the oxidative decarboxylation of isocitrate into αKG and CO2. This reaction occurs concomitantly with the reduction of NADP+ into NADPH, with the exception of IDH3, which generates NADH within the citric acid cycle [91]. Under certain circumstances (e.g. hypoxia), IDH can catalyze the reverse reaction – the reductive carboxylation of αKG into isocitrate, generating NADP+ as a byproduct [92]. Isocitrate is subsequently isomerized into citrate, which is broken down by ATP citrate lyase into acetyl-CoA. Acetyl-CoA can be used for fatty acid biosynthesis under these conditions [93].
Hotspot mutations within the active site of IDH1 (R132X) and IDH2 (R140X or R172X) render the enzymes capable of catalyzing a new reaction - the conversion of αKG into D-2-HG. This is in contrast to the reverse reaction catalyzed by wildtype IDH1 that produces isocitrate. Mutant IDH-driven D-2-HG accumulation is coupled with the oxidation of NADPH into NADP+. Unlike wildtype IDH, mutant IDH consumes both αKG and NADPH, which is likely to impact cellular metabolism and redox homeostasis, respectively.
D-2-HG is classified as an oncometabolite and is almost exclusively produced by IDH mutant cells [14], although there is evidence that other enzymes, including phosphoglycerate dehydrogenase, produce minute amounts of the oncometabolite in cells [94]. Mutant IDH-mediated D-2-HG accumulation is a molecular hallmark of astrocytoma and oligodendroglioma [95]. Importantly, D-2-HG production drives metabolic rewiring in these types of cancer, including but not limited to dysregulation of glucose metabolism, the citric acid cycle, amino acid metabolism, lipid and cholesterol metabolism and redox homeostasis.
Mutant IDH Cells Do Not Perform Aerobic Glycolysis
Perhaps the most well-known example of metabolic reprogramming is the overconsumption of glucose by cancer cells to meet growing energy demands [96]. Cancer cells are also known to produce and secrete copious amounts of lactate as a result of pyruvate metabolism, despite the presence of O2. This phenomenon is better known as aerobic glycolysis or the Warburg effect. Interestingly, IDH mutant glioma do not appear to follow this framework. Glycolytic flux is reduced in IDH1 mutant glioma tumors compared with IDH1 wildtype tumors as a result of dampened expression of the rate-limiting glycolytic enzymes hexokinase and pyruvate kinase [97]. In agreement with this, lower quantities of glycolytic intermediates, including fructose 1,6-bisphosphate, 3-phosphoglycerate and phosphoenolpyruvate, have been observed in IDH1 mutant glioma tissue [95].
IDH mutant glioma tissue and brain tumor stem cells derived from IDH1 mutant glioma tissue show reduced expression of lactate dehydrogenase A (LDHA), the enzyme responsible for converting pyruvate into lactate [98]. Silenced LDHA expression is associated with hypermethylation of the LDHA promoter, which is likely to be a direct result of inhibition of αKG-dependent DNA demethylases by D-2-HG. This relationship, along with other IDH-mutant specific metabolic and epigenetic aberrations, is summarized in Figure 4. This is accompanied by decreased expression of monocarboxylate transporters (MCT) 1 and 4, both of which are involved in the efflux of lactate from the cell. Overall, this suggests that IDH1 mutant cells do not perform aerobic glycolysis. Indeed, lactate levels are undetectable in IDH1 mutant neurospheres [99]. Interestingly, the enzyme LDHB, which is involved in converting lactate to pyruvate, and the lactate importer MCT2 are more abundant in IDH1 mutant glioma tissue compared with wildtype samples [100]. This suggests that lactate may serve as an anaplerotic substrate for pyruvate accumulation.
Citric Acid Cycle Rewiring in IDH Mutant Cells
The citric acid cycle is the cornerstone of cellular metabolism [101]. This pathway is directly involved in energy production and supplies metabolic intermediates for the generation of fatty acids, amino acids and nucleotides. Although IDH3 is the only IDH isozyme that directly participates in the citric acid cycle, both IDH1 and IDH2 help regulate levels of cellular αKG and control redox homeostasis. There is strong agreement among various studies that the citric acid cycle is rewired in IDH1 mutant gliomas. Elevated expression of citric acid cycle enzymes upstream of IDH, including citrate synthase and aconitase, has been reported [97]. In addition, there is decreased expression of enzymes downstream of IDH, including succinate dehydrogenase and fumarase. These findings are supported by studies showing that citric acid cycle intermediates downstream of IDH, including αKG, succinate and fumarate, are decreased [102,103].
Anaplerotic reactions serve to replenish citric acid cycle intermediates, maintaining cycle flux [101]. Three common anaplerotic nutrients are glutamate (converted to αKG by glutamate dehydrogenase), glutamine (converted to glutamate by glutaminase) and pyruvate (converted to oxaloacetate by pyruvate carboxylase). As αKG is consumed by mutant IDH, the metabolism of both glutamate and glutamine would be expected to be particularly important. Indeed, both glutamate and glutamine levels are significantly decreased in U87MG cells expressing the IDH1-R132H mutant, compared with wildtype cells [104]. In partial agreement with this finding, patient-derived IDH1-R132H xenografts showed elevated expression of glutamate dehydrogenase and lower levels of glutamate in mouse brains, suggesting that glutamate metabolism is increased in IDH1 mutant tissue [97,105,106]. However, the authors found no significant difference in the expression of glutaminase or cellular glutamine levels. Somewhat surprisingly, another study found no differences in glutamate or glutamine levels between IDH1 wildtype and IDH1 mutant human glioma tumor samples [97]. Thus, it appears that glutamate versus glutamine usage by IDH mutant cells is context-dependent and requires further exploration.
It is worth noting that pyruvate carboxylase expression was elevated in immortalized human astrocytes expressing the IDH1-R132H mutant and in human glioma tissue [104]. This suggests that regardless of whether αKG is primarily derived from glutamate or glutamine, pyruvate carboxylase may take the driver’s seat as the primary anaplerotic reaction in the citric acid cycle.
Mutant IDH Drives Changes In Amino Acid Metabolism
McBrayer et al. discovered that expression of IDH1-R132H in glioma cell lines elicits an upregulation in glutaminase expression [107]. Interestingly, the authors concluded that enhanced glutamine catabolism was being used to primarily support glutamate production, as opposed to αKG. Glutamate levels were decreased in the mutant cell line as a direct result of branched-chain aminotransferase 1 and 2 (BCAT1/2) inhibition by D-2-HG. As expected, the authors found that the mutant IDH1 cells had increased levels of the branched-chain amino acids (BCAA) valine, isoleucine and leucine. Two additional studies support the finding that BCAT1/2 expression and activity is decreased in IDH mutant tumors compared with wildtype tumors [97,108]. However, Tonjes et al. suggested that reduced activity of BCAT1/2 could be attributed to lower levels of αKG due to mutant IDH activity.
In addition to lower levels of BCAAs, IDH1 mutant glioma tissue contains decreased concentrations of the amino acids glycine and serine [95]. Glycine and serine are integral to one-carbon metabolism, suggesting potential impairments in this metabolic network. In one-carbon metabolism, the interconversion of glycine and serine by the enzyme serine hydroxymethyltransferase is coupled with the folate cycle, the methionine cycle and nucleotide biosynthesis [109]. However, an in-depth analysis of these pathways in IDH mutant glioma is lacking.
Consumption of NADPH by the Mutant IDH Enzyme
NADPH is produced in the canonical reaction catalyzed by wildtype IDH, whereas mutant IDH consumes NADPH in the reaction that produces D-2-HG. As expected, U87MG glioma cells expressing the IDH1-R132H mutant and patient-derived xenografts from IDH1 mutant GBM both showed lower cellular NADPH levels and a decreased [NADPH]/[NADP+] ratio [102,103]. Fack et al. found that [NADH]/[NAD+] was not different between IDH1 wildtype and mutant tumors; however, Biedermann et al. detected less NAD+ in the mutant U87MG cells, suggesting that NAD+ could be used to replenish cellular NADPH. As a potential explanation for how redox homeostasis can be restored in IDH mutant cells, Hollinshead et al. found that human anaplastic oligodendroglioma cells expressing the IDH1-R132H mutant had increased expression of the enzyme proline 5-carboxylase reductase 1 (PYCR1), which is involved in proline biosynthesis [110]. The authors found that not only did the IDH1 mutant cell line accumulate proline as a result of increased PYCR1 activity but NADH consumption in this reaction was a means to uncouple the citric acid cycle from NADH usage in oxidative phosphorylation.
The oxidative pentose phosphate pathway is recognized as the major producer of cytosolic NADPH. NADPH is primarily used for fatty acid biosynthesis and to maintain the cellular pool of reduced glutathione for mitigating ROS. Unfortunately, the impact of reduced NADPH levels in IDH mutant glioma cells specifically is largely lacking. In line with studies performed in glioma cells, HCT116 colon cancer cells expressing the IDH1-R132H mutant consume significantly greater quantities of NADPH compared with wildtype cells [111]. This leads to an increase in the cellular demand for NADPH, amplifying flux through the NADPH-producing pentose phosphate pathway. Interestingly, both IDH1-R132H mutants and IDH2-R172K mutants are more susceptible to oxidative insults. Gelman et al. also found that the IDH1 mutation created competition between the production of D-2-HG and the fatty acid palmitate in colon cancer cells. Fibrosarcoma cells expressing another IDH1 mutant (R132C) consume NADPH at the same rate as de novo lipogenesis [112]. These cells have increased reliance on exogenous lipids for growth.
Dysregulation of Membrane Lipid Biosynthesis
The biosynthesis of fatty acids, membrane lipids and cholesterol is perturbed in IDH1 mutant glioma. Three IDH1 mutant glioma models, including human glioma xenografts in mice, cultured glioma cell lines and human glioma biopsies, showed a distinct phospholipid profile characterized by low levels of phosphoethanolamine and heightened levels of phosphatidylcholine [113]. In contrast, another study showed the phosphatidylcholine levels were decreased in U87MG cells expressing the IDH1-R132H mutation [114].
DNA Damage Repair
Various studies have demonstrated that cells with an IDH mutation have heightened sensitivity to DNA damaging agents. This increased sensitivity is partially mediated by the inhibition of ABH enzymes including ALKBH2 and ALKBH3. ALKBH2/3 remove alkyl adducts from nucleobases via oxidative dealkylation [115], and the inhibition of these enzymes by D-2-HG reduces the repair kinetics of IDH mutant cells resulting in the accretion of DNA damage. Since ABH proteins are αKG-Fe(II)-dependent dioxygenases, the accumulation of D-2-HG impedes normal function, resulting in a 73-88% inhibition of normal DNA repair activities [116]. Ratios of D-2-HG:αKG in patients have been observed at approximately 373:1, while D-2-HG concentrations are dependent upon the source, but in tissues can accumulate up to almost 2 mM [117]. For experiments gauging D-2-HG inhibition of DNA repair activities, Chen et al. utilized a 373:1 ratio of D-2-HG:αKG and concentrations of D-2-HG varying from 0-37 mM. Interestingly, like other dioxygenases that rely upon αKG, this inhibition is reversible if the IDH mutation (and consequently the production of D-2-HG) is lost [118]. The presence of excess αKG can also assist in reversing this inhibition [116].
In addition to ALKBH2/3, IDH mutant cells also demonstrate an impaired ataxia-telangiectasia-mutated (ATM) signaling pathway, which is an essential mediator of cellular response to double stranded breaks (DSB) [119]. Upon the occurrence of a DSB, ATM is recruited to the damage site then initiates DNA damage repair (DDR) complexes by phosphorylating various targets including checkpoint kinases and p53 to begin the repair process [120]. Further investigation into the mechanism of the IDH mutant specific downregulation of ATM has demonstrated that the reduced expression of ATM is due to D-2-HG mediated histone methylation of H3-K9 by KDM4 [121]. The use of pharmaceutical inhibitor AGI-5198 with IDH mutant patient-derived cultured glioma cells has demonstrated the downregulation of essential epigenetic reader Zinc Finger MYND-Type Containing 8 (ZMYND8) [122]. ZMYND8 recognizes modifications such as acetylation and methylation, facilitates DNA repair in the presence of DSB, and may act as either a repressor or enhancer of transcription [123]. KDM5A-dependent H3K4me3 demethylation near DSB is required for the colocalization of ZMYND8 and subunits of the nucleosome remodeling (NuRD) and histone deacetylation complex including HDAC1/2 and chromodomain helicase DNA binding protein 3-5 (CHD3/4/5) [124]. Lending to the known sensitivity of IDH mutant cells to poly(ADP) ribose (PARP) inhibitors [125], it is also of note that ZMYND8/NuRD facilitated repair is dependent upon poly(ADP) ribose. To this end, treatment with PARP inhibitors has been shown to abolish ZMYND8 recruitment in cultured cells [124], and the knockdown of ZMYND8 in IDH mutant patient-derived cultured glioma cells resulted in increased sensitivity to radiotherapy as well as significant phosphorylation of ATM and γH2AX activation in response to ionizing radiation [122].
Despite several known mechanisms for increased vulnerability to DNA damage, variable results exist concerning the response of IDH mutant cells to radiation-induced DNA damage. In some cases IDH mutant glioma cells have demonstrated a decreased sensitivity to radiation-induced DNA damage in comparison to IDH wildtype cells [126]. A potential theory for this unexpected finding posited by the authors is that IDH mutant cells must develop buffering mechanisms against high levels of ROS to survive, thus making them better equipped to survive ROS generated from radiation damage. Despite these findings, other groups have shown the opposite: that IDH mutations increase sensitivity to radiation due to delayed DNA repair [127,128]. For example, when treated with TMZ, IDH mutant cells have an IC50 less than half that of IDH wildtype cells [119]. Since the mechanism of TMZ is to disrupt DNA structure via the addition of alkyl groups, this heightened sensitivity suggests weakened DDR response.
Conflicting results regarding the response of IDH mutant cells to DNA damage could potentially be explained by the genetic complexity found within the tumor environment that varies between patients and is often not accounted for with isogenic knockout models. To this end, several mutations that commonly co-occur with IDH mutations [specifically inactivating TP53 [129] and alpha thalassemia/mental retardation syndrome X-linked gene (ATRX) [130]] have been shown to contribute to genomic stability, enhanced DNA repair, and resistance to genotoxic therapies [131]. Thus, although this relationship has not been extensively explored, it is possible that discrepancies in DDR data are due to co-occurring deletions or other complex genetic factors.
Immunological Impact of IDH Mutations in Glioma
The immunological implications of IDH mutations in glioma has recently been extensively covered [132].
Diagnostics
Efforts to identify and classify biomarkers have yielded valuable targets for diagnostics treatment, with IDH mutant cancers being a key example in glioma. Techniques that may elucidate the IDH mutational status prior to surgical resection remains urgent, as overall survival improves with a maximal surgical resection [133]. Still, CNS malignancies pose unique challenges, with the acquisition of tissue remaining at the forefront. While tissue-based assays remain valuable, many cannot function in a manner rapid enough to provide information capable of guiding the extent of surgical resection. However, strategies leveraging D-2-HG as a surrogate biomarker show great promise for facilitating early detection and monitoring of disease state. Additionally, MRI-based techniques have been highly successful for the early detection of IDH mutant gliomas. Here, we discuss some of the molecular diagnostic techniques developed for the detection of IDH mutations.
Sequencing
The development of assays to successfully detect the IDH mutant genotype has been challenging, particularly due to the heterogeneity of gliomas [134,135,136] and heterozygosity of IDH mutations [137], which collectively lend to low copy number within a sample. Currently, the sequencing of DNA serves as one of the most comprehensive and relied upon methods for characterizing the molecular profile of tumors. Sequencing is particularly beneficial as it is adept at identifying a variety of genetic abnormalities including single nucleotide variants (SNV), indels, and structural variants. The detection of SNVs is particularly relevant for identifying IDH mutations, as traditional PCR or other amplification-based methods struggle to identify the single base changes needed to differentiate between the wildtype and mutant genotype. Furthermore, there is evidence that sequencing-based detection is a more reliable indication of IDH mutational status than IHC [138], which is currently considered a gold standard.
Next generation sequencing (NGS) and third generation sequencing platforms are advantageous as they are high throughput and can successfully identify both known and novel transcripts. However, without precursor targeted enrichment, NGS-based systems may struggle with accurately detecting low-level variants and have reported detection limits between 2-15% variant allele frequency (VAF). Using NGS, Vij et al., found the VAF of IDH1-R132H mutations to be 0.8%, a value significantly lower than other clinically relevant mutations in glioma such a ATRX [139]. While highly sensitive methods like real-time PCR (as low as 0.0002%) [140] are appealing alternatives for low abundance targets, they typically lack SNV discrimination and lack the high throughput of sequencing-based applications. On the contrary, sequencing-based applications can provide unbiased results for new or previously known SNVs. Additionally, targeted sequencing with user-defined primer sets to enrich specific genes or regions allow for the detection of VAF as low as 0.1-0.2% to be achieved [141]. When using sequencing for diagnostic applications in cancer, whole genome sequencing can be used; however, it often poses a significant time and cost burden. Thus, panels targeting known biomarkers are often employed dependent upon cancer type. Panels are beneficial for saving time and costs, but also allow for increased sequencing depth [142] and thus, more reliable results. For gliomas, these panels generally target key biomarkers such as IDH1, TP53, telomerase reverse transcriptase (TERT), and ATRX [143,144,145,146,147]. While methods using NGS techniques such as ion-torrent [144,146,147,148] and Illumina [145,149,150] have been developed to detect IDH mutations, third generation techniques such as oxford nanopore technology (ONT) [151,152,153] have recently gained traction as well. These ONT platforms have demonstrated variant detection limits within the general range of those seen in NGS-based systems (3.3% and
DNA sequencing is most commonly used for diagnostic purposes; however, RNA sequencing (RNAseq) assays have also been developed to include IDH mutations [154]. RNAseq offers some advantages over DNA sequencing, such as the direct detection of rare splice variants, accurate measurements of gene expression, and detection of non-coding RNA species [155]. RNAseq also serves as a valuable approach toward transcriptome profiling and can provide single-cell resolution [156] and spatial information [157]. RNAseq has the potential to uncover cell-type specific treatment responses, better understand disease mechanisms, and provide quantitative transcriptome-wide data from tissue sections [157]. Using single-cell RNAseq (scRNAseq) of GBM samples, Couturier et al., generated a hierarchal map to uncover therapeutic targets of progenitor cancer stem cells [156]. Specific to IDH mutant glioma, scRNAseq has been utilized to explore the identity of progenitor cells for astrocytoma vs oligodendroglioma, as well as identify differences in their respective tumor microenvironments. For example, the data of Venteicher et al., suggest a common progenitor cell type for astrocytoma and oligodendroglioma IDH mutant glioma, [158]. While high cost will likely hinder the widespread implementation of RNAseq in a clinical setting in the immediate future, basic research with these techniques may yield valuable targets for novel therapeutics to improve overall survival.
Sequencing is commonly employed with DNA purified from fresh frozen tissues or formalin-fixed paraffin embedded tissues (FFPE) [143,144,145,146,147,148,149]. Additionally, sequencing for the detection of IDH mutations has been successfully performed on cell-free circulating DNA (cfDNA) derived from CSF [159]. Genetic characterization via liquid biopsies has been particularly challenging in gliomas, with low copy number attributed to the blood-brain barrier. Current data supports that the quantity of cfDNA increases with proximity to the tumor [160], with CSF yielding the highest quantity and fluids like blood [161] and urine proving to be more challenging sample types. To this end, amplification-based library preparation methods such as multiplex PCR can be helpful in enriching samples of interest prior to sequencing. Additionally, others have applied Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) based systems toward the targeted enrichment of regions of interest. An example of this is nanopore Cas9 Targeted Sequencing (nCATS), which selectively ligates sequencing adapters to CRISPR-Cas cut sites by dephosphorylating DNA ends before cleavage occurs, then preferentially ligating to the freshly cut/phosphorylated DNA ends at the cleavage site. nCATS has been utilized to simultaneously determine the IDH mutational status and Methyl Guanine Methyl Transferase (MGMT) methylation status in fresh glioma biopsies [162]. CRISPR-based detection of IDH mutations has also been utilized directly with CRISPR-Cas12a [163,164]. CRISPR-Cas12a binds DS DNA with high specificity, which then induces non-specific (collateral) cleavage of single stranded DNA. Various groups have exploited the unique properties of Cas12a by including single stranded DNA probes that emit fluorescence when cleaved [165,166]. Despite the broad applications of CRISPR-based diagnostics, evidence exists to indicate prevalent nonspecific cleavage of DS DNA by Cas12a [167], and some groups have even attempted to generate variants with more stringent recognition [168].
Epigenetic Detection
As the epigenetic effects of IDH mutations ultimately lead to G-CIMP [29], several groups have identified methylome profiling as an alternative method of diagnosis [169,170,171,172,173]. In an analysis of mixed tumor samples, a pairwise similarity heatmap yielded two major clusters- gliomas with and without IDH mutations [174], demonstrating that the characteristic nature of global methylation patterns can be used to differentiate between the IDH wildtype and IDH mutant genome. Epigenetics can also serve as an indication of changes to disease state, as the overall methylation level has been found to decrease during progression [171]. Another benefit to methylation profiling is that it can inclusively recognize oncogenic variants of IDH1/2 and can be further subclassed into tumor type based on epigenetic marks (for example, astrocytoma or oligodendroglioma) [170,175]. Methylation status has been interrogated using Nanopore sequencing [173,176] and methylation profiling arrays [170,171,172,173] and can be performed using snap frozen or FFPE tissue. Nanopore sequencing for the determination of methylation status is particularly advantageous because it does not require a precursor bisulfite treatment, which is associated with DNA degradation [177] and is rapid enough to adapt for intraoperative use [176]. As discussed later, a more aggressive surgical resection for glioma patients with an IDH mutation is associated with greater survival benefit [178,179,180], making simple assays capable of performing intraoperatively valuable tools for clinicians seeking to use this information to guide the extent of resection.
Amplification-Based Detection
Amplification methods for the detection of IDH mutations are frequently adapted to discriminate between SNVs while retaining the sensitivity necessary for detecting relatively low copy number in a high background of non-target nucleic acids. PCR-based amplification is highly characterized, broadly available, and has long been considered a gold standard in clinical practice. The PCR-based detection of IDH mutations has been accomplished with several variations and/or adaptations, including digital droplet PCR (ddPCR) [181] qRT-PCR [182], and multiplex PCR coupled to a SNaPshot assay [183]. Another modification of PCR, Beads, Emulsion, Amplification, Magnetics (BEAMing) RT-PCR has also been adapted for the detection of IDH mutations [184]. BEAMing PCR is an adaptation of emulsion PCR that meets the need for detection of low frequency alleles in a high background of non-target DNA with a sensitivity of as little as 0.01%. This assay has shown promise in detecting IDH1 mutant mRNA in CSF-derived extracellular vesicles. Through the development of BEAMing-PCR, it was found that the copy number of IDH1 mRNA was significantly elevated in patients with an IDH mutation as opposed to IDH wildtype control samples [184], suggesting that a greater focus on mRNA-based detection could be beneficial.
Relatedly, ddPCR has demonstrated exceptional sensitivity compared to other PCR variations such as qRT-PCR [182,185], and has been employed for the detection of cfDNA in CSF [186] and blood [187,188]. ddPCR is particularly advantageous for the detection of IDH mutations due to the water-oil emulsion partitions that allow for high sensitivity in a high background of non-target DNA. A TaqMan-based allele specific qPCR has also been employed to detect IDH mutations in FFPEs and blood [189], further contributing to the possibility of liquid biopsies for patients to infer the IDH mutational status prior to surgical resection. Due to the similar survival benefit imparted by IDH driver mutations, inclusivity for all known variants with PCR-based assays would be ideal but has proven difficult. Consequently, IDH1-R132H has historically been the primary focus as it is the most encountered mutation. Recently, a cartridge-based RT-PCR assay kit (Idylla) has been developed which qualitatively detects five of the most common codon changes for IDH1 (R132H/C/G/S/L) and nine codon changes in IDH2 (R140Q/L/G/W and R172K/M/G/S/W). The Idylla assay functions with an input of FFPE, has 97% agreement with sequencing results, and requires limited hands-on time for laboratory technicians [190]. With the success of Vorasidenib on less common variants such as IDH1-R132C, it has become increasingly urgent for assays to be inclusive and easily implemented.
In addition to PCR, isothermal amplification techniques have also been developed for the identification and characterization of IDH mutations. Loop-Mediated Isothermal Amplification (LAMP) is among the most popular isothermal amplification techniques due to its ability to amplify DNA or RNA in crude cell lysates, eliminating the need for a nucleic acid extraction step. Additionally, LAMP can facilitate the simple and visual interpretation of data through turbidity or colorimetric means. LAMP coupled with a peptide nucleic acid (PNA) clamping probe has been shown to facilitate the detection of IDH1-R132H in tumor samples within approximately 1 hour [191]. Additionally, a LAMP-based genotyping panel for IDH1 that uses primers modified with Locked Nucleic Acids (LNA) to mediate SNV specificity has been shown to successfully identify the specific IDH1 variant present (R132H/L/C/G/S) within 35 minutes and can be easily interpreted through absorbance or visual interpretation of colorimetric changes [192]. LAMP is particularly advantageous for the detection of IDH mutations because of its ability to rapidly amplify DNA in crude cell lysates, making it a viable candidate for intraoperative use.
Histological Detection
IHC using Hematoxylin and Eosin (H&E) stained tissue sections treated with monoclonal antibodies targeting IDH1-R132H is commonly employed for the diagnosis of IDH1 mutations in clinical practice. Samples are typically FFPE or frozen sections; however, frozen sections have been found to yield false positives in some cases [193]. While H09 is the most widely used antibody, others exist [194,195,196], including a more recently developed antibody MRQ-67 which promises comparable sensitivity and specificity but less background [197,198]. Although IDH1 mutations in glioma are most commonly the IDH1-R132H variant, alternative IDH1-R132 SNVs, including R132S, R132L, R132G, and R132C have been observed in the molecular analysis of primary tumor samples [20,199] (Table 1). While available monoclonal antibodies specifically target IDH1-R132H, cross reactivity has been observed for other IDH SNVs [200]. For example, MsMab-1 recognizes IDH1-R132H/S/G and IDH2-R132S/G, while MsMab-2 recognizes IDH1-R132L and IDH2-R172M [196]. This cross reactivity is not necessarily detrimental, as oncogenic variants of IDH1/2 mutant are often treated in a similar manner; however, an antibody that is cross-reactive with all oncogenic IDH1/2 variants without exhibiting a positive result for the wildtype would be ideal. IHC is a particularly convenient method of diagnosis for IDH mutations from a clinical perspective as the antibody can be applied and assessed alongside routine histological analysis often utilized to gain information about nuclear atypia, mitotic activity, microvascular proliferation, and necrosis within the tumor [201]. More recently, artificial intelligence-based applications such as machine learning and deep learning have advanced the accuracy of histological analysis. To this end, these applications have been highly successful in differentiating between IDH wildtype and IDH mutant gliomas using digitalized whole slide images [202,203,204,205,206].
D-2-HG as a Surrogate Marker
D-2-HG is generated by hydroxyacid-oxoacid transhydrogenase and/or 3-phosphoglycerate dehydrogenase through side reactions with low catalytic efficiency [208] or as part of an anti-inflammatory response [209]. D-2-HG is subsequently broken down by D-2-HG dehydrogenase [210], maintaining relatively low levels in healthy individuals. Elevated levels of D-2-HG in body fluids can be attributed to the metabolic disorder D-2-Hydroxyglutaric aciduria [211] or oncogenic mutations to IDH1/2. To this end, elevated levels of D-2-HG may be utilized as a surrogate marker as an alternative to the genetic or histological elucidation of IDH mutational status.
D-2-HG can accumulate in IDH mutant tumors at a median concentration of 1,965.8 µM [117] and at elevated levels in patient CSF [212,213,214] (up to 109.0 µM), and blood [212,215,216,217] (up to 10.9 µM). Data obtained from currently available studies show a clear and positive correlation between D-2-HG elevation in patients with IDH mutants in comparison to their IDH wildtype counterparts; however, the ideal sample type has been a topic of some debate. The ability to quantify D-2-HG in body fluids as a surrogate marker of IDH mutational status could revolutionize the current standard of care and allow for preoperative, intraoperative, and/or postoperative characterization. Harnessing D-2-HG for diagnostics could also facilitate remote testing for patients, as it is highly stable following exposure to excess heat [218] or several freeze-thaw cycles [213]. A current limitation of this approach is the availability of a simple, rapid, and user-friendly method for quantification. At this time, D-2-HG can be detected by liquid chromatography-mass spectroscopy (LC-MS) [212,214], gas chromatography-mass spectroscopy (GC-MS )[219] or magnetic resonance spectroscopy (MRS) [220]. Fluorometric and colorimetric kits are also commercially available for this purpose [221], but hands-on time and price are likely limiting factors for clinical implementation. A fluorescent resonance energy transfer (FRET) based biosensor for the detection of D-2-HG has been developed [222] but is limited by its dynamic range and use at a physiologically impossible pH of 10. A more recent FRET-based biosensor has been validated using glioma tumor samples and contrived clinical specimens, demonstrating quantification of D-2-HG within a clinically relevant range (~300 nM-100 µM) at physiological pH [223]. FRET-based sensors are promising diagnostic tools; however, they have not yet been evaluated in a clinical setting.
While highly useful for diagnostic purposes, the use of D-2-HG as a surrogate marker can also provide valuable insight towards the effectiveness of pharmaceutical mutant IDH inhibitors such as Ivosidenib [224,225]. This utility becomes an even greater point of interest with the outstanding efficacy of Vorasidenib [10], which is expected to receive a fast track for approval by the Food and Drug Administration (FDA). A convenient, accurate, and highly characterized tool for the quantification of D-2-HG could also provide novel insights toward the relationship between D-2-HG with IDH mutant cancers such as the correlation between disease burden and D-2-HG level. Additionally, the use of D-2-HG as a surrogate marker would offer significant benefits for earlier implementation of treatment as it could alert clinicians of an IDH mutation without the need for a tissue biopsy. Another benefit to a liquid biopsy approach is that it would be expected to indirectly identify the presence of all oncogenic IDH1/2 mutations due to the conserved accumulation of D-2-HG across variants [226]; however, data in this respect is scarce due to the rarity of non IDH1-R132H variants in glioma and relatively small sample sizes of currently available studies. Future studies to validate the consistency of D-2-HG as a surrogate marker of IDH variants would be highly desirable in determining its value as a comprehensive indicator of disease. Moreover, surveying the levels of D-2-HG in a large cohort of IDH mutant glioma patients in various body fluids preoperatively, postoperatively, and through the process of treatment is needed to determine correlations, if any, with D-2-HG levels and disease burden.
In addition to liquid biopsy approaches, MRS has also shown great promise for the non-invasive detection of 2-HG, which we discuss below.
MRI
Conventional MRI is an important standard of care for gliomas, and radiologic features such as contrast enhancement and multifocality can be useful in determining the molecular characteristics of a tumor [227]. Contrast enhancement is utilized to visualize the vascularity of a structure through the administration of a contrast agent, while multifocality is the presence of multiple distinct lesions. Imaging characteristics including a higher percentage of non-contrast enhancing tumor, larger tumor size, presence of cysts, and presence of satellites has been shown to predict the presence of an IDH mutation with 97.5% accuracy [228]. Furthermore, IDH mutant tumors typically grow in a single lobe, with the most common being the frontal [229] or temporal lobe, whereas IDH wildtype tumors are frequently distributed between lobes [230]. Overall, the use of MRI for the elucidation of IDH mutational status is beneficial due to its non-invasive nature, allowing for earlier diagnosis than methods such as sequencing or IHC which require a tissue biopsy. T2-weighted imaging is one of the most common contrast sequences in MRI, and T2-weighted Fluid-attenuated inversion recovery (T2-FLAIR) is an advantageous approach because it can be performed using only standard MRI sequences to differentiate between IDH wildtype and IDH mutant astrocytomas. T2-FLAIR MRI sequences enhance the contrast between gray matter and white matter to improve the visibility of lesions [231], and the use of T2-FLAIR mismatch for detecting IDH mutant gliomas is based on T2 complete (or mostly complete) homogeneous hyperintense signal and attenuation of FLAIR signal intensity with a bright peripheral rim [232,233]. Additionally, the non-contrast enhancing properties of IDH mutant gliomas make the T2-FLAIR mismatch sign a useful indication of IDH mutational status [234].
Perfusion weighted MRI (PWI) has recently gained attention for facilitating a more accurate determination of tumor grade in comparison to conventional MRI alone [235]. PWI provides information about tissue vascularization and angiogenesis [236], and the relative cerebral blood volume (rCBV). rCBV is a value that can be calculated from PWIs by determining the volume of blood in a specific quantity of brain tissue. rCBV values can serve as a powerful indication of IDH wildtype vs IDH mutant gliomas, as recent studies have found these values to be 2-2.5 higher in IDH wildtype glioma samples than their IDH mutant counterparts [237]. PWI has also been combined with dynamic susceptibility contrast-enhanced MRI (DSC) [229]. DSC utilizes signal loss induced by paramagnetic contrast agents like gadolinium-based compounds on T2-weighted images to determine additional parameters such as relative cerebral blood flow (rCBF), and mean transit time (MTT). These parameters are subsequently used to assess regional perfusion and have been used in combination with PWI to determine the apparent diffusion coefficient (ADC). To this end, the minimum/relative ADC [229] and mean ADC [238] have been found to be significantly elevated in IDH mutant astrocytoma (grade II and III) compared to IDH wildtype tumors. These findings are supported with the use of diffusion tensor imaging (DTI) to determine the ADC of gliomas, which found that fractional anisotropy and ADC from DTI can successfully determine the IDH1 mutational status in gliomas [239]. Diffusion weighted MRI (DWI) techniques allow for the observation of the cellular architecture of tumors and surrounding tissue [240] by assessing the Brownian motion of water molecules and have shown promise in differentiating between IDH wildtype and IDH mutant gliomas [239,241]. DWI and PWI have also been used to assess the response of IDH1 mutant gliomas to pharmaceutical IDH mutant inhibitors, where an increase in the normalized rCBV and ADC were found to be a useful indicator of antitumor response within a timeframe of 2-4 months [242]. Recently, artificial intelligence applications such as machine learning and deep learning have become valuable tools for highly accurate differentiation between IDH wildtype and IDH mutant gliomas in MRI-based applications [243,244,245,246,247,248]. Carosi et al., have also extensively covered MRI-based techniques for the detection of IDH mutant gliomas and other solid tumors [249].
MRS
Due to the unique accumulation of D-2-HG, IDH mutations may also be detected with the use of MRS through the measurement of total 2-HG [250,251,252,253,254,255]. With an in vivo sensitivity of approximately 1 mM and an ex vivo sensitivity for intact biopsies of 0.1-0.01 mM [256], MRS offers localized 2-HG quantification directly in the lesion and is well poised to differentiate between IDH wildtype and IDH mutant gliomas. D-2-HG is known to accumulate in IDH mutant gliomas at a median concentration of 1,965.8 µM, a value heavily contrasted by IDH wildtype gliomas which yield a median value of 14.0 µM [117]. 2-HG MRS has demonstrated impressive sensitivity and specificity that outperform conventional MRI as well as DWI and PWI [251]. Additionally, as D-2-HG is known to deplete in response to treatment with pharmaceutical inhibitors such as Ivosidenib [224,225] or Vorasidenib [10], MRS is currently being explored for its ability to gauge the effectiveness of mutant IDH inhibitors [257]. While MRS is both highly sensitive and specific, this method may be limited by a low signal-to-noise ratio which requires lesions to be at least several milliliters in volume and sufficiently distant from fluid-brain or air-fluid interfaces. Furthermore, MRS cannot discriminate between D-2-HG and its naturally occurring enantiomer, L-2-HG. L-2-HG is not a useful indication of disease state; thus, enantiomer discrimination would add a greater level of confidence to MRS-based applications. However, analysis of total 2-HG within glioma tissues has shown a median value of 1,971.5 µM for IDH mutant, compared to a median value of 27.0 µM for IDH wildtype [117]. These results still provide a clear indication of disease state, however including larger patient populations in these types of studies are necessary to determine how much variability exists in enantiomer ratios and 2-HG concentrations.
Clinical Implications of IDH Mutations
Clinical Classification of Gliomas
Despite advances in diagnosis and treatment of glioma, the prognosis for the disease is poor [258]. To aid in curbing the lack of improvement in life expectancy, the medical community has increased reliance on molecular analysis of tumors. In addition to the traditional histological and immunohistological methodologies used to characterize brain tumors, the 2021 WHO CNS5 integrated molecular diagnostics for the further classification of tumors [259]. As a result, there are six new families in the classification of gliomas to include: (1) Adult-type diffuse gliomas, (2) Pediatric-type diffuse low-grade gliomas, (3) Pediatric-type diffuse high-grade gliomas, (4) Circumscribed astrocytic gliomas, (5) Glioneuronal and neuronal, and (6) Ependymomas. The most common family of tumors, adult type diffuse gliomas, includes GBM and mutant IDH gliomas [260]. With this molecular classification, IDH mutant gliomas have been identified as a biologically distinct group of tumors. In cases of adult-type diffuse glioma, the most significant molecular prognostic indicator is IDH mutational status, where IDH mutant gliomas are less aggressive than their wildtype counterparts [261]. Molecular diagnosis is imperative as IDH1/2 mutations are associated with a prolonged survival benefit of approximately 4-fold when molecular identification is combined with surgical resection [262,263]. Identification of IDH status stratifies adult-type diffuse gliomas into separate classifications where GBM is exclusively characterized as IDH wildtype. Further molecular classification for wildtype GBM includes identification of one or more of the following: mutations found in the TERT promoter, amplification of epidermal growth factor receptor (EGFR) and chromosome 7 gain (partial or complete) / chromosome 10 loss. [264,265,266,267]
Separate from IDH wildtype GBM, mutant IDH gliomas are molecularly categorized into oligodendroglioma or astrocytoma. The hallmark of oligodendroglioma includes the 1p/19q codeletion, whereas ATRX and p53 mutations differentiate mutant IDH gliomas into astrocytomas. Among gliomas with ATRX loss, 89% retained IDH1/2 mutations while ATRX retention in IDH1/2 mutants was strongly associated with 1p/19q loss, making this a differentiating feature associated with oligodendroglioma [268]. Further molecular characterization of the mutant IDH astrocytoma include detection of the presence or absence of the CDKN2A/B gene. Homozygous deletion of the CDKN2A/B gene characterizes the tumor as grade 4. Grading of mutant IDH astrocytomas retaining the CDKN2A/B gene relies on histological analysis to be differentiated into grade 2 and 3 mutant IDH astrocytoma. It is worth noting that astrocytomas can also be categorized as wildtype IDH but also retain one or more of the TERT promoter mutations, EGFR amplification and/or chromosome 7/10 aneuploidy. The classification of gliomas has also been extensively covered in a recent review by Weller et al. [269].
Influence of Mutational Status on the Production of D-2-HG
Mutant IDH astrocytoma has an incidence rate of 0.44 per 100,000 individuals with approximately 3,000 cases identified in the United States, making up 11% of all diffuse gliomas [260]. From a clinical perspective, IDH mutant gliomas have a significant survival advantage over wildtype gliomas. As identified earlier in this review, IDH1/2 mutations lead to the accumulation of D-2-HG which has pleiotropic oncogenic effects that result in prolonged life expectancy and delayed therapeutic interventions. The overwhelming majority of this data is based on the IDH1-R132H mutation. However, very little is known regarding the cellular accumulation of D-2-HG for the less common mutations of IDH1 such as R132C/G/S/L. A recent publication by Pusch et. al. performed a study evaluating the enzymatic activity of recombinant IDH1 variants on isocitrate/αKG substrate and found that the prevalence rate of IDH1-R132X variants (Table 1) found in patients is inversely proportional to their respective affinities suggesting that selective pressure (ie. D-2-HG toxicity) favor the common IDH1-R132H variant [270]. Based on these data, a favorable clinical outcome would be seen due to increased production of the oncometabolite, D-2-HG. Natsumeda et. al. demonstrated that elevated 2-HG had a better overall survival than lower 2-HG [271]. However, this analysis was performed using MRS in the evaluation of total 2-HG. As seen in multiple studies, IDH1 mutant clinical patient outcomes are variable [261,272,273,274,275]. Of significance to the prognostic implications of IDH mutants is the presence of sub-clonal populations with mosaic expression [276,277,278,279,280,281,282,283], confounding interpretation of the role of D-2-HG production. As a result, quantitative analysis of sub clonal populations may provide more clarity for the treatment of IDH1 mutant gliomas.
Pharmaceutical Treatment of IDH Mutant Gliomas
Mutant IDH gliomas and subsequent D-2-HG production provide a highly druggable target due to its role in glioma formation and progression [284]. Early IDH inhibition is especially important, as tolerable drugs could potentially delay the long-term neurocognitive toxicities of standard treatment which has been identified as detrimental factor in employment and quality of life [285,286]. Mutant IDH inhibitors have been investigated in glioma for approximately ten years [287,288]. Two inhibitors, Ivosidenib and Enasidenib, have been approved by the FDA for the treatment of IDH-mutant leukemia. The two most studied drugs in glioma are Ivosidenib (AG-120), a mutant IDH1 inhibitor, and Vorasidenib (AG-881) an IDH1/2 inhibitor [10]. Ivosidenib is a specific, reversible, allosteric competitive inhibitor of mutant IDH1, and has shown clinical utility in treating IDH1-mutant gliomas [10]. Vorasidenib, a pan-IDH1/IDH2 inhibitor, displays CNS penetration and successfully demonstrated itself as a potential treatment for IDH-mutant glioma pending FDA approval [289]. The recent phase 3 INDIGO trial evaluating Vorasidenib demonstrated significantly prolonged disease stability as well as the ability to delay additional standard therapeutic interventions [10]. More recently, a a dual-inhibitor of both nicotinamide phosphoribosyl transferase (NAMPT) and mutant IDH1 has been developed which has the ability to cross the BBB and demonstrates potent efficacy in vivo [290]. Carosi et al., have recently provided an in-depth analysis of pharmaceuticals targeting IDH-mutant gliomas and other solid tumors [249].
In addition to pharmaceuticals that directly inhibit the mutant IDH protein, inhibitors that interact with proteins associated with epigenetic marks also make excellent targets. Specifically focusing on the acetylation and methylation state of chromatin, HDACs and Jumonji class demethylases are chromatin erasers that are of clinical interest. In IDH mutant glioma, the downstream effect of D-2-HG production inhibits αKG-dependent DNA demethylases rendering chromatin hypermethylated. HDACs are enriched in hypermethylated regions of chromatin, potentially making mutant IDH cells more susceptible to HDAC therapy. The HDAC inhibitor Panobinostat (Farydak) was an FDA-approved drug for the treatment of multiple myeloma that had been recognized as a potential IDH mutant inhibitor for gliomas [291]. Following treatment with Panobinostat, IDH1 mutant glioma cells demonstrated increased cytotoxicity and inhibited proliferation [45]. Panobinostat was also found to compound to inhibit growth in IDH1 mutant glioma lines [46]. Panibostat was recently withdrawn from USA markets due to incomplete post-approval studies and inability to confirm the clinical benefits within the given constraints [292]. Of significance to Jumonji class demethylases, KDM5 has been shown to be a target of 2-HG production resulting in inhibition of lysine demethylase activity and contributes to cellular transformation in mutant IDH glioma [293]. Nie et. al. has described a family of pyrazolyl pyridines that have demonstrated potent activity targeting KDM5A/B resulting in an increase in H3K4me3 epigenetic marks in cancer cell lines [294].
Molecular Basis for the Improved Prognosis of IDH Mutant Gliomas
The survival advantage for gliomas retaining IDH mutations is poorly understood; however, potential mechanisms for this benefit are slowly being elucidated. IDH mutant cells demonstrate a greater degree of hypermethylation in undifferentiated neural progenitor cells than in mature astrocytes, suggesting that the epigenetic modifications within the IDH mutant genome are dependent upon cellular context [90]. Pursuant to this point, a recent publication identified that D-2-HG reduces glioma cell growth by inhibiting the m6A epi transcriptome regulator, FTO [81]. The aforementioned enzyme is responsible for m6A hypermethylation for a specific set of mRNA transcripts to include ATF5 (Activating Transcription Factor 5) leading to increased cellular apoptosis. Interestingly, inhibition of FTO led to the growth characteristics of wildtype IDH gliomas to be more consistent with IDH mutant growth phenotype. Within IDH mutant astrocytomas, global DNA methylation status and CDKN2A homozygous deletion were found to be significant prognostic indicators [295]. Non-canonical mutations also play a role as a prognostic tool for gliomas. Interestingly, non-canonical IDH1-R132 mutations have an improved prognostic outcome compared to the canonical IDH1-R132H mutation in gliomas [296]. This lends support for the development of diagnostic tools capable of readily differentiating the mutational status in patients.
Clinical Trials
Conclusions
IDH is the most common metabolic mutation associated with oncogenesis, and the production of D-2-HG yields a unique cancer phenotype to include a characteristic epigenetic profile. IDH serves as a critical biomarker in hematological malignancies as well as solid tumors such as glioma, chondrosarcoma, and cholangiosarcoma. While we focus solely on gliomas in this review, Carosi et al. recently covered the role of IDH mutations shared by or unique to these solid tumors. The epigenetic, immunological, and metabolic characteristics are highlighted, as well as selected diagnostics and pharmaceuticals [249]. In this review, we expand upon this work to provide a highly detailed analysis of the molecular mechanism and consequences of IDH mutant gliomas with a special focus on the role of metabolism. D-2-HG is essential for the development and maintenance of IDH mutant glioma. Its inhibition of αKG-dependent dioxygenases yields a unique epigenetic profile/transcriptome reliant upon its continued production and accumulation. We provide an extensive review of current data highlighting the specific role of D-2-HG in various pathways, including m6A modification, DNA damage repair, metabolic rewiring, and epigenetics. In addition to the molecular mechanism of IDH mutations in glioma, we also extensively cover the past, present, and future directions of diagnostics that have been developed for the detection of IDH mutant gliomas. With the recent success of Vorasidenib [10], innovative methods capable of early detection will be increasingly imperative for improving patient prognosis.
Author Contributions
All authors contributed to the writing, revision, and editing of this manuscript.
Conflicts of interest
The authors declare that no conflicts of interest.
Abbreviation
| Abbreviation | Definition |
| 5-mC | 5-methylcytosine |
| ABH | AlkB homologs |
| ADC | Apparent diffusion coefficient |
| αKG | α-ketoglutarate |
| AML | Acute myeloid leukemia |
| ATF | Activating transcription factor |
| ATM | Ataxia-telangiectasia-mutated |
| ATRX | Alpha thalassemia/mental retardation syndrome X-linked gene |
| BBB | Blood-brain barrier |
| BCAA | Branched-chain amino acids |
| BCAT (1/2) | Branched-chain aminotransferase (1 or 2) |
| BEAMing | Beads, emulsion, amplification, magnetics |
| CPE | Carboxypeptidase |
| CDC20 | Cell division cycle 20 |
| cfDNA | Cell-free circulating DNA |
| CHD (3-5) | Chromodomain helicase DNA binding protein (3-5) |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CSF | Cerebral spinal fluid |
| D-2-HG | D-2-Hydroxyglutarate |
| ddPCR | Digital droplet PCR |
| DDR | DNA damage repair |
| DBC1 | Deleted in breast cancer 1 |
| DSB | Double-stranded break |
| DSC | Dynamic susceptibility contrast-enhanced MRI |
| DTI | Diffusion tensor imaging |
| DWI | Diffusion weighted MRI |
| EN2 | Engrailed 2 |
| EGFR | Epidermal growth factor receptor |
| FDA | Food and drug administration |
| FFPE | Formalin-fixed paraffin embedded tissue |
| FRET | Fluorescence resonance energy transfer |
| FTO | Fat mass and obesity-associated protein |
| G-CIMP | Glioma CpG island methylator phenotype |
| GBM | Glioblastoma multiforme |
| GC-MS | Gas chromatography mass-spectroscopy |
| H&E | Hematoxylin and Eosin |
| HDAC | Histone deacetylase |
| HIF | Hypoxia inducible factor |
| IDH | Isocitrate dehydrogenase |
| JmjC | Jumonji-C |
| KDM | Histone lysine demethylase |
| L-2-HG | L-2-Hydroxyglutarate |
| LAMP | Loop-mediated isothermal amplification |
| LC-MS | Liquid chromatography mass-spectroscopy |
| LDH (A/B) | Lactate dehydrogenase A or B |
| LNA | Locked nucleic acid |
| m6A | N6-methyladenosine |
| MAPK | Mitogen-activated protein kinase |
| MCT | Monocarboxylate transporters |
| MGMT | Methyl guanine methyl transferase |
| MRI | Magnetic resonance imaging |
| MRS | Magnetic resonance spectroscopy |
| MTT | Mean transit time |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| nCATS | Nanopore Cas9 targeted sequencing |
| NGS | Next generation sequencing |
| NuRD | Nucleosome remodeling |
| ONT | Oxford nanopore technology |
| PARP | Poly(ADP) ribose |
| PDB | Protein data bank |
| PFKP | Phosphofructokinase platelet |
| PHD | Prolyl hydroxylase |
| PNA | Peptide nucleic acid |
| PWI | Perfusion weighted MRI |
| PYCR1 | Proline 5-carboxylase reductase 1 |
| qRT-PCR | Quantitative real-time PCR |
| rCBF | Relative cerebral blood flow |
| rCBV | Relative cerebral blood volume |
| RNAseq | RNA sequencing |
| SNV | Single nucleotide variant |
| ssRNAseq | Single cell RNA sequencing |
| STAT | Signal transducer and activator of transcription |
| T2-FLAIR | T2 fluid-attenuated inversion recovery |
| TERT | Telomerase reverse transcriptase |
| TET | Ten-eleven translocation |
| TMZ | Temozolomide |
| VAF | Variant allele frequency |
| VIM | Vimentin |
| WHO CNS5 | World Health Organization central nervous system 5 |
| WASF3 | Wiskott-Aldrich syndrome protein family |
| ZMYND8 | Zinc finger MYND-type containing 8 |
References
- Ijzerman-Korevaar, M.; Snijders, T.J.; de Graeff, A.; Teunissen, S.C.C.M.; de Vos, F.Y.F. Prevalence of symptoms in glioma patients throughout the disease trajectory: a systematic review. J. Neuro-Oncology 2018, 140, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Weller, Michael, Wolfgang Wick, Ken Aldape, Michael Brada, Mitchell Berger, Stefan M. Pfister, Ryo Nishikawa, Mark Rosenthal, Patrick Y. Wen, Roger Stupp, and Guido Reifenberger. "Glioma." Nature Reviews Disease Primers 1, no. 1 (2015): 15017.
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Boisselier, B.; Marie, Y.; Labussière, M.; Ciccarino, P.; Desestret, V.; Wang, X.; Capelle, L.; Delattre, J.-Y.; Sanson, M. COLD PCR HRM: a highly sensitive detection method for IDH1 mutations. Hum. Mutat. 2010, 31, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Takano, Shingo, Wei Tian, Masahide Matsuda, Tetsuya Yamamoto, Eiichi Ishikawa, Mika Kato Kaneko, Kentaro Yamazaki, Yukinari Kato, and Akira Matsumura. "Detection of Idh1 Mutation in Human Gliomas: Comparison of Immunohistochemistry and Sequencing." Brain Tumor Pathology 28, no. 2 (2011): 115-23.
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Bent, M.J.v.D.; Blumenthal, D.T.; Touat, M.; Peters, K.B.; Clarke, J.; Mendez, J.; Yust-Katz, S.; Welsh, L.; Mason, W.P.; et al. Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma. New Engl. J. Med. 2023, 389, 589–601. [Google Scholar] [CrossRef]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O'Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Borger, D. R., L. Goyal, T. Yau, R. T. Poon, M. Ancukiewicz, V. Deshpande, D. C. Christiani, H. M. Liebman, H. Yang, H. Kim, K. Yen, J. E. Faris, A. J. Iafrate, E. L. Kwak, J. W. Clark, J. N. Allen, L. S. Blaszkowsky, J. E. Murphy, S. K. Saha, T. S. Hong, J. Y. Wo, C. R. Ferrone, K. K. Tanabe, N. Bardeesy, K. S. Straley, S. Agresta, D. P. Schenkein, L. W. Ellisen, D. P. Ryan, and A. X. Zhu. "Circulating Oncometabolite 2-Hydroxyglutarate Is a Potential Surrogate Biomarker in Patients with Isocitrate Dehydrogenase-Mutant Intrahepatic Cholangiocarcinoma." Clin Cancer Res 20, no. 7 (2014): 1884-90.
- Fathi, A.T.; Sadrzadeh, H.; Comander, A.H.; Higgins, M.J.; Bardia, A.; Perry, A.; Burke, M.; Silver, R.; Matulis, C.R.; Straley, K.S.; et al. Isocitrate Dehydrogenase 1 (IDH1) Mutation in Breast Adenocarcinoma Is Associated With Elevated Levels of Serum and Urine 2-Hydroxyglutarate. Oncol. 2014, 19, 602–607. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 462, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, V.; Ciotti, G.; Gottardi, M.; Ciccarese, F. 2-Hydroxyglutarate in Acute Myeloid Leukemia: A Journey from Pathogenesis to Therapies. Biomedicines 2022, 10, 1359. [Google Scholar] [CrossRef]
- Malta, T.M.; de Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro-Oncology 2017, 20, 608–620. [Google Scholar] [CrossRef]
- Cui, D., J. Ren, J. Shi, L. Feng, K. Wang, T. Zeng, Y. Jin, and L. Gao. "R132h Mutation in Idh1 Gene Reduces Proliferation, Cell Survival and Invasion of Human Glioma by Downregulating Wnt/β-Catenin Signaling." Int J Biochem Cell Biol 73 (2016): 72-81.
- Wang, H.-Y.; Tang, K.; Liang, T.-Y.; Zhang, W.-Z.; Li, J.-Y.; Wang, W.; Hu, H.-M.; Li, M.-Y.; Wang, H.-Q.; He, X.-Z.; et al. The comparison of clinical and biological characteristics between IDH1 and IDH2 mutations in gliomas. J. Exp. Clin. Cancer Res. 2016, 35, 1–9. [Google Scholar] [CrossRef]
- Liu, S.; Cadoux-Hudson, T.; Schofield, C.J. Isocitrate dehydrogenase variants in cancer — Cellular consequences and therapeutic opportunities. Curr. Opin. Chem. Biol. 2020, 57, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Flanagan, S.; Li, C.C.; Lee, M.; Shivalingham, B.; Maleki, S.; Wheeler, H.R.; E Buckland, M. Expanding the spectrum of IDH1 mutations in gliomas. Mod. Pathol. 2013, 26, 619–625. [Google Scholar] [CrossRef]
- Haider, A.S.; Ene, C.I.; Palmisciano, P.; Haider, M.; Rao, G.; Ballester, L.Y.; Fuller, G.N. Concurrent IDH1 and IDH2 mutations in glioblastoma: A case report. Front. Oncol. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1andIDH2Mutations in Tumorigenesis: Mechanistic Insights and Clinical Perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef]
- Dow, Jonathan, and Peter M. Glazer. "Chapter 11 - Oncometabolites, Epigenetic Marks, and DNA Repair." In Epigenetics and DNA Damage, edited by Miriam Galvonas Jasiulionis, 191-202: Academic Press, 2022.
- Myllyharju, J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 2003, 22, 15–24. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Larson, P.E.; Woods, S.M.; Cai, L.; Eriksson, P.; Robinson, A.E.; Lupo, J.M.; Vigneron, D.B.; Nelson, S.J.; Pieper, R.O.; et al. Hyperpolarized [1-13C] Glutamate: A Metabolic Imaging Biomarker of IDH1 Mutational Status in Glioma. Cancer Res. 2014, 74, 4247–4257. [Google Scholar] [CrossRef] [PubMed]
- Kickingereder, P.; Sahm, F.; Radbruch, A.; Wick, W.; Heiland, S.; von Deimling, A.; Bendszus, M.; Wiestler, B. IDH mutation status is associated with a distinct hypoxia/angiogenesis transcriptome signature which is non-invasively predictable with rCBV imaging in human glioma. Sci. Rep. 2015, 5, 16238. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.-J.; Liu, Y.; Lang, F.; Yang, C. D-2-Hydroxyglutarate in Glioma Biology. Cells 2021, 10, 2345. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Sarah, L.; Fujimori, D.G. Recent developments in catalysis and inhibition of the Jumonji histone demethylases. Curr. Opin. Struct. Biol. 2023, 83, 102707–102707. [Google Scholar] [CrossRef] [PubMed]
- Manni, W.; Jianxin, X.; Weiqi, H.; Siyuan, C.; Huashan, S. JMJD family proteins in cancer and inflammation. Signal Transduct. Target. Ther. 2022, 7, 1–22. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, G.W.; Yoo, J.; Lee, S.W.; Jeon, Y.H.; Kim, S.Y.; Kang, H.G.; Kim, D.-H.; Chun, K.-H.; Choi, J.; et al. Histone demethylase KDM4C controls tumorigenesis of glioblastoma by epigenetically regulating p53 and c-Myc. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-J.; Zhu, M.-H.; Lu, X.-J.; Liu, Y.-J.; Lu, J.-F.; Leung, C.-H.; Ma, D.-L.; Chen, J. The emerging role of KDM5A in human cancer. J. Hematol. Oncol. 2021, 14, 1–18. [Google Scholar] [CrossRef]
- Losman, J.-A.; Koivunen, P.; Kaelin, W.G. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 2020, 20, 710–726. [Google Scholar] [CrossRef]
- Zhang, Lan, Yao Chen, Zhijia Li, Congcong Lin, Tongtong Zhang, and Guan Wang. "Development of Jmjc-Domain-Containing Histone Demethylase (Kdm2-7) Inhibitors for Cancer Therapy." Drug Discovery Today 28, no. 5 (2023): 103519.
- Gunn, K., M. Myllykoski, J. Z. Cao, M. Ahmed, B. Huang, B. Rouaisnel, B. H. Diplas, M. M. Levitt, R. Looper, J. G. Doench, K. L. Ligon, H. I. Kornblum, S. K. McBrayer, H. Yan, C. Duy, L. A. Godley, P. Koivunen, and J. A. Losman. "(R)-2-Hydroxyglutarate Inhibits Kdm5 Histone Lysine Demethylases to Drive Transformation in Idh-Mutant Cancers." Cancer Discov 13, no. 6 (2023): 1478-97.
- Guerra-Calderas, L.; González-Barrios, R.; Herrera, L.A.; de León, D.C.; Soto-Reyes, E. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2015, 208, 215–224. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, H.; Zhao, E.; Cui, H. The Diverse Roles of Histone Demethylase KDM4B in Normal and Cancer Development and Progression. Front. Cell Dev. Biol. 2022, 9, 790129. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Tong, K.I.; Goto, K.; Tomida, S.; Komuro, A.; Wang, Z.; Nishio, K.; Okada, H. The H3K27 demethylase, Utx, regulates adipogenesis in a differentiation stage-dependent manner. PLOS ONE 2017, 12, e0173713. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Miller, J.L.; Grant, P.A. The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. In Epigenetics: Development and Disease; Kundu, T.K., Ed.; Springer: Dordrecht, The Netherlands, 2013; pp. 289–317. [Google Scholar] [CrossRef]
- Laukka, T.; Myllykoski, M.; Looper, R.E.; Koivunen, P. Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J. Mol. Biol. 2018, 430, 3081–3092. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone Deacetylases and Mechanisms of Regulation of Gene Expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef]
- Feng, Q. , and Y. Zhang. "The Mecp1 Complex Represses Transcription through Preferential Binding, Remodeling, and Deacetylating Methylated Nucleosomes." Genes Dev 15, no. 7 (2001): 827-32.
- Sears, T.K.; Horbinski, C.M.; Woolard, K.D. IDH1 mutant glioma is preferentially sensitive to the HDAC inhibitor panobinostat. J. Neuro-Oncology 2021, 154, 159–170. [Google Scholar] [CrossRef]
- Garrett, M.C.; Albano, R.; Carnwath, T.; Elahi, L.; Behrmann, C.A.; Pemberton, M.; Woo, D.; O’brien, E.; VanCauwenbergh, B.; Perentesis, J.; et al. HDAC1 and HDAC6 are essential for driving growth in IDH1 mutant glioma. Sci. Rep. 2023, 13, 1–12. [Google Scholar] [CrossRef]
- Zhang, Xinchao, Yue Zhang, Chaofu Wang, and Xu Wang. "Tet (Ten-Eleven Translocation) Family Proteins: Structure, Biological Functions and Applications." Signal Transduction and Targeted Therapy 8, no. 1 (2023): 297.
- Gerecke, C.; Rodrigues, C.E.; Homann, T.; Kleuser, B. The Role of Ten-Eleven Translocation Proteins in Inflammation. Front. Immunol. 2022, 13, 861351. [Google Scholar] [CrossRef] [PubMed]
- Bogdanović, O.; Smits, A.H.; Mustienes, E.d.l.C.; Tena, J.J.; Ford, E.; Williams, R.; Senanayake, U.; Schultz, M.D.; Hontelez, S.; van Kruijsbergen, I.; et al. Active DNA demethylation at enhancers during the vertebrate phylotypic period. Nat. Genet. 2016, 48, 417–426. [Google Scholar] [CrossRef]
- Sanstead, P.J.; Ashwood, B.; Dai, Q.; He, C.; Tokmakoff, A. Oxidized Derivatives of 5-Methylcytosine Alter the Stability and Dehybridization Dynamics of Duplex DNA. J. Phys. Chem. B 2020, 124, 1160–1174. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Godley, L.A. 5-hydroxymethylcytosine in cancer: significance in diagnosis and therapy. Cancer Genet. 2015, 208, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Houseman, E.A.; King, J.E.; von Herrmann, K.M.; Fadul, C.E.; Christensen, B.C. 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat. Commun. 2016, 7, 13177. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Saito, K.; Aihara, K.; Nagae, G.; Yamamoto, S.; Tatsuno, K.; Ueda, H.; Fukuda, S.; Umeda, T.; Tanaka, S.; et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-C.; Chou, S.-C.; Liu, C.-Y.; Chen, C.-Y.; Hou, H.-A.; Kuo, Y.-Y.; Lee, M.-C.; Ko, B.-S.; Tang, J.-L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef]
- Bray, J.K.; Dawlaty, M.M.; Verma, A.; Maitra, A. Roles and Regulations of TET Enzymes in Solid Tumors. Trends Cancer 2021, 7, 635–646. [Google Scholar] [CrossRef]
- Figueroa, M. E., O. Abdel-Wahab, C. Lu, P. S. Ward, J. Patel, A. Shih, Y. Li, N. Bhagwat, A. Vasanthakumar, H. F. Fernandez, M. S. Tallman, Z. Sun, K. Wolniak, J. K. Peeters, W. Liu, S. E. Choe, V. R. Fantin, E. Paietta, B. Löwenberg, J. D. Licht, L. A. Godley, R. Delwel, P. J. Valk, C. B. Thompson, R. L. Levine, and A. Melnick. "Leukemic Idh1 and Idh2 Mutations Result in a Hypermethylation Phenotype, Disrupt Tet2 Function, and Impair Hematopoietic Differentiation." Cancer Cell 18, no. 6 (2010): 553-67.
- Huang, Y.; Wei, J.; Huang, X.; Zhou, W.; Xu, Y.; Deng, D.-H.; Cheng, P. Comprehensively analyze the expression and prognostic role for ten-eleven translocations (TETs) in acute myeloid leukemia. Transl. Cancer Res. 2020, 9, 7259–7283. [Google Scholar] [CrossRef]
- Laukka, T.; Mariani, C.J.; Ihantola, T.; Cao, J.Z.; Hokkanen, J.; Kaelin, W.G., Jr.; Godley, L.A.; Koivunen, P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J. Biol. Chem. 2016, 291, 4256–4265. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef]
- Belle, R.; Saraç, H.; Salah, E.; Bhushan, B.; Szykowska, A.; Roper, G.; Tumber, A.; Kriaucionis, S.; Burgess-Brown, N.; Schofield, C.J.; et al. Focused Screening Identifies Different Sensitivities of Human TET Oxygenases to the Oncometabolite 2-Hydroxyglutarate. J. Med. Chem. 2024, 67, 4525–4540. [Google Scholar] [CrossRef]
- Yue, X.; Rao, A. TET family dioxygenases and the TET activator vitamin C in immune responses and cancer. Blood 2020, 136, 1394–1401. [Google Scholar] [CrossRef]
- Gerecke, C.; Schumacher, F.; Berndzen, A.; Homann, T.; Kleuser, B. Vitamin C in combination with inhibition of mutant IDH1 synergistically activates TET enzymes and epigenetically modulates gene silencing in colon cancer cells. Epigenetics 2019, 15, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Kusi, M.; Zand, M.; Lin, L.-L.; Chen, M.; Lopez, A.; Lin, C.-L.; Wang, C.-M.; Lucio, N.D.; Kirma, N.B.; Ruan, J.; et al. 2-Hydroxyglutarate destabilizes chromatin regulatory landscape and lineage fidelity to promote cellular heterogeneity. Cell Rep. 2022, 38, 110220–110220. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.C.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef]
- Frost, J.; Frost, M.; Batie, M.; Jiang, H.; Rocha, S. Roles of HIF and 2-Oxoglutarate-Dependent Dioxygenases in Controlling Gene Expression in Hypoxia. Cancers 2021, 13, 350. [Google Scholar] [CrossRef]
- D'Ignazio, L., M. Batie, and S. Rocha. "Hypoxia and Inflammation in Cancer, Focus on Hif and Nf-κb." Biomedicines 5, no. 2 (2017).
- Rocha, S. Gene regulation under low oxygen: holding your breath for transcription. Trends Biochem. Sci. 2007, 32, 389–397. [Google Scholar] [CrossRef]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Böttcher, M.; Renner, K.; Berger, R.; Mentz, K.; Thomas, S.; Cardenas-Conejo, Z.E.; Dettmer, K.; Oefner, P.J.; Mackensen, A.; Kreutz, M.; et al. D-2-hydroxyglutarate interferes with HIF-1α stability skewing T-cell metabolism towards oxidative phosphorylation and impairing Th17 polarization. OncoImmunology 2018, 7, e1445454. [Google Scholar] [CrossRef]
- Kuiper, C.; Dachs, G.U.; Currie, M.J.; Vissers, M.C. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free. Radic. Biol. Med. 2014, 69, 308–317. [Google Scholar] [CrossRef]
- Miles, S.L.; Fischer, A.P.; Joshi, S.J.; Niles, R.M. Ascorbic acid and ascorbate-2-phosphate decrease HIF activity and malignant properties of human melanoma cells. BMC Cancer 2015, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Selak, M. A., S. M. Armour, E. D. MacKenzie, H. Boulahbel, D. G. Watson, K. D. Mansfield, Y. Pan, M. C. Simon, C. B. Thompson, and E. Gottlieb. "Succinate Links Tca Cycle Dysfunction to Oncogenesis by Inhibiting Hif-Alpha Prolyl Hydroxylase." Cancer Cell 7, no. 1 (2005): 77-85.
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef]
- Hong, J.; Xu, K.; Lee, J.H. Biological roles of the RNA m6A modification and its implications in cancer. Exp. Mol. Med. 2022, 54, 1822–1832. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Chen, J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Tang, F.; Pan, Z.; Wang, Y.; Lan, T.; Wang, M.; Li, F.; Quan, W.; Liu, Z.; Wang, Z.; Li, Z. Advances in the Immunotherapeutic Potential of Isocitrate Dehydrogenase Mutations in Glioma. Neurosci. Bull. 2022, 38, 1069–1084. [Google Scholar] [CrossRef]
- Pianka, S.T.; Li, T.; Prins, T.J.; Eldred, B.S.; Kevan, B.M.; Liang, H.; Rinonos, S.Z.; Kornblum, H.I.; Nathanson, D.A.; Pellegrini, M.; et al. D-2-HG Inhibits IDH1mut Glioma Growth via FTO Inhibition and Resultant m6A Hypermethylation. Cancer Res. Commun. 2024, 4, 876–894. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ouyang, X.; Zuo, L.; Xiao, Y.; Sun, Y.; Chang, C.; Qin, X.; Yeh, S. R-2HG downregulates ERα to inhibit cholangiocarcinoma via the FTO/m6A-methylated ERα/miR16-5p/YAP1 signal pathway. Mol. Ther. - Oncolytics 2021, 23, 65–81. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell 2018, 172, 90–105. [Google Scholar] [CrossRef]
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m6A/PFKP/LDHB axis. Mol. Cell 2021, 81, 922–939. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Sidoli, S.; Warneford-Thomson, R.; Tatomer, D.C.; Wilusz, J.E.; Garcia, B.A.; Bonasio, R. High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Mol. Cell 2016, 64, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Delatte, B.; Wang, F.; Ngoc, L.V.; Collignon, E.; Bonvin, E.; Deplus, R.; Calonne, E.; Hassabi, B.; Putmans, P.; Awe, S.; et al. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 2016, 351, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Zhang, Q.; Shi, Y.; Shi, Q.; Jiang, Y.; Gu, Y.; Li, Z.; Li, X.; Zhao, K.; Wang, C.; et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 2018, 554, 123–127. [Google Scholar] [CrossRef]
- Tran, Paul M. H., Lynn K. H. Tran, John Nechtman, Bruno dos Santos, Sharad Purohit, Khaled Bin Satter, Boying Dun, Ravindra Kolhe, Suash Sharma, Roni Bollag, and Jin-Xiong She. "Comparative Analysis of Transcriptomic Profile, Histology, and Idh Mutation for Classification of Gliomas." Scientific Reports 10, no. 1 (2020): 20651.
- Cheng, W.; Ren, X.; Zhang, C.; Cai, J.; Han, S.; Wu, A. Gene Expression Profiling Stratifies IDH1-Mutant Glioma with Distinct Prognoses. Mol. Neurobiol. 2016, 54, 5996–6005. [Google Scholar] [CrossRef]
- Unruh, D.; Zewde, M.; Buss, A.; Drumm, M.R.; Tran, A.N.; Scholtens, D.M.; Horbinski, C. Methylation and transcription patterns are distinct in IDH mutant gliomas compared to other IDH mutant cancers. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Alzial, G.; Renoult, O.; Paris, F.; Gratas, C.; Clavreul, A.; Pecqueur, C. Wild-type isocitrate dehydrogenase under the spotlight in glioblastoma. Oncogene 2021, 41, 613–621. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef]
- Fan, J.; Teng, X.; Liu, L.; Mattaini, K.R.; Looper, R.E.; Vander Heiden, M.G.; Rabinowitz, J.D. Human Phosphoglycerate Dehydrogenase Produces the Oncometabolite d-2-Hydroxyglutarate. ACS Chem. Biol. 2015, 10, 510–516. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2018, 18, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.-M. 100 years of the Warburg effect: A cancer metabolism endeavor. Cell 2024, 187, 3824–3828. [Google Scholar] [CrossRef] [PubMed]
- Khurshed, M.; Molenaar, R.J.; Lenting, K.; Leenders, W.P.; van Noorden, C.J. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget 2017, 8, 49165–49177. [Google Scholar] [CrossRef] [PubMed]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro-Oncology 2013, 16, 686–695. [Google Scholar] [CrossRef]
- Chaumeil, M.M.; Radoul, M.; Najac, C.; Eriksson, P.; Viswanath, P.; Blough, M.D.; Chesnelong, C.; Luchman, H.A.; Cairncross, J.G.; Ronen, S.M. Hyperpolarized 13 C MR imaging detects no lactate production in mutant IDH1 gliomas: Implications for diagnosis and response monitoring. NeuroImage: Clin. 2016, 12, 180–189. [Google Scholar] [CrossRef]
- Dekker, L.J.M.; Wu, S.; Jurriëns, C.; Mustafa, D.A.N.; Grevers, F.; Burgers, P.C.; Smitt, P.A.E.S.; Kros, J.M.; Luider, T.M. Metabolic changes related to the IDH1 mutation in gliomas preserve TCA-cycle activity: An investigation at the protein level. FASEB J. 2020, 34, 3646–3657. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W. Regulation and function of the mammalian tricarboxylic acid cycle. J. Biol. Chem. 2022, 299, 102838. [Google Scholar] [CrossRef]
- Biedermann, J.; Preussler, M.; Conde, M.; Peitzsch, M.; Richter, S.; Wiedemuth, R.; Abou-El-Ardat, K.; Krüger, A.; Meinhardt, M.; Schackert, G.; et al. Mutant IDH1 Differently Affects Redox State and Metabolism in Glial Cells of Normal and Tumor Origin. Cancers 2019, 11, 2028. [Google Scholar] [CrossRef]
- Fack, F., S. Tardito, G. Hochart, A. Oudin, L. Zheng, S. Fritah, A. Golebiewska, P. V. Nazarov, A. Bernard, A. C. Hau, O. Keunen, W. Leenders, M. Lund-Johansen, J. Stauber, E. Gottlieb, R. Bjerkvig, and S. P. Niclou. "Altered Metabolic Landscape in Idh-Mutant Gliomas Affects Phospholipid, Energy, and Oxidative Stress Pathways." EMBO Mol Med 9, no. 12 (2017): 1681-95.
- Izquierdo-Garcia, J.L.; Cai, L.M.; Chaumeil, M.M.; Eriksson, P.; Robinson, A.E.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Glioma Cells with the IDH1 Mutation Modulate Metabolic Fractional Flux through Pyruvate Carboxylase. PLOS ONE 2014, 9, e108289. [Google Scholar] [CrossRef]
- Lenting, K.; Khurshed, M.; Peeters, T.H.; Heuvel, C.N.A.M.v.D.; van Lith, S.A.M.; de Bitter, T.; Hendriks, W.; Span, P.N.; Molenaar, R.J.; Botman, D.; et al. Isocitrate dehydrogenase 1–mutated human gliomas depend on lactate and glutamate to alleviate metabolic stress. FASEB J. 2018, 33, 557–571. [Google Scholar] [CrossRef]
- Dekker, L.J.M.; Verheul, C.; Wensveen, N.; Leenders, W.; Lamfers, M.L.M.; Leenstra, S.; Luider, T.M. Effects of the IDH1 R132H Mutation on the Energy Metabolism: A Comparison between Tissue and Corresponding Primary Glioma Cell Cultures. ACS Omega 2022, 7, 3568–3578. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Tonjes, M., S. Barbus, Y. J. Park, W. Wang, M. Schlotter, A. M. Lindroth, S. V. Pleier, A. H. C. Bai, D. Karra, R. M. Piro, J. Felsberg, A. Addington, D. Lemke, I. Weibrecht, V. Hovestadt, C. G. Rolli, B. Campos, S. Turcan, D. Sturm,..., and B. Radlwimmer. "Bcat1 Promotes Cell Proliferation through Amino Acid Catabolism in Gliomas Carrying Wild-Type Idh1." Nat Med 19, no. 7 (2013): 901-08.
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Hollinshead, K. E. R., H. Munford, K. L. Eales, C. Bardella, C. Li, C. Escribano-Gonzalez, A. Thakker, Y. Nonnenmacher, K. Kluckova, M. Jeeves, R. Murren, F. Cuozzo, D. Ye, G. Laurenti, W. Zhu, K. Hiller, D. J. Hodson, W. Hua, I. P. Tomlinson,..., and D. A. Tennant. "Oncogenic Idh1 Mutations Promote Enhanced Proline Synthesis through Pycr1 to Support the Maintenance of Mitochondrial Redox Homeostasis." Cell Rep 22, no. 12 (2018): 3107-14.
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG Synthesis Increases Pentose Phosphate Pathway Flux and Sensitizes Cells to Oxidative Stress. Cell Rep. 2018, 22, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Badur, M.G.; Muthusamy, T.; Parker, S.J.; Ma, S.; McBrayer, S.K.; Cordes, T.; Magana, J.H.; Guan, K.-L.; Metallo, C.M. Oncogenic R132 IDH1 Mutations Limit NADPH for De Novo Lipogenesis through (D)2-Hydroxyglutarate Production in Fibrosarcoma Cells. Cell Rep. 2018, 25, 1680–1680. [Google Scholar] [CrossRef]
- Esmaeili, M.; Hamans, B.C.; Navis, A.C.; van Horssen, R.; Bathen, T.F.; Gribbestad, I.S.; Leenders, W.P.; Heerschap, A. IDH1 R132H Mutation Generates a Distinct Phospholipid Metabolite Profile in Glioma. Cancer Res. 2014, 74, 4898–4907. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Viswanath, P.; Eriksson, P.; Chaumeil, M.M.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Metabolic Reprogramming in Mutant IDH1 Glioma Cells. PLOS ONE 2015, 10, e0118781. [Google Scholar] [CrossRef]
- Fedeles, B. I., V. Singh, J. C. Delaney, D. Li, and J. M. Essigmann. "The Alkb Family of Fe(Ii)/α-Ketoglutarate-Dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond." J Biol Chem 290, no. 34 (2015): 20734-42.
- Chen, F.; Bian, K.; Tang, Q.; Fedeles, B.I.; Singh, V.; Humulock, Z.T.; Essigmann, J.M.; Li, D. Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem. Res. Toxicol. 2017, 30, 1102–1110. [Google Scholar] [CrossRef]
- Sim, H. W., R. Nejad, W. Zhang, F. Nassiri, W. Mason, K. D. Aldape, G. Zadeh, and E. X. Chen. "Tissue 2-Hydroxyglutarate as a Biomarker for Isocitrate Dehydrogenase Mutations in Gliomas." Clin Cancer Res 25, no. 11 (2019): 3366-73.
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.-L.; Ye, D.; et al. Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef]
- Lin, L.; Cai, J.; Tan, Z.; Meng, X.; Li, R.; Li, Y.; Jiang, C. Mutant IDH1 Enhances Temozolomide Sensitivity via Regulation of the ATM/CHK2 Pathway in Glioma. Cancer Res. Treat. 2021, 53, 367–377. [Google Scholar] [CrossRef]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Carney, S. V., K. Banerjee, A. Mujeeb, B. Zhu, S. Haase, M. L. Varela, P. Kadiyala, C. E. Tronrud, Z. Zhu, D. Mukherji, P. Gorla, Y. Sun, R. Tagett, F. J. Núñez, M. Luo, W. Luo, M. Ljungman, Y. Liu, Z. Xia, A. Schwendeman, T. Qin, M. A. Sartor, J. F. Costello, D. P. Cahill, P. R. Lowenstein, and M. G. Castro. "Zinc Finger Mynd-Type Containing 8 (Zmynd8) Is Epigenetically Regulated in Mutant Isocitrate Dehydrogenase 1 (Idh1) Glioma to Promote Radioresistance." Clin Cancer Res 29, no. 9 (2023): 1763-82.
- Jia, P.; Li, X.; Wang, X.; Yao, L.; Xu, Y.; Hu, Y.; Xu, W.; He, Z.; Zhao, Q.; Deng, Y.; et al. ZMYND8 mediated liquid condensates spatiotemporally decommission the latent super-enhancers during macrophage polarization. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Spruijt, C.G.; Luijsterburg, M.S.; Menafra, R.; Lindeboom, R.G.; Jansen, P.W.; Edupuganti, R.R.; Baltissen, M.P.; Wiegant, W.W.; Voelker-Albert, M.C.; Matarese, F.; et al. ZMYND8 Co-localizes with NuRD on Target Genes and Regulates Poly(ADP-Ribose)-Dependent Recruitment of GATAD2A/NuRD to Sites of DNA Damage. Cell Rep. 2016, 17, 783–798. [Google Scholar] [CrossRef]
- Majd, N.; Yap, T.A.; Yung, W.K.A.; de Groot, J. The Promise of Poly(ADP-Ribose) Polymerase (PARP) Inhibitors in Gliomas. J. Immunother. Precis. Oncol. 2020, 3, 157–164. [Google Scholar] [CrossRef]
- Garrett, M.; Sperry, J.; Braas, D.; Yan, W.; Le, T.M.; Mottahedeh, J.; Ludwig, K.; Eskin, A.; Qin, Y.; Levy, R.; et al. Metabolic characterization of isocitrate dehydrogenase (IDH) mutant and IDH wildtype gliomaspheres uncovers cell type-specific vulnerabilities. Cancer Metab. 2018, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhou, H.; Sun, W.; Hou, L.; Wang, Y.; Wang, H.; Lv, Z.; Xue, X. IDH1R132H mutation increases radiotherapy efficacy and a 4-gene radiotherapy-related signature of WHO grade 4 gliomas. Sci. Rep. 2023, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Güttler, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. IDH1R132H mutation causes a less aggressive phenotype and radiosensitizes human malignant glioma cells independent of the oxygenation status. Radiother. Oncol. 2015, 116, 381–387. [Google Scholar] [CrossRef]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 1–7. [Google Scholar] [CrossRef]
- Jiao, Y., P. J. Killela, Z. J. Reitman, A. B. Rasheed, C. M. Heaphy, R. F. de Wilde, F. J. Rodriguez, S. Rosemberg, S. M. Oba-Shinjo, S. K. Nagahashi Marie, C. Bettegowda, N. Agrawal, E. Lipp, C. Pirozzi, G. Lopez, Y. He, H. Friedman, A. H. Friedman, G. J. Riggins, M. Holdhoff, P. Burger, R. McLendon, D. D. Bigner, B. Vogelstein, A. K. Meeker, K. W. Kinzler, N. Papadopoulos, L. A. Diaz, and H. Yan. "Frequent Atrx, Cic, Fubp1 and Idh1 Mutations Refine the Classification of Malignant Gliomas." Oncotarget 3, no. 7 (2012): 709-22.
- Núñez, F.J.; Mendez, F.M.; Kadiyala, P.; Alghamri, M.S.; Savelieff, M.G.; Garcia-Fabiani, M.B.; Haase, S.; Koschmann, C.; Calinescu, A.-A.; Kamran, N.; et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Cai, M.; Zhao, J.; Ding, Q.; Wei, J. Oncometabolite 2-hydroxyglutarate regulates anti-tumor immunity. Heliyon 2024, 10, e24454. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Bansal, A.; Young, E.; Batchala, P.; Patrie, J.; Lopes, M.; Jain, R.; Fadul, C.; Schiff, D. Extent of Surgical Resection in Lower-Grade Gliomas: Differential Impact Based on Molecular Subtype. Am. J. Neuroradiol. 2019, 40, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Gill, B. J., D. J. Pisapia, H. R. Malone, H. Goldstein, L. Lei, A. Sonabend, J. Yun, J. Samanamud, J. S. Sims, M. Banu, A. Dovas, A. F. Teich, S. A. Sheth, G. M. McKhann, M. B. Sisti, J. N. Bruce, P. A. Sims, and P. Canoll. "Mri-Localized Biopsies Reveal Subtype-Specific Differences in Molecular and Cellular Composition at the Margins of Glioblastoma." Proc Natl Acad Sci U S A 111, no. 34 (2014): 12550-5.
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Blanco-Carmona, E.; Narayanan, A.; Hernandez, I.; Nieto, J.C.; Elosua-Bayes, M.; Sun, X.; Schmidt, C.; Pamir, N.; Özduman, K.; Herold-Mende, C.; et al. Tumor heterogeneity and tumor-microglia interactions in primary and recurrent IDH1-mutant gliomas. Cell Rep. Med. 2023, 4, 101249. [Google Scholar] [CrossRef]
- Pirozzi, C.J.; Yan, H. The implications of IDH mutations for cancer development and therapy. Nat. Rev. Clin. Oncol. 2021, 18, 645–661. [Google Scholar] [CrossRef]
- Śledzińska, P.; Bebyn, M.; Szczerba, E.; Furtak, J.; Harat, M.; Olszewska, N.; Kamińska, K.; Kowalewski, J.; Lewandowska, M.A. Glioma 2021 WHO Classification: The Superiority of NGS Over IHC in Routine Diagnostics. Mol. Diagn. Ther. 2022, 26, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Vij, M.; Yokoda, R.T.; Rashidipour, O.; Tran, I.; Vasudevaraja, V.; Snuderl, M.; Yong, R.L.; Cobb, W.S.; Umphlett, M.; Walker, J.M.; et al. The prognostic impact of subclonal IDH1 mutation in grade 2–4 astrocytomas. Neuro-Oncology Adv. 2023, 5, vdad069. [Google Scholar] [CrossRef]
- Singh, Rajesh R. "Next-Generation Sequencing in High-Sensitive Detection of Mutations in Tumors: Challenges, Advances, and Applications." The Journal of Molecular Diagnostics 22, no. 8 (2020): 994-1007.
- Pei, X.M.; Yeung, M.H.Y.; Wong, A.N.N.; Tsang, H.F.; Yu, A.C.S.; Yim, A.K.Y.; Wong, S.C.C. Targeted Sequencing Approach and Its Clinical Applications for the Molecular Diagnosis of Human Diseases. Cells 2023, 12, 493. [Google Scholar] [CrossRef]
- Qin, D. "Next-Generation Sequencing and Its Clinical Application." Cancer Biol Med 16, no. 1 (2019): 4-10.
- Nikiforova, Marina N., Abigail I. Wald, Melissa A. Melan, Somak Roy, Shan Zhong, Ronald L. Hamilton, Frank S. Lieberman, Jan Drappatz, Nduka M. Amankulor, Ian F. Pollack, Yuri E. Nikiforov, and Craig Horbinski. "Targeted Next-Generation Sequencing Panel (Glioseq) Provides Comprehensive Genetic Profiling of Central Nervous System Tumors." Neuro-Oncology 18, no. 3 (2015): 379-87.
- Zacher, A.; Kaulich, K.; Stepanow, S.; Wolter, M.; Köhrer, K.; Felsberg, J.; Malzkorn, B.; Reifenberger, G. Molecular Diagnostics of Gliomas Using Next Generation Sequencing of a Glioma-Tailored Gene Panel. Brain Pathol. 2016, 27, 146–159. [Google Scholar] [CrossRef]
- Higa, N.; Akahane, T.; Yokoyama, S.; Yonezawa, H.; Uchida, H.; Takajo, T.; Kirishima, M.; Hamada, T.; Matsuo, K.; Fujio, S.; et al. A tailored next-generation sequencing panel identified distinct subtypes of wildtype IDH and TERT promoter glioblastomas. Cancer Sci. 2020, 111, 3902–3911. [Google Scholar] [CrossRef]
- Guarnaccia, M.; Guarnaccia, L.; La Cognata, V.; Navone, S.E.; Campanella, R.; Ampollini, A.; Locatelli, M.; Miozzo, M.; Marfia, G.; Cavallaro, S. A Targeted Next-Generation Sequencing Panel to Genotype Gliomas. Life 2022, 12, 956. [Google Scholar] [CrossRef] [PubMed]
- Tirrò, E.; Massimino, M.; Broggi, G.; Romano, C.; Minasi, S.; Gianno, F.; Antonelli, M.; Motta, G.; Certo, F.; Altieri, R.; et al. A Custom DNA-Based NGS Panel for the Molecular Characterization of Patients With Diffuse Gliomas: Diagnostic and Therapeutic Applications. Front. Oncol. 2022, 12, 861078. [Google Scholar] [CrossRef] [PubMed]
- de Biase, D.; Acquaviva, G.; Visani, M.; Marucci, G.; De Leo, A.; Maloberti, T.; Sanza, V.; Di Oto, E.; Franceschi, E.; Mura, A.; et al. Next-Generation Sequencing Panel for 1p/19q Codeletion and IDH1-IDH2 Mutational Analysis Uncovers Mistaken Overdiagnoses of 1p/19q Codeletion by FISH. J. Mol. Diagn. 2021, 23, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, J.; Rothhammer-Hampl, T.; Zoubaa, S.; Bumes, E.; Pukrop, T.; Kölbl, O.; Corbacioglu, S.; Schmidt, N.O.; Proescholdt, M.; Hau, P.; et al. A comprehensive DNA panel next generation sequencing approach supporting diagnostics and therapy prediction in neurooncology. Acta Neuropathol. Commun. 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Yaltirik, Cumhur Kaan, Seda Gulec Yilmaz, Selcuk Ozdogan, Ezel Yaltirik Bilgin, Zerrin Barut, Ugur Ture, and Turgay Isbir. "Determination of ≪Em≫Idh1, Idh2, Mgmt, Tert≪/Em≫ and ≪Em≫Atrx≪/Em≫ Gene Mutations in Glial Tumors." In Vivo 36, no. 4 (2022): 1694.
- Wongsurawat, T.; Jenjaroenpun, P.; Anekwiang, P.; Arigul, T.; Thongrattana, W.; Jamshidi-Parsian, A.; Boysen, G.; Suriyaphol, P.; Suktitipat, B.; Srirabheebhat, P.; et al. Exploiting nanopore sequencing for characterization and grading of IDH-mutant gliomas. Brain Pathol. 2023, 34, e13203. [Google Scholar] [CrossRef]
- Mimosa, Mashiat L., Wafa Al-ameri, Jared T. Simpson, Michael Nakhla, Karel Boissinot, David G. Munoz, Sunit Das, Harriet Feilotter, Ramzi Fattouh, and Rola M. Saleeb. "A Novel Approach to Detect Idh Point Mutations in Gliomas Using Nanopore Sequencing: Test Validation for the Clinical Laboratory." The Journal of Molecular Diagnostics 25, no. 3 (2023): 133-42.
- Patel, Areeba, Helin Dogan, Alexander Payne, Elena Krause, Philipp Sievers, Natalie Schoebe, Daniel Schrimpf, Christina Blume, Damian Stichel, Nadine Holmes, Philipp Euskirchen, Jürgen Hench, Stephan Frank, Violaine Rosenstiel-Goidts, Miriam Ratliff, Nima Etminan, Andreas Unterberg, Christoph Dieterich, Christel Herold-Mende, Stefan M. Pfister, Wolfgang Wick, Matthew Loose, Andreas von Deimling, Martin Sill, David T. W. Jones, Matthias Schlesner, and Felix Sahm. "Rapid-Cns2: Rapid Comprehensive Adaptive Nanopore-Sequencing of Cns Tumors, a Proof-of-Concept Study." Acta Neuropathologica 143, no. 5 (2022): 609-12.
- Shirai, Y.; Ueno, T.; Kojima, S.; Ikeuchi, H.; Kitada, R.; Koyama, T.; Takahashi, F.; Takahashi, K.; Ichimura, K.; Yoshida, A.; et al. The development of a custom RNA-sequencing panel for the identification of predictive and diagnostic biomarkers in glioma. J. Neuro-Oncology 2024, 167, 75–88. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Couturier, Charles P., Shamini Ayyadhury, Phuong U. Le, Javad Nadaf, Jean Monlong, Gabriele Riva, Redouane Allache, Salma Baig, Xiaohua Yan, Mathieu Bourgey, Changseok Lee, Yu Chang David Wang, V. Wee Yong, Marie-Christine Guiot, Hamed Najafabadi, Bratislav Misic, Jack Antel, Guillaume Bourque, Jiannis Ragoussis, and Kevin Petrecca. "Single-Cell Rna-Seq Reveals That Glioblastoma Recapitulates a Normal Neurodevelopmental Hierarchy." Nature Communications 11, no. 1 (2020): 3406.
- Piwecka, M.; Rajewsky, N.; Rybak-Wolf, A. Single-cell and spatial transcriptomics: deciphering brain complexity in health and disease. Nat. Rev. Neurol. 2023, 19, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef]
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M.A.; López-Bigas, N.; Cajal, S.R.Y.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin. Cancer Res. 2018, 24, 2812–2819. [Google Scholar] [CrossRef]
- Mair, R.; Mouliere, F. Cell-free DNA technologies for the analysis of brain cancer. Br. J. Cancer 2021, 126, 371–378. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839–8839. [Google Scholar] [CrossRef] [PubMed]
- Wongsurawat, Thidathip, Piroon Jenjaroenpun, Annick De Loose, Duah Alkam, David W. Ussery, Intawat Nookaew, Yuet-Kin Leung, Shuk-Mei Ho, John D. Day, and Analiz Rodriguez. "A Novel Cas9-Targeted Long-Read Assay for Simultaneous Detection of Idh1/2 Mutations and Clinically Relevant Mgmt Methylation in Fresh Biopsies of Diffuse Glioma." Acta Neuropathologica Communications 8, no. 1 (2020): 87.
- Feng, Z.; Kong, D.; Jin, W.; He, K.; Zhao, J.; Liu, B.; Xu, H.; Yu, X.; Feng, S. Rapid detection of isocitrate dehydrogenase 1 mutation status in glioma based on Crispr-Cas12a. Sci. Rep. 2023, 13, 1–12. [Google Scholar] [CrossRef]
- Yu, D.; Zhong, Q.; Xiao, Y.; Feng, Z.; Tang, F.; Feng, S.; Cai, Y.; Gao, Y.; Lan, T.; Li, M.; et al. Combination of MRI-based prediction and CRISPR/Cas12a-based detection for IDH genotyping in glioma. npj Precis. Oncol. 2024, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhong, M.; Liu, Y.; Ma, P.; Dang, L.; Meng, Q.; Wan, W.; Ma, X.; Liu, J.; Yang, G.; et al. Rapid and sensitive detection of COVID-19 using CRISPR/Cas12a-based detection with naked eye readout, CRISPR/Cas12a-NER. Sci. Bull. 2020, 65, 1436–1439. [Google Scholar] [CrossRef] [PubMed]
- Tsou, J.-H.; Leng, Q.; Jiang, F. A CRISPR Test for Rapidly and Sensitively Detecting Circulating EGFR Mutations. Diagnostics 2020, 10, 114. [Google Scholar] [CrossRef]
- Murugan, K., A. S. Seetharam, A. J. Severin, and D. G. Sashital. "Crispr-Cas12a Has Widespread Off-Target and Dsdna-Nicking Effects." J Biol Chem 295, no. 17 (2020): 5538-53.
- Zhou, J.; Chen, P.; Wang, H.; Liu, H.; Li, Y.; Zhang, Y.; Wu, Y.; Paek, C.; Sun, Z.; Lei, J.; et al. Cas12a variants designed for lower genome-wide off-target effect through stringent PAM recognition. Mol. Ther. 2021, 30, 244–255. [Google Scholar] [CrossRef]
- Kloosterhof, N.K.; de Rooi, J.J.; Kros, M.; Eilers, P.H.C.; Smitt, P.A.E.S.; Bent, M.J.v.D.; French, P.J. Molecular subtypes of glioma identified by genome-wide methylation profiling. Genes, Chromosom. Cancer 2013, 52, 665–674. [Google Scholar] [CrossRef]
- Capper, D.; Stichel, D.; Sahm, F.; Jones, D.T.W.; Schrimpf, D.; Sill, M.; Schmid, S.; Hovestadt, V.; Reuss, D.E.; Koelsche, C.; et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018, 136, 181–210. [Google Scholar] [CrossRef]
- Bai, H.; Harmancı, A.S.; Erson-Omay, E.Z.; Li, J.; Coşkun, S.; Simon, M.; Krischek, B.; Özduman, K.; Omay, S.B.; Sorensen, E.A.; et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat. Genet. 2016, 48, 59–66. [Google Scholar] [CrossRef]
- Schenkel, Laila C., Joseph Mathew, Hal Hirte, John Provias, Guillaume Paré, Michael Chong, Daria Grafodatskaya, and Elizabeth McCready. "Evaluation of DNA Methylation Array for Glioma Tumor Profiling and Description of a Novel Epi-Signature to Distinguish Idh1/Idh2 Mutant and Wild-Type Tumors." Genes 13, no. 11 (2022): 2075.
- Euskirchen, P.; Bielle, F.; Labreche, K.; Kloosterman, W.P.; Rosenberg, S.; Daniau, M.; Schmitt, C.; Masliah-Planchon, J.; Bourdeaut, F.; Dehais, C.; et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol. 2017, 134, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Willscher, E.; Hopp, L.; Kreuz, M.; Schmidt, M.; Hakobyan, S.; Arakelyan, A.; Hentschel, B.; Jones, D.T.W.; Pfister, S.M.; Loeffler, M.; et al. High-Resolution Cartography of the Transcriptome and Methylome Landscapes of Diffuse Gliomas. Cancers 2021, 13, 3198. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.; Carén, H. Methylation Profiling in Diffuse Gliomas: Diagnostic Value and Considerations. Cancers 2022, 14, 5679. [Google Scholar] [CrossRef]
- Djirackor, L.; Halldorsson, S.; Niehusmann, P.; Leske, H.; Capper, D.; Kuschel, L.P.; Pahnke, J.; Due-Tønnessen, B.J.; A Langmoen, I.; Sandberg, C.J.; et al. Intraoperative DNA methylation classification of brain tumors impacts neurosurgical strategy. Neuro-Oncology Adv. 2021, 3, vdab149. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, S.; Xu, J.; Liu, H.; Wan, S. Cancer Biomarkers Discovery of Methylation Modification With Direct High-Throughput Nanopore Sequencing. Front. Genet. 2021, 12. [Google Scholar] [CrossRef]
- Beiko, J.; Suki, D.; Hess, K.R.; Fox, B.D.; Cheung, V.; Cabral, M.; Shonka, N.; Gilbert, M.R.; Sawaya, R.; Prabhu, S.S.; et al. IDH1 mutant malignant astrocytomas are more amenable to surgical resection and have a survival benefit associated with maximal surgical resection. Neuro-Oncology 2013, 16, 81–91. [Google Scholar] [CrossRef]
- Cahill, D. P. "Extent of Resection of Glioblastoma: A Critical Evaluation in the Molecular Era." Neurosurg Clin N Am 32, no. 1 (2021): 23-29.
- Jakola, A.S.; Pedersen, L.K.; Skjulsvik, A.J.; Myrmel, K.; Sjåvik, K.; Solheim, O. The impact of resection in IDH-mutant WHO grade 2 gliomas: a retrospective population-based parallel cohort study. J. Neurosurg. 2022, 137, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Wolter, Marietta, Jörg Felsberg, Bastian Malzkorn, Kerstin Kaulich, and Guido Reifenberger. "Droplet Digital Pcr-Based Analyses for Robust, Rapid, and Sensitive Molecular Diagnostics of Gliomas." Acta Neuropathologica Communications 10, no. 1 (2022): 42.
- Wang, J.; Zhao, Y.-Y.; Li, J.-F.; Guo, C.-C.; Chen, F.-R.; Su, H.-K.; Zhao, H.-F.; Long, Y.-K.; Shao, J.-Y.; To, S.-S.T.; et al. IDH1 mutation detection by droplet digital PCR in glioma. Oncotarget 2015, 6, 39651–39660. [Google Scholar] [CrossRef]
- Perizzolo, M., B. Winkfein, S. Hui, W. Krulicki, J. A. Chan, and D. J. Demetrick. "Idh Mutation Detection in Formalin-Fixed Paraffin-Embedded Gliomas Using Multiplex Pcr and Single-Base Extension." Brain Pathol 22, no. 5 (2012): 619-24.
- Chen, W.W.; Balaj, L.; Liau, L.M.; Samuels, M.L.; Kotsopoulos, S.K.; A Maguire, C.; LoGuidice, L.; Soto, H.; Garrett, M.; Zhu, L.D.; et al. BEAMing and Droplet Digital PCR Analysis of Mutant IDH1 mRNA in Glioma Patient Serum and Cerebrospinal Fluid Extracellular Vesicles. Mol. Ther. - Nucleic Acids 2013, 2, e109. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, N.; Lv, J.; Zhu, P.; Pan, X.; Hu, J.; Wu, W.; Li, S.; Li, H. Comparing the performance of conventional PCR, RTQ-PCR, and droplet digital PCR assays in detection of Shigella. Mol. Cell. Probes 2020, 51, 101531. [Google Scholar] [CrossRef]
- Orzan, Francesca, Francesca De Bacco, Elisabetta Lazzarini, Giovanni Crisafulli, Alessandra Gasparini, Angelo Dipasquale, Ludovic Barault, Marco Macagno, Pasquale Persico, Federico Pessina, Beatrice Bono, Laura Giordano, Pietro Zeppa, Antonio Melcarne, Paola Cassoni, Diego Garbossa, Armando Santoro, Paolo M. Comoglio, Stefano Indraccolo, Matteo Simonelli, and Carla Boccaccio. "Liquid Biopsy of Cerebrospinal Fluid Enables Selective Profiling of Glioma Molecular Subtypes at First Clinical Presentation." Clinical Cancer Research 29, no. 7 (2023): 1252-66.
- Crucitta, S.; Pasqualetti, F.; Gonnelli, A.; Ruglioni, M.; Luculli, G.I.; Cantarella, M.; Ortenzi, V.; Scatena, C.; Paiar, F.; Naccarato, A.G.; et al. IDH1 mutation is detectable in plasma cell-free DNA and is associated with survival outcome in glioma patients. BMC Cancer 2024, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jones, Jordan J, Hong Nguyen, Stephen Q Wong, James Whittle, Josie Iaria, Stanley Stylli, James Towner, Thomas Pieters, Frank Gaillard, Andrew H Kaye, Katharine J Drummond, and Andrew P Morokoff. "Plasma Ctdna Liquid Biopsy of Idh1, Tertp, and Egfrviii Mutations in Glioma." Neuro-Oncology Advances 6, no. 1 (2024).
- Husain, A.; Mishra, S.; Siddiqui, M.H.; Husain, N. Detection of IDH1 Mutation in cfDNA and Tissue of Adult Diffuse Glioma with Allele-Specific qPCR. Asian Pac. J. Cancer Prev. 2023, 24, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.P.; Munoz-Zuluaga, C.; Slocum, C.; Dillard, A.; Cong, L.; Wang, J.; Lindeman, N.; Kluk, M.; Liechty, B.; Pisapia, D.; et al. Evaluation of the rapid Idylla IDH1-2 mutation assay in FFPE glioma samples. Diagn. Pathol. 2024, 19, 1–9. [Google Scholar] [CrossRef]
- Choate, K.A.; Raack, E.J.; Line, V.F.; Jennings, M.J.; Belton, R.J.; Winn, R.J.; Mann, P.B. Rapid extraction-free detection of the R132H isocitrate dehydrogenase mutation in glioma using colorimetric peptide nucleic acid-loop mediated isothermal amplification (CPNA-LAMP). PLOS ONE 2023, 18, e0291666. [Google Scholar] [CrossRef]
- Choate, Kristian A, Edward J Raack, Paul B Mann, Evan A Jones, Robert J Winn, and Matthew J Jennings. "Rapid Idh1-R132 Genotyping Panel Utilizing Locked Nucleic Acid Loop-Mediated Isothermal Amplification (Lna-Lamp)." Biology Methods and Protocols (2024).
- Yoshida, A.; Satomi, K.; Ohno, M.; Matsushita, Y.; Takahashi, M.; Miyakita, Y.; Hiraoka, N.; Narita, Y.; Ichimura, K. Frequent false-negative immunohistochemical staining with IDH1 (R132H)-specific H09 antibody on frozen section control slides: a potential pitfall in glioma diagnosis. Histopathology 2018, 74, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Zentgraf, H.; Balss, J.; Hartmann, C.; von Deimling, A. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009, 118, 599–601. [Google Scholar] [CrossRef]
- Kato, Y., G. Jin, C. T. Kuan, R. E. McLendon, H. Yan, and D. D. Bigner. "A Monoclonal Antibody Imab-1 Specifically Recognizes Idh1r132h, the Most Common Glioma-Derived Mutation." Biochem Biophys Res Commun 390, no. 3 (2009): 547-51.
- Kato, Y. "Specific Monoclonal Antibodies against Idh1/2 Mutations as Diagnostic Tools for Gliomas." Brain Tumor Pathol 32, no. 1 (2015): 3-11.
- Copaciu, R., J. Rashidian, J. Lloyd, A. Yahyabeik, J. McClure, K. Cummings, and Q. Su. "Characterization of an Idh1 R132h Rabbit Monoclonal Antibody, Mrq-67, and Its Applications in the Identification of Diffuse Gliomas." Antibodies (Basel) 12, no. 1 (2023).
- Rashidian, J.; Copaciu, R.; Su, Q.; Merritt, B.; Johnson, C.; Yahyabeik, A.; French, E.; Cummings, K. Generation and Performance of R132H Mutant IDH1 Rabbit Monoclonal Antibody. Antibodies 2017, 6, 22. [Google Scholar] [CrossRef]
- Lu, V.M.; McDonald, K.L. Isocitrate dehydrogenase 1 mutation subtypes at site 132 and their translational potential in glioma. CNS Oncol. 2018, 7, 41–50. [Google Scholar] [CrossRef]
- Agarwal, S.; Sharma, M.C.; Jha, P.; Pathak, P.; Suri, V.; Sarkar, C.; Chosdol, K.; Suri, A.; Kale, S.S.; Mahapatra, A.K. Comparative study of IDH1 mutations in gliomas by immunohistochemistry and DNA sequencing. Neuro-Oncology 2013, 15, 718–726. [Google Scholar] [CrossRef]
- Zhang, S.; William, C. Educational Case: Histologic and Molecular Features of Diffuse Gliomas. Acad. Pathol. 2020, 7. [Google Scholar] [CrossRef]
- Nakagaki, R.; Debsarkar, S.S.; Kawanaka, H.; Aronow, B.J.; Prasath, V.S. Deep learning-based IDH1 gene mutation prediction using histopathological imaging and clinical data. Comput. Biol. Med. 2024, 179, 108902. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Shah, Z.; Sav, A.; Russo, C.; Berkovsky, S.; Qian, Y.; Coiera, E.; Di Ieva, A. Isocitrate dehydrogenase (IDH) status prediction in histopathology images of gliomas using deep learning. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liechty, B.; Xu, Z.; Zhang, Z.; Slocum, C.; Bahadir, C.D.; Sabuncu, M.R.; Pisapia, D.J. Machine learning can aid in prediction of IDH mutation from H&E-stained histology slides in infiltrating gliomas. Sci. Rep. 2022, 12, 1–12. [Google Scholar] [CrossRef]
- Chunduru, P.; Phillips, J.J.; Molinaro, A.M. Prognostic risk stratification of gliomas using deep learning in digital pathology images. Neuro-Oncology Adv. 2022, 4, vdac111. [Google Scholar] [CrossRef]
- Cui, D.; Liu, Y.; Liu, G.; Liu, L. A Multiple-Instance Learning-Based Convolutional Neural Network Model to Detect the IDH1 Mutation in the Histopathology Images of Glioma Tissues. J. Comput. Biol. 2020, 27, 1264–1272. [Google Scholar] [CrossRef]
- Arita, H., Y. Narita, A. Yoshida, N. Hashimoto, T. Yoshimine, and K. Ichimura. "Idh1/2 Mutation Detection in Gliomas." Brain Tumor Pathol 32, no. 2 (2015): 79-89.
- Yang, J.; Zhu, H.; Zhang, T.; Ding, J. Structure, substrate specificity, and catalytic mechanism of human D-2-HGDH and insights into pathogenicity of disease-associated mutations. Cell Discov. 2021, 7, 1–17. [Google Scholar] [CrossRef]
- de Goede, K.E.; Harber, K.J.; Gorki, F.S.; Verberk, S.G.; Groh, L.A.; Keuning, E.D.; Struys, E.A.; van Weeghel, M.; Haschemi, A.; de Winther, M.P.; et al. d-2-Hydroxyglutarate is an anti-inflammatory immunometabolite that accumulates in macrophages after TLR4 activation. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2022, 1868, 166427. [Google Scholar] [CrossRef]
- Achouri, Y.; Noël, G.; Vertommen, D.; Rider, M.H.; Veiga-Da-Cunha, M.; van Schaftingen, E. Identification of a dehydrogenase acting on D-2-hydroxyglutarate. Biochem. J. 2004, 381, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Struys, E.A. D-2-Hydroxyglutaric aciduria: Unravelling the biochemical pathway and the genetic defect. J. Inherit. Metab. Dis. 2006, 29, 21–29. [Google Scholar] [CrossRef]
- Tuna, G., N. E. Dal-Bekar, A. Akay, M. Rükşen, S. İşlekel, and G. H. İşlekel. "Minimally Invasive Detection of Idh1 Mutation with Cell-Free Circulating Tumor DNA and D-2-Hydroxyglutarate, D/L-2-Hydroxyglutarate Ratio in Gliomas." J Neuropathol Exp Neurol 81, no. 7 (2022): 502-10.
- Kalinina, J., J. Ahn, N. S. Devi, L. Wang, Y. Li, J. J. Olson, M. Glantz, T. Smith, E. L. Kim, A. Giese, R. L. Jensen, C. C. Chen, B. S. Carter, H. Mao, M. He, and E. G. Van Meir. "Selective Detection of the D-Enantiomer of 2-Hydroxyglutarate in the Csf of Glioma Patients with Mutated Isocitrate Dehydrogenase." Clin Cancer Res 22, no. 24 (2016): 6256-65.
- Fujita, Y.; Nunez-Rubiano, L.; Dono, A.; Bellman, A.; Shah, M.; Rodriguez, J.C.; Putluri, V.; Kamal, A.H.M.; Putluri, N.; Riascos, R.F.; et al. IDH1 p.R132H ctDNA and D-2-hydroxyglutarate as CSF biomarkers in patients with IDH-mutant gliomas. J. Neuro-Oncology 2022, 159, 261–270. [Google Scholar] [CrossRef]
- Lee, C. L., G. M. O'Kane, W. P. Mason, W. J. Zhang, P. Spiliopoulou, A. R. Hansen, R. C. Grant, J. J. Knox, T. L. Stockley, G. Zadeh, and E. X. Chen. "Circulating Oncometabolite 2-Hydroxyglutarate as a Potential Biomarker for Isocitrate Dehydrogenase (Idh1/2) Mutant Cholangiocarcinoma." Mol Cancer Ther 23, no. 3 (2024): 394-99.
- Delahousse, J.; Verlingue, L.; Broutin, S.; Legoupil, C.; Touat, M.; Doucet, L.; Ammari, S.; Lacroix, L.; Ducreux, M.; Scoazec, J.-Y.; et al. Circulating oncometabolite D-2-hydroxyglutarate enantiomer is a surrogate marker of isocitrate dehydrogenase-mutated intrahepatic cholangiocarcinomas. Eur. J. Cancer 2018, 90, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Strain, Shinji K., Morris D. Groves, and Mark R. Emmett. "Enantioseparation and Detection of (R)-2-Hydroxyglutarate and (S)-2-Hydroxyglutarate by Chiral Gas Chromatography–Triple Quadrupole Mass Spectrometry." In Metabolomics, edited by Paul L. Wood, 89-100. New York, NY: Springer US, 2021.
- Voelxen, N.F.; Walenta, S.; Proescholdt, M.; Dettmer, K.; Pusch, S.; Mueller-Klieser, W. Quantitative Imaging of D-2-Hydroxyglutarate in Selected Histological Tissue Areas by a Novel Bioluminescence Technique. Front. Oncol. 2016, 6, 46–46. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Galán, E.; Massana, N.; Parra-Robert, M.; Hidalgo, S.; Casals, G.; Esteve, J.; Jiménez, W. Validation of a routine gas chromatography mass spectrometry method for 2-hydroxyglutarate quantification in human serum as a screening tool for detection of idh mutations. J. Chromatogr. B 2018, 1083, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef]
- Balss, J.; Pusch, S.; Beck, A.-C.; Herold-Mende, C.; Krämer, A.; Thiede, C.; Buckel, W.; Langhans, C.-D.; Okun, J.G.; von Deimling, A. Enzymatic assay for quantitative analysis of (d)-2-hydroxyglutarate. Acta Neuropathol. 2012, 124, 883–891. [Google Scholar] [CrossRef]
- Xiao, D.; Xu, X.; Gao, K.; Wang, M.; Zhang, W.; Lü, C.; Wang, X.; Wang, Q.; Xu, P.; Ma, C.; et al. A Förster resonance energy transfer-based d-2-hydroxyglutarate biosensor. Sensors Actuators B: Chem. 2023, 385. [Google Scholar] [CrossRef]
- Choate, Kristian Alissa, Wren W. L. Konickson, Matthew James Jennings, Paul Barrie Mann, Robert James Winn, David O. Kamson, and Evan P. S. Pratt. "A Genetically Encoded Fluorescent Sensor Enables Sensitive and Specific Detection of Idh Mutant Associated Oncometabolite D-2-Hydroxyglutarate." bioRxiv (2024): 2024.09.25.615072.
- Fan, B.; Dai, D.; DiNardo, C.D.; Stein, E.; de Botton, S.; Attar, E.C.; Liu, H.; Liu, G.; Lemieux, I.; Agresta, S.V.; et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib in patients with advanced hematologic malignancies with an IDH1 mutation. Cancer Chemother. Pharmacol. 2020, 85, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Fan, B., I. K. Mellinghoff, P. Y. Wen, M. A. Lowery, L. Goyal, W. D. Tap, S. S. Pandya, E. Manyak, L. Jiang, G. Liu, T. Nimkar, C. Gliser, M. Prahl Judge, S. Agresta, H. Yang, and D. Dai. "Clinical Pharmacokinetics and Pharmacodynamics of Ivosidenib, an Oral, Targeted Inhibitor of Mutant Idh1, in Patients with Advanced Solid Tumors." Invest New Drugs 38, no. 2 (2020): 433-44.
- Pusch, S.; Schweizer, L.; Beck, A.-C.; Lehmler, J.-M.; Weissert, S.; Balss, J.; Miller, A.K.; von Deimling, A. D-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol. Commun. 2014, 2, 19–19. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, K.; Li, S.; Wang, J.; Ma, J.; Jiang, T.; Dai, J. Patterns of Tumor Contrast Enhancement Predict the Prognosis of Anaplastic Gliomas withIDH1Mutation. Am. J. Neuroradiol. 2015, 36, 2023–2029. [Google Scholar] [CrossRef]
- Carrillo, J. A., A. Lai, P. L. Nghiemphu, H. J. Kim, H. S. Phillips, S. Kharbanda, P. Moftakhar, S. Lalaezari, W. Yong, B. M. Ellingson, T. F. Cloughesy, and W. B. Pope. "Relationship between Tumor Enhancement, Edema, Idh1 Mutational Status, Mgmt Promoter Methylation, and Survival in Glioblastoma." AJNR Am J Neuroradiol 33, no. 7 (2012): 1349-55.
- Xing, Z., X. Yang, D. She, Y. Lin, Y. Zhang, and D. Cao. "Noninvasive Assessment of Idh Mutational Status in World Health Organization Grade Ii and Iii Astrocytomas Using Dwi and Dsc-Pwi Combined with Conventional Mr Imaging." AJNR Am J Neuroradiol 38, no. 6 (2017): 1138-44.
- Li, Y.; Qin, Q.; Zhang, Y.; Cao, Y. Noninvasive Determination of the IDH Status of Gliomas Using MRI and MRI-Based Radiomics: Impact on Diagnosis and Prognosis. Curr. Oncol. 2022, 29, 6893–6907. [Google Scholar] [CrossRef]
- Kates, R.; Atkinson, D.; Brant-Zawadzki, M. Fluid-attenuated inversion recovery (FLAIR): clinical prospectus of current and future applications. . 1996, 8, 389–96. [Google Scholar] [CrossRef] [PubMed]
- Patel, S. H., L. M. Poisson, D. J. Brat, Y. Zhou, L. Cooper, M. Snuderl, C. Thomas, A. M. Franceschi, B. Griffith, A. E. Flanders, J. G. Golfinos, A. S. Chi, and R. Jain. "T2-Flair Mismatch, an Imaging Biomarker for Idh and 1p/19q Status in Lower-Grade Gliomas: A Tcga/Tcia Project." Clin Cancer Res 23, no. 20 (2017): 6078-85.
- Pinto, C.; Noronha, C.; Taipa, R.; Ramos, C. T2-FLAIR mismatch sign: a roadmap of pearls and pitfalls. Br. J. Radiol. 2021, 95, 20210825. [Google Scholar] [CrossRef] [PubMed]
- Jain, R., D. R. Johnson, S. H. Patel, M. Castillo, M. Smits, M. J. van den Bent, A. S. Chi, and D. P. Cahill. ""Real World" Use of a Highly Reliable Imaging Sign: "T2-Flair Mismatch" for Identification of Idh Mutant Astrocytomas." Neuro Oncol 22, no. 7 (2020): 936-43.
- Shoaib, Y.; Nayil, K.; Makhdoomi, R.; Asma, A.; Ramzan, A.; Shaheen, F.; Wani, A. Role of diffusion and perfusion magnetic resonance imaging in predicting the histopathological grade of gliomas - A prospective study. Asian J. Neurosurg. 2019, 14, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Sonay, Erdem Fatihoğlu, Pınar Nercis Koşar, and Elif Ergün. "Perfusion and Permeability Mri in Glioma Grading." Egyptian Journal of Radiology and Nuclear Medicine 51, no. 1 (2020): 2.
- Álvarez-Torres, M.d.M.; Fuster-García, E.; Juan-Albarracín, J.; Reynés, G.; Aparici-Robles, F.; Ferrer-Lozano, J.; García-Gómez, J.M. Local detection of microvessels in IDH-wildtype glioblastoma using relative cerebral blood volume: an imaging marker useful for astrocytoma grade 4 classification. BMC Cancer 2022, 22, 1–13. [Google Scholar] [CrossRef]
- Feraco, P.; Bacci, A.; Ferrazza, P.; Hauwe, L.v.D.; Pertile, R.; Girlando, S.; Barbareschi, M.; Gagliardo, C.; Morganti, A.G.; Petralia, B. Magnetic Resonance Imaging Derived Biomarkers of IDH Mutation Status and Overall Survival in Grade III Astrocytomas. Diagnostics 2020, 10, 247. [Google Scholar] [CrossRef]
- Tan, W.L.; Huang, W.Y.; Yin, B.; Xiong, J.; Wu, J.S.; Geng, D.Y. Can Diffusion Tensor Imaging Noninvasively Detect IDH1 Gene Mutations in Astrogliomas? A Retrospective Study of 112 Cases. Am. J. Neuroradiol. 2014, 35, 920–927. [Google Scholar] [CrossRef]
- Kurokawa, R.; Baba, A.; Kurokawa, M.; Capizzano, A.; Ota, Y.; Kim, J.; Srinivasan, A.; Moritani, T. Perfusion and diffusion-weighted imaging parameters: Comparison between pre- and postbiopsy MRI for high-grade glioma. Medicine 2022, 101, e30183. [Google Scholar] [CrossRef]
- Gihr, G.; Horvath-Rizea, D.; Kohlhof-Meinecke, P.; Ganslandt, O.; Henkes, H.; Härtig, W.; Donitza, A.; Skalej, M.; Schob, S. Diffusion Weighted Imaging in Gliomas: A Histogram-Based Approach for Tumor Characterization. Cancers 2022, 14, 3393. [Google Scholar] [CrossRef]
- Cho, N.S.; Hagiwara, A.; Eldred, B.S.C.; Raymond, C.; Wang, C.; Sanvito, F.; Lai, A.; Nghiemphu, P.; Salamon, N.; Steelman, L.; et al. Early volumetric, perfusion, and diffusion MRI changes after mutant isocitrate dehydrogenase (IDH) inhibitor treatment in IDH1-mutant gliomas. Neuro-Oncology Adv. 2022, 4, vdac124. [Google Scholar] [CrossRef]
- Stadlbauer, A.; Nikolic, K.; Oberndorfer, S.; Marhold, F.; Kinfe, T.M.; Meyer-Bäse, A.; Bistrian, D.A.; Schnell, O.; Doerfler, A. Machine Learning-Based Prediction of Glioma IDH Gene Mutation Status Using Physio-Metabolic MRI of Oxygen Metabolism and Neovascularization (A Bicenter Study). Cancers 2024, 16, 1102. [Google Scholar] [CrossRef]
- Yuan, J.; Siakallis, L.; Li, H.B.; Brandner, S.; Zhang, J.; Li, C.; Mancini, L.; Bisdas, S. Structural- and DTI- MRI enable automated prediction of IDH Mutation Status in CNS WHO Grade 2–4 glioma patients: a deep Radiomics Approach. BMC Med Imaging 2024, 24, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, X.; Zhang, W.; Zhang, L.; Wen, M.; Gao, J.; Yang, J.; Kan, Y.; Yang, X.; Wen, Z.; et al. A fusion model integrating magnetic resonance imaging radiomics and deep learning features for predicting alpha-thalassemia X-linked intellectual disability mutation status in isocitrate dehydrogenase–mutant high-grade astrocytoma: a multicenter study. Quant. Imaging Med. Surg. 2024, 14, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.-T.; Su, C.-Q.; Lin, J.; Xia, Z.-W.; Lu, S.-S.; Hong, X.-N. T2-FLAIR mismatch sign and machine learning-based multiparametric MRI radiomics in predicting IDH mutant 1p/19q non-co-deleted diffuse lower-grade gliomas. Clin. Radiol. 2024, 79, e750–e758. [Google Scholar] [CrossRef] [PubMed]
- Santinha, J.; Katsaros, V.; Stranjalis, G.; Liouta, E.; Boskos, C.; Matos, C.; Viegas, C.; Papanikolaou, N. Development of End-to-End AI–Based MRI Image Analysis System for Predicting IDH Mutation Status of Patients with Gliomas: Multicentric Validation. J. Imaging Informatics Med. 2024, 37, 31–44. [Google Scholar] [CrossRef]
- Pasquini, L.; Napolitano, A.; Tagliente, E.; Dellepiane, F.; Lucignani, M.; Vidiri, A.; Ranazzi, G.; Stoppacciaro, A.; Moltoni, G.; Nicolai, M.; et al. Deep Learning Can Differentiate IDH-Mutant from IDH-Wild GBM. J. Pers. Med. 2021, 11, 290. [Google Scholar] [CrossRef]
- Carosi, F.; Broseghini, E.; Fabbri, L.; Corradi, G.; Gili, R.; Forte, V.; Roncarati, R.; Filippini, D.M.; Ferracin, M. Targeting Isocitrate Dehydrogenase (IDH) in Solid Tumors: Current Evidence and Future Perspectives. Cancers 2024, 16, 2752. [Google Scholar] [CrossRef]
- Iwahashi, H.; Nagashima, H.; Tanaka, K.; Uno, T.; Hashiguchi, M.; Maeyama, M.; Somiya, Y.; Komatsu, M.; Hirose, T.; Itoh, T.; et al. 2-Hydroxyglutarate magnetic resonance spectroscopy in adult brainstem glioma. J. Neurosurg. 2023, 139, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Suh, C.H.; Kim, H.S.; Jung, S.C.; Choi, C.G.; Kim, S.J. 2-Hydroxyglutarate MR spectroscopy for prediction of isocitrate dehydrogenase mutant glioma: a systemic review and meta-analysis using individual patient data. Neuro-Oncology 2018, 20, 1573–1583. [Google Scholar] [CrossRef]
- Bhandari, A.; Sharma, C.; Ibrahim, M.; Riggs, M.; Jones, R.; Lasocki, A. The role of 2-hydroxyglutarate magnetic resonance spectroscopy for the determination of isocitrate dehydrogenase status in lower grade gliomas versus glioblastoma: a systematic review and meta-analysis of diagnostic test accuracy. Neuroradiology 2021, 63, 1823–1830. [Google Scholar] [CrossRef]
- Branzoli, F.; Di Stefano, A.L.; Capelle, L.; Ottolenghi, C.; Valabrègue, R.; Deelchand, D.K.; Bielle, F.; Villa, C.; Baussart, B.; Lehéricy, S.; et al. Highly specific determination of IDH status using edited in vivo magnetic resonance spectroscopy. Neuro-Oncology 2017, 20, 907–916. [Google Scholar] [CrossRef]
- Zhou, M.; Zhou, Y.; Liao, H.; Rowland, B.C.; Kong, X.; Arvold, N.D.; A Reardon, D.; Wen, P.Y.; Lin, A.P.; Huang, R.Y. Diagnostic accuracy of 2-hydroxyglutarate magnetic resonance spectroscopy in newly diagnosed brain mass and suspected recurrent gliomas. Neuro-Oncology 2018, 20, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Tietze, A.; Choi, C.; Mickey, B.; Maher, E.A.; Ulhøi, B.P.; Sangill, R.; Lassen-Ramshad, Y.; Lukacova, S.; Østergaard, L.; von Oettingen, G. Noninvasive assessment of isocitrate dehydrogenase mutation status in cerebral gliomas by magnetic resonance spectroscopy in a clinical setting. J. Neurosurg. 2018, 128, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Andronesi, O.C.; Rapalino, O.; Gerstner, E.; Chi, A.; Batchelor, T.T.; Cahill, D.P.; Sorensen, A.G.; Rosen, B.R. Detection of oncogenic IDH1 mutations using magnetic resonance spectroscopy of 2-hydroxyglutarate. J. Clin. Investig. 2013, 123, 3659–3663. [Google Scholar] [CrossRef]
- Andronesi, O.C.; Arrillaga-Romany, I.C.; Ly, K.I.; Bogner, W.; Ratai, E.M.; Reitz, K.; Iafrate, A.J.; Dietrich, J.; Gerstner, E.R.; Chi, A.S.; et al. Pharmacodynamics of mutant-IDH1 inhibitors in glioma patients probed by in vivo 3D MRS imaging of 2-hydroxyglutarate. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Lv, X.; Lu, C.; Ye, X.; Chen, X.; Fu, J.; Luo, C.; Zhao, Y. Prognostic factors of patients with Gliomas – an analysis on 335 patients with Glioblastoma and other forms of Gliomas. BMC Cancer 2020, 20, 1–7. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology, v96. [CrossRef]
- Shirahata, M., T. Ono, D. Stichel, D. Schrimpf, D. E. Reuss, F. Sahm, C. Koelsche, A. Wefers, A. Reinhardt, K. Huang, P. Sievers, H. Shimizu, H. Nanjo, Y. Kobayashi, Y. Miyake, T. Suzuki, J. I. Adachi, K. Mishima, A. Sasaki,..., and A. von Deimling. "Novel, Improved Grading System(S) for Idh-Mutant Astrocytic Gliomas." Acta Neuropathol 136, no. 1 (2018): 153-66.
- Parsons, D. W., S. Jones, X. Zhang, J. C. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, I. M. Siu, G. L. Gallia, A. Olivi, R. McLendon, B. A. Rasheed, S. Keir, T. Nikolskaya, Y. Nikolsky, D. A. Busam, H. Tekleab, L. A. Diaz, Jr.,..., and K. W. Kinzler. "An Integrated Genomic Analysis of Human Glioblastoma Multiforme." Science 321, no. 5897 (2008): 1807-12.
- Yan, H., D. W. Parsons, G. Jin, R. McLendon, B. A. Rasheed, W. Yuan, I. Kos, I. Batinic-Haberle, S. Jones, G. J. Riggins, H. Friedman, A. Friedman, D. Reardon, J. Herndon, K. W. Kinzler, V. E. Velculescu, B. Vogelstein, and D. D. Bigner. "Idh1 and Idh2 Mutations in Gliomas." N Engl J Med 360, no. 8 (2009): 765-73.
- Lopez-Gines, C.; Cerda-Nicolas, M.; Gil-Benso, R.; Pellin, A.; A Lopez-Guerrero, J.; Callaghan, R.; Benito, R.; Roldan, P.; Piquer, J.; Llacer, J.; et al. Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. . 2005, 24, 209–18. [Google Scholar]
- Eckel-Passow, Jeanette E., Daniel H. Lachance, Annette M. Molinaro, Kyle M. Walsh, Paul A. Decker, Hugues Sicotte, Melike Pekmezci, Terri Rice, Matt L. Kosel, Ivan V. Smirnov, Gobinda Sarkar, Alissa A. Caron, Thomas M. Kollmeyer, Corinne E. Praska, Anisha R. Chada, Chandralekha Halder, Helen M. Hansen, Lucie S. McCoy, Paige M. Bracci,..., and Robert B. Jenkins. "Glioma Groups Based on 1p/19q, Idh, and Tert Promoter Mutations in Tumors." New England Journal of Medicine 372, no. 26 (2015): 2499-508.
- Killela, P. J., Z. J. Reitman, Y. Jiao, C. Bettegowda, N. Agrawal, L. A. Diaz, Jr., A. H. Friedman, H. Friedman, G. L. Gallia, B. C. Giovanella, A. P. Grollman, T. C. He, Y. He, R. H. Hruban, G. I. Jallo, N. Mandahl, A. K. Meeker, F. Mertens, G. J. Netto,..., and H. Yan. "Tert Promoter Mutations Occur Frequently in Gliomas and a Subset of Tumors Derived from Cells with Low Rates of Self-Renewal." Proc Natl Acad Sci U S A 110, no. 15 (2013): 6021-6.
- Spiegl-Kreinecker, S.; Lötsch, D.; Ghanim, B.; Pirker, C.; Mohr, T.; Laaber, M.; Weis, S.; Olschowski, A.; Webersinke, G.; Pichler, J.; et al. Prognostic quality of activating TERT promoter mutations in glioblastoma: interaction with the rs2853669 polymorphism and patient age at diagnosis. Neuro-Oncology 2015, 17, 1231–1240. [Google Scholar] [CrossRef]
- Ebrahimi, A.; Skardelly, M.; Bonzheim, I.; Ott, I.; Mühleisen, H.; Eckert, F.; Tabatabai, G.; Schittenhelm, J. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol. Commun. 2016, 4, 60. [Google Scholar] [CrossRef]
- Weller, Michael, Patrick Y. Wen, Susan M. Chang, Linda Dirven, Michael Lim, Michelle Monje, and Guido Reifenberger. "Glioma." Nature Reviews Disease Primers 10, no. 1 (2024): 33.
- Pusch, S.; Schweizer, L.; Beck, A.-C.; Lehmler, J.-M.; Weissert, S.; Balss, J.; Miller, A.K.; von Deimling, A. D-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol. Commun. 2014, 2, 19–19. [Google Scholar] [CrossRef]
- Natsumeda, M., H. Igarashi, T. Nomura, R. Ogura, Y. Tsukamoto, T. Kobayashi, H. Aoki, K. Okamoto, A. Kakita, H. Takahashi, T. Nakada, and Y. Fujii. "Accumulation of 2-Hydroxyglutarate in Gliomas Correlates with Survival: A Study by 3.0-Tesla Magnetic Resonance Spectroscopy." Acta Neuropathol Commun 2 (2014): 158.
- Brennan, C. W., R. G. Verhaak, A. McKenna, B. Campos, H. Noushmehr, S. R. Salama, S. Zheng, D. Chakravarty, J. Z. Sanborn, S. H. Berman, R. Beroukhim, B. Bernard, C. J. Wu, G. Genovese, I. Shmulevich, J. Barnholtz-Sloan, L. Zou, R. Vegesna, S. A. Shukla,..., and L. Chin. "The Somatic Genomic Landscape of Glioblastoma." Cell 155, no. 2 (2013): 462-77.
- Ceccarelli, M., F. P. Barthel, T. M. Malta, T. S. Sabedot, S. R. Salama, B. A. Murray, O. Morozova, Y. Newton, A. Radenbaugh, S. M. Pagnotta, S. Anjum, J. Wang, G. Manyam, P. Zoppoli, S. Ling, A. A. Rao, M. Grifford, A. D. Cherniack, H. Zhang,..., and R. G. Verhaak. "Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma." Cell 164, no. 3 (2016): 550-63.
- Aoki, Kosuke, Hideo Nakamura, Hiromichi Suzuki, Keitaro Matsuo, Keisuke Kataoka, Teppei Shimamura, Kazuya Motomura, Fumiharu Ohka, Satoshi Shiina, Takashi Yamamoto, Yasunobu Nagata, Tetsuichi Yoshizato, Masahiro Mizoguchi, Tatsuya Abe, Yasutomo Momii, Yoshihiro Muragaki, Reiko Watanabe, Ichiro Ito, Masashi Sanada,..., and Atsushi Natsume. "Prognostic Relevance of Genetic Alterations in Diffuse Lower-Grade Gliomas." Neuro-Oncology 20, no. 1 (2017): 66-77.
- Yang, R. R., Z. F. Shi, Z. Y. Zhang, A. K. Chan, A. Aibaidula, W. W. Wang, J. S. H. Kwan, W. S. Poon, H. Chen, W. C. Li, N. Y. Chung, G. Punchhi, W. C. Chu, I. S. Chan, X. Z. Liu, Y. Mao, K. K. Li, and H. K. Ng. "Idh Mutant Lower Grade (Who Grades Ii/Iii) Astrocytomas Can Be Stratified for Risk by Cdkn2a, Cdk4 and Pdgfra Copy Number Alterations." Brain Pathol 30, no. 3 (2020): 541-53.
- Johnson, B. E., T. Mazor, C. Hong, M. Barnes, K. Aihara, C. Y. McLean, S. D. Fouse, S. Yamamoto, H. Ueda, K. Tatsuno, S. Asthana, L. E. Jalbert, S. J. Nelson, A. W. Bollen, W. C. Gustafson, E. Charron, W. A. Weiss, I. V. Smirnov, J. S. Song,..., and J. F. Costello. "Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma." Science 343, no. 6167 (2014): 189-93.
- Suzuki, H., K. Aoki, K. Chiba, Y. Sato, Y. Shiozawa, Y. Shiraishi, T. Shimamura, A. Niida, K. Motomura, F. Ohka, T. Yamamoto, K. Tanahashi, M. Ranjit, T. Wakabayashi, T. Yoshizato, K. Kataoka, K. Yoshida, Y. Nagata, A. Sato-Otsubo,..., and S. Ogawa. "Mutational Landscape and Clonal Architecture in Grade Ii and Iii Gliomas." Nat Genet 47, no. 5 (2015): 458-68.
- Favero, F., N. McGranahan, M. Salm, N. J. Birkbak, J. Z. Sanborn, S. C. Benz, J. Becq, J. F. Peden, Z. Kingsbury, R. J. Grocok, S. Humphray, D. Bentley, B. Spencer-Dene, A. Gutteridge, M. Brada, S. Roger, P. Y. Dietrich, T. Forshew, M. Gerlinger,..., and C. Swanton. "Glioblastoma Adaptation Traced through Decline of an Idh1 Clonal Driver and Macro-Evolution of a Double-Minute Chromosome." Ann Oncol 26, no. 5 (2015): 880-87.
- Lopez, G. Y., N. A. Oberheim Bush, J. J. Phillips, J. P. Bouffard, Y. A. Moshel, K. Jaeckle, B. K. Kleinschmidt-DeMasters, M. K. Rosenblum, A. Perry, and D. A. Solomon. "Diffuse Midline Gliomas with Subclonal H3f3a K27m Mutation and Mosaic H3.3 K27m Mutant Protein Expression." Acta Neuropathol 134, no. 6 (2017): 961-63.
- Luo, S.; Zhu, S.; Liao, J.; Zhang, Y.; Hou, X.; Luo, T.; Zhao, E.; Xu, J.; Pang, L.; Liang, X.; et al. IDH clonal heterogeneity segregates a subgroup of non-1p/19q codeleted gliomas with unfavourable clinical outcome. Neuropathol. Appl. Neurobiol. 2020, 47, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.G.; Fine, H.A. Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discov. 2021, 11, 575–590. [Google Scholar] [CrossRef]
- Morgan, K.M.; Danish, S.; Xiong, Z. Diffuse astrocytoma with mosaic IDH1-R132H-mutant immuno-phenotype and low subclonal allele frequency. Intractable Rare Dis. Res. 2022, 11, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Wang, X.; Zhang, H.; Wang, J.; Lyaysan, G.; Xu, S.; Tian, K.; Wang, T.; Li, J.; Wang, N.; et al. Dissecting and analyzing the Subclonal Mutations Associated with Poor Prognosis in Diffuse Glioma. BioMed Res. Int. 2022, 2022, 1–19. [Google Scholar] [CrossRef]
- Voisin, M.R.; Gui, C.; Patil, V.; Gao, A.F.; Zadeh, G. Methylation Profiling Identifies Stability of Isocitrate Dehydrogenase Mutation Over Time. Can. J. Neurol. Sci. / J. Can. des Sci. Neurol. 2023, 51, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Senft, C.; Behrens, M.; Lortz, I.; Wenger, K.; Filipski, K.; Seifert, V.; Forster, M.-T. The ability to return to work: a patient-centered outcome parameter following glioma surgery. J. Neuro-Oncology 2020, 149, 403–411. [Google Scholar] [CrossRef]
- Loon, E.; Ernens, W.; Heijenbrok-Kal, M.; Horemans, H.; Ribbers, G.; Bent, M. Long-term employment status and the association with fatigue in patients with grade II glioma. J. Rehabilitation Med. 2021, 53, jrm00198. [Google Scholar] [CrossRef]
- Birendra, K.; DiNardo, C.D. Evidence for Clinical Differentiation and Differentiation Syndrome in Patients With Acute Myeloid Leukemia and IDH1 Mutations Treated With the Targeted Mutant IDH1 Inhibitor, AG-120. Clin. Lymphoma Myeloma Leuk. 2016, 16, 460–465. [Google Scholar] [CrossRef]
- Fujii, T.; Khawaja, M.R.; DiNardo, C.D.; Atkins, J.T.; Janku, F. Targeting Isocitrate Dehydrogenase (IDH) in Cancer. 2016, 21, 373–380.
- Mellinghoff, I. K., M. J. van den Bent, D. T. Blumenthal, M. Touat, K. B. Peters, J. Clarke, J. Mendez, S. Yust-Katz, L. Welsh, W. P. Mason, F. Ducray, Y. Umemura, B. Nabors, M. Holdhoff, A. F. Hottinger, Y. Arakawa, J. M. Sepulveda, W. Wick, R. Soffietti,..., and Indigo Trial Investigators. "Vorasidenib in Idh1- or Idh2-Mutant Low-Grade Glioma." N Engl J Med 389, no. 7 (2023): 589-601.
- Wen, F.; Gui, G.; Wang, X.; Qin, A.; Ma, T.; Chen, H.; Li, C.; Zha, X. Discovery of Novel Dual Inhibitors Targeting Mutant IDH1 and NAMPT for the Treatment of Glioma with IDH1Mutation. J. Med. Chem. 2024, 67, 8667–8692. [Google Scholar] [CrossRef]
- Moore, D. Panobinostat (Farydak): A Novel Option for the Treatment of Relapsed Or Relapsed and Refractory Multiple Myeloma. Pharm. Ther. 2016, 41, 296–300. [Google Scholar]
- Pan, D.; Mouhieddine, T.H.; Upadhyay, R.; Casasanta, N.; Lee, A.; Zubizarreta, N.; Moshier, E.; Richter, J. Outcomes with panobinostat in heavily pretreated multiple myeloma patients. Semin. Oncol. 2023, 50, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Gunn, Kathryn, Matti Myllykoski, John Z. Cao, Manna Ahmed, Bofu Huang, Betty Rouaisnel, Bill H. Diplas, Michael M. Levitt, Ryan Looper, John G. Doench, Keith L. Ligon, Harley I. Kornblum, Samuel K. McBrayer, Hai Yan, Cihangir Duy, Lucy A. Godley, Peppi Koivunen, and Julie-Aurore Losman. "(R)-2-Hydroxyglutarate Inhibits Kdm5 Histone Lysine Demethylases to Drive Transformation in Idh-Mutant Cancers." Cancer Discovery 13, no. 6 (2023): 1478-97.
- Nie, Z.; Shi, L.; Lai, C.; O'Connell, S.M.; Xu, J.; Stansfield, R.K.; Hosfield, D.J.; Veal, J.M.; Stafford, J.A. Structure-based design and discovery of potent and selective KDM5 inhibitors. Bioorganic Med. Chem. Lett. 2018, 28, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Rautajoki, K. J., S. Jaatinen, A. Hartewig, A. M. Tiihonen, M. Annala, I. Salonen, M. Valkonen, V. Simola, E. M. Vuorinen, A. Kivinen, M. J. Rauhala, R. Nurminen, K. K. Maass, S. L. Lahtela, A. Jukkola, O. Yli-Harja, P. Helén, K. W. Pajtler, P. Ruusuvuori,..., and M. Nykter. "Genomic Characterization of Idh-Mutant Astrocytoma Progression to Grade 4 in the Treatment Setting." Acta Neuropathol Commun 11, no. 1 (2023): 176.
- Weller, M.; Felsberg, J.; Hentschel, B.; Gramatzki, D.; Kubon, N.; Wolter, M.; Reusche, M.; Roth, P.; Krex, D.; Herrlinger, U.; et al. Improved prognostic stratification of patients with isocitrate dehydrogenase-mutant astrocytoma. Acta Neuropathol. 2024, 147, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Mallela, A.N.; Shi, D.D.; Tang, L.W.; Abou-Al-Shaar, H.; Gersey, Z.C.; Zhang, X.; McBrayer, S.K.; Abdullah, K.G. Isocitrate dehydrogenase mutations in gliomas: A review of current understanding and trials. Neuro-Oncology Adv. 2023, 5, vdad053. [Google Scholar] [CrossRef]
- Kayabolen, A., E. Yilmaz, and T. Bagci-Onder. "Idh Mutations in Glioma: Double-Edged Sword in Clinical Applications?" Biomedicines 9, no. 7 (2021).
Figure 1.
Examples of neuroglial cells and glioma types resulting from malignant transformation. Glial cells support neuronal processes and maintain the surrounding environment. This figure was created with biorender.com.
Figure 1.
Examples of neuroglial cells and glioma types resulting from malignant transformation. Glial cells support neuronal processes and maintain the surrounding environment. This figure was created with biorender.com.
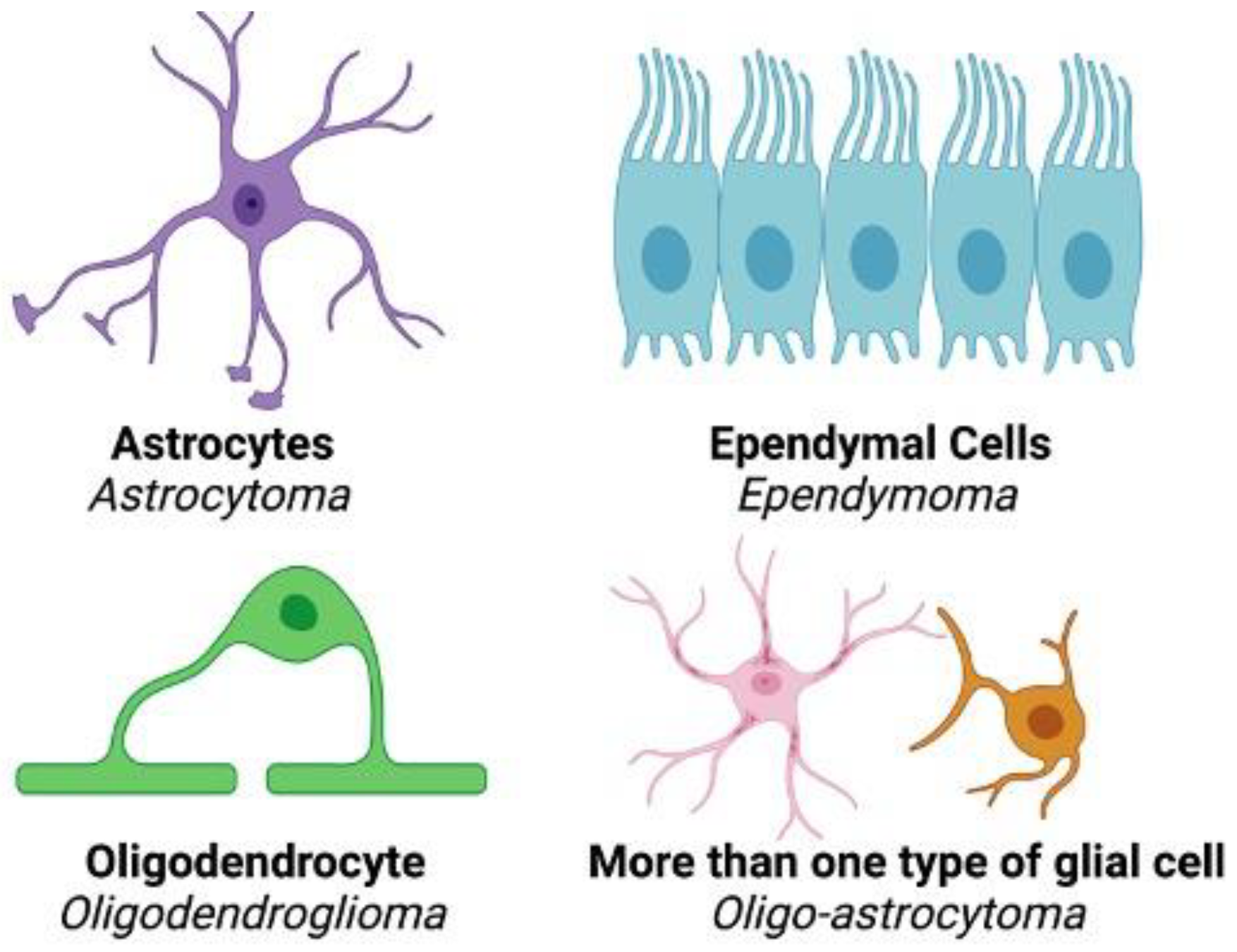
Figure 2.
The difference in roles between wildtype and mutant IDH within the citric acid cycle. Wildtype IDH catalyzes the conversion of isocitrate to αKG. Mutations to conserved residues within the catalytic site of IDH elicit a gain-of-function that results in the aberrant conversion of αKG to D-2-HG. This figure was created with biorender.com.
Figure 2.
The difference in roles between wildtype and mutant IDH within the citric acid cycle. Wildtype IDH catalyzes the conversion of isocitrate to αKG. Mutations to conserved residues within the catalytic site of IDH elicit a gain-of-function that results in the aberrant conversion of αKG to D-2-HG. This figure was created with biorender.com.
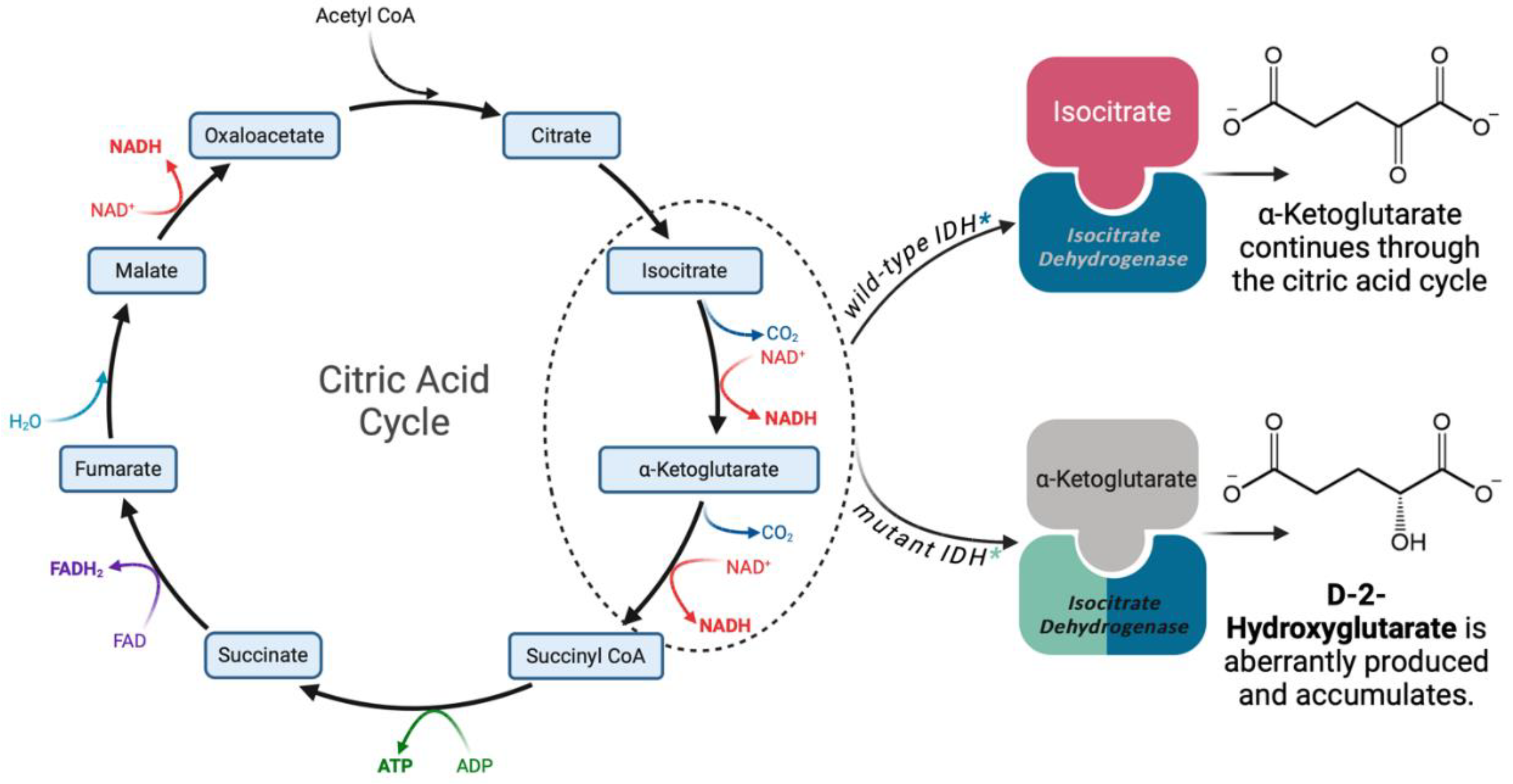
Figure 3.
D-2-HG competitively inhibits various αKG-dependent dioxygenases. From left to right: YTHDC1 with N6-Methyladenosine (m6A), PHD2, TET2 in complex with DNA, the Jumonji domain of human Jumonji domain containing 1C protein. Crystal structures were sourced from Protein Data Bank (PDB); this figure was created using Biorender.com.
Figure 3.
D-2-HG competitively inhibits various αKG-dependent dioxygenases. From left to right: YTHDC1 with N6-Methyladenosine (m6A), PHD2, TET2 in complex with DNA, the Jumonji domain of human Jumonji domain containing 1C protein. Crystal structures were sourced from Protein Data Bank (PDB); this figure was created using Biorender.com.
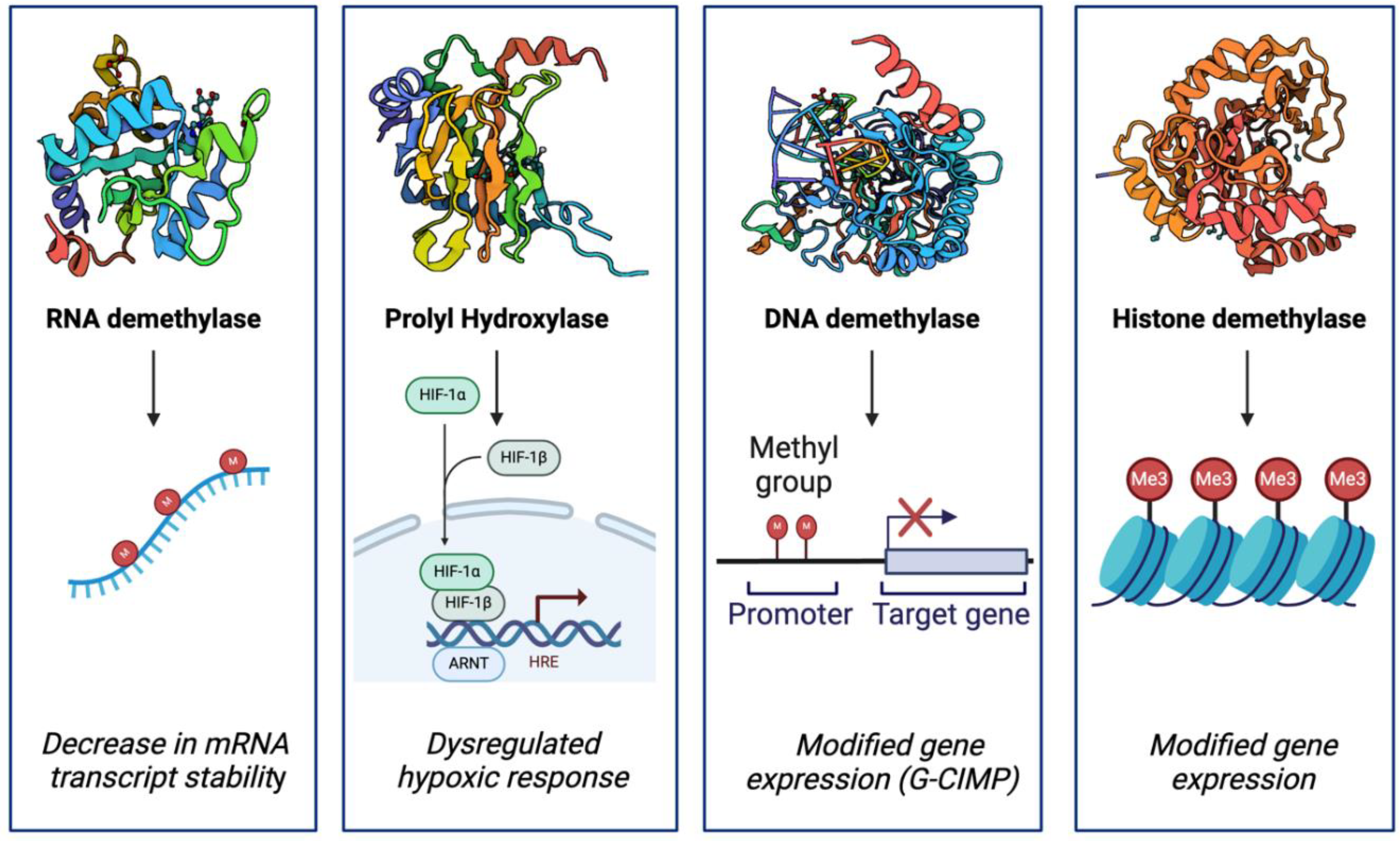
Figure 4.
The molecular impacts of IDH mutations discussed in this review. Metabolic rewiring and D-2-HG mediated inhibition of αKG-Fe(II)-dependent dioxygenase play important roles in the unique phenotype of IDH mutant cells. This figure was created with Biorender.
Figure 4.
The molecular impacts of IDH mutations discussed in this review. Metabolic rewiring and D-2-HG mediated inhibition of αKG-Fe(II)-dependent dioxygenase play important roles in the unique phenotype of IDH mutant cells. This figure was created with Biorender.
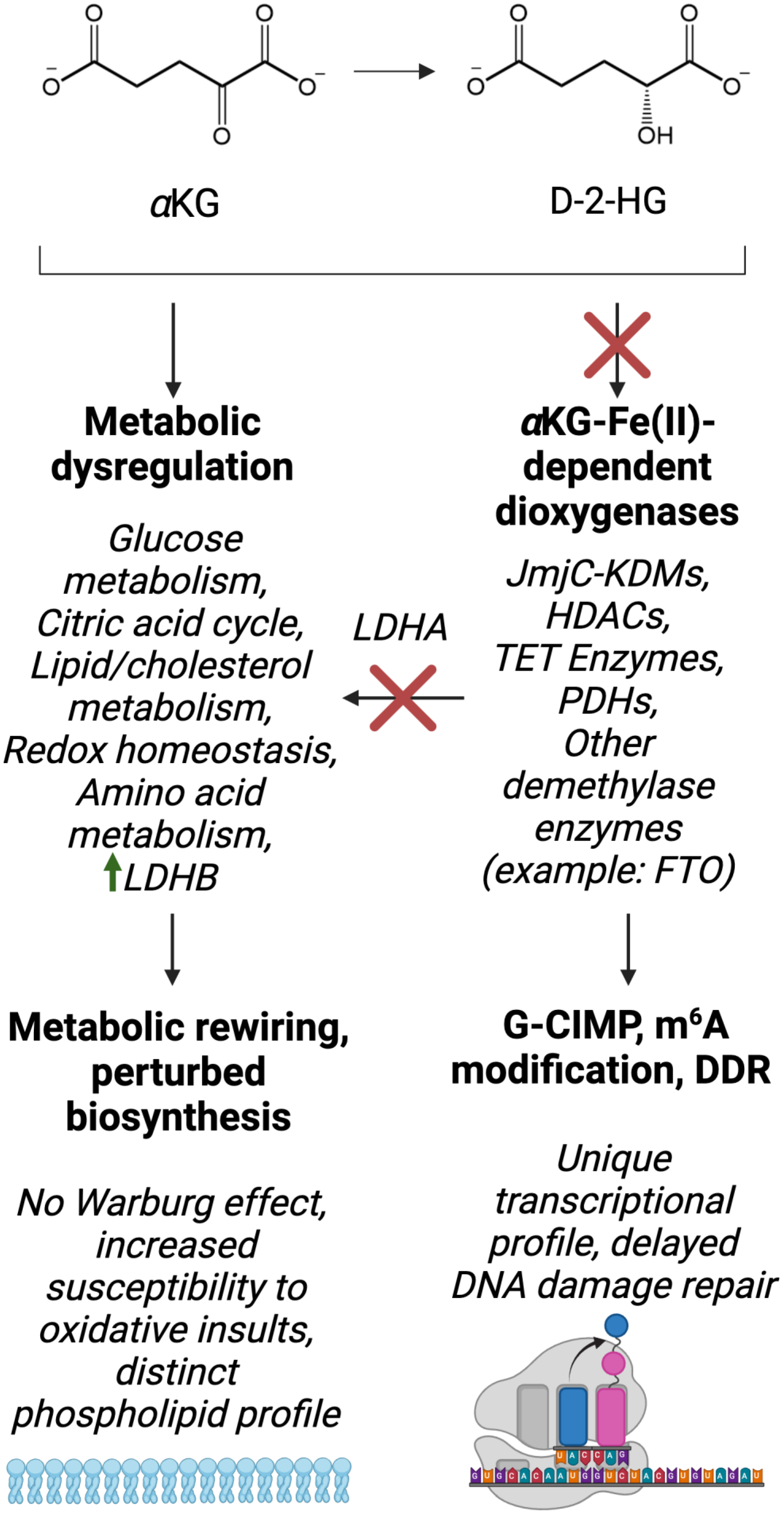

Table 1.
Frequency of IDH variants in low-grade glioma based on values reported in current literature [199,207].
| IDH variant | Frequency in low-grade glioma (%) |
|---|---|
| IDH1-R132H | 62.0-93.0 |
| IDH1-R132C | 2.9-4.3 |
| IDH1-R132G | 1.0-2.5 |
| IDH1-R132S | 1.1-2.2 |
| IDH1-R132L | 0.2-0.6 |
| IDH2-R172K | 2.8-3.0 |
| IDH2-R172W | 0.6 |
| IDH2-R172M | 0.8 |
| IDH2-R172S | 0.2 |
| IDH1-R132PIDH1-R132VIDH2-R172T | Extremely rare |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



