
Submitted:
05 October 2024
Posted:
07 October 2024
You are already at the latest version
Abstract
Efflorescence is a major concern in alkali-activated materials (AAMs), affecting their potential practical applications and challenging the preservation of the material in a time-frozen state for post-characterisation purposes. Efflorescence is a time-dependent phenomenon that may stop with complete water removal. Therefore, this study mixed chemically and physically different secondary raw (slag, fly ash, glass wool, and rock wool) and nonwaste (metakaolin) materials with Na-silicate solution in ratios that facilitated efflorescence and mixtures that could prevent it. The material was cured at 40 °C for 6 days. On the 7th day, half of the intact samples were additionally treated with low-power microwaves (2.45 GHz at 100 W) until complete dehydration to rapidly and gently influence the chemical reaction. The impact of microwave-irradiation-induced dehydration on the specimens was thoroughly examined on day 7, both chemically and physically, and compared with that of a set of samples that were not subjected to dehydration. Chemical alterations were examined using Fourier transform infrared spectroscopy, which revealed the removal of water. Further, X-ray diffraction revealed no changes in crystallinity upon irradiation. The irradiation-increased porosity was evaluated through Mercury Intrusion Porosimetry, whereas the mechanical resilience of the materials was evaluated through compressive strength tests and synchronised with the porosity development. Pores in the irradiated samples that required higher temperatures or longer curing times (glass wool, fly ash, and metakaolin) were spherical; otherwise, dehydration resulted in elongated cracks. This result provided side knowledge on the determination of finalisation of the curing of AAM, which cannot be determined by any surface-penetrating test. Efflorescence formation was evaluated on 1-year-old dehydrated samples, maintained at room temperature, before and after exposure to a water droplet using a low-vacuum scanning electron microscope. This explained the reason why the surface of AAMs exposed to water became slippery and the effect of the water droplet on the surface. The microwave treatment method successfully accomplished the complete dehydration of AAMs within a few minutes, effectively inhibiting reactions in the AAMs, terminating further development of the aluminosilicate network, and partially removing unbonded alkali elements from the chemically unfinished AAM system. Therefore, microwave irradiation can be used to evaluate the chemical compositions of time-evolving materials during dehydration. If the dehydrated samples are stored in a waterless environment, post-characterisation of the AAMs is possible at any later time. The procedure of stopping the reactions offers significant advantages over conventional techniques which require physical processing, such as grinding or milling, the use of solvents, and more time.
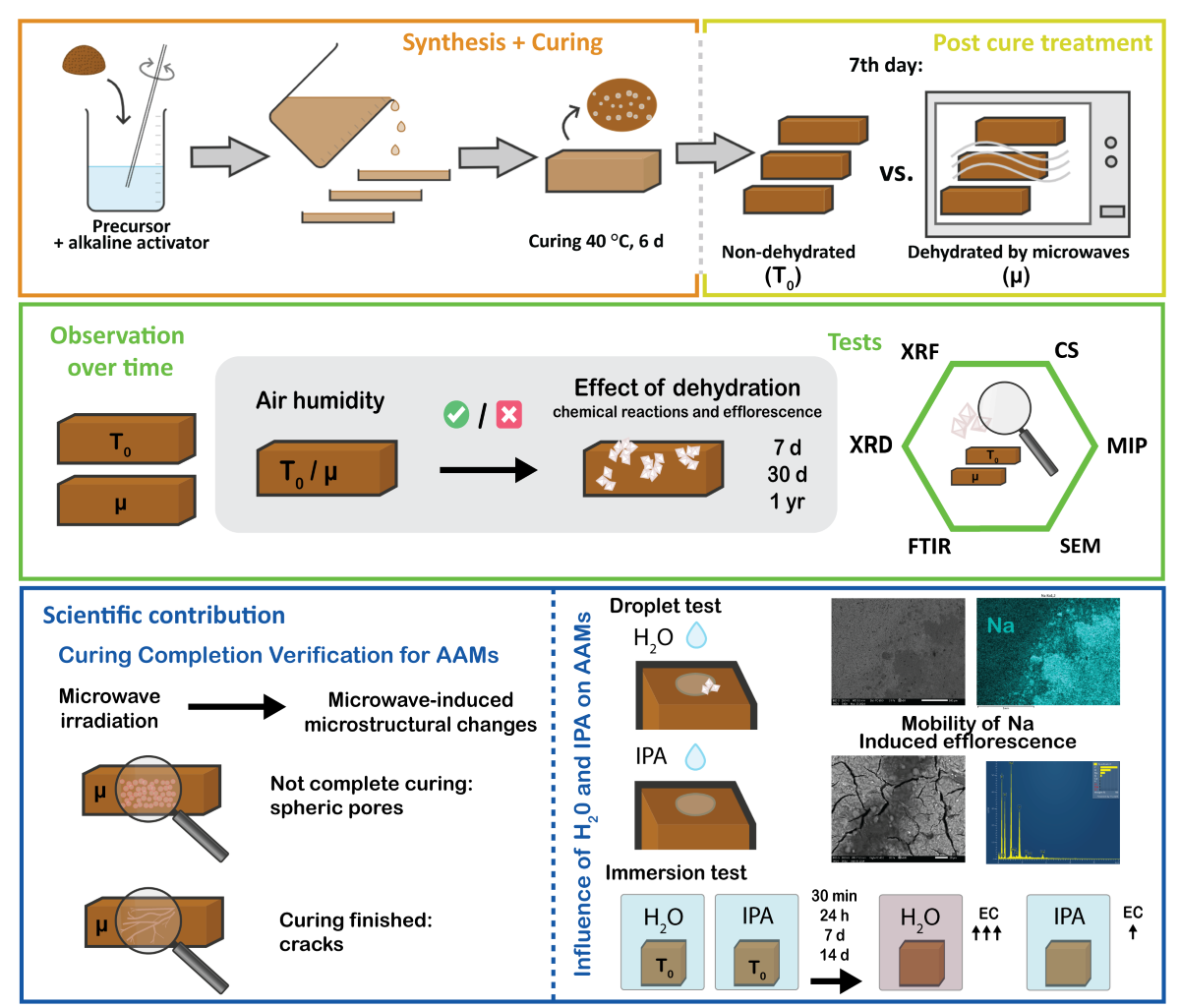
Keywords:
Alkali-activated materials
; Efflorescence
; Dehydration
; Microwave irradiation
; Stopping chemical reactions
; Curing completion
1. Introduction
The construction and civil engineering sectors, in conjunction with the cement production industry, are major contributors to global emissions. The prevailing linear economic model has resulted in a surge in natural resource consumption, global emissions, and waste accumulation. Cement, the most used manmade material in the world and the predominant material for the building and civil engineering sectors, contributes to approximately 8% of the world's total CO2 emissions through its manufacturing processes [1]. The estimated trend of increased percentage of urbanisation and building material usage [2] demands the research and development of a new generation of sustainable materials, with a specific focus on achieving a sustainable construction industry.
In recent decades, there has been a notable increase in the research dedicated to alkali-activated materials (AAMs), which are emerging as promising alternatives to ordinary Portland cement and industrial ceramics [3]. AAMs are cementitious materials formed from solid aluminosilicate precursors with sufficient amounts of Si and Al in amorphous content, which are then alkali-activated using an alkali silicate solution or hydroxides [4]. This alternative material, which can support the objectives of a circular economy, offers economic and environmental benefits, particularly through the reuse of waste materials. As a source of precursors, industrial or agricultural wastes, such as slags, rock wool, glass wool, fly ash, waste glass powder residues, or biomass ashes, can be used to reduce the usage of raw materials in building and civil engineering production [4,5]. AAMs offer several advantages over conventional materials such as simple preparation, sustainability, potential lower cost, vast and versatile access to local precursor materials, lower production energy consumption, low hydration heat, freezing resistance [6], chemical resistance, strong durability, high strength [7], and excellent high-temperature stability. However, a major drawback of AAMs is their inclination toward the time-dependent evolution of the chemical-mineral composition, where the negative outcome is usually observed as efflorescence.
Efflorescence is an intrinsic phenomenon characterised by microstructural changes owing to leaching and carbonation of unreacted alkali cations, leaving behind salts. In the context of AAMs, this phenomenon is attributable to either excess alkali serving as an activator or the dissolution of loosely bound materials, which facilitates the movement of soluble alkalis and the subsequent crystallisation of salts on the surface of the materials [8]. This natural process occurs under normal environmental conditions because of the high alkalinity and tendency of alkalis to migrate within these substances [9]. Visible (common) white deposits, which are characteristic of efflorescence in AAMs, are a typical result of the carbonation of mobile alkalis. However, the presence of efflorescence in AAMs is undesirable because the leaching of calcium salts can reduce the mechanical strength and durability owing to structural degradation [10,11]. However, salts in AAMs typically contain sodium (Na) and are crystalline (thermonatrite, pirssonite, and different sodium carbonate salts). This is because Na alkalis are the most used alkalis in AAM research [12]. Na salts also deteriorate the homogeneity and integrity of AAMs [13] thus, salts containing Na are not desirable in AAMs.
Therefore, products that cause efflorescence are likely to have no future. However, salt formation in construction materials can be mitigated using various methods. Lowering the alkali content in a material can diminish both the solubility and rate at which CO2 is absorbed from the environment [14]. Choosing precursors or cement with a lower alkali content, that is, with a lower number of free alkali cations, can also minimise the risk of efflorescence [15]. Further, the optimisation of the curing conditions by adjusting the temperature and humidity can influence the release of active components and carbonation resistance [16], ultimately contributing to a reduction in efflorescence. Appropriate mix design and curing conditions can reduce the amount of free water available for the migration of alkali elements and salt formation. Further, to avoid efflorescence, the molar amount of elements accessible for the alkali activation reaction, found in the amorphous content, should follow the molar ratio M:Al≤1, where M represents the elements of the first group of the periodic system (typically Na and K), provided the alkalis from the second group of the periodic system are neglected and all amorphous elements dissolve in alkali [17].
However, the theoretical prediction of an ideal mixture is experimentally difficult. Therefore, efflorescence as a time-dependent reaction, although delayed, occurs constantly and changes the chemical-mineralogical composition of the AAM. These changes are owing to the presence of water, which facilitates motion on the ionic-elemental scale in AAM. To hinder or even stop the reactions, the water content must be minimised or completely removed. This is even more crucial when the chemical and mineralogical time evolution of the material under development is followed because with certain techniques (owing to their apparatus characteristics), all samples should be preferably measured simultaneously (aging of the X-ray tube when performing X-ray diffraction analysis [18]) and also re-measured any time later. Therefore, stopping the evolution of materials at preselected times is crucial to understand the design and control of materials in research.
The removal of water from the system should not be complete but should be sufficiently rapid without the introduction of additional mechanical steps (such as milling) and solvents (such as isopropanol). This is to facilitate a decrease in the time required to reach the time-frozen state and avoid a potential influence on the chemistry and mineralogy of the material. In addition, if the water removal technique from the material is sufficiently rapid, different materials can be time-frozen at the same age. A comparison of the methods for dehydration or stopping hydration has mostly been performed using Portland cement. Solvent exchange with isopropanol is recommended by The International Union of Laboratories and Experts in Construction Materials, Systems and Structures (RILEM) as a stoppage method for phase assemblages in cement [19,20]. Isopropanol is weakly aggressive and may interact with the matrix. The interaction depends on the solubility of the material, its porosity, and the interaction between isopropanol and the alkali components of the matrix. Interactions between the material phases in cement and organic solvents have been reported [21,22,23]. With the recent increased interest in more environmentally friendly cementitious materials that exhibit chemical properties different from Portland cement, new research is required to assess proper drying methods. The isopropanol exchange method has been used in only a few studies to dry AAMs with inconsistent results [24], while still requiring grinding of the material and prolonged time or heating to complete the evaporation of isopropanol [25].
Another technique for water removal is the induction of evaporation by heating the sample. Conventional industrial ovens use electric heaters located at the top, bottom, or sides of the apparatus to increase the temperature in the chamber, where heat is conducted into the interior of the sample through the outer edges, resulting in uneven heating owing to the temperature gradient in the material. Indirect heating through conduction and convection, which involves heating the air inside an oven and transferring heat to the sample, can result in higher energy consumption and may take longer depending on the sample size. The longer the exposure time, the longer the time for changes in the chemical-mineral composition of the material. In the worst-case scenario, the outer layer of the heated bulk material may form a crustal structure that would hinder complete dehydration or lead to material explosion, owing to the inner pressures formed when water changes its state from liquid to gas. The procedure of dehydration to hinder unwanted reactions caused by the presence of water in the sample can be accomplished rapidly using a microwave oven where
- The electromagnetic waves in the microwave region heat the sample volumetrically by influencing the dipoles of water molecules that are attempting to align with the rapidly changing electromagnetic field. This causes the water molecules to constantly rub the surrounding material, which increase the temperature owing to molecule-scale friction inside the bulk material [26].
The process of heating a material with electromagnetic irradiation is volumetric on a large scale, with many randomly created heat sources inside the material on the molecular scale. By contrast, conventional heating in a convection oven involves surface heating, which requires more energy and time for dehydration.
The application of microwaves in the civil engineering sector has been demonstrated in several studies and exhibited a successful decrease in water content in building materials using microwave irradiation. This has highlighted the potential application of microwave technology in building materials such as wood, bricks, and soil [31,32,33,34,35]. Nonetheless, research on the impact of microwaves on cement and AAMs remains limited, although the influence of microwaves on the curing process and mechanical strength has been investigated [17,36,37,38,39,40,41,42]. However, the effect of post-curing microwave processing on AAMs and their dehydration has not yet been reported.
This study aimed to evaluate the effectiveness of post-curing low-power microwave irradiation in inducing dehydration in various types of intact AAMs samples, considering the hypothesis that microwave irradiation causing water removal can ‘freeze-in-time‘ chemical reactions without manual destruction of the samples. To test this hypothesis, five different types of precursors, including secondary waste materials such as slag, fly ash, glass, and rock wool, along with the non-waste material metakaolin, were used at different mass ratios in the Na-silicate solution. To evaluate the effects of microwave dehydration during the post-curing phase on the (non)irradiated mechanically intact bulk AAMs, the following physical and chemical properties were analysed: mechanical strength and porosity (mercury intrusion porosimetry (MIP)), chemical (Fourier-transform infrared spectroscopy (FTIR)), and mineralogical (X-ray diffraction (XRD)) composition. The porosity was also evaluated using a scanning electron microscope (SEM), where the pore shape defined whether the AAM was completely cured or would require more time or a higher curing temperature. To the best of our knowledge, this was reported for the first time and cannot be replaced by any conventional surface method. In addition to irradiation influencing porosity through increased inner pressure, newly formed cracks became escape routes for water, owing to which non-bonded, still dissolved, or (at the moment of dehydration) excessive alkali elements were observed on the surface. Only completely cured samples, designed to avoid efflorescence, did not exhibit any early signs of salt after dehydration. However, the presence of salts on one-year-old dehydrated AAMs (compared to their non-dehydrated counterparts) was evaluated along with aged dehydrated samples before and after exposure to a water droplet. Exposure to water demonstrated that excess Na was instantly dragged onto the surface, which is why AAMs with the potential for efflorescence become slippery when in contact with water. In this study, we demonstrated that low-power microwave irradiation can be used to stop chemical reactions and efflorescence provided the sample is not exposed to water or moisture. Although this type of efflorescence hindering is not useful for load-bearing building industry products to be used in real life because of the increase in porosity and decrease in mechanical strength, it can be utilised for (post)characterisation purposes in material development.
2. Materials and Method
The precursors were chemically and mineralogically analysed to theoretically predict the efflorescence ability of AAMs in the prepared mixtures.
2.1. Chemical and Mineralogical Characterisation of Precursors
Five different precursors were used for alkali activation: slag (SA), glass wool (GW), rock wool (RW), fly ash (FA), and metakaolin (MK). Except for MK, which is most commonly used in alkali activation and can be adopted as a standard, all the precursors represent locally obtained industrial waste. All precursors were dried (heating chamber WTB Binder, 70 °C for 24 h), milled (vibrating disk mill Siebtechnik), and sieved below 63 µm for X-ray fluorescence (XRF), XRD, and FTIR analyses.
The mineralogical characteristics of the precursors were determined via XRD measurements (Empyrean PANalytical X-ray diffractometer, Cu X-ray source) in clean room conditions for angles 4–70° with a step size of 0.0263°. Powder samples were prepared in a back-loaded cylindrical sample holder with a diameter of 27 mm. Mineral analysis was performed using Rietveld refinement with the PANalytical X’Pert High Score Plus diffraction software v. 4.8. The amorphous content was evaluated employing an external standard method, as outlined by Madsen et al. [43], using Al2O3 powder (corundum, NIST676a) as the designated reference material.
Loss on ignition (LOI) was determined for dried powder samples by heating in a furnace (Nabertherm B 150) at 550 °C for 2 h in an ambient atmosphere to remove organic compounds. Consequently, samples were heated again at 950 °C and for another 2 h to remove carbonates. The results were presented as mass loss relative to the initial mass of the sample before ignition.
The resulting ignited precursor samples were mixed with Fluxana(s) (FX-X50-2, Li-tetraborate and Li-metaborate mixture, mass ratio 1:1) at a mass ratio of 1:10, respectively, to lower the melting temperature during the formation of the molten discs. To avoid glueing the melt to the Pt crucible, LiBr(l) (50 ml H2O and 7.5 g of LiBr(s) from Acros Organics) was added to the mixture. XRF analysis of the chemical composition was performed on the melted discs using a Thermo Scientific ARL Perform’X Sequential XRF spectrometer, and the data were analysed using UniQuant 5 software.
FTIR (PerkinElmer Spectrum Two) was performed in ATR mode in the range of 380–4000 cm−1 on the precursors to identify the presence of chemical bonds.
2.2. Preparation and Evaluation of Precursors for Synthesis
To synthesise the AAMs, the MK and FA precursors were used as received without any additional treatment. However, RW and GW were processed by milling and sieving to achieve a particle size below 63 µm. Further, SA was milled in large mass (2 kg) for 3 h in a ball mill (ball diameter 5 cm), and sieved to below 400 µm.
The particle size distribution (PSD) of the precursors were determined using laser diffraction granulometry (Microtrack MRB, Sync+TurboSync laser grain size analyser) under a dry-dispersion measurement setting.
The dried precursors without a conductive layer were evaluated using SEM (Jeol JSM-IT500) under low-vacuum conditions at an accelerating voltage of 20 kV to determine the particle shape of the precursors.
2.3. Preparation of AAMs and Microwave Dehydration Treatment
Three mixtures were formulated for each waste precursor (SA, GW, RW, and FA), and for the nonwaste MK, only two (workability of the slurry with the smallest amount of activator (MK 5.1.) exhibited increased rigidity, which impeded the mixing and embedding of the mixture into moulds). Precursors were mixed with an alkali activator Na-silicate solution (Geosil, 344/7, Woelner; SiO2 27.5 m%, Na2O 16.9 m%, H2O 55.6 m%; where m% stand for mass percentage) in ratios presented in Table 1. Mixtures were prepared by experimentally adjusting the activator content to achieve different workability levels of the slurry, from potentially too low to too high, to determine the optimal ratio (mixtures x.3 in Table 1) to avoid buoyancy and gain high compressive strength. Because buoyancy was not controlled solely by water, but also by the Na-silicate solution, the tendency for efflorescence was also affected.
Workability was evaluated by measuring the viscosity (η; Table 1) using viscometer (Haake PK 100, VT 500 with detector PK2 1.0° at 25 °C, with plate-plate geometry of diameter 2 cm and distance between plates of 0.3 mm; with measurement protocol: 60 s upwards ramp (shear rate from 0.13 to 400 s-1), 30 s hold time (shear rate 400 s-1), and 60 s downwards ramp (shear rate from 400 to 0.13 s-1). The lower the amount of liquid, the higher the viscosity. In selected cases, viscosity was not measurable (torque overload: the apparatus stopped when the torque value exceeded 115% of the maximum torque to protect against overload). In the case of mixture SA 2.1, the measured viscosity was 0 owing to severe foaming (when the amount of liquid alkali was higher, slag expressed self-foaming ability).
The slurries of AAM mixtures were moulded in parallels in rubber-silicone 80 × 20 × 20 mm3 moulds and cured in a heating chamber (WTB Binder) at 40 °C for 6 days. The prisms were demoulded after cooling to room temperature. On the 7th day, half of the cured samples were irradiated by microwaves in an inverter microwave oven (Gorenje microwave oven MO 17DV; frequency 2.45 GHz, magnetron source) at 100 W. The dehydration process was conducted at 1-min intervals, with the total dehydration time adjusted based on visual observations of water condensation on a glass beaker covering the sample. The dehydration process was terminated when, after several repetitions of irradiation, no further condensation was observed on the beaker, indicating that the samples were fully dehydrated. Further, the presence of water was evaluated by FTIR measurement. The complete dehydration times of the samples with the percentage mass loss are shown in Supplementary Table S1.
2.4. Characterisation of AAMs
All AAM prisms, non-irradiated and microwave-dehydrated (before and after irradiation), were weighed, and their physical dimensions (height, width, and length) were measured on day 7. The 7-day-old geometric density of the AAMs was calculated by dividing the measured mass by the volume (Figure S2 (a)).
The effect of irradiation was determined as loss of mass in the irradiated material (water evaporation), change in volume, and geometrical density, all calculated as the difference between the mass/volume/geometrical density (Δm/ΔV/ΔGeometrical density) of AAMs after and before dehydration, and normalised to mass/volume/geometrical density before dehydration (results are shown in Supplementary Table S1 and Figure S3).
The compressive strengths of the prisms were measured on day 7 using a mechanical strength testing machine (ToniTechnik ToniNORM).
Destructed pieces of AAMs prisms were milled (vibrating disk mill Siebtechnik) and sieved below 63 µm for FTIR and XRD analysis (same instrument and setup as described in 2.1.). Measurements were performed on 14-day-old and 1-year-old samples to observe the early irradiation effect, long-term evolution, and affinity of the material to water.
Non-destroyed pieces of the AAM prisms were further analysed by MIP (Micromeritics Autopore IV 9500) to evaluate the influence of forced microwave dehydration on the pore size distribution evolution. All samples were dried and kept until MIP measurement in a heating chamber (WTB Binder) at 70 °C.
Microstructural investigation focused on the presence of efflorescence was performed on non-coated, non-destroyed pieces of AAMs: before and after dehydration on 7-day-old and 1-year-old samples, and before and after the application of a water droplet on 1-year-old-samples, using SEM under low vacuum mode at an accelerating voltage of 20 kV. Energy-dispersive X-ray spectroscopy (EDXS, Oxford Instruments, Link Pentafet) was used to evaluate the (potential) salt formation.
Selected 1-month old pieces of the most chemically favourable mixture for the development of efflorescence were additionally exposed to a drop of distilled water and a drop of isopropyl alcohol, for a short-lasting macroscopic development investigation (Macro 5 MP 1/5″ sensor, f/2.4-aperture lens, 4 K, 60 fps video) of changes on the surface. The samples were also immersed in distilled water and isopropyl alcohol (LabExpert, 99.9 %), and the pH and electrical conductivity (Mettler Toledo SevenDirect SD23, pH: InLab Expert PRO-ISM, electrical conductivity (EC): InLab 731-ISM) of the solution were measured after 30 min, 24 h, and 14 days to determine the dissolution in different media.
3. Results and Discussion
The precursors used in this study differed in all aspects to evaluate the validity of the effect of microwave irradiation until the complete dehydration of AAMs and draw overall conclusions.
3.1. Characterisation of the Precursor
The SEM micrographs of the precursors, taken at magnifications of 100 × and 500 ×, and the different sizes, shapes, and morphological aspects of the particles were visualised using SEM and are presented together with the results of PSD in Figure 1.
The shapes of the precursors observed by SEM can provide valuable insights into their microstructural characteristics. The SA particles exhibited dense, irregular, and angular shapes with coarse surfaces as a result of grinding. The GW and RW exhibited an elongated structure with a smooth texture, which is typical for both fibrous insulation materials. The FA particles presented a wide distribution of spherical shapes of various sizes, indicating relatively uniform but diverse particulates. The surfaces of these particles were mostly smooth, with certain degrees of pitting. The MK exhibited a uniform distribution of fine particles with a relatively rough texture and angular shape.
Comparatively, the PSD analysis revealed that SA particles were the largest, followed in descending order by FA, GW, and RW, with the smallest particles observed in the case of MK. The mean diameters and main percentiles are listed in Supplementary Table S2.
Therefore, the trend from larger to smaller average particle sizes is based on the PSD as:
SA > FA > GW > RW > MK
The XRD patterns of the precursors used for the alkali activation and their mineral and amorphous content analyses are shown in Figure 2. The goodness of fit, ranging as 2.73–5.68, surpassed the ideal value of 1. However, this outcome was anticipated, considering that all the precursors comprised waste materials, with the exception of non-waste MK, which was not synthesised in the laboratory from pure substances. Therefore, the evaluation of minerals in the secondary raw material as well as in the raw material exhibited difficulties in (i) not being able to explicitly determine the minerals (certain were barely detectable and certain exhibited overlapping peaks) and (ii) a significant amorphous halo.
The GW and RW patterns exhibited a notably pronounced halo with a few smaller and less pronounced peaks. Thus, the GW and RW were almost completely amorphous (100 and 99.6 %, respectively). The overall determined amorphous content was as follows: GW ≈ RW > FA ≈ MK > SA (Figure 2). The minerals present in the precursors, their mass proportions, and chemical formulas are listed in Supplementary Table S3.
Table 2 presents the LOI results for the five tested (dry) precursors at temperatures of 550 °C (decomposition of an organic compound) and 950 °C (decomposition of carbonates). SA, GW, and RW had LOI values at 550 °C of approximately 5 %. This suggested a higher content of organic matter. FA and MK exhibited relatively low LOI values at 550 °C (0.7 % and 1.3 %, respectively), which was a consequence of the exposure to high temperatures during their production. SA had a significantly higher LOI at 950 °C (7.9 %) than at 550 °C, which was attributed to the decomposition of carbonates (more than 10 % of calcite was detected by XRD; Figure 3) [44]. GW and RW exhibited a decrease in LOI at 950 °C compared to that at 550 °C, which was attributed to the oxidation process (LOI was performed in the non-inert atmosphere). Further, FA and MK exhibited only a slight increase in LOI at 950 °C compared to that at 550 °C, which indicates that FA and MK were exposed to higher temperatures than 950 °C during their ‘synthesis’.
The chemical composition of the amorphous content of all precursors is presented in Table 2 and was determined from the XRF (all elements) and XRD (elements that are present in minerals) results according to the methodology described in a previous work [45]. The elements present in minerals (XRD) were deduced from elements measured by XRF.
The FTIR spectra of precursors are presented in Figure 3. Precursor SA exhibited a strong peak at 1417 cm−1, which was attributed to the asymmetric stretching vibrations of the carbonate ions (CO₃²⁻) in calcite (presence of calcite was determined also by XRD, Figure 2). The peaks at 874 and 712 cm−1 were also owing to calcite [46,47]. The peak observed at approximately 959 cm⁻¹ was attributed to the T–O–Si asymmetric stretching vibration [48], where T can be either silicon (Si) or aluminium (Al). The presence of this non-sharp peak indicated that the slag also contained amorphous silicates/aluminosilicates required for alkali activation.
The GW spectrum broad and less sharp peaks owing to the amorphous nature of the precursor (as indicated by the XRD pattern in Figure 2). The broad peak at approximately 946 cm-1 was associated with the T-O-Si asymmetric stretching vibrations, which are characteristic of silica- or aluminosilica-based materials [48]. The peak at 774 cm-1 could be related to the bending vibrations of Si-O bonds, further suggesting the presence of silica structure [49].
The FTIR spectrum of RW exhibited two distinct broad absorption peaks at wavenumbers 880 and 699 cm−1. The peak at 880 cm-1 was associated with the T-Si-O asymmetric stretching vibration [50] and that at 699 cm-1 was related to the bending vibration of Si-O [51].
The most prominent peak in FA, with a minimum at 1047 cm-1 was characteristic of the Si-O-Si asymmetric stretching vibrations. In the interval of 1253–826 cm-1, a broad vibration of the T-O bond was observed, where the mullite, quartz, and glassy phases overlapped. A less prominent peak at 797 cm-1 was associated with the Si-O-Si bending vibrations [49,52]. The peaks at 675 and 555 cm-1 were related to the bending vibrations of the T-O-Si bonds [52,53], which further confirmed the existence of silicate or aluminosilicate structures in the FA.
The MK spectrum exhibited a well-defined peak with a minimum at 1067, 797, and 727 cm⁻¹ representing Si-O-Si asymmetric stretching vibrations [54]. The peak at 550 cm-1 and the bands at 1253 and 826 cm-1 could be associated with the bending modes of the T-O-Si bonds [53,55], which is expected in metakaolin, a dehydroxylated form of kaolinite that contains aluminosilicate (mullite presence indicated by XRD, Figure 2).
Therefore, the only common parameter for all precursors is that they contain amorphous Al and Si, which indicates that they are potential sources for alkali activation synthesis.
The ingredient used in all experiments with all precursors was a Na-silicate solution, which had, in addition to water peaks (3334 and 1630 cm-1), a peak at 976 cm-1. Because the solution did not contain Al, the peak at 976 cm-1 was owing to Si-O asymmetric stretching vibrations.
3.2. Characterisation of AAMs
The theoretical potential of the mixtures presented in Table 1 to promote or prevent efflorescence was thoroughly examined.
3.2.1. Mixture design chemistry
According to the calculated molar ratios of the elements presented in Table 3 (containing only amorphous elements from the precursor and elements from the alkali-silicate solution), mixtures of 1.3, 3.1, 4.1, 4.3, and 5.2 did not theoretically facilitate the development of efflorescence, considering that the 2nd group of periodic systems does not contribute to the reaction. If the 2nd group participates in the reaction, the only mixture that had the theoretical potential to avoid efflorescence was mixture 5.2. All the remaining mixtures had excess elements of the 1st (and 2nd) group of the periodic system (PS).
3.2.2. Macroscopic Observations of Efflorescence Formation and Response to Microwave Dehydration
Photographs of the 7-day-old non-dehydrated and microwave-dehydrated prisms are presented in Figure 4. The untreated alkali-activated prisms, which were not subjected to microwave dehydration, did not exhibit visible signs of efflorescence on day 7 (one day after the end of the curing treatment conducted at 40 °C). However, the prisms subjected to rapid complete dehydration through microwave irradiation displayed significant morphological alterations compared to their untreated counterparts. The changes were manifested as noticeable curvature, swelling, and surface cracking, where larger ‘surface’ cracks penetrated deep into the interior of the prisms (as in case of μ 5.2. in Figure 4). The only exceptions wherein the external deformation was not significantly pronounced were the mixtures of rock wool prisms. The deformation of the microwave-treated samples was caused by internal pressures formed by the transformation of water from a liquid to a gaseous state, followed by rapid water removal from the prisms (and a decrease in internal pressure)through newly formed cracks.
The non-dehydrated prisms did not exhibit any visible signs of efflorescence after 7 days because of insufficient time for their development. Surface examination of the microwave-dehydrated prisms revealed bright spots formed when the unreacted alkali elements, which had failed to bind to the aluminosilicate network of the AAM over a given curing period of 7 days, were released by the transportation of steam out of the prism. These spots were most prominent on samples with a molar ratio of 1st PS/Al > 1, particularly in the mixtures denoted as μ1.1., μ2.1., μ2.2., μ2.3., μ3.2., μ3.3., μ4.2., and μ5.3. (Figure 4).
When the curing treatment at 40 °C for 6 days ended, water content was still present (Supplementary Table S1), and further reactions were still possible. In the case of microwave dehydration, removing the moisture immediately stopped any Brownian motion, freezing the reactions in time, and removing (part of) the unreacted alkali from the surface.
Photographs of the stored fragments exposed to ambient conditions for 30 days are presented in Figure S1. No additional efflorescence developed on the microwave-dehydrated samples after 30 days. This indicated the successful mitigation of efflorescence formation over a short-term period of 30 days under ambient condition storage.
3.3. Mechanical Properties
The mechanical strengths of the untreated and microwave-dehydrated alkali-activated prisms are presented in Figure 5. A comparative examination of the compressive strengths of the various precursors used in this study revealed that GW and RW exhibited superior strengths, followed by FA, MK, and SA (general trend GW > RW > FA > MK > SA).
Under uniform curing conditions, the particle size of the precursors is an important factor influencing the compressive strength of AAMs samples. The positive impact of smaller raw material particles used in alkali activation has a positive impact on the increased compressive strength [53], which is attributed to the larger reactive surface area in comparison to the volume. Consequently, a higher reaction rate of the precursor and more reacted final material is obtained. This is attributed to the larger reactive surface area in comparison to the volume, leading to a higher reaction rate of the precursor and more reacted final material [54]. Although the particle size is an important factor in the reactivity of AAMs, our observations indicated that other factors, such as the chemical composition of the amorphous content, physical properties, and mixture design, play critical roles in determining the compressive strength of the final product. The amorphous content of the precursor material is critical for AAM synthesis, as it directly affects the reactivity, dissolution, and subsequent formation of the aluminosilicate network. Therefore, the amorphous content of the precursor is a crucial parameter that significantly affects the mechanical properties of the AAMs. In general, the presence of larger amounts of amorphous silica and alumina facilitates higher compressive strength of AAMs [55]. Nonetheless, the versatility of the results indicates that the only common physical parameter is the presence or absence of microwave irradiation.
In all the specimens, a visible decrease in compressive strength was observed upon post-curing microwave-induced dehydration treatment, with the noteworthy exception of mixture 1.1. and 3.3. The compressive strength values remained comparable and exhibited a marginally less significant increase. The decline in compressive strength across the dehydrated samples was ascribed to the generation of cracks and the physical deterioration of the material structure during the heating reaction [44].
As represented in the contour plots in Figure 5 (right column), the optimum ratio between the chemistry of the material and the amount of alkali needed to dissolve the precursor (proper chemical mixture design avoiding efflorescence and aiming for the highest compressive strength [56]) was required to achieve the highest strength for each precursor. This is clearly evident in the case of FA (Figure 5 (d2)), whereas all the others had the lowest ratio leading to efflorescence and the lowest amount of water. The decrease in compressive strength with an increase in microwave irradiation power was determined using Delauney triangulation up to 100 W, depending on the amount of liquid alkali (water and alkali content). In case of less water content, the decrease in compressive strength is smaller, which is logical; less water to be removed from the system implies the production of fewer cracks in the system and fewer imperfections that lead to lower mechanical performance.
The geometrical densities of untreated and microwave-dehydrated AAMs, are presented in Figure 5 (1) as a colour scale and in Figure S2. Mechanical property analysis revealed a reduction in the compressive strength of the microwave-treated samples, primarily attributed to the decrease in geometric density, which is a consequence of newly formed cracks/pores in the irradiated AAMs.
Previous studies on the application of low-power microwave irradiation in the early stages of alkali activation typically resulted in enhanced compressive strength of AAMs owing to the instant increase in the temperature of the slurry and, therefore, the dissolution rate [17,38,57]. However, certain studies have demonstrated an increase in the compressive strength of alkali-activated fly ash with irradiation after conventional curing [58], with no explanation of the microwave used (the source of the microwaves, functioning of microwave in cyclic condition or in continuous mode (i.e., the real-time of irradiation), the mass of irradiated sample, how much water it contained prior to irradiation, etc., which are all crucial parameters for repeating the experiment). Moreover, tests were performed only on one precursor without any definition for when a sample is ‘completely cured’ and material cannot develop further. In addition, there have been certain misleading conclusions on use of microwave power. For instance, that is, 100 W was not sufficient and that 300 W was the ‘dangerous’ power causing cracks; and the proper overall conclusion that lower microwave power required longer time to reach chosen temperature (and ‘level of completeness’ of curing) and higher power required less time, both depending on the mass and size of the specimen, and complete mass of the molecules that interact with the electromagnetic field.
Nonetheless, the present study showed that continuous microwave irradiation of hardened AAMs (that still contain a certain amount of water, 5–20 m%; Table S1) till the point when there is no Brownian motion possible (water is removed from the system; Table S1 and FTIR evaluation in chapter 3.5) decreased the mechanical performance (Figure 5) owing to volumetrically induced porosity in the AAMs (Figure 4). This was further evaluated by MIP and SEM; even with only 100 W we achieved destruction of the sample provided the irradiation was ‘sufficiently long’ (Figure 6).
3.4. Microstructural Investigation
Figure 6. shows micrographs (magnification 50) of the selected mixture samples of each precursor, untreated and microwave-dehydrated, with the molar ratio that is most favourable for efflorescence development (molar ratio 1stPS/Al > 1; see Table 3), along with the corresponding pore size distributions determined by MIP. The pore distributions for all remaining samples are provided in Supplementary Figure S4, whereas the bulk and skeletal densities (also related to the compressive strength) are shown in Supplementary Figure S2.
While MIP analysis detected (entrances of) pores from 1–380 μm, further investigation through scanning microscopy unveiled the shape of formed pores and cracks, and the presence of larger pores and visible cracks within the microstructure of the microwave-treated samples. The total porosity increased with microwave irradiation, particularly when the mixture contained more water (Figure S4; less in mixtures 1.1., 1.2., and more severely in mixtures 2.2., 2.3., 4.2., 5.2., and 5.3.). Considering that every MIP measurement had an error of fragment selection, a small percentage of the difference in the total porosity was not relevant, unless the sample is inhomogeneous. However, from the MIP pore size distribution curve, it was only possible to conclude whether there was still any water present in the system attempting to break out owing to the irradiation-induced pressure unless there were no overall changes in the shape of the curve and total porosity (a few percentages of difference can be assigned to the choice of the fragment), as in the case of alkali-activated RW.
The geometric densities of the samples were lower than or comparable to those after dehydration. When the geometrical density was low, the samples were bloated and reshaped after irradiation, which increased the error in the measurement of dimensions (Figure S3), but could still be used as an indicator of ‘completeness’ of curing. If volume increased after irradiation (Figure S3, green bars), the sample was not ‘completely’ cured (mixtures 2.1. (not that severe), 2.2., 2.3., 5.2., and 5.3.).
However, neither MIP nor the geometrical density can be used to truly evaluate whether the samples were ‘completely’ cured (similarly, no Vicat test or any other tactile surface or surface-penetrating method used to evaluate the ‘rate of the reaction’). Nonetheless, as shown in Figure 6 (only the samples with the highest amounts of liquid alkali per precursor), SEM can reveal the shape and size of the pores, even those that are excessively large for MIP. In the microwave-treated samples, the newly formed microwave-induced pores manifested either as elongated cracks or spherical pores. Spherical pores can form only in the material that is still ‘soft’ and can be ‘squeezed’ on the inside without the ‘straight’ penetration to the surface. Spherical pores were present in samples 2.2., 4.2., and 5.3., for which the MIP indicated a significant increase in the total porosity. This yielded the conclusion that these materials required longer curing times or higher curing temperatures. In contrast, sample 3.2. demonstrated no changes under SEM or MIP, indicating that it was ‘completely’ cured. However, sample 1.2 also did not show significant changes under SEM and total porosity; however, the pore size distribution changed from a monomodal to bimodal curve with the new peak ranging up to 10 µm. Whereas, the peak in irradiated and non-irradiated samples (ranging as 10–300 µm) decreased. This indicated that the water present in the ‘completely’ cured sample exited the aluminosilicate network through newly created smaller-sized cracks. They slightly enlarged the volume of the aluminosilicate network ranging between larger cracks present in the non-irradiated sample, and pushed those bigger cracks together. This can be confirmed through comparisons of SEM micrographs in Figure 6 (a1–2). However, newly formed cracks and pores in microwave-irradiated AAMs negatively affect the mechanical properties and prevent the use of these materials for applications as load-bearing elements in construction. However, they have the potential to be used as insulating materials if the new porosity is in the shape of closed spheres and not open cracks.
3.5. Influence of microwave dehydration on mineralogy and chemical bonds within the AAMs along with material development
Several 7-day-old microwave-irradiated samples exhibited signs of alkaline mobility (as was also reported by [58]; Figure 4) on the surface as bright spots after irradiation (mixtures 1.1., 2.1., 2.2., 2.3., 3.2., 3.3., 4.2., and 5.3., which all have the highest amount of alkali liquid in the mixtures per precursor; Figure 4). However, no signs of salt crystallisation or crystal phase changes owing to the evaporation of water were detected by XRD in the 14-day-old samples. This indicated that the local deposits were either amorphous or below the XRD-detection limit, as shown in Figure S5 (left) in the Supplementary Material.
The FTIR spectra of 14 -day-old AAMs depicted in Figure 7 (left) revealed no distinct changes in the structural chemistry of the aluminosilicate peak [59] of AAMs caused by the post-curing MW microwave treatment on day 7 (green square in Figure 7). The band in the range of 950–980 cm⁻¹, associated to the asymmetric stretching vibration band of T-O-Si (T=Al/Si), was similar for all samples. The major difference was in the broadband spanning 3000–3600 cm⁻¹ (blue square in Figure 7). This region was assigned to O-H stretching vibrations [60], which were indicative of water molecules bound within the material matrix. Similarly, the peak at approximately 1640 cm⁻¹ corresponded to the bending vibration of H-O-H [60], also indicative of water molecules (Figure 3). The absence of this band in the microwave-dehydrated samples indicated complete dehydration, suggesting that irradiation effectively reduced the water content within the materials. Consistent with broadband reduction and disappearance, the treated samples exhibited diminished peak intensities, which reinforced the suggestion of effective dehydration.
The band at 1420 cm⁻¹ indicated the asymmetric stretching vibrations of C–O–C bonds in the carbonate ions (CO32−). In the AAM samples made of the SA precursor, this band is particularly prominent, suggesting the significant presence of the carbonate phase (confirmed by XRD, Figure 2). Along with the peak at 874 cm⁻¹ [61], which is also characteristic of carbonate vibrations, FTIR additionally proved the presence of calcite within the sample (measured by XRD, Figure 2).
However, there was a difference between the irradiated and non-irradiated 14-days-old samples made from SA in the band at 1420 cm⁻¹ (Figure 7 (a1)). The non-irradiated samples exhibited two peaks, irradiated only one (the same peak exhibited by SA). Considering that SA has the highest ratio of amorphous elements from the 2nd group of PS normalised to amorphous Al (Table 3) among all the used precursors (coupled with the fact that mixture 1.1. and 1.2. also have alkali elements in favour of efflorescence), and that non-dehydrated samples still contain water (allowing the movement of ions), the FTIR revealed early efflorescence development in the case of non-dehydrated 14-day-old SA (all mixtures, Figure 7 (a1)). Considering that the non-dehydrated alkali-activated SA contained salts on its surface on day 30 (Figure S1. (a1)), particularly sample 1.2., which had the largest left peak in the band at 1420 cm⁻¹ (Figure 7 (a1)) among non-dehydrated samples at 14 days, this additional peak was attributed to the efflorescence that could not be observed by the naked eye. Because the 14-day-old dehydrated counterparts did not have the ‘efflorescence’ peak (similar to no efflorescence observed on the 30-day-old irradiated SA samples), dehydration efficiently stopped efflorescence formation; that is, at least the rate of efflorescence formation in samples maintained at room temperature.
However, after one year, pulverised samples maintained at room temperature exhibited visible changes in the form of new peaks in the range of 1410–1500 cm-1 and at approximately 860 cm-1 (Figure 7, dashed squares). The peaks were more prominent in mixtures with higher alkali contents and were more pronounced in non-dehydrated samples. In contrast, in the microwave-dehydrated samples, the peaks are less pronounced and more commonly visible as smaller peaks (as in the case of μ1.1. and μ1.2.) or enlarged bands (μ1.3., μ3.3., and μ4.3.). Following prolonged exposure of the samples to ambient conditions, moisture from the air and unreacted alkali allowed the local formation of crystalline efflorescence salts, as confirmed by XRD of the 1-year-old samples in Figure S5. In contrast the 14-day-old samples did not show any newly formed crystalline phases.
Microwave dehydration successfully removed water from AAMs, as indicated by the absence of the spectral range between 3000–3600 cm⁻¹ in the FTIR analysis presented in Figure 7. Together with the XRD analyses, no changes in the chemical bonds or formation of new crystalline phases were observed following the short term of 14 days (Figure S5 in the supplementary material), rendering dehydration potentially useful for stopping the mineralogical reactions after 14 days.
3.6. Long-Term Observation of Dehydrated Samples and Induced Efflorescence Experiment
The 30-day-old non-dehydrated samples that were chemically inclined towards efflorescence showed naked-eye-visible efflorescence (Figure S1), and FTIR of 1-year-old alkali-activated SA samples (all mixtures) exposed to room temperature conditions (constant presence of moisture) showed clear development of the salts (Figure 7 (a2)), regardless of the presence of dehydration. The inner structures were thoroughly examined using SEM, and the surfaces of the dehydrated samples were exposed to water droplets to observe the microstructural potential of salt formation (migration of alkali elements) (Figure 8).
Although signs of efflorescence were not macroscopically visible on microwave dehydrated samples after 1 year, ‘inner’ efflorescence, characterised by acicular or needle-like crystalline habitus, was observed in dehydrated mixtures 2.2., 4.2., and 5.3. Owing to rapid dehydration, the formation of microwave-induced cracks facilitated the permeability of moisture and air through the material; therefore, it was possible to locally determine the formation of Na salts (according to the EDXS results, they are part of the repository collection). This indicates that microwave dehydration is not an effective long-term method for preventing efflorescence formation under ambient storage conditions, because atmospheric moisture can rehydrate the samples, leading to salt formation inside the material. For the prolonged inhibition of efflorescence, it is recommended to store the samples in a vacuum- or humidity-free environment.
Upon reintroducing water onto the microwave-dehydrated AAM samples, Na instantly accumulated on the hydration sites, circularly on the surface of the hydrophobic MK mixture (irradiated 5.3., Figure 7 (e4); which is the first SEM micrograph of the hydrophobicity of the surface), in the surface cracks, which instantly absorbed water (irradiated mixtures from SA, 1.2., and FA, 4.2.), and spread on the surface to the points of water contact (irradiated GW, 2.2., and RW, 3.2.). The immediate appearance of Na on the AAM surface may cause slipperiness upon contact with water, rendering AAMs with improper mixture designs and without proper measures unsuitable for use as walking surfaces (pavements). Because (instant) slipperiness was also observed in our previous study on a geopolymer (made from MK and Na-silicate solution) with a mixture that theoretically did not lead to efflorescence [62], Na-alkali may not be the proper choice for alkalis in alkali-activated synthesis. Na is the smallest atom among the alkalis, and therefore loosely bonded in the structure easily migrates in the aluminosilicate network. However, it also requires only one bond to compensate for its outer electron layer, whereas alkalis from the 2nd group of the periodic system require two bonds, which immediately restrict the movement of larger alkali elements. Therefore, efflorescence and slipperiness can be avoided.
The experiment on induced efflorescence by hydration demonstrates that, despite arresting the reaction through microwave dehydration, the occurrence of efflorescence cannot be entirely prevented if the chemistry of the material remains unfavourable in the presence of moisture or water in the environment.
SEM-EDXS mapping for elements of the 1st and 2nd groups of PS showed exclusive Na excretion at the surface upon induced efflorescence on the dehydrated samples after one year, as shown in Figure S6 in the Supplementary Material.
Time observations of efflorescence induced by the addition of water to the outer surface of the 1-month-old most chemically favourable mixture for the development of efflorescence (2.2. according to the theoretical ratio of 1st group PS/Al Table 3) are presented in Figure 9a. The addition of excessive alkali to the mixture resulted in an excess (free alkali). Consequently, a significantly higher level of alkalinity leaching was observed, which also indicated a greater tendency for efflorescence. The chemistry of solutions in AAMs, where the Na molar ratio exceeded that of Al, facilitated an excess of Na ions that were not bound to the structure [63]. This could be observed by the naked eye 30 min after water droplet introduction (Figure 9a,c).
When a drop of isopropanol (IPA) was added, rapid evaporation was observed, leaving white traces on the surface in the area of droplet contact after 60 s (Figure 9b,c). However, the effects of IPA on AAMs and geopolymers have not yet been thoroughly studied. The white traces may be the result of the formation of the first stages of efflorescence or the mobilisation of pre-existing efflorescence in the outermost layer of the overburden, which cannot yet be detected.
A detailed investigation of the visible edges of the dried droplets of distilled water and IPA (Figure 9c) is presented in Figure S7 in the Supplementary Material.
Higher amounts of free alkalis also indicate a higher electrical conductivity (EC) of the solution [63], as shown in Figure 10, where the immersion of 2.2. (with excess alkali) in distilled water and IPA increased the EC over the immersion time. This increase was more pronounced in the case of distilled water, whereas it was still present after IPA immersion. Pure IPA, such as the 99.9 % pure IPA used in the study, does not conduct electricity, as shown in Figure 8a. However, after immersion of the samples in IPA, the solution contained a small amount of ions, which can conduct to a certain extent. Distilled water, which has low conductivity of approximately 3 µS/cm (where deionised water has EC 0 µS/cm), dissolves free alkalis and unbonded ions from the AAM sample. This results in a significant increase of EC and water solution colour intensity over time.
Detailed data on the electrical resistivity and temperature of the EC and pH probes are presented in Supplementary Table S4.
4. Conclusions
This study aimed to evaluate the influence of low-power microwave irradiation on conventionally cured few-day-old AAMs, with a focus on long-term efflorescence in worst-case scenario mixtures. To draw an overall conclusion in the field of AAMs, the precursors used differed in all aspects: particle size and shape, chemistry, mineralogy, and amorphous content. In addition to using various precursors, AAMs were prepared with different mass ratios of precursor to Na-silicate solution to vary the chemistry (through the addition of Na and Si) and amount of water.
Although low-power microwave irradiation in the early stages of alkali activation usually boosts the compressive strength of AAMs owing to the instant increase in temperature and, therefore, the dissolution rate, the impact of irradiation on the compressive strength of hardened AAMs while boosting volumetric dehydration is aversive (up to 72 % decrease in the case of GW 2.3.) and a non-reversible effect through an increase in porosity (up to a 29 % increase in the case of MK 5.3.). However, the microwave-induced porosity had two types of pore shapes: (i) elongated cracks and (ii) spheres. The first is formed in fully cured and hardened AAM, and the second is formed in AAM, which required additional time or a higher curing temperature. Therefore, microwave irradiation of AAMs can be used to determine whether AAM is completely cured, as well as to foam AAM, while avoiding the buoyancy of gases, which can result in volumetrically homogenous porosity and potentially useful building material as an insulator. However, AAMs that end with elongated cracks cannot be used for any purpose in building and civil engineering, not for load-bearing products (low mechanical strength) or for insulation (large, elongated, connected pores), regardless of whether the mixture had the potential to be efflorescence-free.
Nonetheless, the foaming of slow-curing AAMs can be performed only on AAMs that have alkali elements lower than the limit value to avoid salt formation. In addition, all crucial reactions should be near completion, implying that an increase in temperature provides the final boost to the curing procedure. However, the dehydration for foaming should not be complete to avoid the formation of elongated cracks.
Instant removal of water was achieved through pressure-created cracks using a volumetric heating approach. Along with water migrating out from the heated material, unreacted and unbonded alkali existed, which was observed as white stains on the surface of the AAM. The more efflorescence-favourable the mixture, the more obvious and prominent the white stains, which are the first signs of stopping chemical reactions. With time, salts formed on the surface of non-dehydrated highly efflorescence-favourable mixtures (slag with the highest amount of alkali) after 1 month (detected by FTIR after 14 days), which further evolved in the powder form regardless of the (non)dehydration. Dehydrated samples did not show efflorescence that would be visible to the naked eye even after one year when kept under room conditions (closed in plastic containers, but not air-tight); however, FTIR and XRD of 1-year old samples exhibited efflorescence. To perform chemical analysis on old, dehydrated samples, the samples should be kept under moisture-free conditions.
Because rapid dehydration opened the structure of AAM through elongated cracks, the ‘outer-surface’ of AAM was now also on the inside of the AAM, easily approachable by air and moisture. Therefore, in 1-year-old samples made with excessive alkali (dehydrated mixtures 2.2., 4.2., 5.3.), the formation of Na salts in the inner structure was observed. However, these salts were difficult to find, thus they formed only locally.
Nonetheless, when water was introduced back into the dehydrated system that had the highest amount of alkali (as the droplet on surface of the AAM), ‘efflorescence’ appeared as Na accumulated on the spot where water was applied. If the surface was hydrophobic, the Na stains were small circles; otherwise, they spread around the surface to the point where the water reached. If the surface had cracks, water penetrated the AAM and exposed Na in the cracks. Thus, the reactions were stopped; however, efflorescence could still form if conditions allowed (moisture in the environment or water).
The instant appearance of Na on the surface of the AAMs may be the cause of the instant slipperiness created when the AAM comes into contact with water. Thus, AAMs might not be useful for the walking surface (pavement) if proper measures are not introduced: a rough surface on the walking side of the AAM, sand throughout the entire volume or at least on the surface of AAM, AAM so porous that water penetrates instantly (heavy metal and toxic element leaching should be avoided), rubber on the surface of AAM, or any other type of surface modification. In addition, these results may require the replacement of Na-alkalis in AAMs with alkali elements from the 2nd group of the periodic system, which are larger as atoms, need two bonds, and are therefore less mobile in the aluminosilicate network.
Therefore, the removal of water from intact bulk AAM prisms using volumetric heating can quickly and efficiently stop the chemical and mineralogical evolution of the material, whereas the irradiation itself does not influence the chemistry and mineralogy of AAM. In materials research, where the time evolution of samples follows, being able to physically freeze material-in-time is crucial for quality research.
Induced efflorescence and immersion tests with distilled water indicated that an improper mix design led to efflorescence and leaching. While IPA droplets and immersion may promote small changes in the material chemistry of AAMs, other methods of chemical reaction hindering, such as microwave dehydration, should be further investigated. However, polishing of AAM samples for SEM investigation should not be performed with water, as also IPA can induce changes on the surface of the polished sample.
Moreover, microwave-irradiation-induced dehydration instantly stopped efflorescence, which might represent a promising, time-efficient, cost-effective, solvent-free, and environmentally friendly alternative to the conventional mitigation of efflorescence (using organic solvents such as acetone and isopropanol) in other cement-based materials. However, the irradiated samples should be maintained in a waterless environment.
Author Contributions
Conceptualization, A.T. and B.H.; data curation, A.T. and B.H.; formal analysis, A.T. and B.H.; funding acquisition, A.T. and B.H.; investigation, A.T. and B.H.; methodology, A.T. and B.H.; validation, A.T. and B.H.; visualization, A.T. and B.H.; writing-original draft, A.T.; writing-review and editing, A.T. and B.H. All authors have read and agreed to the published version of the manuscript.
Funding
This study is part of Dr Barbara Horvat's ARIS project and was financially supported by the Slovenian Research Agency under Grant No. J2-3035. Anže Tesovnik was supported by the Young Researcher Program within the Slovenian Research Agency Program Group P2-0273.
Data availability status
The original data presented in the study are openly available in the repository DiRROS at http://hdl.handle.net/20.500.12556/DiRROS-18221.
Acknowledgments
This study is part of Dr Barbara Horvat's ARIS project and was performed at the Slovenian National Building and Civil Engineering Institute while Dr Horvat was still employed there, for which Dr Horvat is thanking. Dr Horvat is thanking also the Milan Vidmar Electric Power Research Institute for offering peer review, proofreading, and evaluation of the work by the committee. Anže Tesovnik is supported by the Young Researcher Program within ARIS Program Group.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAM | Alkali-activated material |
| SA | Electric arc furnace slag |
| RW | Rock wool |
| GW | Glass wool |
| FA | Fly ash |
| MK | Metakaolin |
| µ | Dehydrated sample, with microwave irradiation |
References
- J.S. Damtoft, J. Lukasik, D. Herfort, D. Sorrentino, E.M. Gartner, Sustainable development and climate change initiatives, Cement and Concrete Research 38 (2008) 115–127. [CrossRef]
- K. Kourtit, P. Nijkamp, H. Scholten, The Future of the New Urban World, International Planning Studies 20 (2015) 4–20. [CrossRef]
- J.S. Damtoft, J. Lukasik, D. Herfort, D. Sorrentino, E.M. Gartner, Sustainable development and climate change initiatives, Cement and Concrete Research 38 (2008) 115–127. [CrossRef]
- J.L. Provis, Alkali-activated materials, Cement and Concrete Research 114 (2018) 40–48. [CrossRef]
- S. Zhang, A. Keulen, K. Arbi, G. Ye, Waste glass as partial mineral precursor in alkali-activated slag/fly ash system, Cement and Concrete Research 102 (2017) 29–40. [CrossRef]
- B. Chen, J. Wang, Experimental Study on the Durability of Alkali-Activated Slag Concrete after Freeze-Thaw Cycle, Advances in Materials Science and Engineering 2021 (2021) e9915639. [CrossRef]
- L. Jin, G. Huang, Y. Li, X. Zhang, Y. Ji, Z. Xu, Positive Influence of Liquid Sodium Silicate on the Setting Time, Polymerization, and Strength Development Mechanism of MSWI Bottom Ash Alkali-Activated Mortars, Materials 14 (2021) 1927. [CrossRef]
- M.A. Longhi, E.D. Rodríguez, B. Walkley, Z. Zhang, A.P. Kirchheim, Metakaolin-based geopolymers: Relation between formulation, physicochemical properties and efflorescence formation, Composites Part B: Engineering 182 (2020). [CrossRef]
- Z. Zhang, J.L. Provis, X. Ma, A. Reid, H. Wang, Efflorescence and subflorescence induced microstructural and mechanical evolution in fly ash-based geopolymers, Cement and Concrete Composites 92 (2018) 165–177. [CrossRef]
- H.-J. Kim, S.-P. Kang, G. Choe, Effect of Red Mud Content on Strength and Efflorescence in Pavement using Alkali-Activated Slag Cement, International Journal of Concrete Structures and Materials 12 (2018). [CrossRef]
- X. Yao, T. Yang, Z. Zhang, Compressive strength development and shrinkage of alkali-activated fly ash–slag blends associated with efflorescence, Mater Struct 49 (2016) 2907–2918. [CrossRef]
- A. Cousture, N. Renault, K. Ndiaye, J.-L. Gallias, New binder resulting from alkali-activation of calcareous components, (n.d.).
- R. Jia, Q. Wang, T. Luo, Deterioration and mitigation of efflorescence of alkali-activated phosphorus slag, Construction and Building Materials 407 (2023) 133500. [CrossRef]
- C. Dow, F.P. Glasser, Calcium carbonate efflorescence on Portland cement and building materials, Cement and Concrete Research 33 (2003) 147–154. [CrossRef]
- M.A. Longhi, Z. Zhang, B. Walkley, E.D. Rodríguez, A.P. Kirchheim, Strategies for control and mitigation of efflorescence in metakaolin-based geopolymers, Cement and Concrete Research 144 (2021) 106431. [CrossRef]
- M. Nedeljković, B. Ghiassi, S. van der Laan, Z. Li, G. Ye, Effect of curing conditions on the pore solution and carbonation resistance of alkali-activated fly ash and slag pastes, Cement and Concrete Research 116 (2019) 146–158. [CrossRef]
- B. Horvat, M. Pavlin, V. Ducman, Influence of microwaves in the early stage of alkali activation on the mechanical strength of alkali-activated materials, Ceramics International 49 (2023) 24246–24258. [CrossRef]
- Common X-Ray Tube Failure Modes, (n.d.). https://www.spellmanhv.com/en/Technical-Resources/Application-Notes-X-Ray-Generators/AN-02 (accessed April 25, 2024).
- R. Snellings, J. Chwast, Ö. Cizer, N. De Belie, Y. Dhandapani, P. Durdzinski, J. Elsen, J. Haufe, D. Hooton, C. Patapy, M. Santhanam, K. Scrivener, D. Snoeck, L. Steger, S. Tongbo, A. Vollpracht, F. Winnefeld, B. Lothenbach, RILEM TC-238 SCM recommendation on hydration stoppage by solvent exchange for the study of hydrate assemblages, Mater Struct 51 (2018) 172. [CrossRef]
- R. Snellings, J. Chwast, Ö. Cizer, N. De Belie, Y. Dhandapani, P. Durdzinski, J. Elsen, J. Haufe, D. Hooton, C. Patapy, M. Santhanam, K. Scrivener, D. Snoeck, L. Steger, S. Tongbo, A. Vollpracht, F. Winnefeld, B. Lothenbach, Report of TC 238-SCM: hydration stoppage methods for phase assemblage studies of blended cements—results of a round robin test, Mater Struct 51 (2018) 111. [CrossRef]
- G. Kakali, S. Tsivilis, E. Aggeli, M. Bati, Hydration products of C3A, C3S and Portland cement in the presence of CaCO3, Cement and Concrete Research 30 (2000) 1073–1077. [CrossRef]
- L. Mitchell, J. Margeson, The effects of solvents on C–S–H as determined by thermal analysis, Journal of Thermal Analysis and Calorimetry 86 (2006) 591–594. [CrossRef]
- J.J. Beaudoin, P. Gu, J. Marchand, B. Tamtsia, R.E. Myers, Z. Liu, Solvent Replacement Studies of Hydrated Portland Cement Systems: The Role of Calcium Hydroxide, Advanced Cement Based Materials 8 (1998) 56–65. [CrossRef]
- Z. Zhang, G.W. Scherer, Physical and chemical effects of isopropanol exchange in cement-based materials, Cement and Concrete Research 145 (2021) 106461. [CrossRef]
- K. Yang, C.E. White, Multiscale pore structure determination of cement paste via simulation and experiment: The case of alkali-activated metakaolin, Cement and Concrete Research 137 (2020) 106212. [CrossRef]
- N.J. English, J.M.D. MacElroy, Molecular dynamics simulations of microwave heating of water, The Journal of Chemical Physics 118 (2003) 1589–1592. [CrossRef]
- P. Marracino, M. Liberti, G. d’Inzeo, F. Apollonio, Water response to intense electric fields: A molecular dynamics study, Bioelectromagnetics 36 (2015) 377–385. [CrossRef]
- M. Drab, E. Gongadze, L. Mesarec, S. Kralj, V. kralj-iglic, The internal and external dipole moment of a water molecule and orientational ordering of water dipoles in an electric double layer, 2018. [CrossRef]
- M. Qin, L. Zhang, H. Wu, Dielectric Loss Mechanism in Electromagnetic Wave Absorbing Materials, Advanced Science 9 (2022) 2105553. [CrossRef]
- P. Lunkenheimer, S. Emmert, R. Gulich, M. Köhler, M. Wolf, M. Schwab, A. Loidl, Electromagnetic-radiation absorption by water, Phys. Rev. E 96 (2017) 062607. [CrossRef]
- M. Novotný, K. Šuhajda, J. Sobotka, J. Gintar, Use of EMW Radiation in the Building Industry, Advanced Materials Research 1041 (2014) 297–302. [CrossRef]
- J. Sobotka, R. Smolka, Use of EMW radiation in the building industry at defects in buildings, MATEC Web Conf. 93 (2017) 01008. [CrossRef]
- J. Sobotka, R. Kolář, Drying of the Basement Spaces of the Faculty of Arts in Brno, Applied Mechanics and Materials 861 (2017) 295–302. [CrossRef]
- M. Jastrzębska, PRACTICAL APPLICATION OF THE MICROWAVE OVEN IN THE GEOTECHNICAL LABORATORY, Architecture, Civil Engineering, Environment 12 (2019) 91–104. [CrossRef]
- J. Sobotka, R. Smolka, Thermal performance of wooden elements exposed to microwave radiation during treatment of building defects, MATEC Web Conf. 279 (2019) 02012. [CrossRef]
- S. Hong, H. Kim, Robust synthesis of coal bottom ash-based geopolymers using additional microwave heating and curing for high compressive strength properties, Korean J. Chem. Eng. 36 (2019) 1164–1171. [CrossRef]
- H. S, K. H, Rapid compressive strength achievement of coal bottom ash based geopolymer by employing microwave energy, TechConnect Briefs 2 (2018) 215–218.
- S. Hong, H. Kim, Effects of Microwave Energy on Fast Compressive Strength Development of Coal Bottom Ash-Based Geopolymers, Sci Rep 9 (2019) 15694. [CrossRef]
- X. Guan, W. Luo, S. Liu, A.G. Hernandez, H. Do, B. Li, Ultra-high early strength fly ash-based geopolymer paste cured by microwave radiation, Developments in the Built Environment 14 (2023) 100139. [CrossRef]
- S.K. Saxena, M. Kumar, N.B. Singh, Influence of alkali solutions on properties of pond fly ash-based geopolymer mortar cured under different conditions, Advances in Cement Research 30 (2018) 1–7. [CrossRef]
- A. Graytee, J.G. Sanjayan, A. Nazari, Development of a high strength fly ash-based geopolymer in short time by using microwave curing, Ceramics International 44 (2018) 8216–8222. [CrossRef]
- S. Onutai, S. Jiemsirilers, P. Thavorniti, T. Kobayashi, Fast microwave syntheses of fly ash based porous geopolymers in the presence of high alkali concentration, Ceramics International 42 (2016) 9866–9874. [CrossRef]
- I.C. Madsen, N.V.Y. Scarlett, A. Kern, Description and survey of methodologies for the determination of amorphous content via X-ray powder diffraction, Zeitschrift Für Kristallographie 226 (2011) 944–955. [CrossRef]
- O. Heiri, A.F. Lotter, G. Lemcke, Loss on ignition as a method for estimating organic and carbonate content in sediments: reproducibility and comparability of results, Journal of Paleolimnology 25 (2001) 101–110. [CrossRef]
- B. Horvat, V. Ducman, Potential of Green Ceramics Waste for Alkali Activated Foams, Materials 12 (2019) 3563. [CrossRef]
- S. Hajji, T. Turki, A. Boubakri, M. Ben Amor, N. Mzoughi, Study of cadmium adsorption onto calcite using full factorial experiment design, Desalination and Water Treatment 83 (2017) 222–233. [CrossRef]
- F.B. Reig, J.V.G. Adelantado, M.C.M. Moya Moreno, FTIR quantitative analysis of calcium carbonate (calcite) and silica (quartz) mixtures using the constant ratio method. Application to geological samples, Talanta 58 (2002) 811–821. [CrossRef]
- X. Gao, Q.L. Yu, H.J.H. Brouwers, Reaction kinetics, gel character and strength of ambient temperature cured alkali activated slag–fly ash blends, Construction and Building Materials 80 (2015) 105–115. [CrossRef]
- M. Criado, A. Fernández-Jiménez, A. Palomo, Alkali activation of fly ash: Effect of the SiO2/Na2O ratio, Microporous and Mesoporous Materials 106 (2007) 180–191. [CrossRef]
- A. Agarwal, M. Tomozawa, Correlation of silica glass properties with the infrared spectra, Journal of Non-Crystalline Solids 209 (1997) 166–174. [CrossRef]
- I. Prasad, A. Chandorkar, Spectroscopy of silicon dioxide films grown under negative corona stress, Journal of Applied Physics 94 (2003) 2308–2310. [CrossRef]
- Z. Zhang, H. Wang, J.L. Provis, Quantitative study of the reactivity of fly ash in geopolymerization by FTIR, Journal of Sustainable Cement-Based Materials 1 (2012) 154–166. [CrossRef]
- B. Walkley, R.S. Nicolas, M.-A. Sani, J.D. Gehman, J.S.J. van Deventer, J.L. Provis, Phase evolution of Na2O–Al2O3–SiO2–H2O gels in synthetic aluminosilicate binders, Dalton Trans. 45 (2016) 5521–5535. [CrossRef]
- M. Sitarz, W. Mozgawa, M. Handke, Vibrational spectra of complex ring silicate anions — method of recognition, Journal of Molecular Structure 404 (1997) 193–197. [CrossRef]
- L. Gao, Y. Zheng, Y. Tang, J. Yu, X. Yu, B. Liu, Effect of phosphoric acid content on the microstructure and compressive strength of phosphoric acid-based metakaolin geopolymers, Heliyon 6 (2020) e03853. [CrossRef]
- P. Duxson, S.W. Mallicoat, G.C. Lukey, W.M. Kriven, J.S.J. van Deventer, The effect of alkali and Si/Al ratio on the development of mechanical properties of metakaolin-based geopolymers, Colloids and Surfaces A: Physicochemical and Engineering Aspects 292 (2007) 8–20. [CrossRef]
- Y. Sun, P. Zhang, J. Hu, B. Liu, J. Yang, S. Liang, K. Xiao, H. Hou, A review on microwave irradiation to the properties of geopolymers: Mechanisms and challenges, Construction and Building Materials 294 (2021) 123491. [CrossRef]
- F. Khaleel, C. Atiş, U. Durak, S. İlkentapar, O. Karahan, The Effect of Microwave Curing on the Strength Development of Class-F Fly Ash-Based Geopolymer Mortar, Erciyes Üniversitesi Fen Bilimleri Enstitüsü Fen Bilimleri Dergisi 37 (2021) 118–129.
- R.K. Vempati, A. Rao, T.R. Hess, D.L. Cocke, J. Lauer, Fractionation and characterization of Texas lignite class F fly ash by XRD, TGA, FTIR and SFM, Cement and Concrete Research; (United States) 24:6 (1994). [CrossRef]
- B. Mojet, S. Ebbesen, L. Lefferts, ChemInform Abstract: Light at the Interface: The Potential of Attenuated Total Reflection Infrared Spectroscopy for Understanding Heterogeneous Catalysis in Water, Chemical Society Reviews 39 (2010) 4643–55. [CrossRef]
- G.-B. Cai, S. Chen, L. Liu, J. Jiang, H.-B. Yao, A.-W. Xu, S.-H. Yu, 1,3-Diamino-2-hydroxypropane-N,N,N′,N′-tetraacetic acid stabilized amorphous calcium carbonate: Nucleation, transformation and crystal growth, Crystengcomm 12 (2010). [CrossRef]
- B. Horvat, B. Mušič, Waste rubber incorporated in the alkali-activated metakaolin’s aluminosilicate network enhanced by microwave irradiation, in: 6th International Conference on Technologies & Business Models for Circular Economy, University of Maribor, University Press, Faculty of Chemistry and Chemical Engineering, 2024: pp. 19–51. [CrossRef]
- D. Kioupis, S. Tsivilis, Alkali leaching control of construction and demolition waste based geopolymers, MATEC Web Conf. 149 (2018) 01064. [CrossRef]
Figure 1.
SEM micrographs of precursors at two magnifications: 100 (left column) and 500 (centre column), displaying the morphological differences between SA, GW, RW, FA, and MK particles. On the right, the results of the particle size distribution (PSD) of the precursor is presented based on distribution histogram q3 and cumulative curve Q3.
Figure 1.
SEM micrographs of precursors at two magnifications: 100 (left column) and 500 (centre column), displaying the morphological differences between SA, GW, RW, FA, and MK particles. On the right, the results of the particle size distribution (PSD) of the precursor is presented based on distribution histogram q3 and cumulative curve Q3.
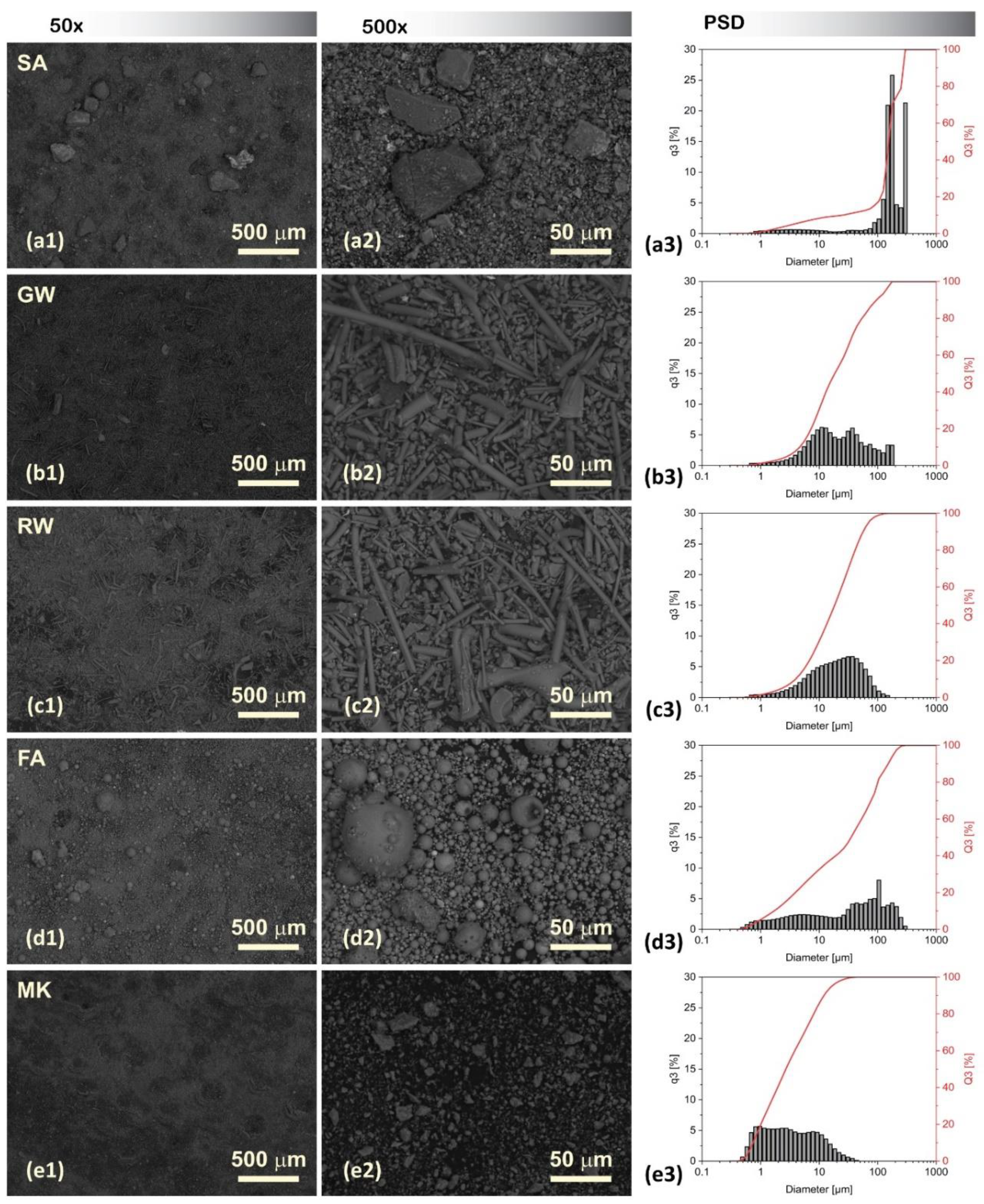
Figure 2.
XRD patterns of precursors used in the study with determined minerals.
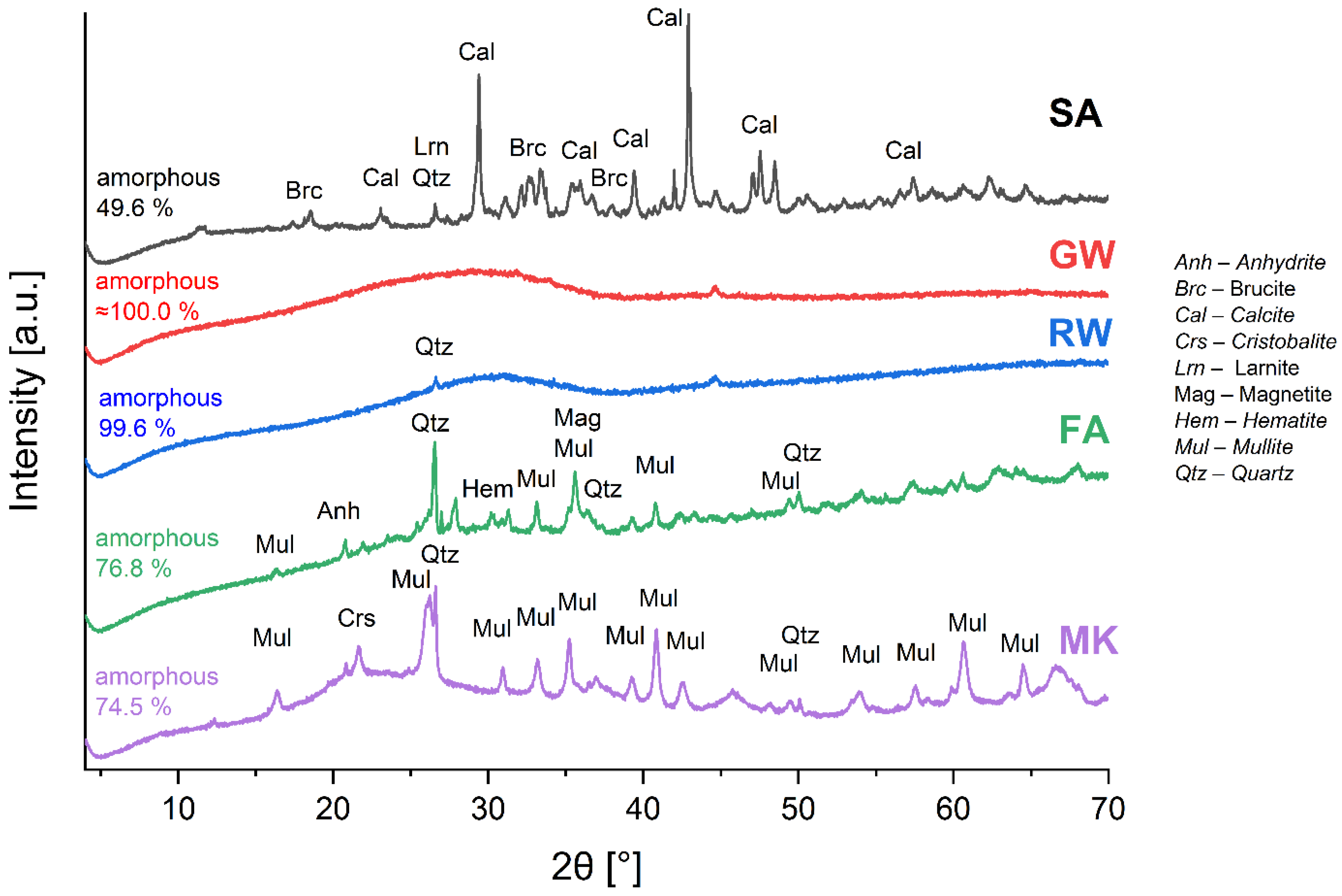
Figure 3.
FTIR spectral analysis of precursors used in alkali activation. The dashed line shows the Na-silicate solution and the distilled water measurement to identify water peaks.
Figure 3.
FTIR spectral analysis of precursors used in alkali activation. The dashed line shows the Na-silicate solution and the distilled water measurement to identify water peaks.
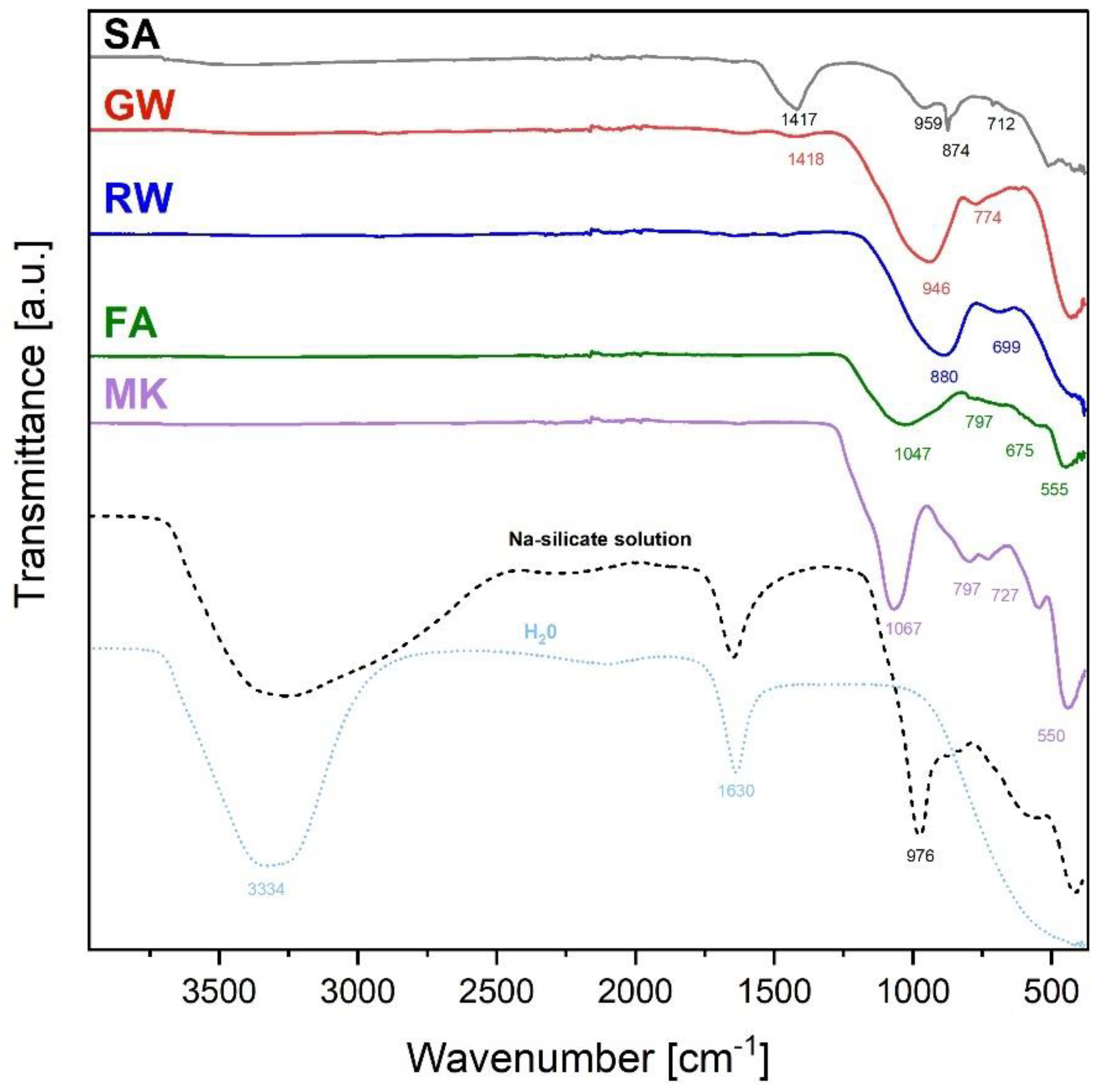
Figure 4.
Photographs of the untreated (left column) and microwave dehydrated prisms (right column) at day 7.
Figure 4.
Photographs of the untreated (left column) and microwave dehydrated prisms (right column) at day 7.

Figure 5.
(Left) Compressive strengths and corresponding geometric densities of AAMs made of different precursors (SA, GW, RW, FA, and MK). Each bar represents the compressive strength of a specific mixture, while the bar colour indicates the geometric density of tested samples. Samples dehydrated with microwaves are marked with μ. (Right) Contour plots showing predicted compressive strengths with varying levels of low power irradiation (1 on y-axis is 100 W) and alkali content.
Figure 5.
(Left) Compressive strengths and corresponding geometric densities of AAMs made of different precursors (SA, GW, RW, FA, and MK). Each bar represents the compressive strength of a specific mixture, while the bar colour indicates the geometric density of tested samples. Samples dehydrated with microwaves are marked with μ. (Right) Contour plots showing predicted compressive strengths with varying levels of low power irradiation (1 on y-axis is 100 W) and alkali content.
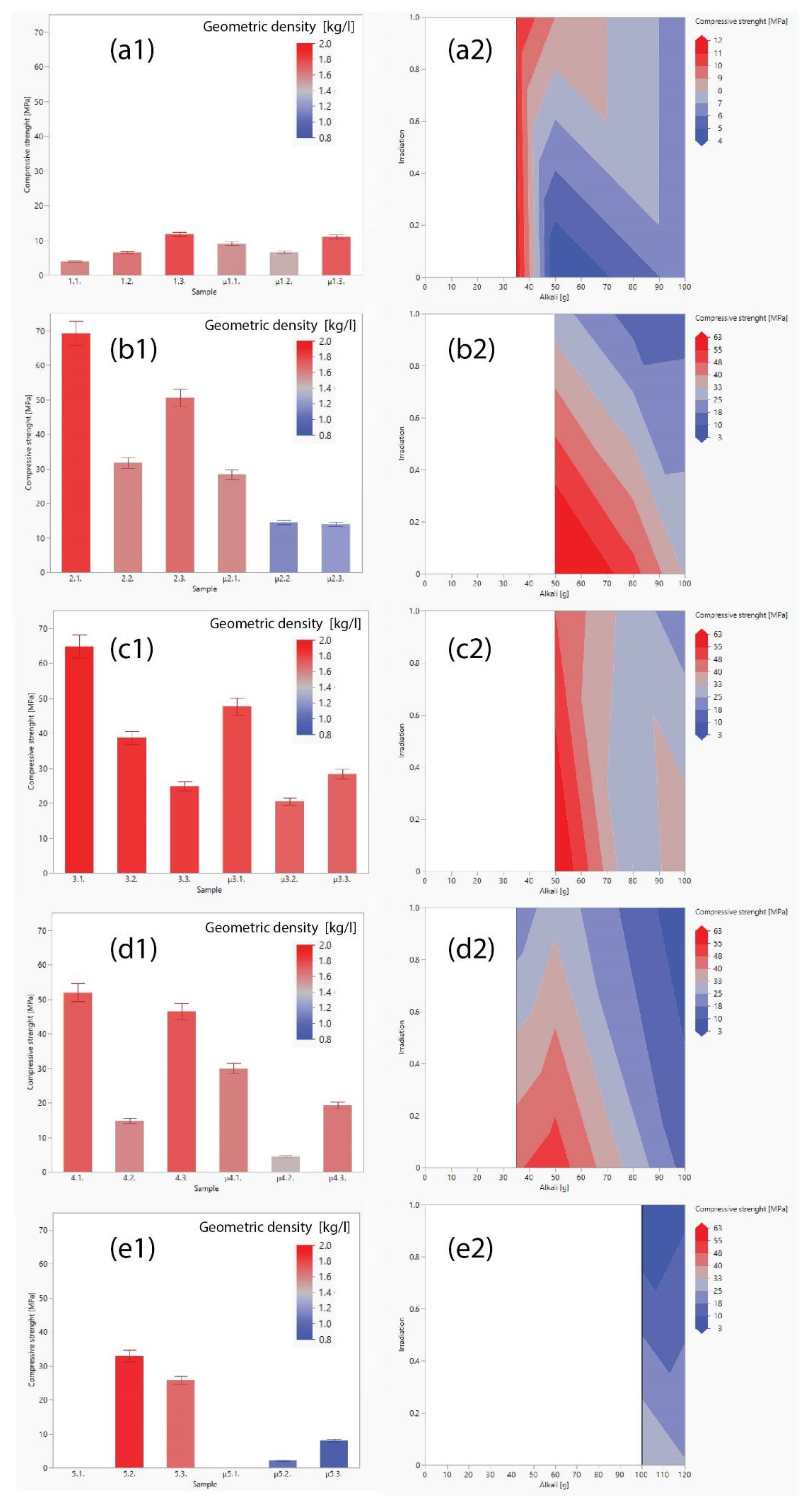
Figure 6.
SEM micrographs (magnification 50) of non-dehydrated (left column) and microwave dehydrated (centre column, μ) samples with the highest amount of alkali solution per precursor and highest probability for the development of efflorescence, along with pore size distribution of selected samples (right column).
Figure 6.
SEM micrographs (magnification 50) of non-dehydrated (left column) and microwave dehydrated (centre column, μ) samples with the highest amount of alkali solution per precursor and highest probability for the development of efflorescence, along with pore size distribution of selected samples (right column).
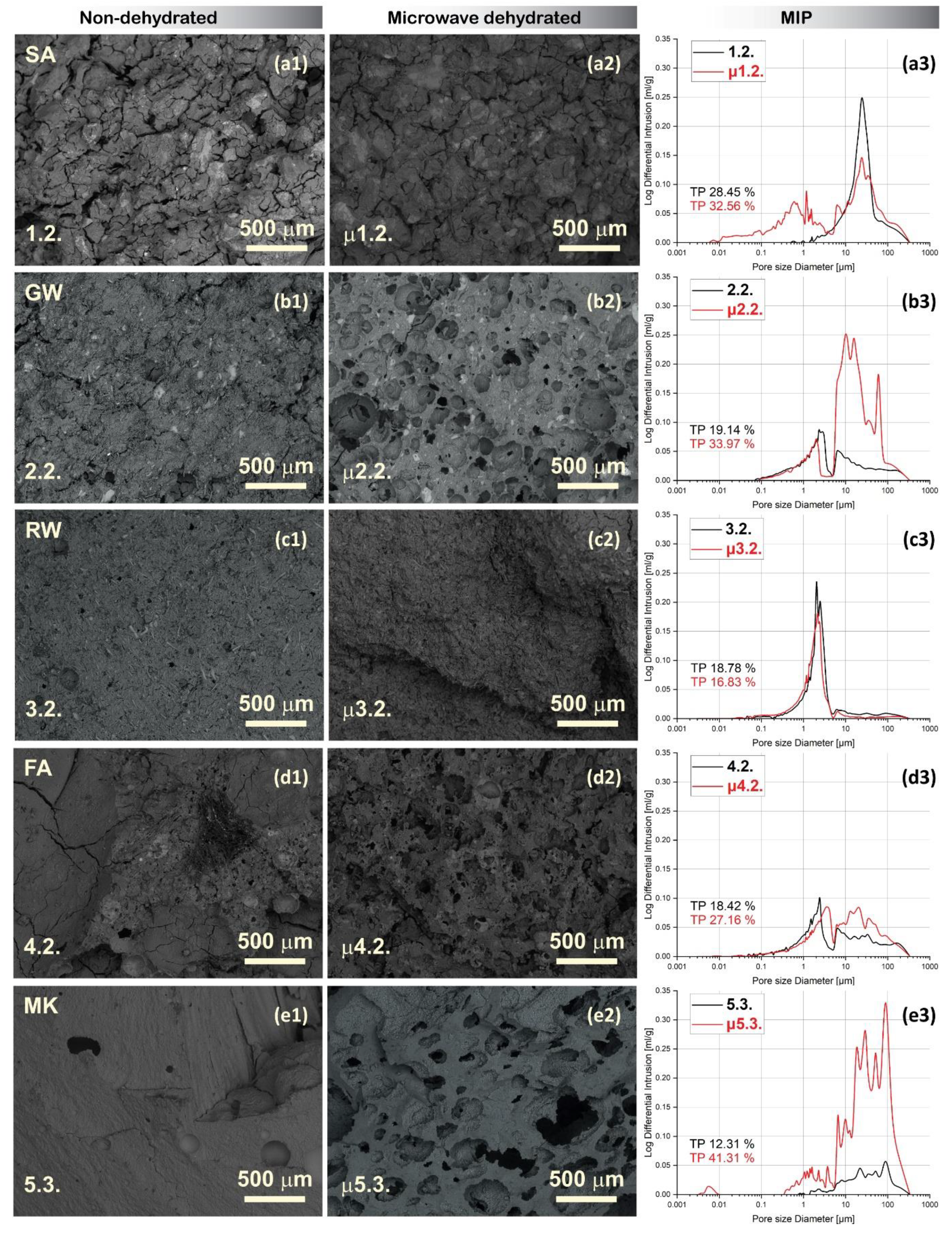
Figure 7.
FTIR spectra of 14-day-old (left) and 1-year-old (right) AAMs made of: a) SA, b) GW, c) RW, d) FA, and e) MK. Samples dehydrated with microwaves are marked with μ. The dashed black line represents the precursor used for alkali activation, and the dashed grey line represents salt formed on the sample's surface after 1 year. The dashed square indicates visible differences indicating efflorescence formation.
Figure 7.
FTIR spectra of 14-day-old (left) and 1-year-old (right) AAMs made of: a) SA, b) GW, c) RW, d) FA, and e) MK. Samples dehydrated with microwaves are marked with μ. The dashed black line represents the precursor used for alkali activation, and the dashed grey line represents salt formed on the sample's surface after 1 year. The dashed square indicates visible differences indicating efflorescence formation.
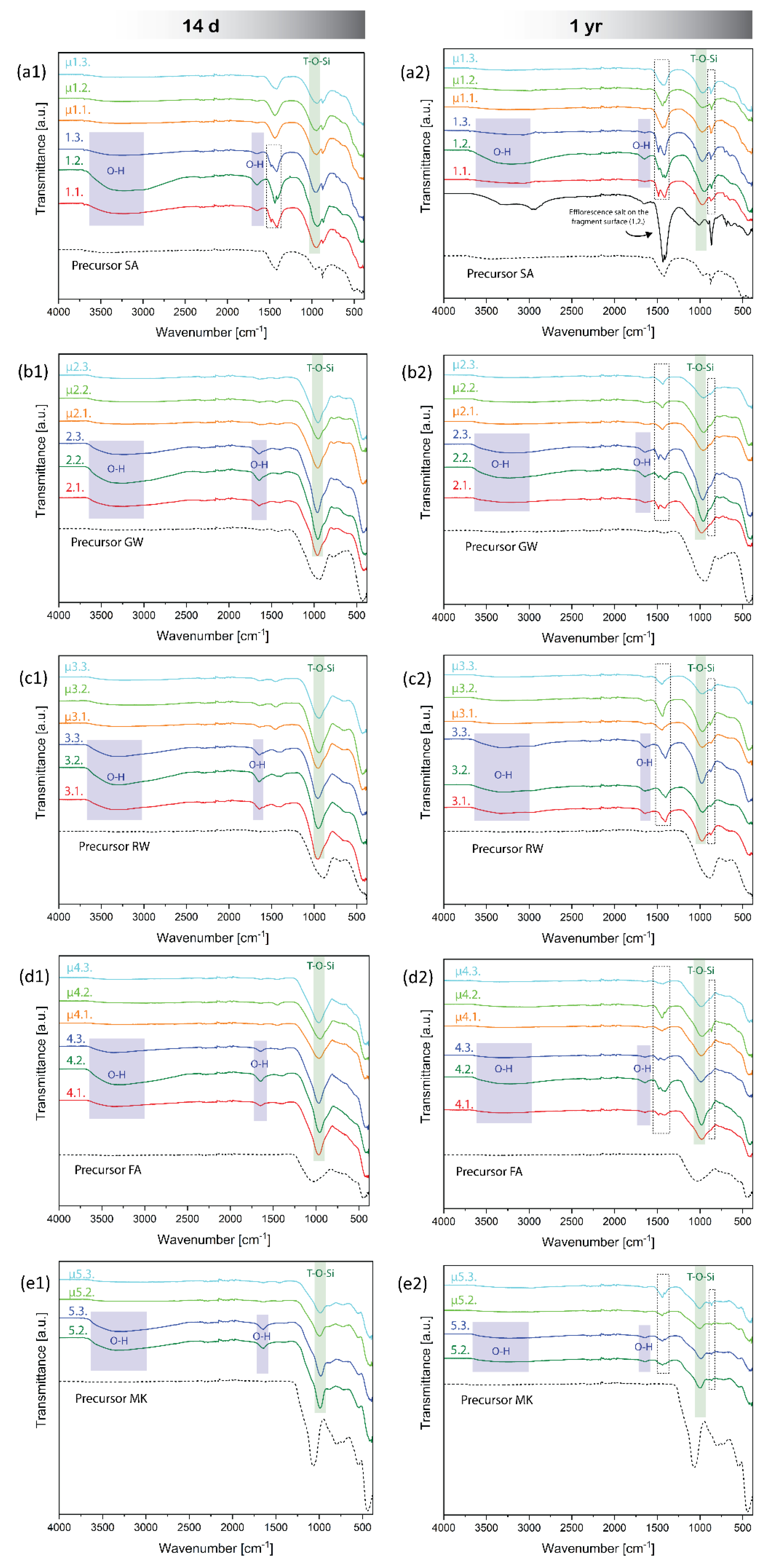
Figure 8.
SEM micrographs of non-dehydrated (first column) and microwave-dehydrated (second column) samples with the highest amount of alkali solution per precursor and highest probability for the development of efflorescence (magnification 2000). In the third column are micrographs of microwave dehydrated samples (magnification 50), where a water droplet induced Na-appearance (column 4, EDXS map).
Figure 8.
SEM micrographs of non-dehydrated (first column) and microwave-dehydrated (second column) samples with the highest amount of alkali solution per precursor and highest probability for the development of efflorescence (magnification 2000). In the third column are micrographs of microwave dehydrated samples (magnification 50), where a water droplet induced Na-appearance (column 4, EDXS map).

Figure 9.
(a) Induced efflorescence formation over time after adding a drop of distilled water to the outer surface of sample 2.2. (b) Experiment with the addition of IPA to the surface of 2.2. (c) Inspection of the surface where distilled water and IPA have been added, with the edge of the dried droplet visible remaining.
Figure 9.
(a) Induced efflorescence formation over time after adding a drop of distilled water to the outer surface of sample 2.2. (b) Experiment with the addition of IPA to the surface of 2.2. (c) Inspection of the surface where distilled water and IPA have been added, with the edge of the dried droplet visible remaining.

Figure 10.
Immersion of sample 2.2 in different media over time: IPA left cup, distilled water right cup. In the case of immersion in water, the colour change of the solution is visible.
Figure 10.
Immersion of sample 2.2 in different media over time: IPA left cup, distilled water right cup. In the case of immersion in water, the colour change of the solution is visible.


Table 1.
Mixture design of AAMs from precursors SA, GW, RW, FA, and MK with the mass ratio of precursor and activator, and viscosity of the fresh slurry (η).
Table 1.
Mixture design of AAMs from precursors SA, GW, RW, FA, and MK with the mass ratio of precursor and activator, and viscosity of the fresh slurry (η).
| Precursor label | Mixture label | mprecursor | mNa-silicate solution | Mass ratio precursor to activator |
η |
| [g] | [g] | [/] | [Pa·s] | ||
| SA | 1.1. | 100 | 50 | 1 : 0.50 | 1.08 |
| SA | 1.2. | 100 | 100 | 1 : 1.00 | 0 (severe foaming) |
| SA | 1.3. | 100 | 35 | 1 : 0.35 | Torque overload |
| GW | W 2.1. | 100 | 50 | 1 : 0.50 | Torque overload |
| GW | 2.2. | 100 | 100 | 1 : 1.00 | 3.56 |
| GW | 2.3. | 100 | 80 | 1 : 0.80 | 9.25 |
| RW | 3.1. | 100 | 50 | 1 : 0.50 | Torque overload |
| RW | 3.2. | 100 | 100 | 1 : 1.00 | 1.43 |
| RW | 3.3. | 100 | 80 | 1 : 0.80 | 11.99 |
| FA | 4.1. | 100 | 50 | 1 : 0.50 | 6.47 |
| FA | 4.2. | 100 | 100 | 1 : 1.00 | 2.27 |
| FA | 4.3. | 100 | 35 | 1 : 0.35 | 31.36 |
| MK | 5.1. | - | - | - | - |
| MK | 5.2. | 100 | 100 | 1 : 1.00 | 3.01 |
| MK | 5.3. | 100 | 120 | 1 : 1.20 | 1.96 |
Table 2.
Mass percentage of elements measured with XRF, elements in the crystalline form with XRD, and elements in the amorphous content (XRF-XRD).
Table 2.
Mass percentage of elements measured with XRF, elements in the crystalline form with XRD, and elements in the amorphous content (XRF-XRD).
| Precursor | Elements [m% ] | Na | K | Mg | Ca | Al | Si | LOI550 °C | LOI950 °C |
| SA | XRF | 0 | 0.03 | 8.22 | 27.53 | 5.92 | 9.15 | 4.75 | 7.86 |
| XRD | 0 | 0 | 2.86 | 15.32 | 0.48 | 4.26 | |||
| XRF-XRD | 0 | 0.03 | 5.36 | 12.22 | 5.44 | 4.89 | |||
| GW | XRF | 10.01 | 0.39 | 3.39 | 6.2 | 3.21 | 29.14 | 5.74 | 3.85 |
| XRD | 0.24 | 0.1 | 0 | 0 | 0 | 0.65 | |||
| XRF-XRD | 9.77 | 0.29 | 3.39 | 6.2 | 3.21 | 28.49 | |||
| RW | XRF | 1.59 | 0.64 | 7.34 | 11.71 | 10.61 | 18.44 | 4.63 | 4.16 |
| XRD | 0.12 | 0.05 | 0 | 0 | 0.16 | 0.28 | |||
| XRF-XRD | 1.47 | 0.59 | 7.34 | 11.71 | 10.45 | 18.16 | |||
| FA | XRF | 0.68 | 2.04 | 1.53 | 6.34 | 14.08 | 20.4 | 0.74 | 0.82 |
| XRD | 0.26 | 0 | 0 | 0.89 | 4.63 | 5.68 | |||
| XRF-XRD | 0.42 | 2.04 | 1.53 | 5.45 | 9.45 | 14.72 | |||
| MK | XRF | 0 | 0.9 | 0.18 | 0.27 | 24.08 | 24.04 | 1.29 | 2.05 |
| XRD | 0 | 0 | 0 | 0 | 7.19 | 5.13 | |||
| XRF-XRD | 0 | 0.9 | 0.18 | 0.27 | 16.89 | 18.91 |
Table 3.
Molar ratios of elements potentially involved in alkali activation normalised to Al.
| Precursor label | Mixture label | 1st PS/Al | 2nd PS/Al | Si/Al |
| [mol/mol] | [mol/mol] | [mol/mol] | ||
| SA | 1.1. | 1.36 | 2.61 | 2 |
| SA | 1.2. | 2.71 | 2.61 | 3.14 |
| SA | 1.3. | 0.95 | 2.61 | 1.66 |
| GW | 2.1. | 5.92 | 2.48 | 10.47 |
| GW | 2.2. | 8.21 | 2.48 | 12.4 |
| GW | 2.3. | 7.3 | 2.48 | 11.63 |
| RW | 3.1. | 0.91 | 1.54 | 2.27 |
| RW | 3.2. | 1.61 | 1.54 | 2.86 |
| RW | 3.3. | 1.33 | 1.54 | 2.62 |
| FA | 4.1. | 0.98 | 0.57 | 2.16 |
| FA | 4.2. | 1.76 | 0.57 | 2.81 |
| FA | 4.3. | 0.75 | 0.57 | 1.96 |
| MK | 5.1. | - | - | - |
| MK | 5.2. | 0.91 | 0.02 | 1.81 |
| MK | 5.3. | 1.08 | 0.02 | 1.96 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



