
Submitted:
06 October 2024
Posted:
07 October 2024
You are already at the latest version
Abstract
An ellagitannin-derived metabolite Urolithin A (UA) has emerged as a potential therapeutic agent for metabolic disorders due to its antioxidant, anti-inflammatory, and mitochondrial function-improving properties, but its efficacy in protecting against ER stress remains underexplored. The endoplasmic reticulum (ER) is a cellular organelle involved in protein folding, lipid synthesis, and calcium regulation. Perturbations in these functions can lead to ER stress, which contributes to the development and progression of metabolic disorders such as metabolic-associated fatty liver disease (MAFLD). In this study, we identified a novel target protein of UA and elucidated its mechanism for alleviating palmitic acid (PA)-induced ER stress. CETSA-LC-MS/MS analysis revealed that UA binds directly to the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA), an important regulator of calcium homeostasis in mitochondria-associated ER membranes (MAMs). As an agonist of SERCA, UA attenuates abnormal calcium fluctuations and ER stress in PA-treated liver cells, thereby contributing to cell survival. In addition, UA inhibits PA-induced mitochondrial ROS and lipid accumulation, further supporting its protective role. The lack of UA activity in SERCA knockdown cells suggests that UA regulates cellular homeostasis through its interaction with SERCA. Collectively, our results demonstrate that UA protects against PA-induced ER stress and enhances cell survival by regulating calcium homeostasis in MAM through SERCA. This study highlights the potential of UA as a therapeutic agent for metabolic disorders associated with ER stress.
Keywords:
Urolithin A
; ER stress
; MAM
; CETSA-LC-MS/MS
; SERCA
; MAFLD
1. Introduction
Metabolic disorders such as metabolic associated fatty liver disease (MAFLD), type 2 diabetes, and obesity are major health problems worldwide. One of the key factors in the development and progression of these metabolic disorders is endoplasmic reticulum (ER) stress. ER stress occurs when the function of the ER is disrupted by protein misfolding or calcium imbalance [1] . This stress triggers a series of responses known as the unfolded protein response (UPR) to restore ER function [2]. However, if ER stress persists and becomes severe, it can lead to cellular dysfunction.
Several proteins and small molecules have been investigated for their role in regulating ER stress, including agents that promote protein folding, maintain ER calcium homeostasis, activate UPR pathways, and alleviate oxidative stress [3,4,5]. In particular, naturally derived compounds such as Urolithin A (UA), the metabolite of ellagitannin, the flavonoid Quercetin, and the polyphenol Resveratrol have been shown to alleviate ER stress [6,7,8]. A previous study showed that UA inhibits ER stress and inflammatory responses induced by glucose toxicity in pancreatic β cells [6]. UA is a well-known mitophagy inducer that removes damaged mitochondria, and also has significant antioxidant and anti-inflammatory effects [9,10,11] . Because of these activities, UA holds great promise as a natural treatment for metabolic diseases. However, its target proteins and detailed mechanisms of action remains to be fully elucidated.
In this study, we investigated the potential of UA to alleviate ER stress associated with MAFLD. Recently, MAFLD has been highlighted as a metabolic disease that includes fatty liver disease and is closely related to insulin resistance and obesity [12]. Using Cellular Thermal Shift Assay (CETSA) [13,14,15,16], we identified sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) as a novel binding protein of UA.
SERCA uses energy from ATP hydrolysis to pump cytosolic calcium ions into the ER or SR [17]. Dysfunction of SERCA is associated with diseases such as muscle disorders and heart failure [18,19]. The development of modulators of SERCA may be a promising strategy to maintain cellular homeostasis due to SERCA dysfunction. Our results show that UA, as an agonist of SERCA, alleviates palmitic acid-induced ER stress in liver cells and protects cells from ER stress. This study provides a new understanding of how UA regulates ER stress and highlights its potential role in the treatment of metabolic disorders.
2. Materials and Methods
2.1. Materials
Urolithin A (SML1791), Dimethyl sulfoxide (D2650), Palmitic acid (P5585), Duolink® In Situ Red Starter Kit (DUO92101), Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). Rhod2-AM (R1244), Fluo4-AM (F14201), Mag-Fluo-4 AM (M14206), MitoSOX (M36008), Hoechst33342 (H3570), BODIPY 493/503 (D3922), lipofectamine RNAimax (13778075), lipofectamine 3000 (2757100), protease and phosphatase inhibitor solution (78441), DMEM (11995065), fetal bovine serum (FBS) (16000044), bovine serum albumin (A2153), antibiotics were purchased from Thermo-Fisher Scientific (Waltham, MA).
2.2. Cell Culture and Treatment
HepG2 were grown in DMEM, supplemented 10% FBS and 1% antibiotics. All cell cultures were maintained at pH 7.4 in a humidified incubator at 37°C under 5% CO2 in air. When cells were treated with PA, DMEM containing 2% BSA was used.
2.3. Western Blot
Soluble proteins were harvested from cells by using SDS lysis buffer (50 mM Tris-HCl, pH 6.8 containing 10% glycerol, 2% SDS, 10 mM dithiothreitol, 0.005% bromophenol blue). Equal amounts of proteins were analyzed by 8 %, 10 %, 12.5 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes. The blots were then blocked and immunolabeled with primary antibodies against GRP78/Bip (Proteintech, 66574-1-Ig), PERK (CST, C33E10), p-PERK (Abclonal, AP1501), eiF2α (CST, L57A5), p-eiF2α (CST, D9G8), CHOP (Proteintech, 15204-1-AP), β-tubulin (Abcam, ab6040), β-actin (Abcam, ab6276), SERCA (Proteintech, 67248-1-Ig), Flag (Proteintech, 20543-1-AP) overnight at 4°C. Immunolabeling was visualized using an enhanced chemiluminescence kit (Bio-Rad Laboratories, 170-5061) according to the manufacturer’s instructions. Images were quantified using Image J software. All band intensities are proportional to the amount of the target protein on the membrane within the linear range of detection.
2.4. CETSA
Cell suspensions at a concentration of 300×104 cells/15 mL were aliquoted into each conical tube. DMSO (control) or UA was treated at 37 °C for 1 hour with gentle mixing. After centrifugation, the pellet was washed with PBS and resuspended in 1 mL of PBS (with protease inhibitor) and aliquoted into PCR tubes (100 μL per tube) and heated between 40-64 °C for 3 minutes and cooled at 25 °C for another 3 minutes in the thermal cycler. The tubes were centrifuged to pellet the cells and the supernatant was discarded and replaced with 0.4% NP-40 (in PBS) supplemented with protease inhibitors to facilitate solubilization of hydrophobic proteins. The cell suspension was subjected to two freeze-thaw cycles in liquid nitrogen and centrifuged at 20,000 g for 20 minutes at 4 °C. The supernatants (soluble proteins) were collected and used for Western blotting or LC-MS/MS analysis
2.5. Sample Preparation for LC-MS/MS Analysis
CETSA samples were denatured in 8 M urea and reduced with 500 mM TCEP for 1 hour at room temperature, followed by alkylation with 500 mM iodoacetamide for 1 hour. The buffer was then changed to 200 mM triethylammonium bicarbonate using a 10 K centrifugal filter. Proteins were digested with MS grade trypsin (1:40) at 37 °C for 16 hours. The resulting peptides were labeled with a 6-plex TMT reagent (Thermo-Fisher Scientific) according to manufacturer’ protocols, and the reaction was quenched with 5% hydroxylamine. TMT labeled samples (TMT-126; 55°C DMSO, TMT-128; 55°C UA, TMT-129; 55°C DMSO, TMT-131; 60°C UA) were pooled, vacuum dried, dissolved in 0.5% formic acid, and desalted using a C18 macro spin column. After desalting, the samples were vacuum dried and kept -80 °C freezer until HPLC fractionation.
2.6. High pH Reversed-Phase Liquid Chromatography for Peptide Fractionation
The dissolved TMT 6 plex labeled sample fractionated using a XBridge BEH Shield RP18 Column (130Å, 2.5㎛, 4.6x150mm, Waters) on NexeraXR HPLC (Shimadzu, Kyoto, Japan) with a 70 min gradient from 5% to 95% mobile phases B at a flow rate of 1.0 mL/min. Mobile phase A consisted of 5 mM ammonium formate in 100% water and mobile phase B consisted of 5 mM ammonium formate in 95% acetonitrile; both buffers were adjusted to a pH of 10 with ammonium hydroxide. A total of 40 fractions were collected using an FRC-10A fraction collector (Shimadzu), after the elution started with an interval of 1 min for each fraction. The 40 fractions were concentrated into 10 fractions. The concatenated fractions were dried and kept -80 °C freezer until LC-MS/MS analysis.
2.7. Liquid Chromatography Tandem Mass Spectrometry
Ten fractions were analyzed using a LC-MS/MS system consisting of an UltiMate U3000 RSLCnano (Thermo Fisher scientific) and an Exploris 480 mass spectrometer with a nano-electrospray source (EASY-Spray Sources). Peptides were first trapped in a precolumn (C18, 75 μm × 2 cm, nanoViper, Acclaim PepMap100, Thermo Fisher Scientific) and then applied to an analytical column (C18, 75 μm × 50 cm PepMap RSLC, Thermo Fisher Scientific) at a flow rate of 250 nL/min. The mobile phases were composed of 100% water (A) and 100% acetonitrile (B), each containing 0.1% formic acid. The LC gradient began with 5% B, maintained with 5% B over 8min, ramped to 25% B over 101 min, followed by 50% B over 10 min and increased to 95% B for 1min, and then was held constant for 8 min, and ended with 5% B over 1 min. After a gradient, the column was re-equilibrated with 5% B for 10 min before the next run. The voltage applied to produce an electrospray was 1800 V. The Exploris 480 mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS with a 2 sec cycle time. Full scan MS spectra (400-1600 m/z) were acquired with an auto maximal injection time mode at a resolution of 120,000 and an automatic gain control (AGC) target value of 1.0× 106. MS/MS spectra were acquired from 110 m/z at a resolution of 30,000 with a high energy collision dissociation (HCD) of 38% normalized collision energy within 1.2 Da isolation window. AGC target value was 1.25× 105 with an auto maximal injection time mode. The exclusion time for previously fragmented ions was 60 s within 10ppm.
2.8. Protein Identification and Quantitation
The Integrated Proteomics Pipeline using built-in search engines (IP2, version 6.5.5, Integrated Proteomics) was utilized for data analysis with UniProt human protein database (January, 2023, Reviewed 20,404 proteins). The reversed sequences of all proteins were appended into the database for calculation of false discovery rate (FDR). ProLucid [20] was used to identify the peptides, a precursor mass error of 5 ppm, and a fragment ion mass error of 50 ppm. Trypsin was selected as the enzyme, with two potential missed cleavages. TMT modification (+ 229.1629) at the N-terminus and lysine residue by the labeling reagent and carbamidomethylation at cysteine were chosen as static modifications. Oxidation at methionine was chosen as variable modification. Reporter ions were extracted from small windows (±20 ppm) around their expected m/z in the HCD spectrum. The output data files were filtered and sorted to compose the protein list using the DTASelect (The Scripps Research Institute, La Jolla, CA) with two and more peptides assignments for a protein identification and a false positive rate less than 0.01 [21].
Quantitative analysis was performed using Census in the IP2 pipeline (Integrated Proteomics, San Diego, CA) using only the unique peptides. The intensity at a reporter ion channel for a protein was calculated as the sum of the intensities of that reporter ion from all constituent peptides of the identified protein [22]. Reverse and potential contaminant proteins were removed. The protein intensities summed by the reporter ion intensities of all identified TMT-labeled peptides were uploaded to the Perseus platform (version 1.6.14.0). The data were normalized by subtracting the median values based on columns after being transformed as log2 values. Quantitative differences in protein levels (log2 fold change, FC) were then calculated as log2 [intensity of proteins in the UA sample] – log2 [intensity of proteins in the DMSO sample] at both 55°C and 60°C, and significant proteins were then selected.
2.9. In Silico Docking Study
Molecular docking analysis was performed using Discovery Studio 2018 software. The structure of the SERCA was obtained from the Protein Data Bank (PDB: 7E7S). For ligands docking, DOCKER (a grid-based molecular docking method using CHARM forcefield) was used. Binding sites are defined by receptor cavities. The ligand was docked to the binding site of the protein and the top 10 hits were generated. The binding energy (CDOCKER energy) was calculated.
2.10. ATPase Activity Assay
The ER Enrichment Kit (Invent Biotechnologies, Minneapolis, MN) was used to isolate ER proteins from liver cells (LX-2), according to the manufacturer’s instructions. The ER fraction was solubilized in 0.5% Triton X-100 buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 1 mM EGTA). ER protein at 1 μg/μL was pre-incubated with various concentrations of UA for 30 minutes, followed by measurement of ATPase activity. The ATPase activity was determined using an ATPase assay kit (Abcam, ab234055) according to the manufacturer’s instructions.
2.11. Lipid Droplet Staining
Cells were seeded in 12-well plates and co-treated with 0.5 mM PA and drugs for 24 hours, followed by treatment with BODIPY 493/503. Images were captured using an LSM980 confocal microscope at 400X magnification. Green fluorescence intensity was quantified using Image J software.
2.12. Calcium Analysis
For mitochondrial or cytosolic calcium analysis, HepG2 cells were grown in 8-well chamber slides. Cells were co-treated with 0.5 mM PA and drugs for 24 hours and then trated with either Rhod-2/AM (Invitrogen, R1244), Mag-Fluo-4/AM (Invitrogen, M14206) or Fluo-4/AM (Invitrogen, F14201) for 30 minutes. Cells were washed with Ca2+-free KRH buffer and live imaged using an LSM980 confocal microscope at 400X magnification. Fluorescence signal intensity was quantified using Image J software.
2.13. Mitochondrial ROS Measurement
Mitochondria ROS levels were assessed using red fluorescent mitochondrial superoxide indicator MitoSOX (Invitrogen, M36008). Cells were treated with drugs for 4 hours, followed by incubation with 5 μM MitoSOX and Hoechst33342 for 10 minutes. The cells were then washed with PBS and live imaged using an LSM980 confocal microscope at 400 x magnification. The intensity of the red fluorescence was quantified using Image J software.
2.14. Transfection
Cells were transfected with the 100 nM SERCA siRNA (Bioneer, Daejeon, South Korea) for 24 hours using Lipofectamine RNAiMAX reagent, according to the manufacturer’s instructions. A SERCA (FLAG-DYK-tagged) human ORF clone was used to generate the mutant SERCA vector. Cells were transfected with the SERCA plasmid using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s instructions. To measure ER calcium, cells were transfected with CMV-ER-LAR-GECO1 (Addgene, #61244) for 48 hours using Lipofectamine 3000 reagent and the images were captured using an LCM980 confocal microscope.
2.15. Statistical Analysis
Statistical analysis was performed using GraphPad Prism 9.0. All results are presented as the mean ± standard deviation (SD) values. Statistical significance was determined using Student’s t-tests (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
3. Results
3.1. UA Alleviates Cellular Stress Caused by Palmitic Acid
Palmitic acid (PA), a saturated fatty acid, induces ER stress when excessively accumulated in cells, which is associated with pathological conditions such as MAFLD [23]. We established a metabolic disease model by treating the HepG2 with 0.5 mM PA for 24 hours to induce lipid accumulation and ER stress. We then investigated whether UA could protect PA-treated cells. To determine the optimal concentration of UA, MTT assay was performed, and the IC50 value at 72 hours was determined to be approximately 40 µM (Figure 1A). Based on this, we found that 20 and 40 µM of UA increased both the proliferation (Figure 1B) and viability (Figure 1C) of PA-treated HepG2 cells in a dose-dependent manner. CHOP is a key transcription factor involved in ER stress-induced cell death. Using a cell model expressing the fluorescent reporter mCherry under the control of the CHOP gene promoter [24], we confirmed that UA reduced ER stress in PA-treated cells. (Figure 1D). In addition, UA treatment reduced the elevated lipid levels induced by PA (Figure 1E). These results demonstrate that UA enhances cell survival in PA-treated HepG2 cells by alleviating cellular stress.
3.2. Target Identification of UA by CETSA-LC-MS/MS
To elucidate the mechanism by which UA reduces ER stress, we performed target identification using a cellular thermal shift assay (CETSA)-LC-MS/MS. CETSA is a method used to assess changes in the thermal stability of a protein caused by binding of a compound to the protein without chemical labeling of the compound. HEK293 cells were treated with DMSO (control) or UA, heated at 55°C and 60°C, and the cells were lysed to obtain proteins. We performed TMT labeling on the CETSA samples to allow multiplex quantification, followed by mass spectrometry analysis (Figure 2A). A total of 5,175 proteins were detected in our samples by MS analysis. Of these, 271 proteins exhibited more than 1.5-fold resistance to heat denaturation (log2 FC = (log2 [the intensity of proteins in the UA sample at 60°C] – log2 [the intensity of proteins in the DMSO sample at 60°C]). (Figure 2B). Analysis of the subcellular location of these candidate target proteins revealed that, interestingly, most of them were located in the ER or mitochondria (Figure 2B).
Through functional analysis of 17 ER proteins and 26 mitochondrial protein candidates, we identified that the target candidates are involved in calcium regulation, protein processing, phospholipid and lipid metabolism, energy metabolism, and oxidative reactions (Figure 2C). The ER is the largest calcium reservoir in the cell, and the calcium concentration within the ER is critical for various physiological activities and signal transduction. Because calcium imbalance increases cellular stress and affects cell survival, we focused on calcium-regulating proteins; SERCA (Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase), ESYT1 (Extended Synaptotagmin 1), and CCDC47 (Coiled-Coil Domain Containing 47) when selecting candidate target proteins for UA. Since SERCA directly regulates ER calcium levels by pumping calcium into the ER, targeting SERCA could have a direct effect on ER calcium balance. Therefore, we considered SERCA as a primary target candidate for UA (Figure 2C).
3.3. Validation of Target Protein of UA
To validate the binding of UA and SERCA by CETSA analysis, UA-treated cells were exposed to heat in the range of 40°C to 64°C and the stability pattern was confirmed (Figure 3A). In addition, isothermal CETSA was performed with different concentrations of UA, using a constant heat of 52°C (Figure 3B). Under these conditions, the EC50 value for UA binding to SERCA was approximately 26 µM (Figure 3B). We also performed a binding assay with CCDC47, one of the target candidates for UA, and obtained a higher EC50 value (approximately 38 µM) compared to SERCA.
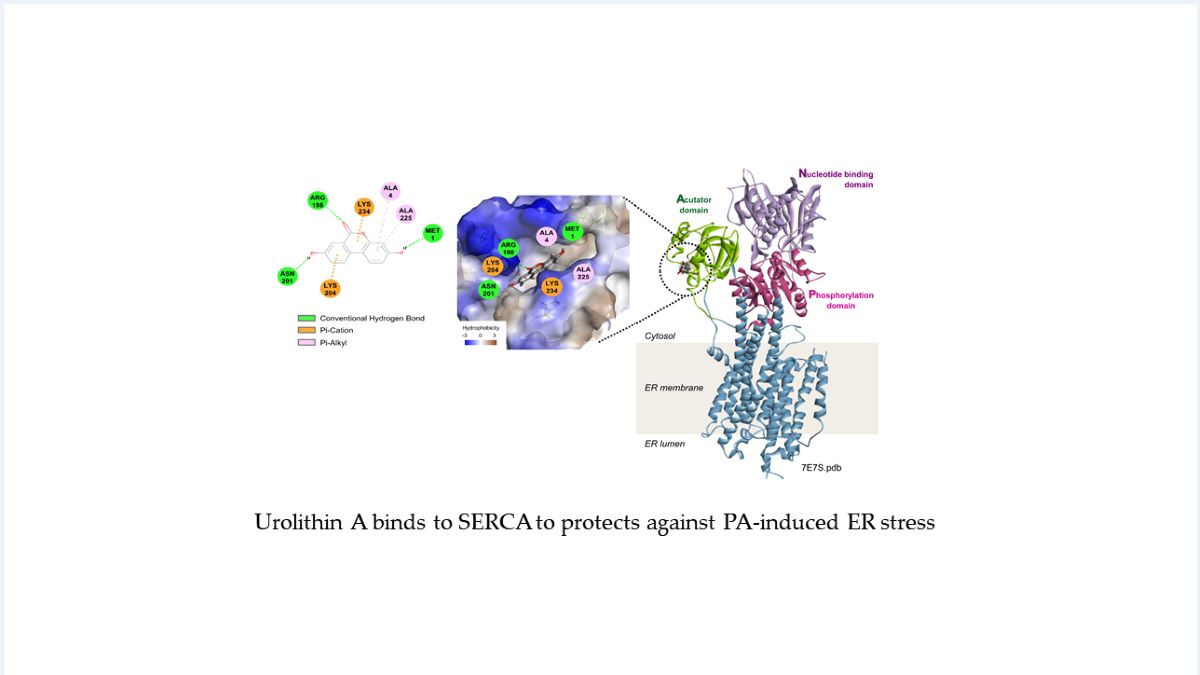
To explore the binding site of UA on SERCA, an in silico docking simulation was performed (Figure 3E). When the binding of UA to the 3D structure of SERCA (PDB: 7E7S) was analyzed, the CDOCKER energy value, which represents the binding affinity of the ligand-protein complex, was -20.57 kcal/mol. SERCA is composed of three main domains; acutator domain, nucleotide domain, and phosphorylation domain [25]. The actuator do-main is responsible for the structural changes of SERCA related to calcium ion transport [26], and the predicted binding pocket for UA is located within this domain (Figure 3D,E). Specifically, the oxygen of UA is expected to form a hydrogen bond with N201 and R198, while the ring structure of UA is predicted to form a Pi-cation interaction with K204 and K234 (Figure 3C). These results suggest that UA may modulate SERCA function by stabilizing critical domains that are involved in structural transitions.
In addition, to validate the predicted binding sites, we constructed mutant versions of the vector by substituting alanine for each site using a FLAG-tagged SERCA expression vector. HEK293 cells were then transfected with either wild-type (WT) or alanine mutant (R198A, K234A) vectors, followed by a CETSA assay. The significantly increased stability of SERCA by UA was only observed in cells transfected with WT SERCA vectors (Figure 3F), highlighting that R198 and K234 play critical roles in the binding of UA to SERCA. To determine whether UA acts as an agonist or antagonist on SERCA, we next performed an ATPase activity assay with isolated ER protein. ER proteins fractionated from liver cells were preincubated with UA, followed by the measurement of ATPase activity levels. Treatment with UA increased ATPase levels in a dose-dependent manner, whereas treatment with thapsigargin (TG, SERCA inhibitor) decreased ATPase levels. (Figure 3G). Taken together, our in vitro and in silico assays support that UA regulates SERCA function by binding directly to SERCA.
3.4. UA Regulates Calcium Levels within Cellular Organelles
To monitor changes in ER calcium levels upon UA treatment, HepG2 cells were transfected with a LAR-GECO1 vector, a fluorescent protein-based calcium indicator [27]. The cells were then treated with DMSO, UA, CDN11163 (CDN, a known agonist of SERCA) [28], and thapsigargin (TG, a known antagonist of SERCA) [29]. UA treatment significantly increased ER calcium levels, similar to CDN-treated cells, whereas TG treatment decreased ER calcium levels, suggesting that UA acts as a SERCA agonist to enhance calcium ion transport to the ER (Figure 4A). PA has been shown to cause hyperactivation of the ER calcium channel IP3R, leading to mitochondrial calcium overload and dysfunction [30]. In PA-treated cells, ER calcium levels decreased by more than 50%. However, UA treatment, similar to CDN treatment, restored ER calcium levels (Figure 4B, Supplementary Figure 1A). PA increased cytosolic calcium, but both UA and CDN treatments reduced the elevated cytosolic calcium (Figure 4C, Supplementary Figure 1B). PA treatment also increased mitochondrial calcium, which was subsequently reduced by UA (Figure 4D). These results suggest that UA alleviates the disrupted intracellular calcium homeostasis induced by PA.
3.5. Regulation of Ca2+ Homeostasis by UA Contributes to Alleviation of ER Stress
To investigate whether the regulation of intracellular calcium homeostasis via SERCA is related to ER stress, we examined representative ER stress markers. PA treatment increased Bip at 3 hours and elevated CHOP at 12 hours (Figure 5A). The ER stress response is divided into three main pathways. We examined molecular markers related to the PERK-eIF2α pathway [31], which is involved in cellular stress responses and survival, after treatment with UA. PA increased the Bip, phosphorylation of PERK, and the phosphorylation of eIF2a at an early time point (6 h) (Figure 5B). Treatment with UA and CDN attenuated ER stress markers, whereas treatment with TG further increased them (Figure 5A). In addition, at the 24 hour time point, PA treatment increased the levels of CHOP, a downstream marker of ER stress. Consistent with the previous results in Figure 1D, UA reduced the elevated CHOP levels (Figure 5C).
In addition, we investigated how UA-mediated regulation of calcium homeostasis in cellular organelles affects other cellular functions. UA reduced PA-induced mROS (Supplementary Figure 2A), which is consistent with the effects of UA to reduce the excess calcium accumulated in mitochondria by PA.
3.6. Knockdown of SERCA reduces the activity of UA
To determine whether the physiological effects of UA are mediated by interaction with its target protein, SERCA, we used siRNA to inhibit SERCA gene expression. After knocking down SERCA, we treated cells with PA and UA to determine whether the effect of UA was abolished. Similar to the previous results in Figure 4, when non-transfected cells were treated with PA and UA, there was an increase in ER calcium, a decrease in cytosolic calcium, and a decrease in mitochondrial calcium. However, in SERCA-depleted cells, there was no change in ER, cytosolic, and mitochondrial calcium despite treatment with UA (Figure 5A–C). This result confirms that UA is involved in calcium regulation within cell organelles by regulating SERCA. In addition, UA treatment did not affect Bip, p-PERK, and p-eIF2a levels in SERCA-deleted cells (Figure 5D), suggesting that UA contributes to alleviate ER stress by interacting with SERCA. Similarly, UA was unable to reduce mROS in SERCA-depleted cells (Supplementary Figure 2B). Our results strongly suggest that the physiological effects of UA on calcium homeostasis, ER stress reduction, and mitochondrial ROS levels are dependent on its interaction with SERCA, highlighting SERCA as a critical mediator of the protective effects of UA.
4. Discussion
Urolithin A (UA) is a natural compound metabolized by gut microbiota from ellagi tannin and ellagic acid [10]. UA prevents the accumulation of damaged mitochondria by inducing mitophagy, enhances fatty acid oxidation and glycogen synthesis by activating the AMPK signaling pathway, and inhibits inflammatory responses by suppressing the NF-κB pathway [6,9,32]. Despite the diverse activities of UA, research on its target proteins are still poorly understood. Using CETSA-LC-MS/MS, a method for identifying target proteins using chemically unmodified compounds, SERCA was identified as a potential target of UA. Palmitic acid (PA) treatment causes lipid accumulation and inflammation in the liver, similar to non-alcoholic fatty liver disease. Intracellular calcium homeostasis was disrupted in PA-treated liver cells and regulation of SERCA by UA restored calcium levels in the ER, cytoplasm, and mitochondria to their original state. Recently, gene therapy aimed at increasing SERCA expression has been investigated for the treatment of heart failure [33]. This supports the importance of SERCA as a target in diseases where ER calcium regulation is impaired.
SERCA undergoes structural transitions between four major states (E1, E1∙Ca²⁺, E2∙Ca²⁺, and E2), that facilitate ATP hydrolysis and calcium ion transport [25]. Thapsigargin (TG), a non-competitive inhibitor of SERCA, binds to the E2 state, inhibiting ATPase activity and preventing calcium ion transport into the ER [34]. Crystal structures have shown that TG binds to SERCA at a pocket formed by the TM helices, with F256 identified as a key residue involved in this binding [35]. Our in silico docking analysis suggests that UA binds to the A (actuator) domain of SERCA, providing a new small molecule tool with a binding site distinct from TG. However, our current studies have not yet confirmed whether UA induces structural changes in SERCA. Investigation of potential structural changes caused by UA binding remains for future research.
Recently, mitochondria-associated ER membranes (MAMs) have been shown to play an important role in maintaining cellular calcium homeostasis and regulating metabolic processes [36]. Lee et al. revealed a mechanism by which UA disrupts the contact between the ER and mitochondria by suppressing the expression of the TGM2 protein in the MAM, thereby inhibiting calcium influx into the mitochondria and subsequently reducing mROS levels [37]. In line with this, we investigated the interactions between the ER and mitochondria in our HepG2 metabolic disorder model. The PLA assay between IP3R and VDAC1 showed that these two organelles are closer together in PA-treated cells and that this interaction is reduced upon UA treatment (Supplementary Figure 3). UA's ability to regulate organelle interactions helps reduce oxidative stress by preventing excessive calcium influx into mitochondria, making it a promising option for treating metabolic disorders. However, further research is needed to elucidate the detailed mechanisms by which UA regulates ER-mitochondria interaction and maintains cellular homeostasis.
Consistent with previous study that UA reduces lipid accumulation [38], we observed that UA reduced lipid droplets increased by PA. Future research should investigate how the reduction of ER stress by UA is related to lipid levels, whether UA inhibits lipid uptake, and whether it can promote the degradation of lipid droplets via autophagy. This will help us better understand the mechanisms of UA regulation of cellular homeostasis and provide insight into the treatment of related diseases.
While further research is needed to fully understand the mechanism of UA, our study is significant because it provides the first evidence that UA acts as an activator of SERCA. This finding highlights the potential role of UA in protecting against ER stress-induced metabolic disfunction.
5. Conclusions
This is the first study to identify UA as an activator of SERCA, which is responsible for organelle calcium regulation activity in hepatocytes undergoing ER stress by PA (Figure 7). Furthermore, we demonstrated that SERCA, a direct binding partner of UA, plays a critical role in the regulation of calcium flux in MAM. Therefore, these results highlight the potential of UA in the treatment of ER stress-induced metabolic dysfunction.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
G.R., M.K., and H.J.K. designed the study. G.R., M.K., S.L., S.I.P., J-Y.C., J.Y.L., and J.Y.K. performed the experiments and analyzed the data. G.R., M.K., and H.J.K wrote the manuscript. HJK supervised the study and secured funding. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by grants from the National Research Foundation of Korea that was funded by the government of the Republic of Korea (MSIP; 2021R1A3B1077371) and the Brain Korea 21 Plus Four Project of the Republic of Korea and ICONS (Institute of Convergence Science), Yonsei University and Research Program of the National Research Council of Science & Technology (CRC22021-100).
Data Availability Statement
The raw data and search results are available at MassIVE : MSV000096011
Acknowledgments
The authors thank Dr. Hye-Ryung Park (Rochester University, NY) for generous gift of the CHOP-mCherry reporter stable cell line.
Conflicts of Interest
The authors declare no conflicts of interest.
The list of abbreviations
CETSA: Cellular Thermal Shift Assay
CDN: CDN1163
LC-MS/MS: Liquid chromatography-mass spectrometry/mass spectrometry
PA: Palmitic acid
TG: Thapsigargin
MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide
IC50: Half maximal inhibitory concentration
EC50: Half maximal effective concentration
References
- Groenendyk, J.; Agellon, L.B.; Michalak, M. Calcium signaling and endoplasmic reticulum stress. International Review of Cell and Molecular Biology 2021, 363, 1-20. [CrossRef]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic reticulum stress in liver diseases. Hepatology 2023, 77, 619-639. [CrossRef]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nature Reviews Drug Discovery 2022, 21, 115-140. [CrossRef]
- Ma, Z.; Du, X.; Sun, Y.; Jia, Y.; Liang, X.; Gao, Y. Attenuation of PM2. 5-Induced Lung Injury by 4-Phenylbutyric Acid: Maintenance of [Ca2+] i Stability between Endoplasmic Reticulum and Mitochondria. Biomolecules 2024, 14, 1135. [CrossRef]
- Im, J.-Y.; Kim, S.J.; Park, J.-L.; Han, T.-H.; Kim, W.-i.; Kim, I.; Ko, B.; Chun, S.-Y.; Kang, M.-J.; Kim, B.-K. CYB5R3 functions as a tumor suppressor by inducing ER stress-mediated apoptosis in lung cancer cells via the PERK-ATF4 and IRE1α-JNK pathways. Experimental & Molecular Medicine 2024, 56, 235-249. [CrossRef]
- Zhang, Y.; Aisker, G.; Dong, H.; Halemahebai, G.; Zhang, Y.; Tian, L. Urolithin A suppresses glucolipotoxicity-induced ER stress and TXNIP/NLRP3/IL-1β inflammation signal in pancreatic β cells by regulating AMPK and autophagy. Phytomedicine 2021, 93, 153741. [CrossRef]
- Sang, A.; Wang, Y.; Wang, S.; Wang, Q.; Wang, X.; Li, X.; Song, X. Quercetin attenuates sepsis-induced acute lung injury via suppressing oxidative stress-mediated ER stress through activation of SIRT1/AMPK pathways. Cellular Signalling 2022, 96, 110363. [CrossRef]
- Chen, Y.; Zhang, H.; Chen, Y.; Zhang, Y.; Shen, M.; Jia, P.; Ji, S.; Wang, T. Resveratrol alleviates endoplasmic reticulum stress–associated hepatic steatosis and injury in mice challenged with tunicamycin. Molecular Nutrition & Food Research 2020, 64, 2000105. [CrossRef]
- Qiu, J.; Chen, Y.; Zhuo, J.; Zhang, L.; Liu, J.; Wang, B.; Sun, D.; Yu, S.; Lou, H. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson's disease model. Neuropharmacology 2022, 207, 108963. [CrossRef]
- Zhang, Y.; Wei, S.; Zhang, H.; Jo, Y.; Kang, J.-S.; Ha, K.-T.; Joo, J.; Lee, H.J.; Ryu, D. Gut microbiota-generated metabolites: missing puzzles to hosts’ health, diseases, and aging. BMB Reports 2024, 57, 207. [CrossRef]
- Jung, Y.H.; Chae, C.W.; Han, H.J. The potential role of gut microbiota-derived metabolites as regulators of metabolic syndrome-associated mitochondrial and endolysosomal dysfunction in Alzheimer’s disease. Experimental & Molecular Medicine 2024, 56, 1691–1702. [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of insulin resistance in MAFLD. International Journal of Molecular Sciences 2021, 22, 4156. [CrossRef]
- Savitski, M.M.; Reinhard, F.B.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Molina, D.M.; Jafari, R.; Dovega, R.B.; Klaeger, S. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [CrossRef]
- Kawatkar, A.; Schefter, M.; Hermansson, N.-O.; Snijder, A.; Dekker, N.; Brown, D.G.; Lundbäck, T.; Zhang, A.X.; Castaldi, M.P. CETSA beyond soluble targets: a broad application to multipass transmembrane proteins. ACS Chemical Biology 2019, 14, 1913-1920. [CrossRef]
- Chang, J.; Kim, Y.; Kwon, H.J. Advances in identification and validation of protein targets of natural products without chemical modification. Natural Product Reports 2016, 33, 719-730. [CrossRef]
- Ko, M.; Jung, H.-Y.; Lee, D.; Jeon, J.; Kim, J.; Baek, S.; Lee, J.Y.; Kim, J.Y.; Kwon, H.J. Inhibition of chloride intracellular channel protein 1 (CLIC1) ameliorates liver fibrosis phenotype by activating the Ca2+-dependent Nrf2 pathway. Biomedicine & Pharmacotherapy 2023, 168, 115776. [CrossRef]
- Lipskaia, L.; Chemaly, E.R.; Hadri, L.; Lompre, A.-M.; Hajjar, R.J. Sarcoplasmic reticulum Ca2+ ATPase as a therapeutic target for heart failure. Expert Opinion on Biological Therapy 2010, 10, 29-41. [CrossRef]
- Zhihao, L.; Jingyu, N.; Lan, L.; Michael, S.; Rui, G.; Xiyun, B.; Xiaozhi, L.; Guanwei, F. SERCA2a: a key protein in the Ca 2+ cycle of the heart failure. Heart Failure Reviews 2020, 25, 523-535. [CrossRef]
- MacLennan, D.H. Ca2+ signalling and muscle disease. European Journal of Biochemistry 2000, 267, 5291-5297. [CrossRef]
- Carvalho, P.C.; Xu, T.; Han, X.; Cociorva, D.; Barbosa, V.C.; Yates III, J.R. YADA: a tool for taking the most out of high-resolution spectra. Bioinformatics 2009, 25, 2734-2736. [CrossRef]
- Tabb, D.L.; McDonald, W.H.; Yates III, J.R. DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. Journal of Proteome Research 2002, 1, 21-26. [CrossRef]
- Raso, C.; Cosentino, C.; Gaspari, M.; Malara, N.; Han, X.; McClatchy, D.; Park, S.K.; Renne, M.; Vadalà, N.; Prati, U. Characterization of breast cancer interstitial fluids by TmT labeling, LTQ-Orbitrap Velos mass spectrometry, and pathway analysis. Journal of Proteome Research 2012, 11, 3199-3210. [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Experimental & Molecular Medicine 2017, 49, e291-e291. [CrossRef]
- Panganiban, R.A.; Park, H.-R.; Sun, M.; Shumyatcher, M.; Himes, B.E.; Lu, Q. Genome-wide CRISPR screen identifies suppressors of endoplasmic reticulum stress-induced apoptosis. Proceedings of the National Academy of Sciences 2019, 116, 13384-13393. [CrossRef]
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 2002, 32, 279-305. [CrossRef]
- Mueller, B.; Zhao, M.; Negrashov, I.V.; Bennett, R.; Thomas, D.D. SERCA structural dynamics induced by ATP and calcium. Biochemistry 2004, 43, 12846-12854. [CrossRef]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proceedings of the National Academy of Sciences 2019, 116, 25322-25328. [CrossRef]
- Dahl, R. A new target for Parkinson’s disease: small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorganic & Medicinal Chemistry 2017, 25, 53-57. [CrossRef]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. Journal of Biological Chemistry 1991, 266, 17067-17071.
- Xiao, W.c.; Zhang, J.; Chen, S.l.; Shi, Y.j.; Xiao, F.; An, W. Alleviation of palmitic acid-induced endoplasmic reticulum stress by augmenter of liver regeneration through IP3R-controlled Ca2+ release. Journal of Cellular Physiology 2018, 233, 6148-6157. [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Current Molecular Medicine 2016, 16, 533-544. [CrossRef]
- Lin, J.; Zhuge, J.; Zheng, X.; Wu, Y.; Zhang, Z.; Xu, T.; Meftah, Z.; Xu, H.; Wu, Y.; Tian, N. Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway. Free Radical Biology and Medicine 2020, 150, 109-119. [CrossRef]
- Hayward, C.; Banner, N.R.; Morley-Smith, A.; Lyon, A.R.; Harding, S.E. The current and future landscape of SERCA gene therapy for heart failure: a clinical perspective. Human Gene Therapy 2015, 26, 293-304. [CrossRef]
- Inesi, G.; Hua, S.; Xu, C.; Ma, H.; Seth, M.; Prasad, A.M.; Sumbilla, C. Studies of Ca2+ ATPase (SERCA) inhibition. Journal of Bioenergetics and Biomembranes 2005, 37, 365-368. [CrossRef]
- Xu, C.; Ma, H.; Inesi, G.; Al-Shawi, M.K.; Toyoshima, C. Specific structural requirements for the inhibitory effect of thapsigargin on the Ca2+ ATPase SERCA. Journal of Biological Chemistry 2004, 279, 17973-17979. [CrossRef]
- An, G.; Park, J.; Song, J.; Hong, T.; Song, G.; Lim, W. Relevance of the endoplasmic reticulum-mitochondria axis in cancer diagnosis and therapy. Experimental & Molecular Medicine 2024, 56, 40-50. [CrossRef]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Lim, J.R.; Kim, S.Y.; Yoon, J.H.; Cho, J.H.; Lee, S.-J. Urolithin A suppresses high glucose-induced neuronal amyloidogenesis by modulating TGM2-dependent ER-mitochondria contacts and calcium homeostasis. Cell Death & Differentiation 2021, 28, 184-202. [CrossRef]
- Cisneros-Zevallos, L.; Bang, W.Y.; Delgadillo-Puga, C. Ellagic acid and urolithins A and B differentially regulate fat accumulation and inflammation in 3T3-L1 adipocytes while not affecting adipogenesis and insulin sensitivity. International Journal of Molecular Sciences 2020, 21, 2086. [CrossRef]
Figure 1.
UA protects hepatocytes from palmitic acid (PA)-induced cellular stress. (A) The MTT assay was performed on HepG2 cells treated with various concentrations of UA (1-100 μM) for 24, 48, and 72 hours to evaluate cell proliferation. (B) The MTT assay was used to measure the cell proliferation of HepG2 cells treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours. (C) The Trypan blue exclusion assay was performed to evaluate the cell viability of HepG2 cells treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours. (D) mCherry-CHOP stable HEK293 cells were treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours, and mCherry fluorescence signals were measured. The expression of CHOP was visualized as a fluorescence signal and observed using confocal microscopy (scale bar: 10 μM). (E) HepG2 cells were treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours, followed by staining of intracellular lipid droplets with BODIPY and observation via confocal microscopy (scale bar: 20 μM). (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
Figure 1.
UA protects hepatocytes from palmitic acid (PA)-induced cellular stress. (A) The MTT assay was performed on HepG2 cells treated with various concentrations of UA (1-100 μM) for 24, 48, and 72 hours to evaluate cell proliferation. (B) The MTT assay was used to measure the cell proliferation of HepG2 cells treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours. (C) The Trypan blue exclusion assay was performed to evaluate the cell viability of HepG2 cells treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours. (D) mCherry-CHOP stable HEK293 cells were treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours, and mCherry fluorescence signals were measured. The expression of CHOP was visualized as a fluorescence signal and observed using confocal microscopy (scale bar: 10 μM). (E) HepG2 cells were treated with PA (0.5 mM) and UA (20, 40 μM) for 24 hours, followed by staining of intracellular lipid droplets with BODIPY and observation via confocal microscopy (scale bar: 20 μM). (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
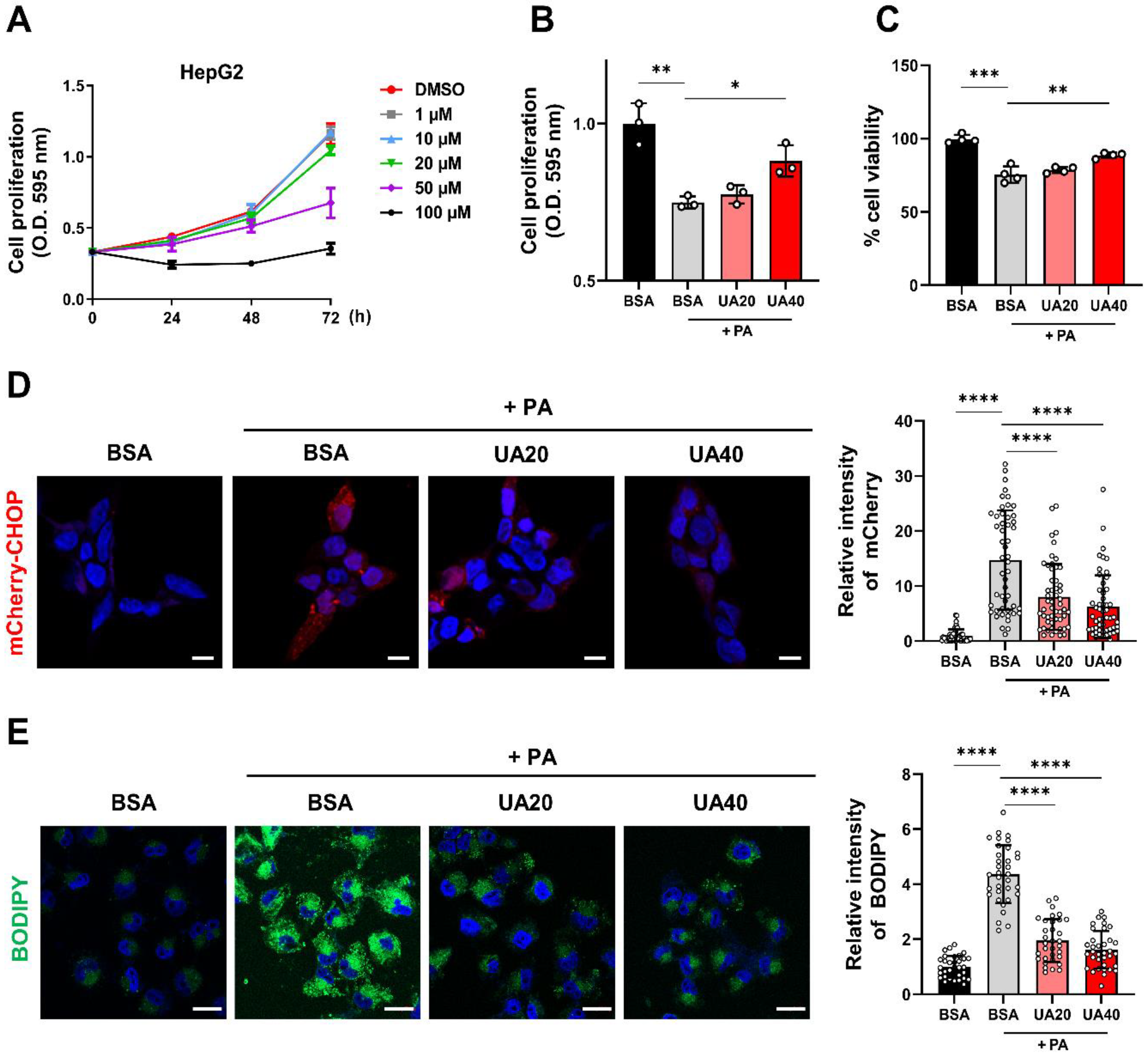
Figure 2.
CETSA-LC-MS/MS for the identification of target protein of UA. (A) Overview of the CETSA-LC-MS/MS method. HEK293 cells were treated with DMSO (control) or UA (20 μM) and subjected to thermal treatment (55°C or 60°C). Proteins were then extracted, digested with trypsin, and labeled with TMT reagents. The labeled peptides were analyzed by HPLC and LC-MS/MS. (B) Schematic diagram of the target selection identification criteria of the UA process. (C) Heatmap of target candidates of UA localized in the ER and mitochondria. Proteins are clustred by their functions, as indicated on the left side of the heatmap.
Figure 2.
CETSA-LC-MS/MS for the identification of target protein of UA. (A) Overview of the CETSA-LC-MS/MS method. HEK293 cells were treated with DMSO (control) or UA (20 μM) and subjected to thermal treatment (55°C or 60°C). Proteins were then extracted, digested with trypsin, and labeled with TMT reagents. The labeled peptides were analyzed by HPLC and LC-MS/MS. (B) Schematic diagram of the target selection identification criteria of the UA process. (C) Heatmap of target candidates of UA localized in the ER and mitochondria. Proteins are clustred by their functions, as indicated on the left side of the heatmap.
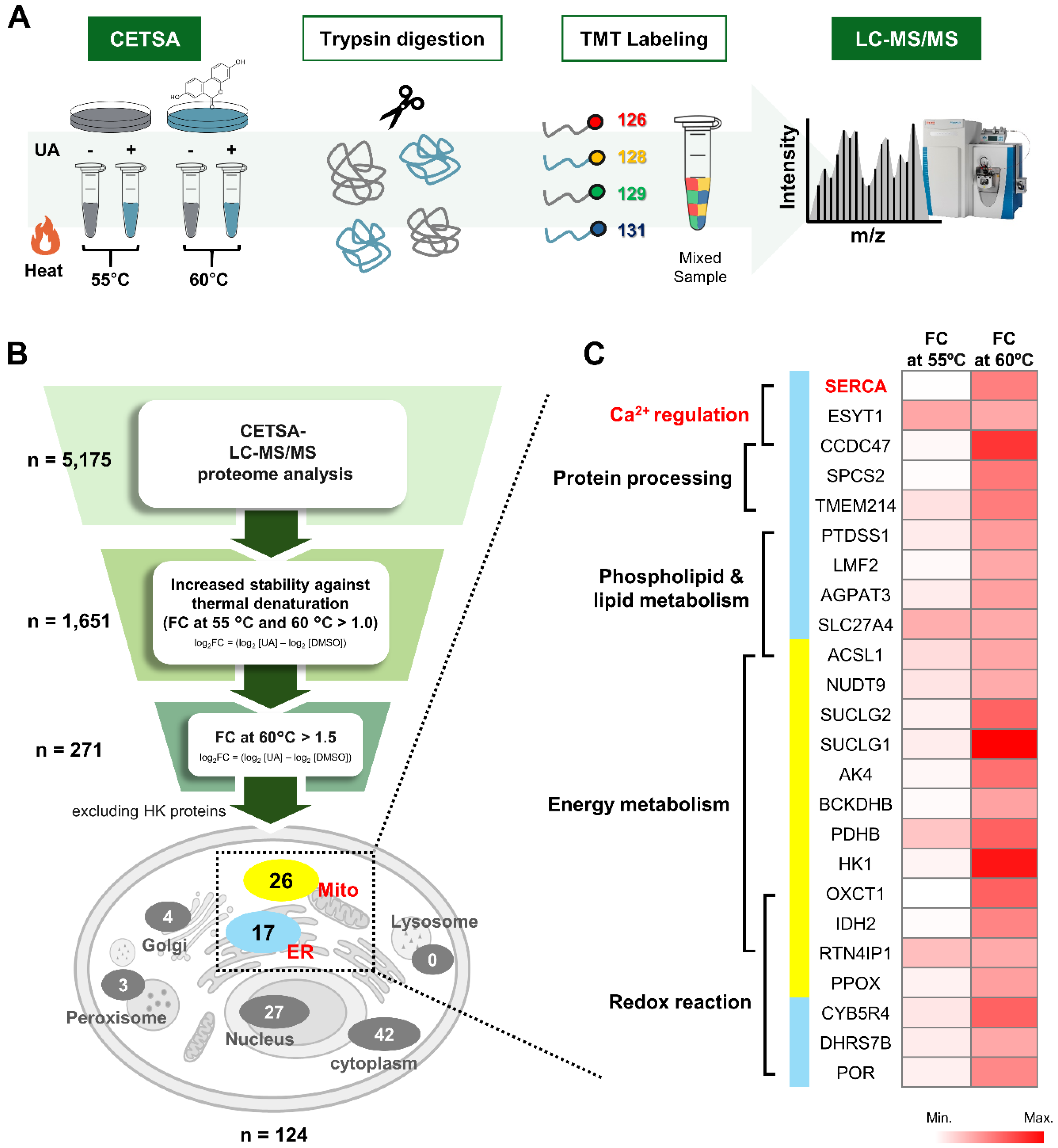
Figure 3.
Validation of UA binding to SERCA. (A) Western blot analysis to evaluate the thermal stability of SERCA in HepG2 cells treated with UA (40 μM) (B) Comparison of the binding affinity between UA and two calcium regulating target proteins; SERCA and CCDC47. HepG2 cells were treated with various concentrations of UA (1-100 μM), followed by isothermal CETSA at 52°C. (C) 2D diagram represents amino acids involved in UA binding to SERCA. (D) Predicted binding of UA to the actuator domain of SERCA, as visualized using Discovery Studio software (-CDOCKER Energy: -20.57 kcal/mol). (E) 3D structure of the full-length SERCA protein (PDB: 7E7S), highlighting its four domains. (F) HEK293 cells were transfected with FLAG-SERCA(WT), FLAG-SERCA(R198A), or FLAG-SERCA(K234A) for 48 hours and then treated with UA (40 μM). After heat treatment at 52°C for 3 minutes, proteins were extracted and analyzed by Western blot. (G) Measurement of ATPase activity of ER proteins extracted from LX2 cells. UA (20, 40 μM) and thapsigargin (0.1 μM) were each treated for 30 minutes before ATPase activity was assessed.
Figure 3.
Validation of UA binding to SERCA. (A) Western blot analysis to evaluate the thermal stability of SERCA in HepG2 cells treated with UA (40 μM) (B) Comparison of the binding affinity between UA and two calcium regulating target proteins; SERCA and CCDC47. HepG2 cells were treated with various concentrations of UA (1-100 μM), followed by isothermal CETSA at 52°C. (C) 2D diagram represents amino acids involved in UA binding to SERCA. (D) Predicted binding of UA to the actuator domain of SERCA, as visualized using Discovery Studio software (-CDOCKER Energy: -20.57 kcal/mol). (E) 3D structure of the full-length SERCA protein (PDB: 7E7S), highlighting its four domains. (F) HEK293 cells were transfected with FLAG-SERCA(WT), FLAG-SERCA(R198A), or FLAG-SERCA(K234A) for 48 hours and then treated with UA (40 μM). After heat treatment at 52°C for 3 minutes, proteins were extracted and analyzed by Western blot. (G) Measurement of ATPase activity of ER proteins extracted from LX2 cells. UA (20, 40 μM) and thapsigargin (0.1 μM) were each treated for 30 minutes before ATPase activity was assessed.
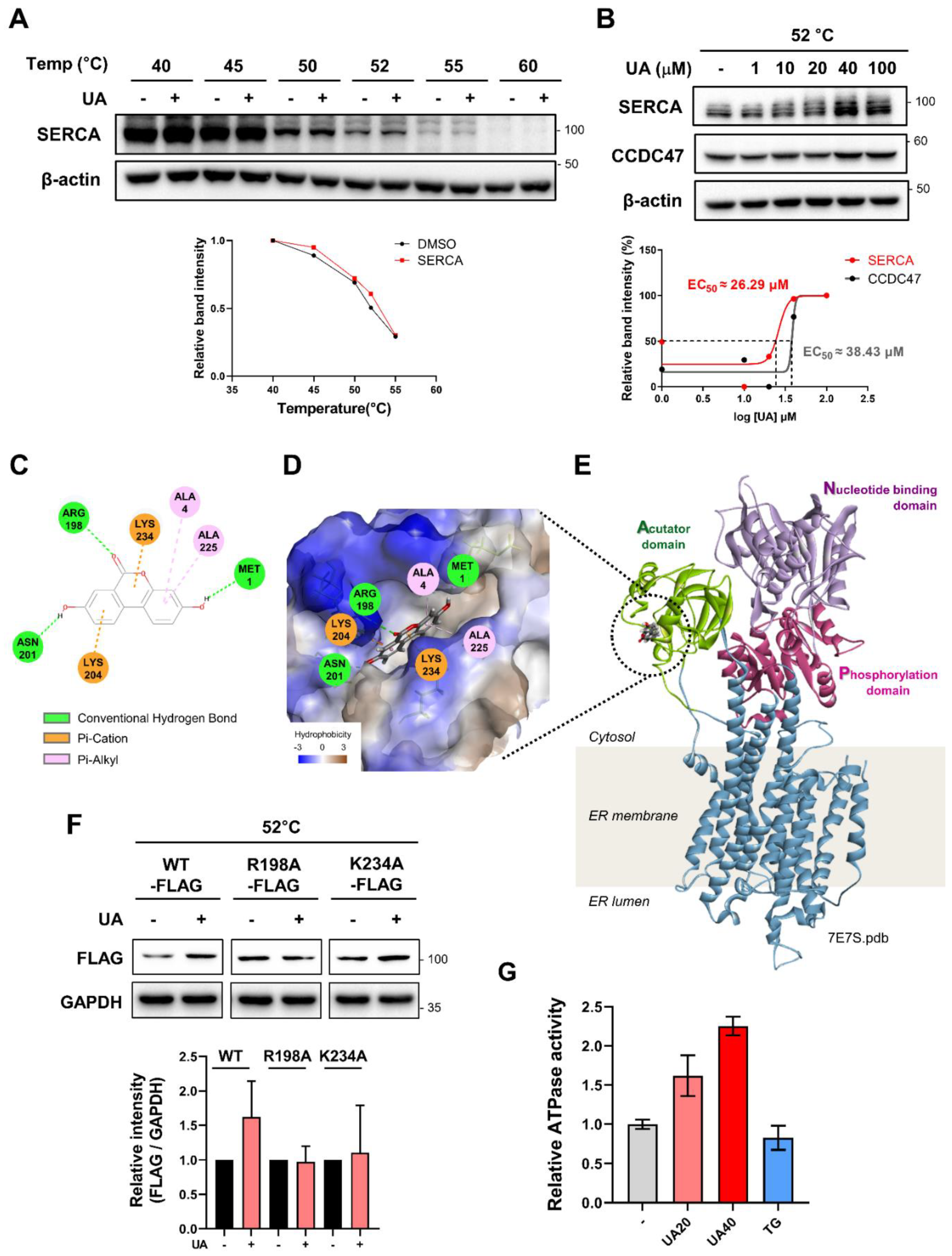
Figure 4.
UA prevents PA-induced disruption of intracellular calcium homeostasis. (A) HepG2 cells were transfected with the ER calcium indicator ER-LAR-GECO vector for 48h, followed by treatment with UA (40 μM), CDN1163 (10 μM), or TG (0.1 μM) for 6 h (scale bar: 20 μm). (B) ER calcium levels were measured in HepG2 cells treated with PA (500 μM) for 6 h, either alone or co-treated with UA (40 μM) and CDN1163 (10 μM) (scale bar: 10 μm). (C) Cytosolic calcium levels were assessed using the Fluo-4-AM after 6 hours of treatment with PA, either alone or co-treated with UA (40 μM), and CDN1163 (10 μM) (scale bar: 20 μm). (D) Mitochondrial calcium levels were determined using the Rhod-2-AM after 24 hours of treatment with PA, either alone or co-treated with UA (40 μM), and CDN1163 (10 μM) (scale bar: 20 μm). (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
Figure 4.
UA prevents PA-induced disruption of intracellular calcium homeostasis. (A) HepG2 cells were transfected with the ER calcium indicator ER-LAR-GECO vector for 48h, followed by treatment with UA (40 μM), CDN1163 (10 μM), or TG (0.1 μM) for 6 h (scale bar: 20 μm). (B) ER calcium levels were measured in HepG2 cells treated with PA (500 μM) for 6 h, either alone or co-treated with UA (40 μM) and CDN1163 (10 μM) (scale bar: 10 μm). (C) Cytosolic calcium levels were assessed using the Fluo-4-AM after 6 hours of treatment with PA, either alone or co-treated with UA (40 μM), and CDN1163 (10 μM) (scale bar: 20 μm). (D) Mitochondrial calcium levels were determined using the Rhod-2-AM after 24 hours of treatment with PA, either alone or co-treated with UA (40 μM), and CDN1163 (10 μM) (scale bar: 20 μm). (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
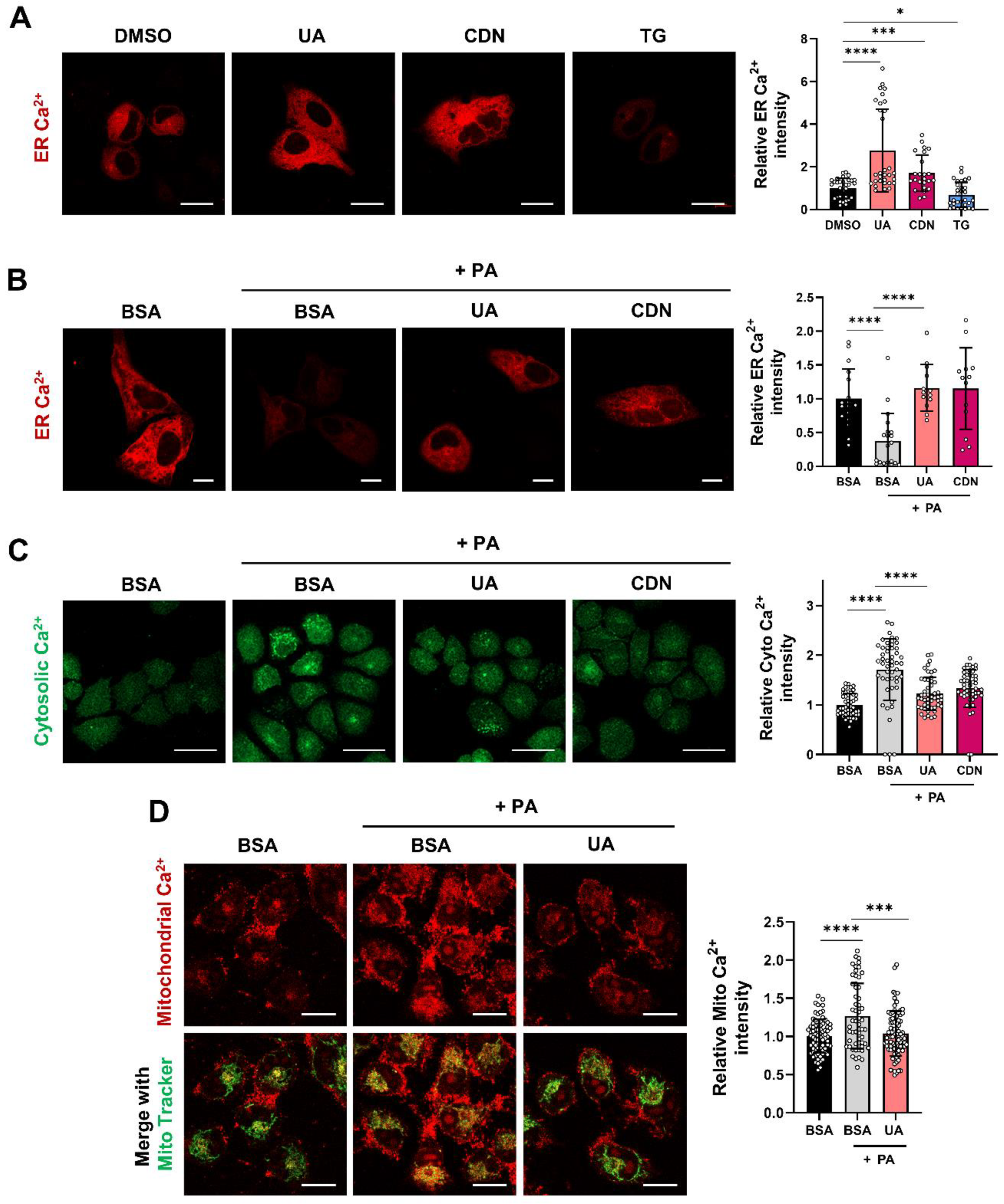
Figure 5.
UA mitigates PA-induced ER Stress. (A) Western blot results showing changes in ER stress markers in HepG2 cells treated with PA. (B) HepG2 cells were treated with PA for 6 h in the absence or presence of UA (40μM), CDN1163 (10 μM) and Thapsigargin (0.1 μM). UA down-regulated the level of ER stress-related proteins. (C) HepG2 cells were treated with PA for 24 h in the absence or presence of UA (40μM), CDN1163 (10 μM) and Thapsigargin (0.1 μM). UA down-regulated the level of CHOP. (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
Figure 5.
UA mitigates PA-induced ER Stress. (A) Western blot results showing changes in ER stress markers in HepG2 cells treated with PA. (B) HepG2 cells were treated with PA for 6 h in the absence or presence of UA (40μM), CDN1163 (10 μM) and Thapsigargin (0.1 μM). UA down-regulated the level of ER stress-related proteins. (C) HepG2 cells were treated with PA for 24 h in the absence or presence of UA (40μM), CDN1163 (10 μM) and Thapsigargin (0.1 μM). UA down-regulated the level of CHOP. (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
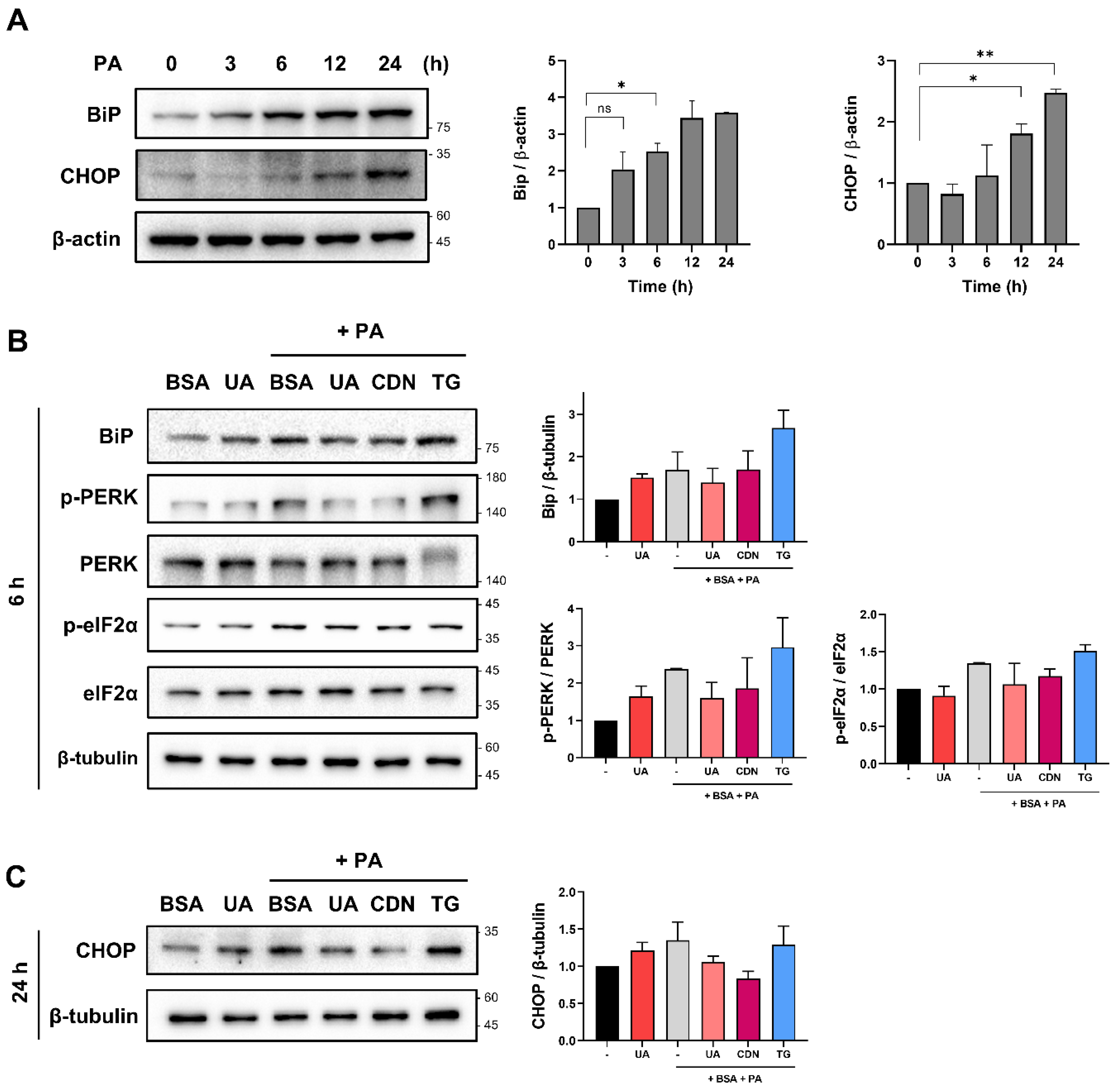
Figure 6.
The activity of UA is suppressed in cells where SERCA is knocked down. (A-C) Following SERCA knockdown with si-SERCA for 24 hours, cells were co-treated with PA and UA (40 μM) for 6 hours. ER, cytosolic, and mitochondrial calcium levels were then measured using Mag-Fluo-4 AM, Fluo-4 AM, and Rhod-2 AM, respectively (scale bar: 40 μm). (D) SERCA knockdown using si-SERCA for 24 hours confirmed the effect of UA (40 μM) on PA-induced ER stress protein levels, as shown by Western blot. (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
Figure 6.
The activity of UA is suppressed in cells where SERCA is knocked down. (A-C) Following SERCA knockdown with si-SERCA for 24 hours, cells were co-treated with PA and UA (40 μM) for 6 hours. ER, cytosolic, and mitochondrial calcium levels were then measured using Mag-Fluo-4 AM, Fluo-4 AM, and Rhod-2 AM, respectively (scale bar: 40 μm). (D) SERCA knockdown using si-SERCA for 24 hours confirmed the effect of UA (40 μM) on PA-induced ER stress protein levels, as shown by Western blot. (*p < 0.05, * *p < 0.01, * **p < 0.001, * ** *p < 0.0001).
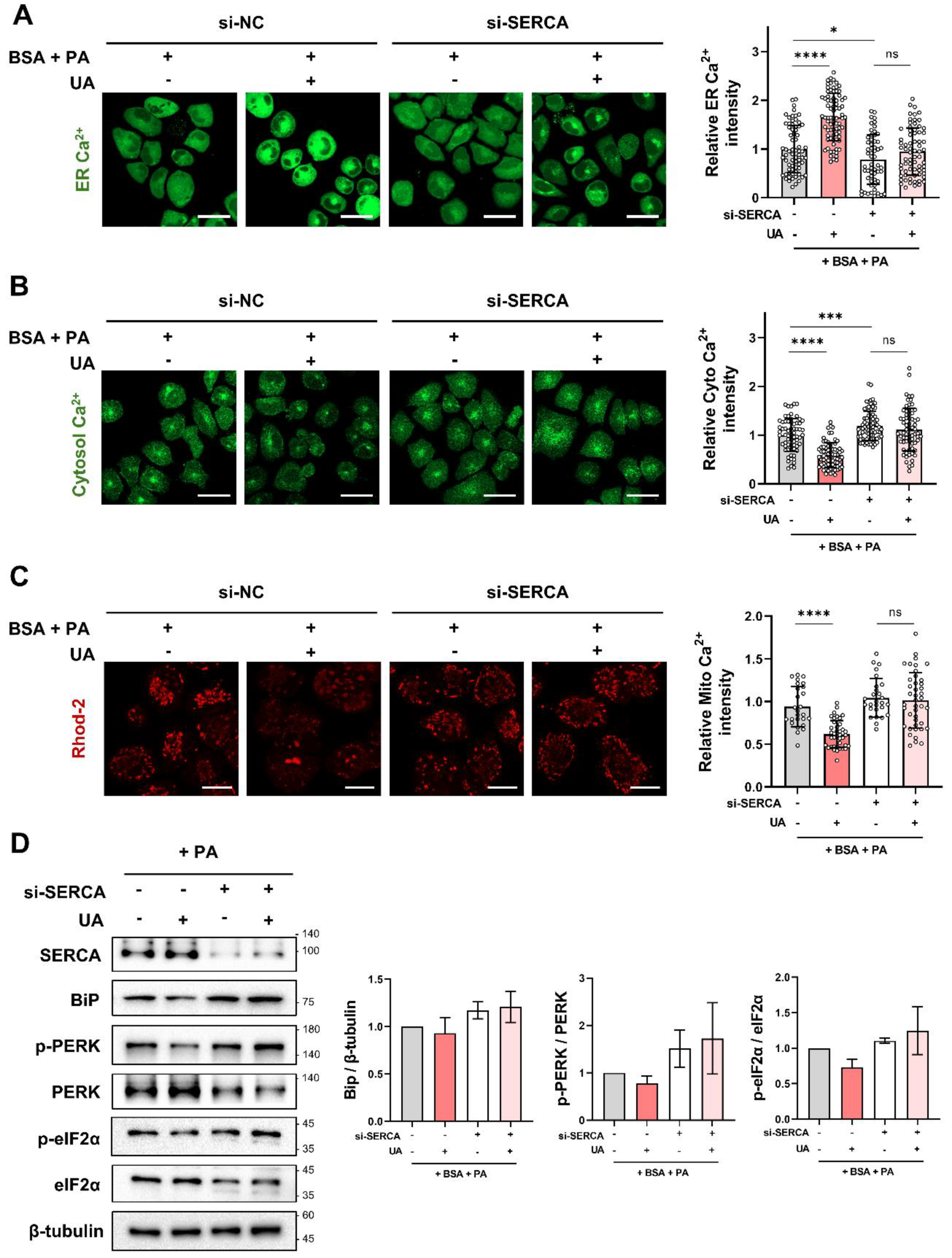
Figure 7.
Schematic summary of the target proteins and mechanisms of action of UA. HepG2 cells stimulated with PA release ER calcium through IP3R, leading to ER stress. UA binds to SERCA, the ER calcium pump, replenishing ER calcium levels and maintaining calcium homeostasis. This mechanism helps protect the cells from stress-induced damage.
Figure 7.
Schematic summary of the target proteins and mechanisms of action of UA. HepG2 cells stimulated with PA release ER calcium through IP3R, leading to ER stress. UA binds to SERCA, the ER calcium pump, replenishing ER calcium levels and maintaining calcium homeostasis. This mechanism helps protect the cells from stress-induced damage.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



