
Submitted:
05 October 2024
Posted:
07 October 2024
You are already at the latest version
Abstract
This perspective describes the current status and future prospects of developing long- to ultralong-acting anti-obesity peptides. First, we review the current status of lipidation, PEGylation, and Fc fusion technologies to obtain long-acting peptides administered once weekly. Next, we describe the approach and current results of using macromolecular peptide prodrugs with pre-programmed releasable linkers to achieve longer-acting peptides that can be administered weekly or monthly. Finally, we posit novel modifications of the latter technology that could provide ultralong half-lives of over one month by advantageously exploiting the clearance rates of released peptides. As examples, we posit that prodrugs of low-clearance GLP-1 agonists derived from peptides already modified by lipidation, PEGylation, and Fc fusion could produce ultralong-acting agonists with dosing intervals reaching 3 to even 6 months.
Keywords:
GLP-1
; hydrogel
; microspheres
; obesity
; semaglutide
Introduction
There is a renaissance of interest in incretin-based peptides as treatments for obesity and other conditions. As an example, peptidic GLP-1 receptor agonists (RAs) are mainstays of treatments for T2D and obesity, and are potential therapies for MAFLD (previously called MASH) and age-related diseases such as Parkinson’s and Alzheimer’s. However, a problem is that all peptides have short in vivo half-lives of minutes to hours and require some form of half-life extension to make them practical for use as therapeutics. The most common half-life extension technologies used for peptides include lipidation, PEGylation, and Fc fusions, all of which allow only weekly administration [1]. Since the half-life is limited by the technology’s mechanism of half-life extension, it is unlikely that longer-acting peptides can be achieved using these approaches per se [1].
It has been well established that persistence and adherence in anti-obesity drug use are low and is a very large problem [2,3]. In part, this is caused by the adverse GI effects that are so common with GLP-1 receptor agonists. Persistence and compliance in anti-obesity drug use can be significantly increased by reducing the dosing frequency [4,5]. Also, the GI toxicity of GLP-1 agonists is related to dosing frequencies in the order twice daily (BID) > daily (QD) > weekly (QWk), which is thought to reflect the higher Cmax or Cmax/Cmin of BID and QD drugs [4]. Hence, GI effects are less common with long-acting than short-acting compounds, and the earlier shorter-acting peptide agonists have been displaced by those that can be administered weekly. For the future, it would be beneficial to obtain peptides that have optimized pharmacokinetic profiles that allow dosing intervals longer than one week, such as once-monthly (QMo)and perhaps even longer than one month.
Achieving an optimal pharmacokinetic profile requires a balance between half-life and dosing. To keep a peptide above its minimal therapeutic level, Cmin, the optimal half-life is about equal to the dosing interval, and the optimal dose maintains plasma concentrations above Cmin while minimizing Cmax/Cmin. With peptides having a shorter half-life than the dosing interval, it is often common practice to maintain the concentration above Cmin by increasing its dose and risking Cmax-related toxicities. For example, AMG133 – a GLP-1 agonist/GIP antagonist mAb – has a half-life of 14 days yet is dosed every month; here, administering sufficient drug to maintain Cmin for two half-lives appears not to cause adverse effects. However, at some point, increasing the dose to achieve longer-acting agents will undoubtedly cause toxicities.
It would be timely and impactful to develop approaches to obtain longer-acting and ultralong-acting peptides that allow less frequent dosing. As examples, longer acting GLP-1 agonists should increase convenience and decrease some toxicities. Advanced systems for drug delivery would also mitigate many common impediments to adherence [5], which is a major problem with GLP-1RAs [3].
Here, we review the technologies to obtain long-acting peptides that are administered once weekly, our approach to create longer-acting peptides administered once monthly, and a new approach we posit will allow ultralong-acting peptides that can be administered at even less frequent intervals of 3 months or maybe even 6 months.
Long-Acting Peptides: Lipidation, PEGylation and Fc Fusions
Following are brief descriptions of the three major technologies currently used for half-life extension of peptides that allow QWk administration – lipidation, PEGylation and Fc fusion. First, lipidation covalently connects a fatty acid to a peptide. The fatty acid moiety tightly and reversibly binds to serum albumin, and converts the parent peptide’s half-life (t1/2) from about one hour to about one week by piggybacking on the long-lived albumin [6,7]. The intense efforts of pharmaceutical companies to optimize lipidation has likely achieved the practical upper limit of half-life extension using this technology [6]. But, the price paid for half-life extension by lipidation is the high dose of peptide needed to bind the albumin sink yet provide a sufficient amount of active free lipidated peptide to exert its intended activity. Second, PEGylation permanently connects a high molecular weight PEG polymer – optimal at about 40 kDa – to the peptide to retard renal filtration. However, 40 kDa PEG itself has an elimination half-life of about 6 d in humans, limiting half-life extension so PEGylated peptides and proteins require at least QWk administration [1,8]. Finally, fusions of peptides to the Fc fragment of an IgG show a longer half-life because of the approximately 50 kDa increase in size and Fc recycling [9]. Many scientists who are unfamiliar with specifics of the technology assume Fc fusions have half-lives similar to IgG or therapeutic antibodies, and are surprised to learn that the average half-life of Fc fusions is just 4- to 5 d which best supports QWk administration [9]. In summary, the three most frequently used technologies for half-life extension of peptides require QWk administration and there are no obvious approaches to increase dosing intervals other than through excessive dosing and risk of Cmax-related toxicities. We argue that, if a drug has a tight pharmacokinetic-pharmacodynamic relationship, using dosing intervals much longer than the half-life of a peptide could be problematic.
Regardless, it is asserted that many such agonists can be used for once-monthly (QMo) dosing. Table 1 shows GLP-1RA-containing anti-obesity agents with half-lives of 5- to 14 days that have been posited to be suitable for QMo dosing. Also shown are the estimated dose and Cmax increases that would be necessary to keep the drug over a therapeutic Cmin for 1 Mo compared to the dose needed and ensuant CMax for dosing intervals equal to the half-life. It can be seen that the monthly doses are inversely related to the half-life of the drug: the shorter the half-life, the higher the dose and the higher the risk for Cmax-related toxicities. Hence, while some peptides might tolerate overdosing to achieve QMo dosing intervals, many would likely breach their tolerance barrier and cause unacceptable toxicities. A much better solution would be to develop technologies that could overcome the barriers to QMo administration.
Longer-Acting Peptides: Prolonged Drug Release from Microsphere (MS)-Peptide Prodrugs
We have developed a general approach for half-life extension of therapeutics that theoretically allows the achievement of any desirable half-life [10,11]. Practically, we have increased peptide half-lives to over 2 Mo, and we believe that achieving temporal drug exposure is not the limitation of the technology [12,13]. Rather, the limitation is imposed by the potency and stability of the peptide and the dose needed to supply sufficient drug over the desired dosing interval – we target injections of 1- to 2 mL containing ~5 μmol drug/mL. In our approach, a drug is covalently tethered to a long-lived carrier by a linker that cleaves in a base-catalyzed β-elimination rate-determining step, k1 (Figure 1) [14]. All subsequent steps are faster, so the rate of drug release directly reflects that of this first step. The cleavage rate of the linker is controlled by the nature of an electron-withdrawing “modulator” (Mod) which regulates the acidity of an adjacent carbon-hydrogen bond. Since the in vitro and in vivo cleavage rates follow a tight structure-activity relationship with the electron-withdrawing ability of the modulator, cleavage rates are predictable and tunable, and the in vitro-in-vivo correlation is high [15]. After release of the active drug, the drug is cleared from the system by elimination rate k2 which, by design, is faster than cleavage k1. Also, the releasable linkers are not affected by enzymes and are extraordinarily stable when stored at lower pH and temperature [10,16].
With this system, the pharmacokinetics can be easily and accurately simulated and modeled [12,15]. This is because there are only two relevant processes that define the pharmacokinetics: k1, the slow cleavage rate of the linker, and k2, the faster elimination rate of the released drug. The rate-determining linker cleavage manifests as the in vivo half-life of the released drug, t1/2,1, but – perhaps unintuitively – the elimination half-life, t1/2,2, of the released drug manifests in the steady-state concentration of the drug.
An important differentiating feature of this half-life extension technology from others is that drug release is chemically controlled, so in vivo half-lives are species-independent [10,12,17]; they can be determined in the mouse and be translated to man. In contrast, half-lives of lipidated, PEGylated and Fc fusion peptides, are species-dependent and have shorter half-lives in rodents than humans; translation from preclinical models to humans requires oft-risky allometric scaling.
One carrier we use is a mesoporous tetra-PEG hydrogel to which a drug can be tethered via a releasable linker [11,18]. These hydrogels – fabricated as uniform ~50 μm microspheres (MS) [19,20] – are injected subcutaneously (SC) through a small-bore needle where they sit at the injection site as a stationary SC depot and slowly release the drug to the systemic circulation. We also incorporate slower-cleaving β-eliminative linkers in crosslinks of these polymers with half-lives longer than the linker used for drug release, so gel degradation occurs in vivo after drug release [19].
Proofs of concept for the effectiveness of the technology for peptides reside in its success in providing longer-acting MS prodrugs of [Gln28]exenatide [12], semaglutide [21], and octreotide [13] that show ≥30 d half-lives. To our knowledge, other than a few peptides delivered from polymeric depots and devices, these are the only peptides thus far reported with half-lives of one month or longer.
Expectedly, there is a strong preference among patients for QWk vs. QD GLP-1 agonists. However, since there are no incretin-based peptide agonists approved or in clinical trials with half-lives longer than ~1 Wk, there are no real-world data on benefits or detriments of longer-acting peptides. However, if advantages of longer-acting agonists mirror those of QD vs. QWk agonists [5], we project positive effects on patient’s preference, persistence and compliance. Also, QWk dosing of GLP-1RAs give lower side effects than BID and QD dosing due to Cmax effects [4]. Once-monthly or longer dosing intervals using the optimal half-life will give even lower Cmax and Cmax/Cmin values, and could lower associated toxicities even more. This is nicely exemplified by comparing the C vs. t profiles of multiple dosing of QWk semaglutide to a simulation of the recently described QMo semaglutide (Figure 2) [21,22]. Here, QMo MS~semaglutide maintains the therapeutic Cmin of QWk semaglutide with only 75% of the Cmax, and a lower Cmax/Cmin. Hence, we posit that compared to QD or QWk dosing, a QMo peptide should improve tolerability – e.g., reduce GI side effects – and/or allow higher dosing.
Ultralong-Acting Peptides: Using Drug Clearance (CL) to Increase Peptide Concentration and Dosing Interval
Since the technology described here can confer almost any half-life to a drug, it should be able to achieve dosing intervals for peptides of even longer than one month – “ultralong-acting” peptides. However, delivering the amount of drug needed for longer periods in a suitable dosing volume can be a major impediment. For example, MS conjugates of the same peptide, e.g., exenatide, with half-lives optimized to 1 and 3 months could be problematic in a dose-limiting situation since the Q3M conjugate would require at least 3-fold increase in dose. We recently recognized that an approach to over-ride this problem and achieve blood concentrations that mitigate problematic dose limitations is simply to use an agonist with decreased drug clearance, CL (k2*Vd; where Vd is the volume of distribution). While the half-life is governed by the release rate, k1, the plasma concentration of the released peptide is governed by the balance between the slow rate of peptide released into the circulation (k1) and the rate it is cleared from the circulation (k2). So, all else constant, the CL is inversely proportional to the peptide concentration (eq 1); hence, lowering the CL has the effect of increasing the plasma concentration.
Ct = Dose*k1/CL * e-k1t
From eq 1, when the release half-life is adjusted to equal the dosing interval, τ, the dose required to maintain plasma levels above Cmin is given as eq 2, and the comparative doses of two different MS~peptide conjugates, A and B, is given by eq 3:
Dose = Cmin*CL/k1*2
DoseB/DoseA = Cmin,B/Cmin,A * CLB/CLA * 𝜏B/𝜏A
A plentiful source of peptide agonists with reduced clearance is the collection already modified to provide QWk dosing; we posit that many of these can be converted from long-acting to ultralong-acting therapeutics. As examples of how lowered clearance can alleviate dose limitations and be used to obtain effective ultralong-acting drugs, we compare the pharmacokinetics of MS conjugates of unmodified peptides with MS conjugates of peptides modified by lipidation, PEGylation, or Fc fusion.
First, we compare the pharmacokinetics of a MS conjugate of an unmodified GLP-1 peptide to MS~semaglutide, which releases the lipidated peptide with a lowered CL (Figure 3). PLX039 is a MS~[Gln28]exenatide that confers a 30-day half-life to the released peptide in multiple species [12,17]. In humans, exenatide has a rapid elimination t1/2,2 of 2.5 h and CL of 9.1 L/h, and semaglutide has a lower CL of 0.05 L/h. For the same linker release rate k1 and dose, the plasma concentrations reflect CLEx/CLSema (eq 3), and the semaglutide concentration will be 180-fold higher than exenatide. This higher concentration should be – and has recently been shown to be [21] – sufficient to prolong the dosing interval to at least 1 Mo; however, the high ~50 nM Cmin required to satisfy albumin binding requires such high doses that achieving longer dosing intervals would be challenging.
Similarly, we compare MS~exenatide to a MS~PEGylated exenatide (Figure 3). Pegloxenatide is a PEGylated exenatide [23] with a therapeutic target concentration, Cmin, similar to exenatide, but a much lower CL of 0.014 L/h vs. 9.1 L/h [24]. The difference in CL values means that – all else being equal – the plasma concentrations reflect CLEx/CLPEGlox (eq 2), and the Pegloxenatide concentration will be 650-fold higher than exenatide (SI Figure 1). Thus, with an optimized dose and linker it should be possible to produce a much longer acting conjugate of Pegloxenatide than with exenatide. Indeed, pharmacokinetic simulations (SI Figure 1A) indicate a MS~Pegloxenatide conjugate having a linker with a t1/2,1 similar to the dosing interval would allow Q3Mo, and maybe even Q6Mo dosing in an injection volume of less than 1 mL.
Finally, ultra-long dosing frequencies could be achieved with other peptidic drugs that are sufficiently potent and have low CL, such as Fc fusions (Figure 3). Fc fusions typically have half-lives of 4- to 5 d [5], which, assuming similar Vds, reflect CL. Thus, when attached to MSs the conjugates should provide higher concentrations of the fused than the non-fused peptide. And, if not too extreme, reduced potencies of Fc fusions compared to non-fused peptides can be overcome by their lower CL (eq. 3). For example, the potent Fc GLP-1 fusion, dulaglutide, with a t1/2,2 of 5 d and CL of 0.11 L/h, has a ~1 nM Cmin that is about 10-fold higher than exenatide [25]. Nevertheless, a MS~dulaglutide with a linker release half-life of 30 d should give a ~80-fold higher concentration than the same dose of MS~exenatide, which translates to a longer-acting GLP-1 agonist. Our simulations (SI Figure 2) indicate that a MS~dulaglutide conjugate having a linker with a release half-life similar to the dosing interval would allow at least Q3Mo dosing with an injection volume of less than 1 mL.
Summary
Currently, three technologies are commonly used to convert short-acting peptides to long-acting peptides that can be administered once weekly – lipidation, PEGylation and Fc fusions. However, other than overdosing, options to use these technologies per se to increase dosing intervals beyond one week are limited. Using MS carriers and β-eliminative cleavable linkers, active peptides with in vivo half-lives of ~one-month can be achieved. Furthermore, using MS conjugates of peptide analogs with low clearance – e.g., lipidated, PEGylated or Fc fusion peptides – increase steady-state concentrations of released peptides to allow ultralong-acting peptides with dosing intervals of up to 3- or even 6 months. There could be huge practical impacts of using MS conjugates of potent peptides/proteins with low clearance. As examplles, MS~conjugates of potent lipidated, PEGylated and Fc fusion peptides with moderate half-lives of ~5- to 7 days could be converted to ultralong-acting peptides that could maintain therapeutic levels for up to 3 or even 6 months. Additionally, there are over 50 lipidated peptides, 100 PEGylated drugs and a similar number of Fc fusions that are approved or in clinical trials (GlobalData.com, Sept 2, 2024); it is likely that some significant number of these would be suitable for developing bio-better longer- or ultralong-acting therapeutics.
Materials and Methods. Pharmacokinetic simulations were performed as reported [15] and are provided in the Supplementary Information.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author contributions
ELS designed/performed research; JAH designed/performed research; GWA designed/performed research; and DVS. designed research/wrote the paper.
Data and materials availability
All data are available in the main text or the supplementary materials.
Acknowledgements
We thank David Parkes and Matthias Urmann for helpful suggestions.
Competing interests
All authors have Prolynx shares and/or options
References
- Yu, M.; Benjamin, M.M.; Srinivasan, S.; Morin, E.E.; Shishatskaya, E.I.; Schwendeman, S.P.; Schwendeman, A. Battle of GLP-1 delivery technologies. Adv Drug Deliv Rev 2018, 130, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Gasoyan, H.; Pfoh, E.R.; Schulte, R.; Le, P.; Rothberg, M.B. Early- and later-stage persistence with antiobesity medications: A retrospective cohort study. Obesity (Silver Spring) 2024, 32, 486–493. [Google Scholar] [CrossRef]
- Palanca, A.; Ampudia-Blasco, F.J.; Calderon, J.M.; Sauri, I.; Martinez-Hervas, S.; Trillo, J.L.; Redon, J.; Real, J.T. Real-World Evaluation of GLP-1 Receptor Agonist Therapy Persistence, Adherence and Therapeutic Inertia Among Obese Adults with Type 2 Diabetes. Diabetes Ther 2023, 14, 723–736. [Google Scholar] [CrossRef]
- Filippatos, T.D.; Panagiotopoulou, T.V.; Elisaf, M.S. Adverse Effects of GLP-1 Receptor Agonists. Rev Diabet Stud 2014, 11, 202–230. [Google Scholar] [CrossRef] [PubMed]
- Baryakova, T.H.; Pogostin, B.H.; Langer, R.; McHugh, K.J. Overcoming barriers to patient adherence: the case for developing innovative drug delivery systems. Nat Rev Drug Discov 2023, 22, 387–409. [Google Scholar] [CrossRef]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front Endocrinol (Lausanne) 2019, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, Y. Prediction of Half-Life Extension of Peptides via Serum Albumin Binding: Current Challenges. Eur J Drug Metab Pharmacokinet 2021, 46, 163–172. [Google Scholar] [CrossRef]
- Sharda, N.; Khandelwal, P.; Zhang, L.; Caceres-Cortes, J.; Marathe, P.; Chimalakonda, A. Pharmacokinetics of 40 kDa Polyethylene glycol (PEG) in mice, rats, cynomolgus monkeys and predicted pharmacokinetics in humans. Eur J Pharm Sci 2021, 165, 105928. [Google Scholar] [CrossRef]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef]
- Santi, D.V.; Schneider, E.L.; Reid, R.; Robinson, L.; Ashley, G.W. Predictable and tunable half-life extension of therapeutic agents by controlled chemical release from macromolecular conjugates. Proc Natl Acad Sci U S A 2012, 109, 6211–6216. [Google Scholar] [CrossRef]
- Ashley, G.W.; Henise, J.; Reid, R.; Santi, D.V. Hydrogel drug delivery system with predictable and tunable drug release and degradation rates. Proc Natl Acad Sci U S A 2013, 110, 2318–2323. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.L.; Hearn, B.R.; Pfaff, S.J.; Reid, R.; Parkes, D.G.; Vrang, N.; Ashley, G.W.; Santi, D.V. A Hydrogel-Microsphere Drug Delivery System That Supports Once-Monthly Administration of a GLP-1 Receptor Agonist. ACS Chem Biol 2017, 12, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.L.; Henise, J.; Reid, R.; Ashley, G.W.; Santi, D.V. Subcutaneously Administered Self-Cleaving Hydrogel-Octreotide Conjugates Provide Very Long-Acting Octreotide. Bioconjug Chem 2016, 27, 1638–1644. [Google Scholar] [CrossRef] [PubMed]
- Hearn, B.R.; Fontaine, S.D.; Pfaff, S.J.; Schneider, E.L.; Henise, J.; Ashley, G.W.; Santi, D.V. Primary deuterium kinetic isotope effects prolong drug release and polymer biodegradation in a drug delivery system. J Control Release 2018, 278, 74–79. [Google Scholar] [CrossRef]
- Schneider, E.L.; Henise, J.; Reid, R.; Ashley, G.W.; Santi, D.V. Hydrogel Drug Delivery System Using Self-Cleaving Covalent Linkers for Once-a-Week Administration of Exenatide. Bioconjug Chem 2016, 27, 1210–1215. [Google Scholar] [CrossRef]
- Henise, J.; Yao, B.; Ashley, G.W.; Santi, D.V. Autoclave sterilization of tetra-polyethylene glycol hydrogel biomaterials with β-eliminative crosslinks. Engineering Reports 2020, 2, e12091. [Google Scholar] [CrossRef]
- Schneider, E.L.; Reid, R.; Parkes, D.G.; Lutz, T.A.; Ashley, G.W.; Santi, D.V. A once-monthly GLP-1 receptor agonist for treatment of diabetic cats. Domest Anim Endocrinol 2020, 70, 106373. [Google Scholar] [CrossRef]
- Henise, J.; Hearn, B.R.; Ashley, G.W.; Santi, D.V. Biodegradable tetra-PEG hydrogels as carriers for a releasable drug delivery system. Bioconjug Chem 2015, 26, 270–278. [Google Scholar] [CrossRef]
- Henise, J.; Yao, B.; Hearn, B.R.; Schneider, E.L.; Ashley, G.W.; Santi, D.V. High-throughput, aseptic production of injectable Tetra-PEG hydrogel microspheres for delivery of releasable covalently bound drugs. Engineering Reports 2020, 2, e12213. [Google Scholar] [CrossRef]
- Henise, J.; Yao, B.; Ashley, G.W.; Santi, D.V. Facile preparation of tetra-polyethylene glycol hydrogel microspheres for drug delivery by cross-flow membrane emulsification. Engineering Reports 2021, 3, e12412. [Google Scholar] [CrossRef]
- Schneider, E.L.; Hangasky, J.A.; Ashley, G.W.; Santi, D.V. The limitation of lipidation: conversion of semaglutide from once-weekly to once-monthly dosing. 2024. [Google Scholar]
- WEGOVY (semaglutide) injection, f.s.u.I.U.S.A. 2017.
- Cai, H.; Chen, Q.; Duan, Y.; Zhao, Y.; Zhang, X. Short-term effect of polyethylene glycol loxenatide on weight loss in overweight or obese patients with type 2 diabetes: An open-label, parallel-arm, randomized, metformin-controlled trial. Front Endocrinol (Lausanne) 2023, 14, 1106868. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, J.; Li, W.; Tang, S.; Sun, J.; Zhang, X.; Liu, J.; Yi, B.; Liu, J.; Zhang, X.; et al. Polyethylene glycol loxenatide (PEX168) in subjects with renal impairment: A pharmacokinetic study. Br J Clin Pharmacol 2019, 85, 2714–2720. [Google Scholar] [CrossRef] [PubMed]
- 2Geiser, J.S.; Heathman, M.A.; Cui, X.; Martin, J.; Loghin, C.; Chien, J.Y.; de la Pena, A. Clinical Pharmacokinetics of Dulaglutide in Patients with Type 2 Diabetes: Analyses of Data from Clinical Trials. Clin Pharmacokinet 2016, 55, 625–634. [Google Scholar] [CrossRef]
Figure 1.
β-eliminative release of a drug from linker.
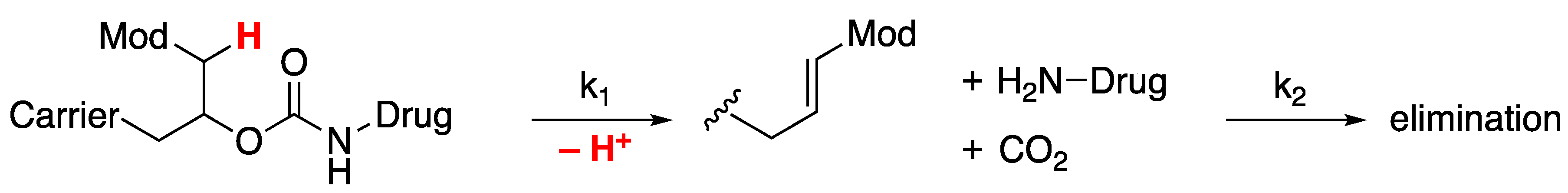
Figure 2.
Simulated C vs. T of semaglutide vs. QMo MS~semaglutide in humans. QWk semaglutide dose escalation uses pharmacokinetic data for the recommended 4 wks each of 0.25 mg, 0.5 mg, 1 mg, and 1.7 mg followed by 2.4 mg per week [22]. QMo MS~semaglutide shows drug concentrations for single doses of 2.5 mg, 5 mg over two months followed by 9 mg/month. .
Figure 2.
Simulated C vs. T of semaglutide vs. QMo MS~semaglutide in humans. QWk semaglutide dose escalation uses pharmacokinetic data for the recommended 4 wks each of 0.25 mg, 0.5 mg, 1 mg, and 1.7 mg followed by 2.4 mg per week [22]. QMo MS~semaglutide shows drug concentrations for single doses of 2.5 mg, 5 mg over two months followed by 9 mg/month. .
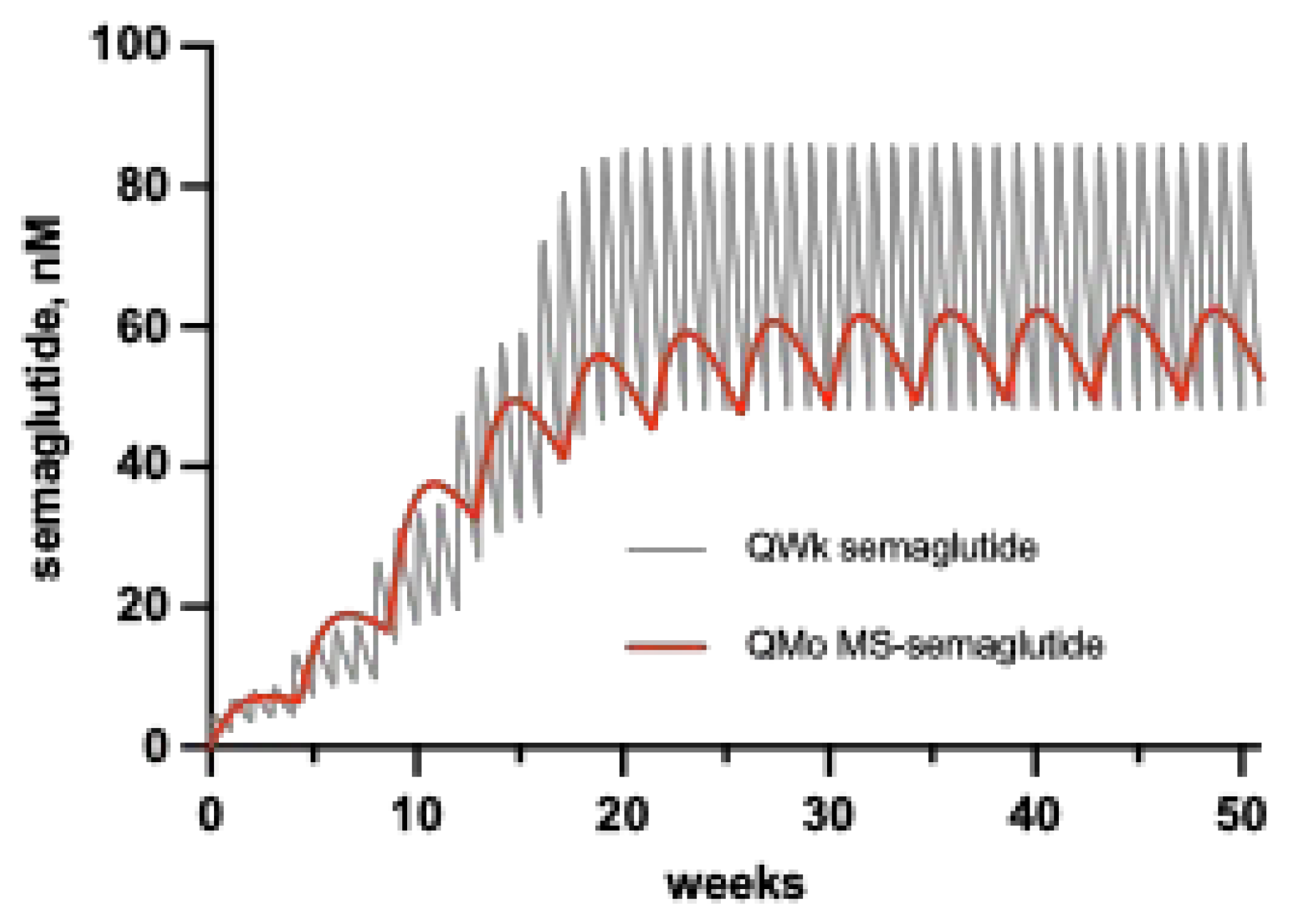
Figure 3.
Depiction of the cleavage of MS prodrugs and clearance of released drugs.
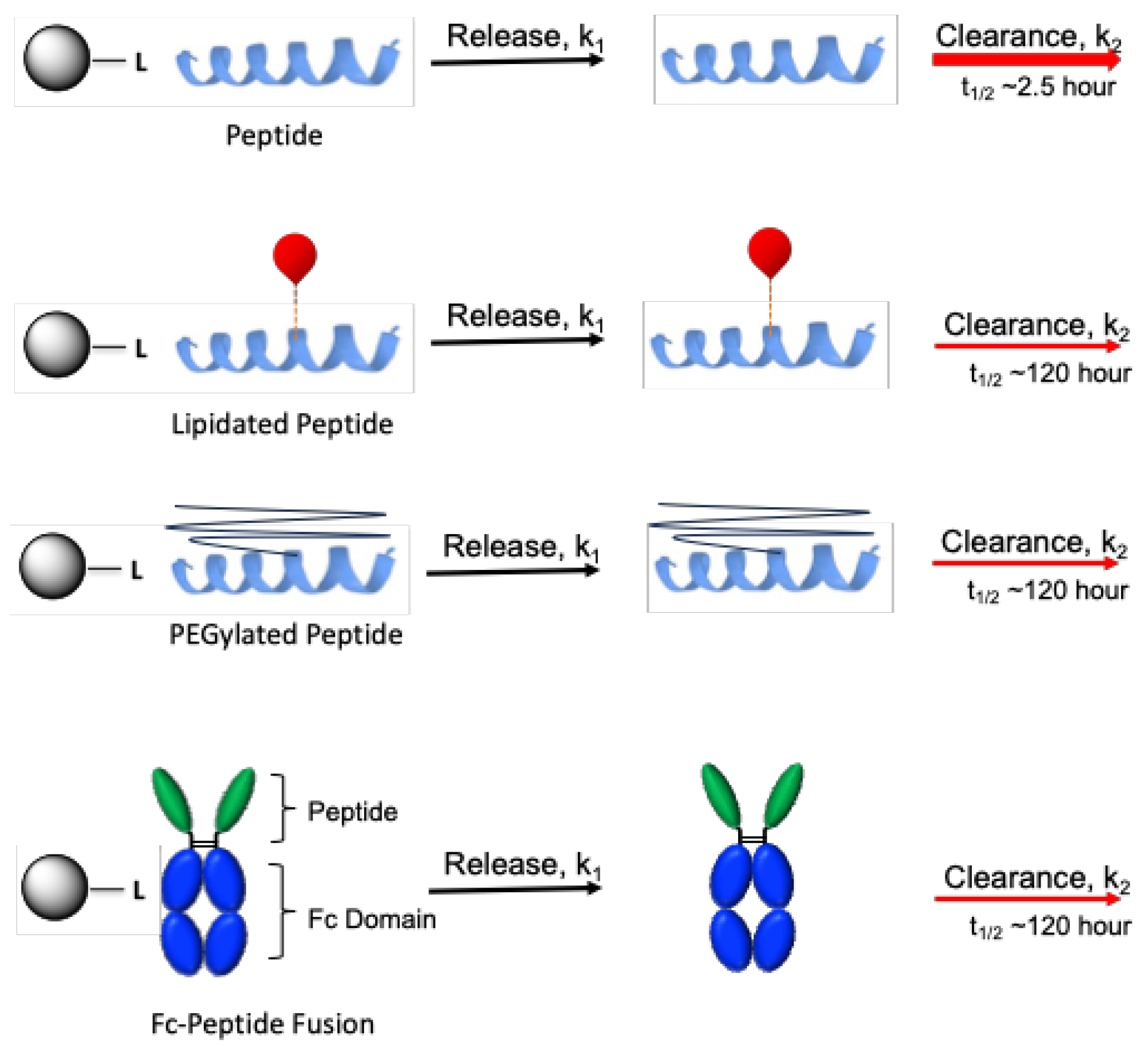

Table 1.
GLP-1 receptor agonists that have been proposed to be suitable for QMo dosing *.
| Company | Product | Receptor target | Drug type | Half-life, d*** |
current dosing interval, Wk | Fold dose & Cmax increase for QMo dosing **** |
|---|---|---|---|---|---|---|
| Progen | PG-102 | GLP-1/GLP-2 | Fc fusion | 5 | 1 | 32 |
| Hanmi | efpeglenatide | GLP-1/GLP-2 | Fc fusion | 6 | 1 | 13 |
| MBX | MBX4291 | GLP1/GIPR | acyl-peptide ** | 7 | 1 | 10 |
| Viking | VK2735 | GLP1/GIPR | acyl-peptide ** | 9 | 1 | 5 |
| QL Bio | ZT002 | GLP-1 | acyl-peptide ** | 11 | 1 | 3 |
| Amgen | AMG133 | GIP(ant)/GLP-1 | mAb | 14 | 4 | 2 |
* data sources: Progen, Diabetes 2024;73 (Supp1):1859-LB; Hanmi, Diabetes Care 2022, 45, 1592; MXB, SEC 08/023/24; Viking, Investor’s Business Daily 08/27/2024; QL Bio, Diabetes 2024;73(Supp. 1):119-OR; Amgen, Nat Metab 2, 290, 2024. ** lipidated peptide. ***median t1/2 if range is reported. **** calculated as 2^(30/t1/2)/2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



