
Submitted:
07 October 2024
Posted:
08 October 2024
You are already at the latest version
Abstract
The blood-brain barrier (BBB) serves as an interface between the bloodstream and central nervous system. It limits the movement of molecules and immune cells, regulates the entry of nutrients, and removes waste products from the brain. The dysfunction of the BBB has been identified in Parkinson´s disease (PD) but the role of the BBB and endothelial cells (ECs) has not been well studied. LRRK2 G2019S mutation is the most common PD causing mutation with similar pathophysiology than in sporadic cases. How the mutation affects EC function has not been investigated previously in patient’s cells. In the study, we used iPSC-derived ECs from PD patients with the LRRK2 mutation as well as cells from healthy individuals. We report that PD ECs have higher levels of α-synuclein, altered mitochondrial respiration and response to inflammatory exposure, especially to TNFα. In addition, transcriptomic analysis showed upregulation of fatty acid synthesis related pathways in PD ECs and downregulation of lncRNA MEG3, both of which have been associated with PD. Altogether, PD ECs manifest some of the PD related hallmarks and are likely to contribute to the pathogenesis of PD.
Keywords:
endothelial cell
; blood-brain barrier
; Parkinson´s disease
; iPSC-cells
; inflammation
; MEG3
1. Introduction
Parkinson´s disease (PD) is the fastest-growing human neurological disease due to an aging population, with a prediction to affect almost 2.5 million people in Europe by 2040 [1]. It is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta and the presence of Lewy bodies [2]. The loss of dopaminergic neurons is considered to be the reason for the motor symptoms (tremor, rigidity, and slowness with walking), but there are several non-motor symptoms that frequently occur and may precede the onset of the motor symptoms. Although most of the PD cases are sporadic, around 10% have a familial background. So far 20 disease-associated genes have been identified, including mutations in LRRK2, SNCA, PINK1, Parkin, and GBA [3]. The G2019S mutation in LRRK2 (leucine-rich repeat kinase 2) accounts for 5-13% of the familial cases [4]. This mutation results in excessive activation of the kinase domain, and the clinical pathophysiology closely mimics that of sporadic cases. LRRK2 has been shown to be expressed and is known to be localized in mitochondria, endoplasmic reticulum, Golgi apparatus, endolysosomal system, and synaptic vesicles. The physiological function of LRRK2 has been linked to neurite outgrowth, vesicle trafficking, regulation of the autophagy pathway, cytoskeleton dynamics, mitochondrial function, mRNA translation, and the immune system.
While several molecular mechanisms are identified in PD, including α-synuclein pathology, neuroinflammation, mitochondrial dysfunction, and impaired protein degradation, the exact cause remains unknown [5]. Furthermore, disruption of the blood-brain barrier (BBB) has been identified in PD patients and in animal models [6]. The BBB is a semipermeable membrane that separates the central nervous system from the periphery. The BBB is mainly formed by brain endothelial cells (ECs), pericytes, and astrocytes that, together with neurons and microglia, present a functional unit called the neurovascular unit. The BBB maintains the brain homeostasis, regulates delivery of oxygen and important nutrients to the brain, protects the brain from changes in the periphery, and removes carbon dioxide and toxic metabolites from the brain. Altogether, proper function of the BBB is crucial for maintenance of healthy brain tissue.
Previous studies have identified decreased P-glycoprotein expression, increased leakiness of the BBB in basal ganglia, accumulation of serum proteins, and EC degeneration in PD patients [7,8,9,10,11,12,13]. 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) animal models have identified BBB leakage, increased P-gp immunoreactivity, and infiltration of immune cells [14,15,16,17]. Moreover, activation of astrocytes in PD can enhance release of pro-inflammatory cytokines, increasing neuronal death and affecting the function of the BBB [18]. Although BBB impairment is recognized in PD, there have been limited investigations on this topic, and the underlying mechanism is incompletely understood.
Here, we report for the first time how the LRRK2 G2019S mutation affects human induced pluripotent stem cell (hiPSC)-derived ECs. The impact was examined at the transcriptome and functional levels under basal and inflammatory-exposed conditions.
2. Results
2.1. Differentiation and Characterization of hiPSC-Derived Endothelial Cells
The differentiation of ECs was adopted from a previously published protocol [19] with minor modifications. ECs were differentiated to vascular progenitor cells through mesoderm. The progenitors were then sorted and cultured for two passages before being used for experiments (Figure 1a,b). The expression levels of EC-related genes CDH5, CLDN5, OCLN, and TJP1 were compared to the negative fraction obtained from the MACS separations (Figure 1c). Both healthy and PD ECs exhibited elevated expression levels of the genes of interest in comparison to the MACS negative fraction. Healthy and PD ECs also expressed CD31, ZO1, VE cadherin, and claudin 5 at the protein level (Figure 1d, Figure S1). To evaluate the barrier formation by the ECs, we carried out permeability tests with 4 kDa dextran and Lucifer yellow (LY, 0.4 kDa). ECs showed size selectiveness as permeability was lower for 4 kDa dextran than LY (Figure 1e). There were no observed differences between healthy and PD ECs. LRRK2 protein expression was confirmed in ECs using western blot. (Figure 1f, Figure S2).
2.2. LRRK2 Mutation induces Moderate Changes in Transcriptomics of Endothelial Cells
RNA sequencing was conducted to investigate the impact of the LRRK2 mutation on the transcriptome of ECs. Due to the limited sample size, p-value <0.05 was used to identify differentially expressed genes (DEGs). A total of 336 genes were downregulated and 96 genes were upregulated in PD ECs when compared to healthy controls (Figure 2a). Among the top downregulated DEGs in PD ECs were pluripotency genes (KLF4, SOX2 and POU5F1), long non-coding RNAs (MEG3, MEG8) and EC function-associated genes (KLF4, VCL, CYP1B1) (Figure 2b). To validate the expression levels of downregulated genes in ECs, we employed qPCR and confirmed that the levels of KLF4 and MEG3 were significantly decreased when compared to healthy ECs (Figure 2c). We also confirmed that the expression of pluripotency genes SOX2 and POU5F1 was significantly lower in ECs compared to hiPSCs (Figure 2d). Up- and down-regulated DEGs were analyzed with Enrichr to identify the pathways that were altered between healthy and PD ECs. The pathway analysis (KEGG) revealed that the genes that were elevated in PD ECs were involved with metabolic processes of arachidonic and linoleic acid (Figure 2e,f). On the other hand, the downregulated genes were found to be linked to the TGFβ and hippo signaling pathways (Figure 2e). The upregulated gene ontology (GO) biological processes in PD ECs included positive regulation of unsaturated fatty acid and prostaglandin biosynthesis (Figure 2g). This finding was consistent with results of the KEGG analysis. The processes that were downregulated were specifically associated with axonogenesis and axon guidance (Figure. 2g). We also examined if EC function related pathways were altered. In PD ECs, blood vessel morphogenesis and vascular transport/transport across the BBB were reduced (Figure. 2g,i).
2.3. LRRK2 Mutant Endothelial Cells Have Higher Levels of A-Synuclein
This Given that α-synuclein accumulation is one of the hallmarks of PD, we quantified the amounts of α-synuclein in ECs. The presence of α-synuclein was detected in ECs by immunostaining (Figure 3a) and measured using ELISA. The amount of α-synuclein in LRRK2 mutant ECs was significantly higher compared to the healthy cells (Figure 3b). No differences in secreted α-synuclein were observed in media (Figure 3c).
2.4. LRRK2 Mutant Endothelial Cells Have Altered Mitochondrial Respiration
In order to investigate the mitochondrial function of ECs, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were quantified using the Seahorse XF assay. The OCR values for basal respiration, maximal, and spare respiratory capacity, proton leak and ATP linked respiration were determined (Fig. 4a-e). The levels of maximal respiration and ATP linked respiration were significantly decreased in PD ECs (Fig. 4c,e). A declining pattern was also noted in basal respiration and spare respiratory capacity although the differences were not significant (Fig. 4d,e). The proton leak remains constant in both healthy and PD ECs. The OCR/ECAR ratio computed from the basal level exhibited a minor decrease in PD ECs but not significantly (Fig. 4f). There were no observed differences in glycolysis, maximal glycolytic or spare glycolytic capacity between healthy and PD LRRK2 groups (Figure 4g-i).
2.4. RNA Transcriptomics Reveal More Prominent effect in PD Endothelial Cells After Tnfα Exposure
We used rna sequencing to compare the impact of 4 h TNFα and TNFα+IL-1β exposures on the transcriptomics of healthy and PD ECs. Both healthy and PD ECs responded to inflammatory stimuli, however the reaction was more pronounced when they were exposed to a combination of stimuli (Figure 3S a-i). For both exposures, pathway analysis revealed a distinct increase in inflammatory related pathways and processes, including TNF and NF-κB signaling pathways as well as inflammatory response (Figure 3S c,e,h,i). While both healthy and PD ECs showed a comparable response to exposures, the number of DEGs was greater in PD ECs (Figure 3S a,f). This phenomenon was particularly observed in cells exposed to TNFα, suggesting a modified response in cells affected by PD.
Given that the ECs of both genotypes responded to inflammatory stimuli (Figure 3S), next we wanted to examine the differences between healthy and PD ECs after being exposed to inflammation. The exposure to TNFα altered the gene expression between healthy and PD ECs. Specifically, 609 genes were found to be downregulated and 522 upregulated (Figure 5a). The pluripotency genes SOX2 and POU5F1 were shown to be reduced in PD ECs but their expression levels were similar to those in non-exposed cells (raw data). As in non-exposed, MEG3 expression was decreased in PD ECs, and this finding was further validated using qPCR (Figure 5b). The KEGG Pathway analysis revealed that upregulated genes were connected to steroid biosynthesis, p53 pathway and biosynthesis of unsaturated fatty acids whereas downregulated genes were linked to cell cycle (Figure 5c,d). The GO pathways were associated with cell migration and vascular endothelial growth factor production for upregulated genes and EC proliferation for downregulated genes (Figure 5c). The exposure to TNFα+IL-1β resulted in upregulation of 91 genes and downregulation of 277 genes which was in the same range as in the non-exposed condition (Figure 5e). Once again, pluripotency genes SOX2 and POU5F1 were downregulated in PD ECs but the levels were unchanged from the non-exposed (raw data). The expression of MEG3 was found to be downregulated in PD ECs as it was seen in TNFα and non-exposed cells. The gene expression was confirmed with qPCR (Figure 5f). Altered pathways of upregulated genes were related to mast cell activation, regulation of unsaturated fatty acid and prostaglandin biosynthesis processes in KEGG (Figure 5g). While GO pathways were linked to blood vessel morphogenesis, vasculature development and vascular transport in downregulated genes (Figure 5g). Altogether, the changes between healthy and PD ECs were more prominent with TNFα alone whereas the TNFα+IL-1β combination did not lead to additional patient specific changes when compared to the unexposed condition (Figure 5h).
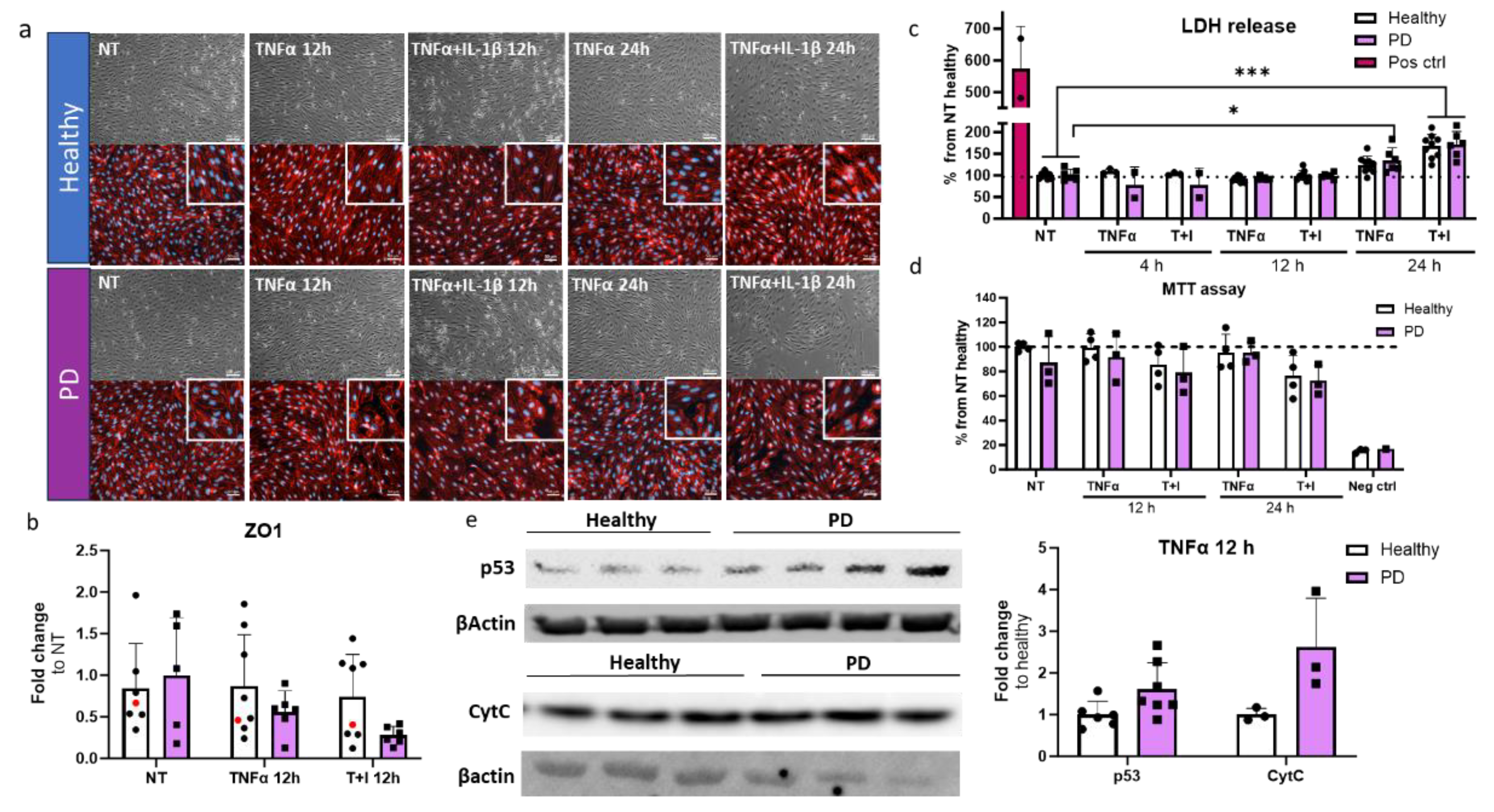
2.5. Prolonged Inflammatory Environment Affects Endothelial Cells Viability
Next, we investigated the cell viability and cytotoxicity in the ECs after inflammatory stimuli, especially TNFα, which generated alterations in transcription levels related to the p53 signaling pathway. For this, we utilized longer stimulation times of 12 and 24 h. First, we examined whether longer inflammatory stimuli affect cell viability. The viability was evaluated with brightfield imaging and subsequent immunostaining for tight junction protein ZO1 (Figure 6a). In PD ECs, TNFα+IL-1β exposure for 24 h resulted in cell number decrease while ZO1 expression was disrupted in PD ECs with all exposures. On the other hand, in healthy ECs, ZO1 disruption was only evident in combined exposures. Quantitative analysis of the immunocytochemistry showed a 1.45-fold decrease in ZO1 expression in PD ECs after 12 h TNFα exposure (Figure 6b). A 1.7-fold decrease was observed after combination exposure of TNFα+IL-1β (Figure 6b). We then assessed the release of LDH, a cytotoxicity marker. Elevated LDH levels by 1.7-fold were observed with 24 h of TNFα+IL-1β (TI) exposure in both healthy and PD ECs, as compared to non-exposed cells (Figure 6c). In PD ECs, 24 h TNFα exposure also significantly increased the LDH release by 1.35-fold which was not seen in healthy ECs. The viability and metabolic activity of cells were assessed using MTT assay (Figure 6d). The combined exposure led to a modest decrease in activity in both healthy and PD ECs compared to the unexposed group, although the change was not statistically significant. TNFα had no effect on viability.
Transcriptome analysis revealed that PD ECs exhibited the activation of p53 pathway and a reduction in cell cycle after being exposed to TNFα for 4 h. In order to determine whether the RNA expression translates to protein levels, p53 and Cytochrome C, a downstream target of p53, expression was analyzed by western blot for TNFα exposed cells (Figure 6e, Figure S4). As expected, the expression of p53 and Cytochrome C was elevated in PD ECs.
2.6. Endothelial cells Show Aberrant Phenotype In Pro-Inflammatory Conditions
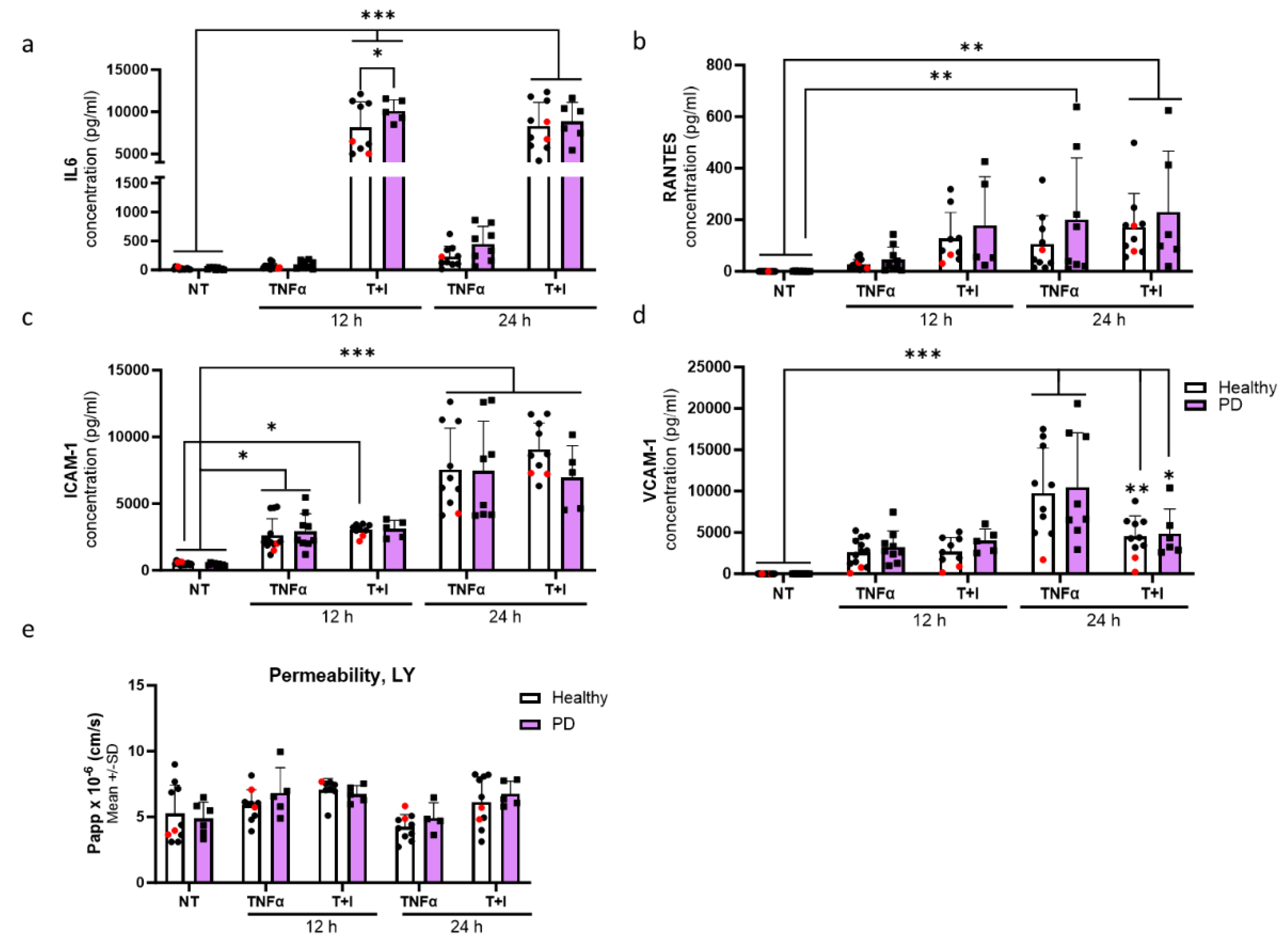
To assess the effect of inflammatory exposures on secretion of cytokines from endothelial cells, we first exposed the cells to TNFα or a combination of TI for either 12 or 24 h. IL-6 release was especially increased after combination exposures. At 12 h, the release was significantly higher in PD ECs with no difference at later time point when compared to healthy ECs (Figure 7a). An elevation in the release of RANTES was observed at 12 h in both healthy and PD ECs following TI exposure (Figure 7b). This elevation persisted at 24 h with no differences between genotypes, likely due to large variability among the lines that responded. The levels of ICAM1 gradually increased over time. However, various exposures did not have a significant impact on the response of both healthy and PD ECs (Figure 7c). Although, a decrease in ICAM1 levels was observed in PD ECs after 24 h combination exposure. The release of VCAM1 was highest after 24 h TNFα exposure but no differences were seen between healthy and PD ECs (Figure 7d). We further investigated the permeability using LY under the same inflammatory stimulating condition (Figure 7e). No changes in permeability were detected between healthy and PD ECs. However, the inflammatory conditions slightly increase the permeability at 12 h, still this change was not statistically significant.
Figure 6.
Aberrant phenotype of endothelial cells in pro-inflammatory conditions. (a-d) Release of (a) IL6, (b) RANTES, (c) ICAM1 and (d) VCAM1 quantified from media after 12 and 24 h TNFα and TNFα+IL-1β (TI) exposures measured with CBA. n= 10-13 (healthy), 1-2 (isogenic), 5-9 (PD LRRK2) from three independent experiments. (e) Permeability for Lucifer yellow (LY) in non-exposed, 12, and 24 h TNFα and TNFα+IL-1β (TI) exposed ECs. n=6-8 (healthy), 1-2 (isogenic), 4-6 (PD LRRK2) from two independent experiments. All data shown as mean +/-SD, Two-way ANOVA with Tukey´s multiple comparison test. p*<0.05, p**<0.01, p***<0.001.
Figure 6.
Aberrant phenotype of endothelial cells in pro-inflammatory conditions. (a-d) Release of (a) IL6, (b) RANTES, (c) ICAM1 and (d) VCAM1 quantified from media after 12 and 24 h TNFα and TNFα+IL-1β (TI) exposures measured with CBA. n= 10-13 (healthy), 1-2 (isogenic), 5-9 (PD LRRK2) from three independent experiments. (e) Permeability for Lucifer yellow (LY) in non-exposed, 12, and 24 h TNFα and TNFα+IL-1β (TI) exposed ECs. n=6-8 (healthy), 1-2 (isogenic), 4-6 (PD LRRK2) from two independent experiments. All data shown as mean +/-SD, Two-way ANOVA with Tukey´s multiple comparison test. p*<0.05, p**<0.01, p***<0.001.
3. Discussion
In the past, PD research concentrated mainly on neurons; but a current interest in studying other types of brain cells has shifted this viewpoint. While the involvement of astrocytes [20,21] and microglia [22] in PD pathogenesis is now well-established, there have been limited research focused on investigating the role of ECs, or the BBB, in this context. A recent study using single cell RNA sequencing from PD and healthy individuals suggested that ECs may have a significant impact on cell-cell communication in PD. The study identified ECs as the core cell cluster involved in intracellular communications [23]. Nevertheless, the effect of PD-related mutations on EC function remains unexplored.
Although the overall impact of the LRRK2 mutation on the ECs transcriptome was moderate, several noteworthy genes were identified. Kruppel-like factor 4 (KLF4) and maternally expressed gene 3 (MEG3) were downregulated in PD ECs. KLF4 has an important role in regulating several biological processes, including embryogenesis, proliferation, differentiation, neuroinflammation, oxidative stress, and apoptosis [24]. In ECs, KLF4 has been shown to promote vascular integrity and maintain vascular health [25]. In stroke models, KLF4 has a protective effect on microvascular ECs [26,27]. In addition, KLF4 has been shown to regulate EC activation after pro-inflammatory stimuli. Moreover, the overexpression of KLF4 induces the expression of several anti-inflammatory and anti-thrombotic factors [28]. In contrast to ECs, knockdown of KLF4 has shown beneficial effects on neuronal survival in several PD models [24]. This indicates that KLF4 has different roles in various cell types and downregulation might have a negative effect on LRRK2 mutated ECs.
MEG3 is a long noncoding RNA, and its role has been studied especially in cancer. Recent studies have identified MEG3 levels as being altered in PD patients. Two studies have reported decreased levels in plasma, and one has increased [29,30,31]. Huang et al. also studied the effect of MEG3 in SH-SY5Y cells treated with MPP+ [29]. Overexpression of MEG3 increased LRRK2 expression, improved cell viability, and decreased apoptosis, indicating a connection between MEG3 and LRRK2. In ECs, MEG3 has been shown to prevent vascular EC senescence, control VEGFA-mediated angiogenesis, and regulate proliferation and migration [32,33,34]. Knockdown of MEG3 in HUVECs decreased cell viability, increased apoptotic cell rate, and impaired migration function [35]. In addition, MEG3 knockdown has shown to induce cellular senescence, which is characterized by increased senescence-associated β–galactosidase activity, increased levels of endogenous superoxide, impaired mitochondrial structure and function, and impaired autophagy [36]. Taken together, these studies indicate that MEG3 has an important role in EC function and that the loss of expression in PD cells could be linked to pathogenesis.
Accumulation of α-synuclein is one of the hallmarks of PD. α-synuclein is a small 14 kDa protein highly expressed in neurons but also expressed in peripheral cells and ECs [6]. It can be transported bi-directionally across the BBB. Higher plasma levels of exosomal α-synuclein have been found in PD patients and post-mortem studies have associated α-synuclein aggregates with endothelial degeneration and decreased P-gp expression [9,37,38]. Our findings demonstrate that LRRK2 mutant ECs have higher levels of α-synuclein, but the released levels remained the same compared to healthy cells. This could indicate failure in exporting or degradation of α-synuclein in PD ECs, although this would require more extensive studies.
The transcriptomics analysis revealed alterations in pathways related to unsaturated fatty acids and their biosynthesis in PD ECs. Fatty acids have several important roles in the body, as they serve as a source of energy, are major components of cell membranes, and regulate inflammatory responses [39]. Excessive intake of fatty acids is associated with obesity, energy overload, and increased brain inflammation. Polyunsaturated fatty acids can exert a direct cytotoxic effect on α-synuclein. Polyunsaturated fatty acids can bind to α-synuclein and promote oligomerization, and high concentrations of DHA increase intracellular accumulation of soluble and insoluble α-synuclein and neuronal injury in A53T mice [40,41,42]. In addition, a higher intake of arachidonic acid has also been linked to an increased risk of PD pathogenesis [43]. Besides fatty acids themselves, fatty acid binding proteins (FABPs) 3 and 5 have been linked to α-synuclein accumulation and aggregation, and mitochondrial dysfunction, and increased apoptosis [39]. The RNA expression of both genes was elevated in PD ECs after TNFα exposure. Increased expression of FABPs combined with increased fatty acid biosynthesis might exacerbate the toxic effect of α-synuclein in ECs.
Mitochondrial dysfunction is one of the key mechanisms linked to PD pathogenesis. Higher oxidative stress, reduced mitochondria membrane potential, decreased ATP production, mitochondria DNA damage, elongated mitochondria, mitochondrial fragmentation and mitophagy have been observed in postmortem human tissues and animal and cellular models of LRRK2 [44]. PD ECs with the LRRK2 mutation showed altered mitochondrial function, including decreased maximal respiration and ATP-linked respiration. Alterations in oxygen consumption have been found in other cell types, including hiPSC-derived neurons [45] and astrocytes [20] which carry the LRRK2 G2019S mutation. This could indicate a similar effect of the mutation, regardless of the cell type.
One common hallmark of several neurodegenerative diseases, including PD, is neuroinflammation. The effect of inflammatory stimuli on PD ECs was studied on a transcriptomic and functional level. While TNFα+IL-1β exposure elicited a more pronounced response in both healthy and PD ECs compared to TNFα alone, the combination did not cause a substantial genotypic difference in transcriptomics. This could suggest that PD ECs have an altered response only to specific stimuli. At the functional level, the LRRK2 mutation did not cause major differences. ECs responded to inflammatory exposures by increasing the release of cytokines, but the variation across different cell lines was substantial. This could suggest that the response is rather individual than LRRK2-related. Although we did detect increased IL6 secretion after 12 h TNFα+IL-1β exposure, elevated levels of IL6 have been detected in PD patients and linked to disease mortality [46,47,48,49].
Upon longer inflammatory exposure, the cell death was increased in ECs. This was seen in both healthy and PD ECs after 24 h of combination exposure, but already in PD ECs after 24 h of TNFα exposure. We also detected elevated levels of p53 and cytochrome C in PD ECs after 12 h TNFα exposure. The p53 signaling pathway was also linked to upregulated genes in PD ECs after 4 h TNFα exposure. P53 is involved in several processes which are related to PD, like neuronal oxidative stress, apoptosis, and abnormal protein aggregation [50]. Moreover, high expression of p53 has been found in PD cell models and in the substantia nigra region in PD patients and animal models [51,52,53]. In addition, p53 can induce apoptosis through mitochondrial cytochrome C release and activation of caspases [54]. Interestingly, MEG3 has been shown to regulate p53 signaling, but the response seems to be cell specific. For example, in neurons, MEG3 activates p53 and mediates neuronal death in stroke [55], whereas in cardiac fibroblasts, MEG3 does not alter the p53 response [56]. The role of MEG3 and p53 in HUVECs was studied by Ali et al. [57]. After 4 h TNFα exposure, MEG3 knockdown induced p53 activation by increasing the expression of p53 target genes, promoting apoptosis, and inhibiting proliferation. Whether LRRK2 mutation is linked to MEG3 and p53 activation is still unclear and would need further studies.
Limitations of the study
This study focused on the LRRK2 G2019S mutation in ECs only. It is important to keep in mind that these results represent LRRK2 related cases and may not directly translate into sporadic ones. We haven´t checked additional mutations from the LRRK2 mutated cells and there is a small possibility that additional mutations or risk variants might influence the results. Although the dysfunction of the BBB and ECs is recognized in PD, its role probably is not as substantial as, for example, in Alzheimer's disease, and individual variation might play a part. Our results showed variation between healthy and PD individuals especially in transcriptomics and inflammatory responses. Of course, a low number of cell lines could mask the differences and ideally, more lines should be included. One way to reduce the variation is to use isogenic lines. We included one isogenic line, but this was absent from the transcriptomic studies. It would be important to repeat the RNA sequencing with additional lines and isogenic controls. Additionally, we used only 2D monoculture, and the remaining BBB cell types were missing. The BBB is a multicellular unit, including astrocytes and pericytes, and their presence could change the properties of ECs and thus also the effect of the LRRK2 mutation. Within this study, we used a protocol for deriving vascular ECs as opposed to brain ECs. Because of this, some BBB specific changes might have been overlooked in our study. Given our emphasis on inflammation, we aimed to ensure that cells respond to inflammatory stimuli. While there are existing protocols published for obtaining brain-like ECs, these protocols have major limitations. It has been shown that brain-like EC protocols produce cells with more epithelial cell phenotype and lack the EC-specific response to inflammatory stimuli [58,59,60]. Furthermore, one common feature of all hiPSC-ECs is the absence of P-glycoprotein expression and more importantly, its functionality [61]. Similarly, we observed very low levels in our cells. Decreased expression of P-glycoprotein has been linked to PD and investigation of this would require a different model system. Advances in different 3D models, like organ-on-chips or vascularized brain organoids, could provide more accurate models in the future.
4. Materials and Methods
4.1. Cell Culture
Human iPSCs (hiPSC) lines used in this study included six healthy controls, three lines with G2019S mutation in the LRRK2 gene and one isogenic line (Table S1). Healthy lines 4-6 and PD LRRK2 line 1 were generated and characterized in the Stem Cell Laboratory, at the University of Eastern Finland [62]. Healthy lines 1-3 were obtained from Takara Bio (Y00275, Y00305, and Y00325) and PD LRRK2 lines 2 and 3 and isogenic line from NIBSC.
4.2. Endothelial Cell Differentiation
Two differentiation protocols were used in this study (referred to as protocols 1 and 2). Detailed information of lines and protocols used in different experiments are listed in Table S2. In both protocols, ECs are differentiated first into EC progenitors and then vascular ECs with small differences in small molecules. Protocol 1 was more efficient and for that reason it was chosen for most of the experiments. Protocol 2 was used to measure α-synuclein levels with ELISA and metabolic function with Seahorse assay.
For protocol 1, ECs were differentiated according to the protocol published by Harding et al. 2017 with minor modifications. To start the differentiation, hiPSCs were plated to Matrigel-coated dishes at a density of 15-25 k/cm2 in E8 media with 5 µM of ROCK inhibitor (Y-27632 dihydrochloride, Selleckchem). The next day (day 1), the media was changed to Stemdiff APEL2 media (Stemcell Technologies) supplemented with 6 µM CHIR-99021 (Cayman). On day 3, the media was changed to APEL2 media with an additional 25 ng/ml of BMP4 (Peprotech), 10 ng/ml of FGF (Peprotech), and 50 ng/ml of VEGF (Peprotech). Cells were cultured in this medium for three days, changing media on day 5. On day 6, ECs were sorted by magnetic separation (Miltenyi Biotec) using CD144 microbeads according to the manufacture`s protocol. CD144 positive cells were plated on Matrigel-coated dishes in Endothelial Growth Media MV2 (ECGM-MV2, PromoCell) with an additional 20 ng/ml of VEGF for the first passage. ECs were expanded, and cells in passage three were used for the experiments.
For protocol 2, ECs were differentiated using a protocol published by Patch et al. [63]. Briefly, the hiPSCs were dissociated using EDTA (Invitrogen) and seeded at a density of 300 000 cells/35 mm plate in E8 medium supplemented with ROCK inhibitor. The next day, the induction of the lateral mesoderm was started by replacing the media with N2B27 medium supplemented with 8 µM CHIR-99021 and 25 ng/ml BMP4 and continued for three days. The EC induction was started by replacing the N2B27 media with StemPro-34 SFM medium (Life Technologies) supplemented with 200 ng/ml VEGF and 2 µM Forskolin (Sigma). The cells were incubated in StemPro medium for two to four days, changing the medium every day. When the areas of differentiation were visible, the ECs were sorted by magnetic separation using CD144 microbeads according to the manufacture`s protocol. CD144 positive cells were plated on 6 cm Matrigel-coated plates in StemPro medium supplemented with 20 ng/ml VEGF.
4.3. Immunocytochemistry
ECs were fixed with 4 % formaldehyde (VWR) for 15 min in room temperature (RT) or methanol (VWR) for 15 min at 4°C followed by washes with PBS. For intracellular antigens, the ECs were permeabilized with 0.2 % Triton X-100 in PBS (Sigma) for 20 min at RT. The cells were blocked with 5 % normal goat serum (NGS, Vector) or 10 % horse serum (HS, Gibco) in PBS at RT for 1 h and then incubated with primary antibodies in 5 % NGS or 10 % HS at 4 °C overnight. For unconjugated antibodies, the cells were incubated in 5 % NGS or 10 % HS in PBS containing a secondary antibody for 1 h at RT. The cells were washed with PBS, and the nuclei were visualized with DAPI (ThermoFisher). The cells were imaged with the Zen Observer Z1 or Zen Imager AX10. The antibodies used in this study are listed in Table S3.
4.4. RT-qPCR
RNA was extracted using the RNA easy MiniKit (Qiagen) and RNA concentration was measured with NanoDrop or DS-11 FX Spectrophotometer/Fluorometer (Denovix). 500-1000 ng of the RNA were converted to cDNA (Maxima Reverse Transcriptase, ThermoFisher). RT-qPCR was conducted using the Maxima probe/ROX qPCR master mix (ThermoFisher) to quantify the relative expression of genes of interest utilizing TaqMan assays (Table S4) with Step One Plus (Applied Biosciences). The Ct mean value was normalized to internal Ct mean value of β-actin, and the relative expression is presented as a fold change compared to the control group
4.5. α-Synuclein ELISA
The amount of endogenous α-synuclein was determined from ECs using a human α-synuclein ELISA kit (Invitrogen and Biolegend). For the assay, cells were lysed with M-PER (Invitrogen) or cell extraction buffer (Invitrogen) and diluted 1 to 10 in reagent diluent. The luminescence or absorbance 450 nm was measured with PerkinElmer`s VICTOR2 multilabel plate reader. The α-synuclein concentration from the cells was standardized to total protein concentration using the Pierce™ BCA Protein Assay Kit (ThermoFisher). Media samples were collected from ECs cultured for three days without media change. Samples were stored at -70 °C before analysis. Human α-synuclein ELISA kit (AssayGene) was used to measure the α-synuclein levels from media. The luminescence or absorbance at 450 nm was measured with PerkinElmer`s VICTOR2 multilabel plate reader.
4.6. Seahorse
The oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR), indicators of mitochondrial respiration and glycolysis, respectively, were measured in real time from live cells with the Seahorse Extracellular Flux (XF) 24 or 96 Analyzer. First, the ECs were plated on Matrigel-coated plates in StemPro34 or ECGM-MV2 medium depending on protocol used and incubated overnight. Before the assay, the medium was changed to Seahorse XF Assay medium (Seahorse DMEM supplemented with 2 mM Glutamax) and incubated for 1 h at 37 °C without CO2 before adding the compounds. The final concentrations of D-glucose and sodium pyruvate were 10 mM, and 0.5 mM, respectively. For oligomycin, FCCP, antimycin, and rotenone the concentrations were 1.0 µM. The assay was performed as follows: 2 min mix time, 2 min wait time, and 3 min measure cycle. After the assay, the results were normalized to protein concentrations using Pierce™ BCA Protein Assay Kit (ThermoFisher). The results were analyzed with Wave program. Wells with negative values or aberrant OCR or ECAR patterns were excluded from each of the individual experiments.
4.7. Inflmammatory Exposures
To assess the inflammatory response, ECs were exposed to TNFα or a combination of TNFα (Peprotech) and IL-1β (Peprotech). Detailed information on duration, concentrations, and assays are listed in Table S5. Briefly, ECs were plated in ECGM-MV2 medium, and the next day, the media was changed to human Endothelial Serum Free Media (ESFM, Gibco) with additional 1X B27 (Gibco) and 10 ng/ml FGF (Peprotech). The following day, we initiated inflammatory exposures using the same media as described above.
4.8. Total RNA Sequencing
Cells were lysed on ice (Buffer RLT + β-mercaptoethanol 10 µl/ml), and RNA was extracted directly after lysis with the Qiagen RNeasy Mini Kit (Qiagen) with DNase I digestion (Qiagen). RNA concentration was measured with DS-11 FX Spectrophotometer/Fluorometer (Denovix). 1-4 µg of RNA were sent for library preparation and sequencing (performed by Azenta). Quality was evaluated with FastQC, and the mean quality score for the samples was 35.30. The library preparation workflow included ribosomal RNA depletion, RNA fragmentation and random priming, cDNA synthesis, end repair, 5´ phosphorylation and dA-tailing, adaptor ligation and PCR enrichment. Sequencing was done with Illumina NovaSeq, PE 2x150. Sequence reads were trimmed by using Trimmomatic v.0.36 to remove possible adaptor sequences and nucleotides of poor quality. Trimmed reads were mapped to the Homo sapiens GRCh38 reference genome available on ENSEMBL using the STAR aligner v.2.5.2b. Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2 and used for downstream differential expression analysis (DESeq2). The Wald test was used to generate p-values and log2 fold changes, and the Benjamini-Hochberg for adjusted p-values. Genes with a p-value < 0.05 and absolute log2 fold change > 1 were called as differentially expressed genes (DEGs) for healthy vs. PD LRRK2 and genes with an adjusted p-value < 0.05 and absolute log2 fold change > 1 when comparing exposures within the group. Pathway enrichment analysis (KEGG and gene ontology biological processes, GO BP) was conducted on DEGs using Enrichr.
4.9. LDH Cytotoxicity Test
To assess the toxicity of TNFα and IL-1β exposures, the release of LDH was measured from ECs media samples at different time points (Table S5) using the CyQuant LDH Cytotoxicity assay (Invitrogen). The samples were analyzed with a Wallac Victor2 1420 Multilabel Counter (Perkin Elmer).
4.10. MTT Assay
ECs were plated to 96-well plate and treated as previously described (Table S5). After inflammatory exposure, a part of the cells was exposed to 1 % Triton-X for 5 min to induce cell death. This was used as a negative control. Then fresh media was changed with an additional 1.2 mM of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (VWR) and cells were incubated for 2 h at 37 °C. After the incubation, media was removed, and DMSO was added to the cells and incubated at RT overnight. The next day, 100 µl of sample was transferred to a new 96-well plate and absorbance was measured at 595 nm with PerkinElmer`s Victor2 multilabel plate reader.
4.11. Western Blotting
Protein was extracted from ECs using in-house made RIPA buffer (50 mM Tris, 1% Triton-X 100, 0.5% Sodium deoxycholate, 0.1% SDS, 150 mM NaCl) with Pierce™ protease inhibitor (ThermoFisher). Total protein amount was measured with Pierce™ BCA Protein Assay Kit (ThermoFisher). Samples were mixed with Laemmli sample buffer (60 mM Tris, 10% glycerol, 2% SDS, 1% Bromophenol blue, with 5% 2-Mercaptoethanol added just before use) and incubated at 95°C for 5 min. 15 µg (P53, Cytochrome C) or 30 µg (LRRK2) of protein was resolved with 12 % gels (P53, Cytochrome C) or 4%-12% gradient gel in Tris-Glycine running buffer.
Proteins were transferred to 0.2 µm PVDF pre-cut membrane transfer pack (Bio-rad) and transferred with Trans-Blot Turbo® transfer system (Bio-Rad, Hercules, CA, USA). Membranes were blocked in 5% Milk powder in TBST (20 mM Trish, 150 mM NaCl, 0.1% Tween-20, pH 7.6) and the membranes were incubated overnight at 4°C with primary antibodies diluted in 5% BSA in TBST (1:1000): p53, (1:1000): Cytochrome C, (1:10,000): LRRK2. Membranes were washed with TBST and incubated with HRP-conjugated antibodies for 2 hours at room temperature. After washing, detection was performed with SuperSignal™ West Pico PLUS Chemiluminescent Substrate (ThermoFisher) and imaged with ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA, USA). For normalization, membranes were incubated with anti β-actin primary antibody (1:1000) diluted in 5% BSA in TBST overnight at 4°C. Cy3-conjugated secondary antibody was diluted in 1:1000 in TBST and incubated for 2 h at room temperature. After washing, the membrane was imaged with the same equipment as previously. The intensity of the target bands was quantified with ImageLab (version 5.1, Bio-Rad Laboratories). Detailed information about antibodies is found in Table S3.
4.12. Quantification of Immunofluorescence
ECs were fixed and stained as previously described. Images were taken with Zen Imager AX10 and minimum of two images per sample. Fluorescence was analyzed with Fiji (ImageJ). IntDen of each sample was normalized to number and intensity of DAPI.
4.13. Cytometric Bead Assay
Smount of released cytokines was measured with the cytometric bead array (CBA) Human Soluble Protein Master Buffer Kit (BD). Before analysis, media samples from non-exposed and exposed samples were collected and stored at -70 °C. Capture beads used were: Human IL-6, Human RANTES, Human ICAM1, Human VCAM1. The samples were analyzed with CytoflexS (Becman Coultier). At least 300 events per cytokine were measured. Excitation was done with 638 nm red laser, and the bead clusters were detected with 660/20 BP (APC) and 780/60 BP filters (APC-A750). Cytokine reporter PE was excited with 561 nm yellow laser and filter 585/42 BP was used for detection. Data were analyzed using FCAP Array 2.0 (SoftFlow, Hungary) and cytokine concentrations were calculated by regression analysis from known standard concentrations.
4.14. Permeability Assay
ECs were plated to Matrigel-coated 24 well inserts (0.4 µm pore size, Sarstedt) at a density of 100 k/cm2 in ECGM-MV2. The next day, the media was changed to human Endothelial Serum Free Media (ESFM, Gibco) with additional 1X B27 (Gibco) and 10 ng/ml FGF (Peprotech). The following day, media was refreshed, or inflammatory exposures were started (Table S5). Permeability was measured with 4 kDa FITC Dextran (Merck) and Lucifer yellow (LY, Merck) from unexposed ECs. LY was used to measure permeability from inflammatory stimulated ECs. 150 µl of dextran (500 µg/ml) or LY (100 µg/ml) solution was added to the apical side and 800 µl of fresh media to basolateral side. Basal ESFM was used for the assay. Samples were collected from the wells at timepoints of 20, 40 and 60 min and the volume was replaced with fresh media. The fluorescence values were measured with PerkinElmer`s Victor2 multilabel plate reader. The background was reduced, and corrected fluorescence values were calculated. Standards were used to calculate the accumulated amounts over time and plotted against time. The apperent permeability coefficient (Papp) was calculated according to the equation (1), where dQ/dt (µg/s) is the dextran/LY flux (liner range) across the barrier, A is the insert membrane area (cm2) and C0 is the initial dextran/LY concentration (µg/cm3).
4.15. Data Analysis
The data were analyzed with GraphPad Prism (version 10.0.3). When comparing healthy vs. PD LRRK2, unpaired t-test was used. When comparing exposures, a two-way ANOVA with Tukey´s multiple comparison test was used. Levels of significance *p < 0.05; **p < 0.01; ***p < 0.001 were used. Outliers were tested with the GraphPad Grubbs' test.
The visualization of RNA transcriptomic data was done by using a free online platform for data analysis and visualization, https://www.bioinformatics.com.cn/en. Images were modified with Inkscape 1.2.
5. Conclusions
Overall, LRRK2 G2019S causes alterations in hiPSC-derived ECs. These include increased α-synuclein expression, decreased mitochondrial respiration, and altered transcriptome profile, especially upregulation of fatty acid related pathways, and altered responses to TNFα.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Expression of EC markers in hiPSC-derived ECs; Figure S2: Full length Western blot images of LRRK2 and βActin in PD and healthy ECs; Figure S3: Endothelial cells respond to inflammatory stimuli; Figure S4: Full length Western blot images of Cytochrome C (CytC), p53 and βActin in PD and healthy ECs; Table S1: Information of the patient; Table S2: Information of cell lines used in different assays; Table S3: Information of antibodies used in immunocytochemistry and western blotting; Table S4: List of primers used in RT-qPCR; Table S5: Inflammatory exposures for ECs.
Author Contributions
Conceptualization, T-M.S. and Š.L.; methodology, T-M.S.; validation, T-M.S.; formal analysis, T-M.S.; investigation, T-M.S., S.P., J.N.; resources, J.K.; data curation, T-M.S.; writing—original draft preparation, T-M.S. and Š.L.; writing—review and editing, Š.L. and R.M.; visualization, T-M.S.; supervision, Š.L. and R.M.; project administration, T-M.S. and Š.L; funding acquisition, Š.L., R.M., T-M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Sigrid Juselius Foundation (Š.L.), Doctoral Programme for Molecular Medicine (T-M.S.), Finnish Cultural foundation (T-M.S) and Business Finland (Š.L and R.H.H).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Hospital District of Northern Savo (#42//2010 and #123//2016).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request through Zenodo (doi: 10.5281/zenodo.10809110 (H1), 10.5281/zenodo.10817195 (H2), 10.5281/zenodo.10821851 (H3), 10.5281/zenodo.10829887 (PD1), 10.5281/zenodo.10837057 (PD2), 10.5281/zenodo.10837072 (PD3).
Acknowledgments
We would like to thank the Biocenter Kuopio services, including the Stem Cell Center. We thank Laila Kaskela and Eila Korhonen for their outstanding technical assistance and Sanna Ryytty for her help with the Seahorse and western blot analysis.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J Parkinsons Dis 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s Disease. J Neural Transm (Vienna) 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Piramanayagam, S. Impact of Gene Mutation in the Development of Parkinson’s Disease. Genes Dis 2019, 6, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr Neuropharmacol 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current Understanding of the Molecular Mechanisms in Parkinson’s Disease: Targets for Potential Treatments. Transl Neurodegener 2017, 6, 28. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Sonninen, T.-M.; Peltonen, S.; Koistinaho, J.; Lehtonen, Š. Blood–Brain Barrier and Neurodegenerative Diseases—Modeling with IPSC-Derived Brain Cells. Int J Mol Sci 2021, 22, 7710. [Google Scholar] [CrossRef]
- Bartels, A.L.; Willemsen, A.T.M.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.H.; Portman, A.; Leenders, K.L. Decreased Blood-Brain Barrier P-Glycoprotein Function in the Progression of Parkinson’s Disease, PSP and MSA. J Neural Transm (Vienna) 2008, 115, 1001–1009. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.H.; Vaalburg, W.; Bart, J.; Willemsen, A.T.M.; Hendrikse, N.H. Blood-Brain Barrier Dysfunction in Parkinsonian Midbrain in Vivo. Ann Neurol 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Kim, H.; Shin, J.-Y.; Lee, Y.-S.; Yun, S.P.; Maeng, H.-J.; Lee, Y. Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. Int J Mol Sci 2020, 21, 8538. [Google Scholar] [CrossRef]
- Ham, J.H.; Yi, H.; Sunwoo, M.K.; Hong, J.Y.; Sohn, Y.H.; Lee, P.H. Cerebral Microbleeds in Patients with Parkinson’s Disease. J Neurol 2014, 261, 1628–1635. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Vidyasagar, R.; Emsley, H.; Parkes, L. PO071 Mri Assessment of Neurovascular Changes in Idiopathic Parkinson’s Disease. J Neurol Neurosurg Psychiatry 2017, 88, A30–A30. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal Blood-Brain Barrier Permeability in Parkinson’s Disease. J Cereb Blood Flow Metab 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Pienaar, I.S.; Lee, C.H.; Elson, J.L.; McGuinness, L.; Gentleman, S.M.; Kalaria, R.N.; Dexter, D.T. Deep-Brain Stimulation Associates with Improved Microvascular Integrity in the Subthalamic Nucleus in Parkinson’s Disease. Neurobiol Dis 2015, 74, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-Induced Alterations in Blood-Brain Barrier Permeability. Eur J Neurosci 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Olmedo-Díaz, S.; Estévez-Silva, H.; Orädd, G.; Af Bjerkén, S.; Marcellino, D.; Virel, A. An Altered Blood-Brain Barrier Contributes to Brain Iron Accumulation and Neuroinflammation in the 6-OHDA Rat Model of Parkinson’s Disease. Neuroscience 2017, 362, 141–151. [Google Scholar] [CrossRef]
- Chung, Y.C.; Kim, Y.-S.; Bok, E.; Yune, T.Y.; Maeng, S.; Jin, B.K. MMP-3 Contributes to Nigrostriatal Dopaminergic Neuronal Loss, BBB Damage, and Neuroinflammation in an MPTP Mouse Model of Parkinson’s Disease. Mediators Inflamm 2013, 2013, 370526. [Google Scholar] [CrossRef]
- Zhao, C.; Ling, Z.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-Alpha Knockout and Minocycline Treatment Attenuates Blood-Brain Barrier Leakage in MPTP-Treated Mice. Neurobiol Dis 2007, 26, 36–46. [Google Scholar] [CrossRef]
- Wong, D.; Dorovini-Zis, K.; Vincent, S.R. Cytokines, Nitric Oxide, and CGMP Modulate the Permeability of an in Vitro Model of the Human Blood-Brain Barrier. Exp Neurol 2004, 190, 446–455. [Google Scholar] [CrossRef]
- Harding, A.; Cortez-Toledo, E.; Magner, N.L.; Beegle, J.R.; Coleal-Bergum, D.P.; Hao, D.; Wang, A.; Nolta, J.A.; Zhou, P. Highly Efficient Differentiation of Endothelial Cells from Pluripotent Stem Cells Requires the MAPK and the PI3K Pathways. Stem Cells 2017, 35, 909–919. [Google Scholar] [CrossRef]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic Alterations in Parkinson’s Disease Astrocytes. Sci Rep 2020, 10, 14474. [Google Scholar] [CrossRef]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific IPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Reports 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Ohtonen, S.; Giudice, L.; Jäntti, H.; Fazaludeen, M.F.; Shakirzyanova, A.; Gómez-Budia, M.; Välimäki, N.-N.; Niskanen, J.; Korvenlaita, N.; Fagerlund, I.; et al. Human IPSC-Derived Microglia Carrying the LRRK2-G2019S Mutation Show a Parkinson’s Disease Related Transcriptional Profile and Function. Sci Rep 2023, 13, 22118. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, L.; Qin, L.; Huang, H.; Li, X. Single-Cell Transcriptomics Uncovers Cellular Heterogeneity, Mechanisms, and Therapeutic Targets for Parkinson’s Disease. Front Genet 2022, 13, 686739. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, M.Y.; Golmohammadi, M.; Amin, R.S.; Bustani, G.S.; Romero-Parra, R.M.; Zabibah, R.S.; Oz, T.; Jalil, A.T.; Soltani, A.; Kujawska, M. Therapeutic Targeting of Krüppel-Like Factor 4 and Its Pharmacological Potential in Parkinson’s Disease: A Comprehensive Review. Mol Neurobiol 2023. [Google Scholar] [CrossRef]
- Tugal, D.; Jain, M.K.; Simon, D.I. Endothelial KLF4: Crippling Vascular Injury? Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease 2014, 3. [Google Scholar] [CrossRef]
- Huang, T.; Yin, J.; Ren, S.; Zhang, X. Protective Effects of KLF4 on Blood-Brain Barrier and Oxidative Stress after Cerebral Ischemia-Reperfusion in Rats through the Nrf2/Trx1 Pathway. Cytokine 2023, 169, 156288. [Google Scholar] [CrossRef]
- Yang, H.; Xi, X.; Zhao, B.; Su, Z.; Wang, Z. KLF4 Protects Brain Microvascular Endothelial Cells from Ischemic Stroke Induced Apoptosis by Transcriptionally Activating MALAT1. Biochemical and Biophysical Research Communications 2018, 495, 2376–2382. [Google Scholar] [CrossRef]
- Hamik, A.; Lin, Z.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerszten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like Factor 4 Regulates Endothelial Inflammation*. Journal of Biological Chemistry 2007, 282, 13769–13779. [Google Scholar] [CrossRef]
- Huang, H.; Zheng, S.; Lu, M. Downregulation of LncRNA MEG3 Is Involved in Parkinson’s Disease. Metab Brain Dis 2021, 36, 2323–2328. [Google Scholar] [CrossRef]
- Quan, Y.; Wang, J.; Wang, S.; Zhao, J. Association of the Plasma Long Non-Coding RNA MEG3 With Parkinson’s Disease. Front Neurol 2020, 11, 532891. [Google Scholar] [CrossRef]
- Honarmand Tamizkar, K.; Gorji, P.; Gholipour, M.; Hussen, B.M.; Mazdeh, M.; Eslami, S.; Taheri, M.; Ghafouri-Fard, S. Parkinson’s Disease Is Associated With Dysregulation of Circulatory Levels of LncRNAs. Front Immunol 2021, 12, 763323. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Li, Y.-J.; Li, D.-J.; Li, P.; Wang, J.-Y.; Diao, Y.-P.; Ye, G.-D.; Li, Y.-F. Long Noncoding RNA MEG3 Prevents Vascular Endothelial Cell Senescence by Impairing MiR-128-Dependent Girdin Downregulation. American Journal of Physiology-Cell Physiology 2019, 316, C830–C843. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Zhao, F.; Zhao, S.; Zhang, L.; Shi, L.; Pang, T. Knockdown of Long Noncoding RNA MEG3 Impairs VEGF-Stimulated Endothelial Sprouting Angiogenesis via Modulating VEGFR2 Expression in Human Umbilical Vein Endothelial Cells. Gene 2018, 649, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.-Z.; Tian, W.; Fu, H.-T.; Li, C.-P.; Liu, B. Long Noncoding RNA-MEG3 Is Involved in Diabetes Mellitus-Related Microvascular Dysfunction. Biochem Biophys Res Commun 2016, 471, 135–141. [Google Scholar] [CrossRef]
- Song, J.; Huang, S.; Wang, K.; Li, W.; Pao, L.; Chen, F.; Zhao, X. Long Non-Coding RNA MEG3 Attenuates the Angiotensin II-Induced Injury of Human Umbilical Vein Endothelial Cells by Interacting With P53. Front Genet 2019, 10, 78. [Google Scholar] [CrossRef]
- Cheng, X.; Shihabudeen Haider Ali, M.S.; Moran, M.; Viana, M.P.; Schlichte, S.L.; Zimmerman, M.C.; Khalimonchuk, O.; Feinberg, M.W.; Sun, X. Long Non-Coding RNA Meg3 Deficiency Impairs Glucose Homeostasis and Insulin Signaling by Inducing Cellular Senescence of Hepatic Endothelium in Obesity. Redox Biol 2021, 40, 101863. [Google Scholar] [CrossRef]
- Yang, P.; Min, X.-L.; Mohammadi, M.; Turner, C.; Faull, R.; Waldvogel, H.; Dragunow, M.; Guan, J. Endothelial Degeneration of Parkinson’s Disease Is Related to Alpha-Synuclein Aggregation. J Alzheimers Dis Parkinsonism 2017, 7. [Google Scholar] [CrossRef]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma Exosomal α-Synuclein Is Likely CNS-Derived and Increased in Parkinson’s Disease. Acta Neuropathol 2014, 128, 639–650. [Google Scholar] [CrossRef]
- Kawahata, I.; Fukunaga, K. Pathogenic Impact of Fatty Acid-Binding Proteins in Parkinson’s Disease—Potential Biomarkers and Therapeutic Targets. International Journal of Molecular Sciences 2023, 24, 17037. [Google Scholar] [CrossRef]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Exposure to Long Chain Polyunsaturated Fatty Acids Triggers Rapid Multimerization of Synucleins. J Biol Chem 2001, 276, 41958–41962. [Google Scholar] [CrossRef]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. Alpha-Synuclein Occurs in Lipid-Rich High Molecular Weight Complexes, Binds Fatty Acids, and Shows Homology to the Fatty Acid-Binding Proteins. Proc Natl Acad Sci U S A 2001, 98, 9110–9115. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The Formation of Highly Soluble Oligomers of Alpha-Synuclein Is Regulated by Fatty Acids and Enhanced in Parkinson’s Disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; Kawamura, N.; et al. Dietary Fat Intake and Risk of Parkinson’s Disease: A Case-Control Study in Japan. J Neurol Sci 2010, 288, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and Mitochondria: Recent Advances and Current Views. Brain research 2019, 1702, 96. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Familial Parkinson’s Disease IPSCs Show Cellular Deficits in Mitochondrial Responses That Can Be Pharmacologically Rescued. Sci Transl Med 2012, 4, 141ra90. [Google Scholar] [CrossRef]
- Diaz, K.; Kohut, M.L.; Russell, D.W.; Stegemöller, E.L. Peripheral Inflammatory Cytokines and Motor Symptoms in Persons with Parkinson’s Disease. Brain, Behavior, & Immunity - Health 2022, 21, 100442. [Google Scholar] [CrossRef]
- Hofmann, K.W.; Schuh, A.F.S.; Saute, J.; Townsend, R.; Fricke, D.; Leke, R.; Souza, D.O.; Portela, L.V.; Chaves, M.L.F.; Rieder, C.R.M. Interleukin-6 Serum Levels in Patients with Parkinson’s Disease. Neurochem Res 2009, 34, 1401–1404. [Google Scholar] [CrossRef]
- Dufek, M.; Rektorova, I.; Thon, V.; Lokaj, J.; Rektor, I. Interleukin-6 May Contribute to Mortality in Parkinson’s Disease Patients: A 4-Year Prospective Study. Parkinsons Dis 2015, 2015, 898192. [Google Scholar] [CrossRef]
- Scalzo, P.; Kümmer, A.; Cardoso, F.; Teixeira, A.L. Serum Levels of Interleukin-6 Are Elevated in Patients with Parkinson’s Disease and Correlate with Physical Performance. Neuroscience Letters 2010, 468, 56–58. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.; Li, H.; Pan, X.; Wang, Y. New Insights into the Roles of P53 in Central Nervous System Diseases. International Journal of Neuropsychopharmacology 2023, 26, 465–473. [Google Scholar] [CrossRef]
- Li, D.-W.; Li, G.-R.; Zhang, B.-L.; Feng, J.-J.; Zhao, H. Damage to Dopaminergic Neurons Is Mediated by Proliferating Cell Nuclear Antigen through the P53 Pathway under Conditions of Oxidative Stress in a Cell Model of Parkinson’s Disease. Int J Mol Med 2016, 37, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Kondo, T.; Mizuno, Y.; Nagatsu, T. P53 Protein, Interferon-Gamma, and NF-KappaB Levels Are Elevated in the Parkinsonian Brain. Neurosci Lett 2007, 414, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Sekar, S.; Taghibiglou, C. Nuclear Accumulation of GAPDH, GluA2 and P53 in Post-Mortem Substantia Nigral Region of Patients with Parkinson’s Disease. Neurosci Lett 2020, 716, 134641. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. P53 Induces Apoptosis by Caspase Activation through Mitochondrial Cytochrome c Release. J Biol Chem 2000, 275, 7337–7342. [Google Scholar] [CrossRef]
- Yan, H.; Yuan, J.; Gao, L.; Rao, J.; Hu, J. Long Noncoding RNA MEG3 Activation of P53 Mediates Ischemic Neuronal Death in Stroke. Neuroscience 2016, 337, 191–199. [Google Scholar] [CrossRef]
- Piccoli, M.-T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.L.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the Cardiac Fibroblast-Enriched LncRNA Meg3 Prevents Cardiac Fibrosis and Diastolic Dysfunction. Circ Res 2017, 121, 575–583. [Google Scholar] [CrossRef]
- Shihabudeen Haider Ali, M.S.; Cheng, X.; Moran, M.; Haemmig, S.; Naldrett, M.J.; Alvarez, S.; Feinberg, M.W.; Sun, X. LncRNA Meg3 Protects Endothelial Function by Regulating the DNA Damage Response. Nucleic Acids Res 2019, 47, 1505–1522. [Google Scholar] [CrossRef]
- Nishihara, H.; Gastfriend, B.D.; Soldati, S.; Perriot, S.; Mathias, A.; Sano, Y.; Shimizu, F.; Gosselet, F.; Kanda, T.; Palecek, S.P.; et al. Advancing Human Induced Pluripotent Stem Cell-Derived Blood-Brain Barrier Models for Studying Immune Cell Interactions. FASEB J 2020, 34, 16693–16715. [Google Scholar] [CrossRef]
- Delsing, L.; Dönnes, P.; Sánchez, J.; Clausen, M.; Voulgaris, D.; Falk, A.; Herland, A.; Brolén, G.; Zetterberg, H.; Hicks, R.; et al. Barrier Properties and Transcriptome Expression in Human IPSC-Derived Models of the Blood-Brain Barrier. Stem Cells 2018, 36, 1816–1827. [Google Scholar] [CrossRef]
- Lu, T.M.; Houghton, S.; Magdeldin, T.; Durán, J.G.B.; Minotti, A.P.; Snead, A.; Sproul, A.; Nguyen, D.-H.T.; Xiang, J.; Fine, H.A.; et al. Pluripotent Stem Cell-Derived Epithelium Misidentified as Brain Microvascular Endothelium Requires ETS Factors to Acquire Vascular Fate. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar] [CrossRef]
- Ozgür, B.; Puris, E.; Brachner, A.; Appelt-Menzel, A.; Oerter, S.; Balzer, V.; Holst, M.R.; Christiansen, R.F.; Hyldig, K.; Buckley, S.T.; et al. Characterization of an IPSC-Based Barrier Model for Blood-Brain Barrier Investigations Using the SBAD0201 Stem Cell Line. Fluids and Barriers of the CNS 2023, 20, 96. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Lehtonen, Š.; Chumarina, M.; Puttonen, K.A.; Azevedo, C.; Lebedeva, O.; Ruponen, M.; Oksanen, M.; Djelloul, M.; Collin, A.; et al. Creation of a Library of Induced Pluripotent Stem Cells from Parkinsonian Patients. npj Parkinson’s Disease 2016, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of Vascular Endothelial and Smooth Muscle Cells from Human Pluripotent Stem Cells. Nat Cell Biol 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Differentiation and characterization of hiPSC-derived endothelial cells. (a) A schematic illustration of the differentiation of hiPSC-ECs. HiPSCs are differentiated first to mesoderm and then vascular progenitors. VE cadherin (CD144) positive cells are MACS separated and ECs are expanded before experiments. (b) Representative bright field images of different stages of EC differentiation. Scale bar 50 µm. (c) Relative gene expression of CDH5, CLDN5, OCLN, and TJP1 in ECs compared to CD144 negative cells from MACS separation. n= 2 (MACS neg), 3 (healthy), 3 (PD LRRK2) (d) Representative fluorescent images of ECs stained with CD31, ZO1, VE cadherin, and claudin 5. Nuclei stained with DAPI. Scale bar 50 µm. (e) Permeability (Papp) of 4 kDa Dextran and Lucifer yellow. n= 8-14 (healthy), 1-2 (isogenic), 6-13 (PD LRRK2) from four (4kDa Dextran) or two (Lucifer yellow) independent experiments. (F) Western blot analysis of LRRK2 (230 kDa). Normalization to β-actin (42 kDa) protein levels. Representative blots and quantification of the data. n=8 (healthy), 2 (isogenic), 8 (PD LRRK2). All data shown as mean +/-SD.
Figure 1.
Differentiation and characterization of hiPSC-derived endothelial cells. (a) A schematic illustration of the differentiation of hiPSC-ECs. HiPSCs are differentiated first to mesoderm and then vascular progenitors. VE cadherin (CD144) positive cells are MACS separated and ECs are expanded before experiments. (b) Representative bright field images of different stages of EC differentiation. Scale bar 50 µm. (c) Relative gene expression of CDH5, CLDN5, OCLN, and TJP1 in ECs compared to CD144 negative cells from MACS separation. n= 2 (MACS neg), 3 (healthy), 3 (PD LRRK2) (d) Representative fluorescent images of ECs stained with CD31, ZO1, VE cadherin, and claudin 5. Nuclei stained with DAPI. Scale bar 50 µm. (e) Permeability (Papp) of 4 kDa Dextran and Lucifer yellow. n= 8-14 (healthy), 1-2 (isogenic), 6-13 (PD LRRK2) from four (4kDa Dextran) or two (Lucifer yellow) independent experiments. (F) Western blot analysis of LRRK2 (230 kDa). Normalization to β-actin (42 kDa) protein levels. Representative blots and quantification of the data. n=8 (healthy), 2 (isogenic), 8 (PD LRRK2). All data shown as mean +/-SD.
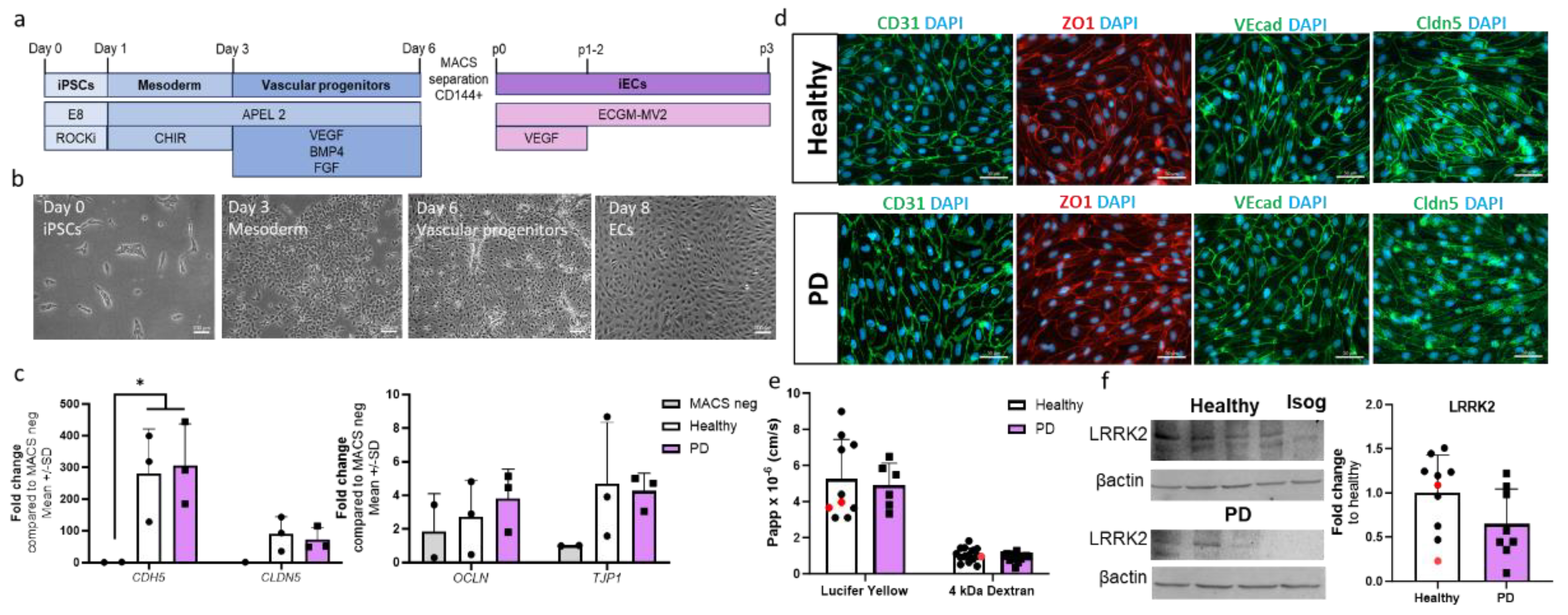
Figure 2.
RNA expression analysis of hiPSC-derived endothelial cells from healthy donors and PD patients. (a) Volcano plot of up- and down-regulated DEGs between healthy and PD ECs based on p-value (<0.05) and absolute log2 Fold >1. (b) Cluster heatmap of the top 30 DEGs between healthy and PD ECs based on the p-value (<0.05). (c) Gene expression levels of POU5F1 and SOX-2 in healthy and PD ECs compared to hiPSCs, assessed by qPCR. n= 2 (hiPSCs), n=6 (healthy), n=5 (PD LRRK2), two-way ANOVA. (d) Gene expression of KLF4 and MEG3 in healthy and PD ECs quantified with qPCR compared to healthy. KLF4 n=7 (healthy), n=6 (PD LRRK2), MEG3 n=6 (healthy), n=5 (PD LRRK2). Unpaired t-test. (e) KEGG pathway of up- and down-regulated genes in PD ECs compared to healthy. (f) Heatmap showing genes related to arachidonic acid metabolism in healthy and PD ECs. (g) GO pathway of up- and down-regulated genes in PD ECs compared to healthy. (h) GO biological processes related to vascular or EC function of up- and down-regulated genes in PD ECs compared to healthy. (i) Heatmap showing genes related to Blood vessel morphogenesis and vascular transport/transport across BBB in healthy and PD ECs. KEGG, the Kyoto Encyclopedia of Genes and Genomes; GO BP, gene ontology biological processes; PD1-3, patients carrying a mutation in LRRK2 gene (G2019S.) Data shown as mean +/-SD.
Figure 2.
RNA expression analysis of hiPSC-derived endothelial cells from healthy donors and PD patients. (a) Volcano plot of up- and down-regulated DEGs between healthy and PD ECs based on p-value (<0.05) and absolute log2 Fold >1. (b) Cluster heatmap of the top 30 DEGs between healthy and PD ECs based on the p-value (<0.05). (c) Gene expression levels of POU5F1 and SOX-2 in healthy and PD ECs compared to hiPSCs, assessed by qPCR. n= 2 (hiPSCs), n=6 (healthy), n=5 (PD LRRK2), two-way ANOVA. (d) Gene expression of KLF4 and MEG3 in healthy and PD ECs quantified with qPCR compared to healthy. KLF4 n=7 (healthy), n=6 (PD LRRK2), MEG3 n=6 (healthy), n=5 (PD LRRK2). Unpaired t-test. (e) KEGG pathway of up- and down-regulated genes in PD ECs compared to healthy. (f) Heatmap showing genes related to arachidonic acid metabolism in healthy and PD ECs. (g) GO pathway of up- and down-regulated genes in PD ECs compared to healthy. (h) GO biological processes related to vascular or EC function of up- and down-regulated genes in PD ECs compared to healthy. (i) Heatmap showing genes related to Blood vessel morphogenesis and vascular transport/transport across BBB in healthy and PD ECs. KEGG, the Kyoto Encyclopedia of Genes and Genomes; GO BP, gene ontology biological processes; PD1-3, patients carrying a mutation in LRRK2 gene (G2019S.) Data shown as mean +/-SD.
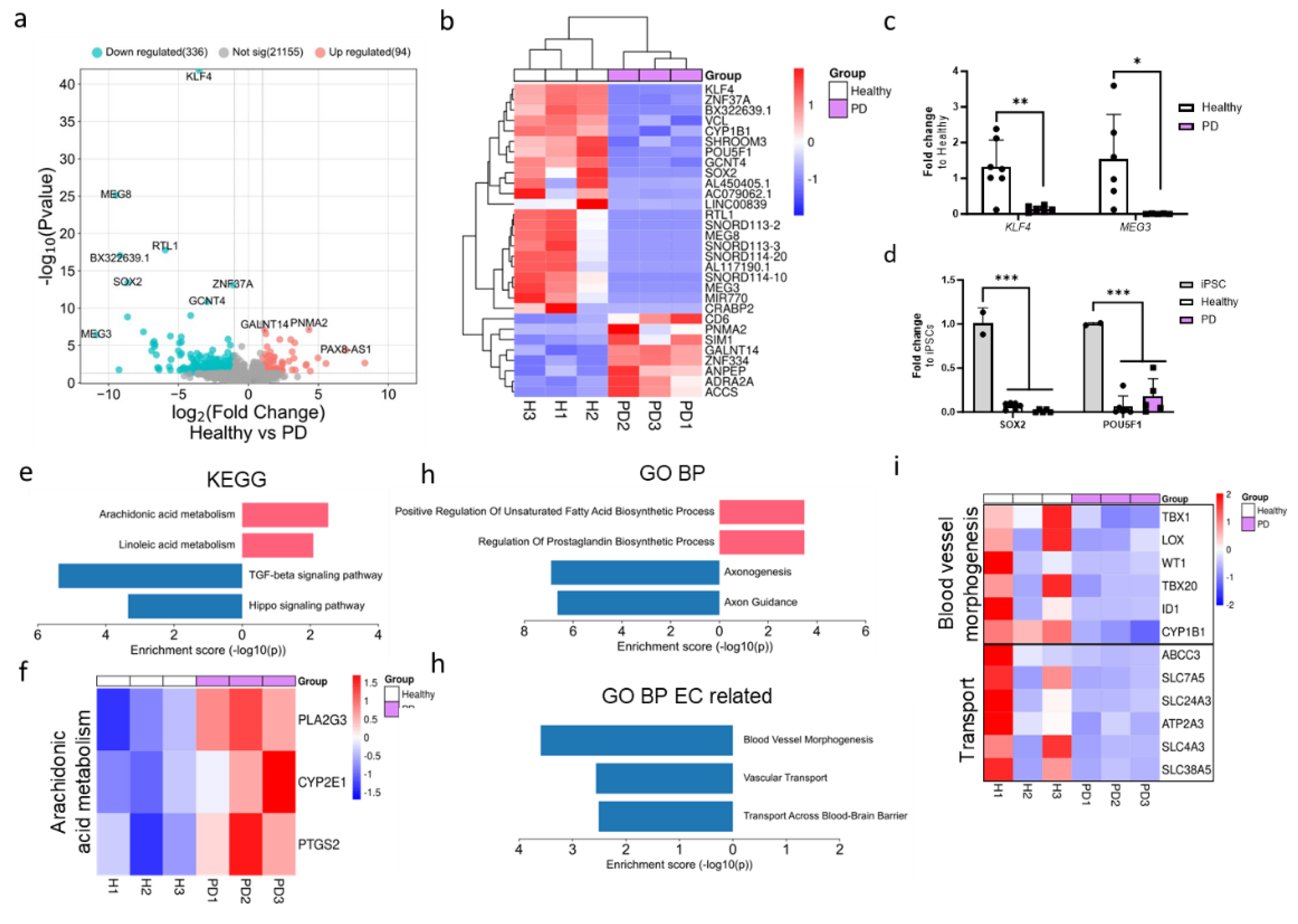
Figure 3.
Alpha-synuclein protein expression is increased in LRRK2 mutant endothelial cells. (a) Representative fluorescent images of ECs stained with ZO1 and α-synuclein (αsyn). Nuclei stained with DAPI. Scale bar 50 µm. (b) Protein expression of α-synuclein in ECs measured with ELISA. Data shown as fold change to healthy + isogenic (marked as red) and results are normalized to total protein amount. n= 16 (healthy), 1 (isogenic), 14 (PD LRRK2), from five independent experiments. (c) Released amount of α-synuclein in media measured with ELISA. Data shown as fold change to healthy + isogenic. n= 14 (healthy), 2 (isogenic), 12 (PD LRRK2) from five independent experiments. Data shown as mean +/- SD. Unpaired t-test, ** p< 0.01.
Figure 3.
Alpha-synuclein protein expression is increased in LRRK2 mutant endothelial cells. (a) Representative fluorescent images of ECs stained with ZO1 and α-synuclein (αsyn). Nuclei stained with DAPI. Scale bar 50 µm. (b) Protein expression of α-synuclein in ECs measured with ELISA. Data shown as fold change to healthy + isogenic (marked as red) and results are normalized to total protein amount. n= 16 (healthy), 1 (isogenic), 14 (PD LRRK2), from five independent experiments. (c) Released amount of α-synuclein in media measured with ELISA. Data shown as fold change to healthy + isogenic. n= 14 (healthy), 2 (isogenic), 12 (PD LRRK2) from five independent experiments. Data shown as mean +/- SD. Unpaired t-test, ** p< 0.01.
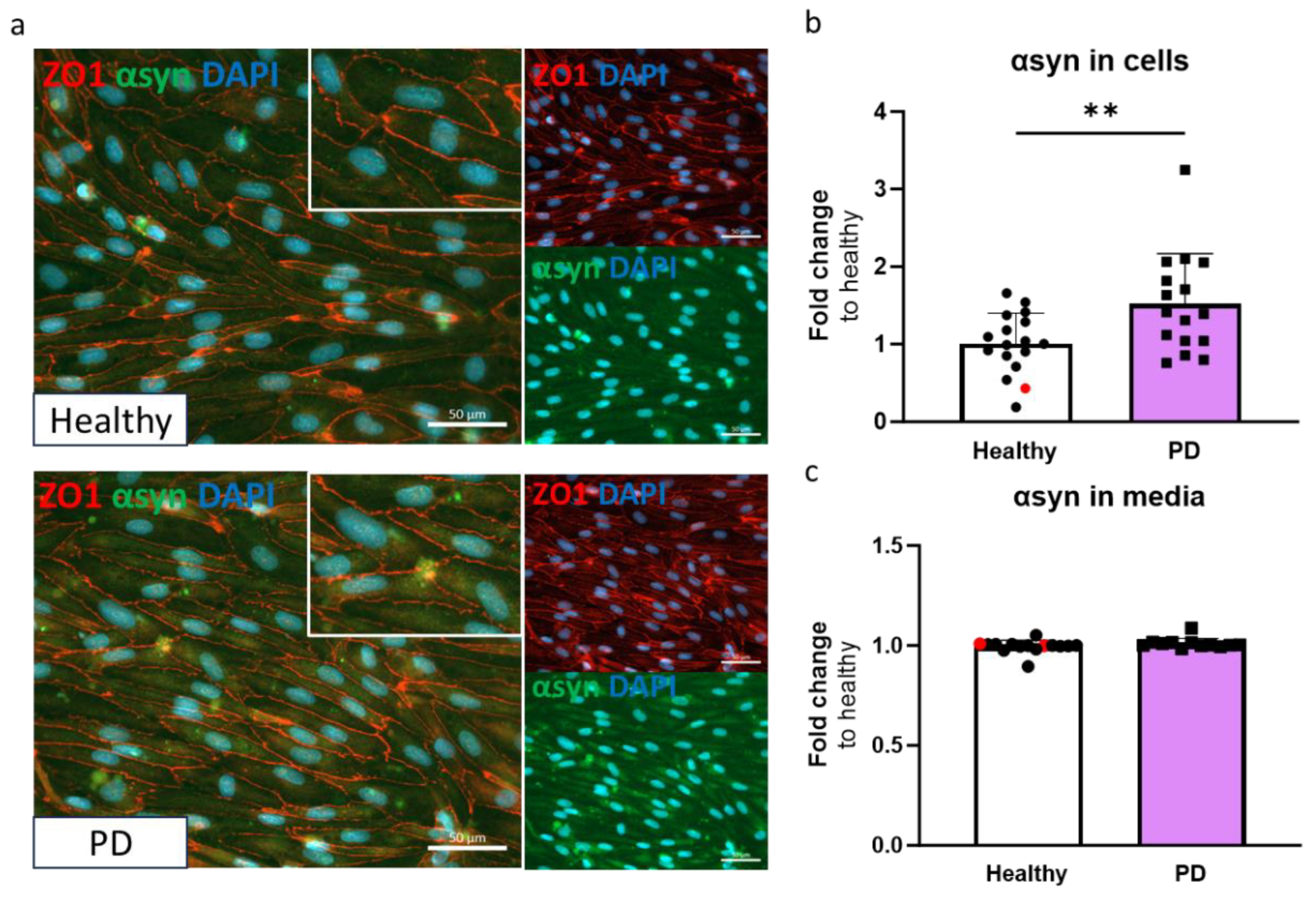
Figure 4.
The altered metabolic profile in PD endothelial cells. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured with Seahorse XF assay. (a-e) Calculated OCR values of basal respiration, proton leak, maximal respiration, spare respiratory capacity, and ATP linked respiration, respectively. (f) OCR/ECAR ratio from basal level. (g-i) Calculated ECAR values of glycolysis, maximal glycolytic capacity, and spare glycolytic capacity, respectively. Data shown as fold change to healthy + isogenic. n= 11-12 (healthy), 1 (isogenic), 10-11 (PD LRRK2) from five independent experiments. Data shown as mean +/- SD, Unpaired t-test, * p< 0.05.
Figure 4.
The altered metabolic profile in PD endothelial cells. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured with Seahorse XF assay. (a-e) Calculated OCR values of basal respiration, proton leak, maximal respiration, spare respiratory capacity, and ATP linked respiration, respectively. (f) OCR/ECAR ratio from basal level. (g-i) Calculated ECAR values of glycolysis, maximal glycolytic capacity, and spare glycolytic capacity, respectively. Data shown as fold change to healthy + isogenic. n= 11-12 (healthy), 1 (isogenic), 10-11 (PD LRRK2) from five independent experiments. Data shown as mean +/- SD, Unpaired t-test, * p< 0.05.
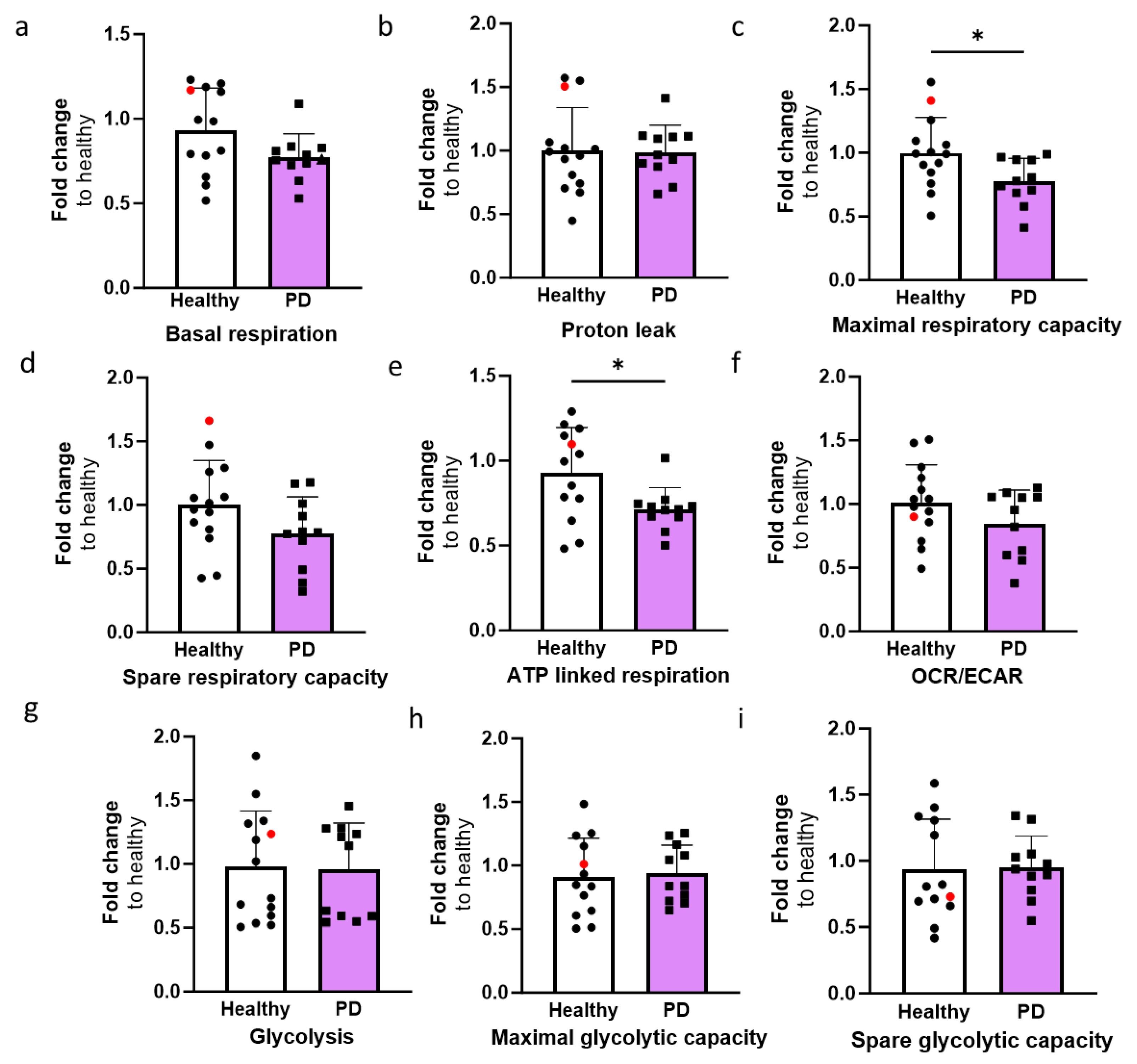
Figure 5.
Transcriptional profile of PD endothelial cells after inflammatory stimuli. (a)Volcano plot of up- and down-regulated DEGs between healthy and PD ECs after TNFα exposure based on p-value (<0.05) and absolute log2 Fold >1. (b) Gene expression of MEG3 in healthy and PD ECs quantified with qPCR. n=5 (healthy), 6 (PD LRRK2). Two independent experiments. (c) KEGG and GO pathways of up- and down-regulated DEGs in PD ECs compared to healthy. (d) Heatmap showing genes related to p53 signalling pathway and steroid biosynthesis in healthy and PD ECs. (e) Volcano plot of up- and down-regulated DEGs between healthy and PD ECs after TNFα+IL-1β exposure based on p-value (<0.05) and absolute log2 Fold >1. (f) Gene expression of MEG3 in healthy and PD ECs. n=6 (healthy), 6 (PD LRRK2). Two independent experiments. (g) KEGG and GO pathways of up- and down-regulated genes in PD ECs compared to healthy. (h) Venn diagram of DEGs between healthy and PD ECs in unexposed, TNFα and TNFα+IL-1β exposed conditions. All exposures lasted for 4 h and the concentrations were: 10 ng/ml TNFα or 10 ng/ml TNFα+ IL-1β. Data shown as mean +/-SD, n= 3, unpaired t-test ** p< 0.01.
Figure 5.
Transcriptional profile of PD endothelial cells after inflammatory stimuli. (a)Volcano plot of up- and down-regulated DEGs between healthy and PD ECs after TNFα exposure based on p-value (<0.05) and absolute log2 Fold >1. (b) Gene expression of MEG3 in healthy and PD ECs quantified with qPCR. n=5 (healthy), 6 (PD LRRK2). Two independent experiments. (c) KEGG and GO pathways of up- and down-regulated DEGs in PD ECs compared to healthy. (d) Heatmap showing genes related to p53 signalling pathway and steroid biosynthesis in healthy and PD ECs. (e) Volcano plot of up- and down-regulated DEGs between healthy and PD ECs after TNFα+IL-1β exposure based on p-value (<0.05) and absolute log2 Fold >1. (f) Gene expression of MEG3 in healthy and PD ECs. n=6 (healthy), 6 (PD LRRK2). Two independent experiments. (g) KEGG and GO pathways of up- and down-regulated genes in PD ECs compared to healthy. (h) Venn diagram of DEGs between healthy and PD ECs in unexposed, TNFα and TNFα+IL-1β exposed conditions. All exposures lasted for 4 h and the concentrations were: 10 ng/ml TNFα or 10 ng/ml TNFα+ IL-1β. Data shown as mean +/-SD, n= 3, unpaired t-test ** p< 0.01.
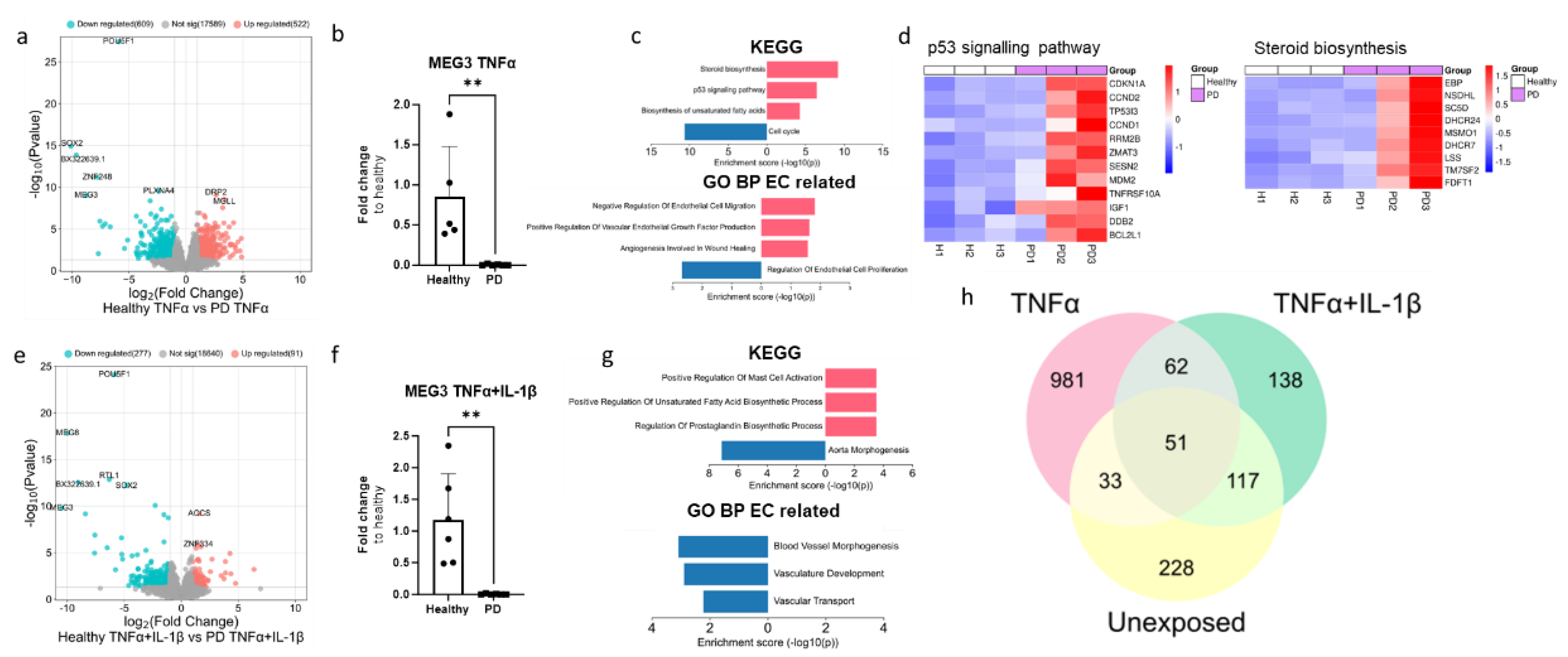
Figure 6.
Cell viability and cytotoxicity in endothelial cells after inflammatory stimuli. (a) Representative bright field (upper panel) and immunofluorescence images stained for ZO1 (lower panel) of ECs, non-exposed, 12 h TNFα and TNFα+IL-1β exposed, 24 h TNFα and TNFα+IL-1β exposed. Scale bar 50 µm. (b) Quantification of the immunocytochemistry. Fluorescence normalized to DAPI. n=6-7 (Healthy), 1 (isogenic),5-6 (PD LRRK2) from two independent experiments. (c) LDH release from ECs of non-exposed and after 4, 12 and 24 h TNFα and TNF+IL-1β (TI) exposure. n= 3-9 (healthy), 2-6 (PD LRRK2) from two independent experiments. (d) MTT assay of non-treated and 12 and 24 h TNFα and TNF+IL-1β treated ECs. n= 2-4 (healthy), 1-3 (PD LRRK2. (e) Western blot analysis of p53 and Cytochrome C (CytC) in healthy and PD EC normalized to β-actin protein levels. Representative images and quantification of the data. n=3-6 (healthy), 3-7 (PD LRRK2). Data shown as mean +/-SD. Two-way ANOVA with Tukey´s multiple comparison test, p*<0.05, p**<0.01, p***<0.001.
Figure 6.
Cell viability and cytotoxicity in endothelial cells after inflammatory stimuli. (a) Representative bright field (upper panel) and immunofluorescence images stained for ZO1 (lower panel) of ECs, non-exposed, 12 h TNFα and TNFα+IL-1β exposed, 24 h TNFα and TNFα+IL-1β exposed. Scale bar 50 µm. (b) Quantification of the immunocytochemistry. Fluorescence normalized to DAPI. n=6-7 (Healthy), 1 (isogenic),5-6 (PD LRRK2) from two independent experiments. (c) LDH release from ECs of non-exposed and after 4, 12 and 24 h TNFα and TNF+IL-1β (TI) exposure. n= 3-9 (healthy), 2-6 (PD LRRK2) from two independent experiments. (d) MTT assay of non-treated and 12 and 24 h TNFα and TNF+IL-1β treated ECs. n= 2-4 (healthy), 1-3 (PD LRRK2. (e) Western blot analysis of p53 and Cytochrome C (CytC) in healthy and PD EC normalized to β-actin protein levels. Representative images and quantification of the data. n=3-6 (healthy), 3-7 (PD LRRK2). Data shown as mean +/-SD. Two-way ANOVA with Tukey´s multiple comparison test, p*<0.05, p**<0.01, p***<0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



