
Submitted:
08 October 2024
Posted:
08 October 2024
You are already at the latest version
Abstract
Naegleria fowler, also known as brain-eating amoeba is a thermophilic microorganism that causes primary amoebic encephalitis (PAM) having >98% mortality rate. To date, there are no specific drugs identified to treat this disease. N. fowleri expresses only one form of hexokinase i.e. glucokinase (nGLK) unlike humans that express four isozymes of hexokinase including glucokinase (hexokinase IV) or hGLK that is critical for the first step of glycolysis. Until now, there have not been many studies on glucokinase as a drug target to treat PAM, hence, this study provides an exhaustive analysis of this relatively novel drug target. Thus, it was hypothesized that amoebic glucokinase can arrest carbohydrate metabolism and assist regulating its virulence. To find suitable antagonists to inhibit the activity of nGLK, we first tested the present recommended drugs by the CDC on their ability to bind nGLK. Interestingly our studies show that many of the present recommended drugs can inhibit both amoebic (nGLK) and human (hGLK) homologs of glucokinase. We identified new inhibitors for nGLK and further explored suitable hGLK antagonists for the potential treatment of diabetes. Despite significant structural differences between nGLK and hGLK, it was interesting to note that all our screened inhibitors bound with both nGLK and hGLK. Levomefolic acid; Ziprasidone; and Risperidone were found to be more suited to treat PAM while, Simeprevir; Indacaterol; Crizotinib and Mocetinostat were found to be suited for treating diabetes. The selections were based on multiple criteria including binding affinity; ADME scores; absorption; and its ability to cross the blood brain barrier. This study identifies multiple drugs that have potential not only to treat PAM but also to treat diabetes and also use for lab based biochemical assays for glycolysis inhibitors.
Keywords:
Naegleria fowleri
; Glucokinase
; Hexokinase
; type 2 diabetes
; Levomefolic acid
; Ziprasidone
; Risperidone
; Simeprevir
; Indacaterol
; Crizotinib
; Mocetinostat
1. Introduction
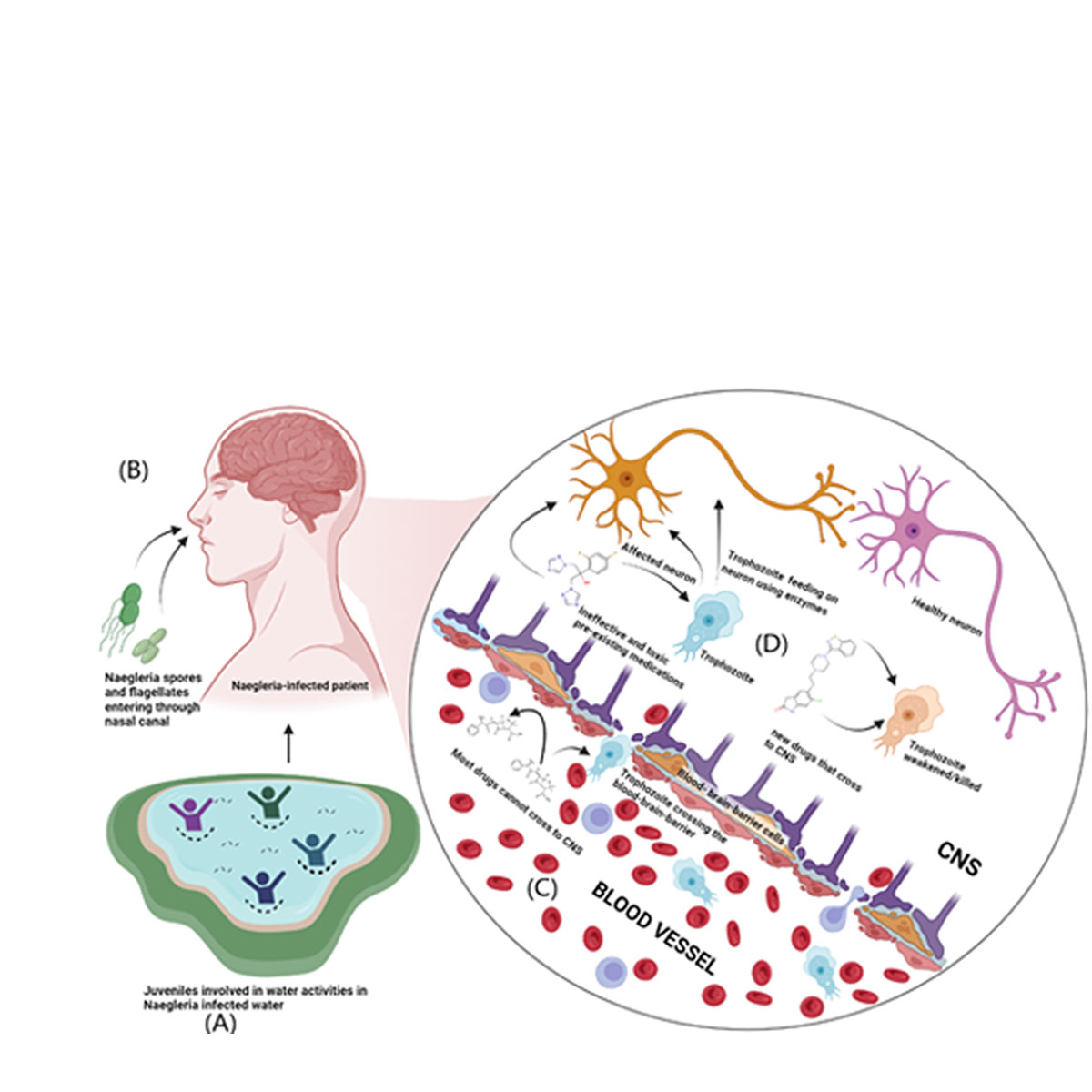
Naegleria fowleri is a free-living, pathogenic amoeba that acts as an opportunistic pathogen causing primary amoebic meningoencephalitis, with a mortality rate of above 97% especially children who are the most vulnerable. This infection begins when the pathogen gets into the nasal cavity and enters the central nervous system through the cribriform plate, leading to death in most cases within a few days as there are no effective treatments available for Naegleria fowleri infection [1].
This pathogenic amoeba has unique traits and an unconventional life cycle as they can transform into a cyst, flagellate, or trophozoite depending on the environment. The trophozoite stage is the feeding stage that can turn into a flagellate depending on the scarcity of nutrients or change in pH of the medium. Cysts of Naegleria fowleri develop when unfavorable conditions like elevated temperature and acidic or high salt environments are present[1,2,3]. Water activities are the major cause of exposure to the pathogen[4,5]. When a pathogen enters the nasal canal, the pathogen migrates to the olfactory nerves through the porous cribriform bone. Once the pathogen enters the central nervous system, it releases cytolytic enzymes such as acid hydrolases, phospholipases, neuraminidases, and cysteine proteases causing primary amoebic meningoencephalitis. Figure 1 (A-D)
Repurposed antibacterial and antifungal medications such as amphotericin B, rifampicin, miltefosine, corifungin, and azithromycin, in addition to experimental therapies like induced hypothermia, are the only recommended treatments[6]. Early detection and diagnosis improve the likelihood of the treatment to be effective. As previously mentioned, these medications have serious adverse effects such chills, fever, and nephrotoxicity and have been repurposed to treat Naegleria fowleri[7,8]. To treat Naegleria fowleri with fewer side effects and more accessibility, novel drugs need to be explored[9].
Naegleria fowleri like most other organisms that uses carbon as an energy source derives its energy from lipids and carbohydrate. Previously, we identified various natural and semi-synthetic compounds that can potentially inhibit the sterol 24-C methyltransferase enzyme involved in ergosterol metabolism and are vital for the survival of Naegleria fowleri[10]. In this paper, we targeted glucokinase and identified potential drugs that could inhibit the enzyme and, in the process, arrest critical pathway of carbohydrate metabolism.
Glucose is the simplest carbohydrate used as an energy source by organisms. A large concentration of glucose is present in the brain because it is one of the most metabolically active organs in the body[11]. Naegleria fowleri has only one enzyme involved in the transfer of phosphate group to glucose, namely glucokinase. This phosphorylation step ensures that the glucose molecule proceeds towards future catabolic steps of glycolysis instead of escaping from the cell. Cases reveal that there is a decrease in cerebrospinal glucose after Naegleria fowleri infection, indicating that the pathogen is using glucose in its metabolism when infecting the host [12]. Thus, we hypothesized that amoebic glucokinase could serve as a promising drug target as it will arrest carbohydrate metabolism in amoeba.
Homo sapiens have glucokinase, hexokinase, and other isoforms of glucokinases that carry out the addition of phosphate to glucose for carbohydrate metabolism. Human glucokinase, also known as hexokinase IV, is one of the major isoenzymes found in hepatocytes and pancreatic islets[13]. It is responsible for catalyzing the phosphorylation of glucose and its activity is directly correlated to the plasma concentration of glucose. The activity of insulin secretion of the beta cells is also related to the presence of glucose-6-phosphate, a product of glucokinase activity. Thus, glucokinase acts as a primary target to control insulin release and related metabolic disorders like diabetes[13]. A recent study showed that the high Km value of this enzyme is due to the structural difference between glucose-bound and unbound enzyme conformation. In our study, we chose human glucokinase structure 3FGU, which was bound to beta-d-glucose, phosphoaminophosphonic acid-adenylate ester, magnesium ion, and potassium ion. Previously, the action of activators on the active confirmation of glucokinase was shown[14]. Our study would give insight into the activity of inhibitors against the active conformation of human glucokinase.
Initially, we screened Naegleria fowleri drugs against nGLK and hGLK to identify the effects of these drugs on the carbohydrate metabolism of both the host and the pathogen. Then, we screened clinical and FDA-approved drugs against pathogen and human homologs of glucokinase to identify how pathogen-specific drugs interact with human homolog. The comparative study revealed how these new drugs interact with the two glucokinases. In addition, it led to the repurposing of drugs against both glucokinases. Unlike humans, absence of any isoforms of hexokinase in N. fowleri makes nGLK a viable and a promising target as its inhibition can arrest amoebic glycolysis completely. Inhibiting glycolysis is predicted to contain amoebic virulence and proliferation due to their depleted energy levels.
2. Results
Glucokinase is an essential component of carbohydrate metabolism and is required for the synthesis of nucleotides via the pentose phosphate pathway[15,16]. The lack of Hexokinase enzyme in Naegleria fowleri species insists that glucokinase enzyme is the major carbohydrate metabolism enzyme in that organism[16].
2.1. Sequence Analysis for Glucokinases
Glucokinase converts glucose to glucose-6-phosphate by transferring a phosphate group from ATP to glucose. The pentose phosphate route is opened up by this initial stage of glycolysis, which also initiates nucleotide synthesis[17]. Both hGLK and nGLK sequences were obtained from the RCSB PDB (Protein Data Bank), and a pairwise alignment was carried out using Emboss Needle. Two protein sequences are 13.9% identical and 24.3% similar. The PDB structure of the nGLK only covers 378 of the 449 amino acid residues, whereas the PDB structure of the hGLK only covers 447 of the 470 amino acid residues.
The Conserved Domain Database (CDD) was used to check for the activity of missing portions of the PDB structure. As shown in Supplementary Figure 1. (a), the sequence analysis of the structure of nGLK did not include the first 30 amino acids and the last 41 amino acids. CDD analysis of the sequence showed that the regions necessary for glucokinase activity were present for both nGLK and hGLK. And, in supplementary Figure 1. (b), the sequence analysis of the structure of human glucokinase did not include the first 9 amino acids and the last 7 amino acids. The comparison between nGLK and hGLK is given in Figure 3. and Figure 4. (a), (b), (c).
Figure 2.
Structural integrity was analyzed via Ramachandran plots for proteins. (a) Ramachandran plot for nGLK (b) Ramachandran plot for hGLK.
Figure 2.
Structural integrity was analyzed via Ramachandran plots for proteins. (a) Ramachandran plot for nGLK (b) Ramachandran plot for hGLK.
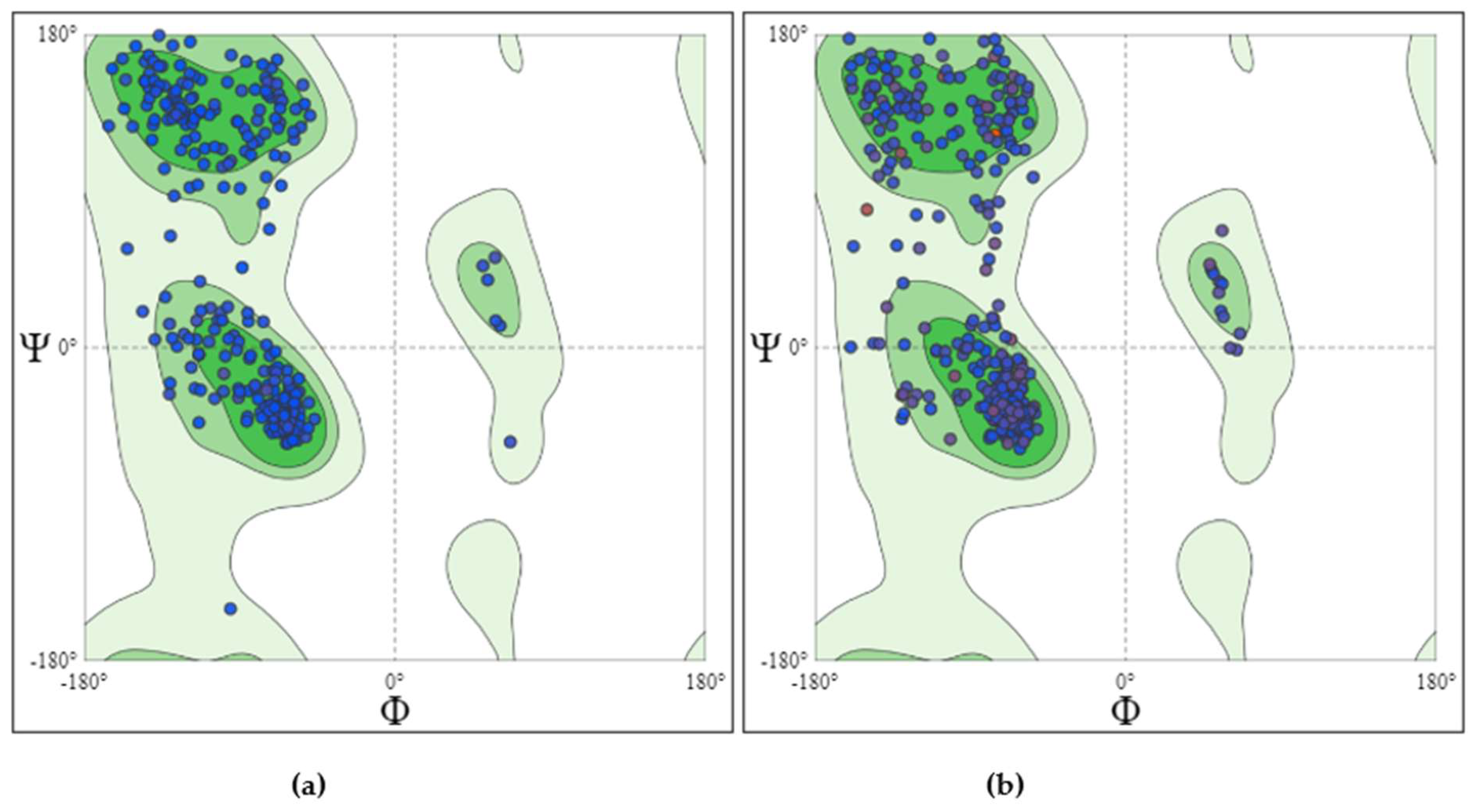
Figure 3.
Sequence analysis of Naegleria fowleri (6DAO.Str) and human glucokinase (3FGU.Str).

Figure 4.
Aligned residues of nGLK (yellow) and hGLK (blue). (a) Pathogenic glucokinase with binding sites for beta-d-glucopyranose(blue sphere) and phosphoaminophosphonic acid-adenylate ester(pale tan). (b) human glucokinase with binding sites for beta-d-glucopyranose(orange-red sphere) and phosphoaminophosphonic acid-adenylate ester(multi-color). (c) overlay of glucokinase structures showing active sites and positions of glucose and phosphoaminophosphonic acid-adenylate ester.
Figure 4.
Aligned residues of nGLK (yellow) and hGLK (blue). (a) Pathogenic glucokinase with binding sites for beta-d-glucopyranose(blue sphere) and phosphoaminophosphonic acid-adenylate ester(pale tan). (b) human glucokinase with binding sites for beta-d-glucopyranose(orange-red sphere) and phosphoaminophosphonic acid-adenylate ester(multi-color). (c) overlay of glucokinase structures showing active sites and positions of glucose and phosphoaminophosphonic acid-adenylate ester.

The amino acid numbering for the structure for nGLK (PDB ID: 6DA0) starts from Proline (PRO1) and ends with Leucine (LEU378), while for the hGLK (PDB ID: 3FGU) it starts from Asparagine (ASP5) and ends with Lysine (LYS458). The 3FGU structure had gaps between PRO66 and SER69, GLU93 and GLY97, LYS140 and LYS143.
After the minimization of the protein, it was necessary to analyze the Ramachandran outliers. nGLK showed no outliers while hGLK showed one outlier, i.e., ALA176. The Molprobity score for Naegleria protein was 0.86 while for the human analog, it is 1.86. More detailed information on the structural assessment of nGLK and hGLK is given in
Supplementary Table (a). and Supplementary Table (b). Protparam analysis of the structural sequence of the two proteins revealed that the nGLK has a -6 charge with an aliphatic index of 81.06 while the hGLK has a charge of -18 with an aliphatic index of 79.98.
2.2. Druggable Site Analysis of nGLK and hGLK Structure
The analysis of binding sites for the nGLK revealed the presence of three major binding pockets through the use of the online tool https://proteins.plus/. The active sites of the two proteins are compared and their role are discussed later in the manuscript.
The DoGSiteScorer function in proteins.plus helped determine whether the protein pockets are druggable[18]. The drug score is predicted by considering global properties (volume and size, shape, depth) and local similarities (nearest neighbor). This helped predict which pocket provides the best binding site for potential drugs. Figure 5. , Figure 6. and Table 1. , Table 2. giving detailed information on the position, size, drug score, hydrophobicity, hydrogen bond donors/acceptors, and amino acids involved in the top pockets of nGLK and hGLK, respectively.
Figure 5.
Image of nGLK binding sites in Yellow, Purple, and Green. (a) Image of nGLK highlighting green and yellow pocket. (b) Image of nGLK highlighting green and purple pocket. (c) Image of nGLK highlighting purple and yellow pocket.
Figure 5.
Image of nGLK binding sites in Yellow, Purple, and Green. (a) Image of nGLK highlighting green and yellow pocket. (b) Image of nGLK highlighting green and purple pocket. (c) Image of nGLK highlighting purple and yellow pocket.
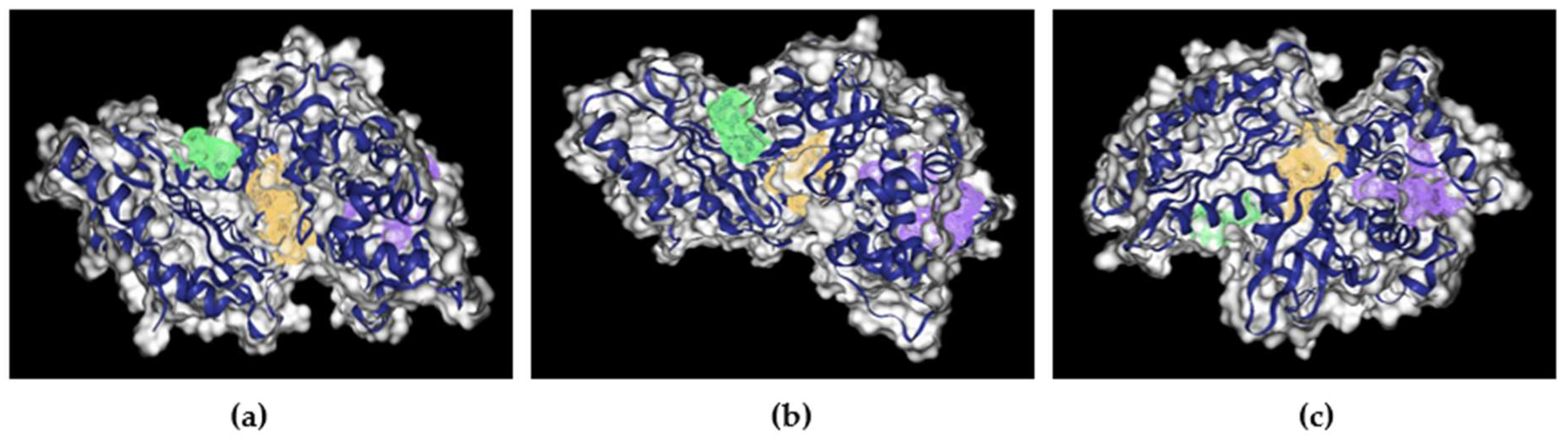
Figure 6.
Image of hGLK binding sites in Yellow, Purple, and Green. (a) Image of hGLK highlighting purple and yellow pocket. (b) Image of nGLK highlighting green and purple pocket. (c) Image of nGLK highlighting green and yellow pocket.
Figure 6.
Image of hGLK binding sites in Yellow, Purple, and Green. (a) Image of hGLK highlighting purple and yellow pocket. (b) Image of nGLK highlighting green and purple pocket. (c) Image of nGLK highlighting green and yellow pocket.
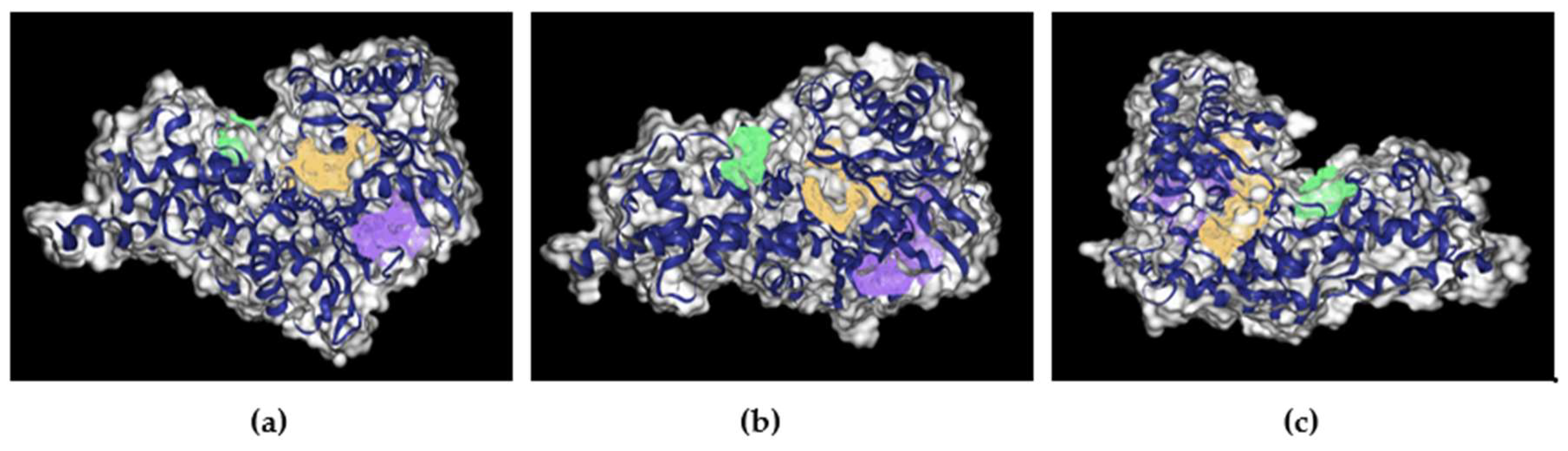

Table 1.
Summary of the binding pocket analysis results showing pocket color, drug score, hydrogen bond donors/acceptors, hydrophobicity ratio, surface area, and involved amino acids in nGLK.
Table 1.
Summary of the binding pocket analysis results showing pocket color, drug score, hydrogen bond donors/acceptors, hydrophobicity ratio, surface area, and involved amino acids in nGLK.
| Pocket Color | Drug Score | H-bond donors/ acceptors | Hydrophobicity ratio | Surface area(Ų) | Amino Acids involved |
|---|---|---|---|---|---|
| Yellow | 0.81 | 28/62 | 0.28 | 950.12 | ASP32, GLY34, ALA35, ARG39, ALA94, GLY95, PRO96, VAL97, THR105, ASN106, ASN133, ASP134, LEU135, GLU136, SER138, CYS139, ILE142, VAL180, ALA182, MET183, GLY184, THR185, GLY186, LEU187, GLY188, THR189, GLY190, LEU191, VAL202, ILE203, PRO204, LEU205, GLU206, ALA207, GLY208, HIS209, GLU246, ALA317, GLY318, ASP319, GLN357, TYR361, ASN362, PHE363, ASN364 |
| Purple | 0.85 | 19/58 | 0.41 | 1131.09 | LEU181, LEU187, THR189, ALA207, GLY208, VAL210, ILE212, ALA213, THR214, PRO215, SER219, GLU220, HIS221, PHE222, GLU224, GLU225, ARG228, ILE229, LEU232, ILE243, TYR245, ILE248, GLU261, GLN288, ILE291, THR292, ARG295, TYR296, LYS299, ALA300, ALA301, GLN302, ASN303, ILE304, VAL306, ILE334, LYS337, GLU338, THR342, HIS343 |
| Green | 0.64 | 6/17 | 0.45 | 403.82 | ILE28, GLN43, ILE45, ASP51, VAL54, MET56, ASN144, TYR361, ASN362, PHE363, VAL365, LYS366, LEU369, GLN370, ARG373 |
Table 2.
Summary of the binding pocket analysis results showing pocket color, drug score, hydrogen bond donors/acceptors, hydrophobicity ratio, surface area, and involved amino acids in hGLK.
Table 2.
Summary of the binding pocket analysis results showing pocket color, drug score, hydrogen bond donors/acceptors, hydrophobicity ratio, surface area, and involved amino acids in hGLK.
| Drug Score | H-bond donors/ acceptors | Hydrophobicity ratio | Surface area(Ų) | Amino Acids involved | |
|---|---|---|---|---|---|
| Yellow | 0.79 | 30/76 | 0.20 | 856.65 | SER54, LYS56, SER76, LEU77, ASP78, LEU79, GLY80, GLY81, THR82, ASN83, ARG85, VAL86, MET87, LYS102, HIS105, GLN106, MET107, THR149, SER151, PHE152, PRO153, LEU165, ASN166, TRP167, THR168, LYS169, ASN204, ASP205, THR206, THR209, ILE225, GLY227, THR228, GLY229, CYS230, ASN231, GLU256, TRP257, GLY258, ALA259, ASP262, GL286, GLN287, GLU290, ASP409, GLY410, GLU443, GLY444, SER445, GLY446 |
| Purple | 0.80 | 10/51 | 0.42 | 840.72 | TYR61, VAL62, ARG63, SER64, PRO66, SER69, VAL91, GLY97, GLN98, TRP99, SER100, ASP158, ILE159, ASP187, ALA201, MET210, ILE211, SER212, CYS213, TYR214, TYR215, GLU216, HIS218, CYS220, GLU221, MET235, ARG250, CYS252, ILE439, GLU440, SER441, ARG447, LEU451, VAL452, ALA454, VAL455, ALA456 |
| Green | 0.47 | 8/22 | 0.35 | 288.84 | VAL227, SER280, SER281, ALA282, LYS291, GLY295, LYS296, TYR297, MET298, GLY299, GLU300, ARG327, GY328, PHE330, GLU33, THR332 |
2.3. Screening of Recommended Drugs to Treat Naegleria fowleri against nGLK and hGLK
None of the currently available medications for Naegleria fowleri targets organism’s glucose metabolism [2,6]. Hence, it is imperative to investigate the activity of existing treatments on the glucose metabolism of both pathogen and host before we looked for potential drugs. Multiple drugs have been used to treat Naegleria fowleri infections. To identify the effectiveness of these drugs against glucokinase enzymes derived from both pathogen (PDBID:6DA0) and its human homolog (PDBID:3FGU), present drugs that are used for treatment such as azithromycin, amphotericin b, Miltefosine, fluconazole, ketoconazole were docked against both glucokinase protein. The structures of the drugs were retrieved from PubChem and ChemSpider. The binding affinities of the drugs to the enzymes revealed that these drugs may have a key role to play in negatively affecting carbohydrate metabolism. Supplementary Figure 2. , and Supplementary Figure 4. show the binding of mentioned drugs around nGLK and hGLK respectively.
Table 3.
Screening of established drugs that are used to treat Naegleria fowleri against nGLK and hGLK.
Table 3.
Screening of established drugs that are used to treat Naegleria fowleri against nGLK and hGLK.
| Name of drug | Binding affinity for N.F(kcal/mol) | Binding affinity for Human |
|---|---|---|
| Azithromycin | -7.0 | -7.4 |
| Amphotericin B | -7.6 | -8.5 |
| Miltefosine | -4.1 | -5.5 |
| Fluconazole | -7.4 | -6.6 |
| ketoconazole | -8.2 | -8.8 |
| Rifampin | -9.2 | -8.3 |
| Dexamethasone | -7.9 | -7.6 |
2.3.1. Activity of Azithromycin against nGLK and hGLK
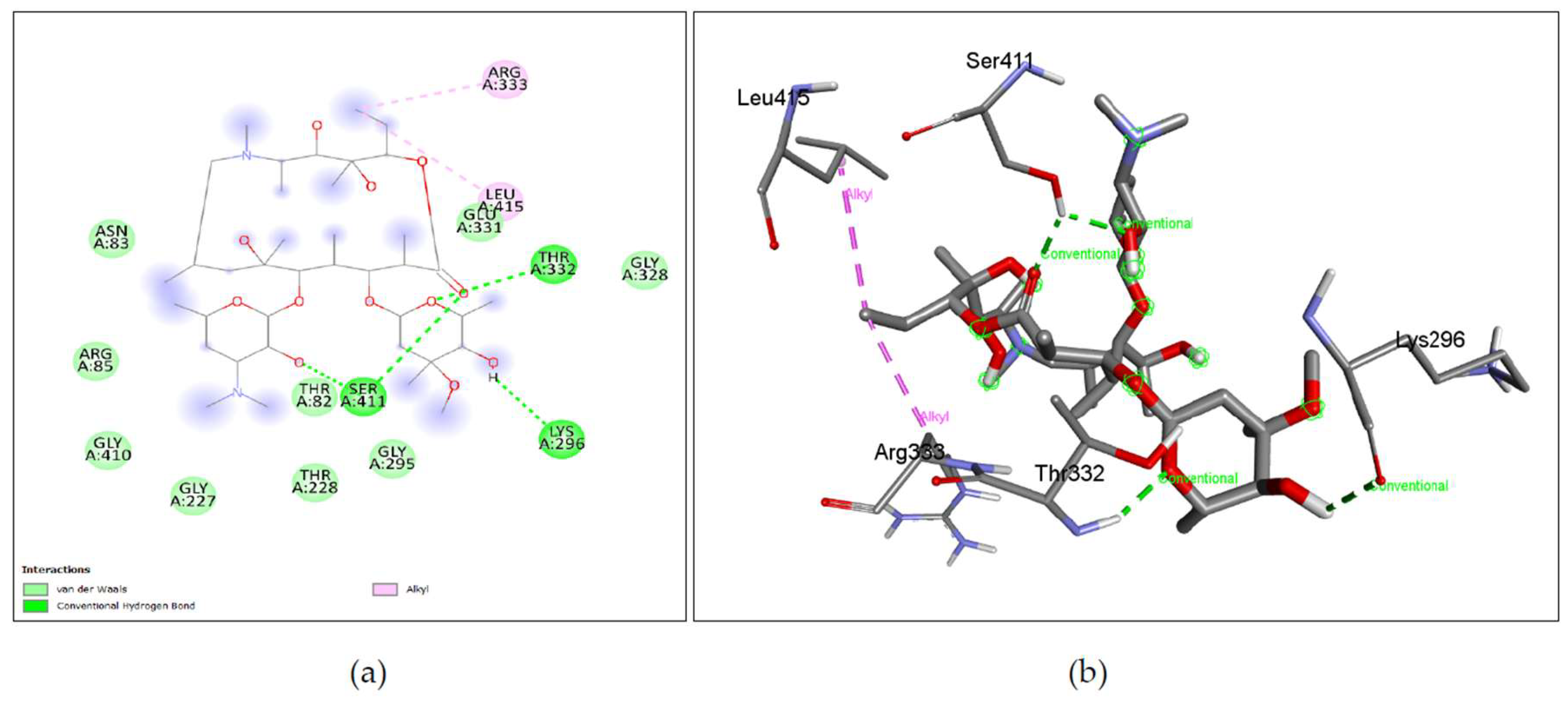
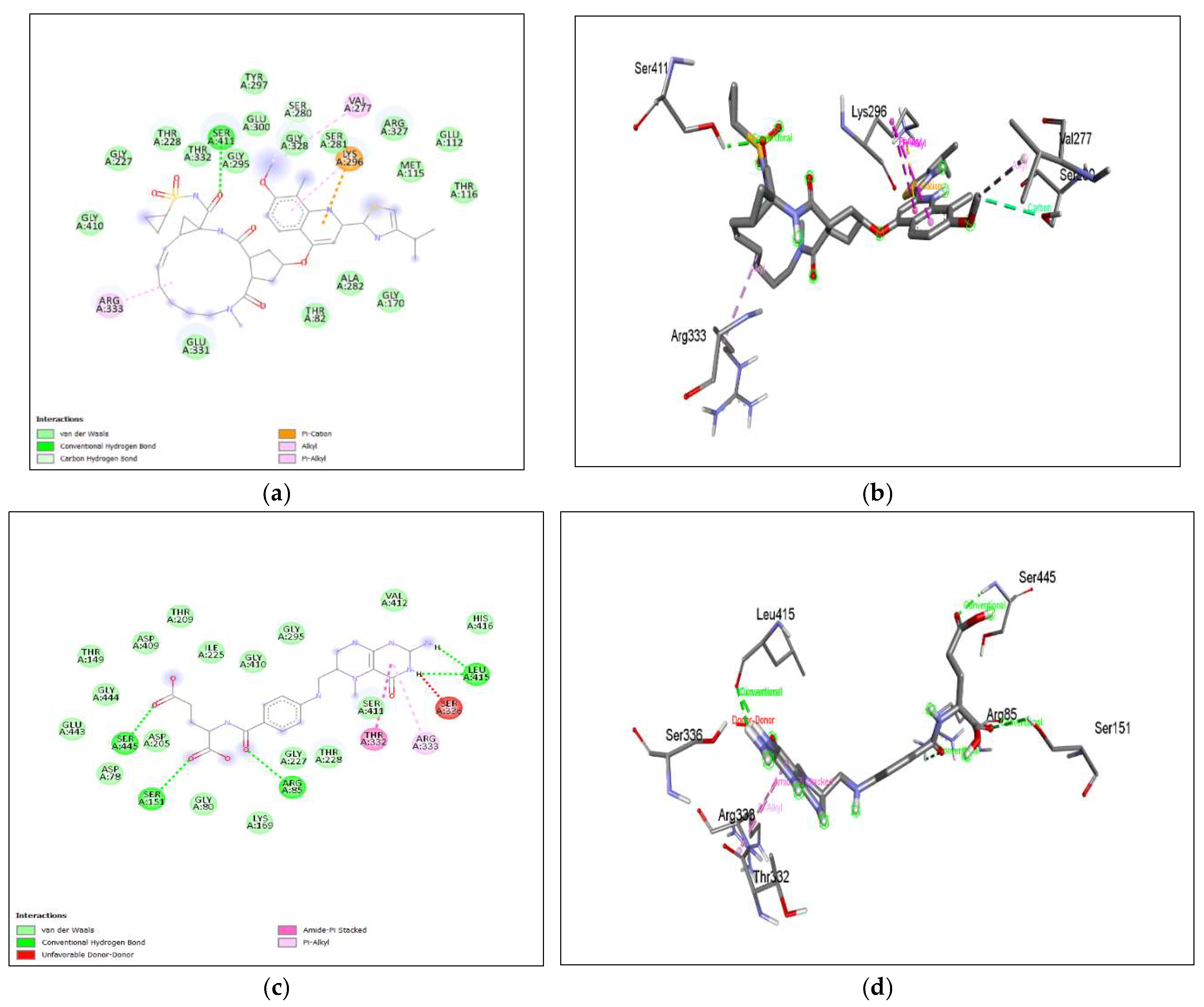
Azithromycin is an FDA-approved drug for the treatment of various bacterial infections. It is a derivative of erythromycin and is administered orally. It prevents protein synthesis by binding to 23S rRNA and inhibits the assembly of bacterial 50S ribosomal subunit [19]. It is used to treat opportunistic infections including pneumonia, acute sinus, bronchitis, throat, and tonsil infection. There are side effects associated with the use of azithromycin [20,21,22]. The docking of azithromycin against glucokinase gave a moderate binding affinity of -7kcal/mol. This indicates the use of azithromycin for the treatment of Naegleria fowleri could have been affecting the carbohydrate metabolism of the pathogen by affecting glucokinase.
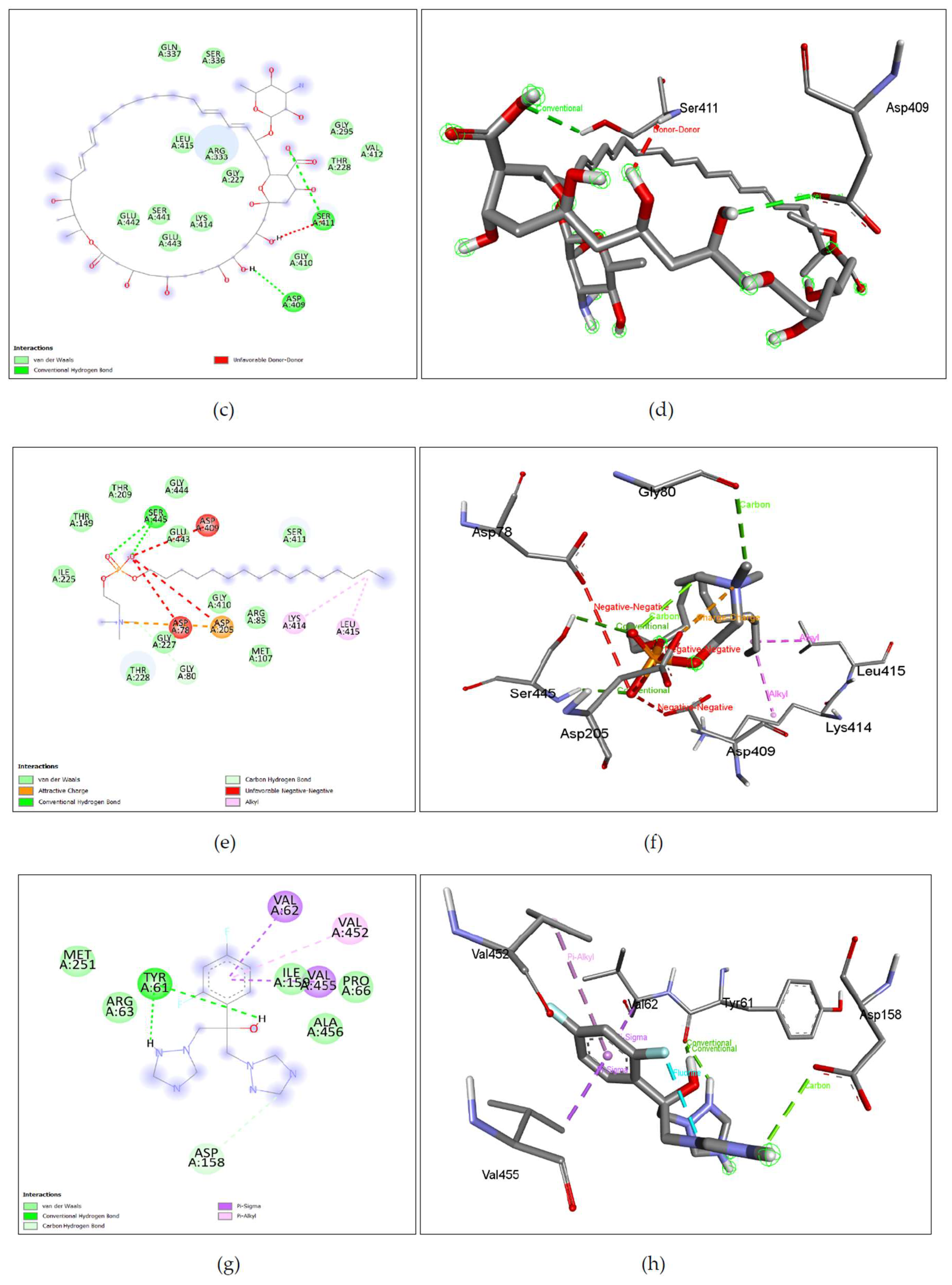
The binding affinity of the drug with the human homolog of glucokinase also gave a moderate binding affinity of -7.4kcalmol, which is even higher than the binding to pathogen glucokinase, indicating that this drug could be involved in affecting human carbohydrate metabolism. As shown in Figure 7. (a), (b) Azithromycin shows binding with nGLK around THR272, SER274, ALA273, GLY251, LEU254, ALA277, PHE323, ARG39, GLY34, ASN37, THR36, MET183, ASP319 and shows binding at ASN320, ASN324 via carbon hydrogen bond and THR185 via conventional hydrogen bond. Also, Figure 8. (a), (b) shows Azithromycin binding with hGLK around ASN83, ARG85, GLY410, GLY227, THR228, GLY295, THR82, GLU331, GLY328 and interacts with SER411, LYS296, THR332 via conventional hydrogen bond and LEU415, ARG333 via alkyl bond. Supplementary Figure 3. (a) and Supplementary Figure 5. (a) show amino acids of nGLK and hGLK within 5 A of azithromycin.
Figure 7.
Lig-plots and 3D images showing interactions between Target nGLK and well-known Naegleria fowleri drugs. (a) Azithromycin interacts with nGLK through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), and conventional hydrogen bond (pale green). (b) Azithromycin interactions visualized in 3D. (c) Amphotericin B interacts with nGLK through Van Der Waals interactions (light green), and conventional hydrogen bond (dark green). (d) Amphotericin B interactions visualized in 3D. (e) Miltefosine interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and alkyl interactions (pale pink). (f) Miltefosine interactions visualized in 3D. (g) Fluconazole interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), halogen interaction(blue), unfavorable bond(red), and pi-alkyl (pale pink). (h) Fluconazole interactions visualized in 3D. (i) Ketoconazole interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (pale green), pi-anion (gold), pi-alkyl (pale pink), and alkyl (pale pink). (j) Ketoconazole interactions visualized in 3D. (k) Rifampin interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (pale green), and attractive charge (gold) interaction. (l) Rifampin interactions visualized in 3D. (m) Dexamethasone interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green). (n) Dexamethasone interactions visualized in 3D.
Figure 7.
Lig-plots and 3D images showing interactions between Target nGLK and well-known Naegleria fowleri drugs. (a) Azithromycin interacts with nGLK through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), and conventional hydrogen bond (pale green). (b) Azithromycin interactions visualized in 3D. (c) Amphotericin B interacts with nGLK through Van Der Waals interactions (light green), and conventional hydrogen bond (dark green). (d) Amphotericin B interactions visualized in 3D. (e) Miltefosine interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and alkyl interactions (pale pink). (f) Miltefosine interactions visualized in 3D. (g) Fluconazole interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), halogen interaction(blue), unfavorable bond(red), and pi-alkyl (pale pink). (h) Fluconazole interactions visualized in 3D. (i) Ketoconazole interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (pale green), pi-anion (gold), pi-alkyl (pale pink), and alkyl (pale pink). (j) Ketoconazole interactions visualized in 3D. (k) Rifampin interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (pale green), and attractive charge (gold) interaction. (l) Rifampin interactions visualized in 3D. (m) Dexamethasone interacts with nGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green). (n) Dexamethasone interactions visualized in 3D.
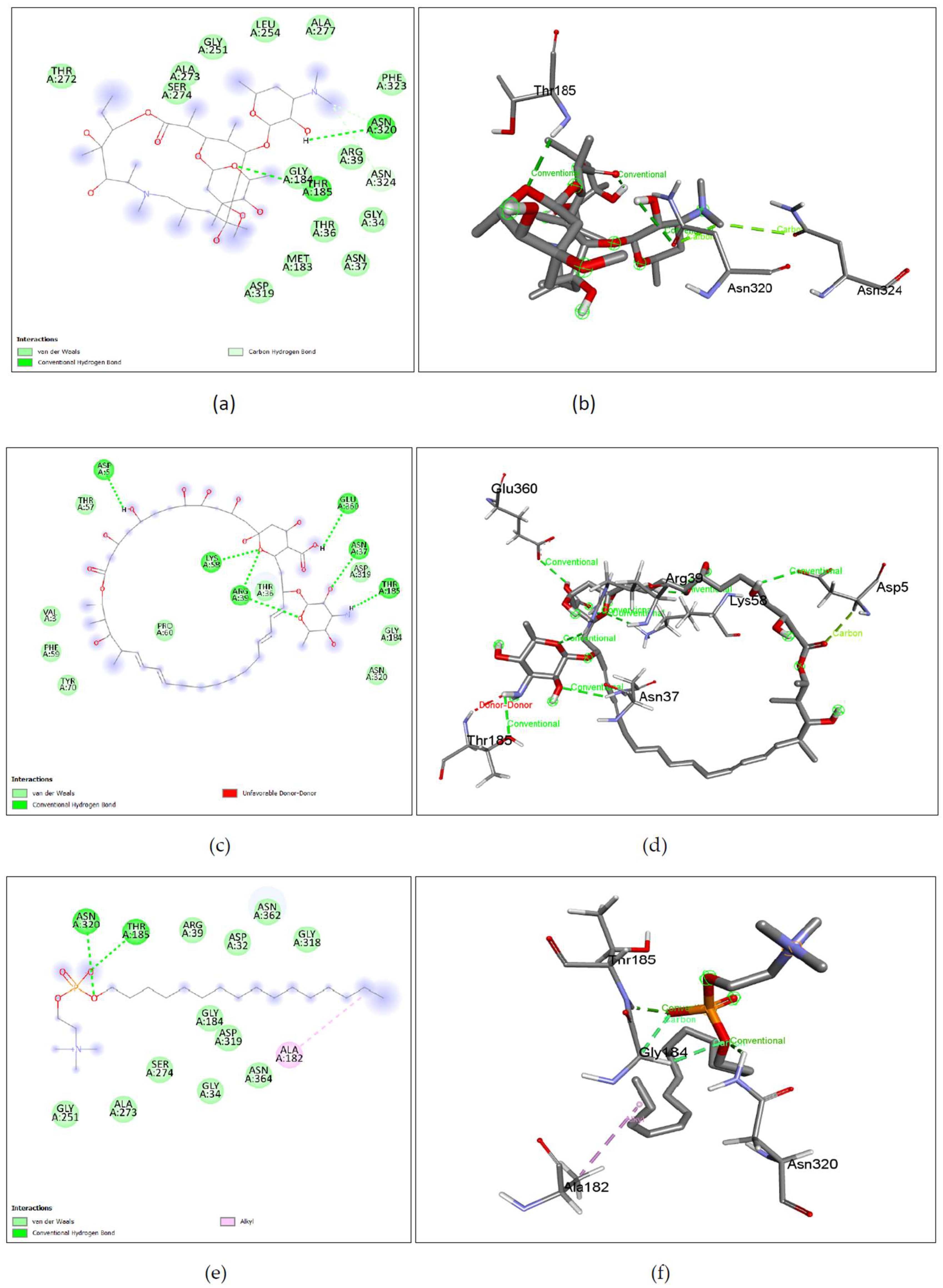
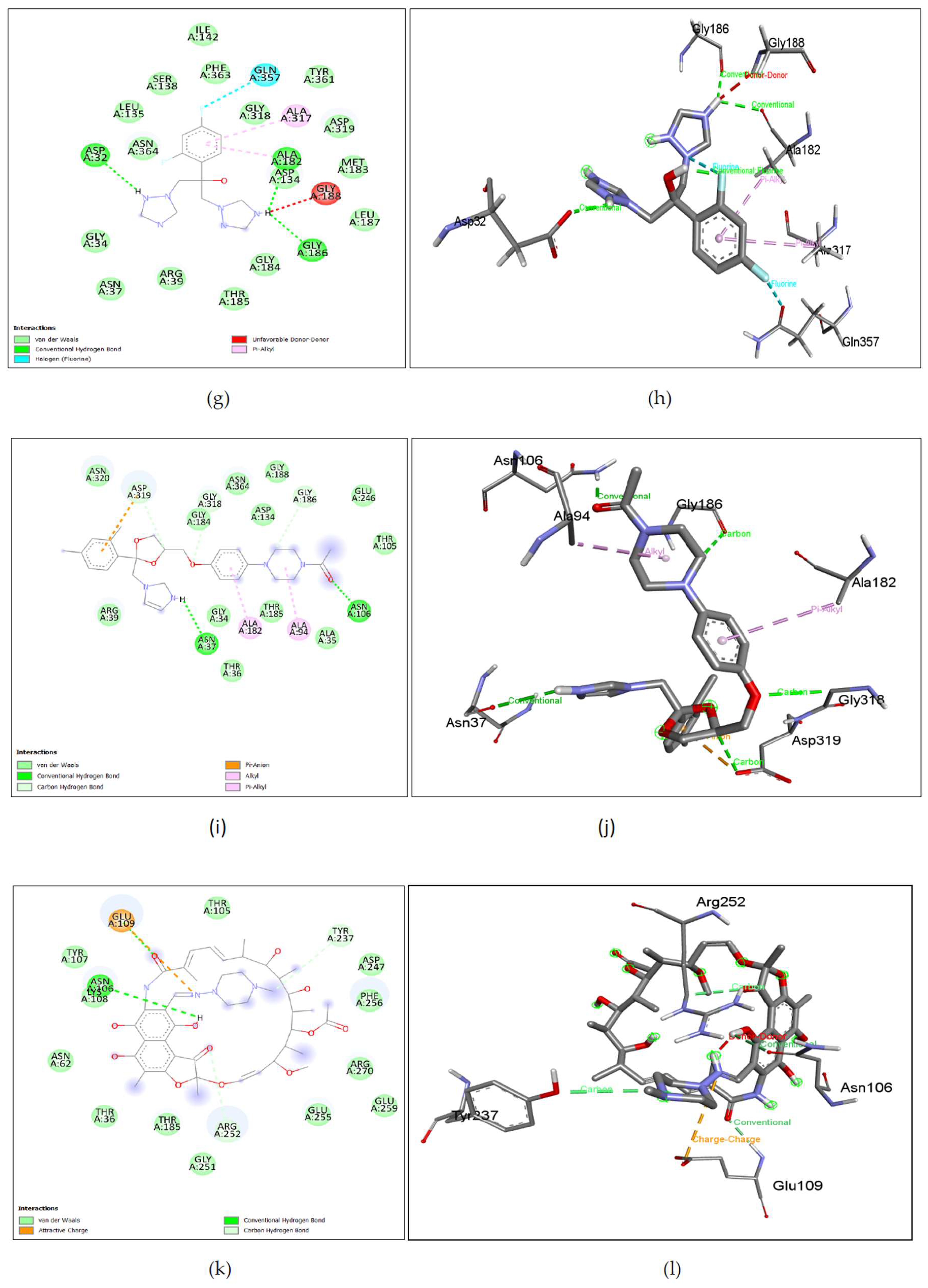
Figure 8.
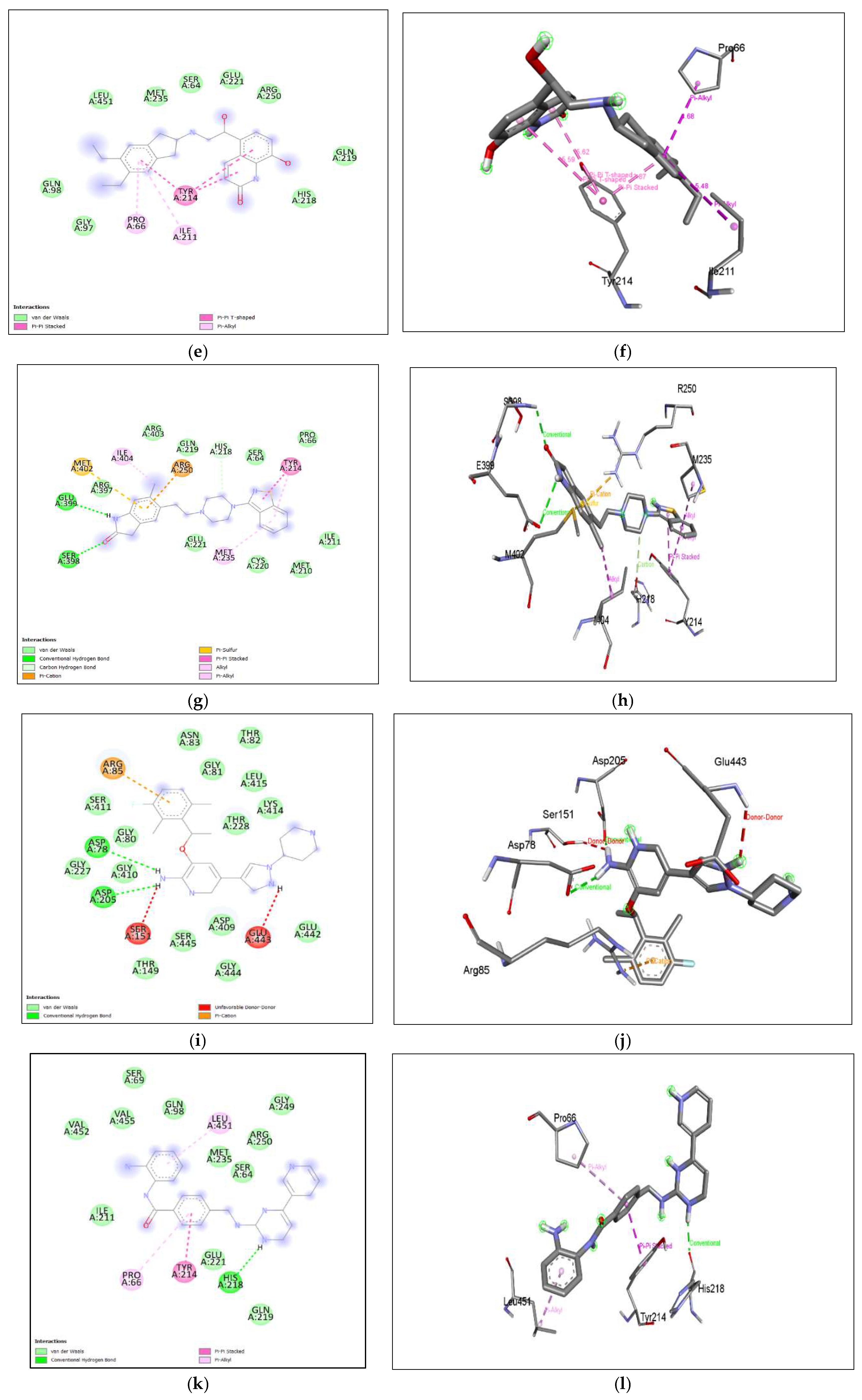
Lig-plots showing interactions between Human glucokinase hGLK and well-known Naegleria fowleri drugs. (a) Azithromycin interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and alkyl interactions. (b) Azithromycin interactions visualized in 3D. (c) Azithromycin B interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and unfavorable bond (red). (d) Amphotericin B interactions visualized in 3D. (e) Miltefosine interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), attractive charge (yellow), carbon hydrogen bond (pale green), alkyl interactions (pale pink), and unfavorable bond (red). (f) Miltefosine interactions visualized in 3D. (g) Fluconazole interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-sigma interactions (purple), and pi-alkyl interactions (pale pink). (h) Fluconazole interactions visualized in 3D. (i) Ketoconazole interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-sigma interactions (purple), alkyl (pale pink), and pi-alkyl interactions (pale pink). (j) Ketoconazole interactions visualized in 3D. (k)) Rifampin interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and pi-cation interactions (yellow). (l) Rifampin interactions visualized in 3D. (m) Dexamethasone interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and halogen interactions (blue). (n) Dexamethasone interactions visualied in 3D.
Figure 8.
Lig-plots showing interactions between Human glucokinase hGLK and well-known Naegleria fowleri drugs. (a) Azithromycin interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and alkyl interactions. (b) Azithromycin interactions visualized in 3D. (c) Azithromycin B interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and unfavorable bond (red). (d) Amphotericin B interactions visualized in 3D. (e) Miltefosine interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), attractive charge (yellow), carbon hydrogen bond (pale green), alkyl interactions (pale pink), and unfavorable bond (red). (f) Miltefosine interactions visualized in 3D. (g) Fluconazole interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-sigma interactions (purple), and pi-alkyl interactions (pale pink). (h) Fluconazole interactions visualized in 3D. (i) Ketoconazole interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-sigma interactions (purple), alkyl (pale pink), and pi-alkyl interactions (pale pink). (j) Ketoconazole interactions visualized in 3D. (k)) Rifampin interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and pi-cation interactions (yellow). (l) Rifampin interactions visualized in 3D. (m) Dexamethasone interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), and halogen interactions (blue). (n) Dexamethasone interactions visualied in 3D.
2.3.2. Activity of Amphotericin B against nGLK and hGLK
Amphotericin B, also known as Fungizone is an FDA-approved antifungal drug used to treat histoplasmosis, cryptococcosis, and opportunistic infections due to HIV. Amphotericin B binds to ergosterol, found on the fungal and Naegleria fowleri membrane. It is reported to induce immune cell stimulation[18,23]. The binding affinity of amphotericin B against glucokinase was -7.6kcal/mol. This score suggests that the binding is moderately strong and could mean that the drug could reduce the efficiency of the glucokinase activity of the pathogen. Amphotericin B has an even higher binding affinity of -8.5kcal/mol to the human homolog of glucokinase, which indicates that the drug could be affecting the carbohydrate metabolism of the host. This result confirm studies that have shown marked difference in carbohydrate metabolism caused by amphotericin B [24,25]. Previously, our study had identified sterol 24-C methyltransferase inhibitors that could inhibit ergosterol biosynthesis in N. gruberi [10]. Amphotericin is also known to inhibit the ergosterol pathway which is essential for fungi. N. fowleri also requires ergosterol in its cell membrane[26]. This docking result could indicate that amphotericin could inhibit both lipid and carbohydrate metabolism. The administration of this drug is limited due to its severe acute and chronic toxicity leading to nausea, vomiting, fever, hypoxia, hypertension, and nephrotoxicity[23]. As shown in Figure 7. (c), (d) Amphotericin B shows binding with nGLK around VAL3, PHE59, TYR70, PRO60, ASN320, GLY184, ASP319, THR57 and interacts with ASP5, LYS58, ARG39, ASN37, GLU360, THR185 via Conventional hydrogen bond, and THR185 via unfavorable interactions. Also, Figure 8. (c), (d) shows amphotericin B binding with human glucokinase around GLN337, SER336, GLY295, VAL412, THR228, GLY410, GLY227, ARG333, LEU415, LYS414, GLU443, GLU442, SER441 and interacts with SER411, ASP 409 via conventional hydrogen bond and SER411 via unfavorable donor-donor interaction. Supplementary Figure 3. (b) and Supplementary Figure 5. (b) show amino acids of nGLK and hGLK within 5 A of amphotericin.
2.3.3. Activity of Miltefosine against nGLK and hGLK
Miltefosine is an antimicrobial drug that was originally used against cancer where it interfered with lipid-dependent cell signaling and induced apoptosis. Miltefosine is an ester of phosphocholine compound and has anti-inflammatory, anti-fungal, antiprotozoal, and antiviral effects [7,27]. It modulates cell membrane permeability, lipid composition, signal transduction, and phospholipid metabolism. It is known to be an inhibitor of MAPK and induces apoptosis by balancing MAPK and JNK pathways[7,27,28]. It also modulates the immune response. The docking of miltefosine against nGLK gave a low binding score of -4.7kcal/mol. It also bound to the hGLK with an affinity of -5.5kcal/mol, indicating this could be affecting carbohydrate metabolism in the host. It is known to be teratogenic, impair fertility and cause fetal death below recommended doses in animals (IMPAVIDO). The administration of these drugs has potentially lasting side effects on patients. It is administered to young adults for the treatment of Naegleria infections. As shown in Figure 7. (e), (f), Mitefosine shows binding with nGLK at ARG39, ASP32, GLY318, ASN362, GLY184, ASP319, GLY34, ASN364, SER274, ALA273, GLY251 and shows interaction with ASN320, THR185 via conventional hydrogen bond and ALA182 via Alkyl interaction against the pathogen’s glucokinase. Also, Figure 8. (e), (f), shows Miltefosine binding with hGLK around THR149, THR209, GLY444, ILE225, SER411, GLY227, THR228, GLY410, ARG85, MET107, GLY80, GLY443 and interacts with SER445 via conventional hydrogen bond, ASP205 via attractive charge interactions, LYS414, LEU415 via alkyl interactions, GLY80 via carbon hydrogen bond and ASP78, ASP205, ASP409 via unfavorable interactions. Supplementary Figure 3. (c) and Supplementary Figure 5. (c) show amino acids of nGLK and hGLK within 5 A of miltefosine.
2.3.4. Activity of Fluconazole against nGLK and hGLK
Fluconazole is an FDA-approved antifungal drug used against esophagus, throat, vaginal infection, meningitis, and other opportunistic infections. It works against superficial fungal infections [29]. It is a synthetic triazole acting against fungal sterol demethylation. It had a moderate binding affinity of -7.4 kcal/mol against glucokinase. The drug may affect the glucokinase enzyme negatively. The drug has a binding affinity of -6.6kcal/mol when docked against hGLK. It is a highly selective inhibitor for the fungal ergosterol synthesis enzyme, lanosterol 14-α demethylase. It is known to cause hallucinations and paranoia and carries a risk of severe hepatic toxicity [30]. As shown in Figure 7. (g), (h), Fluconazole shows binding with nGLK around ASN364, LEU135, SER138, PHE363, ILE142, GLY318, ASP319, MET183, TYR361, LEU187, GLY184, ARG39, THR185, ASN37, GLY34 and interacts with ASP32, ALA182, GLY186 via conventional hydrogen bond and ALA317, ALA182 via pi-alkyl interactions and GLN357 via Halogen interaction. Also, Figure 8. ((g), (h), shows Fluconazole binding with hGLK around MET251, ARG63, ALA456, PRO66, ILE159 and interacts with TYR61 via a conventional hydrogen bond, VAL455, VAL62 via pi-sigma bond, VAL452 via pi-alkyl and ASP158 via carbon hydrogen bond. Supplementary Figure 3. (d) and Supplementary Figure 5. (d) show amino acids of nGLK and hGLK within 5 A of fluconazole.
2.3.5. Activity of Ketoconazole against nGLK and hGLK
Ketoconazole also known as xolegel is a broad-spectrum fungicide for superficial fungal infections. It is used in immunosuppressed individuals. It inhibits ergosterol synthesis by interacting with 14-α-sterol demethylase. It is an imidazole with acute drug-induced liver toxicity and is not used for anti-fungal treatments [31,32]. Ketoconazole is a potent inhibitor of CYP3A4/5 and is known to affect the concentration of saxagalaptin, an orally available drug for type 2 diabetes [33]. The binding affinity of ketoconazole against pathogen glucokinase was -8.2kcal/mol which is a good binding score. It could mean that the treatment administered to Naegleria fowleri patients may have been inhibiting the glucokinase activity and affecting the pathogen negatively. Inhibition of the glucokinase enzyme in Naegleria fowleri was shown to be detrimental to the growth of the pathogen. Ketoconazole also binds to the human homolog of glucokinase with a binding affinity of -8.8kcal/mol. As shown in Figure 7. (i), (j), Ketoconazole shows binding with nGLK around ASN320, ARG39, THR36, ALA35, THR185, THR105, GLU246, GLY188, ASN364, ASP134, GLY318, GLY34, ASN364, ASN320 and interacts with ASP319, GLY184, GLY186 via carbon hydrogen bond, ASN106, ASN37 via conventional hydrogen bond, ALA182, ALA94 via pi-alkyl interactions, ASP319 via pi-anion interaction. Also, Figure 8. (i), (j), shows Ketoconazole binding with hGLK around SER64, GLY97, TYR215, LEU451, TYR61, ILE159, ASP158, ILE211 and interacts with GLN98 via conventional hydrogen bond, PRO66, VAL452, via pi-alkyl, ARG63 via alkyl interactions, TYR214 via pi-pi- stacked and pi-alkyl interactions, VAL455, VAL62 via pi-sigma interactions. Supplementary Figure 3. (e) and Supplementary Figure 5. (e) show amino acids of nGLK and hGLK within 5 A of ketoconazole.
2.3.6. Activity of Rifampin against nGLK and hGLK
Rifampin is also known as rifampicin is a semisynthetic antibiotic that can acts as an RNA polymerase inhibitor, DNA synthesis inhibitor, protein synthesis inhibitor, and well-known anti-tuberculosis drug[34]. It has neuroprotective, antiangiogenic, leprostatic, antiameobic, and antineoplastic activity[35]. Rifampin has been known to eliminate bacteria from noses and throats and inhibits tau progression in Alzheimer’s[36]. This is important considering the route taken by Naegleria fowleri is through the nasal pathway. Docking of rifampin against glucokinase revealed high binding affinity scores of -9.2kcal/mol. This could mean that rifampin could have been hampering carbohydrate metabolism and the pentose phosphate pathway of nucleotide synthesis by binding to glucokinase. The binding affinity of rifampin against human glucokinase is -8.3kcal/mol. As shown in Figure 7. (k), (l), Rifampin shown binding with nGLK around ASN62, THR107, THR105, THR36, THR185, GLY251, GLU255, GLU259, ARG270, PHE256, ASP247, LYS108 and interacts with ASN106, GLU109 via a conventional hydrogen bond, ARG252, TYR237 via carbon hydrogen bond, GLU109 via attractive charge interaction. Also, Figure 8. (k), (l), shows Rifampin binding with hGLK around MET107, LEU415, LYS414, ASP409, GLU443, GLY410, SER445, ARG333, THR332, LYS296, GLY295, ASN83, GLY227, THR228, GLY81, GLY80 and interacts with ASP78, ARG85, THR82 via conventional hydrogen bond, ARG85 via pi-cation interactions and THR82 via unfavorable interactions. Rifampin is also known to enhance the glucose-lowering effects of metformin[37]. Supplementary Figure 3. (f) and Supplementary Figure 5. (f) show amino acids of nGLK and hGLK within 5 A of rifampin.
2.3.7. Activity of Dexamethasone against nGLK and hGLK
Dexamethasone is a synthetic corticosteroid that has an adrenergic, antineoplastic, immunosuppressive, and anti-inflammatory effect. Dexamethasone was used for the treatment of COVID-19 and reduced the deaths of patients on ventilation by one-third and one-fifth for patients on oxygen[38]. It is also used in gastrointestinal, dermatological, and allergic conditions. It can affect the central nervous system[39]. It is known to increase blood glucose in humans[40]. The docking of dexamethasone against pathogen glucokinase gave a moderate binding affinity of -7.9kcal/mol. This could indicate that it may hamper the efficiency of glucokinase in Naegleria fowleri and affect carbohydrate catabolism negatively. It showed a binding affinity of -7.6kcal/mol when docked against hGLK. As shown in Figure 7. (m), (n), Dexamethasone shows binding with nGLK around ASP134, ALA317, PHE363, TYR361, GLY318, ASP319, ARG39, GLY34, ALA35, GLY186, THR185, GLY184, ALA182, LEU135, SER138, ASN362 and interacts with ASN364, ASN37, THR36 via conventional hydrogen bond. Also, Figure 8. (m), (n), shows Dexamethasone binding with hGLK around LEU415, LYS414, GLU443, SER445, THR228, ASP205, GLY80, GLY227, ARG85, ILE225, GLY410, SER411 and interacts with ASP78 via conventional hydrogen bond and ASP409 via halogen interaction. Supplementary Figure 3. (g) and Supplementary Figure 5. (g) show amino acids of nGLK and hGLK within 5 A of dexamethasone.
The binding affinities against human homolog of glucokinase indicated that all these FDA-approved drugs may induce inhibition of carbohydrate metabolism. This could help understand some of the side effects caused by these drugs. Though approved for medical use, these drugs cause severe negative effects. The need for drugs that specifically target the pathogen alone is necessary to decrease the side effects of Naegleria fowleri treatment.
2.3.8. Virtual Screening and Visualization of Ligands against nGLK and hGLK
Since the FDA approved CDC recommended drugs for the treatment of PAM could bind to both glucokinases i.e., nGLK and hGLK, even though the intended targets are different partially explains why there is a drop in blood sugar levels upon treatment with these drugs. Hence, new ligands were screened against both nGLK and hGLK to identify potential drugs that could specifically bind to only the pathogenic nGLK. The compounds represented here are selected based on the specificity of binding of the compounds to the nGLK.
The top compounds selected post screening based on pharmacokinetic properties and drug-likeness. Though most compounds may have a higher binding affinity towards the pathogenic enzyme nGLK, but were disregarded due to poor ADME and pharmacokinetic properties. Most compounds could not cross the blood-brain barrier. The complexity of finding a drug that specifically binds to the target and passes all the criteria was challenging.
Most ligands showed excellent binding to the glucokinase enzymes. It was challenging to find a compound that specifically bonded with nGLK only. The difference in binding affinities between the two proteins was within the range of 3kcal/mol. The binding affinities of the compounds ranged from -4kcal/mol to -10.7kcal/mol. The ligands that had the best binding affinity and largest difference in binding between the nGLK and hGLK were chosen, along with pharmacokinetic properties in mind. Drugs that could not cross the blood-brain barrier would not be suitable for PAM treatment but were further tested to check whether they could potentially inhibit the human homolog hGLK. As mentioned in earlier sections, hGLK plays a critical role in the progression of diabetes and is a prime target for diabetes treatment[13,17,33].
Table 4.
Comparative analysis of binding affinities of compounds against nGLK and hGLK.
| Ligands | Binding affinity with nGLK (Kcal/mol) | Binding affinity with hGLK (Kcal/mol) |
|---|---|---|
| Simeprevir (PCID: 24873435) | -10.7 | -9 |
| Levomefolic acid (PCID: 135398561) | -10.4 | -9 |
| Indacaterol (PCID: 6918554) | -10.4 | -8.1 |
| Ziprasidone (PCID: 60854) | -10.2 | -8.4 |
| Crizotinib (PCID: 11626560) | -9.8 | -7.8 |
| Mocetinostat (PCID: 9865515) | -10 | -8.8 |
| Risperidone (PCID: 5073) | -10 | -8.9 |
Figure 9.
Top potential drugs identified against nGLK and hGLK: (a) Simeprevir (b) Levomefolic acid (c) Indacaterol (d) Ziprasidone, and (e) Crizotinib.
Figure 9.
Top potential drugs identified against nGLK and hGLK: (a) Simeprevir (b) Levomefolic acid (c) Indacaterol (d) Ziprasidone, and (e) Crizotinib.
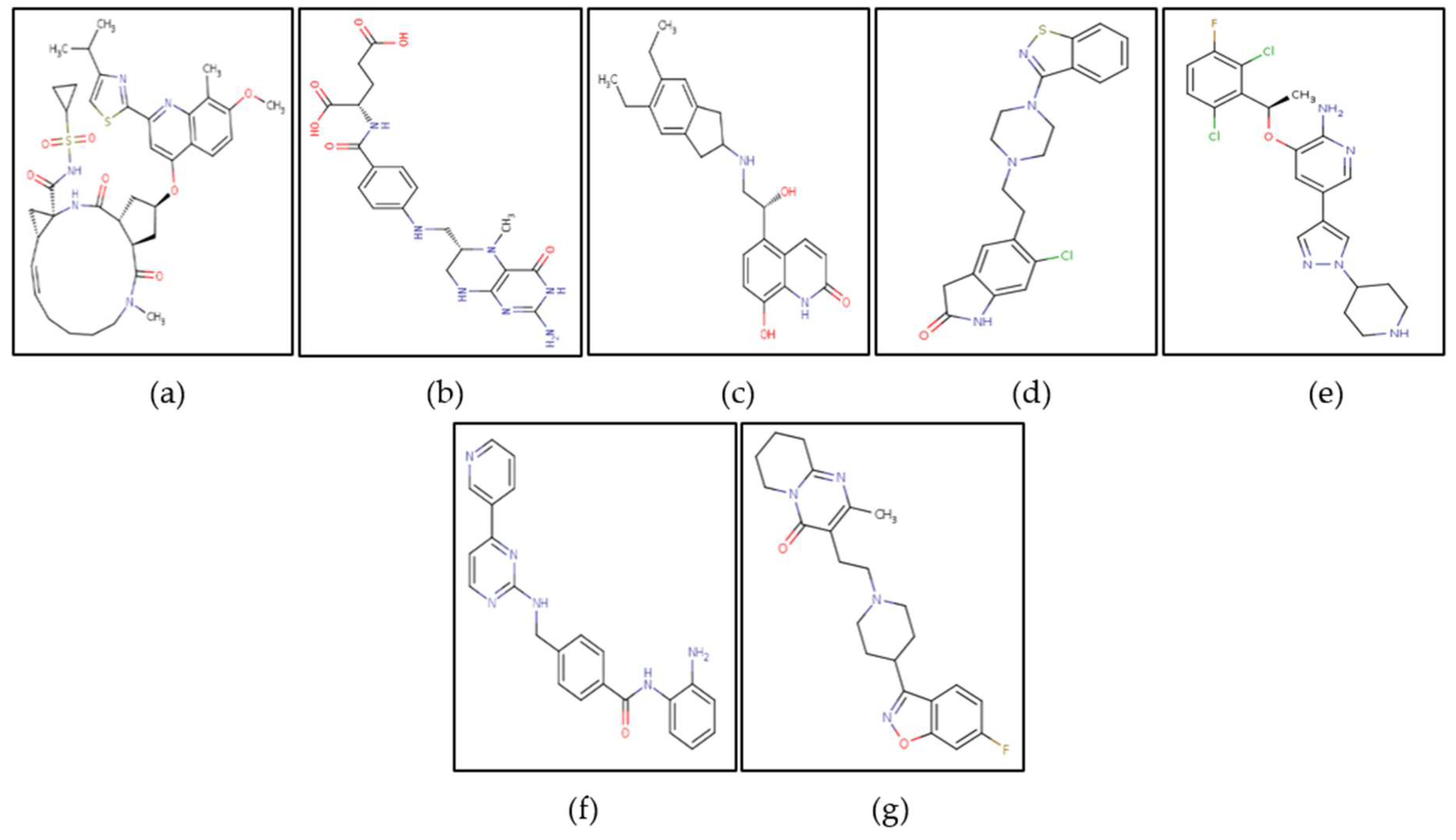
Though the pharmacokinetic studies suggest that Simeprevir and Levomefolic acid have violations of druglikeness and ADME properties, but they are approved by the FDA and have been used for the treatment of various diseases. The pharmacokinetic properties of the compounds shown in Figure. 14 are given in Table 5.
Table 5.
Drug-likeness and ADME properties of top ligands.
| Ligand | Molecular weight (g/mol) | Lipophilicity (Log P o/w (XLOGP3)) | Hydrogen bond acceptors, donors | Water solubility (Log S (ESOL)) |
|---|---|---|---|---|
| Simeprevir | 749.94 | 4.81 | 9,2 | -7.14 Poorly soluble |
| Levomefolic | 459.46 | -0.49 | 7,7 | -1.99 Very soluble |
| Indacaterol | 392.49 | 3.27 | 4,4 | -4.40 Moderately soluble |
| Ziprasidone | 412.94 | 4.02 | 3,1 | -5.07 Moderately soluble |
| Crizotinib | 450.34 | 3.69 | 5,2 | -5.05 Moderately soluble |
| Mocetinostat | 396.44 | 2.76 | 4,3 | -4.17 Moderately soluble |
| Risperidone | 410.48 | 2.72 | 6,0 | -4.20 Poorly soluble |
Table 6.
Pharmacokinetic properties of top ligands.
| Ligand | Druglikeness violations (Lipinski, Ghose, Veber, Egan, Muegge) | Bioavailability | Blood-brain barrier crossing | GI absorption | Skin permeability (cm/s) |
|---|---|---|---|---|---|
| Simeprevir | Yes (Lipinski-2, Ghose-3, Verber-1, Egan-1, Mugge-2) | 0.17 | No | Low | -7.46 |
| Levomefolic acid | Yes (Lipinski-2, Ghose-1, Verber-1, Egan-1, Mugge-2) | 0.11 | Yes | Low | -9.45 |
| Indacaterol | No | 0.55 | No | High | -6.32 |
| Ziprasidone | No | 0.55 | Yes | High | -5.96 |
| Crizotinib | No | 0.55 | No | High | -6.43 |
| Mocetinostat | No | 0.55 | No | High | -6.76 |
| Risperidone | No | 0.55 | Yes | High | -6.87 |
Table 7.
Drugs against nGLK.
| nGLK inhibitors | Drug type |
|---|---|
| Levomefolic acid | Human metabolite |
| Ziprasidone | Antipsychotic |
| Risperidone | Antipsychotic |
Table 8.
Drugs against hGLK.
| hGLK inhibitors | Drug type |
|---|---|
| Simeprevir | Antiviral |
| Indacaterol | Bronchodilator |
| Crizotinib | Anticancer |
| Mocetinostat | Anticancer |
Based on the blood-brain barrier crossing ability, the drugs were divided into two categories as shown in Table 7. and Table 8. The drugs that crossed the blood-brain barrier could serve as potential drugs against Naegleria fowleri while those that could not cross into the CNS could be used as hGLK inhibitors for the treatment of diabetes. Supplementary Figure 6. And Supplementary Figure 8. shows the binding of potential drugs around nGLK and hGLK respectively.
2.3.9. Repurposing of Antiviral Drug Simeprevir against nGLK and Comparing Ligand Interactions against hGLK
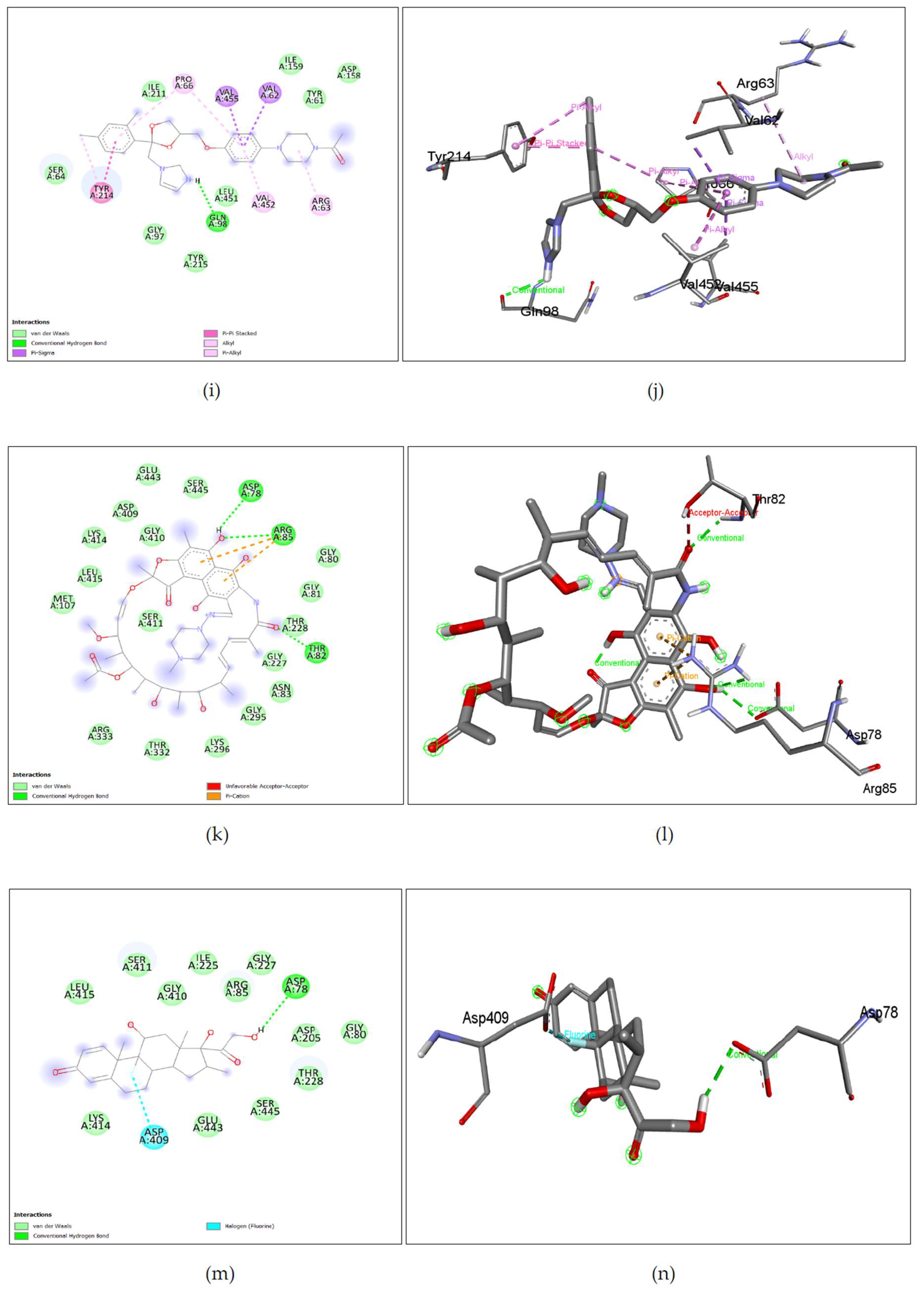
Simeprevir is a NS3/4A inhibitor used against the Hepatitis C virus protease complex, where it binds to the active site and disrupts viral protein processing and replication[41,42]. This azamacrocycle, was approved for use by the FDA in November 2014 and marketed under the name Olysio. It is known to bind albumin and alpha 1 acid glycoprotein which might assist it to cross the blood-brain barrier. It was predicted to cross the blood-brain barrier according to one in-silico study, though, in our ADME analysis and other literature, it showed to not cross the blood-brain barrier [43,44]. It shows binding affinity of -10.7kcl/mol and -9kacl/mol against nGLK and hGLK respectively. As shown in Figure 10. (a), (b) Simeprevir shows binding with nGLK around amino acids ASN37, GLY184, ASN362, TYR361, GLN357, PHE363, SER138, GLY318, ASN364, ALA182, ALA317, LEU135, ALA94, GLY186, LEU187, SER274, ASN320, GLY251, ALA273 and shows interactions with THR36, THR185, GLY188 via conventional hydrogen bond, ASP319, ASP134 via pi-anion bond. The interacting amino acids came under the glucokinase domain of the pathogen’s protein. Also, in Figure 11. (a), (b) Simeprevir shows binding with hGLK around amino acids GLY410, GLU331, THR82, ALA282, GLY170, THR116, MET115, GLU112, ARG327, SER281, GLY328, GLU300, TYR297, GLY295, THR332, THR228, GLY227 and interacts with SER411 via a conventional hydrogen bond, SER280 via carbon hydrogen bond, ARG333, VAL227 via pi-alkyl interaction, LYS296 via alkyl and via pi-cation interaction. The interacting amino acids fall under the glucokinase domain of the human protein. Supplementary Figure 7. (a) and Supplementary Figure 9. (a) show amino acids of nGLK and hGLK within 5 A of Simeprevir.
Figure 10.
Lig-plots and 3D images showing interactions between nGLK and potential drugs. (a) Simeprevir interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bonds (dark green), and pi-anion interactions (yellow). (b) Simeprevir interactions visualized in 3D. (c) Levomefolic acid interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green), pi-donor hydrogen bond (light green), pi-alkyl interaction (light pink). (d) Levomefolic acid interactions visualized in 3D. (e) Indacaterol interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green), pi-donor hydrogen bond (light green), pi-alkyl interaction (light pink), pi-anion interaction (yellow), alkyl interaction (light pink), amide-pi stacked interaction (dark pink), pi-sigma interaction (purple). (f) Indacaterol interactions visualized in 3D. (g) Ziprasidone interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), alkyl interaction (light pink), and pi-sigma interaction (purple). (h) Ziprasidone interactions visualized in 3D. (i) Crizotinib interacts with target glucokinase through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), Halogen interactions (blue), unfavorable donor-donor (red), and pi-anion interaction (yellow). (j) Crizotinib interactions visualized in 3D. (k) Mocetinostat interacts with target glucokinase through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), conventional hydrogen bond (dark green), pi-donor hydrogen bond (light green), pi-anion interaction (yellow), amide-pi stacked (dark pink), pi-alkyl interaction (light pink). (l) Mocetinostat interactions visualized in 3D. (m) Risperidone interacts with target glucokinase through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), conventional hydrogen bond (dark green), pi-anion interaction (yellow), alkyl interaction (light pink), pi-alkyl interaction (dark pink), pi-sigma interaction (purple), halogen interactions (blue). (n) Mocetinostat interactions visualized in 3D.
Figure 10.
Lig-plots and 3D images showing interactions between nGLK and potential drugs. (a) Simeprevir interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bonds (dark green), and pi-anion interactions (yellow). (b) Simeprevir interactions visualized in 3D. (c) Levomefolic acid interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green), pi-donor hydrogen bond (light green), pi-alkyl interaction (light pink). (d) Levomefolic acid interactions visualized in 3D. (e) Indacaterol interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green), pi-donor hydrogen bond (light green), pi-alkyl interaction (light pink), pi-anion interaction (yellow), alkyl interaction (light pink), amide-pi stacked interaction (dark pink), pi-sigma interaction (purple). (f) Indacaterol interactions visualized in 3D. (g) Ziprasidone interacts with target glucokinase through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), alkyl interaction (light pink), and pi-sigma interaction (purple). (h) Ziprasidone interactions visualized in 3D. (i) Crizotinib interacts with target glucokinase through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), Halogen interactions (blue), unfavorable donor-donor (red), and pi-anion interaction (yellow). (j) Crizotinib interactions visualized in 3D. (k) Mocetinostat interacts with target glucokinase through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), conventional hydrogen bond (dark green), pi-donor hydrogen bond (light green), pi-anion interaction (yellow), amide-pi stacked (dark pink), pi-alkyl interaction (light pink). (l) Mocetinostat interactions visualized in 3D. (m) Risperidone interacts with target glucokinase through Van Der Waals interactions (light green), carbon hydrogen bond (dark green), conventional hydrogen bond (dark green), pi-anion interaction (yellow), alkyl interaction (light pink), pi-alkyl interaction (dark pink), pi-sigma interaction (purple), halogen interactions (blue). (n) Mocetinostat interactions visualized in 3D.
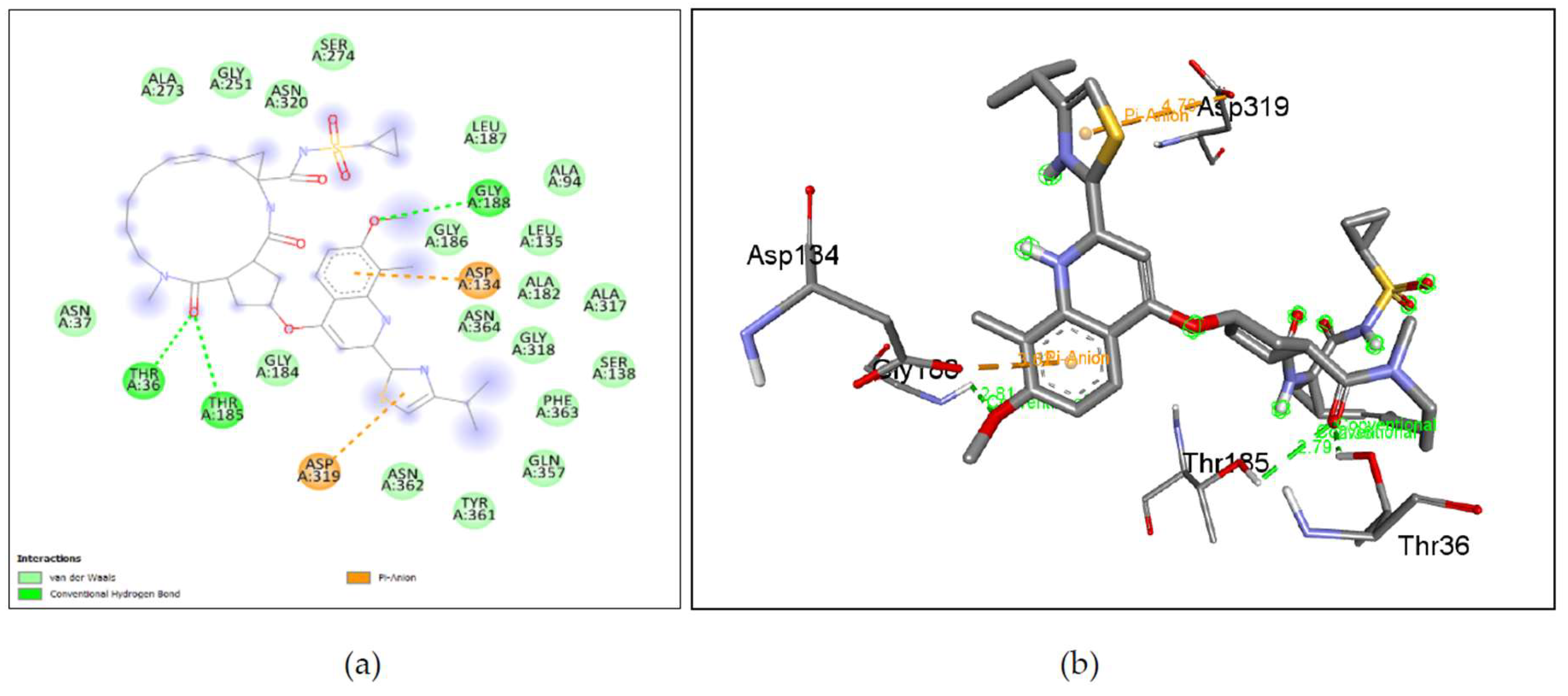
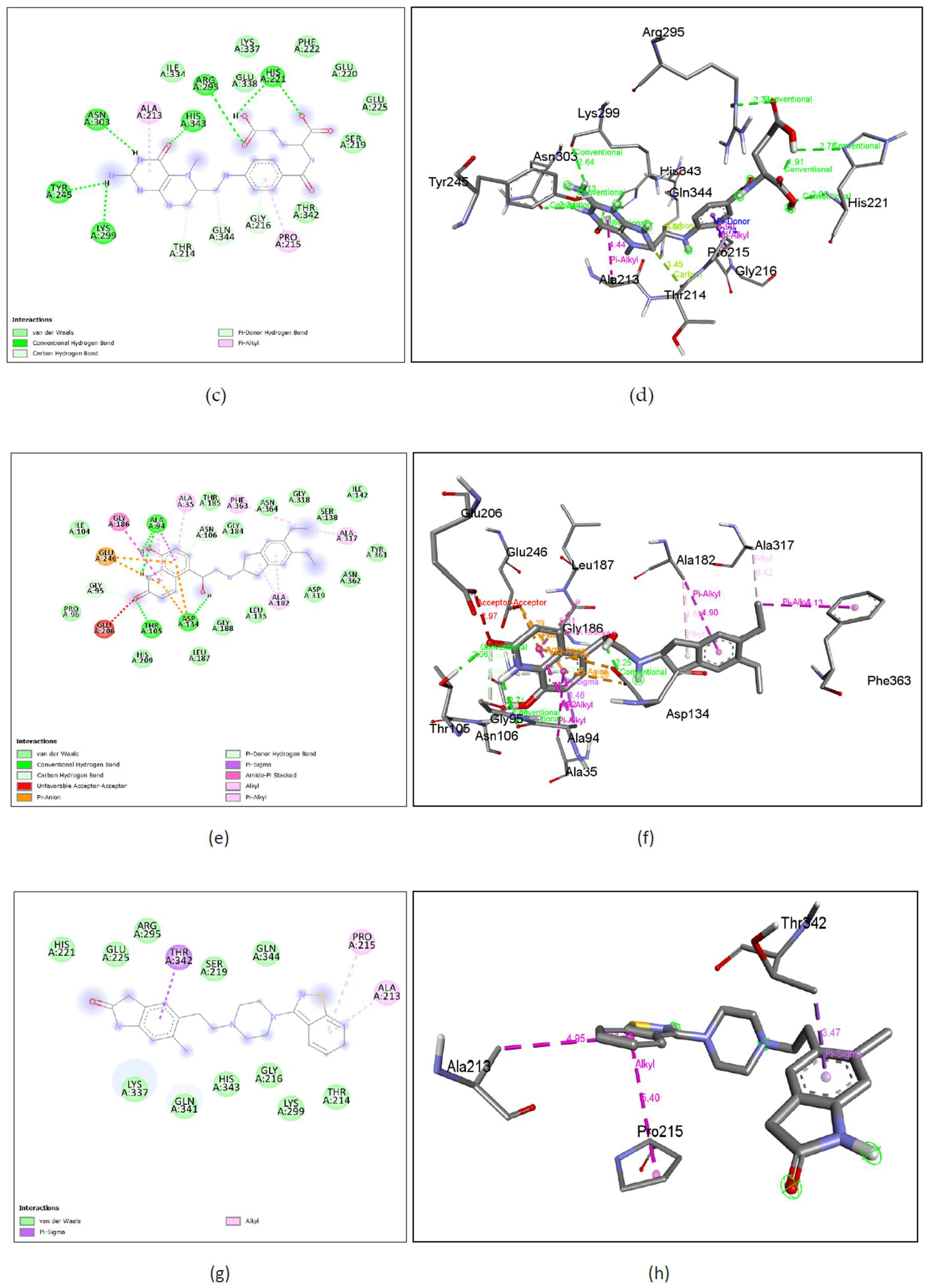
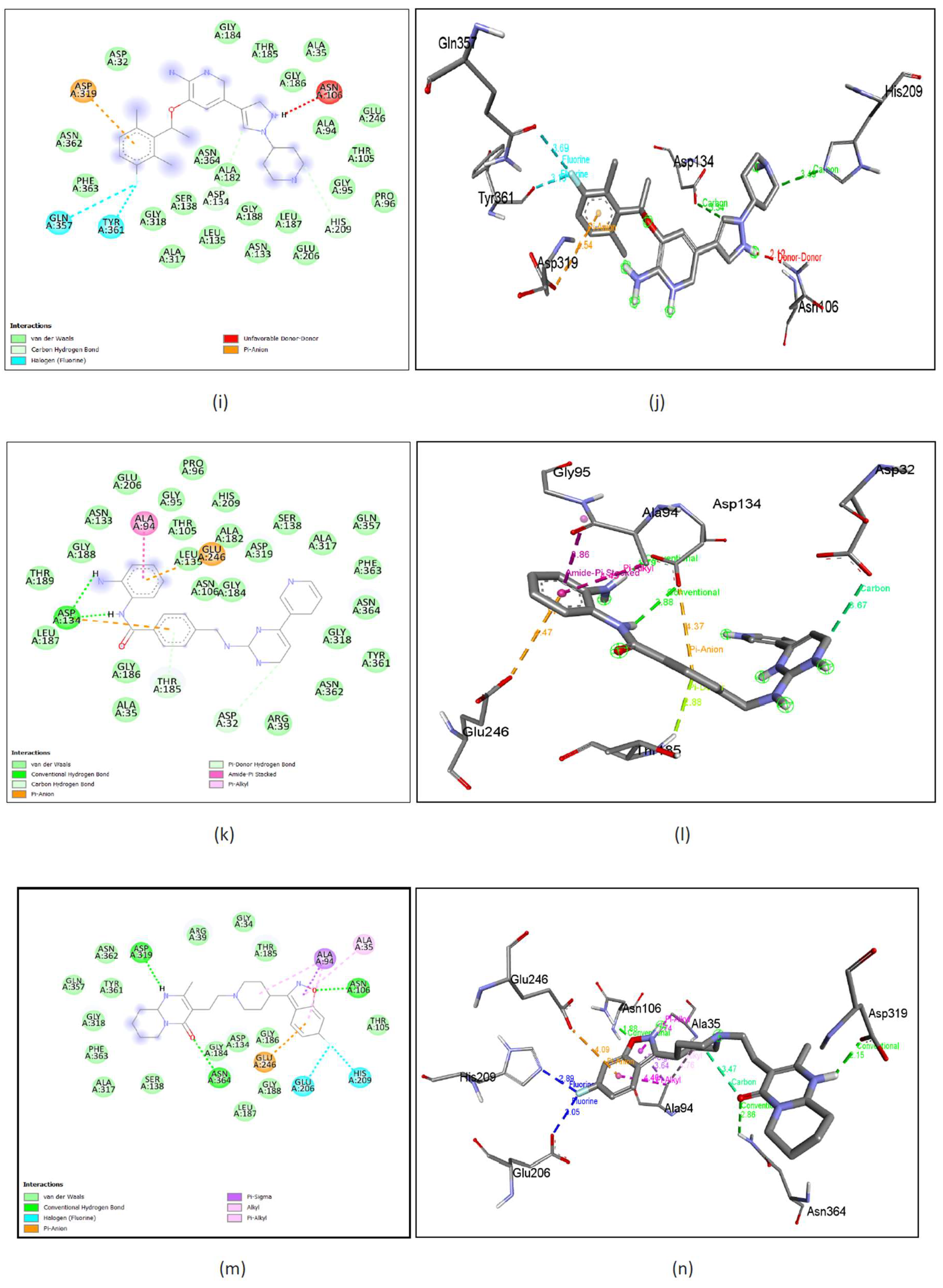
Figure 11.
Lig-plots and 3D images showing interactions between hGLK and potential drugs. (a) Simeprevir interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green) and pi-cation interactions (yellow), pi-alkyl interactions, alkyl interactions (light pink). (b) Simeprevir interactions visualized in 3D. (c) Levomefolic acid interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-alkyl interactions (light pink) and amide-pi stacked interactions (dark pink). (d) Levomefolic acid interactions visualized in 3D. (e) Indacaterol interacts with hGLK through Van Der Waals interactions (light green pi-pi stacked interactions (dark pink), pi-alkyl interactions (light pink), and pi-pi-T shaped interactions (dark pink). (f) Indacaterol interactions visualized in 3D. (g) Ziprasidone interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green) and pi-cation interactions (yellow), pi-sulfur interactions (yellow), pi-pi stacked (dark pink), pi-alkyl interactions (light pink), alkyl interactions (light pink). (h) Ziprasidone interactions visualized in 3D. (i) Crizotinib interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-cation interaction (yellow) and unfavorable donor-donor interactions (red). (j) Crizotinib interactions visualized in 3D. (k) Mocetinostat interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-pi stacked interactions (dark pink), and pi-alkyl interactions (light pink). (l) Mocetinostat interactions visualized in 3D. (m) Risperidone interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon-hydrogen bond (light green), and halogen interactions (light and dark blue). (n) Risperidone interactions visualized in 3D.
Figure 11.
Lig-plots and 3D images showing interactions between hGLK and potential drugs. (a) Simeprevir interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green) and pi-cation interactions (yellow), pi-alkyl interactions, alkyl interactions (light pink). (b) Simeprevir interactions visualized in 3D. (c) Levomefolic acid interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-alkyl interactions (light pink) and amide-pi stacked interactions (dark pink). (d) Levomefolic acid interactions visualized in 3D. (e) Indacaterol interacts with hGLK through Van Der Waals interactions (light green pi-pi stacked interactions (dark pink), pi-alkyl interactions (light pink), and pi-pi-T shaped interactions (dark pink). (f) Indacaterol interactions visualized in 3D. (g) Ziprasidone interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon hydrogen bond (light green) and pi-cation interactions (yellow), pi-sulfur interactions (yellow), pi-pi stacked (dark pink), pi-alkyl interactions (light pink), alkyl interactions (light pink). (h) Ziprasidone interactions visualized in 3D. (i) Crizotinib interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-cation interaction (yellow) and unfavorable donor-donor interactions (red). (j) Crizotinib interactions visualized in 3D. (k) Mocetinostat interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), pi-pi stacked interactions (dark pink), and pi-alkyl interactions (light pink). (l) Mocetinostat interactions visualized in 3D. (m) Risperidone interacts with hGLK through Van Der Waals interactions (light green), conventional hydrogen bond (dark green), carbon-hydrogen bond (light green), and halogen interactions (light and dark blue). (n) Risperidone interactions visualized in 3D.
2.3.10. Repurposing of Human Metabolite/Supplement Levomefolic Acid against nGLK and Comparing Ligand Interactions against hGLK
Levomefolic acid, an active form of folate is a naturally found metabolite in Homo sapiens. Though the SwissADME results indicate that the compound cannot cross the blood-brain barrier, research confirms that it can transfer across all membranes even the blood-brain-barrier[45]. It plays a vital role in DNA synthesis and the cysteine cycle. It is being studied for use as a psychedelic drug[45,46]. It is a food additive and a supplement and is considered an excellent drug due to its ability to cross blood-brain-barrier. The possibility of side effects due to a naturally found metabolite seems unlikely. It showed a binding affinity of -10.4kcal/mol and -9kcal/mol with nGLK and hGLK, respectively. The possibility of toxicity against humans seems minimal as it has a half-life of 3 hours and is eliminated via the renal or fecal route (https://www.caymanchem.com/msdss/21616m.pdf). Levomefolic acid is metabolized into tetrahydrofolate and then converted to polyglutamate form. It has been used to prevent the risk of neural tube defects in pregnant women as a folate supplement. As shown in Figure 10. (c), (d) Levomefolic acid shows binding with nGLK around amino acids ILE334, LYS337, GLU338, PHE222, GLU220, GLU225, SER219, THR342 and shows interactions with ASN303, HIS343, TYR245, LYS299, ARG295, HIS221 via conventional hydrogen bond, THR214, GLN344, GLY216 via carbon hydrogen bond and PRO215, ALA213 via pi-alkyl bond. The interacting amino acids come under the glucokinase domain of the pathogen’s protein. Also, Figure 11. (c), (d) Levomefolic acid shows binding with hGLK around amino acid THR149, ASP409, THR209, ILE225, GLY410, GLY295, VAL412, HIS416, SER411, THR228, GLY227, LYS169, GLY80, ASP205, ASP78, GLU443, GLY444 and interacts with SER445, SER151, ARG85, LEU415 via conventional hydrogen bond, THR332 via amide pi-stacked interactions, ARG333 via pi-alkyl interactions and SER336 via unfavorable interactions. The interacting amino acids come under the glucokinase domain of the human protein. As the only form of folate that can cross the blood-brain barrier, it acts as a cofactor in the production of monoamine neurotransmitters such as dopamine, serotonin, and norepinephrine. Supplementary Figure 7. (b) and Supplementary Figure 9. (b) show amino acids of nGLK and hGLK within 5 A of levomefolic acid.
2.3.11. Repurposing of Bronchodilator Indacaterol against hGLK and Comparing Ligand Interactions against nGLK
Indacaterol is a monohydroxyquinoline that is a potent inhibitor of β (2)-adrenoceptor agonist used for the treatment of asthma and pulmonary disease[47]. It is a bronchodilator and is an approved drug by the European Medicines Agency and FDA. It showed no druglikeness violations, excellent bioavailability, and high gastrointestinal absorption. The binding affinity of indacaterol to the nGLK is -10.4kcal/mol and hGLK is -8.1kcal/mol. As shown in Figure 10. (e), (f) Indacaterol shows binding with nGLK around amino acids ILE104, PRO96, HIS209, LEU187, GLY188, LEU135, ASP319, ASN362, TYR361, SER138, ILE142, GLY318, ASN364, GLY184, THR185 and interacts with ALA94, ASP134, THR105, via conventional hydrogen bond, ASN106 via Pi-donor hydrogen bond, GLY95 via a carbon hydrogen bond, ALA182, PHE363, ALA317, ALA94, ALA35 via pi-alkyl bond, ALA94 via pi-sigma bond, ALA182, ALA94 via alkyl interactions, GLU246, ASP134 via pi-anion bond, GLU206 via unfavorable acceptor-acceptor interactions. The interacting amino acids come under the glucokinase domain of the pathogen’s protein. Also, Figure 11. (e), (f) Indacaterol shows binding with hGLK around amino acids GLY97, GLN98, LEU451, MET235, SER64, GLU221, ARG250, GLN219, HIS218, and interacts with TYR214 via pi-pi-T stacked interactions, PRO66, ILE211 via pi-alkyl interactions. The interacting amino acids come under the glucokinase domain of the human protein. Supplementary Figure 7. (c) and Supplementary Figure 9. (c) show amino acids of nGLK and hGLK within 5 A of indacaterol.
2.3.12. Repurposing of Antipsychotic Ziprasidone against hGLK and Comparing Ligand Interactions against nGLK
Ziprasidone, a piperazine derivative which is a second-generation antipsychotic drug used to treat schizophrenia and bipolar disorder[48]. It functions as an antagonist to dopamine, histamine, muscarinic, and serotonin receptor, and is an inhibitor of synaptic reuptake of serotonin, and norepinephrine. It is used to treat schizophrenia and bipolar disorder[49]. This compound has the best drug-likeness and pharmacokinetic properties to be considered a promising lead drug for inhibiting nGLK. The binding affinity of the ligand against nGLK is -10.2kcal/mol and hGLK is -8.1kcal/mol. The potential of ziprasidone to inhibit hGLK is yet to be reported. A previous study has shown that it has no effect on the blood glucose levels while an epidemiological study found no link between ziprasidone and increased risk of hyperglycemia unlike olanzapine and clozapine [50]. It has a half-life of 6-7 hours and is heavily metabolized in the liver into 12 different metabolites by CYP3A4 and aldehyde oxidase enzymes. As shown in Figure 10. (g), (h), Ziprasidone shows binding to nGLK around amino acids LYS337, GLN341, HIS343, GLY216, LYS299, THR214, GLN344, SER219, ARG295, GLU225, HIS221 and interacts with THR342 via pi-sigma bond, PRO215, ALA213 via alkyl interaction. The interacting amino acids come under the glucokinase domain of the pathogen’s protein. Also, in Figure 11. (g), (h), Ziprasidone shows binding with hGLK around amino acids ARG397, ARG403, GLN219, SER64, PRO66, CYS220, MET210, GLU221 and ILE211 and interacts with SER398, GLU399 via conventional hydrogen bonds, HIS218 via carbon-hydrogen bond, TYR214, ILE404, MET235 via pi-alkyl and alkyl interactions, TYR214 via pi-pi stacked interaction, ARG250 via pi-cation interaction and MET402 via pi-sulfur interaction. The interacting amino acids come under the glucokinase domain of the human protein. Also, multiple antipsychotic drugs are known to influence glucose metabolism[51]. In one study, it was found that olanzapine and clozapine bind strongly to muscarinic M3 and inhibit insulin release[52]. Ziprasidone is known to reverse olanzapine and clozapine-induced hyperglycemia[52,53]. Supplementary Figure 7. (d) and Figure 9. (d) show amino acids of nGLK and hGLK within 5 A of ziprasidone.
2.3.13. Repurposing of Anticancer Crizotinib against hGLK and Comparing Ligand Interactions against nGLK:
Crizotinib is a specific aminopyridine inhibitor of tyrosine kinase anaplastic lymphoma kinase receptor and c-Met/hepatocyte growth factor receptor. It binds to tyrosine kinase anaplastic lymphoma kinase by competing with ATP[54]. Tyrosine kinase anaplastic lymphoma kinase plays a significant role in neuronal development. Crizotinib is known to be used in certain cases against non-small cell lung cancer (PubChem). It is an oral drug and has side effects[55]. The binding affinity of Crizotinib to nGLK is -9.8kcal/mol and hGLK is -7.8kcal/mol. The drug has no drug-likeness violations but must be considered before use as a drug against N. fowleri due to its inability to cross-blood the brain barrier. Though the binding to hGLK is moderately strong, yet, it is unclear whether the side effects are due to the inhibition of hGLK. As shown in Figure 10. (i), (j), Crizotinib shows binding with nGLK around amino acids ASP32, GLY184, THR185, GLY186, ALA35, ALA94, GLU246, THR105, GLY95, PRO96, GLU206, LEU187, GLY188, ASN133, LEU135, ALA182, ASN364, SER138, GLY318, PHE363, ASN362 and interacts with amino acids ASP319 via pi-anion bond, HIS209, ASP134 via carbon hydrogen bond, GLN357, TYR361 via halogen interactions and ASN106 via unfavorable interactions. Also, in Figure 11. (i), (j), Crizotinib shows binding with hGLK around amino acids GLY80, GLY227, GLY410, THR149, SER445, ASP409, GLY444, GLU442, THR228, LYS414, LEU415, GLY81, THR82, ASN83, SER411 and interacts with ASP78, ASP205 via conventional hydrogen bond, ARG85 via pi-cation interaction, SER151, GLU443 via unfavorable interaction. Crizotinib has been shown to moderately decrease blood glucose levels in rats[56]. Supplementary Figure 7. (e) and Supplementary Figure 9. (e) show amino acids of nGLK and hGLK within 5 A of crizotinib.
2.3.14. Repurposing of Anticancer Mocetinostat against hGLK and Comparing Ligand Interactions against nGLK:
Mocetinostat is a. novel, oral drug used to treat tumor cells, causing apoptosis of cells[57,58]. This benzanilide derivative binds to Histone deacetylase 1, 2, 3 and inhibits its activity. It leads to the upregulation of tumor-suppressive genes, and cell cycle arrest inhibition of DNA repairs. Though the exact mechanism is not defined, it is being studied to be used in trials for the treatment of urothelial carcinoma, lymphoma, and metastatic leiomyosarcoma[59]. The binding of mocetinostat is -10kcal/mol and -8.8kcal/mol for nGLK and hGLK, respectively. As shown in Figure 10. (k), (l), Mocetinostat interacts with THR189, GLY188, ASN133, GLU206, GLY95, PRO96, THR105, LEU135, ALA182, HIS209, GLY184, ASP319, ALA317, GLN357, PHE363, ASN364, GLY318, ASN362, ARG39, ALA35, GLY186, LEU187, SER138 and interact with ASP134 via conventional hydrogen bond, GLU246, ASP134 via pi-anion bond, ALA94 via amide-pi stacked, THR185, ASP32 via pi-donor hydrogen bond. Also, in Figure 11. (k), (l), Mocetinostat shows binding with hGLK around amino acids VAL452, VAL455, ILE211, GLU221, GLN219, SER64, MET235, ARG250, GLY249, GLN98, SER69 and interacts with HIS218 via conventional hydrogen bond, TYR214 via pi-pi stacked interaction and PRO66, LEU415 via pi-alkyl interaction. Supplementary Figure 7. (f) and Supplementary Figure 9. (f) show amino acids of nGLK and hGLK within 5 A of mocetinostat.
2.3.15. Repurposing of Antipsychotic Risperidone against nGLK and Comparing Ligand Interactions against hGLK
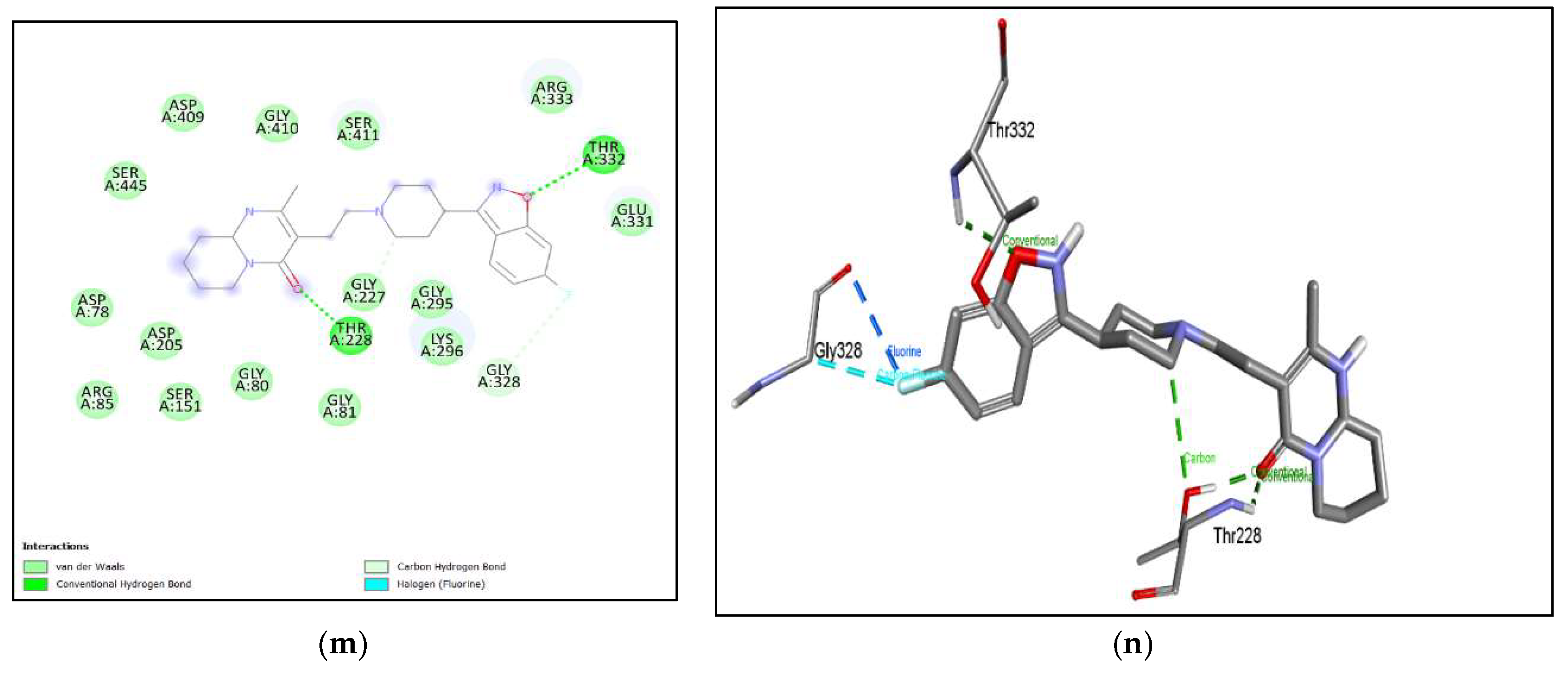
Risperidone is a second-generation antipsychotic drug used in the treatment of hallucinations, delusions, and other psychotic effects associated with schizophrenia and bipolar disorder. It is derived from benzisoxazole and is a pyridopyrimidine. It has an inhibitory action on both dopaminergic D2 receptors and serotonergic 5-HT2A receptors but has a 10-fold higher activity against the latter as compared to D2 receptors in the central nervous system. It is mainly metabolized in the liver with a half-life of 3 hours and is cleared via the kidneys. This drug has also been tested for the treatment of irritability in autistic children[60]. The binding of Risperidone is -10kcal/mol and -8.9kcal/mol for nGLK and hGLK, respectively. As shown in Figure 10. (m), (n), Risperidone shows binding with nGLK around amino acids ALA317, PHE363, GLY318, TYR361, GLN357, ASN362, ARG39, GLY34, THR185, THR105, GLY188, LEU187, ASP134, GLY184, GLY186 and interacts with ASP319, ASN364, ASN106 via conventional hydrogen bond, ALA94, ALA35 via pi-alkyl interactions, ALA94 via alkyl and pi-sigma interactions, GLU246 via pi-anion interactions, GLU206, HIS209 via halogen interactions. Also, in Figure 11. (m), (n), Risperidone shows binding with hGLK around amino acids ASP78, ARG85, ASP205, SER151, GLY81, GLY80, GLY295, LYS296, GLU331, ARG333, SER411, GLY410, ASP409, SER445 and interacts with THR228, THR332 via conventional hydrogen bond, GLY227 via carbon hydrogen bond and GLY328 via halogen interactions. Research showed that mice that were treated with risperidone had elevated fasting blood glucose levels and lower serum insulin, indicating that these drugs do influence glucose metabolism through various mechanisms [61]. Thus, our computational research data is in line with previously published research and adds a new perspective to it by showing that it binds strongly to the glucokinase of both nGLK and hGLK. Supplementary Figure 7. (g) and Supplementary Figure 9. (g) show amino acids of nGLK and hGLK within 5 A of Risperidone.
Though the binding of these drugs is strong, a standard is required to ensure that the numbers are relevant. Another set of docking was done to identify the binding affinity of known inhibitors of hGLK to be used as a standard for comparison. Three known inhibitors of the hGLK enzyme, namely 2-Deoxy-D-glucose, lonidamine, and 3-bromopyruvate were docked against the target. The binding affinities were -5.3kcal/mol, -8.3kcal/mol and -4.5kcal/mol, respectively (data not shown). This indicates that even well-known enzyme inhibitors may have a binding affinity ranging from as low as -4.5kcal/mol.
The binding affinities of these drugs indicate that there is a strong interaction between them and the targets. Several research articles have also attributed some of these drugs to influence glucose metabolism through various mechanisms. According to our results, one mechanism by which these drugs influence our metabolism could be by directly inhibiting the glucokinase enzyme.
3. Discussion
3.1. Comparative Analysis of nGLK and hGLK
Structural analysis of nGLK and hGLK was conducted using PDB Pairwise Structural Alignment that uses jFATCAT-rigid algorithm Figure 4. (c), This algorithm allows flexible protein structure alignment. The RMSD score for the alignment was 3.09, with a TM-score of 0.52 and 226 equivalent residues. The target coverage was 60% and the reference coverage was 70%. In Figure 3.; the sequence overlay of the two glucokinases showed that there is 20.53% identity between the two structures. The main amino acid residues involved in the glucose binding in nGLK includes HIS231, GLU268, GLU228, and ASN115[16]. The phosphoaminophosphonic acid-adenylate ester interacts with LEU276, ALA296, ALA299, ASN346, and ASP342. For hGLK, the amino acid residues the binding of glucose include ASP205, and LYS169, and those around phosphoaminophosphonic acid-adenylate ester, are SER151, ASP78, ASN83, SER411, SER366, THR332, and THR228. These structures are different and can be targeted separately, through further in-depth studies. Despite distinct structural differences, we could not identify any inhibitor that exclusively binds to nGLK and hGLK. The difference in the range of binding affinities of ligands between nGLK and hGLK was approximately -2kcal/mol. Among the seven potential drugs identified, all showed stronger binding towards Naegleria glucokinase, but four of the drugs i.e., Simeprevir; Indacaterol; Crizotinib and Mocetinostat cannot cross the blood-brain barrier and thus will not be effective in the treatment of Naegleria infection. These drugs are predicted to be used for the treatment of diabetes because these act as inhibitors for glucokinase[62]. The drugs that can cross the blood-brain barrier and can strongly bind to both nGLK and the hGLK are Levomefolic acid; Ziprasidone; and Risperidone respectively. in the order of their binding affinity.
The drugs mentioned are more specific towards Naegleria glucokinase but have strong enough binding to influence human analog as well. Comparative analysis of the binding pockets in Table 1 and Table 2. reveals that there is a major difference in size, number of hydrogen bond donors and acceptors, hydrophobicity ratio, drug scores, and amino acids involved. This difference could be another cause for the dissimilarity in the binding affinities of ligands between the two proteins. As shown previously, established drugs that are presently used to treat PAM showed strong binding with both nGLK and hGLK. This is the first comparative study of the interaction of established drugs on nGLK and hGLK.
3.2. Mechanism of Human Glucokinase (hGLK)
Glucokinase (also known as hexokinase IV or hGLK) is essential for maintaining glucose homeostasis. In liver, it assists in removing excess glucose and store as glycogen and in pancreatic beta-cells, it helps sensing the level of free glucose in the bloodstream and ensure release of equivalent amount of insulin. The pancreatic beta cell’s ability to secrete insulin in response to glucose stimulation depends heavily on glucose metabolism[63] . The glucose enters the beta-cell through high capacity insulin independent glucose transporters causing internal and extracellular glucose concentrations to quickly equilibrate. The cytosolic ATP increases with a decrease in cytosolic ADP as a result of subsequent metabolism through glycolysis and mitochondrial metabolism. As a result of these alterations in adenine nucleotides, the potassium channel KATP is inhibited, which causes membrane depolarization that in turn opens the voltage-gated calcium channels, calcium influx, and the exocytosis of insulin granules[64]. The primary step in glucose metabolism is phosphoryaltion which can be initiated by all the four isozymes of hexokinase. Hexokinase IV or hGLK is unique as it has low affinity for glucose and has sigmoidal dependency unlike hyperbolic dependency as the other three hexokinases. Its inflexion point is close to the threshold of insulin secretion and does not saturate at physiological glucose concentrations (Vmax > 20 mM) unlike other three isozymes that saturate at normal fasting concentrations (~5 mM in human beta-cells) [65]. These characteristics allow glucokinase to function as the beta-cell glucose sensor by ensuring that the rate of glucose phosphorylation changes across the physiological range (3–15 mM) and is proportional to the extracellular glucose concentration. hGLK is not allostearically inhibited by the end product (Glucose-6-phosphate)unlike other hexokinases [65].
3.3. hGLK Agonists vs. hGLK Antagonists
The initial prediction was that by regulating metabolic flux in the beta-cell, activating glucokinase might enhance beta-cell metabolism while stimulating insulin production[63,64,66]. However, when hGLK was overexpressed in insulin-secreting cells, it had no effect at low glucose levels and actually decreased glucose uptake at high glucose levels[67,68]. Similarly, adult mice with a specific hGLK mutation experienced increased beta-cell replication, fasting hypoglycemia, and elevated oxygen consumption at a glucose concentration where hGLK activity is typically minimal[69]. Surprisingly, the hypoglycemia peaked after 4 days, however it lasted for a short time before restoring to normal blood sugar levels. Due to double-stranded DNA damage, there was an increase in apoptosis and a loss of beta-cells, that synthesized this. Activation of hGLK altered numerous genes in a manner similar to those observed in islets from diabetic individuals, mice, and beta-cell lines cultured at high glucose levels[70,71,72,73]. These changes were evident during the period of highest hypoglycemia, suggesting that they were induced by increased hGLK activity rather than elevated blood sugar levels.
Initially, it was believed that activating hGLK would increase hepatic glucose uptake and stimulate insulin secretion in people with diabetes, leading to the development of many small molecule hGLK activators[74,75]. These medications activate hGLK by binding to its allosteric activator site and increasing the enzyme’s affinity for glucose and/or its maximum rate of activity.
Early studies in both normal and diabetic mice models demonstrated encouraging effects, with a single oral dose increasing plasma insulin and decreasing blood glucose levels[74,75,76]. Numerous studies using animal models revealed beneficial effects on the progression of hyperglycemia and diabetes[77,78]. Additionally, a limited Phase 1 clinical trial showed decreased blood glucose levels in patients with mild type 2 diabetes (T2D) after using an hGLK activator[75].
However, hGLK activation posed risks, including beta-cell stress caused by excessive activation, hepatic steatosis (fatty liver), and hypoglycemia resulting from excessive insulin secretion at low glucose levels[79,80]. Hypoglycemic episodes were observed in both animal models and T2D patients treated with hGLK activators[75,81]. Alarmingly, patients with T2D experienced vascular hypertension, hepatic steatosis, and hyperlipidemia during a significant 54-week Phase 2 trial. There was only a moderate impact on glucose regulation that was not sustained beyond 3-4 months[80,81]. Blood pressure and plasma lipid levels also increased. Further studies failed to demonstrate long-term improvements in glycemia[77,82]. The decline in the popularity of hGLK activators can be attributed to this loss of potency, highlighting the need for caution when prescribing these drugs[75,80,81]. The effectiveness of hGLK activators for treating decreased insulin secretion in T2D has been questioned due to inconsistent outcomes[74,83,84]. Recent research suggests that the progressive loss of beta-cell function in diabetes is primarily driven by accelerated glucose metabolism rather than glucose itself, indicating that hGLK inhibitors, rather than activators, may have a therapeutic effect on T2D patients[62,85,86].
4. Materials and Methods
4.1. Retrieval of Ligands
The natural, semi-synthetic, and synthetic substances were retrieved from an internet library. The 3D structures for the compounds and ligands were retrieved from PubChem and ChemSpider. Open Babel plug-in of PyRx 0.8 was used to minimize energy and prepare the ligands for screening studies.
The drugs used for the treatment of Naegleria fowleri were docked with the target to check for the possibility of strong binding. Rifampin, azithromycin, dexamethasone, amphotericin, miltefosine, and fluconazole are recommended medications by the CDC for the treatment of Naegleria fowleri. Certain structures lacked 3D structures and were built using the UCSF Chimera tool[87]. The SMILES for azithromycin, rifampin, and amphotericin B were retrieved from the PubChem database. The structure was saved as a PDB and prepared using PyRx[10,88].
4.2. ADME and Toxicity Assays
Pharmacokinetic properties determine the success or failure of a potential drug in clinical trials. A drug must possess a favorable ratio of activity to drug-likeness in order to be effective. Absorption, Distribution, Metabolism, and Excretion is referred to as ADME. The requirements for drug-likeness must be met for any drug to be successful. SwissADME was used for determining the ADME attributes[19].
The blood-brain barrier (BBB) is one of the most exclusive blood tissue barriers found in the brain, testes, and other extremely sensitive organs. Since efflux transporters are present and endothelial cells provide a tight connection between membrane cells, it is difficult to target the central nervous system. BBB crossing capacity was one of the key criteria to select prospective nGLK inhibitor. During screening prospective inhibitors, other variables such as absorbance, oral bioavailability, and minimal drug-likeness violations were taken into account.
4.3. Retrieval of Protein
The structure of the nGLK protein (PDB: 6DA0) and hGLK (PDB: 3FGU) were retrieved from the Protein Data Bank. Using the Discovery Studio Visualizer tool, the ligands and water were removed from both protein structures. The proteins were prepared for docking by using the Dock prep option in USCF Chimera. The proteins were then minimized by keeping the steepest descent steps at 1000, with step size of 0.02 Å and conjugate gradient steps at 500 with step size of 0.02 Å. The prepared proteins were annotated and downloaded for the virtual screening of ligands.
4.4. Virtual Screening and Visualization
Brain-eating amoeba or Naegleria fowleri exclusively codes for glucokinase and not hexokinases[2]. The enzyme glucokinase is crucial for the metabolism of carbohydrates and is required for the production of nucleotides[13]. Using the website https://proteins.plus/, the glucokinase protein was examined for the existence of drug-binding pockets. The two-glucokinase proteins were tested for ligands using the PyRx virtual screening program. Naegleria fowleri glucokinase (nGLK) was used to screen ligands to find the ones with lowest binding energy. The top-scoring ligands were again screened against human glucokinase (hGLK). Autodock Vina plugged into Chimera was used for the virtual screening of the ligands. The grid box was set to cover the entire protein to cover all docking sites. The binding affinity scores of the ligands were saved as CSV files and the interactions were visualized using Discovery studio and UCSF Chimera. The Ligplots were developed using Discovery Studio.
5. Conclusions
Naegleria fowleri causes fatal amebic meningoencephalitis, leading to death in most cases[2]. Most of the prescribed drugs for Naegleria fowleri cause severe side effects. In our study, we first docked the present prescribed drugs against both nGLK and hGLK to examine their binding affinities. The compounds had affinities ranging from -4.1 to -9.2kcal/mol and -5.5kcal/mol to -8.8kcal/mol for both nGLK and hGLK, respectively. Though none of these drugs were previously identified as glucokinase inhibitors, they showed a high binding affinity. This reveals how drugs can affect off-targets and influence them. Though the similarities between the two glucokinases are low, the binding affinities suggest that the many of the present drugs can influence both glucokinases. Multiple researchers have shown that some of these drugs influence glucose metabolism and this study corroborates the finding that could be possible through the inhibition of glucokinase directly.
This study identifies compounds that have inhibitory effects against both glucokinases and may solve two divergent issues i.e., infectious disease and metabolic disorder. Most of the drugs aim at activating glucokinases claiming that the increase in the activity of glucokinase would directly increase the action of release in insulin from pancreatic beta-cells [86]. We have explained why hGLK antagonist holds more promise than hGLK agonist. In our paper, we aimed at inhibiting glucokinase activity which would lead to the weakening of the pathogen by depleting their energy production. This also parallelly led to a discovery of a potential treatment for diabetes. In our earlier study, we identified compounds against SMT for the treatment of Naegleria infections[10]. The possibility of synergistic use of compounds to block both lipid and glucose metabolism needs to be studied.
Among the top 6 ligands against nGLK, Ziprasidone and Risperidone excelled in all criteria of druggability. The two ligands are well-known antipsychotic drugs administered both intramuscularly and orally. These antipsychotic drugs showed no violations and had high binding against nGLK. Blood-brain-barrier crossing was an important criterion for the selection of drugs against nGLK. Levomefolic acid also showed excellent binding and druggability against nGLK. In our study, we were able to identify potential drugs against hGLK, which could be used to treat glucose insensitivity that occurs due to the extensive use of glucokinase activators. Though all the top drugs showed strong binding against both targets, drugs were divided based on CNS permeability. Those that could cross the BBB would be used for inhibiting nGLK and the BBB impermeant drugs would be used as hGLK inhibitors. More specific drugs for each target could be designed by tailoring the side chains based on the amino acid pocket patterns that are mentioned in the paper. The next step would include testing the efficacy and toxicity of these compounds using cell-based assays.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, S.R.; Investigation, J.A., S.R., and N.C. Writing—original draft preparation, J.A. and S.R.; Writing—review and editing, J.A., S.R., and N.C.; Supervision, S.R.; Project administration, S.R.; Funding acquisition, S.R. All authors have read and agreed to the published version of the manuscript.
Funding
NIST grant 60NANB23D215 supported the research & NASA grant 80NSSC22K0869 supported JA. .
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and supplementary material.
Acknowledgments
Special thanks to the Bowie State University, G. Acquaah, G. Ude, and C. Esimai for their constant support to our research without which this manuscript would not have been possible. Special thanks to the CHEM 309 class (Spring 2024) who participated in the initial screening of the inhibitors as a part of the project based learning (PBL) during the coursework.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the writing of the manuscript, or in the decision to publish the results.
References
- M. Jahangeer, Z. Mahmood, N. Munir, U. e. A. Waraich, I.M. Tahir, M. Akram, S.M. Ali Shah, A. Zulfqar, R. Zainab, Naegleria fowleri: Sources of infection, pathophysiology, diagnosis, and management; a review, 2020. [CrossRef]
- L.G. Capewell, A.M. Harris, J.S. Yoder, J.R. Cope, B.A. Eddy, S.L. Roy, G.S. Visvesvara, L.A.M. Fox, M.J. Beach, Diagnosis, clinical course, and treatment of primary amoebic meningoencephalitis in the United States, 1937-2013, J. Pediatric Infect. Dis. Soc. 4 (2015) e68–e75. [CrossRef]
- R. Siddiqui, I.K.M. Ali, J.R. Cope, N.A. Khan, Biology and pathogenesis of Naegleria fowleri, Acta Trop. 164 (2016) 375–394. [CrossRef]
- T.W. Heggie, Swimming with death: Naegleria fowleri infections in recreational waters, Travel Med. Infect. Dis. 8 (2010) 201–206. [CrossRef]
- A. Shariq, F.I. Afridi, B.J. Farooqi, S. Ahmed, A. Hussain, Fatal primary meningoencephalitis caused by Naegleria fowleri, J. Coll. Physicians Surg. Pakistan. 24 (2014) 523–525. [CrossRef]
- CDC, Naegleria fowleri — Primary Amebic Meningoencephalitis (PAM) — Amebic Encephalitis, (2022). https://www.cdc.gov/parasites/naegleria/treatment.html.
- A. Alli, J.F. Ortiz, Á. Morillo Cox, M. Armas, V.A. Orellana, Miltefosine: A Miracle Drug for Meningoencephalitis Caused by Free-Living Amoebas, Cureus. 13 (2021) 1–11. [CrossRef]
- A. Debnath, J.B. Tunac, S. Galindo-Gómez, A. Silva-Olivares, M. Shibayama, J.H. McKerrow, Corifungin, a new drug lead against Naegleria, identified from a high-throughput screen, Antimicrob. Agents Chemother. 56 (2012) 5450–5457. [CrossRef]
- C.A. Rice, B.L. Colon, E. Chen, M. V. Hull, D.E. Kyle, Discovery of repurposing drug candidates for the treatment of diseases caused by pathogenic free-living amoebae, PLoS Negl. Trop. Dis. 14 (2020) 1–21. [CrossRef]
- J. Abraham, N. Chauhan, S. Ray, Virtual Screening of Alkaloid and Terpenoid Inhibitors of SMT Expressed in Naegleria sp., Molecules. (2022).
- J.C. Lamanna, N. Salem, M. Puchowicz, B. Erokwu, S. Koppaka, C. Flask, Z. Lee, Ketones suppress brain glucose consumption, Adv. Exp. Med. Biol. 645 (2009) 301–306. [CrossRef]
- Q. Wang, J. Li, J. Ji, L. Yang, L. Chen, R. Zhou, Y. Yang, H. Zheng, J. Yuan, L. Li, Y. Bi, G.F. Gao, J. Ma, Y. Liu, A case of Naegleria fowleri related primary amoebic meningoencephalitis in China diagnosed by next-generation sequencing, BMC Infect. Dis. 18 (2018) 1–5. [CrossRef]
- F.M. Matschinsky, Glucokinase as glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes, Diabetes. 39 (1990) 647–652. [CrossRef]
- P. Petit, M. Antoine, G. Ferry, J.A. Boutin, A. Lagarde, L. Gluais, R. Vincentelli, L. Vuillard, The active conformation of human glucokinase is not altered by allosteric activators, Acta Crystallogr. Sect. D Biol. Crystallogr. 67 (2011) 929–935. [CrossRef]
- S.B. Green, R.J. Lanier, S.M. Carey, D.R. Morgan, H. Gracz, J. Sherman, A. Rodriguez, E.L. D’Antonio, Synthesis, biochemical, and biological evaluation of C2 linkage derivatives of amino sugars, inhibitors of glucokinase from Trypanosoma cruzi, Bioorganic Med. Chem. Lett. 47 (2021) 128227. [CrossRef]
- J.E. Milanes, J. Suryadi, J. Abendroth, W.C. Van Voorhis, K.F. Barrett, D.M. Dranow, I.Q. Phan, S.L. Patrick, S.D. Rozema, M.M. Khalifa, J.E. Golden, J.C. Morris, Enzymatic and Structural Characterization of the Naegleria fowleri Glucokinase, Antimicrob. Agents Chemother. 63 (2019) 1–16. [CrossRef]
- S.M. Sternisha, B.G. Miller, Molecular and cellular regulation of human glucokinase, Arch. Biochem. Biophys. 663 (2019) 199–213. [CrossRef]
- M. Baginski, J. Czub, Amphotericin B and Its New Derivatives – Mode of Action, Curr. Drug Metab. 10 (2009) 459–469. [CrossRef]
- W.S. Champney, M. Miller, Inhibition of 50S ribosomal subunit assembly in Haemophilus influenzae cells by azithromycin and erythromycin, Curr. Microbiol. 44 (2002) 418–424. [CrossRef]
- M.J. Parnham, V.E. Haber, E.J. Giamarellos-Bourboulis, G. Perletti, G.M. Verleden, R. Vos, Azithromycin: Mechanisms of action and their relevance for clinical applications, Pharmacol. Ther. 143 (2014) 225–245. [CrossRef]
- N.M. Maisch, J.G. Kochupurackal, J. Sin, Azithromycin and the risk of cardiovascular complications, J. Pharm. Pract. 27 (2014) 496–500. [CrossRef]
- F.A. E., A. Sparreboom, R.B. Altman, T.E. Klein, PharmGKB summary: macrolide antibiotic pathway, pharmacokinetics/pharmacodynamics, Pharmacogenet Genomics. 176 (2017) 139–148. [CrossRef]
- R. Laniado-Laborín, M.N. Cabrales-Vargas, Amphotericin B: side effects and toxicity, Rev. Iberoam. Micol. 26 (2009) 223–227. [CrossRef]
- J. Wietzerbin, M. Herve, O. Lebourguais, S. Tran-Dinh, Comparative study of the effects of amphotericin B on the glucose metabolism in Saccharomyces cerevisiae in K+- and Na+-rich media, BBA - Mol. Cell Res. 1136 (1992) 105–112. [CrossRef]
- S. TRAN-DINH, M. HERVÉ, O. LEBOURGUAIS, M. JEROME, J. WIETZERBIN, Effects of amphotericin B on the glucose metabolism in Saccharomyces cerevisiae cells: Studies by 13C-, 1H-NMR and biochemical methods, Eur. J. Biochem. 197 (1991) 271–279. [CrossRef]
- A. Debnath, C.M. Calvet, G. Jennings, W. Zhou, A. Aksenov, M.R. Luth, R. Abagyan, W.D. Nes, J.H. McKerrow, L.M. Podust, CYP51 is an essential drug target for the treatment of primary amoebic meningoencephalitis (PAM), PLoS Negl. Trop. Dis. 11 (2017) 1–24. [CrossRef]
- J.H. Kim, S.Y. Jung, Y.J. Lee, K.J. Song, D. Kwon, K. Kim, S. Park, K. Il Im, H.J. Shin, Effect of therapeutic chemical agents in vitro and on experimental meningoencephalitis due to Naegleria fowleri, Antimicrob. Agents Chemother. 52 (2008) 4010–4016. [CrossRef]
- T.P.C. Dorlo, M. Balasegaram, J.H. Beijnen, P.J. de vries, Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis, J. Antimicrob. Chemother. 67 (2012) 2576–2597. [CrossRef]
- G. Palareti, C. Legnani, B. Cosmi, E. Antonucci, N. Erba, D. Poli, S. Testa, A. Tosetto, A systematic review of fluconazole resistance in clinical isolates of Cryptococcus species, Int. J. Lab. Hematol. 38 (2016) 42–49. [CrossRef]
- Pfizer Inc. Diflucan (fluconazole) U.S. Food and Drug Administration website., (2019). https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/019949s065,020090s047lbl.pdf.
- J.H. Van Tyle, Ketoconazole: Mechanism of action, spectrum of activity, pharmacokinetics, drug interactions, adverse reactions and therapeutic use, Pharmacotherapy. 4 (1984) 343–373. [CrossRef]
- A.K. Gupta, D.C.A. Lyons, The rise and fall of oral ketoconazole, J. Cutan. Med. Surg. 19 (2015) 352–357. [CrossRef]
- M.K. Piya, A.A. Tahrani, A.H. Barnett, Emerging treatment options for type 2 diabetes, Br. J. Clin. Pharmacol. 70 (2010) 631–644. [CrossRef]
- J. Grosset, S. Leventis, Adverse effects of rifampin, Rev. Infect. Dis. 5 (1983) S440–S446. [CrossRef]
- J. Baysarowich, K. Koteva, D.W. Hughes, L. Ejim, E. Griffiths, K. Zhang, M. Junop, G.D. Wright, Rifamycin antibiotic resistance by ADP-ribosylation: Structure and diversity of Arr, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 4886–4891. [CrossRef]
- T. Umeda, R. Uekado, K. Shigemori, H. Eguchi, T. Tomiyama, Nasal Rifampicin Halts the Progression of Tauopathy by Inhibiting Tau Oligomer Propagation in Alzheimer Brain Extract-Injected Mice, Biomedicines. 10 (2022). [CrossRef]
- S.K. Cho, J.S. Yoon, M.G. Lee, D.H. Lee, L.A. Lim, K. Park, M.S. Park, J.Y. Chung, Rifampin enhances the glucose-lowering effect of metformin and increases OCT1 mRNA levels in healthy participants, Clin. Pharmacol. Ther. 89 (2011) 416–421. [CrossRef]
- T. Lammers, A.M. Sofias, R. van der Meel, R. Schiffelers, G. Storm, F. Tacke, S. Koschmieder, T.H. Brümmendorf, F. Kiessling, J.M. Metselaar, Dexamethasone nanomedicines for COVID-19, Nat. Nanotechnol. 15 (2020) 618–621. [CrossRef]
- M. Ciriaco, P. Ventrice, G. Russo, M. Scicchitano, G. Mazzitello, F. Scicchitano, E. Russo, Corticosteroid-related central nervous system side effects, J. Pharmacol. Pharmacother. 4 (2013). [CrossRef]
- D. Abdelmannan, R. Tahboub, S. Genuth, F. Ismail-Beigi, Effect of dexamethasone on oral glucose tolerance in healthy adults, Endocr. Pract. 16 (2010) 770–777. [CrossRef]
- L. Izquierdo, F. Helle, C. François, S. Castelain, G. Duverlie, E. Brochot, Simeprevir for the treatment of hepatitis C virus infection, Pharmgenomics. Pers. Med. 7 (2014) 241–249. [CrossRef]
- Olysio (Simeprevir).”Prescribing information.”, (2013). http://www.accessdata.fda.gov/drugsatfda_docs/ label/2013/205123s001lbl.pdf.
- H.R.A. El-Mageed, D.A. Abdelrheem, S.A. Ahmed, A.A. Rahman, K.N.M. Elsayed, S.A. Ahmed, A.A. EL-Bassuony, H.S. Mohamed, Combination and tricombination therapy to destabilize the structural integrity of COVID-19 by some bioactive compounds with antiviral drugs: insights from molecular docking study, Struct. Chem. 32 (2021) 1415–1430. [CrossRef]
- D. Bhowmik, R. Nandi, R. Jagadeesan, N. Kumar, A. Prakash, D. Kumar, Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches, Infect. Genet. Evol. 84 (2020) 104451. [CrossRef]
- N.S.K. Lam, X.X. Long, X. Li, M. Saad, F. Lim, J.C. Doery, R.C. Griffin, C. Galletly, The potential use of folate and its derivatives in treating psychiatric disorders: A systematic review, Biomed. Pharmacother. 146 (2022) 112541. [CrossRef]
- R.C. Shelton, J. Sloan Manning, L.W. Barrentine, E. V. Tipa, Assessing effects of L-methylfolate in depression management: Results of a real-world patient experience trial, Prim. Care Companion J. Clin. Psychiatry. 15 (2013) 1–6. [CrossRef]
- J. Jiang, L. Li, H. Yin, R. Woessner, C. Emotte, R. Li, S. Khindri, H. Pei, Single- and multiple-dose pharmacokinetics of inhaled indacaterol in healthy Chinese volunteers, Eur. J. Drug Metab. Pharmacokinet. 40 (2015) 203–208. [CrossRef]
- M.L. Martel, B.E. Driver, J.R. Miner, M.H. Biros, J.B. Cole, Randomized Double-blind Trial of Intramuscular Droperidol, Ziprasidone, and Lorazepam for Acute Undifferentiated Agitation in the Emergency Department, Acad. Emerg. Med. 28 (2021) 421–434. [CrossRef]
- A. Davis, Ziprasidone, XPharm Compr. Pharmacol. Ref. 20 (2007) 1–4. [CrossRef]
- M.U. Yood, G. DeLorenze, C.P. Quesenberry, S.A. Oliveria, A.-L. Tsai, V.J. Willey, R. McQuade, J. Newcomer, G. L’Italein, The incidence of diabetes in atypical antipsychotic users differs according to agent—results from a multisite epidemiologic study, Pharmacoepidemiol. Drug Saf. 16 (2007) 228–228. [CrossRef]
- D.K. Arulmozhi, D.S. Dwyer, S.L. Bodhankar, Antipsychotic induced metabolic abnormalities: An interaction study with various PPAR modulators in mice, Life Sci. 79 (2006) 1865–1872. [CrossRef]
- D.E. Johnson, H. Yamazaki, K.M. Ward, A.W. Schmidt, W.S. Lebel, J.L. Treadway, E.M. Gibbs, W.S. Zawalich, H. Rollema, Inhibitory effects of antipsychotics on carbachol-enhanced insulin secretion from perifused rat islets: Role of muscarinic antagonism in antipsychotic-induced diabetes and hyperglycemia, Diabetes. 54 (2005) 1552–1558. [CrossRef]
- A. SPIVAK, S.S. ALAMY, L.F. JARSKOG, B.B. SHEITMAN, J.A. LIEBERMAN, Ziprasidone Alternative for Olanzapine- Induced Hyperglycemia, Am. J. Psychiatry. 159 (2002) 1606. [CrossRef]
- S. Park, E.A. Cho, J.N. Chun, D.Y. Lee, S. Lee, M.Y. Kim, S.M. Bae, S.I. Jo, S.H. Lee, H.H. Park, T.M. Kim, I. So, S.Y. Kim, J.H. Jeon, Crizotinib attenuates cancer metastasis by inhibiting TGFβ signaling in non-small cell lung cancer cells, Exp. Mol. Med. (2022). [CrossRef]
- M.J. Ahn, H.R. Kim, J.C.H. Yang, J.Y. Han, J.Y.C. Li, M.J. Hochmair, G.C. Chang, A. Delmonte, K.H. Lee, R.G. Campelo, C. Gridelli, A.I. Spira, R. Califano, F. Griesinger, S. Ghosh, E. Felip, D.W. Kim, Y. Liu, P. Zhang, S. Popat, D.R. Camidge, Efficacy and Safety of Brigatinib Compared With Crizotinib in Asian vs. Non-Asian Patients With Locally Advanced or Metastatic ALK–Inhibitor-Naive ALK+ Non–Small Cell Lung Cancer: Final Results From the Phase III ALTA-1L Study, Clin. Lung Cancer. (2022) 1–11. [CrossRef]
- K. Hadova, L. Mesarosova, E. Kralova, G. Doka, P. Krenek, J. Klimas, The tyrosine kinase inhibitor crizotinib influences blood glucose and mrna expression of glut4 and ppars in the heart of rats with experimental diabetes, Can. J. Physiol. Pharmacol. 99 (2021) 637–645. [CrossRef]
- H.K. Çakir, O. Eroglu, In vitro anti-proliferative effect of capecitabine (Xeloda) combined with mocetinostat (MGCD0103) in 4T1 breast cancer cell line by immunoblotting, Iran. J. Basic Med. Sci. 24 (2021) 1515–1522. [CrossRef]
- P. Grivas, A. Mortazavi, J. Picus, N.M. Hahn, M.I. Milowsky, L.L. Hart, A. Alva, J. Bellmunt, S.K. Pal, R.M. Bambury, P.H. O’Donnell, S. Gupta, E.A. Guancial, G.P. Sonpavde, D. Faltaos, D. Potvin, J.G. Christensen, R.C. Chao, J.E. Rosenberg, Mocetinostat for patients with previously treated, locally advanced/metastatic urothelial carcinoma and inactivating alterations of acetyltransferase genes, Cancer. 125 (2019) 533–540. [CrossRef]
- J. Liu, J.N. Li, H. Wu, P. Liu, The Status and Prospects of Epigenetics in the Treatment of Lymphoma, Front. Oncol. 12 (2022) 1–23. [CrossRef]
- FDA, RISPERDAL (Risperidone), Interactions. 50 (1998) 1–25.
- G.R. Chang, P.H. Hou, W.C. Yang, C.M. Wang, P.S. Fan, H.J. Liao, T.P. Chen, Risperidone Exacerbates Glucose Intolerance, Nonalcoholic Fatty Liver Disease, and Renal Impairment in Obese Mice, Int. J. Mol. Sci. (2021). [CrossRef]
- K. Omori, A. Nakamura, H. Miyoshi, Y. Yamauchi, S. Kawata, K. Takahashi, N. Kitao, H. Nomoto, H. Kameda, K.Y. Cho, Y. Terauchi, T. Atsumi, Glucokinase Inactivation Paradoxically Ameliorates Glucose Intolerance by Increasing β-Cell Mass in db/db Mice, Diabetes. 70 (2021) 917–931. [CrossRef]
- M. Prentki, F.M. Matschinsky, S.R.M. Madiraju, Metabolic signaling in fuel-induced insulin secretion, Cell Metab. 18 (2013) 162–185. [CrossRef]
- P. Rorsman, F.M. Ashcroft, Pancreatic β-cell electrical activity and insulin secretion: Of mice and men, Physiol. Rev. 98 (2018) 117–214. [CrossRef]
- F.M. Matschinsky, Regulation of Pancreatic B-Cell Glucokinase, 51 (2002).
- F. Matschinsky, Y. Liang, P. Kesavan, L. Wang, P. Froguel, G. Velho, D. Cohen, Glucokinase as Pancreatic ft Cell Glucose Sensor and Diabetes Gene, J. Clin. Invest. 92 (1993).
- H. Wang, P.B. Iynedjian, Modulation of glucose responsiveness of insulinoma β-cells by graded overexpression of glucokinase, Proc. Natl. Acad. Sci. U. S. A. 94 (1997) 4372–4377. [CrossRef]
- H.K. Berman, C.B. Newgard, Fundamental Metabolic Differences between Hepatocytes and Islet -cells Revealed by Glucokinase Overexpression, 2960 (1998) 4543–4552.
- S. Tornovsky-Babeay, D. Dadon, O. Ziv, E. Tzipilevich, T. Kadosh, R. Schyr-Ben Haroush, A. Hija, M. Stolovich-Rain, J. Furth-Lavi, Z. Granot, S. Porat, L.H. Philipson, K.C. Herold, T.R. Bhatti, C. Stanley, F.M. Ashcroft, P. In’T Veld, A. Saada, M.A. Magnuson, B. Glaser, Y. Dor, Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in β cells, Cell Metab. 19 (2014) 109–121. [CrossRef]
- Å. Segerstolpe, A. Palasantza, P. Eliasson, E.M. Andersson, A.C. Andréasson, X. Sun, S. Picelli, A. Sabirsh, M. Clausen, M.K. Bjursell, D.M. Smith, M. Kasper, C. Ämmälä, R. Sandberg, Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes, Cell Metab. 24 (2016) 593–607. [CrossRef]
- A. Haythorne, M. Rohm, M. van de Bunt, M.F. Brereton, A.I. Tarasov, T.S. Blacker, G. Sachse, M. Silva dos Santos, R. Terron Exposito, S. Davis, O. Baba, R. Fischer, M.R. Duchen, P. Rorsman, J.I. MacRae, F.M. Ashcroft, Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells, Nat. Commun. 10 (2019). [CrossRef]
- J. Hou, Z. Li, W. Zhong, Q. Hao, L. Lei, L. Wang, D. Zhao, P. Xu, Y. Zhou, Y. Wang, T. Xu, Temporal transcriptomic and proteomic landscapes of deteriorating pancreatic islets in type 2 diabetic rats, Diabetes. 66 (2017) 2188–2200. [CrossRef]
- M.F. Brereton, M. Rohm, K. Shimomura, C. Holland, S. Tornovsky-Babeay, D. Dadon, M. Iberl, M. V. Chibalina, S. Lee, B. Glaser, Y. Dor, P. Rorsman, A. Clark, F.M. Ashcroft, Hyperglycaemia induces metabolic dysfunction and glycogen accumulation in pancreatic β-cells, Nat. Commun. 7 (2016) 1–15. [CrossRef]
- F.M. Matschinsky, GKAs for diabetes therapy: Why no clinically useful drug after two decades of trying?, Trends Pharmacol. Sci. 34 (2013) 90–99. [CrossRef]
- R.C. Bonadonna, T. Heise, C. Arbet-Engels, C. Kapitza, A. Avogaro, J. Grimsby, J. Zhi, J.F. Grippo, R. Balena, Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: A mechanistic study, J. Clin. Endocrinol. Metab. 95 (2010) 5028–5036. [CrossRef]
- J. Grimsby, R. Sarabu, W.L. Corbett, N.E. Haynes, F.T. Bizzarro, J.W. Coffey, K.R. Guertin, D.W. Hilliard, R.F. Kester, P.E. Mahaney, L. Marcus, L. Qi, C.L. Spence, J. Tengi, M.A. Magnuson, C.A. Chu, M.T. Dvorozniak, F.M. Matschinsky, J.F. Grippo, Allosteric activators of glucokinase: Potential role in diabetes therapy, Science (80-. ). 301 (2003) 370–373. [CrossRef]
- A. Nakamura, H. Shimazaki, S. Ohyama, J. Eiki, Y. Terauchi, Effect of long-term treatment with a small-molecule glucokinase activator on glucose metabolism, lipid profiles and hepatic function, J. Diabetes Investig. 2 (2011) 276–279. [CrossRef]
- M. Futamura, J. Yao, X. Li, R. Bergeron, J.L. Tran, E. Zycband, J. Woods, Y. Zhu, Q. Shao, H. Maruki-Uchida, H. Goto-Shimazaki, R.B. Langdon, M.D. Erion, J. Eiki, Y.P. Zhou, Chronic treatment with a glucokinase activator delays the onset of hyperglycaemia and preserves beta cell mass in the Zucker diabetic fatty rat, Diabetologia. 55 (2012) 1071–1080. [CrossRef]
- R.M. O’Doherty, D.L. Lehman, S. Télémaque-Potts, C.B. Newgard, Metabolic impact of glucokinase overexpression in liver: Lowering of blood glucose in fed rats is accompanied by hyperlipidemia, Diabetes. 48 (1999) 2022–2027. [CrossRef]
- S. Kawata, A. Nakamura, H. Miyoshi, K. Yang, I. Shigesawa, Y. Yamauchi, K. Tsuchida, K. Omori, K. Takahashi, H. Nomoto, H. Kameda, K.Y. Cho, Y. Terauchi, T. Atsumi, Glucokinase activation leads to an unsustained hypoglycaemic effect with hepatic triglyceride accumulation in db/db mice., Diabetes. Obes. Metab. 24 (2022) 391–401. [CrossRef]
- G.E. Meininger, R. Scott, M. Alba, Y. Shentu, E. Luo, H. Amin, M.J. Davies, K.D. Kaufman, B.J. Goldstein, Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes, Diabetes Care. 34 (2011) 2560–2566. [CrossRef]
- A. Nakamura, K. Omori, Y. Terauchi, Glucokinase activation or inactivation: Which will lead to the treatment of type 2 diabetes?, Diabetes, Obes. Metab. 23 (2021) 2199–2206. [CrossRef]
- N.B. Whitticar, C.S. Nunemaker, Reducing Glucokinase Activity to Enhance Insulin Secretion: A Counterintuitive Theory to Preserve Cellular Function and Glucose Homeostasis, Front. Endocrinol. (Lausanne). 11 (2020) 1–10. [CrossRef]
- M.G. Rees, A.L. Gloyn, Small molecular glucokinase activators: Has another new anti-diabetic therapeutic lost favour?, Br. J. Pharmacol. 168 (2013) 335–338. [CrossRef]
- B. Brock, J.H. Mogensen, S. Gregersen, K. Hermansen, Glucose desensitization in INS-1 cells: Evidence of impaired function caused by glucose metabolite(s) rather than by the glucose molecule per se, Metabolism. 51 (2002) 671–677. [CrossRef]
- K.A. Toulis, K. Nirantharakumar, C. Pourzitaki, A.H. Barnett, A.A. Tahrani, Glucokinase Activators for Type 2 Diabetes: Challenges and Future Developments, Drugs. 80 (2020) 467–475. [CrossRef]
- E.F. Pettersen, T.D. Goddard, C.C. Huang, G.S. Couch, D.M. Greenblatt, E.C. Meng, T.E. Ferrin, UCSF Chimera - A visualization system for exploratory research and analysis, J. Comput. Chem. 25 (2004) 1605–1612. [CrossRef]
- S. Dallakyan, A.J. Olson, Small-molecule library screening by docking with PyRx, Methods Mol. Biol. 1263 (2015) 243–250. [CrossRef]
Figure 1.
(A) Juveniles and young adults involved in water activities in Naegleria-infected locations. Water sports/activities that include chances of water gushing into the nasal canal can cause infection when Naegleria is present. (B) Spores and flagellates of Naegleria travel through the nasal cavity when water enters through it. Spores can travel without water gushing into the nasal cavity, while the flagellates infect through contact with water. (C) Naegleria enters the blood vessel fighting off any immune response and crosses into the blood-brain barrier. The majority of drugs cannot cross the blood-brain barrier and thus are ineffective in preventing the progression of infection. (D) Naegleria trophozoites feed on the neurons of the host. Trophozoites feed on neurons by releasing enzymes causing severe inflammation and damage to the central nervous system. Drugs used for treatment also have severe side effects. Effective drugs that specifically target only the pathogen are required.
Figure 1.
(A) Juveniles and young adults involved in water activities in Naegleria-infected locations. Water sports/activities that include chances of water gushing into the nasal canal can cause infection when Naegleria is present. (B) Spores and flagellates of Naegleria travel through the nasal cavity when water enters through it. Spores can travel without water gushing into the nasal cavity, while the flagellates infect through contact with water. (C) Naegleria enters the blood vessel fighting off any immune response and crosses into the blood-brain barrier. The majority of drugs cannot cross the blood-brain barrier and thus are ineffective in preventing the progression of infection. (D) Naegleria trophozoites feed on the neurons of the host. Trophozoites feed on neurons by releasing enzymes causing severe inflammation and damage to the central nervous system. Drugs used for treatment also have severe side effects. Effective drugs that specifically target only the pathogen are required.
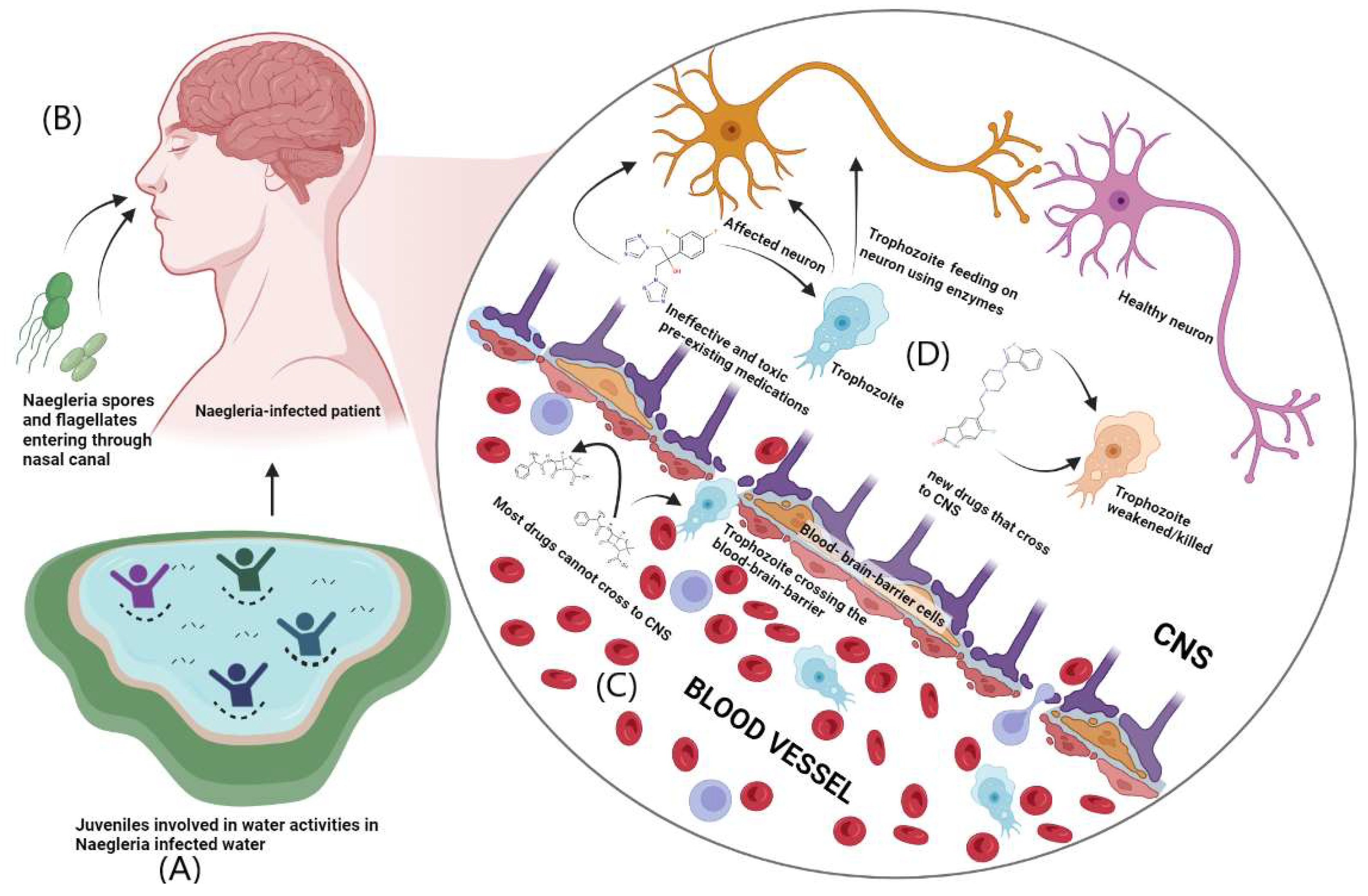
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



