
Submitted:
07 October 2024
Posted:
09 October 2024
You are already at the latest version
Abstract
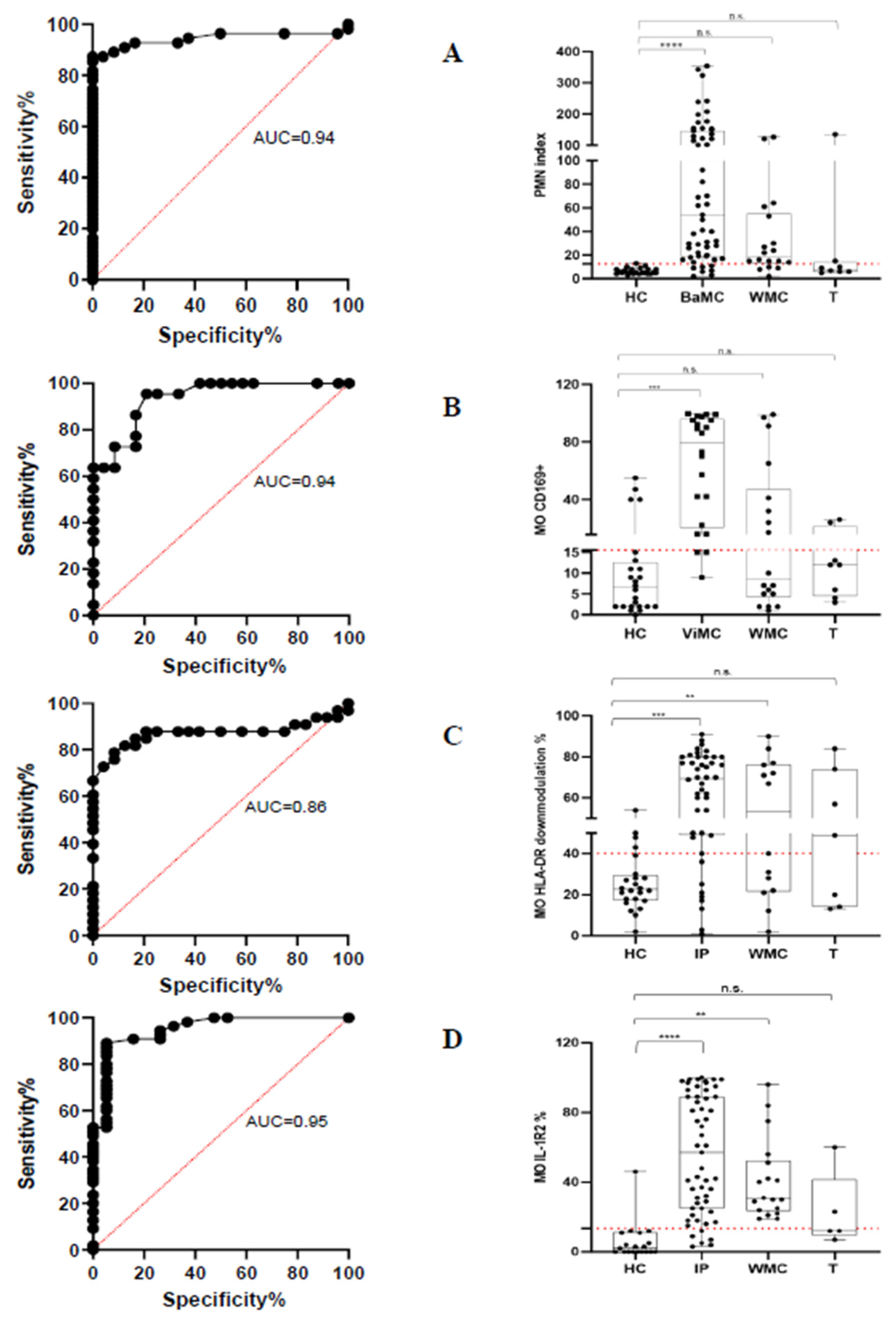
We developed a flow cytometry (FC) assay enabling rapid and accurate identification of bacterial and viral infections using whole blood samples. The streamlined flow cytometry assay is designed to be user-friendly, making it accessible even for operators with limited experience in FC techniques. The key components of the assay focus on the expression levels of specific surface markers—CD64 on polymorphonuclear neutrophils (PMN) as a marker for bacterial infection, and CD169 on monocytes (MO) for viral infection. The strong performance indicated by an area under the receiver operating characteristic (ROC) curve of 0.94 for both PMN CD64 positive predictive value (PPV), 97.96%; negative predictive value (NPV) 76.67% and MO CD169 PPV 82.6% and NPV 86.9% highlights the assay's robustness in differentiating between bacterial and viral infections accurately. The FC assay includes the assessment of immune system status through HLA-DR and IL-1R2 modulation in MO providing useful insight into the patients' immune response. The significant increase in the frequency of MO exhibiting reduced HLA-DR expression and elevated IL-1R2 levels in infected patients (compared to healthy controls) underscores the potential of these markers as indicators of infection severity. Although the overall correlation between HLA-DR and IL-1R2 expression levels was not significant across all patients, there was a trend in patients with more severe disease suggesting that these markers may have the potential to assist in stratifying patient risk. The present FC assay has the potential to become routine in the clinical microbiology laboratory community and to be helpful in guiding clinical decision-making.
Keywords:
1. Introduction
2. Results
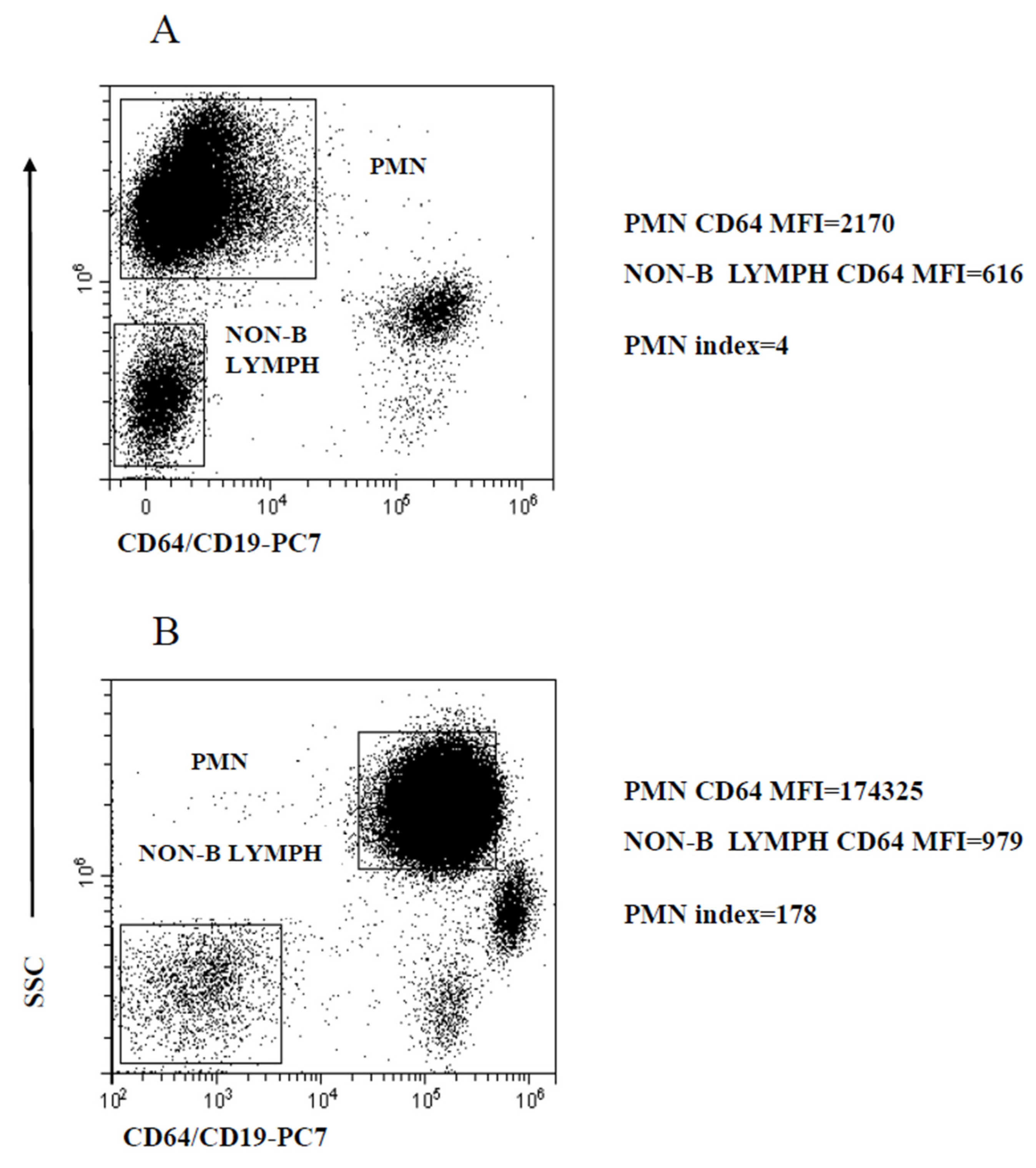
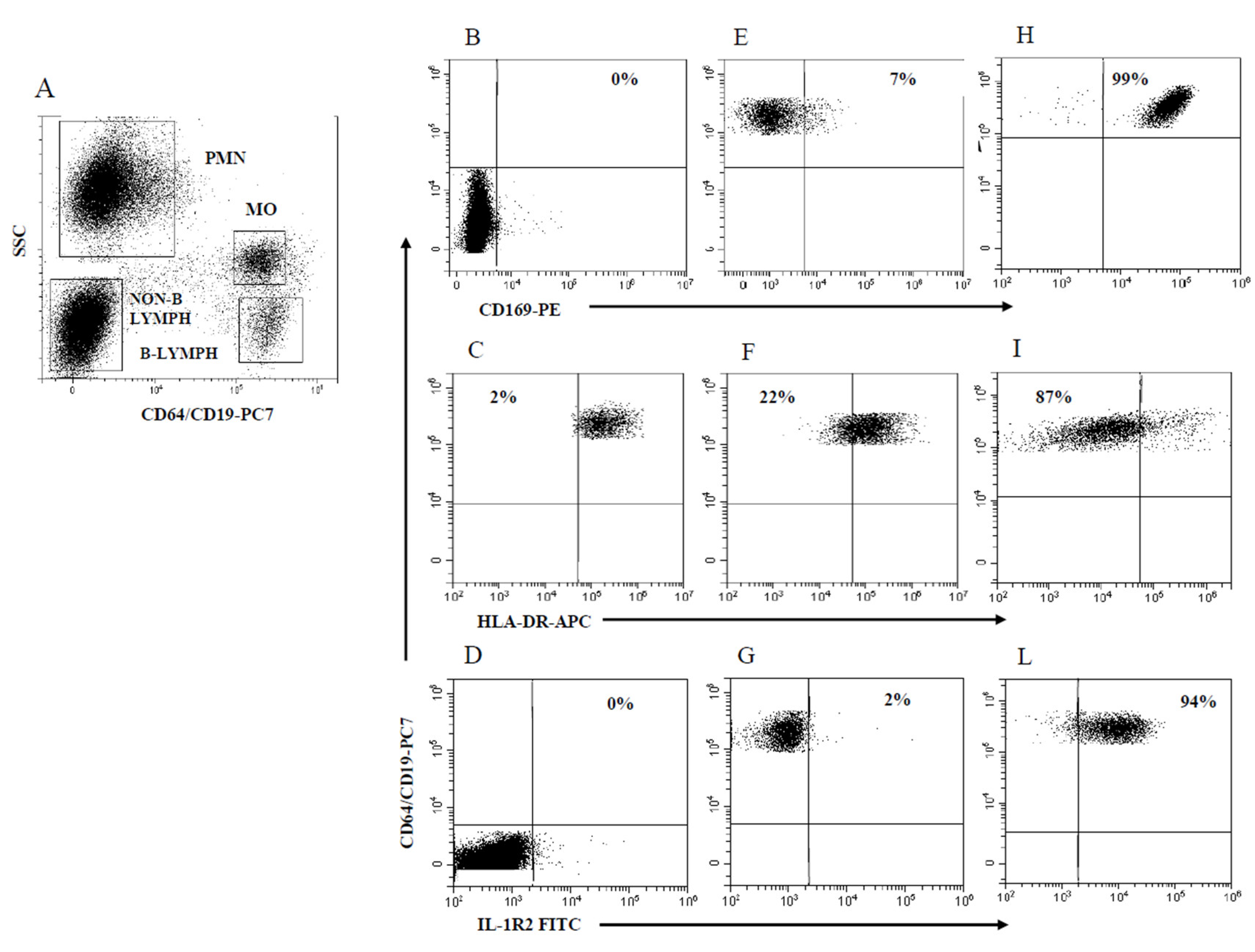
2.1. Gating Strategy and Assessment of PMN and MO Marker Modulation
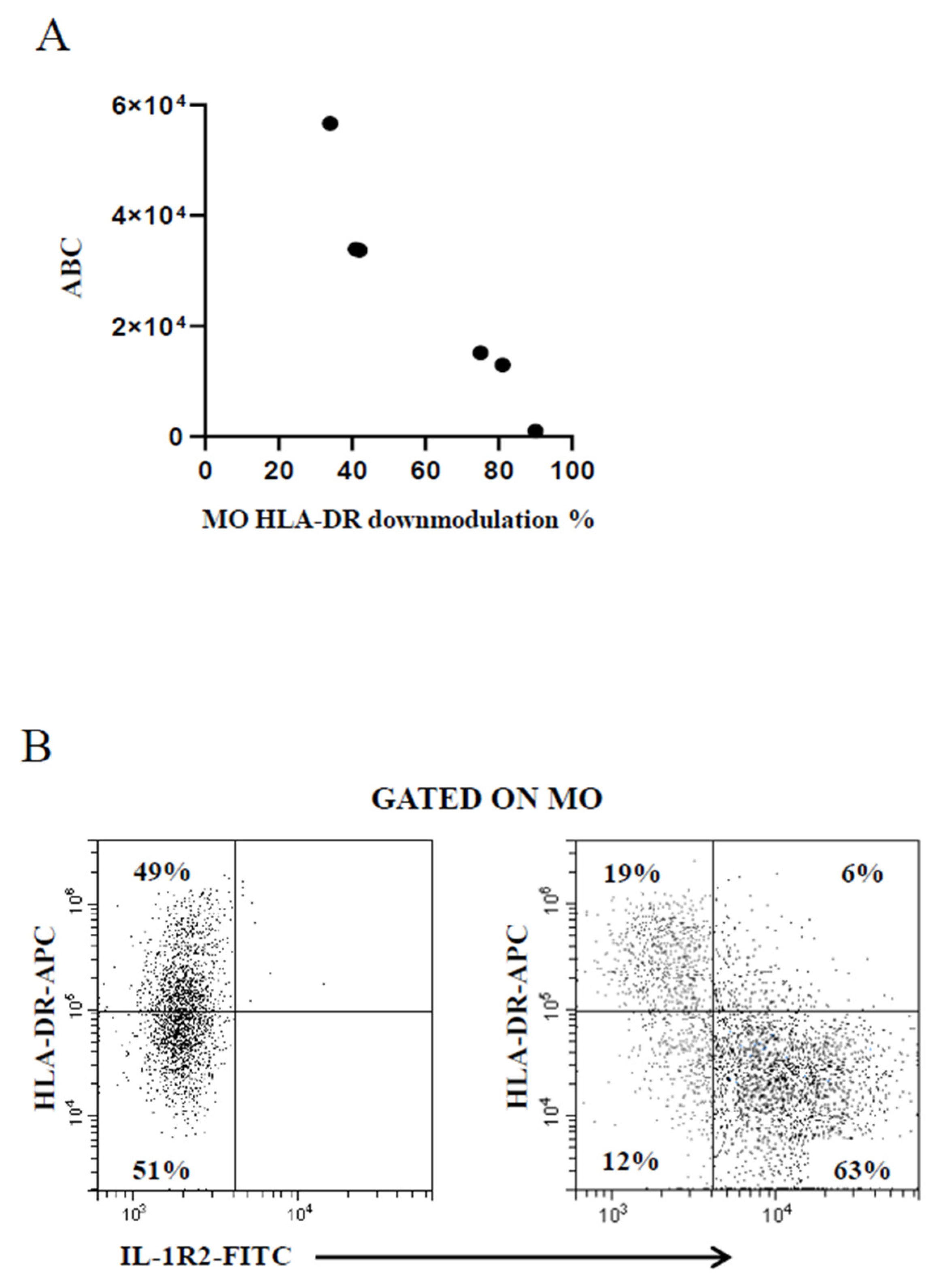
2.2. Modulation of PMN Index and MO Markers
3. Discussion
4. Materials and Methods
4.1. Patient Accrual and Ethics Statement
4.2. Study Population
4.3. Sample Collection
4.4. Monoclonal Antibodies and Staining Procedure
4.5. Statistics
Supplementary Materials
Funding
Institutional Review Board Statement
References
- Baron, E. J.; Miller, J. M.; Weinstein, M. P.; Richter, S. S.; Gilligan, P. H.; Thomson, R. B.; Bourbeau, P.; Carroll, K. C.; Kehl, S. C.; Dunne, W. M.; Robinson-Dunn, B.; Schwartzman, J. D.; Chapin, K. C.; Snyder, J. W.; Forbes, B. A.; Patel, R.; Rosenblatt, J. E.; Pritt, B. S. A Guide to Utilization of the Microbiology Laboratory for Diagnosis of Infectious Diseases: 2013 Recommendations by the Infectious Diseases Society of America (IDSA) and the American Society for Microbiology (ASM)(a). Clin Infect Dis 2013, 57 (4). https://doi.org/10.1093/CID/CIT278.
- Faix, J. D. Biomarkers of Sepsis. Crit Rev Clin Lab Sci 2013, 50 (1), 23. https://doi.org/10.3109/10408363.2013.764490.
- Woodhead, M.; Blasi, F.; Ewig, S.; Garau, J.; Huchon, G.; Ieven, M.; Ortqvist, A.; Schaberg, T.; Torres, A.; van der Heijden, G.; Read, R.; Verheij, T. J. M. Guidelines for the Management of Adult Lower Respiratory Tract Infections--Full Version. Clin Microbiol Infect 2011, 17 Suppl 6 (Suppl 6), E1–E59. https://doi.org/10.1111/J.1469-0691.2011.03672.X.
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A. K. M.; Wertheim, H. F. L.; Sumpradit, N.; Vlieghe, E.; Hara, G. L.; Gould, I. M.; Goossens, H.; Greko, C.; So, A. D.; Bigdeli, M.; Tomson, G.; Woodhouse, W.; Ombaka, E.; Peralta, A. Q.; Qamar, F. N.; Mir, F.; Kariuki, S.; Bhutta, Z. A.; Coates, A.; Bergstrom, R.; Wright, G. D.; Brown, E. D.; Cars, O. Antibiotic Resistance-the Need for Global Solutions. Lancet Infect Dis 2013, 13 (12), 1057–1098. https://doi.org/10.1016/S1473-3099(13)70318-9.
- Machen, A.; Drake, T.; Wang, Y. F. Same Day Identification and Full Panel Antimicrobial Susceptibility Testing of Bacteria from Positive Blood Culture Bottles Made Possible by a Combined Lysis-Filtration Method with MALDI-TOF VITEK Mass Spectrometry and the VITEK2 System. PLoS One 2014, 9 (2). https://doi.org/10.1371/JOURNAL.PONE.0087870.
- Connell, T. G.; Rele, M.; Cowley, D.; Buttery, J. P.; Curtis, N. How Reliable Is a Negative Blood Culture Result? Volume of Blood Submitted for Culture in Routine Practice in a Children’s Hospital. Pediatrics 2007, 119 (5), 891–896. https://doi.org/10.1542/PEDS.2006-0440.
- Rhedin, S.; Lindstrand, A.; Rotzeń-Östlund, M.; Tolfvenstam, T.; Öhrmalm, L.; Rinder, M. R.; Zweygberg-Wirgart, B.; Ortqvist, A.; Henriques-Normark, B.; Broliden, K.; Naucler, P. Clinical Utility of PCR for Common Viruses in Acute Respiratory Illness. Pediatrics 2014, 133 (3). https://doi.org/10.1542/PEDS.2013-3042. 8. Murphy, K.; Weaver, C.; Janeway's Immunobiology, 9th ed.; Garland Science: New York City, United States, 2016.
- Nimmerjahn, F.; Ravetch, J. V. Fcgamma Receptors: Old Friends and New Family Members. Immunity 2006, 24 (1), 19–28. https://doi.org/10.1016/J.IMMUNI.2005.11.010.
- Hoffmeyer, F.; Witte, K.; Schmidt, R. E. The High-Affinity Fc Gamma RI on PMN: Regulation of Expression and Signal Transduction. Immunology 1997, 92 (4), 544. https://doi.org/10.1046/J.1365-2567.1997.00381.X.
- Wagner, C.; Deppisch, R.; Denefleh, B.; Hug, F.; Andrassy, K.; Hänsch, G. M. Expression Patterns of the Lipopolysaccharide Receptor CD14, and the FC gamma Receptors CD16 and CD64 on Polymorphonuclear Neutrophils: Data from Patients with Severe Bacterial Infections and Lipopolysaccharide-Exposed Cells. Shock 2003, 19 (1), 5–12. https://doi.org/10.1097/00024382-200301000-00002.
- Repp, R.; Valerius, T.; Sendler, A.; Gramatzki, M.; Iro, H.; Kalden, J. R.; Platzer, E. Neutrophils Express the High Affinity Receptor for IgG (Fc Gamma RI, CD64) after in Vivo Application of Recombinant Human Granulocyte Colony-Stimulating Factor. Blood 1991, 78 (4), 885–889. https://doi.org/10.1182/BLOOD.V78.4.885.BLOODJOURNAL784885.
- Martijn Kerst, B. J.; de Haas, M.; Ellen van der Schoot, C.; Slaper-Cortenbach, lneke C.; Kleijer, M.; EGKr von dem Borne, A.; van Oers, R. H. Recombinant Granulocyte Colony-Stimulating Factor Administration to Healthy Volunteers: Induction of Immunophenotypically and Functionally Altered Neutrophils via an Effect on Myeloid Progenitor Cells. Blood 1993, 82 (11), 3265–3272. https://doi.org/10.1182/BLOOD.V82.11.3265.3265.
- Uchil, P. D.; Pi, R.; Haugh, K. A.; Ladinsky, M. S.; Ventura, J. D.; Barrett, B. S.; Santiago, M. L.; Bjorkman, P. J.; Kassiotis, G.; Sewald, X.; Mothes, W. A. Protective Role for the Lectin CD169/Siglec-1 against a Pathogenic Murine Retrovirus. Cell Host Microbe 2019, 25 (1), 87-100.e10. https://doi.org/10.1016/J.CHOM.2018.11.011.
- Shinde, P. V.; Xu, H. C.; Maney, S. K.; Kloetgen, A.; Namineni, S.; Zhuang, Y.; Honke, N.; Shaabani, N.; Bellora, N.; Doerrenberg, M.; Trilling, M.; Pozdeev, V. I.; van Rooijen, N.; Scheu, S.; Pfeffer, K.; Crocker, P. R.; Tanaka, M.; Duggimpudi, S.; Knolle, P.; Heikenwalder, M.; Ruland, J.; Mak, T. W.; Brenner, D.; Pandyra, A. A.; Hoell, J. I.; Borkhardt, A.; Häussinger, D.; Lang, K. S.; Lang, P. A. Tumor Necrosis Factor-Mediated Survival of CD169+ Cells Promotes Immune Activation during Vesicular Stomatitis Virus Infection. J Virol 2018, 92 (3). https://doi.org/10.1128/JVI.01637-17.
- Kim, W. K.; McGary, C. M.; Holder, G. E.; Filipowicz, A. R.; Kim, M. M.; Beydoun, H. A.; Cai, Y.; Liu, X.; Sugimoto, C.; Kuroda, M. J. Increased Expression of CD169 on Blood Monocytes and Its Regulation by Virus and CD8 T Cells in Macaque Models of HIV Infection and AIDS. AIDS Res Hum Retroviruses 2015, 31 (7), 696–706. https://doi.org/10.1089/AID.2015.0003.
- van der Kuyl, A. C.; van den Burg, R.; Zorgdrager, F.; Groot, F.; Berkhout, B.; Cornelissen, M. Sialoadhesin (CD169) Expression in CD14+ Cells Is Upregulated Early after HIV-1 Infection and Increases during Disease Progression. PLoS One 2007, 2 (2). https://doi.org/10.1371/JOURNAL.PONE.0000257.
- Michlmayr, D.; Kim, E. Y.; Rahman, A. H.; Raghunathan, R.; Kim-Schulze, S.; Che, Y.; Kalayci, S.; Gümüş, Z. H.; Kuan, G.; Balmaseda, A.; Kasarskis, A.; Wolinsky, S. M.; Suaréz-Fariñas, M.; Harris, E. Comprehensive Immunoprofiling of Pediatric Zika Reveals Key Role for Monocytes in the Acute Phase and No Effect of Prior Dengue Virus Infection. Cell Rep 2020, 31 (4). https://doi.org/10.1016/J.CELREP.2020.107569.
- Bedin, A. S.; Makinson, A.; Picot, M. C.; Mennechet, F.; Malergue, F.; Pisoni, A.; Nyiramigisha, E.; Montagnier, L.; Bollore, K.; Debiesse, S.; Morquin, D.; Veyrenche, N.; Renault, C.; Foulongne, V.; Bret, C.; Bourdin, A.; Le Moing, V.; Van De Perre, P.; Tuaillon, E. Monocyte CD169 Expression as a Biomarker in the Early Diagnosis of COVID-19. J Infect Dis 2021, 223 (4), 562–567. https://doi.org/10.1093/INFDIS/JIAA724.
- Minutolo, A.; Petrone, V.; Fanelli, M.; Iannetta, M.; Giudice, M.; Belkacem, I. A.; Zordan, M.; Vitale, P.; Rasi, G.; Sinibaldi-vallebona, P.; Sarmati, L.; Andreoni, M.; Malergue, F.; Balestrieri, E.; Grelli, S.; Matteucci, C. High CD169 Monocyte/Lymphocyte Ratio Reflects Immunophenotype Disruption and Oxygen Need in COVID-19 Patients. Pathogens 2021, 10 (12). https://doi.org/10.3390/PATHOGENS10121639/S1.
- Comins-Boo, A.; Gutiérrez-Larrañaga, M.; Roa-Bautista, A.; Guiral Foz, S.; Renuncio García, M.; González López, E.; Irure Ventura, J.; Fariñas-Álvarez, M. C.; San Segundo, D.; López Hoyos, M. Validation of a Quick Flow Cytometry-Based Assay for Acute Infection Based on CD64 and CD169 Expression. New Tools for Early Diagnosis in COVID-19 Pandemic. Front Med (Lausanne) 2021, 8. https://doi.org/10.3389/FMED.2021.655785.
- Gatti, A.; Fassini, P.; Mazzone, A.; Rusconi, S.; Brando, B.; Mistraletti, G. Kinetics of CD169, HLA-DR, and CD64 Expression as Predictive Biomarkers of SARS-CoV2 Outcome. Journal of Anesthesia, Analgesia and Critical Care 2023, 3 (1). https://doi.org/10.1186/S44158-023-00090-X.
- Park J, Dean LS, Jiyarom B, Gangcuangco LM, Shah P, Awamura T, Ching LL, Nerurkar VR, Chow DC, Igno F, Shikuma CM, Devendra G. Elevated circulating monocytes and monocyte activation in COVID-19 convalescent individuals. Front Immunol. 2023 Apr 3;14:1151780. doi: 10.3389/fimmu.2023.1151780. PMID: 37077911; PMCID: PMC10106598.
- Affandi, A. J.; Olesek, K.; Grabowska, J.; Nijen Twilhaar, M. K.; Rodríguez, E.; Saris, A.; Zwart, E. S.; Nossent, E. J.; Kalay, H.; de Kok, M.; Kazemier, G.; Stöckl, J.; van den Eertwegh, A. J. M.; de Gruijl, T. D.; Garcia-Vallejo, J. J.; Storm, G.; van Kooyk, Y.; den Haan, J. M. M. CD169 Defines Activated CD14+ Monocytes With Enhanced CD8+ T Cell Activation Capacity. Front Immunol 2021, 12. https://doi.org/10.3389/FIMMU.2021.697840.
- Bourgoin, P.; Biéchelé, G.; Ait Belkacem, I.; Morange, P. E.; Malergue, F. Role of the Interferons in CD64 and CD169 Expressions in Whole Blood: Relevance in the Balance between Viral- or Bacterial-oriented Immune Responses. Immun Inflamm Dis 2020, 8 (1), 106. https://doi.org/10.1002/IID3.289.
- Sakumura, N.; Yokoyama, T.; Usami, M.; Hosono, Y.; Inoue, N.; Matsuda, Y.; Tasaki, Y.; Wada, T. CD169 Expression on Monocytes as a Marker for Assessing Type I Interferon Status in Pediatric Inflammatory Diseases. Clin Immunol 2023, 250. https://doi.org/10.1016/J.CLIM.2023.109329.
- Volk, H. D.; Reinke, P.; Krausch, D.; Zuckermann, H.; Asadullah, K.; Muller, J. M.; Docke, W. D.; Kox, W. J. Monocyte Deactivation--Rationale for a New Therapeutic Strategy in Sepsis. Intensive Care Med 1996, 22 Suppl 4 (4). https://doi.org/10.1007/BF01743727.
- Monneret, G.; Lepape, A.; Voirin, N.; Bohé, J.; Venet, F.; Debard, A. L.; Thizy, H.; Bienvenu, J.; Gueyffier, F.; Vanhems, P. Persisting Low Monocyte Human Leukocyte Antigen-DR Expression Predicts Mortality in Septic Shock. Intensive Care Med 2006, 32 (8), 1175–1183. https://doi.org/10.1007/S00134-006-0204-8.
- Garlanda, C.; Riva, F.; Bonavita, E.; Mantovani, A. Negative Regulatory Receptors of the IL-1 Family. Semin Immunol 2013, 25 (6), 408–415. https://doi.org/10.1016/J.SMIM.2013.10.019.
- Zhang, Y.; Liu, K.; Guo, M.; Yang, Y.; Zhang, H. Negative Regulator IL-1 Receptor 2 (IL-1R2) and Its Roles in Immune Regulation of Autoimmune Diseases. Int Immunopharmacol 2024, 136. https://doi.org/10.1016/J.INTIMP.2024.112400.
- Reyes, M.; Filbin, M. R.; Bhattacharyya, R. P.; Billman, K.; Eisenhaure, T.; Hung, D. T.; Levy, B. D.; Baron, R. M.; Blainey, P. C.; Goldberg, M. B.; Hacohen, N. An Immune-Cell Signature of Bacterial Sepsis. Nat Med 2020, 26 (3), 333–340. https://doi.org/10.1038/S41591-020-0752-4.
- Bourgoin, P.; Soliveres, T.; Ahriz, D.; Arnoux, I.; Meisel, C.; Unterwalder, N.; Morange, P. E.; Michelet, P.; Malergue, F.; Markarian, T. Clinical Research Assessment by Flow Cytometry of Biomarkers for Infectious Stratification in an Emergency Department. Biomark Med 2019, 13 (16), 1373–1386. https://doi.org/10.2217/BMM-2019-0214.
- Bourgoin, P.; Soliveres, T.; Barbaresi, A.; Loundou, A.; Belkacem, I. A.; Arnoux, I.; Bernot, D.; Loosveld, M.; Morange, P. E.; Michelet, P.; Malergue, F.; Markarian, T. CD169 and CD64 Could Help Differentiate Bacterial from CoVID-19 or Other Viral Infections in the Emergency Department. Cytometry A 2021, 99 (5), 435–445. https://doi.org/10.1002/CYTO.A.24314.
- Perry, S. E.; Mostafa, S. M.; Wenstone, R.; Shenkin, A.; McLaughlin, P. J. Is Low Monocyte HLA-DR Expression Helpful to Predict Outcome in Severe Sepsis? Intensive Care Med 2003, 29 (8), 1245–1252. https://doi.org/10.1007/S00134-003-1686-2.
- Hiesmayr, M. J.; Spittler, A.; Lassnigg, A.; Berger, R.; Laufer, G.; Kocher, A.; Artemiou, O.; Boltz-Nitulescu, G.; Roth, E. Alterations in the Number of Circulating Leucocytes, Phenotype of Monocyte and Cytokine Production in Patients Undergoing Cardiothoracic Surgery. Clin Exp Immunol 1999, 115 (2), 315–323. https://doi.org/10.1046/J.1365-2249.1999.00801.X.
- Oczenski, W.; Krenn, H.; Jilch, R.; Watzka, H.; Waldenberger, F.; Köller, U.; Schwarz, S.; Fitzgerald, R. D. HLA-DR as a Marker for Increased Risk for Systemic Inflammation and Septic Complications after Cardiac Surgery. Intensive Care Med 2003, 29 (8), 1253–1257. https://doi.org/10.1007/S00134-003-1826-8.
- Döcke, W. D.; Höflich, C.; Davis, K. A.; Röttgers, K.; Meisel, C.; Kiefer, P.; Weber, S. U.; Hedwig-Geissing, M.; Kreuzfelder, E.; Tschentscher, P.; Nebe, T.; Engel, A.; Monneret, G.; Spittler, A.; Schmolke, K.; Reinke, P.; Volk, H. D.; Kunz, D. Monitoring Temporary Immunodepression by Flow Cytometric Measurement of Monocytic HLA-DR Expression: A Multicenter Standardized Study. Clin Chem 2005, 51 (12), 2341–2347. https://doi.org/10.1373/CLINCHEM.2005.052639.
- Krabbe, J.; Beilmann, V.; Alamzad-Krabbe, H.; Böll, S.; Seifert, A.; Ruske, N.; Kraus, T.; Martin, C. Blood Collection Technique, Anticoagulants and Storing Temperature Have Minor Effects on the Isolation of Polymorphonuclear Neutrophils. Scientific Reports 2020 10:1 2020, 10 (1), 1–10. https://doi.org/10.1038/s41598-020-71500-1.
- Mosiman, V. L.; Patterson, B. K.; Canterero, L.; Goolsby, C. L. Reducing Cellular Autofluorescence in Flow Cytometry: An In Situ Method. https://doi.org/10.1002/(SICI)1097-0320(19970615)30:3.
- Kwok, A. J.; Allcock, A.; Ferreira, R. C.; Cano-Gamez, E.; Smee, M.; Burnham, K. L.; Zurke, Y. X.; Novak, A.; Darwent, M.; Baron, T.; Brown, C.; Beer, S.; Espinosa, A.; Panduro, T.; Georgiou, D.; Martinez, J.; Thraves, H.; Perez, E.; Fernandez, R.; Sobrino, A.; Sanchez, V.; Magallano, R.; Dineen, K.; Wilson, J.; McKechnie, S.; Mentzer, A. J.; Monaco, C.; Udalova, I. A.; Hinds, C. J.; Todd, J. A.; Davenport, E. E.; Knight, J. C. Neutrophils and Emergency Granulopoiesis Drive Immune Suppression and an Extreme Response Endotype during Sepsis. Nature Immunology 2023 24:5 2023, 24 (5), 767–779. https://doi.org/10.1038/s41590-023-01490-5.
- McMahan, C. J.; Slack, J. L.; Mosley, B.; Cosman, D.; Lupton, S. D.; Brunton, L. L.; Grubin, C. E.; Wignall, J. M.; Jenkins, N. A.; Brannan, C. I. A Novel IL-1 Receptor, Cloned from B Cells by Mammalian Expression, Is Expressed in Many Cell Types. EMBO J 1991, 10 (10), 2821–2832. https://doi.org/10.1002/J.1460-2075.1991.TB07831.X.
- Doehn, J. M.; Tabeling, C.; Biesen, R.; Saccomanno, J.; Madlung, E.; Pappe, E.; Gabriel, F.; Kurth, F.; Meisel, C.; Corman, V. M.; Hanitsch, L. G.; Treskatsch, S.; Heim, K.; Stegemann, M. S.; Ruwwe-Glösenkamp, C.; Müller-Redetzky, H. C.; Uhrig, A.; Somasundaram, R.; Spies, C.; von Bernuth, H.; Hofmann, J.; Drosten, C.; Suttorp, N.; Witzenrath, M.; Sander, L. E.; Hübner, R. H. CD169/SIGLEC1 Is Expressed on Circulating Monocytes in COVID-19 and Expression Levels Are Associated with Disease Severity. Infection 2021, 49 (4), 757–762. https://doi.org/10.1007/S15010-021-01606-9.
- Vetter, P.; Eberhardt, C. S.; Meyer, B.; Martinez Murillo, P. A.; Torriani, G.; Pigny, F.; Lemeille, S.; Cordey, S.; Laubscher, F.; Vu, D.-L.; Calame, A.; Schibler, M.; Jacquerioz, F.; Blanchard-Rohner, G.; Siegrist, C.-A.; Kaiser, L.; Didierlaurent, A. M.; Eckerle, I. Daily Viral Kinetics and Innate and Adaptive Immune Response Assessment in COVID-19: A Case Series. mSphere 2020, 5 (6). https://doi.org/10.1128/MSPHERE.00827-20.
- Hotchkiss, R. S.; Monneret, G.; Payen, D. Immunosuppression in Sepsis: A Novel Understanding of the Disorder and a New Therapeutic Approach. Lancet Infect Dis 2013, 13 (3), 260. https://doi.org/10.1016/S1473-3099(13)70001-X.
- Lukaszewicz, A. C.; Grienay, M.; Resche-Rigon, M.; Pirracchio, R.; Faivre, V.; Boval, B.; Payen, D. Monocytic HLA-DR Expression in Intensive Care Patients: Interest for Prognosis and Secondary Infection Prediction. Crit Care Med 2009, 37 (10), 2746–2752. https://doi.org/10.1097/CCM.0B013E3181AB858A.
- Zhuang, Y.; Peng, H.; Chen, Y.; Zhou, S.; Chen, Y. Dynamic Monitoring of Monocyte HLA-DR Expression for the Diagnosis, Prognosis, and Prediction of Sepsis. Front Biosci (Landmark Ed) 2017, 22 (8), 1344–1354. https://doi.org/10.2741/4547.
- Monneret, G.; Venet, F. Sepsis-Induced Immune Alterations Monitoring by Flow Cytometry as a Promising Tool for Individualized Therapy. Cytometry B Clin Cytom 2016, 90 (4), 376–386. https://doi.org/10.1002/CYTO.B.21270.
- Dinarello, C. A. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol Rev 2018, 281 (1), 8–27. https://doi.org/10.1111/IMR.12621.
- Boudousquie, C.; Bossi, G.; Hurst, J. M.; Rygiel, K. A.; Jakobsen, B. K.; Hassan, N. J. Polyfunctional Response by ImmTAC (IMCgp100) Redirected CD8+ and CD4+ T Cells. Immunology 2017, 152 (3), 425–438. https://doi.org/10.1111/IMM.12779.
- Comolli, G.; Torchio, M.; Lenta, E.; Franceschetti, B.; Chiesa, A.; Calarota, S. A.; Baldanti, F.; Scudeller, L.; Marone, P.; Danova, M. Neutrophil CD64 expression: a reliable diagnostic marker of infection in advanced cancer patients? New Microbiol 2015, 38 (3), 427-30.
- Jalava-Karvinen, P.; Hohenthal, U.; Laitinen, I.; Kotilainen, P.; Rajamäki, A.; Nikoskelainen, J.; Lilius, E. M.; Nuutila, J. Simultaneous Quantitative Analysis of FcγRI (CD64) and CR1 (CD35) on Neutrophils in Distinguishing between Bacterial Infections, Viral Infections, and Inflammatory Diseases. Clinical Immunology 2009, 133 (3), 314–323. https://doi.org/10.1016/J.CLIM.2009.08.003.
- Steinbach, F.; Henke, F.; Krause, B.; Thiele, B.; Burmester, G. R.; Hiepe, F. Monocytes from Systemic Lupus Erythematous Patients Are Severely Altered in Phenotype and Lineage Flexibility. Ann Rheum Dis 2000, 59 (4), 283. https://doi.org/10.1136/ARD.59.4.283.
- York, M. R.; Nagai, T.; Mangini, A. J.; Lemaire, R.; Van Seventer, J. M.; Lafyatis, R. A Macrophage Marker, Siglec-1, Is Increased on Circulating Monocytes in Patients with Systemic Sclerosis and Induced by Type I Interferons and Toll-like Receptor Agonists. Arthritis Rheum 2007, 56 (3), 1010–1020. https://doi.org/10.1002/ART.22382.
- Fiori, B.; D’Inzeo, T.; Giaquinto, A.; Menchinelli, G.; Liotti, F. M.; De Maio, F.; De Angelis, G.; Quaranta, G.; Nagel, D.; Tumbarello, M.; Posteraro, B.; Sanguinetti, M.; Spanu, T. Optimized Use of the MALDI BioTyper System and the FilmArray BCID Panel for Direct Identification of Microbial Pathogens from Positive Blood Cultures. J Clin Microbiol 2016, 54 (3), 576–584. https://doi.org/10.1128/JCM.02590-15.
- Bourgoin, P.; Lediagon, G.; Arnoux, I.; Bernot, D.; Morange, P. E.; Michelet, P.; Malergue, F.; Markarian, T. Flow Cytometry Evaluation of Infection-Related Biomarkers in Febrile Subjects in the Emergency Department. Future Microbiol 2020, 15 (3), 189–201. https://doi.org/10.2217/FMB-2019-0256.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



